 Lorena Stannek1†
Lorena Stannek1† Katrin Gunka
Katrin Gunka Rachel A. Care
Rachel A. Care Fabian M. Commichau
Fabian M. Commichau- 1Department of General Microbiology, Georg-August-University Göttingen, Göttingen, Germany
- 2Institute of Microbiology, Ernst-Moritz-Arndt-University Greifswald, Greifswald, Germany
The Gram-positive model bacterium Bacillus subtilis contains two glutamate dehydro genase-encoding genes, rocG and gudB. While the rocG gene encodes the functional GDH, the gudB gene is cryptic (gudBCR) in the laboratory strain 168 due to a perfect 18 bp-long direct repeat that renders the GudB enzyme inactive and unstable. Although constitutively expressed the GudBCR protein can hardly be detected in B. subtilis as it is rapidly degraded within stationary growth phase. Its high instability qualifies GudBCR as a model substrate for studying protein turnover in B. subtilis. Recently, we have developed a visual screen to monitor the GudBCR stability in the cell using a GFP-GudBCR fusion. Using fluorescent microscopy we found that the GFP protein is simultaneously degraded together with GudBCR. This allows us to analyze the stability of GudBCR in living cells. By combining the visual screen with a transposon mutagenesis approach we looked for mutants that show an increased fluorescence signal compared to the wild type indicating a stabilized GFP-GudBCR fusion. We observed, that disruption of the arginine kinase encoding gene mcsB upon transposon insertion leads to increased amounts of the GFP-GudBCR fusion in this mutant. Deletion of the cognate arginine phosphatase YwlE in contrast results in reduced levels of the GFP-GudBCR fusion. Recently, it was shown that the kinase McsB is involved in phosphorylation of GudBCR on arginine residues. Here we show that selected arginine-lysine point mutations of GudBCR exhibit no influence on degradation. The activity of McsB and YwlE, however, are crucial for the activation and inhibition, respectively, of a proteolytic machinery that efficiently degrades the unstable GudBCR protein in B. subtilis.
Introduction
Posttranslational modifications of proteins allow bacteria to control several important cellular processes. Phosphorylation is such a posttranslational modification event that can severely affect the function of a protein, which is targeted by a specific kinase (Pawson and Scott, 2005; Jers et al., 2008; Kobir et al., 2011). In bacteria, phosphorylation of enzymes and of enzyme regulators is important for the re-direction of fluxes through central metabolic pathways (LaPorte and Koshland, 1983; Cozzone and El-Mansi, 2005; Niebisch et al., 2006). Moreover, posttranslational modification of RNA- and DNA-binding transcription factors by phosphorylation may result in induction or repression of gene expression (Bird et al., 1993; Stülke et al., 1997; Jung et al., 2012; Mascher, 2014).
In the past years, several studies revealed that beside serine, threonine, histidine, and cysteine also amino acids like tyrosine and arginine are phosphorylated in bacteria (Meins et al., 1993; Hoch, 2000; Deutscher and Saier, 2005; Macek et al., 2007; Kobir et al., 2011). For instance, the activity of the UDP-glucose dehydrogenase in the Gram-positive model bacterium Bacillus subtilis is controlled by reversible phosphorylation of a tyrosine residue (Mijakovic et al., 2004). Phosphorylation of tyrosine residues has also been shown to be important for controlling the activity of DNA-binding proteins (Mijakovic et al., 2006; Derouiche et al., 2013). Recently, phosphoproteomic studies revealed that phosphorylation of arginine residues is an emerging posttranslational modification, which is implicated in general stress response in B. subtilis (Elsholz et al., 2012; Schmidt et al., 2014; Trentini et al., 2014). The kinase responsible for arginine phosphorylation in B. subtilis was shown to be McsB (Fuhrmann et al., 2009). Under normal growth conditions McsB is bound and inhibited by the ClpC ATPase subunit of the ClpCP protease complex and/or the activator of McsB kinase activity, McsA. At the same time, the DNA-binding transcription factor CtsR represses the genes of the CtsR-regulon (Derré et al., 1999). In contrast, if the bacteria encounter heat stress, ClpC preferentially interacts with misfolded proteins and releases McsB, which finally targets CtsR for degradation (Kirstein et al., 2005). Inactivation of CtsR results in upregulation of genes that encode proteins of a central protein quality network. The proteins of this network include chaperones, proteases, and adaptor proteins that improve the recognition of substrates by proteases (Elsholz et al., 2010a; Battesti and Gottesmann, 2013). Recent findings indicate that the detachment of CtsR from the DNA provoked by heat seems to be mediated by an intrinsic protein domain that senses heat rather than by McsB-dependent phosphorylation of arginine residues (Elsholz et al., 2010b). By contrast, upon oxidative stress, McsA does not longer bind to and inhibit McsB, which subsequently removes CtsR from the DNA (Elsholz et al., 2011). Thus, the way of how the DNA-binding activity of CtsR is controlled by oxidative stress and by heat is strikingly different.
In recent global phosphoproteomic studies using a B. subtilis ywlE mutant strain lacking the cognate phosphatase YwlE of the kinase McsB, several arginine phosphorylation sites were detected (Elsholz et al., 2012; Schmidt et al., 2014; Trentini et al., 2014). Two phosphorylatable arginine residues in the ClpC protein were shown to be important for McsB-dependent activation of the ATPase subunit of the ClpCP protease complex (Elsholz et al., 2012). In the same study it has been shown that the arginine kinase McsB and the cognate phosphatase YwlE may influence the expression of different global regulons. However, the impact of arginine phosphorylation on the physiology of B. subtilis is not yet fully understood. Analyses of the dynamic changes in the arginine phosphoproteome in response to heat and oxidative stress revealed that only a minor fraction of the phosphorylation sites were differentially modified (Schmidt et al., 2014).
We are interested in the regulation of glutamate metabolism in B. subtilis. In addition to de novo synthesis of the important amino group donor glutamate, the bacteria may use glutamate as a source of carbon and nitrogen (for a recent review see Gunka and Commichau, 2012). Utilization of glutamate requires expression of the rocG and gudB genes encoding the catabolically active glutamate dehydrogenases (GDHs) RocG and GudB, respectively (Belitsky and Sonenshein, 1998; Gunka et al., 2013). Some isolates of B. subtilis like the “wild” ancestor strain NCIB3610 indeed synthesize two active GDHs allowing the bacteria to use glutamate as the single source of carbon and nitrogen (Zeigler et al., 2008; unpublished results). In the domesticated B. subtilis strain 168 only the rocG gene encodes a functional GDH (Belitsky and Sonenshein, 1998; Zeigler et al., 2008). In this strain, the gudBCR gene is cryptic (CR) due to a perfect 18 bp-long direct repeat (DR). This occurs in the part of the gene encoding the active center of the enzyme (Belitsky and Sonenshein, 1998). The GudBCR is enzymatically inactive and also subject to rapid proteolytic degradation, especially when the bacteria starve for nutrients, which is the case when bacteria enter stationary phase (Gerth et al., 2008; Gunka et al., 2012, 2013). Although ClpP was shown to slightly affect GudBCR stability (Gerth et al., 2008), other factors that are involved in the recognition and degradation of the protein are unknown. Interestingly, McsB was shown to phosphorylate the inactive GudBCR protein on four arginine residues (Elsholz et al., 2012). It is tempting to speculate that this phosphorylation serves as a label that directs the inactive GudBCR protein to the proteolytic machinery (see below).
In the present study, we apply a visual screen that is based on a GFP-GudBCR fusion to monitor the GudBCR stability in vivo. By applying microscopical and biochemical techniques, we found that GFP and GudBCR are simultaneously degraded. Thus, the visual screen is suitable to analyze the cellular amount of GudBCR. To identify novel factors that are involved in GudBCR degradation, we combined the visual screen with a transposon mutagenesis approach. Afterward we looked for mutants that show an increased fluorescence, indicating increased amounts of the GFP-GudBCR fusion. Among the transposants we found one insertion in the mcsB gene encoding the arginine kinase McsB. Moreover, inactivation of the cognate phosphatase YwlE resulted in a decreased fluorescence of a strain synthesizing the GFP-GudBCR fusion. The possible mechanisms of how the activity of the kinase McsB and the cognate phosphatase YwlE affect the amount of the GudBCR protein are discussed.
Materials and Methods
Chemicals, Media, and DNA Manipulation
The oligonucleotides were purchased from Sigma-Aldrich (Germany) and are listed in Table 1. B. subtilis chromosomal DNA was isolated using the DNeasy Blood & Tissue Kit (Qiagen, Germany). Plasmids were isolated from Escherichia coli using the Nucleospin Extract Kit (Macherey and Nagel, Germany). PCR products were purified using the PCR Purification Kit (Qiagen, Germany). Phusion DNA polymerase, restriction enzymes and T4 DNA ligase were purchased from Thermo Scientific (Germany) and used according to the manufacturer’s instructions. Other chemicals and media were purchased from Sigma-Aldrich (Germany), Carl Roth (Karlsruhe, Germany) and Becton Dickinson (Heidelberg, Germany). Sequencing of DNA was performed by the SeqLab Sequence Laboratories (Göttingen, Germany).
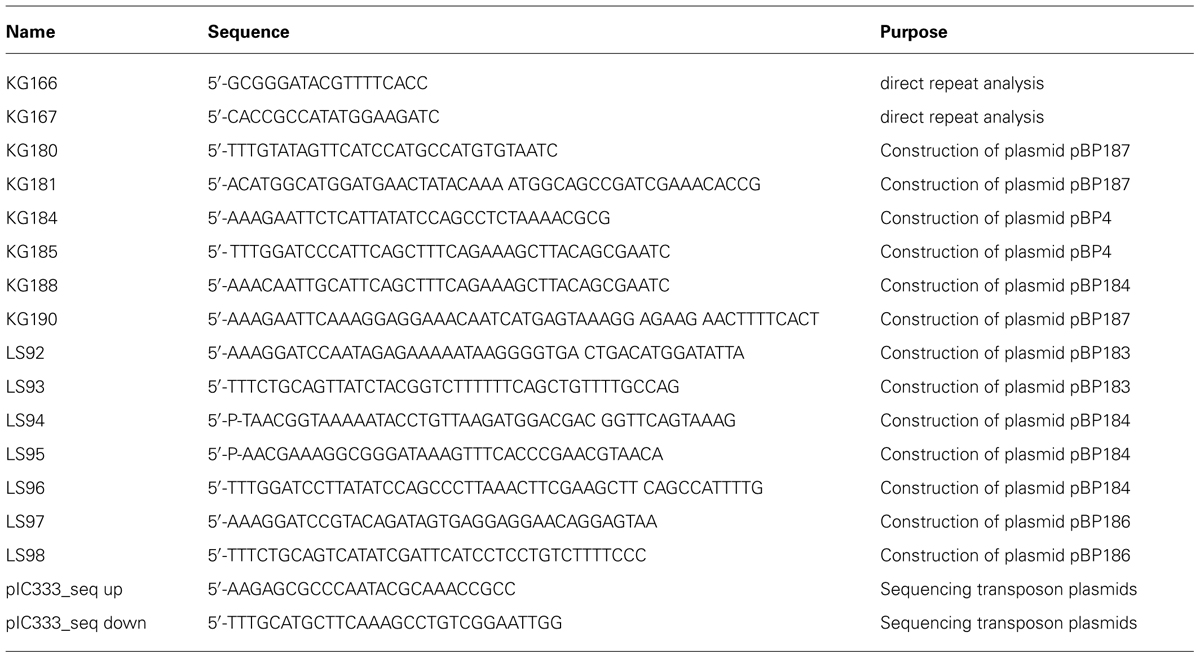
TABLE 1. Oligonucleotides used in this study.
Bacterial Strains, Growth Conditions, and Construction of Mutant Strains
B. subtilis strains (Table 2) were grown in LB and SP medium, respectively. LB and SP plates were prepared by the addition of 17 g agar/l (Roth, Germany) to LB and SP (8 g nutrient broth/l, 1 mM MgSO4, 13 mM KCl, supplemented after sterilization with 2.5 μM ammonium ferric citrate, 500 μM CaCl2, and 10 μM MnCl2), respectively. When required, media were supplemented with antibiotics at the following concentrations: ampicillin (100 μg/ml), kanamycin (10 μg/ml), chloramphenicol (5 μg/ml), lincomycin/erythromycin (25/2 μg/ml), tetracyclin (12.5 μg/ml), and spectinomycin (150 μg/ml). B. subtilis was transformed with plasmid and chromosomal DNA according to a previously described two-step protocol (Kunst and Rapoport, 1995).

TABLE 2. Bacillus subtilis strains used in this study.
Construction of Plasmids
The plasmids for complementation of the ywlE and mcsB mutations in B. subtilis were constructed as follows. The ywlE and mcsB genes were amplified by PCR from chromosomal DNA using the oligonucleotide pairs LS92/LS93 and LS97/LS98, respectively (Table 1). The PCR products were digested with the enzymes BamHI and PstI and ligated to the plasmid pBQ200 that was cut with the same enzymes. The plasmids harboring the ywlE and mcsB genes and their native ribosome-binding sites were designated pBP183 and pBP186, respectively. Expression of the genes is driven by the constitutively active PdegQ promoter (Martin-Verstraete et al., 1994). The quadruple gfp-gudBCR mutant (designated as gfp-gudBCR-mut), encoding the GudBCR protein in which the arginine residues 56, 83, 421, and 423 were replaced by lysine, was constructed by the Multiple-mutation reaction (MMR; Hames et al., 2005). The mutated gudBCR allele was amplified with the oligonucleotide pair KG188/LS96 and the mutagenic oligonucleotides LS94, LS95, and LS96 using plasmid pBP4 as a template. The MMR product was digested with the enzymes MfeI and BamHI, and ligated to the plasmid pAC5 that was cut with the enzymes EcoRI and BamHI. The resulting plasmid was designated as pBP184. This plasmid was used to amplify the promoterless quadruple gudBCR mutant allele by PCR using the oligonucleotide pair KG181/LS96. The gfp gene containing the ribosome-binding site of the B. subtilis gapA gene was amplified by PCR from plasmid pBP8 using the oligonucleotide pair KG180/KG190. The gfp and gudBCR genes were fused by PCR using the external oligonucleotides KG190 and LS96, the PCR product was digested with BamHI and EcoRI, and ligated to the plasmid pBP7 that was cut with the same enzymes. The resulting plasmid pBP187 contains the native gudB promoter and integrates in single copy into the amyE locus. Replacement of the arginine codons in the gfp-gudBCR gene was confirmed by DNA sequencing. All cloning procedures were performed with the E. coli strain DH5α (Sambrook et al., 1989).
Transposon Mutagenesis
For transposon mutagenesis of the B. subtilis strain BP25, we used the mini-Tn10 delivery vector pIC333 (Steinmetz and Richter, 1994) as described previously (Chauvaux et al., 1998). The transposants were grown on SP agar plates for 48 h at 42°C and the intensity of the GFP signal was evaluated by stereo fluorescence microscopy. For the determination of the site of mini-Tn10 insertion, we made use of the fact that the integrated DNA fragment does not contain any EcoRI restriction sites. The chromosomal DNA of the mutants was digested with EcoRI and re-ligated. The ligation mixture was used to transform E. coli DH5α (Sambrook et al., 1989). For all mutants that were further analyzed, we obtained plasmids conferring spectinomycin resistance (Table 3). The insertion sites of the mini-Tn10 transposon were determined by DNA sequencing of the plasmids using the oligonucleotides pIC333_seq up and pIC333_seq down.
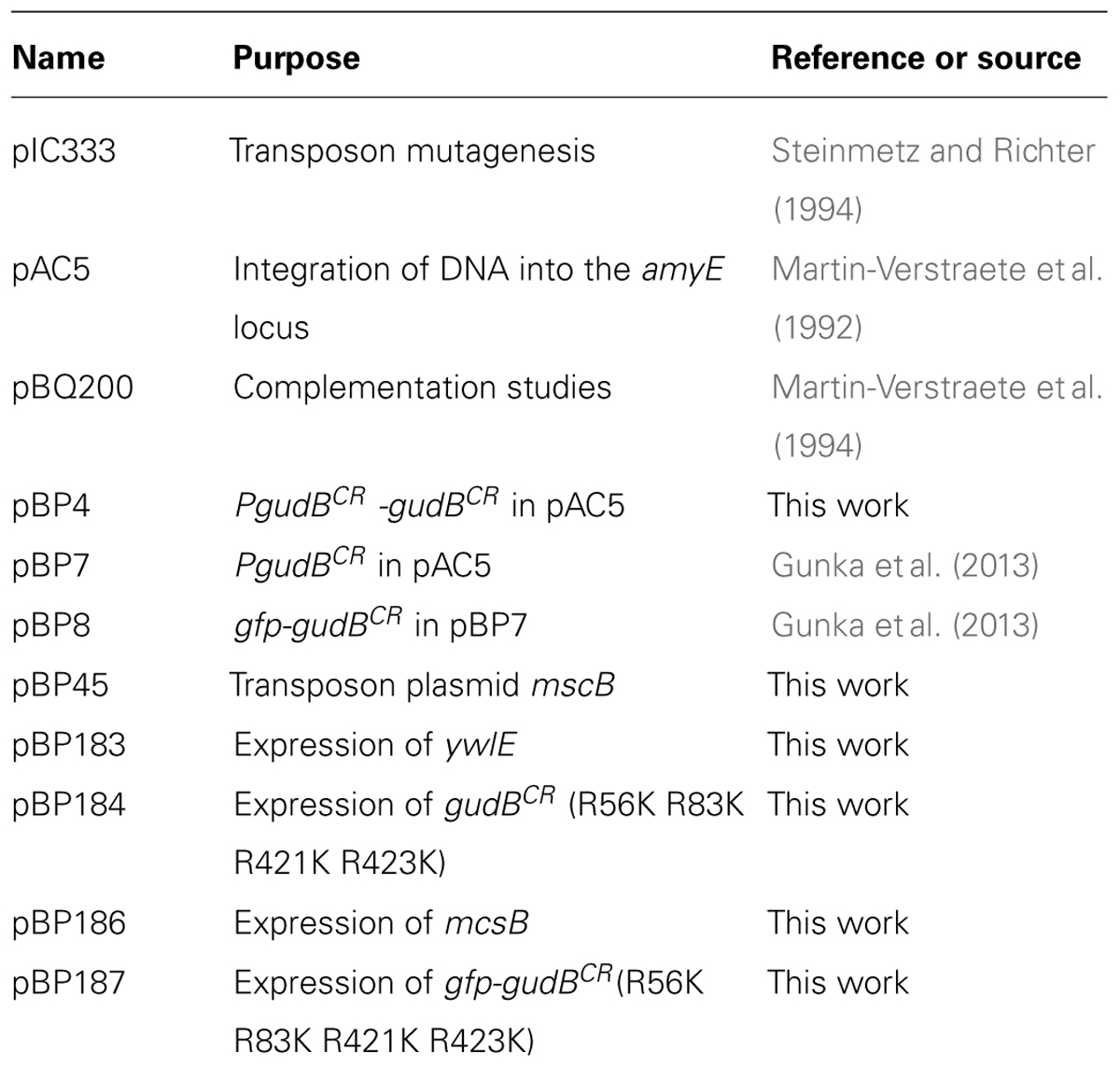
TABLE 3. Plasmids used and constructed in this study.
Western Blotting
For Western blot analyses, proteins present in 20–50 μg of cell free crude extracts were separated by 12.5% SDS PAGE and transferred onto polyvinylidene difluoride membrane (BioRad, Germany) by semi-dry electroblotting. Anti-GFP (PromoKine, Germany; MBL, Japan), anti-YwlE, anti-McsB, and anti-GapA polyclonal antibodies were diluted 1:10.000, 1:1000, 1:5.000, and 1:30.000, respectively, and served as primary antibodies. The antibodies were visualized using anti-rabbit immunoglobulin alkaline phosphatase secondary antibodies (Promega, Germany) and the CDP-Star detection system (Roche Diagnostics, Switzerland) as described previously (Commichau et al., 2007).
Fluorescence Microscopy
For fluorescence microscopy, cells were grown in SP medium to optical densities as indicated, and analyzed on agarose microscopy slides. Fluorescence images were obtained with an Axioskop 40 FL fluorescence microscope, equipped with digital camera AxioCam MRm and AxioVision Rel (version 4.8) software for image processing (Carl Zeiss, Göttingen, Germany) and Neofluar series objective at x 100 primary magnification. The applied filter set was eGFP HC-Filterset (band-pass [BP] 472/30, FT 495, and long-pass [LP] 520/35; AHF Analysentechnik, Tübingen, Germany) for GFP detection. Pictures of B. subtilis colonies were taken with a stereo fluorescence microscope Lumar.V12 (Zeiss, Jena, Germany) equipped with the ZEN lite 2011 (blue edition) software. The applied filter set was Lumar 38 for eGFP detection (Zeiss, Jena, Germany). Images were taken at room temperature and an exposure time of 1 s.
Monitoring GFP-GudBCR Levels in Growing Cultures
Cellular amounts of the GFP-GudBCR fusion protein were determined by monitoring the fluorescence (excitation 489/9.0 nm, emission 509/9.0 nm) in a growing bacterial culture using the Synergy MX II multimode microplate reader (BioTek). For this purpose, 4 ml LB medium were inoculated with the precultures to an OD600 of 0.1. The cultures, that had an approximate OD600 of 1.0, were used to inoculate a 96 well plate (Corning, Sigma) containing 180 μl medium per well. To avoid evaporation, the outermost wells were filled with 200 μl sterile water. The plates were incubated for a maximum of 10 h at 37°C and fast shaking speed. OD600 was measured every 10 min throughout the experiment. Background fluorescence of the parental strains was subtracted from the raw fluorescence of all gfp fusion strains at the same OD600. The cellular amounts of the GFP-GudBCR fusion protein correspond to the fluorescence divided by the OD600 at each time point.
Results
A Stable Screening System for Identifying Factors Involved in GudBCR Degradation
The fact that also the GFP-GudBCR protein is degraded (Gunka et al., 2012, 2013) qualifies it as a substrate to uncover the proteolytic machinery. Before identifying factors that contribute to GudBCR degradation, we constructed the rocG plus strain BP25 that is genetically stable (Gunka et al., 2012) and synthesizes the active GDH RocG as well as the inactive GFP-GudBCR fusion. To test if the GFP-GudBCR fusion protein is degraded in this strain, we compared the fluorescence signal of cells to those of strain BP26 harboring the active gfp-gudB fusion. As shown in Figure 1A, while the bacteria with the active GFP-GudB fusion were strongly fluorescent, the fluorescence signal of bacteria synthesizing the inactive GFP-GudBCR protein was reduced. Thus, the inactive GFP-GudBCR fusion is also degraded in the new strain background. We also tested whether the two strains can be distinguished from each other by monitoring the fluorescence emitted by colonies that were grown on rich medium agar plates. For this purpose, the strains BP25 (gfp-gudBCR) and BP26 (gfp-gudB) were grown in liquid medium, mixed in a 1:1 ratio and appropriate dilutions were propagated on SP plates to allow growth of individual colonies. By visual inspection of the plates using a stereo fluorescence microscope we found several colonies that were grown close to each other and showed different fluorescence signals (Figure 1B). We then re-streaked some of the colonies showing different fluorescence signals on agar plates to obtain individual colonies. Next, we performed colony PCRs and confirmed that the higher and lower fluorescence signals were due to the presence of the gfp-gudB and gfp-gudBCR alleles, respectively. In conclusion, the visual screen seems to be suitable to look for mutants, lacking factors that enhance or decrease proteolytic degradation of GFP-GudBCR.
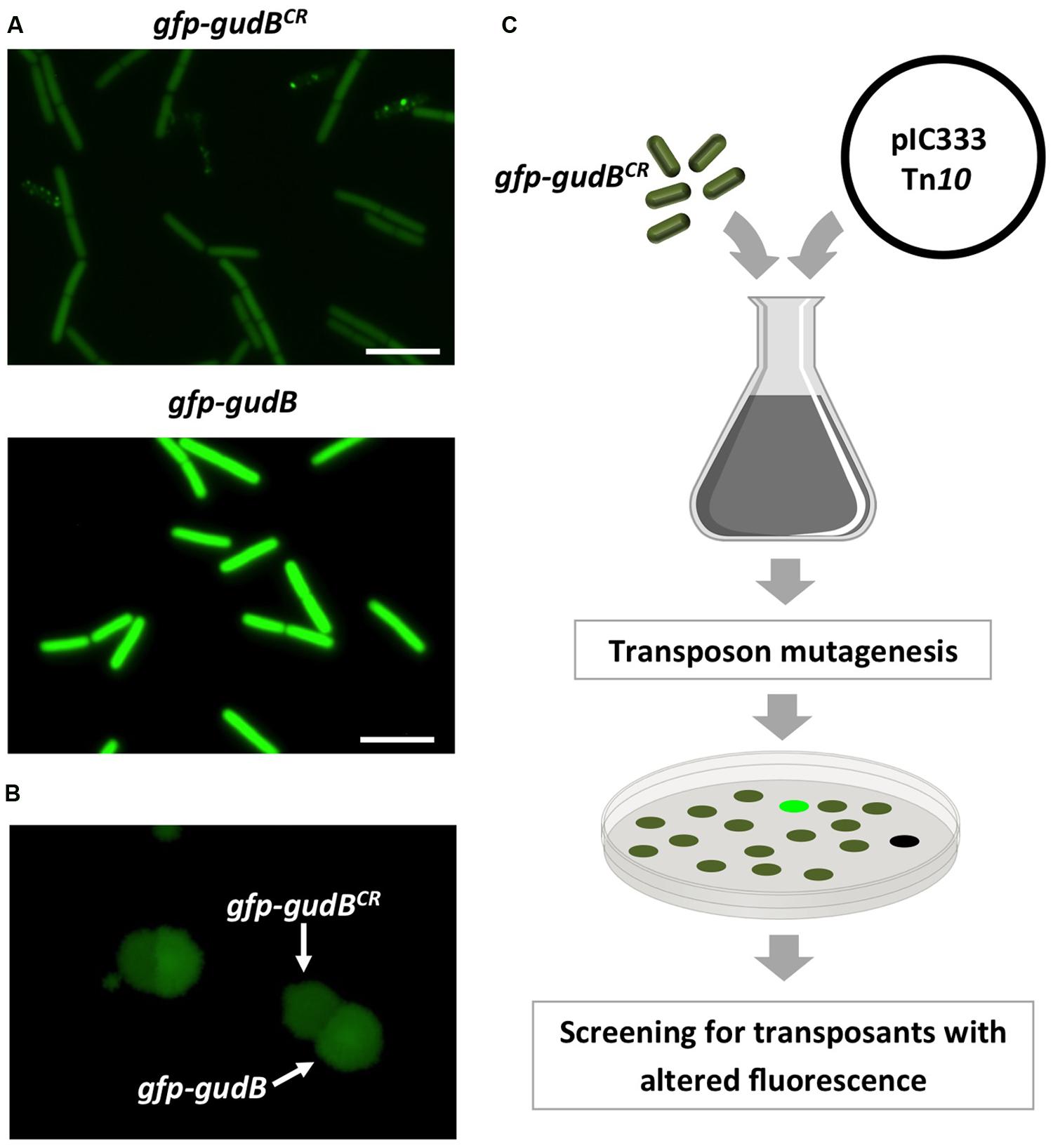
FIGURE 1. Fluorescence of strains BP25 (gfp-gudBCR) and BP26 (gfp-gudB) synthesizing the GFP-GudBCR and GFP-GudB proteins, respectively, at the single cell (A) and at the colony level (B); transposon mutagenesis to identify factors involved in GFP-GudBCR degradation (C). For single cell analysis the bacteria were grown in SP medium. Exposure time, 5 s; scale bar, 5 μm. To monitor fluorescence of colonies the strains were grown in SP medium, mixed and appropriate dilutions were propagated on SP plates, which were incubated for 24 h at 37°C. Exposure time, 1 s.
Identification of MscB Contributing to GudBCR Degradation
To identify factors that are involved in degradation or stabilization of GudBCR, we performed a transposon mutagenesis with strain BP25 (gfp-gudBCR) using the mini-Tn10 delivery vector pIC333 (Steinmetz and Richter, 1994). Afterward, we screened for mutants that show an altered fluorescence signal using a stereo fluorescence microscope (Figure 1C). Appropriate dilutions of the transposants were propagated on SP plates that were incubated for 48 h at 42°C. By visual inspection of about 8000 transposants we could identify one mutant that showed no fluorescence signal, whereas a second mutant showed an increase in fluorescence intensity. While the first mutant had obviously lost the ability to synthesize GFP because the transposon was inserted into the gfp gene, the mutant showing increased fluorescence had integrated the transposon at position 580 into the arginine kinase encoding mcsB gene (Fuhrmann et al., 2009). This transposon mutant was designated as BP69. A re-evaluation of the fluorescence signal of single cells and of a colony of the mcsB transposon mutant revealed that the cellular amount of the GFP-GudBCR fusion was increased when compared to that of the parent strain BP25 (Figures 2A,B). The lack of McsB also resulted in the formation of large aggregates of the GFP-GudBCR fusion protein at the cell poles (Figure 2A), an observation that can be made when aggregation prone proteins are synthesized in bacteria (Rokney et al., 2009; Villar-Pique et al., 2012). In conclusion, using transposon mutagenesis in combination with a visual screen, we identified the arginine kinase McsB being a novel factor that contributes to GudBCR degradation.
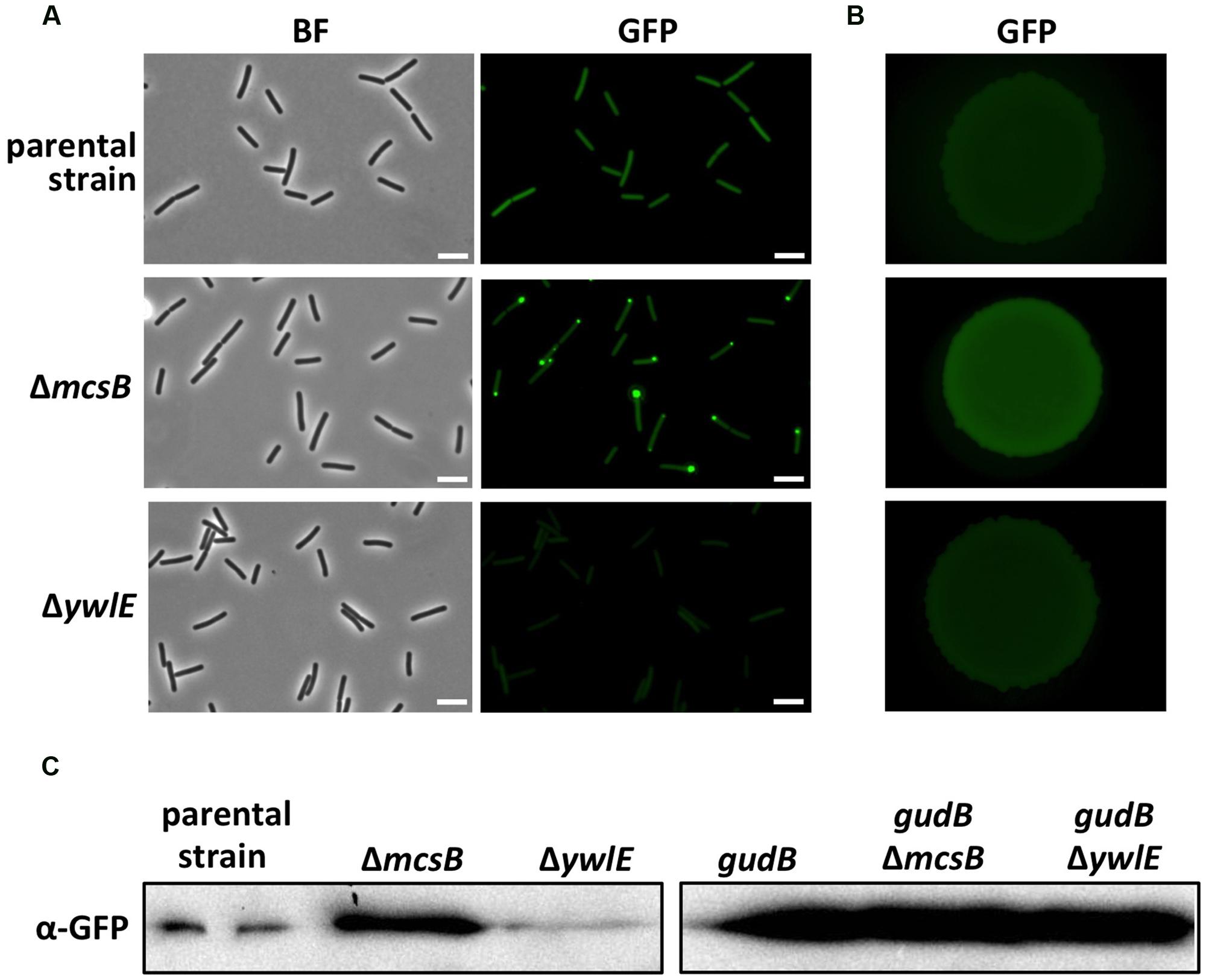
FIGURE 2. Evaluation of the GFP-GudBCR/GFP-GudB levels by fluorescence microscopy and Western blotting. For fluorescence microscopic analyses of single cells (A) the parental strain B25 (gfp-gudBCR) and the strains BP69 (gfp-gudBCR mcsB) and BP74 (gfp-gudBCR ywlE) were grown in SP medium. For bright field and fluorescence microscopy the exposure times were 150 ms and 5 s, respectively. For the evaluation of the GFP-GudBCR level by stereo fluorescence microscopy (B) the bacteria were grown in SP medium until stationary growth phase and 10 μl of cell suspensions with an approximate OD600 of 1 were dropped on a SP plate. The plate was incubated for 24 h at 37°C. Exposure time, 1 s; scale bar, 5 μm. For Western blot analysis (C) the strains B25 (gfp-gudBCR), BP69 (gfp-gudBCR mcsB) and BP74 (gfp-gudBCR ywlE) as well as the isogenic strains BP26 (gfp-gudB), BP311 (gfp-gudB mcsB) and BP75 (gfp-gudB ywlE) expressing the active gfp-gudB fusion were grown in SP medium and 30 μg of the cell free crude extracts were loaded onto a 12.5% SDS PAGE. The fusion proteins were detected using GFP polyclonal antibodies.
McsB and YwlE are Involved in GudBCR Stability
To underpin the role of arginine phosphorylation in the degradation of the GudBCR protein, we inactivated the ywlE gene in the strain BP25 (gfp-gudBCR). In case the arginine phosphatase YwlE counteracts the function of its cognate kinase McsB, we expected to observe that single cells as well as colonies of the ywlE mutant BP74 would show a reduced fluorescence. This was indeed the case for single cells of the ywlE mutant strain in comparison to cells of the mcsB mutant and parent strains BP69 and BP25, respectively (Figure 2A). Although less pronounced, fluorescence of the ywlE mutant was also reduced at the level of single colonies (Figure 2B). However, a quantification of the fluorescence of the GFP-GudBCR fusion protein monitored in growing cultures in the ywlE mutant strain clearly demonstrates that the phosphatase YwlE affects GudBCR stability (see below, Figure 3A).
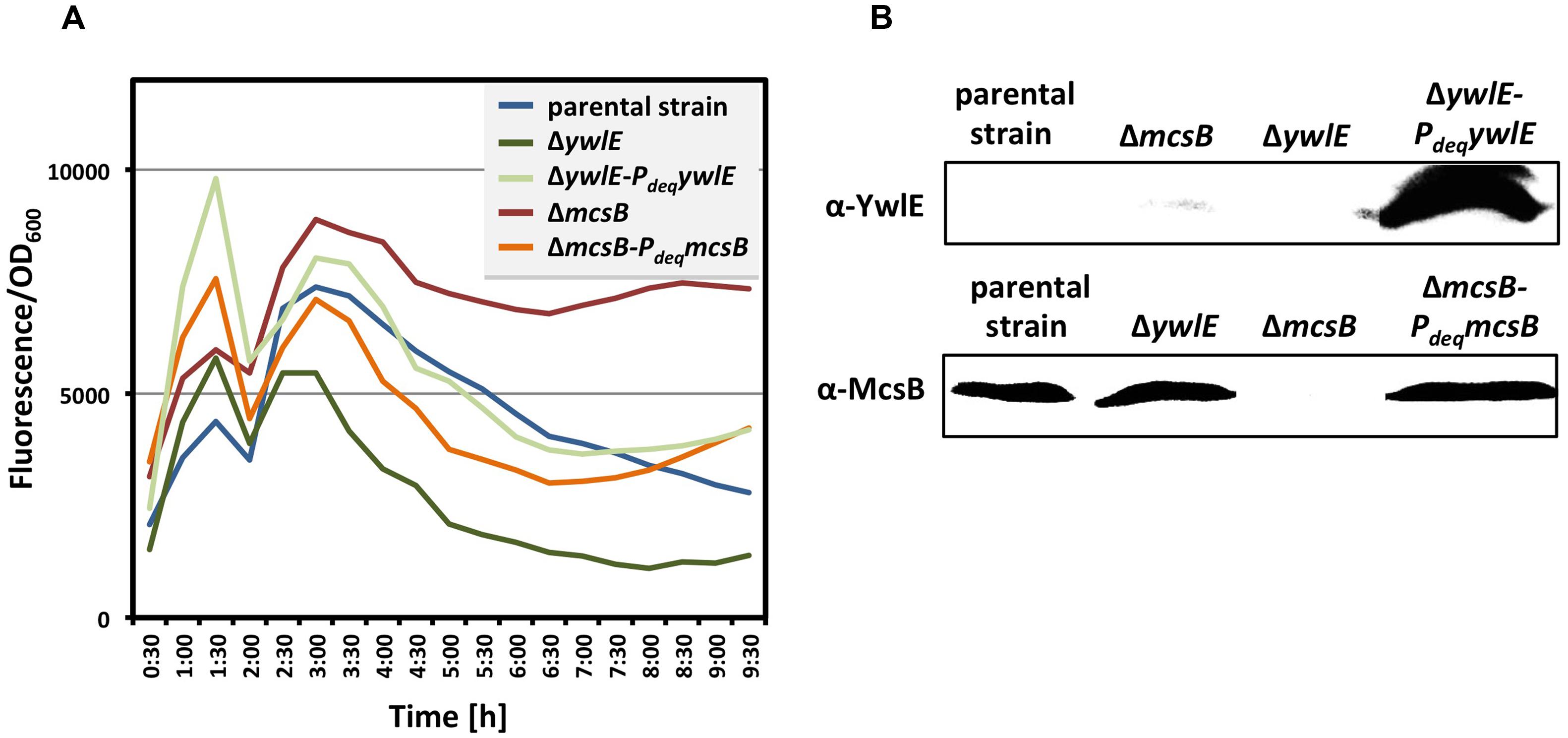
FIGURE 3. Complementation of the mcsB and ywlE mutations. To verify the complementation of the mcsB and the ywlE mutations in vivo (A), the strains BP69 (gfp-gudBCR mcsB) and BP74 (gfp-gudBCR ywlE), and the isogenic strains BP69-pBP186 and BP74-pBP183 expressing mcsB and ywlE from the overexpression vector pBP200 were grown in SP medium and the relative cellular levels of the GFP-GudBCR fusion is reflected by the GFP signal divided by the OD600. The parental strain BP25 (gfp-gudBCR) served as a control. All strains entered stationary phase around 6 h of growth. The maximum deviation of the series of representative data shown here was <30%. For the Western blot analysis (B) the bacteria were cultivated in SP medium. 40 μg and 50 μg of the cell free crude extracts were loaded onto a 12.5% SDS PAGE for the detection of the YwlE and McsB proteins, respectively, using polyclonal antibodies.
Next, we confirmed that the kinase McsB and the phosphatase YwlE affect the cellular levels of the GFP-GudBCR fusion protein. For this purpose, we cultivated the parent strain BP25 as well as the mcsB and ywlE mutant strains BP69 and BP74, respectively, in SP medium until stationary phase (the OD600 was around 3.0) and analyzed the amounts of the GFP-GudBCR fusion protein by Western blotting using GFP-specific antibodies. As shown in Figure 2C, in strain BP69 lacking McsB the cellular amount of the GFP-GudBCR fusion protein was strongly increased. By contrast, the inactivation of the ywlE gene resulted in a decrease of GFP-GudBCR levels. In conclusion, the semi-quantitative Western Blot analyses are in perfect agreement with the fluorescence microscopical studies.
McsB and YwlE Do Not Influence the Cellular Levels of the Active GudB Protein
Subsequently, we wanted to answer the question of whether McsB and YwlE do also influence the cellular amounts of the enzymatically active GFP-GudB fusion protein lacking the duplication of three amino acids in the active center of the enzyme. For this purpose, we cultivated the parent strain BP26 (gfp-gudB) synthesizing the active GFP-GudB fusion and the isogenic mcsB and ywlE mutant strains BP311 and BP75 (Table 2), respectively, in SP medium until stationary phase (OD600 of about 3.0). Afterward, we quantified the amount of the GFP-GudB protein by Western blotting using antibodies specific for GFP. As shown in Figure 2C, irrespective of the absence of either McsB or YwlE all strains synthesized similar amounts of the active GFP-GudB fusion protein. In conclusion, only the cellular amount of the inactive GFP-GudBCR but not that of the active GFP-GudB fusion protein is significantly affected by McsB.
Complementation of the mcsB and ywlE Mutations
For complementation studies of the mcsB and ywlE mutant strains BP69 and BP74, respectively, we constructed the plasmids pBP186 (mcsB) and pBP183 (ywlE). Both plasmids are derivatives of the non-integrative overexpression plasmid pBQ200 and gene expression is driven by the constitutively active PdegQ promoter (Martin-Verstraete et al., 1994). The plasmids pBP186 and pBP183 were introduced into the corresponding mutant strains by transformation. Next, we compared the cellular amounts of the GFP-GudBCR fusion protein in the mcsB and ywlE mutant strains BP69 and BP74, respectively, with those of the isogenic complementation strains by monitoring the fluorescence, which reflects the cellular amounts of the GFP-GudBCR fusion protein during growth of the bacteria. The parent strain BP25 (gfp-gudBCR) served as a control. As shown in Figure 3A, the emitted fluorescence of all cultures was similar during exponentially growth. In the stationary phase the fluorescence signal was much higher in the mcsB mutant strain BP69 when compared to that of the parent strain BP25. By contrast, inactivation of the ywlE resulted in a strong decrease of the fluorescence signal. Overexpression of the mcsB and ywlE genes in the mcsB and ywlE mutant strains BP69 and BP74, respectively, restored the fluorescence signal in the stationary phase almost to the extent of the parent strain. Western blot experiments using antibodies specific for McsB and YwlE confirmed overexpression of the arginine kinase and the phosphatase from the complementation plasmids in the mcsB and ywlE mutant strains BP69 and BP74, respectively (Figure 3B). In conclusion, the cultivation experiments to detect the cellular levels of the GFP-GudBCR fusion protein are in good agreement with the previous experiments showing that the lack of the McsB and YwlE results in elevated and reduced levels, respectively, of the inactive GDH. Moreover, together with the Western blot experiments the cultivation experiments also revealed that the mcsB and ywlE mutations can be complemented by expressing the mcsB and ywlE genes from plasmids.
McsB Seems to Act Independently of ClpC and ClpP on GudBCR Degradation
The mcsB gene lies immediately upstream of the clpC gene in the ctsR mcsA mcsB clpC operon. Since the mcsB mutation can be complemented, it can be ruled out that enhanced cellular levels of the GFP-GudBCR fusion are a consequence of a polar effect of the transposon insertion into the mcsB gene leading to a reduced clpC expression and a lower proteolytic activity. However, the lower proteolytic activity in the mcsB mutant strain might be due to the missing of McsB-dependent activation of the ClpC-ClpP protease complex. To exclude this possibility, we compared the cellular amounts of the GFP-GudBCR fusion in the background of the clpC and clpP mutant strains BP98 and BP99, respectively, to that of the parent strain BP25 (gfp-gudBCR). For this purpose, we grew the bacteria in SP medium and collected samples from exponential and stationary phases and performed Western blot analyses (Figure 4). The strain BP26 (gfp-gudB) as well as the mcsB and ywlE mutant strains BP69 and BP74, respectively, served as controls. As expected, in contrast to the inactive GFP-GudBCR fusion protein the active GFP-GudB variant was more abundant during exponential and stationary phase. Moreover, as observed in the previous experiments, the GFP-GudBCR levels were increased and decreased in the mcsB and ywlE mutants, respectively (see also Figure 2C). Finally, the GFP-GudBCR levels in the clpC and clpP mutant strains BP98 and BP99, respectively, were similar to that of the parent strain BP25 (gfp-gudBCR). Using GapA and GFP antibodies, we show that only the GFP-GudBCR fusion but not GapA was degraded in stationary growth phase samples. Thus, McsB is involved in GudBCR degradation in a rather ClpP and ClpC-independent manner.
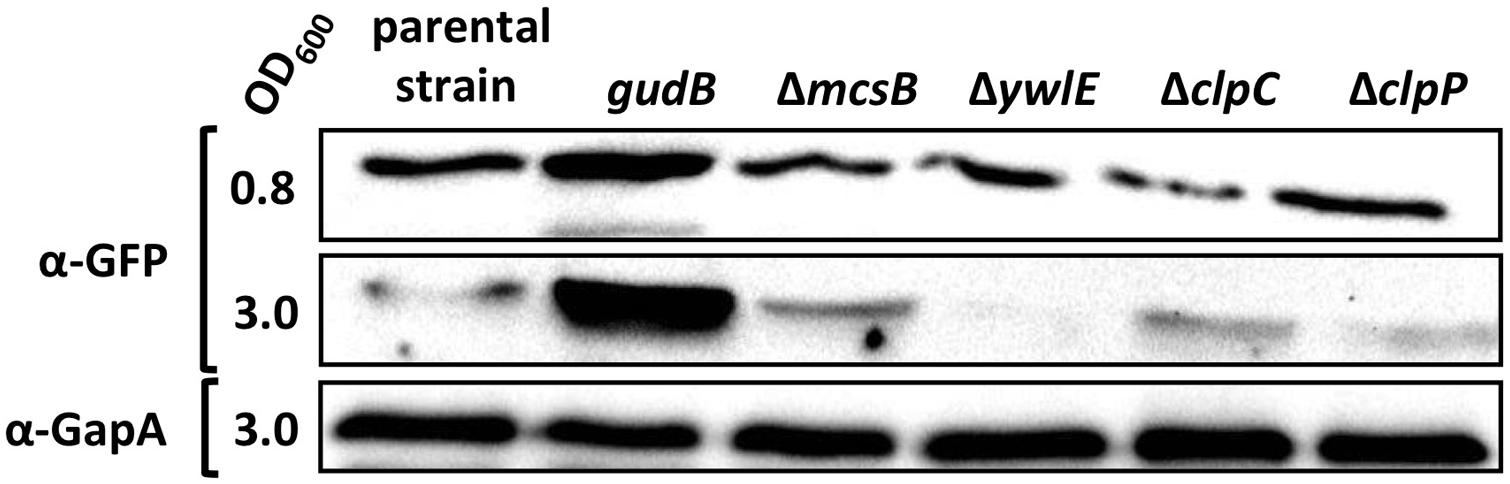
FIGURE 4. McsB acts independently of ClpC and ClpP. For the Western blot analysis the parental strain BP25 (gfp-gudBCR) and the strains BP26 (gfp-gudB), BP69 (gfp-gudBCR mcsB), BP74 (gfp-gudBCR ywlE), BP98 (gfp-gudBCR clpC), and BP99 (gfp-gudBCR clpP) were cultivated in SP medium until the indicated optical densities (OD600). 30 μg of the cell free crude extracts were loaded onto a 12.5% SDS PAGE for the detection of the GFP and GapA proteins using polyclonal antibodies.
Replacement of Phosphorylation Sites Does Not Affect Mcsb-Dependent GudBCR Degradation
In a recent phosphoproteome analysis it has been shown that the inactive GudBCR protein is phosphorylated on the arginine residues at positions 56, 83, 421, and 423 (Elsholz et al., 2012). To evaluate whether phosphorylation of these sites is important for the degradation of the GFP-GudBCR protein, we replaced the arginine by the structurally similar amino acid lysine and monitored the amount of the GudBCR variant in vivo. For this purpose the parent strain BP25 (gfp-gudBCR), the mcsB mutant strain BP69 (mcsBgfp-gudBCR), the quadruple GFP-GudBCR mutant strain BP230 (gfp-gudBCR-mut (R56K R83K R421K R423K)), and the isogenic mcsB mutant strain BP231 (mcsBgfp-gudBCR-mut (R56K R83K R421K R423K)) were cultivated in SP medium. Simultaneously, the cellular levels of the fusion proteins were determined by monitoring the fluorescence during bacterial growth. As shown in Figure 5, the fluorescence measurements revealed that the cellular levels of the fusion proteins in strains BP25 (gfp-gudBCR) and BP230 (gfp-gudBCR-mut (R56K R83K R421K R423K)) was much lower in comparison to those of the isogenic mcsB mutant strains BP69 (mcsB gfp-gudBCR), and BP231 (mcsB gfp-gudBCR-mut (R56K R83K R421K R423K)). In conclusion, these observations suggest that phosphorylation of the arginine residues 56, 83, 421, and 423 sites is rather not important for the degradation of the inactive GudBCR protein.

FIGURE 5. Impact of McsB on the cellular levels of the GFP-GudBCR and GFP-GudBCR(R56K, R83K, R421K, R423K) proteins. The strains BP25 (gfp-gudBCR), BP69 (gfp-gudBCR mcsB), BP230 (gfp-gudBCR-mut), and BP231 (gfp-gudBCR-mut mcsB) were cultivated in SP medium and the relative cellular levels of the GFP-GudBCR fusion is reflected by the GFP signal divided by the OD600. The parental strain BP25 (gfp-gudBCR) served as a control. All strains entered stationary phase around 6 h of growth. The maximum deviation of the series of representative data shown here was <30%.
Discussion
In the present study, we found that the inactivation of the mcsB arginine kinase gene resulted in stabilization of the inactive GDH GudBCR during stationary growth phase of B. subtilis. Thus, beside its role in controlling the degradation of the DNA-binding transcription factor CtsR (Elsholz et al., 2010b) and delocalization of proteins involved in the development of transformability of B. subtilis (Hahn et al., 2009), McsB activity also mediates degradation of GudBCR. Moreover, we found that the arginine phosphatase YwlE counteracts the function of McsB and prevents degradation of GudBCR.
There are several possibilities how McsB and YwlE might stimulate and prevent GudBCR degradation, respectively. As it has been reported previously for the proteolytic degradation of CtsR (Elsholz et al., 2012), McsB-dependent activation of the ATPase subunit ClpC of the ClpCP protease complex by phosphorylation of two specific arginine residues could also be crucial for GudBCR degradation. However, according to our Western blot analysis ClpP and ClpC appear apparently not involved in GudBCR degradation. Recent global phosphoproteomic studies have revealed that in the absence of YwlE several proteins, among them the GudBCR protein are phosphorylated on arginine residues (Elsholz et al., 2012; Schmidt et al., 2014; Trentini et al., 2014). These studies prompted us to address the question of whether the phosphorylation of GudBCR by McsB could serve as a label for proteolysis. However, although the cellular levels of the GudBCR quadruple mutant, in which the arginine residues 56, 83, 421, and 423 were replaced by lysine residues, were slightly increased, the protein was still degraded in a McsB-dependent manner when the bacteria entered stationary phase (see Figure 5). Thus, the degradation of GudBCR seems to be rather indirectly influenced by McsB. Finally, an unknown proteolytic machinery that remains to be identified might be responsible for the degradation of the misfolded and inactive GDH GudBCR. On one hand the activity of the proteolytic machinery might be controlled by McsB-dependent phosphorylation of an unknown adaptor protein that specifically recognizes GudBCR and directs the protein to the protease for degradation. On the other hand McsB could be important for the activation of one of the AAA+ proteases or other unknown proteases that remain to be identified. One could also envision that McsB acts itself as the adaptor that mediates proteolysis of the GudBCR protein. The interaction between McsB and GudBCR could result in coincidental phosphorylation of the GDH. This could also be the case for the other arginine phosphorylations of the B. subtilis proteome (Elsholz et al., 2012).
As described above it is interesting to note that only the domesticated B. subtilis strains 160, 166, and 168, of which the latter one is used worldwide in basic research and in industry, harbor the gudBCR gene that is enzymatically inactive and unstable (Zeigler et al., 2008). It has been suggested that the gudBCR allele appeared as a consequence of X-ray mutagenesis and subsequent adaptation for rapid growth of the bacteria in minimal medium lacking the amino group donor glutamate (Burkholder and Giles, 1947). This hypothesis is supported by the observation that a strain that synthesizes in addition to RocG also the enzymatically active GDH GudB is rapidly outcompeted by the laboratory strain 168 (rocG gudBCR) when exogenous glutamate is not available (Gunka et al., 2013; Stannek et al., 2014). Obviously, the presence of both, RocG and GudB is disadvantageous for the bacteria because the catabolic GDHs degrade the endogenously produced glutamate, which is needed in anabolism. Thus, under laboratory growth conditions a permanent selective pressure must act on the bacteria, which prevents the accumulation of mutants that have spontaneously mutated the cryptic gudBCR gene and synthesize in addition to RocG the functional GDH GudB. Moreover, the selective pressure acting on the B. subtilis strain 168 might be an explanation for the observation that the cryptic gudBCR gene is stably inherited since the bacterium has been domesticated. Recently, it has been shown that bacteria rapidly loose genes and reduce their genome sizes when adapted to specialized environments. This might also be observed in the laboratory by experimental evolution of bacterial cell populations (Koskiniemi et al., 2012; Lee and Marx, 2012). Therefore, it is somewhat surprising that B. subtilis affords to waste energy by permanently synthesizing an inactive enzyme that is subject to rapid degradation. However, under certain growth conditions it must be advantageous for B. subtilis to harbor the cryptic gudBCR gene that, when activated by spontaneous mutagenesis (Gunka et al., 2012), encodes a functional GDH. Indeed, a derivative of the B. subtilis 168 expressing rocG and gudB can use glutamate as a singly source of carbon and nitrogen (Gunka et al., 2013). Thus, under very specific nutritional conditions bacteria that are endowed with high-level GDH activity have a strong selective growth advantage.
In the future it will be interesting to identify additional factors that are involved in the rapid degradation of the enzymatically inactive GDH GudBCR. This goal might be achieved by monitoring the cellular amounts of the GFP-GudBCR fusion protein in a mutant collection that have inactivated all non-essential genes by targeted gene deletion or by a next time saturated transposon mutagenesis. The identification of novel factors that are involved in GFP-GudBCR proteolysis might be facilitated by monitoring growth and fluorescence over time because the fusion protein seems to be preferentially degraded in stationary phase. Moreover, it will be interesting to address the question whether arginine phosphorylation influences the physiological functions of other proteins in B. subtilis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by the grant CO 1139/1-1 of the Deutsche Forschungsgemeinschaft (http://www.dfg.de), the Fonds der Chemischen Industrie (http://www.vci.de/fonds), the Göttingen Centre for Molecular Biology (GZMB), and the Max-Buchner-Forschungsstiftung (http://www.dechema.de/mbf.html; MBFSt-Kennziffer 3381) to Fabian M. Commichau. Work of Katrin Gunka in the authors’ lab was supported by the grant SFB860. Sabine Lentes is acknowledged for expert technical support. We are grateful to Miriam Dormeyer for critical reading of the manuscript and for helpful discussions.
References
Battesti, A., and Gottesmann, S. (2013). Roles of adaptor proteins in regulation of bacterial proteolysis. Curr. Opin. Microbiol. 16, 140–147. doi: 10.1016/j.mib.2013.01.002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Belitsky, B. R., and Sonenshein, A. L. (1998). Role and regulation of Bacillus subtilis glutamate dehydrogenase genes. J. Bacteriol. 180, 6298–6305.
Bird, T. H., Grimsley, J. K., Hoch, J. A., and Spiegelman, G. B. (1993). Phosphorylation of Spo0A activates its stimulation of in vitro transcription from the Bacillus subtilis spoIIG operon. Mol. Microbiol. 9, 741–749. doi: 10.1111/j.1365-2958.1993.tb01734.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Burkholder, P. R., and Giles, N. H. (1947). Induced biochemical mutations in Bacillus subtilis. Am. J. Bot. 34, 345–348. doi: 10.2307/2437147
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chauvaux, S., Paulsen, I. T., and Saier, M. H. Jr. (1998). CcpB, a novel transcription factor implicated in catabolite repression in Bacillus subtilis. J. Bacteriol. 180, 491–497.
Commichau, F. M., Herzberg, C., Tripal, P., Valerius, O., and Stülke, J. (2007). A regulatory protein-protein interaction governs glutamate biosynthesis in Bacillus subtilis: the glutamate dehydrogenase RocG moonlights in controlling the transcription factor GltC. Mol. Microbiol. 65, 642–654. doi: 10.1111/j.1365-2958.2007.05816.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cozzone, A. J., and El-Mansi, M. (2005). Control of isocitrate dehydrogenase catalytic activity by protein phosphorylation in E. coli. J. Mol. Microbiol. Biotechnol. 9, 132–146. doi: 10.1159/000089642
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Derouiche, A., Bidnenko, V., Grenha, R., Pigonneau, N., Ventroux, M., Franz-Wachtel, M.,et al. (2013). Interaction of bacterial fatty-acid-displaced regulators with DNA is interrupted by tyrosine phosphorylation in the helix-turn-helix domain. Nucleic. Acids Res. 41, 9371–9381. doi: 10.1093/nar/gkt709
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Derré, I., Rapoport, G., and Msadek, T. (1999). CtsR, a novel regulator of stress and heat shock response, controls clp and molecular chaperone gene expression in Gram-positive bacteria. Mol. Microbiol. 31, 117–131. doi: 10.1046/j.1365-2958.1999.01152.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Deutscher, J., and Saier, M. H. Jr. (2005). Ser/Thr/Tyr protein phosphorylation in bacteria – for long time neglected, now well established. J. Mol. Microbiol. Biotechnol. 9, 125–131. doi: 10.1159/000089641
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elsholz, A. K., Gerth, U., and Hecker, M. (2010a). Regulation of CtsR activity in low GC, Gram+ bacteria. Adv. Microb. Physiol. 57, 119–144. doi: 10.1016/B978-0-12-381045-8.00003-5
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elsholz, A. K. W., Michalik, S., Zühlke, D., Hecker, M., and Gerth, U. (2010b). CtsR, the Gram-positive master regulator of protein quality control, feels the heat. EMBO J. 29, 3621–3629. doi: 10.1038/emboj.2010.228
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elsholz, A. K., Hempel, K., Pöther, D. C., Becher, D., Hecker, M., and Gerth, U. (2011). CtsR inactivation during thiol-specific stress in low GC, Gram+ bacteria. Mol. Microbiol. 79, 772–785. doi: 10.1111/j.1365-2958.2010.07489.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Elsholz, A. K., Turgay, K., Michalik, S., Hessling, B., Gronau, K., Oertel, D.,et al. (2012). Global impact of protein arginine phosphorylation on the physiology of Bacillus subtilis. Proc. Natl. Acad. Sci. U.S.A. 109, 7451–7456. doi: 10.1073/pnas.1117483109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fuhrmann, J., Schmidt, A., Spiess, S., Lehner, A., Turgay, K., Mechtler, K.,et al. (2009). McsB is a protein arginine kinase that phosphorylates and inhibits the heat-shock regulator CtsR. Science 324, 1323–1326. doi: 10.1126/science.1170088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gerth, U., Kock, H., Kusters, I., Michalik, S., Switzer, R. L., and Hecker, M. (2008). Clp-dependent proteolysis down-regulates central metabolic pathways in glucose-starved Bacillus subtilis. J. Bacteriol. 190, 321–331. doi: 10.1126/science.1170088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gunka, K., and Commichau, F. M. (2012). Control of glutamate homeostasis in Bacillus subtilis: a complex interplay between ammonium assimilation, glutamate biosynthesis and degradation. Mol. Microbiol. 85, 213–224. doi: 10.1111/j.1365-2958.2012.08105.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gunka, K., Stannek, L., Care, R. A., and Commichau, F. M. (2013). Selection-driven accumulation of suppressor mutants in Bacillus subtilis: the apparent high mutation frequency of the cryptic gudB gene and the rapid clonal expansion of gudB(+) suppressors are due to growth under selection. PLoS ONE 8:e66120. doi: 10.1371/journal.pone.0066120
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gunka, K., Tholen, S., Gerwig, J., Herzberg, C., Stülke, J., and Commichau, F. M. (2012). A high-frequency mutation in Bacillus subtilis: requirements for the decryptification of the gudB glutamate dehydrogenase gene. J. Bacteriol. 194, 1036–1044. doi: 10.1128/JB.06470-11
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hahn, J., Kramer, N., Briley, K. Jr., and Dubnau, D. (2009). McsA and B mediate delocalization of competence proteins from cell poles of Bacillus subtilis. Mol. Microbiol. 72, 202–215. doi: 10.1111/j.1365-2958.2009.06636.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hames, C., Halbedel, S., Schilling, O., and Stülke, J. (2005). Multiple-mutation reaction: a method for simultaneous introduction of multiple mutations into the glpK gene of Mycoplasma pneumoniae. Appl. Environ. Microbiol. 71, 4097–4100. doi: 10.1128/AEM.71.7.4097-4100.2005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hoch, J. A. (2000). Two-component and phosphorelay signal transduction. Curr. Opin. Microbiol. 3, 165–170. doi: 10.1016/S1369-5274(00)00070-9
Jers, C., Soufi, B., Grangeasse, C., Deutscher, J., and Mijakovic, I. (2008). Phosphoproteomics in bacteria: towards a systemic understanding of bacterial phosphorylation networks. Expert Rev. Proteomics 5, 619–627. doi: 10.1586/14789450.5.4.619
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jung, K., Fried, L., Behr, S., and Heermann, R. (2012). Histidine kinases and response regulators in networks. Curr. Opin. Microbiol. 15, 118–124. doi: 10.1016/j.mib.2011.11.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kirstein, J., Zühlke, D., Gerth, U., Turgay, K., and Hecker, M. (2005). A tyrosine kinase and its activator control the activity of the CtsR heat shock repressor in B. subtilis. EMBO J. 24, 3435–3445. doi: 10.1038/sj.emboj.7600780
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kobir, A., Shi, L., Boskovic, A., Grangeasse, C., Franjevic, D., and Mijakovic, I. (2011). Protein phosphorylation in bacterial signal transduction. Biochim. Biophys. Acta 1810, 989–994. doi: 10.1016/j.bbagen.2011.01.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Koskiniemi, S., Sun, S., Berg, O. G., and Andersson, D. I. (2012). Selection-driven gene loss in bacteria. PLoS Genet. 8:e1002787. doi: 10.1371/journal.pgen.1002787
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kunst, F., and Rapoport, G. (1995). Salt stress is an environmental signal affecting degradative enzyme synthesis in B. subtilis. J. Bacteriol. 177, 2403–2407. doi: 10.1038/305286a0
LaPorte, D. C., and Koshland, D. E. Jr. (1983). Phosphorylation of isocitrate dehydrogenase as a demonstration of enhanced sensitivity in covalent regulation. Nature 305, 286–290. doi: 10.1038/305286a0
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lee, M. C., and Marx, C. J. (2012). Prepeated, selection-driven genome reduction of accessory genes in experimental populations. PLoS Genet. 8:e1002651. doi: 10.1371/journal.pgen.1002651
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Macek, B., Mijakovic, I., Olsen, J. V., Gnad, F., Kumar, C., Jensen, P. R.,et al. (2007). The serine/threonine/tyrosine phosphoproteome of the model bacterium Bacillus subtilis. Mol. Cell. Proteomics 6, 697–707. doi: 10.1074/mcp.M600464-MCP200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Martin-Verstraete, I., Débarbouillé, M., Klier, A., and Rapoport, G. (1992). Mutagenesis of the Bacillus subtilis “-12, -24” promoter of the levanase operon and evidence for the existence of an upstream activating sequence. J. Mol. Biol. 226, 85–99. doi: 10.1016/0022-2836(92)90126-5
Martin-Verstraete, I., Débarbouillé, M., Klier, A., and Rapoport, G. (1994). Interactions of wild-type and truncated LevR of Bacillus subtilis with the upstream activating sequence of the levanase operon. J. Mol. Biol. 241, 178–192. doi: 10.1006/jmbi.1994.1487
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mascher, T. (2014). Bacterial (intramembrane-sensing) histidine kinases: signal transfer rather than stimulus perseption. Trends Microbiol. 22, 559–565. doi: 10.1016/j.tim.2014.05.006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meins, M., Jenö, P., Müller, D., Richter, W. J., Rosenbusch, J. P., and Erni, B. (1993). Cysteine phosphorylation of the glucose transporter of Escherichia coli. J. Biol. Chem. 268, 11604–11609.
Mijakovic, I., Petranovic, D., and Deutscher, J. (2004). How tyrosine phosphorylation affects the UDP-glucose dehydrogenase activity of Bacillus subtilis YwqF. J. Mol. Microbiol. Biotechnol. 8, 19–25. doi: 10.1159/000082077
Mijakovic, I., Petranovic, D., Macek, B., Cepo, T., Mann, M., Davies, J.,et al. (2006). Bacterial single-stranded DNA-binding proteins are phosphorylated on tyrosine. Nucleic. Acids Res. 34, 1588–1596. doi: 10.1093/nar/gkj514
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Niebisch, A., Kabus, A., Schultz, C., Weil, B., and Bott, M. (2006). Corynebacterial protein kinase G controls 2-oxoglutarate dehydrogenase activity via the phosphorylation status of the OdhI protein. J. Biol. Chem. 281, 12300–12307. doi: 10.1074/jbc.M512515200
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Pawson, T., and Scott, J. D. (2005). Protein phosphorylation in signaling – 50 years and counting. Trends Biochem. Sci. 30, 286–290. doi: 10.1016/j.tibs.2005.04.013
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rokney, A., Shagan, M., Kessel, M., Smith, Y., Rosenshine, I., and Oppenheim, A. B. (2009). E. coli transports aggregated proteins to the poles by a specific and energy-dependent process. J. Mol. Biol. 392, 589–601. doi: 10.1016/j.jmb.2009.07.009
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory.
Schmidt, A., Trentini, D. B., Spiess, S., Fuhrmann, J., Ammerer, G., Mechtler, K.,et al. (2014). Quantitative phophoproteome reveals the role of protein arginine phophorylation in the bacterial stress response. Mol. Cell. Proteomics. 3, 1832–1839. doi: 10.1074/mcp.M113.032292
Stannek, L., Egelkamp, R., Gunka, K., and Commichau, F. M. (2014). Monitoring intraspecies competition in a bacterial cell population by cocultivation of fluorescently labelled strains. J. Vis. Exp. 18:e51196. doi: 10.3791/51196
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Steinmetz, M., and Richter, R. (1994). Easy cloning of mini-Tn10 insertions from the Bacillus subtilis chromosome. J. Bacteriol. 176, 1761–1763.
Stülke, J., Martin-Verstraete, I., Zagorec, M., Rose, M., Klier, A., and Rapoport, G. (1997). Induction of the Bacillus subtilis ptsGHI operon by glucose is controlled by a novel antiterminator, GlcT. Mol. Microbiol. 25, 65–78. doi: 10.1046/j.1365-2958.1997.4351797.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Trentini, D. B., Fuhrmann, J., Mechtler, K., and Clausen, T. (2014). Chasing phosphoarginine proteins: development of a selective enrichment method using a phosphatase trap. Mol. Cell. Proteomics 13, 1953–1964. doi: 10.1074/mcp.O113.035790
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Villar-Pique, A., de Groot, N. S., Sabaté, R., Acebrón, S. P., Celaya, G., Fernàndez-Busquets, X.,et al. (2012). The effect of amyloidogenic peptides on bacterial aging correlates with their intrinsic aggregation propensity. J. Mol. Biol. 421, 270–281. doi: 10.1016/j.jmb.2011.12.014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zeigler, D. R., Prágai, Z., Rodriguez, S., Chevreux, B., Muffler, A., Albert, T.,et al. (2008). The origins of 168, W23, and other Bacillus subtilis legacy strains. J. Bacteriol. 190, 6983–6995. doi: 10.1128/JB.00722-08
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: proteolysis, arginine phosphorylation, protein modification, protein folding, glutamate dehydrogenase
Citation: Stannek L, Gunka K, Care RA, Gerth U and Commichau FM (2015) Factors that mediate and prevent degradation of the inactive and unstable GudB protein in Bacillus subtilis. Front. Microbiol. 5:758. doi: 10.3389/fmicb.2014.00758
Received: 23 November 2014; Accepted: 12 December 2014;
Published online: 07 January 2015.
Edited by:
Ivan Mijakovic, Chalmers University of Technology, SwedenReviewed by:
Sven Halbedel, Robert Koch Institute, GermanyOscar P. Kuipers, University of Groningen, Netherlands
Copyright © 2015 Stannek, Gunka, Care, Gerth and Commichau. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fabian M. Commichau, Department of General Microbiology, Georg-August-University Göttingen, Grisebachstraße 8, 37077 Göttingen, Germany e-mail:ZmNvbW1pYzFAZ3dkZy5kZQ==
†These authors have contributed equally to this work.