 Claire Bertelli1,2
Claire Bertelli1,2 Sébastien Aeby1
Sébastien Aeby1 Bérénice Chassot3
Bérénice Chassot3 James Clulow3
James Clulow3 Olivier Hilfiker3Samuel Rappo3Sébastien Ritzmann3Paolo Schumacher3Céline Terrettaz3Paola Benaglio4
Olivier Hilfiker3Samuel Rappo3Sébastien Ritzmann3Paolo Schumacher3Céline Terrettaz3Paola Benaglio4 Laurent Falquet5,2Laurent Farinelli6
Laurent Falquet5,2Laurent Farinelli6 Walid H. Gharib7,2Alexander Goesmann8Keith Harshman9Burkhard Linke8
Walid H. Gharib7,2Alexander Goesmann8Keith Harshman9Burkhard Linke8 Ryo Miyazaki10Carlo Rivolta4
Ryo Miyazaki10Carlo Rivolta4 Marc Robinson-Rechavi7,2
Marc Robinson-Rechavi7,2 Jan Roelof van der Meer10
Jan Roelof van der Meer10 Gilbert Greub1*
Gilbert Greub1*- 1Center for Research on Intracellular Bacteria, Institute of Microbiology, University Hospital Center and University of Lausanne, Lausanne, Switzerland
- 2SIB Swiss Institute of Bioinformatics, Lausanne, Switzerland
- 3School of Biology, University of Lausanne, Lausanne, Switzerland
- 4Department of Medical Genetics, University of Lausanne, Lausanne, Switzerland
- 5Division of Biochemistry, Department of Biology, University of Fribourg, Fribourg, Switzerland
- 6Fasteris SA, Geneva, Switzerland
- 7Department of Ecology and Evolution, University of Lausanne, Lausanne, Switzerland
- 8Department of Bioinformatics and Systems Biology, Justus-Liebig-University Giessen, Gießen, Germany
- 9Lausanne Genomic Technologies Facility, Center for Integrative Genomics, University of Lausanne, Lausanne, Switzerland
- 10Department of Fundamental Microbiology, University of Lausanne, Lausanne, Switzerland
With the widespread availability of high-throughput sequencing technologies, sequencing projects have become pervasive in the molecular life sciences. The huge bulk of data generated daily must be analyzed further by biologists with skills in bioinformatics and by “embedded bioinformaticians,” i.e., bioinformaticians integrated in wet lab research groups. Thus, students interested in molecular life sciences must be trained in the main steps of genomics: sequencing, assembly, annotation and analysis. To reach that goal, a practical course has been set up for master students at the University of Lausanne: the “Sequence a genome” class. At the beginning of the academic year, a few bacterial species whose genome is unknown are provided to the students, who sequence and assemble the genome(s) and perform manual annotation. Here, we report the progress of the first class from September 2010 to June 2011 and the results obtained by seven master students who specifically assembled and annotated the genome of Estrella lausannensis, an obligate intracellular bacterium related to Chlamydia. The draft genome of Estrella is composed of 29 scaffolds encompassing 2,819,825 bp that encode for 2233 putative proteins. Estrella also possesses a 9136 bp plasmid that encodes for 14 genes, among which we found an integrase and a toxin/antitoxin module. Like all other members of the Chlamydiales order, Estrella possesses a highly conserved type III secretion system, considered as a key virulence factor. The annotation of the Estrella genome also allowed the characterization of the metabolic abilities of this strictly intracellular bacterium. Altogether, the students provided the scientific community with the Estrella genome sequence and a preliminary understanding of the biology of this recently-discovered bacterial genus, while learning to use cutting-edge technologies for sequencing and to perform bioinformatics analyses.
Introduction
Since the onset of pyrosequencing in 2007, ultra-high-throughput sequencing (UHTS) technologies have democratized the access of large-scale sequencing to small laboratories by decreasing the cost and the turnaround time (MacLean et al., 2009). This resulted in a flood of new genome sequences, and especially unfinished genome sequences, particularly in the field of microbiology (Bertelli and Greub, 2013). Large sequence datasets are generated daily not only for genome sequencing, but also for metagenomics studies, RNA-seq or ChIP-seq. The treatment of sequence data has thus become the main bottleneck for many studies in microbiology, and more generally in biology. This underlines the current need for biologists with skills in bioinformatics and for bioinformaticians embedded in wet lab research groups. Thus, it is extremely important to make students in molecular life sciences aware of the main challenges related to the use of UHTS in biology projects.
Several publications or web pages have reported the teaching of genomics to undergraduate students using phages (Jordan et al., 2014), microbes (Kerfeld and Simons, 2007; Drew and Triplett, 2008; Coil1 JGI2), or mammals (Edwards et al., 2013). Microbial genomics has the advantage of providing tractable projects in a reasonable time frame and budget, while providing the students with an opportunity to familiarize themselves with important concepts such as quality control, read filtering, assembly, annotation and genome analysis. The “Sequence a genome” class, a compulsory practical course, has been set up for students enrolled in the Master of Molecular Life Sciences at the School of Biology of the University of Lausanne, Switzerland. Students are provided with bacterial strains of interest to Lausanne research groups, whose genomes are completely unknown. They learn to use state-of-the-art UHTS technologies and bioinformatics tools while producing new knowledge on a specific organism. Here, we report the main idea and concepts of the class, and the results obtained by seven master students who studied the Estrella genome.
Estrella lausannensis is an obligate intracellular bacterium isolated from the Llobregat river water (Barcelona, Spain) by amoebal co-culture (Corsaro et al., 2009), a cell culture system using amoebae as a cell background (Jacquier et al., 2013). The bacterium was recently classified in the Criblamydiaceae family based on phylogenetic analysis of ribosomal RNA, core genes, and MALDI-TOF profiles (Lienard et al., 2011). Like all other members of the Chlamydiales order, this strict intracellular bacterium exhibits two developmental stages: an infectious stage called the elementary body (EB) and a replicative stage named the reticulate body (RB). First electron microscopy showed star-shaped EBs leading to the name Estrella (Lienard et al., 2011), but star shapes are less frequent in E. lausannensis than in the related species Criblamydia sequanensis (Thomas et al., 2006; Rusconi et al., 2013). Although these morphologies probably results from a fixative artifact, they certainly reveal underlying differences in cell wall structure and composition (Rusconi et al., 2013).
A survey of metagenomics sequences available in public databases showed that the Criblamydiaceae family forms a small operational taxonomic units (OTUs) compared to other widely represented OTUs such as the Rhabdochlamydiaceae (Lagkouvardos et al., 2014). However, recent serological studies suggested a common exposure of human to Estrella with a seroprevalence varying from 2.9 to 12.7% (De Barsy et al., 2014). Human exposure and E. lausannensis ability to grow in human macrophages that point to a potential pathogenicity triggered the investigation of E. lausannensis resistance to commonly used antibiotics (De Barsy et al., 2014). When cultured in Vero cells, the bacterium was resistant to beta-lactams and fluoroquinolones, but sensitive to cyclones. In addition, E. lausannensis replicates efficiently in four different species of free-living amoebae, inducing host cell lysis after 48–96 h (Lienard et al., 2011). The bacterium was also shown to grow in two fish cell lines but could not induce cell lysis (Kebbi-Beghdadi et al., 2011). Survival and growth in macrophages and other professional phagocytes is made possible by the presence of a class 3 catalase that degrade reactive oxygen species (Rusconi and Greub, 2013).
All Chlamydiales species, like many intracellular bacteria, lack complete biosynthetic pathways for essential compounds, but the ability of each genus and species varies (reviewed in Omsland et al., 2014). We hypothesized that the wide host range and the rapid growth of E. lausannensis may be linked to wider metabolic abilities compared to other Chlamydia-related bacteria and especially compared to its closest known relative C. sequanensis whose genome has just been released (Bertelli et al., 2014). Thus, E. lausannensis, the type strain of the genus and species, was selected in the first implementation of the “Sequence a genome” course. We present here the results of the sequencing, analysis and annotation of the E. lausannensis genome by the students themselves. The results section of the manuscript is mostly based on their written reports provided as part of the course. Hence, this should be considered a preliminary assessment of this bacterial genome, which illustrates how an undergraduate course can explore genome biology.
Materials and Methods
Strain Culture and Purification
Estrella lausannensis strain CRIB-30 was co-cultured in Acanthamoeba castellanii ATCC 30010 at 32°C in 75-cm2-surface cell culture flasks (Becton Dickinson, Allschwil, Switzerland) with 30 ml of PYG medium as described previously (Greub and Raoult, 2002). Co-cultures were harvested when a complete lysis of the amoebae was observed. Then, Estrella elementary bodies were purified using successive sucrose and gastrografin gradients, as described previously (Bertelli et al., 2010). These steps were not performed by the course students.
Genome Sequencing
DNA extraction and library preparation failed likely due to an insufficient amount of bacteria resulting from cell culture. However, students successfully extracted the DNA from Pseudomonas knackmussii that was performed in parallel and which analysis is published in another paper (Miyazaki et al., 2014). Thus, a set of backup reads obtained previously was used. Therefore, the following lines describe the protocol effectively used and not that used by students during the course.
Estrella lausannensis DNA was purified from the bacterial pellet using the QIAmp DNA extraction kit (Qiagen, Hombrechtikon, Switzerland) and eluted in 100 μl of the provided elution buffer. The library was prepared according to Illumina standard protocols with the addition of a 5 bp-index to allow for sample multiplexing. A 38 bp paired-end (PE) run of the Estrella library was sequenced on a lane of an Illumina GAIIx sequencer at Fasteris (Plan-les-Ouates, Switzerland). The raw data was processed according to the Illumina pipeline and exported as fastq files.
Genome Assembly
The quality of Estrella reads (33 bp without tag, PE, insert size ~300 bp) was controlled using FastQC (http://www.bioinformatics.bbsrc.ac.uk/projects/fastqc/) revealing an excellent quality over their entire length. Thus, no trimming or filtering was performed. Reads were assembled using CLC Genomics Workbench 4 (CLCbio, Aarhus, Denmark), ABySS V1.2.1 (n = 5) (Simpson et al., 2009), Velvet V1.0 (Zerbino and Birney, 2008) and SOAPdenovo (Config file: max_rd_len = 33, avg_ins = 330, reverse_seq = 0, asm_flags = 3, rank = 1, Assembly: −p 2–d 2) combined with GapCloser (−t 2) (Li et al., 2010) with default parameters unless previously mentioned and a k-mer that varied between 19 and 29. Assembly results were compared and the best assembly (Velvet, k-mer = 23) was selected according to the following criteria: minimum number of scaffolds larger than 1000 bp, maximum N50, and maximal total size of assembled nucleotides. All of these steps were performed by the students, on a Linux cluster (http://www.vital-it.ch/), over the autumn semester.
We checked the occurrence of large scaffolds (>1000 bp) with a coverage higher or lower than the mean ± 1.5*standard deviation and investigated their content by BLASTN against the non-redundant database (nt). Large scaffolds were searched for similarity to A. castellanii genome by BLASTN.
Although they assemble the same data, the different softwares used in this analysis implement different methods for graph construction and resolution which results in slightly different contigs. To make students aware of the differences that may exist between the assemblies, the best assemblies of all software were aligned using Mauve (Darling et al., 2004). By doing this, the students recorded cases were the best Velvet assembly provided two contigs for a region that was solved in one contig by another software. These differences enabled to scaffold contigs in Velvet assembly. To further scaffold contigs, a multiple alignment of the best genome assembly of each software was performed using Mauve and the positioning of read pairs on different contigs was analyzed using Phrap and Consed (Gordon et al., 1998). To improve genome assembly, primers were designed between 200 and 400 bp of each contig end using Consed (Gordon et al., 1998), R (Cran, 2010), and in-house scripts. Primer combinations suggested based on the scaffold were tested first by PCR. Positive combinations were sequenced using Sanger technology and resulting reads were reassembled with contigs of the best assembly using Phrap and manually curated to remove spurious errors using Consed. All these steps were performed by the students, with limited improvements by the teachers for failed experiments.
Annotation
The draft genome was submitted to GenDB (Meyer et al., 2003) for automatic annotation. The annotation of genes belonging to selected pathways or particular regions of interest were manually curated by the students under the teachers' supervision, according to guidelines available as Supplementary material. The biocuration was performed on gene name, gene product, E.C. number, a description field (notes), as well as a status of confidence and a GO evidence code. For this purpose, BLAST searches (Altschul et al., 1990) against Swiss-Prot/UniProtKB (UNIPROT, 2014), KEGG (Ogata et al., 1999), the non-redundant database, local databases, as well as HMM searches against Pfam (Finn et al., 2014) and TIGRfam (Selengut et al., 2007) were taken into account. Thanks to the possibilities offered by the GenDB interface, the genes annotated by the students were tagged with a specific status of function. Then, the teacher responsible for each corresponding topic corrected the annotation, added more information if required or answered questions asked by the students. Difficult annotations were discussed directly among students, assistants and professors during the practical courses.
Accession Number
The genome of Estrella lausannensis strain CRIB-30 is available in the European Nucleotide Archive database with the accession number PRJEB7018: http://www.ebi.ac.uk/ena/data/view/PRJEB7018
Results
Course Set-Up
The course entitled “Sequence a genome” was initiated with the launching of the new Master of Life Sciences at the University of Lausanne during the academic year from September 2010 to June 2011 (Figure 1). Fourteen pre-graduate students participated in the class, under the supervision of nine teachers, including Professors, Post-doctoral fellows and PhD/graduate students (“assistants”) during a full academic year.
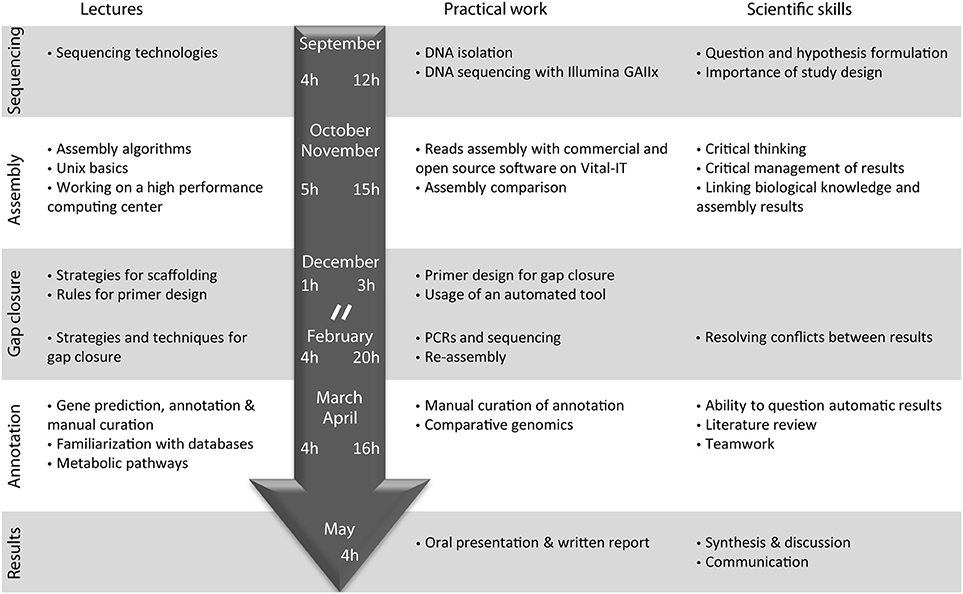
Figure 1. Organization of the course “Sequence a genome” in 2010–2011. Topics of corresponding lectures are detailed on the left. The main steps of genome sequencing performed by the students as well as the main scientific skills acquired by the students are indicated on the right. The central arrow indicates the time (in hours) dedicated to lectures (left) and practical work (right) during the autumn and spring semester of the year 2010–2011. Lectures were immediately followed by the practical work in a 4 h class every 2 weeks. “//” represents the semester break.
After a general introduction on sequencing technologies and on the bacteria selected for analysis, one full day was dedicated to DNA extraction, DNA purity control and a visit to the genome sequencing facility at the start of the course. Subsequently, the class consisted of a 1-h lecture on technical aspects immediately followed by 3-h practical exercises every 2 weeks. The 14 students were split in two groups that worked in parallel on two projects: genome sequencing of three Pseudomonas knackmussii strains that were the subject of another publication (Miyazaki et al., 2014), and E. lausannensis. During the class, the students performed de novo genome assembly using four different software tools and various parameter settings on a Linux cluster.
At the beginning of the spring semester, 3 days were dedicated to gap closure of the genome by performing PCRs, sequencing and reassembly (Figure 1). The course continued by manually curating the annotation of the Estrella genome on the online GenDB platform while deepening our understanding of specific topics. Finally students were evaluated on the basis of a written report and an oral presentation on their annotation topic, as well as on the basis of their practical investment and interest during the course and their understanding of the various steps of the sequencing project.
Genome Sequencing, Assembly and Gap Closure
The sequencing of E. lausannensis genome yielded 7,930,903 paired-end reads of 33 bp, leading to a theoretical coverage of 175 fold for a 3 Mb genome. Velvet (Zerbino and Birney, 2008) produced the de novo assembly with the lowest number of large scaffolds, the highest N50 and a total genome size within the expected range (Table 1). Therefore, the Velvet assembly was selected for gap closure. No large scaffold showed high similarity to A. castellanii genome, which indicates that contaminant reads from the host are scarce.
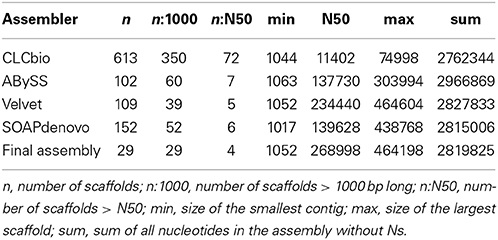
Table 1. Assembly statistics of Estrella lausannensis.
Two contigs attracted our attention due to their 3-fold higher coverage than the rest of the genome. These contigs encode the 16S, 23S and 5S genes. Therefore, we concluded that E. lausannensis possesses three ribosomal operons. Since no phylogenetically closely related organism was available, the sequence of the selected Velvet assembly was compared to the best assembly achieved using each different software. In addition, the position of read pairs in different contigs was analyzed to scaffold contigs. In short, these strategies enabled us to detect one misassembly, to order 15 contigs, and to identify possible combinations around the 3 rRNA sequences and other repeated elements. PCRs resulted in positive amplicons for 20 gaps, which enabled us to solve 9 gaps by a first round of Sanger sequencing. The final assembly included 29 scaffolds made of 35 contigs. Finally, a putative plasmid was assembled differently by the four software tools. This enabled us to resolve its circular sequence in silico (see below).
The final draft genome sequence encompasses 2,820,195 bp for the main chromosome and 9136 bp for the plasmid, which is in the range of other Chlamydiales bacteria (Table 2). The GC content is notably higher than in other members of the Chlamydiales order, reaching 48.2%. This represent a 10% difference in GC content compared to its closest known relative, C. sequanensis, a member of the same family. The prediction of 40 tRNAs and the presence of all Chlamydiales core genes suggest that the draft genome is almost complete and likely lacks only repetitive elements such as mobile genes. All Chlamydiales sequenced so far, including E. lausannensis, do not harbor CRISPR elements.
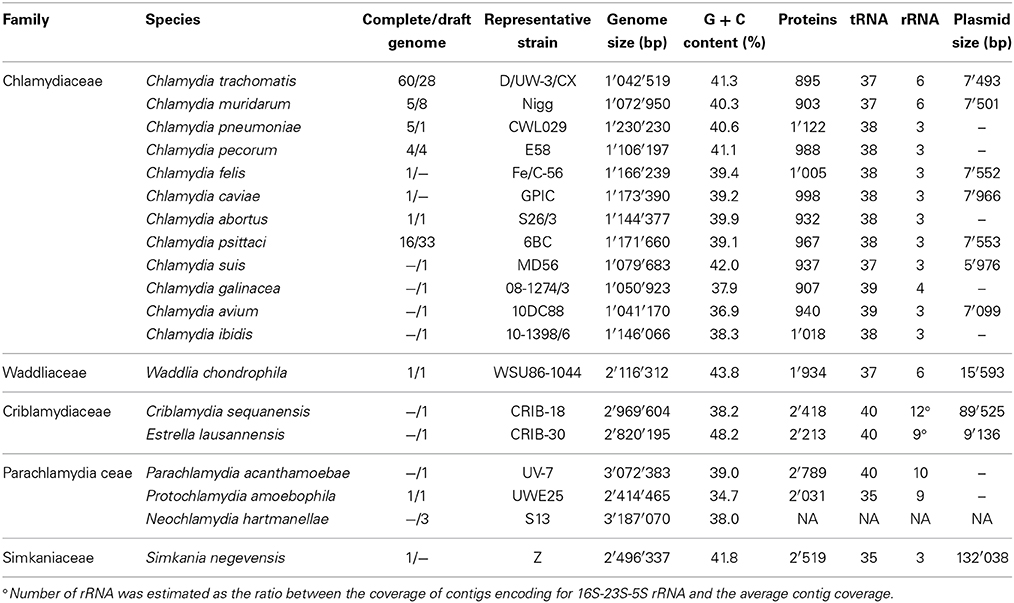
Table 2. Genomic characteristics of members of the Chlamydiales order.
Genome Annotation
Each student was responsible for manually curating the annotation of genes related to a specific topic of interest. Findings had to be summarized in a short paragraph that formed the basis for this article. These final reports, corrected by the professors at the time of the course, are intentionally provided “as such” below with only slight language editing.
Type III secretion system
The type III secretion system (T3SS) is an important bacterial virulence factor that acts as a syringe and allows injection of proteins in the eukaryotic host cell (Cornelis, 2006). This highly conserved system is present in all members of the Chlamydiales order studied so far: Chlamydia spp., Parachlamydia acanthamoebae, Protochlamydia amoebophila, Simkania negevensis and Waddlia chondrophila (Peters et al., 2007; Greub et al., 2009; Bertelli et al., 2010; Collingro et al., 2011). The sequencing of the Estrella genome has confirmed the striking conservation of gene order between the members of four different families (Figure 2). Interestingly, all genes coding for core components and chaperones of the T3SS are located in four different DNA regions. Thus, a T3SS was already present in the common ancestor of all Chlamydiales that diverged more than 700 million years ago (Greub and Raoult, 2003). The T3SS effectors are however poorly conserved and more work will be needed to identify the effectors of E. lausannensis and understand their effects on the eukaryotic cell.
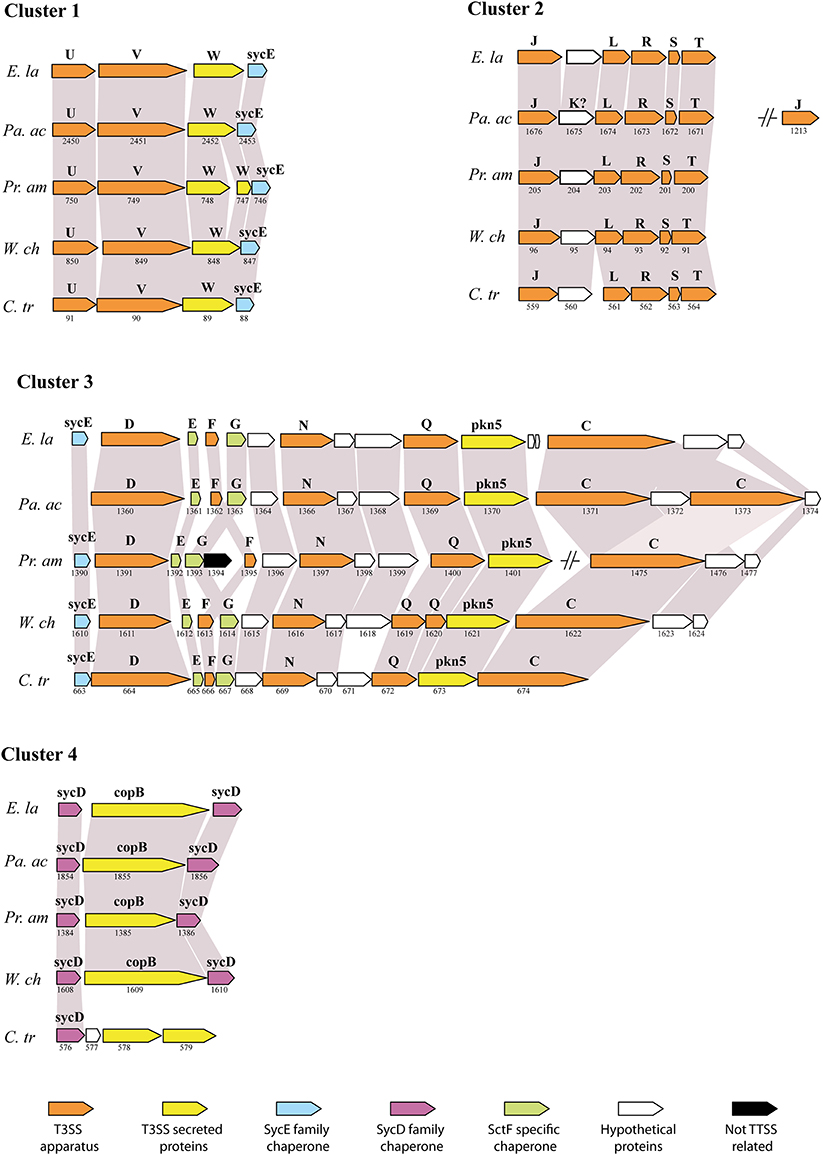
Figure 2. Conservation of the type III secretion system (adapted from Bertelli et al., 2010). Comparison of the genetic clusters encoding for T3SS genes between E. lausannensis (E. la), Parachlamydia acanthamoebae (Pa. ac), Protochlamydia amoebophila (Pr. am), Waddlia chondrophila (W. ch) and Chlamydia trachomatis (C. tr) that belong to 4 different families within the Chlamydiales order. Gray shading indicates the conservation of the genes. Gene names and ORF numbers are respectively indicated above and below each gene. Genes are colored according to their specific functions. Capital letters refer to sct gene names according to the unified nomenclature proposed by Hueck (1998). sycE and sycD are genes encoding for SycE-like and SycD/LcrH-like T3SS chaperones.
Nucleotide biosynthesis
Purines and pyrimidines are heterocyclic aromatic molecules necessary for every living organism. These molecules are required for the biosynthesis of nucleotides and nucleosides that are essential (i) as building blocks for DNA and RNA, (ii) in the form of energy molecules (ATP and GTP), and (iii) for protein biosynthesis. Therefore, every organism possesses tools to metabolize, salvage, degrade and recycle purines and pyrimidines in order to provide sufficient amounts to the organism. Members of the Chlamydiales order are known to be auxotroph for nucleotides, and possess specific or wide-range transporters to scavenge nucleotides from their host (Haferkamp et al., 2004, 2006; Knab et al., 2011; Fisher et al., 2013). We analyzed purine and pyrimidine biosynthetic pathways in Estrella and related species (Figures 3, 4).
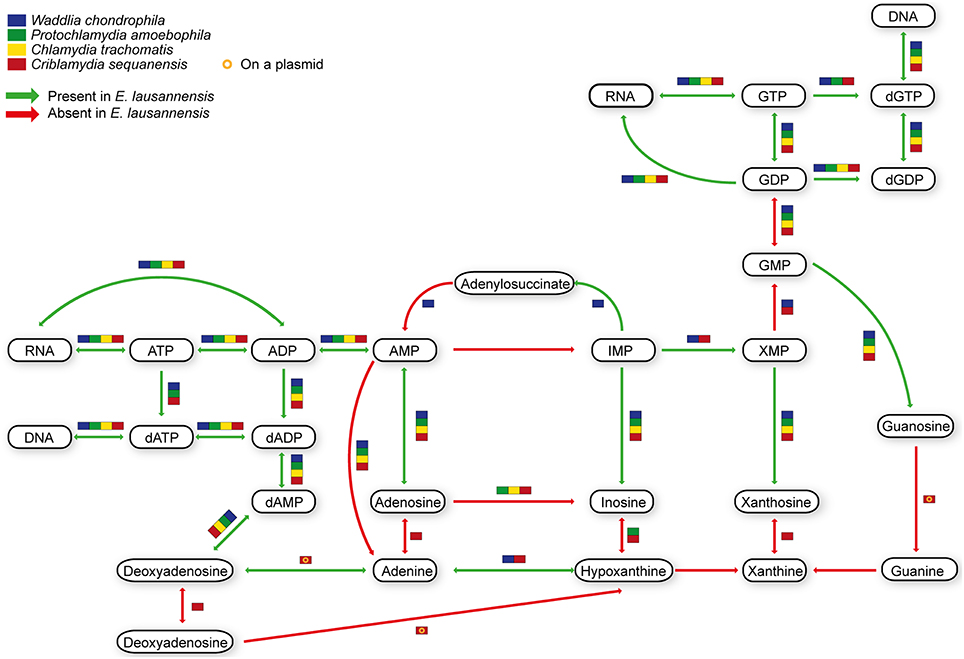
Figure 3. Purine biosynthesis pathways predicted in E. lausannensis and related chlamydia. The pathway for the biosynthesis of IMP from ribose or amino acids appears to be absent in E. lausannensis. Green arrows indicate enzyme reactions predicted to be catalyzed by proteins encoded in the genome of E. lausannensis. Red arrows indicate reactions catalyzed by enzymes that could not be detected in the genome. Colored boxes indicate the presence of an enzyme reaction in four other members of the Chlamydiales order.
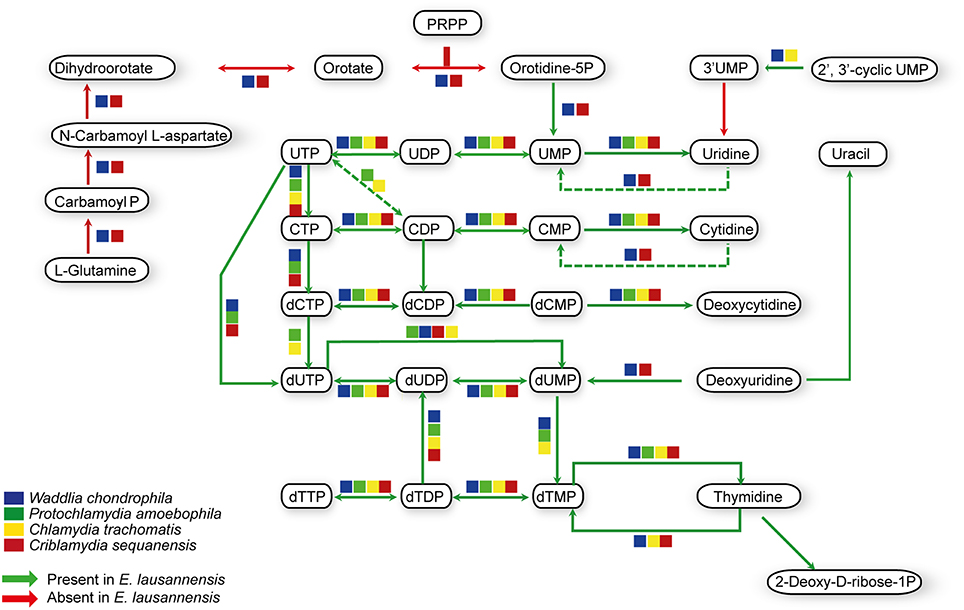
Figure 4. Pyrimidine biosynthesis pathways predicted in E. lausannensis and related chlamydia. The pathway for the biosynthesis of UMP from PRPP and amino-acids appears to be absent in E. lausannensis. Green arrows indicate enzyme reactions predicted to be catalyzed by proteins encoded in the genome of E. lausannensis. Red arrows indicate reactions catalyzed by enzymes that could be discovered in the genome. Colored boxes indicate the presence of an enzyme catalyzing the reaction in four other members of the Chlamydiales order.
Like other members of the Chlamydiales order, E. lausannensis is not able to synthesize inosine monophosphate (IMP), which is the central element of the purine pathway. However as expected, E. lausannensis is equipped with the complete machinery to produce purines and deoxy-purines in different phosphorylated states from ADP or GDP nucleotides. Concerning pyrimidine biosynthesis, E. lausannensis is able to synthesize cytosine, thymine and uracil from uridine monophosphate (UMP). Similar to Chlamydia and Protochlamydia, E. lausannensis is not able to synthesize UMP from amino acid degradation products or from PRPP produced in the pentose phosphate pathway. On the contrary, W. chondrophila and C. sequanensis, seem to be able to generate pyrimidines from L-glutamine. These findings imply that E. lausannensis probably imports core components such as ATP, GTP, UMP or other pyrimidine nucleotides from the host cell using nucleotide transporters. Indeed, five nucleotide transporters were identified which exhibit significant sequence similarity to transporters of Pr. amoebophila and S. negevensis, which have been biochemically characterized (Haferkamp et al., 2004, 2006; Knab et al., 2011).
Amino acid metabolism
Members of the Chlamydiales order exhibit significant differences in the metabolism of amino acids (Figure 5). C. trachomatis is auxotroph for most amino acids, including cysteine, glycine, serine and threonine. All Chlamydia-related bacteria are able to synthesize serine from pyruvate, which can then be transformed into glycine from serine. Cysteine can be synthesized from pyruvate or serine in W. chondrophila, E. lausannensis and C. sequanensis. In all bacteria analyzed, threonine is produced from glycine in a two-step reaction involving L-aminoacetoacetate as an intermediate. Interestingly, C. sequanensis is able to produce threonine from glycine in a one-step reaction using threonine aldolase whereas other Chlamydia-related bacteria encode a two-step reaction catalyzed by glycine C-acetyltransferase and L-threonine 3-dehydrogenase.
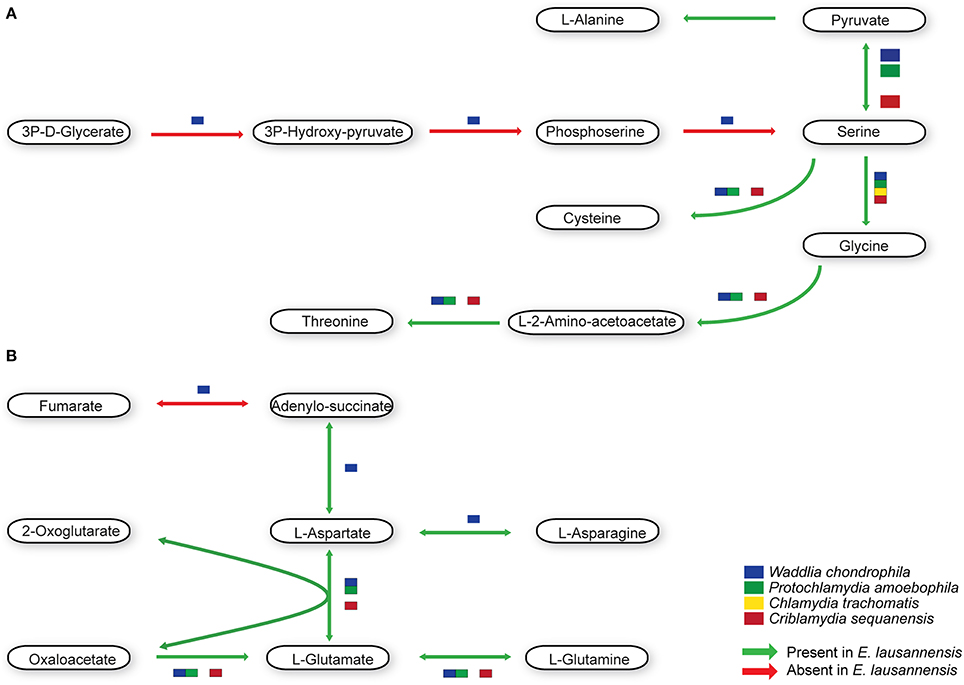
Figure 5. Amino acid metabolism. E. lausannensis is able to synthesize (A) cysteine, glycine, serine, threonine, and alanine from pyruvate as well as (B) aspartate, glutamate and their amidated forms asparagine and glutamine from oxaloacetate. Green arrows indicate enzyme reactions predicted to be catalyzed by proteins encoded in the genome of E. lausannensis. Red arrows indicate reactions catalyzed by enzymes that could not be discovered in the genome. Colored boxes indicate the presence of an enzyme catalyzing the reaction in four other members of the Chlamydiales order.
In contrast with C. trachomatis that lacks all the enzymes needed to produce alanine, aspartate and glutamate independently from the host cell, E. lausannensis has the ability to newly synthesize alanine from pyruvate, through the action of an alanine dehydrogenase. Glutamate synthesis is linked to the citric acid cycle by the action of either a glutamate dehydrogenase, or an aspartate aminotransferase, both providing a link between glutamate and 2-oxoglutarate. The only enzyme shared by all members of the Chlamydiales order studied here is the aspartate aminotransferase, catalyzing the transamination of 2-oxoglutarate and aspartate to form oxaloacetate and glutamate. Almost all enzymes for alanine, glutamate and aspartate metabolism present in E. lausannensis are also conserved in W. chondrophila. However, the latter possesses the additional capacity to synthesize L-aspartate from fumarate through adenylo-succinate lyase and adenylo-succinate synthetase.
Cofactor biosynthesis
Ubiquinone and menaquinone, two interchangeable molecules that share a similar backbone structure but have different side chains, are key players in electron transfer systems. E. lausannensis, W. chondrophila and Pr. amoebophila encode the entire menaquinone pathway whereas C. sequanensis lacks the possibility to convert the 2-succinyl-5-enolpyruvoyl-6-hydroxy-3-cyclohexene-1-carboxylate into (1R,6R)-2-succinyl-6hydroxy-2,4-cyclohexadiene-1-carboxylate (Figure 6). Conversely, C. trachomatis does not encode the classical menaquinone biosynthesis pathway, but seems to encode an alternative route, named the futalosine pathway (Hiratsuka et al., 2008).
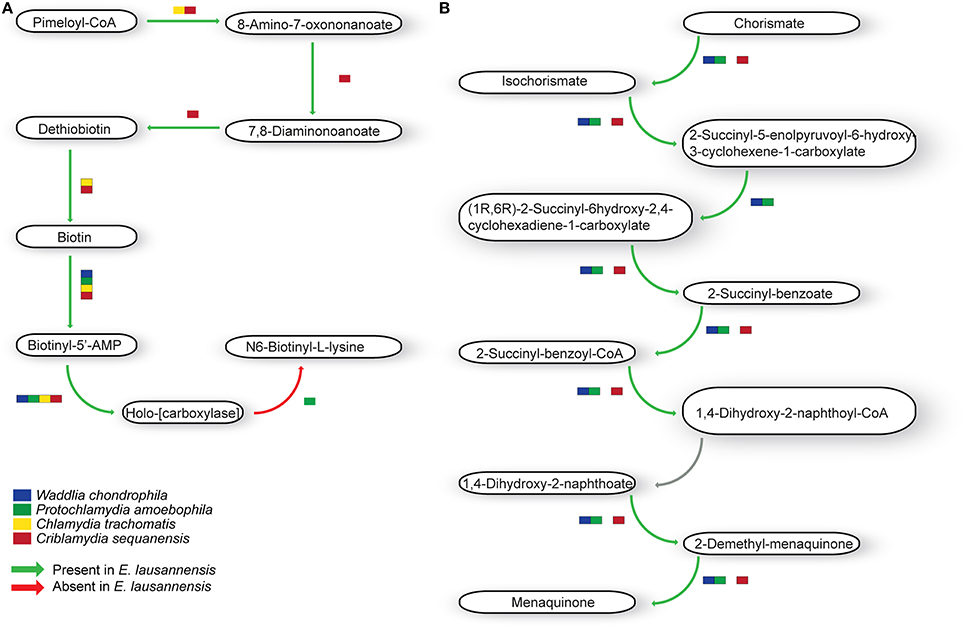
Figure 6. Co-factor metabolism in E. lausannensis and related chlamydia. Predicted intermediate metabolism for biotin (A) and menaquinone (B) biosynthesis. Green arrows indicate enzyme reactions predicted to be catalyzed by proteins encoded in the genome of E. lausannensis. Red arrows indicate reactions catalyzed by enzymes that could not be discovered in the genome. Colored boxes indicate the presence of an enzyme catalyzing the reaction in four other members of the Chlamydiales order.
Biotin (vitamin H) is another important cofactor notably involved in bacterial growth as well as in different regulation networks, including control of toxin production. E. lausannensis, like C. sequanensis, exhibits a complete pathway for the synthesis of biotin from Pimeloyl-CoA (Figure 6). On the contrary, C. trachomatis and P. amoebophila lack several enzymatic steps for the production and conversion of biotin, and may retrieve biotin from the host cell.
Estrella plasmid
E. lausannensis contains a small 9.1 kb plasmid, whose sequence was completely solved. It encodes 15 coding sequences (CDSs). Nine CDSs encode for hypothetical proteins with no assigned function (Figure 7). Genes with assigned function encode a DNA primase, a RelE type toxin-antitoxin module, a putative integrase/recombinase, a putative excisionase, and a putative chromosome partitioning protein; all of which fall loosely into the categories of plasmid maintenance, replication and/or integration. Interestingly, many of the hypothetical proteins with unknown function are conserved in related species including P. acanthamoebae, Pr. amoebophila, and W. chondrophila. Two of these hypothetical genes, ELAC_p0005 and ELAC_p0006, are also found next to each other in P. acanthamoebae suggesting that they may be of importance to amoeba-resisting bacteria.
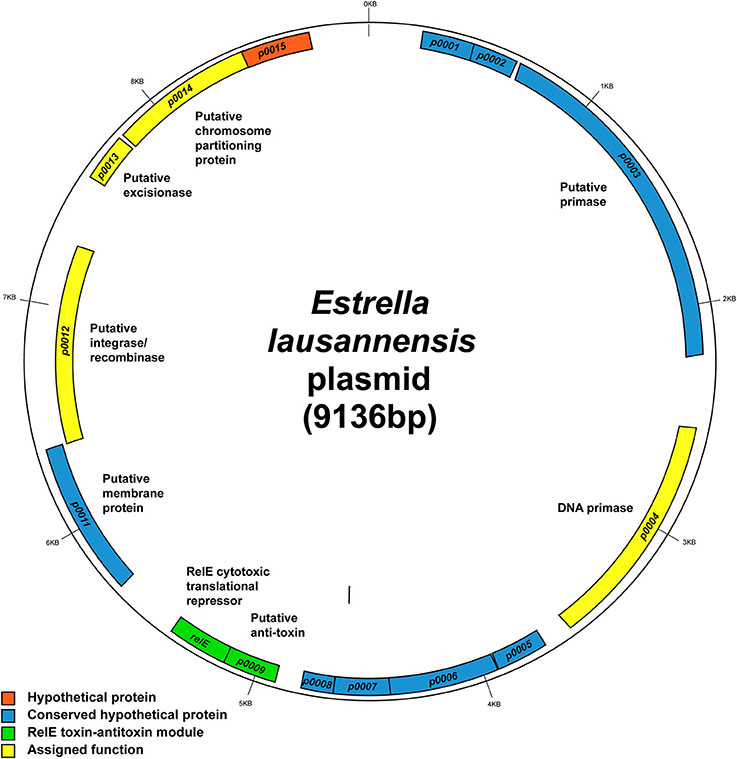
Figure 7. Map of the 9.1-kb Estrella plasmid. The map shows the predicted location of the 14 open reading frames on the plasmid. Hypothetical genes with no known functions are depicted in orange boxes. Blue, hypothetical genes conserved in related species. Green, the predicted RelE toxin-antitoxin module, which is also found in Protochlamydia amoebophila. Yellow, genes with clear functional assignment. Image created using GenDB 2.4 Circular Plot tool.
Also of interest is the RelE family toxin-antitoxin system. RelE is part of a type II (protein-protein) toxin-antitoxin module thought to be involved in plasmid addiction. RelE is a cytotoxic translational repressor that functions alongside an anti-toxic protein RelB (Kamphuis et al., 2007). In this case, the gene adjacent to relE is ELAC_p0009, which did not match to any characterized RelB-like antitoxins and thus could encode a novel antitoxin working in partnership with RelE. Furthermore, the two genes relE (51% identity) and ELAC_p0009 (30% identity) are conserved in Pr. amoebophila, but despite their predicted role in plasmid addiction, they are located on the chromosome of Pr. amoebophila outside the genomic island Pam100G (Greub et al., 2004).
Discussion
This “Sequence a genome” course was made possible by recent advances in sequencing technologies, the commitment of a small group of teachers, and the concentration of competences in several institutions in Lausanne. The class was a success since it enabled all students to acquire theoretical knowledge, scientific skills and practical experience on a real research project dealing with genomic data from the most recent sequencing technologies (at the time of the course). Furthermore, it improved our knowledge of a newly discovered bacterial species, E. lausannensis (this work), as well as of a strain of P. knackmussii (Miyazaki et al., 2014).
During the last decade, several universities started courses aiming at introducing students to genomics while providing novel information on new bacterial strains. Table 3 summarizes some of these initiatives which mostly started with the introduction of high-throughput technologies and the decreasing of costs. As with the variety of biology research, the organisms studied come across the whole range of bacterial diversity, some courses targeting single poorly known organisms (Kerfeld and Simons, 2007 and this course), whereas others sequenced new strains of widely known bacteria (Drew and Triplett, 2008) or a diversity of organisms from a given environment (Edwards et al., 2013). The strategy for the publication of student's results vary from blog posts or website results to short genome announcements or full research paper publication. A main strength of this course is to propose all steps of a bacterial genome project, from bacterial culture to genome analysis and biocuration of annotation. The translation and integration of information relevant to biology (biocuration) in the data provided to the scientific community is essential to raise the standards of data release and facilitate knowledge transfer.
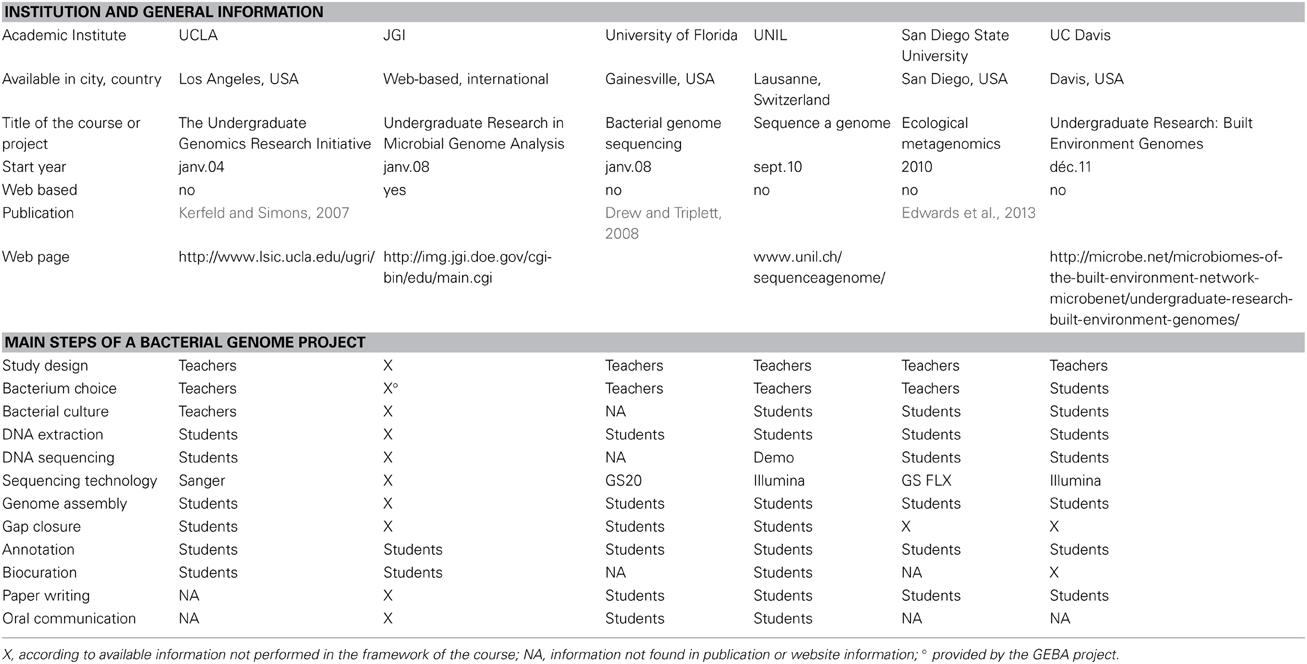
Table 3. Comparison of genomics courses and teaching initiatives.
The availability of the Estrella genome is a first step in the understanding of the biology of this recently-discovered genus of intracellular bacteria. The students performed a targeted analysis of E. lausannensis biosynthetic abilities for essential compounds. They provided information on major differences in metabolism across different members of the Chlamydiales order, including two additional genus compared to a recent review by Omsland et al (Omsland et al., 2014). As a whole, the two sequenced members of the Criblamydiaceae family show variable metabolic potential, with no further abilities in the pathways studied here, than other Chlamydia-related bacteria. Although this might reflect their less homeostatic niche as previously suggested (Omsland et al., 2014), the ability of E. lausannensis to thrive in a variety of different cell lines might be due to other factors such as effectors secreted by the complete T3SS apparatus evidenced in this study. Further analyses through novel metabolomics methods are required to tackle the metabolism of E. lausannensis and other Chlamydia-related bacteria.
At the end of the year, the course was evaluated by the students. They showed a great deal of enthusiasm and involvement into this technical and conceptual adventure on a new and poorly studied microorganism. The students developed their practical knowledge in dealing with UHTS data and a UNIX-like computer environment. These skills are increasingly needed by life scientists to face the flood of genome sequencing projects and other sequence-based projects (RNA-seq, ChIP-seq, etc.). Aware of the future challenges to deal with large-scale data, students appreciated a first travel at the border between bioinformatics and wet lab techniques, while staying in the comfortable environment of a relatively “simple” bacterial genome.
The balance between wet lab experiments and bioinformatics was appreciated. As we could expect, the use of complex bioinformatics tools, a high-performance computing cluster, and major bioinformatics databases such as GenBank, Pfam or KEGG was especially appreciated by a subset of the students. Most students did not intend to continue into bioinformatics and were planning to become wet-lab biologists. Therefore, they lacked a background in UNIX and had only little experience with bioinformatics. However, they liked being introduced to the basic concepts around sequence analysis.
The spring semester provided a stronger link to biology with the annotation of the E. lausannensis genome. Manual curation made them aware of the difficulties and potential errors hidden behind a gene annotation available for any given organism. They learned to be critical about annotations and to verify information from multiple sources and types of evidence.
Many participants enjoyed being enrolled in a real research experience and not a pre-prepared practical course where all answers are already known. Moreover, a major praise was the possibility to participate in all steps from DNA extraction to report preparation with only little intervention of the teachers to provide extra analyses. They reported a gain in autonomy by learning this way. Most students concluded that although the course was very challenging to follow all the way from bioinformatics to the wet lab, it was also highly interesting and rewarding.
In summary, students learned essential scientific skills from study design, hypothesis formulation, critical mind, literature review, to the ability to synthesize information and to communicate the information both verbally and in writing (Figure 1). Moreover, they learned technical competence, knowledge on a variety of technological and methodological aspects related to the sequencing as well as biological background on their organism of interest. This course further raised awareness among the students on how difficult yet powerful it can be to obtain such a central resource—a complete and annotated genome sequence—even in the era of UHTS. The continuous evolution of these technologies forces teachers to stay at the forefront of both experimental and computational aspects. In the following years, several aspects of the class have been improved and regular updates are posted on a blog of the class (http://www.unil.ch/sequenceagenome/).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Teachers of the “Sequence a genome” class received collectively the 2012 “Excellence in teaching award” of the Faculty of Biology and Medicine of the University of Lausanne. The computations were performed at the Vital-IT (http://www.vital-it.ch) Center for high-performance computing of the SIB Swiss Institute of Bioinformatics.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fmicb.2015.00101/abstract
Footnotes
1. ^Coil, D. Undergraduate Research: Built Environment Genomes - microBEnet. Available online at: http://microbe.net/microbiomes-of-the-built-environment-network-microbenet/undergraduate-research-built-environment-genomes/
2. ^JGI. Undergraduate Research in Microbial Genome Analysis. Available online at: http://img.jgi.doe.gov/cgi-bin/edu/main.cgi
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bertelli, C., Collyn, F., Croxatto, A., Rückert, C., Polkinghorne, A., Kebbi-Beghdadi, C., et al. (2010). The waddlia genome: a window into chlamydial biology. PLoS ONE 5:10890. doi: 10.1371/journal.pone.0010890
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bertelli, C., Goesmann, A., and Greub, G. (2014). Criblamydia sequanensis harbors a megaplasmid encoding arsenite resistance. Genome Announc. 2:e00949-14. doi: 10.1128/genomeA.00949-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bertelli, C., and Greub, G. (2013). Rapid bacterial genome sequencing: methods and applications in clinical microbiology. Clin. Microbiol. Infect. 19, 803–813. doi: 10.1111/1469-0691.12217
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Collingro, A., Tischler, P., Weinmaier, T., Penz, T., Heinz, E., Brunham, R. C., et al. (2011). Unity in variety - the pan-genome of the Chlamydiae. Mol. Biol. Evol. 28, 3253–3270. doi: 10.1093/molbev/msr161
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cornelis, G. R. (2006). The type III secretion injectisome. Nat. Rev. Microbiol. 4, 811–825. doi: 10.1038/nrmicro1526
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Corsaro, D., Feroldi, V., Saucedo, G., Ribas, F., Loret, J.-F., and Greub, G. (2009). Novel Chlamydiales strains isolated from a water treatment plant. Environ. Microbiol. 11, 188–200. doi: 10.1111/j.1462-2920.2008.01752.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cran (2010). The comprehensive R archive network. Wiley Interdiscip. Rev. n/a–n/a. doi: 10.1002/wics.1212
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Barsy, M., Bottinelli, L., and Greub, G. (2014). Antibiotic susceptibility of Estrella lausannensis, a potential emerging pathogen. Microbes Infect. 16, 746–754. doi: 10.1016/j.micinf.2014.08.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Drew, J. C., and Triplett, E. W. (2008). Whole genome sequencing in the undergraduate classroom: outcomes and lessons from a pilot course. J. Microbiol. Biol. Educ. 9, 3–11. doi: 10.1128/jmbe.v9.89
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Edwards, R. A., Haggerty, J. M., Cassman, N., Busch, J. C., Aguinaldo, K., Chinta, S., et al. (2013). Microbes, metagenomes and marine mammals: enabling the next generation of scientist to enter the genomic era. BMC Genomics 14:600. doi: 10.1186/1471-2164-14-600
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230. doi: 10.1093/nar/gkt1223
Fisher, D. J., Fernández, R. E., and Maurelli, A. T. (2013). Chlamydia trachomatis transports NAD via the Npt1 ATP/ADP translocase. J. Bacteriol. 195, 3381–3386. doi: 10.1128/JB.00433-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gordon, D., Abajian, C., and Green, P. (1998). Consed: a graphical tool for sequence finishing. Genome Res. 8, 195–202. doi: 10.1101/gr.8.3.195
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greub, G., Collyn, F., Guy, L., and Roten, C.-A. (2004). A genomic island present along the bacterial chromosome of the Parachlamydiaceae UWE25, an obligate amoebal endosymbiont, encodes a potentially functional F-like conjugative DNA transfer system. BMC Microbiol. 4:48. doi: 10.1186/1471-2180-4-48
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greub, G., Kebbi-Beghdadi, C., Bertelli, C., Collyn, F., Riederer, B. M., Yersin, C., et al. (2009). High throughput sequencing and Proteomics to identify immunogenic proteins of a new pathogen: the dirty genome approach. PLoS ONE 4:e8423. doi: 10.1371/journal.pone.0008423
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greub, G., and Raoult, D. (2002). Crescent bodies of Parachlamydia acanthamoeba and its life cycle within Acanthamoeba polyphaga: an electron micrograph study. Appl. Environ. Microbiol. 68, 3076–3084. doi: 10.1128/AEM.68.6.3076-3084.2002
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Greub, G., and Raoult, D. (2003). History of the ADP/ATP-translocase-encoding gene, a parasitism gene transferred from a Chlamydiales ancestor to plants 1 billion years ago. Appl. Environ. Microbiol. 69, 5530–5535. doi: 10.1128/AEM.69.9.5530-5535.2003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haferkamp, I., Schmitz-Esser, S., Linka, N., Urbany, C., Collingro, A., Wagner, M., et al. (2004). A candidate NAD+ transporter in an intracellular bacterial symbiont related to Chlamydiae. Nature 432, 622–625. doi: 10.1038/nature03131
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Haferkamp, I., Schmitz-Esser, S., Wagner, M., Neigel, N., Horn, M., and Neuhaus, H. E. (2006). Tapping the nucleotide pool of the host: novel nucleotide carrier proteins of Protochlamydia amoebophila. Mol. Microbiol. 60, 1534–1545. doi: 10.1111/j.1365-2958.2006.05193.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hiratsuka, T., Furihata, K., Ishikawa, J., Yamashita, H., Itoh, N., Seto, H., et al. (2008). An alternative menaquinone biosynthetic pathway operating in microorganisms. Science 321, 1670–1673. doi: 10.1126/science.1160446
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hueck, C. J. (1998). Type III protein secretion systems in bacterial pathogens of animals and plants. Microbiol. Mol. Biol. Rev. 62, 379–433.
Jacquier, N., Aeby, S., Lienard, J., and Greub, G. (2013). Discovery of new intracellular pathogens by amoebal coculture and amoebal enrichment approaches. J. Vis. Exp. e51055. doi: 10.3791/51055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jordan, T. C., Burnett, S. H., Carson, S., Caruso, S. M., Clase, K., DeJong, R. J., et al. (2014). A broadly implementable research course in phage discovery and genomics for first-year undergraduate students. MBio 5, e01051–e01013. doi: 10.1128/mBio.01051-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kamphuis, M. B., Monti, M. C., van den Heuvel, R. H. H., López-Villarejo, J., Díaz-Orejas, R., and Boelens, R. (2007). Structure and function of bacterial kid-kis and related toxin-antitoxin systems. Protein Pept. Lett. 14, 113–124. doi: 10.2174/092986607779816096
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kebbi-Beghdadi, C., Batista, C., and Greub, G. (2011). Permissivity of fish cell lines to three Chlamydia-related bacteria: Waddlia chondrophila, Estrella lausannensis and Parachlamydia acanthamoebae. FEMS Immunol. Med. Microbiol. 63, 339–345. doi: 10.1111/j.1574-695X.2011.00856.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kerfeld, C. A., and Simons, R. W. (2007). The undergraduate genomics research initiative. PLoS Biol. 5:e141. doi: 10.1371/journal.pbio.0050141
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Knab, S., Mushak, T. M., Schmitz-Esser, S., Horn, M., and Haferkamp, I. (2011). Nucleotide parasitism by Simkania negevensis (Chlamydiae). J. Bacteriol. 193, 225–235. doi: 10.1128/JB.00919-10
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lagkouvardos, I., Weinmaier, T., Lauro, F. M., Cavicchioli, R., Rattei, T., and Horn, M. (2014). Integrating metagenomic and amplicon databases to resolve the phylogenetic and ecological diversity of the Chlamydiae. ISME J. 8, 115–125. doi: 10.1038/ismej.2013.142
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Li, R., Zhu, H., Ruan, J., Qian, W., Fang, X., Shi, Z., et al. (2010). De novo assembly of human genomes with massively parallel short read sequencing. Genome Res. 20, 265–272. doi: 10.1101/gr.097261.109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lienard, J., Croxatto, A., Prod'hom, G., and Greub, G. (2011). Estrella lausannensis, a new star in the Chlamydiales order. Microbes Infect. 13, 1232–1241. doi: 10.1016/j.micinf.2011.07.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
MacLean, D., Jones, J. D. G., and Studholme, D. J. (2009). Application of “next-generation” sequencing technologies to microbial genetics. Nat. Rev. Microbiol. 7, 287–296. doi: 10.1038/nrmicro2088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Meyer, F., Goesmann, A., McHardy, A. C., Bartels, D., Bekel, T., Clausen, J., et al. (2003). GenDB–an open source genome annotation system for prokaryote genomes. Nucleic Acids Res. 31, 2187–2195. doi: 10.1093/nar/gkg312
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Miyazaki, R., Bertelli, C., Benaglio, P., Canton, J., De Coi, N., Gharib, W. H., et al. (2014). Comparative genome analysis of Pseudomonas knackmussii B13, the first bacterium known to degrade chloroaromatic compounds. Environ. Microbiol. 17, 91–104. doi: 10.1111/1462-2920.12498
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ogata, H., Goto, S., Sato, K., Fujibuchi, W., Bono, H., and Kanehisa, M. (1999). KEGG: Kyoto Encyclopedia of genes and genomes. Nucleic Acids Res. 27, 29–34. doi: 10.1093/nar/28.1.27
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Omsland, A., Sixt, B. S., Horn, M., and Hackstadt, T. (2014). Chlamydial metabolism revisited: interspecies metabolic variability and developmental stage-specific physiologic activities. FEMS Microbiol. Rev. 38, 779–801. doi: 10.1111/1574-6976.12059
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Peters, J., Wilson, D. P., Myers, G., Timms, P., and Bavoil, P. M. (2007). Type III secretion à la Chlamydia. Trends Microbiol. 15, 241–251. doi: 10.1016/j.tim.2007.04.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rusconi, B., and Greub, G. (2013). Discovery of catalases in members of the Chlamydiales order. J. Bacteriol. 195, 3543–3551. doi: 10.1128/JB.00563-13
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rusconi, B., Lienard, J., Aeby, S., Croxatto, A., Bertelli, C., and Greub, G. (2013). Crescent and star shapes of members of the Chlamydiales order: impact of fixative methods. Antonie Van Leeuwenhoek 104, 521–532. doi: 10.1007/s10482-013-9999-9
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Selengut, J. D., Haft, D. H., Davidsen, T., Ganapathy, A., Gwinn-Giglio, M., Nelson, W. C., et al. (2007). TIGRFAMs and genome properties: tools for the assignment of molecular function and biological process in prokaryotic genomes. Nucleic Acids Res. 35, D260–D264. doi: 10.1093/nar/gkl1043
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Simpson, J. T., Wong, K., Jackman, S. D., Schein, J. E., Jones, S. J. M., and Birol, I. (2009). ABySS: a parallel assembler for short read sequence data. Genome Res. 19, 1117–1123. doi: 10.1101/gr.089532.108
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thomas, V., Casson, N., and Greub, G. (2006). Criblamydia sequanensis, a new intracellular Chlamydiales isolated from Seine river water using amoebal co-culture. Environ. Microbiol. 8, 2125–2135. doi: 10.1111/j.1462-2920.2006.01094.x
UNIPROT. (2014). Activities at the universal protein resource (UniProt). Nucleic Acids Res. 42, D191–8. doi: 10.1093/nar/gkt1140
Zerbino, D. R., and Birney, E. (2008). Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 18, 821–829. doi: 10.1101/gr.074492.107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: chlamydia, teaching, metabolic pathways, genome sequencing, biocuration, annotation, genomics
Citation: Bertelli C, Aeby S, Chassot B, Clulow J, Hilfiker O, Rappo S, Ritzmann S, Schumacher P, Terrettaz C, Benaglio P, Falquet L, Farinelli L, Gharib WH, Goesmann A, Harshman K, Linke B, Miyazaki R, Rivolta C, Robinson-Rechavi M, van der Meer JR and Greub G (2015) Sequencing and characterizing the genome of Estrella lausannensis as an undergraduate project: training students and biological insights. Front. Microbiol. 6:101. doi: 10.3389/fmicb.2015.00101
Received: 12 November 2014; Accepted: 26 January 2015;
Published online: 19 February 2015.
Edited by:
Eric Altermann, AgResearch Ltd, New ZealandCopyright © 2015 Bertelli, Aeby, Chassot, Clulow, Hilfiker, Rappo, Ritzmann, Schumacher, Terrettaz, Benaglio, Falquet, Farinelli, Gharib, Goesmann, Harshman, Linke, Miyazaki, Rivolta, Robinson-Rechavi, van der Meer and Greub. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Gilbert Greub, Institute of Microbiology, University of Lausanne, Bugnon 48, 1011 Lausanne, Switzerland e-mail:Z2lsYmVydC5ncmV1YkBjaHV2LmNo