 Benoit St-Pierre
Benoit St-Pierre Laura M. Cersosimo
Laura M. Cersosimo Suzanne L. Ishaq
Suzanne L. Ishaq André-Denis G. Wright
André-Denis G. Wright- 1Department of Animal Science, South Dakota State University, Brookings, SD, USA
- 2Department of Animal and Veterinary Sciences, The University of Vermont, Burlington, VT, USA
- 3Department of Animal and Range Sciences, Montana State University, Bozeman, MT, USA
- 4School of Animal and Comparative Biomedical Sciences, University of Arizona, Tucson, AZ, USA
In herbivores, enteric methane is a by-product from the digestion of plant biomass by mutualistic gastrointestinal tract (GIT) microbial communities. Methane is a potent greenhouse gas that is not assimilated by the host and is released into the environment where it contributes to climate change. Since enteric methane is exclusively produced by methanogenic archaea, the investigation of mutualistic methanogen communities in the GIT of herbivores has been the subject of ongoing research by a number of research groups. In an effort to uncover trends that would facilitate the development of efficient methane mitigation strategies for livestock species, we have in this review summarized and compared currently available results from published studies on this subject. We also offer our perspectives on the importance of pursuing current research efforts on the sequencing of gut methanogen genomes, as well as investigating their cellular physiology and interactions with other GIT microorganisms.
Introduction
In herbivores, fermentation of feed by mutualistic gastrointestinal tract (GIT) communities of microorganisms is essential for proper nutrition of their hosts (Hungate, 1966). These microbial communities consist of a great number of species from phylogenetically diverse groups, mainly bacteria, archaea, protozoa, and fungi, that are mutually dependent through complex trophic relationships (Wolin, 1979). As a result of the collective activities of these microorganisms, polysaccharides, proteins, and lipids are metabolized into end products such as volatile fatty acids (VFAs) that are assimilated by their host to fulfill their energy needs.
Certain products of microbial fermentation, such as carbon dioxide and methane, are not absorbed by the host and are released into the environment. There are two main concerns over methane emissions by livestock animals. First, they have a negative impact on animal productivity, as this process results in lost energy from the host, which can range between 2 and 12% of an animal’s energy intake (Johnson and Johnson, 1995). Secondly, methane is a much more potent greenhouse gas than carbon dioxide, thus having a greater effect on climate change (Lashof and Ahuja, 1990). Since the continuous growth of the human population is expected to result in an increase in the number of domesticated ruminants, decreasing methane emissions by livestock has become a priority and an integral part of climate control policies (Thorpe, 2008).
Methane is synthesized by obligate anaerobic archaea that share methanogenesis as part of their energy metabolism (Liu and Whitman, 2008). Many methanogenic archaea, or methanogens, use H2 and CO2 as substrates to synthesize methane, with certain species also capable of metabolizing small organic compounds such as formate, methanol, methylamines, or acetate (Thauer et al., 2008). Although they do not contribute to fulfilling their host’s energy requirements, methanogens play an important role in the GIT of herbivores by maintaining the fermentative performance of the microbial community. By metabolizing H2 generated from fermentation of plant polysaccharides, methanogens function as a sink to maintain a low H2 pressure, which promotes plant fiber digestion by protozoa and bacteria (Wolin, 1982).
As the only producers of enteric methane, methanogens are responsible for the contribution of livestock industries to climate change (Thorpe, 2008), and have thus become the focus of research toward developing mitigation strategies. Variations in methane emissions according to host and/or diet present an important challenge toward achieving this goal. For instance, an early study reported that gray kangaroos emitted less methane than sheep fed the same diet (Kempton et al., 1976). Similarly, lower levels of methane were observed for camelids compared to ruminant livestock (Pinares-Patino et al., 2003; Dittmann et al., 2014), and Franz et al. (2010) found that methane emissions were higher in sheep compared to ponies. Methane production has been found to increase on higher forage/cellulose diets, especially when comparing grass forage to legume forage (McAllister et al., 1996). In growing beef cattle, methane emissions were not affected by the type of grain fed during backgrounding, but they were found to be lower for corn compared to barley during the finishing phase (Beauchemin and McGinn, 2005). In contrast, the addition of high quality feeds, oils, plant secondary compounds, or microbial modifiers can reduce methane emissions (Lovett et al., 2003; Woodward et al., 2004; Carulla et al., 2005; Puchala et al., 2005; Beauchemin et al., 2007, 2009; Grainger et al., 2009, 2010). In most cases, this variation does not appear to be due to differences in methanogen cell density, but rather in the composition of the methanogen community. For instance, it was observed during anti-methanogen vaccination trials that, while methane emissions from immunized animals were decreased in early stages, they returned to control levels after prolonged immunization (Wright et al., 2004). The vaccine was expected to target methanogens that were highly represented in the rumen microbial community, which may have allowed other methanogens that would otherwise be at a disadvantage to increase in abundance in the rumen of immunized animals (Williams et al., 2009). Based on these results, it was hypothesized that GIT methanogen communities may consist of different groups that could vary in their potential for growth and methane production.
Since the composition of a methanogen community represented a likely determinant of its capacity to produce methane, the investigation of mutualistic methanogen communities in the GIT of a variety of host herbivores or in response to different diets has been the subject of active and ongoing research. As with other fields in environmental microbiology, research on GIT methanogens has benefited greatly from the rapid technological improvements of culture-independent experimental approaches. In this review, we have summarized and compared data available from published studies on the composition and representation of methanogens in the GIT of herbivores. While they tend to be distinct, according to a variety of factors including host breed, species, diet and geographical location, and by mechanisms that remain poorly characterized (Kim et al., 2011), the data also indicate that GIT methanogens form phylogenetic clusters that exhibit a certain degree of overlap among different communities.
Prevalent Methanogens in the GIT Communities of Herbivores
Archaea have been identified in a wide range of habitats (Liu and Whitman, 2008), forming a large and diverse prokaryotic domain, not only ecologically but also phylogenetically. The majority of currently known archaeal species have been assigned to the phyla Euryarchaeota or Crenarchaeota, but additional phyla have been proposed to account for the high degree of divergence found in certain archaea, including Thaumarchaeota, Nanoarchaeota, Korarchaeota, Parvarchaeota, and Aigarchaeota (Shin et al., 2004; Allers and Mevarech, 2005; Brochier-Armanet et al., 2008, 2011; Nunoura et al., 2011; Rinke et al., 2013; Petitjean et al., 2014; Raymann et al., 2015). All currently known methanogens belong to the phylum Euryarchaeota, which is divided into seven orders (Methanobacteriales, Methanocellales, Methanococcales, Methanomassiliicoccales, Methanomicrobiales, Methanosarcinales, and Methanopyrales), that include 10 families and 31 genera (Liu and Whitman, 2008; Sakai et al., 2008; Paul et al., 2012; Iino et al., 2013). Methanogens have colonized as a group a wide variety of anaerobic environments, including marine and freshwater sediments, soil, and landfills, and are thus not limited to just the GIT of animals.
The 16S rRNA gene is the most commonly used phylogenetic marker for the characterization of bacterial and methanogen communities (Skillman et al., 2006; Rajendhran and Gunasekaran, 2011). Thus, the data we have selected on methanogen composition from the gut of herbivorous animals was generated using 16S rRNA gene clone libraries or next generation sequencing of amplicons. Typically, a minority of the GIT archaeal 16S rRNA gene sequences identified to date are identical to validly characterized methanogens species, while the remaining majority of sequences exhibit a varying degree of relation to methanogen species. Despite their diversity, GIT methanogens group into very distinct phylogenetic clusters of archaea (Kim et al., 2011). In this section, we aim to present the major groups of methanogens that have been identified in the GIT of herbivores.
Methanobrevibacter-Related Archaea
16S rRNA gene sequences closely related to certain species belonging to the genus Methanobrevibacter (order Methanobacteriales) are among the most frequently found in GIT samples from livestock animals. While representation can vary according to host species, diet, and/or geographical location, dominance of Methanobrevibacter-related archaea reported by a number of different studies is quite striking. Indeed, Methanobrevibacter-related methanogens represented more than ∼80% of 16S rRNA gene sequences from hosts ranging from birds [hoatzin (Wright et al., 2009)] and marsupials [wallaby-May sample (Evans et al., 2009)] to pseudo-ruminants [alpaca (St-Pierre and Wright, 2012), Bactrian camel (Turnbull et al., 2011)] and ruminants [buffalo – Mediterranean breed (Franzolin et al., 2012), cattle-New Zealand (Seedorf et al., 2015), dairy cattle (Hook et al., 2009, 2011; King et al., 2011), goats (Cunha et al., 2011), impala (Cersosimo et al., 2015), reindeer-Norway (Sundset et al., 2009a), sheep-Venezuela (Wright et al., 2008), sheep-Scotland (Snelling et al., 2014), sheep-New Zealand (Seedorf et al., 2015), and yak (An et al., 2005)]. In other studies, they represented a lower, but well represented proportion (27–60%) of identified clones or sequence reads in cattle (Whitford et al., 2001; Skillman et al., 2006), reindeer-Svalbard (Sundset et al., 2009b), white rhinoceroses (Luo et al., 2013), Chinese roe deer (Li et al., 2014), and Mehsani water buffaloes (Singh et al., 2015). Since characterized species of Methanobrevibacter mainly use H2 and CO2 as substrates for methanogenesis, it is hypothesized that uncultured Methanobrevibacter-related methanogens identified by their 16S rRNA gene sequence are also hydrogenotrophic.
Currently, 15 cultured Methanobrevibacter species have been characterized according to the List of Prokaryotic Names with Standing in Nomenclature (LPSN). However, GIT Methanobrevibacter-related methanogens from livestock animals tend to be more closely related to either Methanobrevibacter ruminantium, Methanobrevibacter millerae, Methanobrevibacter gottschalkii, or Methanobrevibacter smithii. While typically found in lower frequency, 16S rRNA sequences with closest identity to either Methanobrevibacter olleyae, Methanobrevibacter thaueri, or Methanobrevibacter wolinii have also been reported in GIT samples. Methanobrevibacter boviskoreani has been the latest addition to the list of cultured rumen methanogens from this group (Lee et al., 2013), with Methanobrevibacter wolinii as its closest relative. To our knowledge, Methanobrevibacter woesei related methanogens have only been reported in chickens, and 16S rRNA gene sequences from the GIT of herbivores that are related to either Methanobrevibacter curvatus, Methanobrevibacter cuticularis, Methanobrevibacter oralis, Methanobrevibacter arboriphilus, Methanobrevibacter filiformis, or Methanobrevibacter acididurans have only rarely if ever been identified in this environment.
SGMT-RO Population Model for Methanobrevibacter-Related Methanogens
While it appears that most Methanobrevibacter-related GIT 16S rRNA gene sequences tend to be closely related to a limited number of valid Methanobrevibacter species, they exhibit a remarkable level of diversity that has been estimated to be in the 100s of species-level operational taxonomic units (OTUs; Kim et al., 2011). Indeed, the level of sequence identity for Methanobrevibacter-related 16S rRNA gene sequences can typically vary between 90 and 100% with their respective closest valid methanogens species. Therefore, although GIT methanogens are from similar phylogenetic groups, they appear to form a continuum of species rather than discrete groups (Janssen and Kirs, 2008). However, only a subset of OTUs are identified in each sample, with typically a few OTUs that tend to be more abundant (Wright et al., 2007, 2009; Sundset et al., 2009a,b; Hook et al., 2011; King et al., 2011; Turnbull et al., 2011; Franzolin et al., 2012; St-Pierre and Wright, 2012; Snelling et al., 2014; Cersosimo et al., 2015; Seedorf et al., 2015). To facilitate the creation of GIT methanogen community structure models from environmental samples, sequence identity cutoffs can be set at specific levels to group 16S rRNA genes from methanogens of the same presumptive species or of the same presumptive genus. The representation of each category in an environmental sample can thus be expressed as a percentage of the total number of clones or sequence reads identified in its corresponding study. Methanogen communities can then be compared between host breeds, species, feed regimens, and/or geographical locations. While there is currently no absolute 16S rRNA gene sequence identity cutoff that has been set to formally distinguish methanogens of the same species or genus from uncultured archaea, it remains a very useful tool to uncover various trends in archaeal community composition.
As a complementary approach, we have also explored the use of phylogenetic analyses of Methanobrevibacter-related GIT 16S rRNA gene sequences to create community structure models. While they appear to form a continuum of species, we observed that Methanobrevibacter-related GIT 16S rRNA gene sequences are mostly distributed between two large clades. One clade consists of sequences that are closely related to Methanobrevibacter smithii, Methanobrevibacter gottschalkii, Methanobrevibacter millerae or Methanobrevibacter thaueri, which we have referred to as the smithii – gottschalkii – millerae – thaurei clade, or simply as the SGMT clade. The other major clade groups Methanobrevibacter ruminantium and Methanobrevibacter olleyae – like sequences, which we have referred to as the ruminantium – olleyae or RO clade.
After re-examining available data by our research team and other research groups to compare the sequence distribution between the SGMT clade and the RO clade, we were able to group samples from a wide variety of sources into more encompassing categories (Table 1). For instance, the SGMT clade was clearly more dominant than the RO clade in impalas (Cersosimo et al., 2015), wallabies (May sample; Evans et al., 2009), in two separate studies involving Holstein dairy cows (Hook et al., 2009, 2011), in alpacas (St-Pierre and Wright, 2012), in water buffaloes (Franzolin et al., 2012), in sheep from Venezuela (Wright et al., 2008), in sheep from Scotland (Snelling et al., 2014), in New Zealand sheep fed two different diets (Seedorf et al., 2015), in Chinese roe deer (Li et al., 2014), and in reindeers (Norway and Svalbard; Sundset et al., 2009a,b). In contrast, the RO clade was distinctively more highly represented than the SGMT clade in the hoatzin (Wright et al., 2009), in an early analysis involving Holstein dairy cows (Whitford et al., 2001), in corn-fed beef cattle (Wright et al., 2007), in Jersey dairy cows (Skillman et al., 2006), and in horses fed a pasture or forage-grain diet (Fernandes et al., 2014). Notably, only a few studies have reported a balanced SGMT:RO, such as from potato-fed beef cattle (Wright et al., 2007).
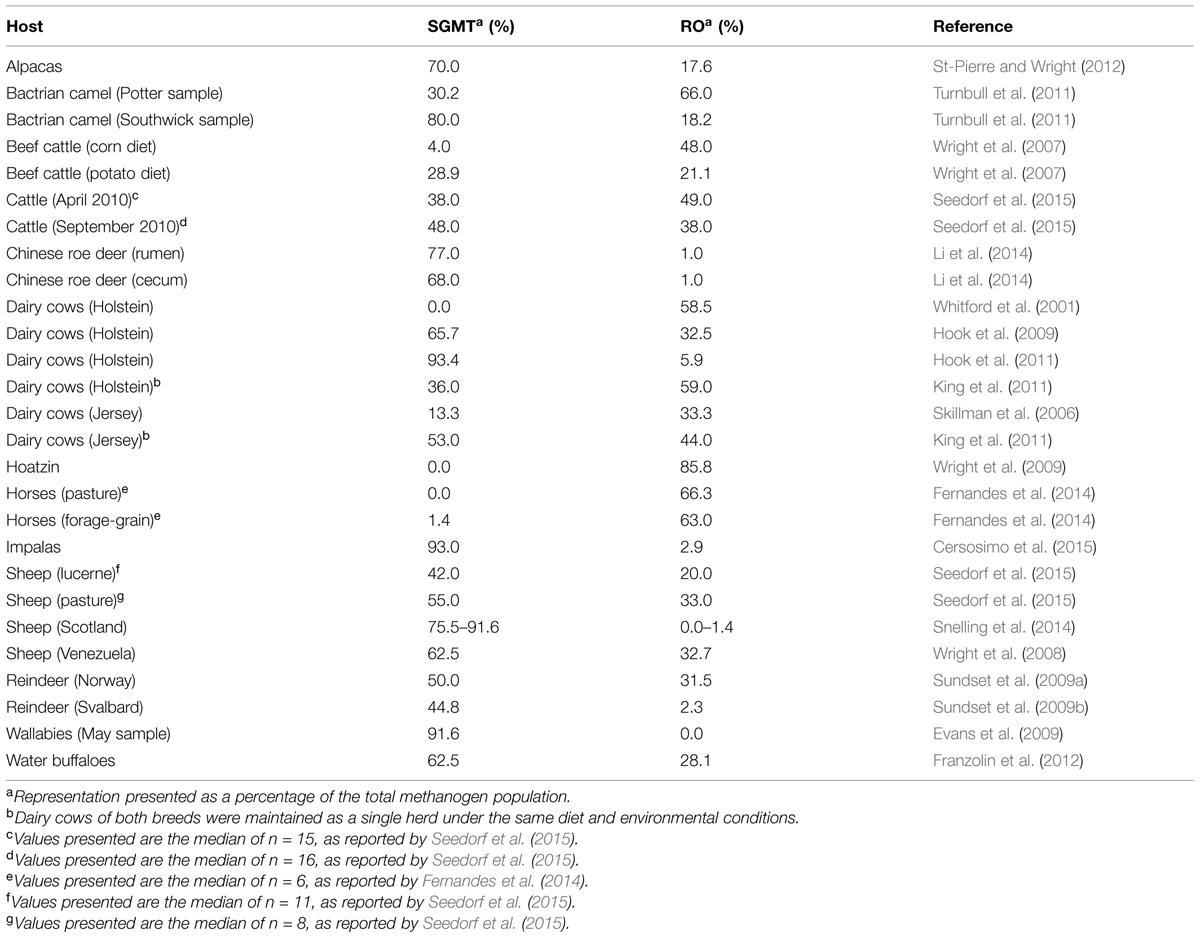
TABLE 1. Representation of SGMT and R0 methanogens in different hosts and diets.
In some reports, comparative studies have revealed opposite SGMT:RO population composition as a function of breeds or as a function of environmental factors within the same breed. This was observed in Holstein and Jersey dairy cows from the same herd maintained under common environmental conditions (King et al., 2011), as well as in cattle from New Zealand sampled at two different time points (Seedorf et al., 2015). In captive Bactrian camels sampled from zoological parks at two different locations in the USA, the SGMT:RO ratio for hindgut methanogens showed an opposite population structure pattern between the two sampled communities (Turnbull et al., 2011).
Dividing sequences between SGMT and RO clades can also help in uncovering differences in community structure between GIT samples that have a similar representation of Methanobrevibacter-related sequences. For instance, while they account for 93.0 and 85.8% of methanogens identified in sheep from Venezuela and in the hoatzin, Methanobrevibacter-related sequences have a completely opposite SGMT:RO distribution in these hosts. While additional studies are required to elucidate the respective contributions of host species genetics and environmental factors in the determination of whether the SGMT or the RO clade will be the most highly represented in a methanogen community, they may represent archaeal groups that thrive in different conditions. For instance, factors such as rumen or forestomach pH, tolerance to toxic compounds, and the rate of passage can act as selection agents, either individually or in combination, by promoting the growth of particular groups of methanogens, thereby affecting the population structure of the archaeal community (Janssen and Kirs, 2008). In this context, the natural division of Methanobrevibacter-like sequences into the SGMT and RO clades allows a higher level of specificity in developing population structure models for GIT methanogens that take into account phylogeny and representation, which can then be tested for methane production under controlled conditions in vivo or in vitro. This strategy could prove to be very valuable in the design of broad range mitigation strategies in the future.
Other Methanogen Groups Commonly Identified in the GIT of Herbivores
In addition to Methanobrevibacter-related methanogens, other archaeal phylogenetic groups have also been frequently reported in herbivore GIT samples. Indeed, members of the order Methanomassiliicoccales (Iino et al., 2013), a group of methanogens also referred to as rice cluster III (Kemnitz et al., 2005), rumen cluster C (Janssen and Kirs, 2008) or Methanoplasmatales (Paul et al., 2012), are also a prominent group of GIT methanogens. Not only are they frequently found in GIT samples from livestock animals, they have also been found to be a highly prevalent type of archaea in the rumen environment. This has been the case in wallabies sampled in November (91.7%; Evans et al., 2009), sheep from Australia (80.8%; Wright et al., 2006), yak from China (79.4%; Huang et al., 2012), Svalbard reindeer (47.4%; Sundset et al., 2009b), and in beef cattle fed either a potato (50.0%) or corn (46.1%) diet (Wright et al., 2007). Rumen methanogens from this taxonomic group have been reported to use methylamines as substrates for methanogenesis (Poulsen et al., 2013). Since compounds such as betaine and choline have been shown to be metabolized by rumen bacteria to produce methylamines (Bradbeer, 1965; Neill et al., 1978; Mitchell et al., 1979; Moller et al., 1986; Eklund et al., 2005), their presence in certain feedstuffs such as molasses and wheat derived products, or their use as feed additives, may favor the prevalence of Methanomassiliicoccales methanogens in a rumen environment. Paul et al. (2012) also reported that uncultured Methanomassiliicoccales methanogens could be enriched from the gut of higher termites when methanol was used as a substrate for methanogenesis.
Since they have been found to be highly prevalent in host species that can also have a high representation of Methanobrevibacter-related methanogens, this information is necessary to generate more comprehensive models for methanogen populations in the GIT of herbivores, such as perhaps be incorporated with the SGMT-RO model. Interestingly, sequences from specific habitats tend to be associated with certain clades (Paul et al., 2012; Seedorf et al., 2014). However, the limited number of isolates or representative 16S rRNA gene sequences that are available may not currently allow the same level of resolution that can be obtained with Methanobrevibacter-related sequences (Seedorf et al., 2014).
While they are in general less abundant than Methanobrevibacter-related or Methanomassiliicoccales sequences, 16S rRNA gene sequences that are more closely related to other methanogen species, such as Methanosphaera stadtmanae and Methanomicrobium mobile, or genera, such as Methanoculleus and Methanosarcina, have also been identified in the GIT of herbivores. While they are usually detected at a low frequency, they have in some studies been shown to be the most prevalent methanogens under certain conditions. For instance, from studies conducted in India, 94.4% of 16S rRNA gene sequences identified in the rumen of Murrah buffaloes were closely related to Methanomicrobium mobile (Chaudhary and Sirohi, 2009), and abundances of 97.1 and 72.3% of the same methanogen group were reported in Surti buffaloes (Singh et al., 2011, 2013). Furthermore, archaea belonging to the order Methanomicrobiales were predominant in the GIT of Japanese local ponies and thoroughbred horses (Lwin and Matsui, 2014). It remains to be determined why these methanogens were so prevalent in these particular conditions while they are usually detected at a much lower frequency. Methanosphaera stadtmanae was found to be the most prevalent methanogen in the hindgut of captive orangutans (Facey et al., 2012). This methanogen species has a limited substrate range for methane synthesis, and is notably unable to use H2 and CO2 for this purpose. Digestion of fruit pectin in frugivores like the orangutan has been hypothesized to increase GIT concentrations of methanol and acetate, which would provide a favorable environment for Methanosphaera stadtmanae methanogens to thrive. Finally, Methanocorpusculum labreanum was found to be the most abundant (59.9%) in the hindgut of captive white rhinoceroses (Luo et al., 2013). The identification and predominance of this type of methanogen in a GIT environment is unusual compared to most other reported studies. Predominance of Methanocorpusculum has also been reported in the fecal microbiota of Irish Thoroughbred racehorses (O’Donnell et al., 2013), but, as pointed out by the authors of that study, the use of the 16S rRNA gene V4 region may have underestimated archaeal diversity. Another study on equine fecal microbiota found that Methanocorpusculum-related methanogens were co-abundant with Methanobrevibacter-related methanogens (Fernandes et al., 2014). Methanocorpusculum archaea were observed at a median of 17.7% in horses fed a forage-grain diet, and at a median of 31.9% in horses maintained on pasture. They were only found to be more abundant than Methanobrevibacter-related methanogens in samples collected 4 days after a transition from a forage-grain diet to pasture had occurred.
Future Perspectives on GIT Methanogen Research in Herbivores
Sequencing of GIT Methanogen Genomes
Progress in biological research is often the result of technological advancements that improve experimental approaches. Numerous investigations of GIT methanogen communities to date have been performed using denaturing gradient gel electrophoresis (DGGE) analyses or Sanger sequencing of clone libraries, which both have intrinsic limitations in scope and resolution. However, next-generation sequencing has greatly improved the scope of microbial ecology studies, providing more comprehensive sequence datasets as well as allowing analysis of more independent samples and replicates (Denman and McSweeney, 2015).
While great strides have been made in characterizing the taxonomic composition of rumen and GIT methanogen communities, there remains a critical need to further our understanding of their metabolism and cellular physiology, particularly for species or candidate species that tend to be the most abundant. This knowledge would greatly contribute to the development of practical mitigation strategies. By revealing the biochemical potential of an organisms through prediction of its proteome, genome sequencing represents an effective strategy to elucidate the physiology of poorly characterized organisms. In terms of methane mitigation, it could for instance allow the identification of enzymes whose activity may be targeted with chemical antagonists, or surface proteins that may be used as antigens for the production of antibodies. Whether the devised strategies directly target methanogenesis, aim at reducing growth rates of methanogens or antagonize interactions with other microorganisms, they each have the potential to reduce enteric methane production.
Representative genomes of methanogens that have been identified in the GIT of livestock are currently limited in number. For instance, Methanobrevibacter ruminantium (Leahy et al., 2010) and Methanobrevibacter smithii (Samuel et al., 2007) are the only GIT Methanobrevibacter for which genomic data and predicted proteomes have been described in peer-reviewed publications. Permanent drafts for Methanobrevibacter boviskoreani and Methanobrevibacter wolinii are available, while efforts to complete the genomes of Methanobrevibacter millerae and Methanobrevibacter olleyae are ongoing. Both Methanobrevibacter gottschalkii and Methanobrevibacter thaurei have been selected to have their genome sequenced [see the Joint Genome Institute (JGI), Genomes Online Database (GOD)1]. As discussed in previous sections, these methanogens together represent the most common or abundant GIT archaea in livestock animals.
For GIT intestinal methanogens belonging to the order Methanomassiliicoccales, three genomes have so far been reported, all from isolates cultured from human feces: Methanomassiliicoccus luminyensis (Dridi et al., 2012), Methanomassiliicoccus alvus (Borrel et al., 2012), and Methanomassiliicoccus intestinalis (Borrel et al., 2013). Once sequence information from Methanomassiliicoccales representatives isolated from livestock become available, it will be of great interest to compare their genome with the human isolates. Available genomes of methanogens that are generally less well represented in GIT environments include species from the genera Methanosarcina (Deppenmeier et al., 2002; Galagan et al., 2002; Maeder et al., 2006), Methanosphaera (Fricke et al., 2006), Methanocorpusculum (Anderson et al., 2009), and Methanomicrobium mobile (see JGI-GOD1).
Analysis of the Methanobrevibacter ruminantium genome is a good example of the information that can be obtained from predicting the proteome of a methanogen (Leahy et al., 2010). For instance, it revealed the ability to use formate in addition to H2 as a substrate for methanogenesis, showed that this organism is unable to synthesize coenzyme M, and provided a metabolic explanation for the requirement of acetate for growth. It also uncovered a large array of genes encoding putative adhesins, and identified loci related to phage genes. In addition, this genomic information can also be used as a reference for metagenomics and metatranscriptomics analyses in GIT environments.
While this technology is providing an unprecedented capacity for genome sequencing, as attested by the increasing number of published microbial genomes, the complete and accurate determination of a prokaryotic genome is not a trivial undertaking and requires research teams adept in technical and bioinformatic skills. In addition, an important limitation in this process is the isolation and cultivation of methanogens, which remain a challenge for many strains. Therefore, genomes to sequence should be strategically selected considering the wide diversity of methanogens that populate the GIT of herbivores. In this context, population structure studies such as summarized in this review that are based on representation and phylogeny provide a critical basis in the selection of methanogens of interest.
In the long term, providing an increased number of available GIT methanogens genomes is essential for the development of effective and comprehensive mitigation strategies. Since the use of entire genome sequences dramatically improves phylogenetic analysis of archaea compared to only using 16S rRNA gene sequences (Brochier-Armanet et al., 2011), this will allow the accurate identification of phylogenetic nodes that are shared by clusters of GIT methanogens, which can be targeted for mitigation. In addition, comparative genome analyses will reveal conserved proteins within phylogenic clusters of methanogens, such as surface molecules that can be targeted by vaccination or intracellular factors that can be targeted for chemical inhibition. Alternatively, metatranscriptomics can also be used to identify mitigation targets. For instance, Shi et al. (2014) recently reported that transcription of methanogenesis pathway genes was elevated in sheep with high methane emissions.
Culture-Based Investigations of GIT Methanogen Microbiology
As highlighted in the previous section, the available community compositions from gut methanogens in herbivores has revealed that, while there can be some overlap between samples, each so far appears to be unique. By mechanisms that are currently unknown, certain methanogens can be prevalent under particular conditions (e.g., host breed, species, diet, or geographical location), while they are detected at a lower frequency in other cases. In order to gain further insight, there needs to be an increase in culture-based microbiological studies of GIT methanogens, which are better suited for mechanistic studies that require a controlled environment. Due to the limited number of GIT methanogen species that have successfully been isolated and grown in vitro (Creevey et al., 2014), direct culturing of GIT samples represents an attractive alternative which would yield valuable insights not only about methanogens, but also of their interactions with other members of the community. The importance of such investigations can be emphasized by reports such as by Popova et al. (2013), where differences in methane production capacity were found between rumen and cecal contents from lambs fed high grain content diets, despite Methanobrevibacter-related methanogens being the most abundant archaea in both environments.
Investigation of Intra-Community Interactions Involving Methanogens
The complexity of GIT microbial communities in herbivores is not simply due to their high cellular density and diversity, but is also a result of intricate networks of inter-species trophic relationships. Methanogens depend on other microorganisms for substrates such as H2 and CO2 to sustain their energy needs through anaerobic respiration and methane synthesis. While methanogens can acquire substrates from their surrounding environment, some can associate intimately with protozoa or fungi. For instance, it was reported that the free-living (FL) and protozoa-associated methanogen (PAM) populations were composed of the same major groups (Methanobrevibacter and Methanomicrobium), but that their composition differed between FL and PAM (Tymensen et al., 2012). In addition, the distribution of species-level OTUs within the same subgroups was found to differ as well. A study by Belanche et al. (2014) also reported that PAMs represented a more variable population than FL methanogens. If such interactions contribute to greater methane production, then their disruption could potentially be used as a mitigation strategy. It will also be of interest to investigate the degree of specificity between partner species that is required for these cell–cell interactions to occur.
The potential of specific trophic relationships between methanogens and bacteria should also be further explored. In studies conducted in sheep as a model ruminant (Morgavi et al., 2012), it was reported that the liquid-associated bacterial and methanogens fraction of animals kept without protozoa for more than 2 years produced more methane than the corresponding rumen fractions from faunated animals or animals defaunated for only a few months. Accordingly, the same study found that animals maintained without protozoa for more than 2 years were higher methane emitters than animals that had been defaunated for a few months.
Concluding Remarks
While great strides have been made in the study of rumen methanogen populations in a variety of hosts and environmental conditions, further investigations still are required in order to gain sufficient insight to develop comprehensive methane mitigation strategies targeting methanogens. It will only be through a sustained effort in combining genomics and cellular analyses that this goal may be reached in the near future.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Allers, T., and Mevarech, M. (2005). Archaeal genetics – the third way. Nat. Rev. Genet. 6, 58–73. doi: 10.1038/nrg1504
An, D., Dong, X., and Dong, Z. (2005). Prokaryote diversity in the rumen of yak (Bos grunniens) and Jinnan cattle (Bos taurus) estimated by 16S rDNA homology analyses. Anaerobe 11, 207–215. doi: 10.1016/j.anaerobe.2005.02.001
Anderson, I. J., Sieprawska-Lupa, M., Goltsman, E., Lapidus, A., Copeland, A., Glavina Del Rio, T., et al. (2009). Complete genome sequence of Methanocorpusculum labreanum type strain Z. Stand. Genomic Sci. 1, 197–203. doi: 10.4056/sigs.35575
Beauchemin, K. A., McAllister, T. A., and McGinn, S. M. (2009). Dietary mitigation of enteric methane from cattle. CAB Rev. 4, 1–18. doi: 10.1079/PAVSNNR20094035
Beauchemin, K. A., and McGinn, S. M. (2005). Methane emissions from feedlot cattle fed barley or corn diets. J. Anim. Sci. 83, 653–661.
Beauchemin, K. A., McGinn, S. M., Martinez, T. F., and McAllister, T. A. (2007). Use of condensed tannin extract from quebracho trees to reduce methane emissions from cattle. J. Anim. Sci. 85, 1990–1996. doi: 10.2527/jas.2006-686
Belanche, A., De La Fuente, G., and Newbold, C. J. (2014). Study of methanogen communities associated with different rumen protozoal populations. FEMS Microbiol. Ecol. 90, 663–677. doi: 10.1111/1574-6941.12423
Borrel, G., Harris, H. M., Parisot, N., Gaci, N., Tottey, W., Mihajlovski, A., et al. (2013). Genome Sequence of “Candidatus Methanomassiliicoccus intestinalis” Issoire-Mx1, a third Thermoplasmatales-related methanogenic archaeon from human feces. Genome Announc. 1, e00453-13. doi: 10.1128/genomeA.00453-13
Borrel, G., Harris, H. M. B., Tottey, W., Mihajlovski, A., Parisot, N., Peyretaillade, E., et al. (2012). Genome sequence of “Candidatus Methanomethylophilus alvus” Mx1201, a methanogenic archaea from the human gut belonging to a seventh order of methanogens. J. Bacteriol. 194, 6944–6945. doi: 10.1128/JB.01867-12
Bradbeer, C. (1965). The clostridial fermentations of choline and ethanolamine. J. Biol. Chem. 240, 4669–4674.
Brochier-Armanet, C., Boussau, B., Gribaldo, S., and Forterre, P. (2008). Mesophilic Crenarchaeota: proposal for a third archaeal phylum, the Thaumarchaeota. Nat. Rev. Microbiol. 6, 245–252. doi: 10.1038/nrmicro1852
Brochier-Armanet, C., Forterre, P., and Gribaldo, S. (2011). Phylogeny and evolution of the Archaea: one hundred genomes later. Curr. Opin. Microbiol. 14, 274–281. doi: 10.1016/j.mib.2011.04.015
Carulla, J. E., Kreuzer, M., Machmüller, A., and Hess, H. D. (2005). Supplementation of Acacia mearnsii tannins decreases methanogenesis and uninary nitrogen in forage-feed sheep. Aust. J. Agr. Res. 56, 961–970. doi: 10.1071/AR05022
Cersosimo, L. M., Lachance, H., St-Pierre, B., Van Hoven, W., and Wright, A.-D. G. (2015). Examination of the rumen bacteria and methanogenic archaea of wild impalas (Aepyceros melampus melampus) from Pongola, South Africa. Microb. Ecol. 69, 577–585. doi: 10.1007/s00248-014-0521-3
Chaudhary, P. P., and Sirohi, S. K. (2009). Dominance of Methanomicrobium phylotype in methanogen population present in Murrah buffaloes (Bubalus bubalis). Lett. Appl. Microbiol. 49, 274–277. doi: 10.1111/j.1472-765X.2009.02654.x
Creevey, C. J., Kelly, W. J., Henderson, G., and Leahy, S. C. (2014). Determining the culturability of the rumen bacterial microbiome. Microb. Biotechnol. 7, 467–479. doi: 10.1111/1751-7915.12141
Cunha, I. S., Barreto, C. C., Costa, O. Y. A., Bomfim, M. A., Castro, A. P., Kruger, R. H., et al. (2011). Bacteria and archaea community structure in the rumen microbiome of goats (Capra hircus) from the semiarid region of Brazil. Anaerobe 17, 118–124. doi: 10.1016/j.anaerobe.2011.04.018
Denman, S. E., and McSweeney, C. S. (2015). The early impact of genomics and metagenomics on ruminal microbiology. Annu. Rev. Anim. Biosci. 3, 9.1–9.19. doi: 10.1146/annurev-animal-022114-110705
Deppenmeier, U., Johann, A., Hartsch, T., Merkl, R., Schmitz, R. A., Martinez-Arias, R., et al. (2002). The genome of Methanosarcina mazei: evidence for lateral gene transfer between bacteria and archaea. J. Mol. Microbiol. Biotechnol. 4, 453–461.
Dittmann, M. T., Runge, U., Lang, R. A., Moser, D., Galeffi, C., Kreuzer, M., et al. (2014). Methane emissions by camelids. PLoS ONE 9:e94363. doi: 10.1371/journal.pone.0094363
Dridi, B., Fardeau, M. L., Ollivier, B., Raoult, D., and Drancourt, M. (2012). Methanomassiliicoccus luminyensis gen. nov., sp. nov., a methanogenic archaeon isolated from human faeces. Int. J. Syst. Evol. Microbiol. 62, 1902–1907. doi: 10.1099/ijs.0.033712-0
Eklund, M., Bauer, E., Wamatu, J., and Mosenthin, R. (2005). Potential nutritional and physiological functions of betaine in livestock. Nutr. Res. Rev. 18, 31–48. doi: 10.1079/NRR200493
Evans, P. N., Hinds, L. A., Sly, L. I., McSweeney, C. S., Morrison, M., and Wright, A.-D. G. (2009). Community composition and density of methanogens in the foregut of the Tammar wallaby (Macropus eugenii). Appl. Environ. Microbiol. 75, 2598–2602. doi: 10.1128/AEM.02436-08
Facey, H. V., Northwood, K. S., and Wright, A.-D. G. (2012). Molecular diversity of methanogens in fecal samples from zoo captive Sumatran orangutans (Pongo abelii). Am. J. Primat. 74, 408–413. doi: 10.1002/ajp.21992
Fernandes, K. A., Kittelmann, S., Rogers, C. W., Gee, E. K., Bolwell, C. F., Bermingham, E. N., et al. (2014). Faecal microbiota of forage-fed horses in New Zealand and the population dynamics of microbial communities following dietary change. PLoS ONE 9:e112846. doi: 10.1371/journal.pone.0112846
Franz, R., Soliva, C. R., Kreuzer, M., Steuer, P., Hummel, J., and Clauss, M. (2010). Methane production in relation to body mass of ruminants and equids. Evol. Ecol. Res. 12, 727–738.
Franzolin, R., St-Pierre, B., Northwood, K., and Wright, A.-D. G. (2012). Analysis of rumen methanogen diversity in water buffaloes (Bubalus bubalis) under three different diets. Microb. Ecol. 64, 131–139. doi: 10.1007/s00248-012-0007-0
Fricke, W. F., Seedorf, H., Henne, A., Krüer, M., Liesegang, H., Hedderich, R., et al. (2006). The genome sequence of Methanosphaera stadtmanae reveals why this human intestinal archaeon is restricted to methanol and H2 for methane formation and ATP synthesis. J. Bacteriol. 188, 642–658. doi: 10.1128/JB.188.2.642-658.2006
Galagan, J. E., Nusbaum, C., Roy, A., Endrizzi, M. G., Macdonald, P., FitzHugh, W., et al. (2002). The genome of M. acetivorans reveals extensive metabolic and physiological diversity. Genome Res. 12, 532–542. doi: 10.1101/gr.223902
Grainger, C., Clarke, T., Auldist, M. J., Beauchemin, K. A., McGinn, S. M., Waghorn, G. C., et al. (2009). Mitigation of greenhouse gas emissions from dairy cows fed pasture and grain through supplementation with Acacia mearnsii tannins. Can. J. Anim. Sci. 89, 241–251. doi: 10.4141/CJAS08110
Grainger, C., Williams, R., Clarke, T., Wright, A.-D. G., and Eckard, R. J. (2010). Supplementation with whole cottonseed causes long-term reduction of methane emissions from lactating dairy cows offered a forage and cereal grain diet. J. Dairy Sci. 93, 2612–2619. doi: 10.3168/jds.2009-2888
Hook, S. E., Northwood, K. S., Wright, A.-D. G., and McBride, B. W. (2009). Long-term monensin supplementation does not significantly affect the quantity or diversity of methanogens in the rumen of the lactating dairy cow. Appl. Environ. Microbiol. 75, 374–380. doi: 10.1128/AEM.01672-08
Hook, S. E., Steele, M. A., Northwood, K. S., Wright, A.-D. G., and McBride, B. W. (2011). Impact of high-concentrate feeding and low ruminal pH on methanogens and protozoa in the rumen of dairy cows. Microb. Ecol. 62, 94–105. doi: 10.1007/s00248-011-9881-0
Huang, X. D., Tan, H. Y., Long, R., Liang, J. B., and Wright, A.-D. G. (2012). Comparison of methanogen diversity of yak (Bos grunniens) and cattle (Bos taurus) from the Qinghai-Tibetan plateau, China. BMC Microbiol. 12:237. doi: 10.1186/1471-2180-12-237
Iino, T., Tamaki, H., Tamazawa, S., Ueno, Y., Ohkuma, M., Suzuki, K., et al. (2013). Candidatus Methanogranum caenicola: a novel methanogen from the anaerobic digested sludge, and proposal of Methanomassiliicoccaceae fam. nov. and Methanomassiliicoccales ord. nov., for a methanogenic lineage of the class Thermoplasmata. Microbes Environ. 28, 244–250. doi: 10.1264/jsme2.ME12189
Janssen, P. H., and Kirs, M. (2008). Structure of the archaeal community of the rumen. Appl. Environ. Microbiol. 74, 3619–3625. doi: 10.1128/AEM.02812-07
Johnson, K. A., and Johnson, D. E. (1995). Methane emissions from cattle. J. Anim. Sci. 73, 2483–2492.
Kemnitz, D., Kolb, S., and Conrad, R. (2005). Phenotypic characterization of rice cluster III archaea without prior isolation by applying quantitative polymerase chain reaction to an enrichment culture. Environ. Microbiol. 7, 553–565. doi: 10.1111/j.1462-2920.2005.00723.x
Kempton, T. J., Murray, R. M., and Leng, R. A. (1976). Methane production and digestibility measurements in the grey kangaroo and sheep. Aust. J. Biol. Sci. 29, 209–214.
Kim, M., Morrison, M., and Yu, Z. (2011). Status of the phylogenetic diversity census of ruminal microbiomes. FEMS Microbiol. Ecol. 76, 49–63. doi: 10.1111/j.1574-6941.2010.01029.x
King, E. E., Smith, R. P., St-Pierre, B., and Wright, A.-D. G. (2011). Differences in the rumen methanogen populations of lactating Jersey and Holstein dairy cows under the same diet regimen. Appl. Environ. Microbiol. 77, 5682–5687. doi: 10.1128/AEM.05130-11
Lashof, D. A., and Ahuja, D. (1990). Relative contributions of greenhouse gas emissions to global warming. Nature 344, 529–531. doi: 10.1038/344529a0
Leahy, S. C., Kelly, W. J., Altermann, E., Ronimus, R. S., Yeoman, C. J., Pacheco, D. M., et al. (2010). The genome sequence of the rumen methanogen Methanobrevibacter ruminantium reveals new possibilities for controlling ruminant methane emissions. PLoS ONE 5:e8926. doi: 10.1371/journal.pone.0008926
Lee, J. H., Kumar, S., Lee, G. H., Chang, D. H., Rhee, M. S., Yoon, M. H., et al. (2013). Methanobrevibacter boviskoreani sp. nov., isolated from the rumen of Korean native cattle. Int. J. Syst. Evol. Microbiol. 63, 4196–4201. doi: 10.1099/ijs.0.054056-0
Li, Z., Zhang, Z., Xu, C., Zhao, J., Liu, H., Fan, Z., et al. (2014). Bacteria and methanogens differ along the gastrointestinal tract of Chinese roe deer (Capreolus pygargus). PLoS ONE 9:e114513. doi: 10.1371/journal.pone.0114513
Liu, Y., and Whitman, W. B. (2008). Metabolic, phylogenetic, and ecological diversity of the methanogenic archaea. Ann. N. Y. Acad. Sci. 1125, 171–189. doi: 10.1196/annals.1419.019
Lovett, D., Lovell, S., Stack, L., Callan, J., Finlay, M., Conolly, J., et al. (2003). Effect of forage/concentrate ratio and dietary coconut oil level on methane output and performance of finishing beef heifers. Livest. Prod. Sci. 84, 135–146. doi: 10.1016/j.livprodsci.2003.09.010
Luo, Y. H., Wright, A.-D. G., Li, Y. L., Li, H., Yang, Q. H., Luo, L. J., et al. (2013). Diversity of methanogens in the hindgut of captive white rhinoceroses, Ceratotherium simum. BMC Microbiol. 13:207. doi: 10.1186/1471-2180-13-207
Lwin, K. O., and Matsui, H. (2014). Comparative analysis of the methanogen diversity in horse and pony by using mcrA gene and archaeal 16S rRNA gene clone libraries. Archaea 2014, 1–11. doi: 10.1155/2014/483574
Maeder, D. L., Anderson, I., Brettin, T. S., Bruce, D. C., Gilna, P., Han, C. S., et al. (2006). The Methanosarcina barkeri genome: comparative analysis with Methanosarcina acetivorans and Methanosarcina mazei reveals extensive rearrangement within methanosarcinal genomes. J. Bacteriol. 188, 7922–7931. doi: 10.1128/JB.00810-06
McAllister, T. A., Okine, E. K., Mathison, G. W., and Cheng, K.-J. (1996). Dietary, environmental and microbiological aspects of methane production in ruminants. Can. J. Anim. Sci. 76, 231–243. doi: 10.4141/cjas96-035
Mitchell, A. D., Chappell, A., and Knox, K. L. (1979). Metabolism of betaine in the ruminant. J. Anim. Sci. 49, 764–774.
Moller, B., Hippe, H., and Gottschalk, G. (1986). Degradation of various amine compounds by mesophilic clostridia. Arch. Microbiol. 145, 85–90. doi: 10.1007/BF00413032
Morgavi, D. P., Martin, C., Jouany, J. P., and Ranilla, M. J. (2012). Rumen protozoa and methanogenesis: not a simple cause-effect relationship. Br. J. Nutr. 107, 388–397. doi: 10.1017/S0007114511002935
Neill, A. R., Grime, D. W., and Dawson, R. M. C. (1978). Conversion of choline methyl groups through trimethylamine into methane in the rumen. Biochem. J. 170, 529–535.
Nunoura, T., Takaki, Y., Kakuta, J., Nishi, S., Sugahara, J., Kazama, H., et al. (2011). Insights into the evolution of Archaea and eukaryotic protein modifier systems revealed by the genome of a novel archaeal group. Nucleic Acids Res. 39, 3204–3223. doi: 10.1093/nar/gkq1228
O’Donnell, M. M., Harris, H. M. B., Jeffery, I. B., Claesson, M. J., Younge, B., O’ Toole, P. W., et al. (2013). The core faecal bacterial microbiome of Irish Thoroughbred racehorses. Lett. Appl. Microbiol. 57, 492–501. doi: 10.1111/lam.12137
Paul, K., Nonoh, J. O., Mikulski, L., and Brune, A. (2012). “Methanoplasmatales,” Thermoplasmatales-related archaea in termite guts and other environments, are the seventh order of methanogens. Appl. Environ. Microbiol. 78, 8245–8253. doi: 10.1128/AEM.02193-12
Petitjean, C., Deschamps, P., López-García, P., and Moreira, D. (2014). Rooting the domain archaea by phylogenomic analysis supports the foundation of the new kingdom Proteoarchaeota. Genome Biol. Evol. 7, 191–204. doi: 10.1093/gbe/evu274
Pinares-Patino, C. S., Ulyatt, M. J., Waghorn, G. C., Lassey, K. R., Barry, T. N., Holmes, C. W., et al. (2003). Methane emission by alpaca and sheep fed on lucerne hay or grazed on pastures of perennial ryegrass/white clover or birdsfoot trefoil. J. Agric. Sci. 140, 215–226. doi: 10.1017/S002185960300306X
Popova, M., Morgavi, D. P., and Martin, C. (2013). Methanogens and methanogenesis in the rumens and ceca of lambs fed two different high-grain-content diets. Appl. Environ. Microbiol. 79, 1777–1786. doi: 10.1128/AEM.03115-12
Poulsen, M., Schwab, C., Jensen, B. B., Engberg, R. M., Spang, A., Canibe, N., et al. (2013). Methylotrophic methanogenic Thermoplasmata implicated in reduced methane emissions from bovine rumen. Nat. Commun. 4:1428. doi: 10.1038/ncomms2432
Puchala, R., Min, B. R., Goetsch, A. L., and Sahlu, T. (2005). The effect of a condensed tannin containing forage on methane emission by goats. J. Anim. Sci. 83, 182–186.
Rajendhran, J., and Gunasekaran, P. (2011). Microbial phylogeny and diversity: small subunit ribosomal RNA sequence analysis and beyond. Microbiol. Res. 166, 99–110. doi: 10.1016/j.micres.2010.02.003
Raymann, K., Brochier-Armanet, C., and Gribaldo, S. (2015). The two-domain tree of life is linked to a new root for the Archaea. Proc. Natl. Acad. Sci. U.S.A. 112, 6670–6675. doi: 10.1073/pnas.1420858112
Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N. N., Anderson, I. J., Cheng, J. F., et al. (2013). Insights into the phylogeny and coding potential of microbial dark matter. Nature 499, 431–437. doi: 10.1038/nature12352
Sakai, S., Imachi, H., Hanada, S., Ohashi, A., Harada, H., and Kamagata, Y. (2008). Methanocella paludicola gen. nov., sp. nov., a methane-producing archaeon, the first isolate of the lineage ‘Rice Cluster I’, and proposal of the new archaeal order Methanocellales ord. nov. Int. J. Syst. Evol. Microbiol. 58, 929–936. doi: 10.1099/ijs.0.65571-0
Samuel, B. S., Hansen, E. E., Manchester, J. K., Coutinho, P. M., Henrissat, B., Fulton, R., et al. (2007). Genomic and metabolic adaptations of Methanobrevibacter smithii to the human gut. Proc. Natl. Acad. Sci. U.S.A. 104, 10643–10648. doi: 10.1073/pnas.0704189104
Seedorf, H., Kittelmann, S., Henderson, G., and Janssen, P. H. (2014). RIM-DB: a taxonomic framework for community structure analysis of methanogenic archaea from the rumen and other intestinal environments. PeerJ 2:e494. doi: 10.7717/peerj.494
Seedorf, H., Kittelmann, S., and Janssen P. H. (2015). Few highly abundant operational taxonomic units dominate within rumen methanogenic archaeal species in New Zealand sheep and cattle. Appl. Environ. Microbiol. 81, 986–995. doi: 10.1128/AEM.03018-14
Shi, W., Moon, C. D., Leahy, S. C., Kang, D., Froula, J., Kittelmann, S., et al. (2014). Methane yield phenotypes linked to differential gene expression in the sheep rumen microbiome. Genome Res. 24, 1517–1525. doi: 10.1101/gr.168245.113
Shin, E. C., Choi, B. R., Lim, W. J., Hong, S. Y., An, C. L., Cho, K. M., et al. (2004). Phylogenetic analysis of archaea in three fractions of cow rumen based on the 16S rDNA sequence. Anaerobe 10, 313–319. doi: 10.1016/j.anaerobe.2004.08.002
Singh, K. M., Patel, A. K., Shah, R. K., Reddy, B., and Joshi, C. G. (2015). Potential functional gene diversity involved in methanogenesis and methanogenic community structure in Indian buffalo (Bubalus bubalis) rumen. J. Appl. Genet. doi: 10.1007/s13353-015-0270-0 [Epub ahead ofprint].
Singh, K. M., Tripathi, A. K., Pandya, P. R., Parnerkar, S., Kothari, R. K., and Joshi, C. G. (2013). Molecular genetic diversity and quantification of methanogen in ruminal fluid of Buffalo (Bubalus bubalis) fed ration (wheat straw and concentrate mix). Genet. Res. Int. 2013, 1–7. doi: 10.1155/2013/980191
Singh, K. M., Tripathi, A. K., Pandya, P. R., Parnerkar, S., Rank, D. N., Kothari, R. K., et al. (2011). Methanogen diversity in the rumen of Indian Surti buffalo (Bubalus bubalis), assessed by 16S rDNA analysis. Res. Vet. Sci. 92, 451–455. doi: 10.1016/j.rvsc.2011.03.022
Skillman, L. C., Evans, P. N., Strompl, C., and Joblin, K. N. (2006). 16S rDNA directed PCR primers and detection of methanogens in the bovine rumen. Lett. Appl. Microbiol. 42, 222–228. doi: 10.1111/j.1472-765X.2005.01833.x
Snelling, T. J., Genç, B., McKain, N., Watson, M., Waters, S. M., Creevey, C. J., et al. (2014). Diversity and community composition of methanogenic archaea in the rumen of Scottish upland sheep assessed by different methods. PLoS ONE 9:e106491. doi: 10.1371/journal.pone.0106491
St-Pierre, B., and Wright, A.-D. G. (2012). Molecular analysis of methanogenic archaea in the forestomach of the alpaca (Vicugna pacos). BMC Microbiol. 12:1. doi: 10.1186/1471-2180-12-1
Sundset, M. A., Edwards, J. E., Cheng, Y. F., Senosiain, R. S., Fraile, M. N., Northwood, K. S., et al. (2009a). Molecular diversity of the rumen microbiome of Norwegian reindeer on natural summer pasture. Microb. Ecol. 57, 335–348. doi: 10.1007/s00248-008-9414-7
Sundset, M. A., Edwards, J. E., Cheng, Y. F., Senosiain, R. S., Fraile, M. N., Northwood, K. S., et al. (2009b). Rumen microbial diversity in Svalbard reindeer, with particular emphasis on methanogenic archaea. FEMS Microbiol. Ecol. 70, 553–562. doi: 10.1111/j.1574-6941.2009.00750.x
Thauer, R. K., Kaster, A.-K., Seedorf, H., Buckel, W., and Hedderich, R. (2008). Methanogenic archaea: ecologically relevant differences in energy conservation. Nat. Rev. Microbiol. 6, 579–591. doi: 10.1038/nrmicro1931
Thorpe, A. (2008). Enteric fermentation and ruminant eructation: the role (and control?) of methane in the climate change debate. Climatic Change 93, 407–431. doi: 10.1007/s10584-008-9506-x
Turnbull, K. L., Smith, R. P., St-Pierre, B., and Wright, A.-D. G. (2011). Molecular diversity of methanogens in fecal samples from Bactrian camels (Camelus bactrianus) at two zoos. Res. Vet. Sci. 93, 246–249. doi: 10.1016/j.rvsc.2011.08.013
Tymensen, L. D., Beauchemin, K. A., and Mcallister, T. A. (2012). Structures of free-living and protozoa-associated methanogen communities in the bovine rumen differ according to comparative analysis of 16S rRNA and mcrA genes. Microbiology 158, 1808–1817. doi: 10.1099/mic.0.057984-0
Whitford, M. F., Teather, R. M., and Forster, R. J. (2001). Phylogenetic analysis of methanogens from the bovine rumen. BMC Microbiol. 1:1–5. doi: 10.1186/1471-2180-1-5
Williams, Y. J., Popovski, S., Rea, S. M., Skillman, L. C., Toovey, A. F., Northwood, K. S., et al. (2009). A vaccine against rumen methanogens can alter the composition of archaeal populations. Appl. Environ. Microbiol. 75, 1860–1866. doi: 10.1128/AEM.02453-08
Wolin, M. J. (1979). “The rumen fermentation: a model for microbial interactions in anaerobic ecosystems,” in Advances in Microbial Ecology, Vol. 3, ed. M. Alexander (New York: Planum Press), 49–77.
Wolin, M. J. (1982). “Hydrogen transfer in microbial communities,” in Microbial Interactions and Communities, Vol. 1, eds A. T. Bull and J. H. Slater (London: Academic Press), 323–356.
Woodward, S. L., Waghorn, G. C., and Laboyrie, P. (2004). Condensed tannins in birdsfoot trefoil (Lotus corniculatus) reduced methane emission from dairy cows. Proc. N. Z. Soc. Anim. Prod. 64, 160–164.
Wright, A.-D. G., Auckland, C. H., and Lynn, D. H. (2007). Molecular diversity of methanogens in feedlot cattle from Ontario and Prince Edward Island, Canada. Appl. Environ. Microbiol. 73, 4206–4210. doi: 10.1128/AEM.00103-07
Wright, A.-D. G., Kennedy, P., O’Neill, C. J., Toovey, A. F., Popovski, S., Rea, S. M., et al. (2004). Reducing methane emissions in sheep by immunization against rumen methanogens. Vaccine 22, 3976–3985. doi: 10.1016/j.vaccine.2004.03.053
Wright, A.-D. G., Ma, X., and Obispo, N. E. (2008). Methanobrevibacter phylotypes are the dominant methanogens in sheep from Venezuela. Microb. Ecol. 56, 390–394. doi: 10.1007/s00248-007-9351-x
Wright, A.-D. G., Northwood, K. S., and Obispo, N. E. (2009). Rumen-like methanogens identified from the crop of the folivorous South American bird, the hoatzin (Opisthocomus hoazin). ISME 3, 1120–1126. doi: 10.1038/ismej.2009.41
Keywords: methanogens, 16S rRNA analysis, herbivores, rumen microbiology, methane mitigation
Citation: St-Pierre B, Cersosimo LM, Ishaq SL and Wright A-DG (2015) Toward the identification of methanogenic archaeal groups as targets of methane mitigation in livestock animals. Front. Microbiol. 6:776. doi: 10.3389/fmicb.2015.00776
Received: 07 May 2015; Accepted: 14 July 2015;
Published: 30 July 2015.
Edited by:
Emilio M. Ungerfeld, Instituto de Investigaciones Agropecuarias – Carillanca, ChileReviewed by:
Henning Seedorf, AgResearch Ltd., New ZealandGunjan Goel, Jaypee University of Information Technology, India
Copyright © 2015 St-Pierre, Cersosimo, Ishaq and Wright. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: André-Denis G. Wright, School of Animal and Comparative Biomedical Sciences, University of Arizona, 1117 E. Lowell Street, Tucson, AZ 85721, USA,YWR3cmlnaHRAZW1haWwuYXJpem9uYS5lZHU=