Abstract
Helicobacter pylori is a Gram-negative spiral-shaped bacterium that colonizes over half of the world's population. Chronic H. pylori infection is associated with increased risk for numerous disease outcomes including gastritis, dysplasia, neoplasia, B-cell lymphoma of mucosal-associated lymphoid tissue (MALT lymphoma), and invasive adenocarcinoma. The complex interactions that occur between pathogen and host are dynamic and exquisitely regulated, and the relationship between H. pylori and its human host are no exception. To successfully colonize, and subsequently persist, within the human stomach H. pylori must temporally regulate numerous genes to ensure localization to the gastric lumen and coordinated expression of virulence factors to subvert the host's innate and adaptive immune response. H. pylori achieves this precise gene regulation by sensing subtle environmental changes including host-mediated alterations in nutrient availability and responding with dramatic global changes in gene expression. Recent studies revealed that the presence or absence of numerous metal ions encountered in the lumen of the stomach, or within host tissues, including nickel, iron, copper and zinc, can influence regulatory networks to alter gene expression in H. pylori. These expression changes modulate the deployment of bacterial virulence factors that can ultimately influence disease outcome. In this review we will discuss the environmental stimuli that are detected by H. pylori as well as the trans regulatory elements, specifically the transcription regulators and transcription factors, that allow for these significant transcriptional shifts.
Introduction
Helicobacter pylori colonizes over half of the world's population making it arguably the most successful bacterial pathogen (Atherton and Blaser, 2009). H. pylori is uniquely adapted to colonize the human stomach and is the dominant microorganism within the gastric niche (Bik et al., 2006; Cover and Blaser, 2009). Nearly all individuals that are persistently colonized with H. pylori will experience chronic gastritis, however, in a subset of individuals, H. pylori infection results in more severe disease outcomes including peptic and duodenal ulcers, neoplasia, dysplasia, B-cell lymphoma of mucosal-associated lymphoid tissue (MALT lymphoma), and invasive gastric adenocarcinoma (Cover and Blaser, 2009). For successful colonization of the host pathogenic bacteria must sense subtle changes in their environment, and rapidly respond with alterations in their transcriptional profile and H. pylori is no exception to this paradigm (Sharma et al., 2010). These environmental changes include the low pH characteristic of the gastric niche, alterations in nutrient availability including divalent cations, fluctuations in osmolarity, and the presence of the human immune system (Algood and Cover, 2006; de Bernard and Josenhans, 2014; Haley and Gaddy, 2015).
Transition metals participate in a multitude of critical biological processes, and are incorporated into numerous metalloproteins; making them a vital nutrient for all living organisms (Foster et al., 2014). The human host exploits this requirement for metals by restricting bacterial access to them in a dynamic process termed nutritional immunity (Hood and Skaar, 2012; Diaz-Ochoa et al., 2014). It is postulated that access to these nutrients within the gastric niche is incredibly variable, and therefore H. pylori likely experiences periodic abundances of critical metals followed by episodes of extreme depletion mediated by alterations in environmental pH, which can influence metal solubility, and expression of host derived metal-binding proteins such as the calgranulin proteins (calprotectin and S100A12) and iron-binding proteins like lactoferrin (Belzer et al., 2007; Senkovich et al., 2010; Gaddy et al., 2014; Haley et al., 2015). Much of the infectious potential of H. pylori is dependent on detecting fluctuations in metal availability and external ion concentrations, and responding with the expression of virulence factors and metal acquisitions systems (Danielli and Scarlato, 2010). For example, the cytotoxin-associated gene A (CagA) and the vacuolating cytotoxin (VacA) are both implicated in perturbing host cell iron trafficking, and both of these toxins are critical for host persistence (Salama et al., 2000; Tan et al., 2011). Detecting alterations in environmental metal quantities allows H. pylori to appropriately respond to changes in the host environment while simultaneously ensuring that its repertoire of metalloenzymes, which are required for essential processes within the cell, remain adequately metallated (Ge et al., 2013; Diaz-Ochoa et al., 2014). Many prokaryotes sense and respond to metals via two-component system (TCS) and signal-transduction networks (Silver and Walderhaug, 1992; Groisman, 2001; Eguchi and Utsumi, 2008). The typical H. pylori genome encodes remarkably few TCS, indicating metal sensing and response is likely done in novel ways (Panthel et al., 2003). Furthermore, it is likely that there are redundant and overlapping mechanisms that govern these regulons (Table 1). In this review we will highlight the complex regulatory networks, mediated through trans-regulatory elements including transcriptional regulators (TRs) and transcription factors, utilized by H. pylori to sense and react to subtle changes in extracellular metal concentrations within the human stomach.
Table 1
| Gene | Metal associated with regulation | Regulation mechanism | References |
|---|---|---|---|
| fecA1 | Iron | Fe-Fur (repression) | Ernst et al., 2005a; Danielli et al., 2006, 2009; Pich et al., 2012 |
| fecA2 | Iron | Fe-Fur (repression) | Fassbinder et al., 2000; Ernst et al., 2005a; Danielli et al., 2006, 2009; Pich et al., 2012 |
| frpB1 | Iron | Fe-Fur (repression) | Delany et al., 2001; Ernst et al., 2005a; Danielli et al., 2006; Pich et al., 2012 |
| feoB | Iron | Fe-Fur (repression) | Ernst et al., 2005a |
| flaB | Iron | Fe-Fur (activation) | Danielli et al., 2006 |
| fliY | Iron | Fe-Fur (activation) | Danielli et al., 2006 |
| flgK | Iron | Fe-Fur (activation) | Danielli et al., 2006 |
| cheA | Iron | Fe-Fur (activation) | Danielli et al., 2006 |
| exbBD-tonB | Iron, Nickel, Copper | Fe-Fur (repression), Ni-NikR (repression), Copper (induction) | Contreras et al., 2003; Danielli et al., 2006 |
| frpB4 | Iron, Nickel | Fe-Fur (repression), Nik-R (repression) | Danielli et al., 2006; Ernst et al., 2006; Romagnoli et al., 2011 |
| frpB2 | Iron, Nickel | Fe-Fur (repression), Ni-NikR (repression) | Danielli et al., 2006; Muller et al., 2011 |
| fecD | Iron | Fe-Fur (repression) | Danielli et al., 2006 |
| yaeE | Iron | Fe-Fur (repression) | Danielli et al., 2006 |
| pdxJ | Iron | Fe-Fur (repression) | Pich et al., 2012; Carpenter et al., 2013 |
| pdxA | Iron | Fe-Fur (repression) | Carpenter et al., 2013 |
| amiE | Iron | Fe-Fur (repression) | Pich et al., 2012 |
| Hpn2 | Iron, Nickel | Fe-Fur (repression), Ni-NikR (activation) | Contreras et al., 2003; Danielli et al., 2006 |
| c553 | Iron | Apo-Fur (repression) | Carpenter et al., 2013 |
| hydAB* | Iron, Nickel | Apo-Fur (repression), Ni-NikR (repression) | Contreras et al., 2003; Carpenter et al., 2013 |
| serB | Iron | Apo-Fur (repression) | Carpenter et al., 2013 |
| pfr* | Iron, Copper, Zinc, Nickel, Manganese | Apo-Fur (repression), Fur-dependent (repression) | Waidner et al., 2005; Danielli et al., 2006; Carpenter et al., 2013; Zhao and Drlica, 2014 |
| cagA | Iron | Fe-Fur (activation), apo-Fur (repression) | Odenbreit et al., 2000; Pich et al., 2012 |
| oorDABC* | Iron | Fe-Fur (activation) | Gancz et al., 2006 |
| ribBA | Iron | Fe-Fur (repression) | Worst et al., 1998; Fassbinder et al., 2000 |
| sodB | Iron | Apo-Fur (repression) | Bereswill et al., 2000; Carpenter et al., 2013 |
| fecA3 | Nickel, Iron | Ni-NikR (repression), Fe-Fur (repression) | Ernst et al., 2006; Danielli et al., 2009 |
| ceuA | Nickel | Ni-NikR (repression) | Muller et al., 2011 |
| nixA | Nickel | Ni-NikR (repression) | Muller et al., 2011 |
| nikR | Nickel | Ni-NikR (repression) | Muller et al., 2011 |
| hspA | Nickel | Ni-NikR (activation) | Contreras et al., 2003 |
| hpn | Nickel | Ni-NikR (activation) | Contreras et al., 2003 |
| ureA | Nickel | Ni-NikR (activation), Mua (repression) | Benoit and Maier, 2011 |
| fliS | Copper | Copper (activation) | Waidner et al., 2002 |
| rlmA | Copper | Copper (activation) | Waidner et al., 2002 |
| nadC | Copper | Copper (activation) | Waidner et al., 2002 |
| trpA | Copper | Copper (activation) | Waidner et al., 2002 |
| HP1255 | Copper | Copper (activation) | Waidner et al., 2002 |
| HP1516 | Copper | Copper (activation) | Waidner et al., 2002 |
| HP0733 | Copper | Copper (activation) | Waidner et al., 2002 |
| HP0994 | Copper | Copper (activation) | Waidner et al., 2002 |
| hpylM | Copper | Copper (activation) | Waidner et al., 2002 |
| nadC | Copper | Copper (activation) | Waidner et al., 2002 |
| proC | Copper | Copper (activation) | Waidner et al., 2002 |
| crdAB* | Copper | CrdRS (activation) | Waidner et al., 2005 |
| czcAB* | Copper, Zinc | CrdRS (activation), Zinc (activation) | Waidner et al., 2005; Stähler et al., 2006 |
| copAB | Copper | CopP (activation) | Waidner et al., 2005 |
| cznABC | Zinc | Zinc (activation) | Stähler et al., 2006 |
Summary of genes regulated by metals and their corresponding regulatory mechanisms and references to the associated publication demonstrating these interactions.
Indicates gene(s) located in an operon that are co-regulated.
Iron
The use of iron in a number of critical metabolic pathways including electron transport, DNA replication, and amino acid synthesis, as well as its role as a cofactor within iron sulfur clusters and heme, makes it a necessity for nearly every living organism including H. pylori (Becker and Skaar, 2014). The human body exploits this need for iron by limiting access to this critical micronutrient through nutritional immunity (Cassat and Skaar, 2013). Iron is bound within host molecules such as lactoferrin, transferrin, heme, hemoglobin, and haptoglobin, making it relatively unavailable to pathogens (Yen et al., 2011; Haley and Skaar, 2012). Within the gastric niche H. pylori has access to host dietary iron, however, it must compete with the host for this limited nutrient (Figure 1). In conditions of restricted iron availability, H. pylori can utilize alternate sources of nutrient iron including hemoglobin, transferrin, heme, and lactoferrin (Dhaenens et al., 1999; Senkovich et al., 2010). Consequently, H. pylori responds to iron limitation by upregulating an arsenal of molecular pathways devoted to diverse iron acquisition functions; therefore, the ability to detect and respond to environmental iron concentrations is critical to the survival of H. pylori.
Figure 1
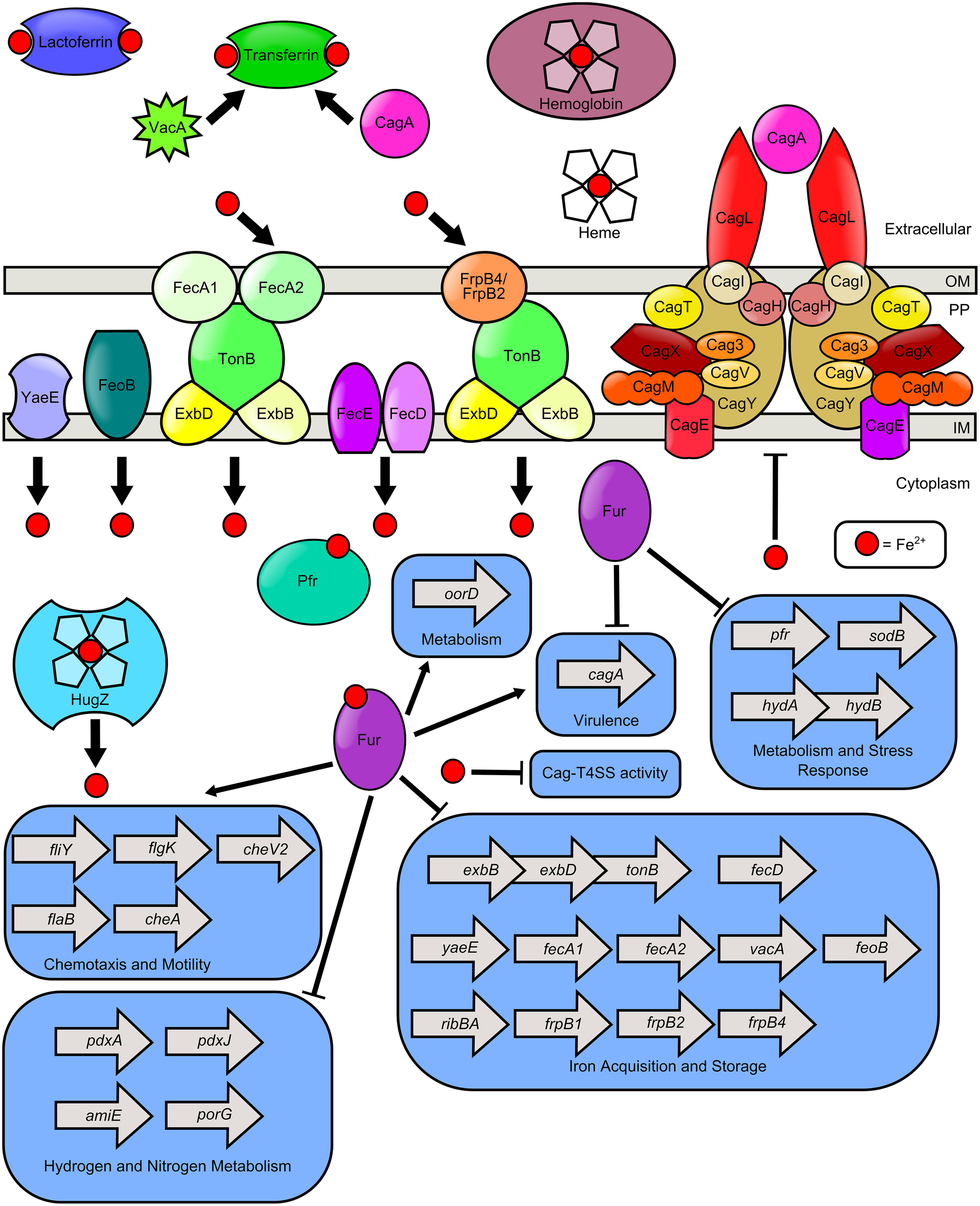
Model of iron transport and iron-dependent gene regulation in H. pylori.H. pylori can use numerous sources of nutrient iron including hemoglobin, holo-lactoferrin, holo-transferrin, and heme, which is degraded in the cytoplasm by the heme oxygenase HugZ. Host cell transferrin can be trafficked to the apical cell surface by the cytotoxins VacA and CagA, produced by H. pylori. FecA1, FecA2, FrpB4, and FrpB2 are implicated as outer membrane iron receptors which transport iron through the TonB-ExbB-ExbD complex. FeoB and YaeE are putative permeases which shuttle iron from the periplasm, across the inner membrane to the cytoplasm. The FecD/FecE system also shuttles iron from the periplasm to the cytoplasm. Once in the cytoplasm, iron binds to Fur and promotes expression of the fliY, flgK, flab, and cheA genes which encode flagellar and chemotactic components. Fe-Fur also represses the expression of numerous genes involved in iron homeostasis (including exbB-exbD-tonB, fecD, yaeE, fecA1, fecA2, vacA, feoB, ribBA, frpB1, frpB2, frpB4) and metabolism of hydrogen or nitrogen (including pdxA, pdxJ, amiE, and porG). Furthermore, apo-Fur represses pfr, sodB, hydAB, and cagA expression. Iron is stored by bacterial ferritin (Pfr) and free cytoplasmic iron can repress elaboration of Cag-T4SS pili and activity of the T4SS.
The number of TRs utilized by H. pylori is relatively small for a genome of its size (~1600 kb), and consequently the TRs of H. pylori display a more diverse array of functions than their counterparts in other bacteria (Pich et al., 2012; Troxell and Hassan, 2013). The broad activity of H. pylori TRs is best exemplified by the ferric uptake regulator, Fur. Canonically Fur binds its ferrous iron cofactor to form holo-Fur (Fe-Fur) which then mediates Fur binding to conserved DNA sequences, specifically a 7-1-7 motif with dyad symmetry (5′-TAATAATnATTATTA-3′) within the promoter region of regulated genes resulting in the repression of their expression (Pich et al., 2012). While holo-Fur repression is the best characterized Fur mediated regulation, it is not the only way in which Fur modifies transcriptional rates. Specifically, within H. pylori, holo-Fur has been shown to function as an activator, and apo-Fur, which is not bound to an iron cofactor, has also been shown to regulate gene transcription (Gancz et al., 2006; Carpenter et al., 2013). This unique flexible transcriptional regulation facilitated by Fur indicates that Fur functions as a global regulator within H. pylori; therefore, minute alterations in iron availability can lead to significant changes in the transcriptome of H. pylori.
Holo-fur
Many of the H. pylori genes which are regulated by Fur in the traditional manner are involved in iron acquisition. This allows H. pylori, upon experiencing iron starvation, to upregulate a complex network of proteins that facilitate the acquisition and trafficking of this vital nutrient. For example, upon experiencing iron restriction Fur is no longer able to bind to the promoter regions of the genes encoding the high affinity iron transporters fecA1, fecA2, frpB1, and feoB, thus resulting in a dramatic increase in transcriptional rates of these genes and subsequent iron import into the cell (Delany et al., 2001; Ernst et al., 2005a; Danielli et al., 2009). In iron replete conditions, Fe-Fur represses a variety of iron transport genes including the exbB-exbD-tonB operon, frpB4, and frpB2, which encode outer membrane iron transporters, fecD, and yaeE, which encode inner membrane iron permeases (Fassbinder et al., 2000; Danielli et al., 2006). The Fur-regulon also includes genes which encode proteins involved in nitrogen and hydrogen metabolism, including amiE, the gene encoding pyruvate ferrodoxin oxidoreductase (porG), and genes implicated in pyridoxal phosphate biosynthesis (pdxJ and pdxA) (Gancz et al., 2006; Carpenter et al., 2013). Together, these findings indicate that H. pylori exploits Fur to regulate and enable metabolic flexibility which helps the cell circumnavigate the stress imposed by changes in iron availability. In addition to increasing expression of iron transporters upon iron starvation H. pylori also increases binding to the host chelating proteins lactoferrin, and transferrin presumably by upregulating expression of the receptors for these proteins (Senkovich et al., 2010). Thus, the Fur-mediated iron regulation of these iron acquisition systems enables H. pylori to rapidly respond to the host-imposed iron limitation by increasing intracellular iron import.
An additional strategy employed by H. pylori to alleviate iron starvation is to increase the biosynthesis of flavins, which are used within ferric iron reductases to mediate the reduction of extracellular iron within iron-containing compounds. Reduction of iron to its more soluble ferrous form increases the concentration of available iron while decreasing the affinity of the iron for some host ligands (Worst et al., 1998). Expression of the enzyme responsible for the initial rate limiting step in riboflavin synthesis, RibBA, is repressed by holo-Fur. Consequently, iron reductases are indirectly Fur-regulated, such that upon encountering iron restriction, H. pylori increases transcription of ribBA resulting in an increase in flavin production that can then be used by the ferric iron reductases (Worst et al., 1998).
Iron-bound Fur has also been shown to directly activate gene expression including fliY, flgK, flaB, and cheA which encode flagellar and chemotactic components (Danielli et al., 2006). Additionally, holo-Fur has been shown to activate the oorDABC operon which encodes for a 2-oxoglutarate oxidoreductase, an enzyme that catalyzes the formation of succinyl-CoA, a major intermediate in carbon metabolism (Gilbreath et al., 2012). This non-canonical activity of Fur, which is commonly thought to be directly involved in gene repression, highlights the flexibility of this molecule within H. pylori; a characteristic that likely aids in persistence in the gastric niche.
Apo-fur
While iron acquisition is critical for H. pylori colonization, excessive cellular levels of iron can be detrimental to the cell as it can lead to the formation of reactive oxygen species through the Fenton reaction. Reactive oxygen species can lead to DNA, protein, and lipid membrane damage (Aguirre and Culotta, 2012; Zhao and Drlica, 2014). To mitigate this damage, H. pylori increases expression of the bacterial ferritin Pfr, an iron storage protein, in response to high iron levels. Iron regulation of pfr is facilitated by Fur, however, this regulation occurs through non-canonical Fur regulation such that apo-Fur represses pfr transcription (Bereswill et al., 2000; Carpenter et al., 2013). An additional mechanism for coping with oxidative stress induced by high iron levels is through the activity of superoxide dismutases as it catalyzes the conversion of superoxide species into oxygen and hydrogen peroxide (Ernst et al., 2005b). Not surprisingly, the only identified superoxide dismutase in H. pylori, SodB, is transcriptionally regulated by Fur, such that apo-Fur binds to the promoter region of sodB, occluding RNA polymerase binding (Ernst et al., 2005b). The combinatorial result of apo-Fur regulation of both sodB and pfr is H. pylori can appropriately respond to high iron levels with a set of proteins designed to alleviate iron toxicity. Interestingly, apo-Fur has been shown to repress hydABCDE, an operon which encodes a Ni/Fe hydrogenase, indicating that apo-Fur is also implicated in the regulation of hydrogen metabolism in H. pylori. It is also postulated that apo-Fur-dependent repression of genes (including sodB and those encoded in the hydABCDE operon) evolved because their protein products utilize iron as a cofactor, and repression by apo-Fur ensures these proteins are produced only when sufficient levels of that cofactor are available (Ernst et al., 2005b).
The iron restrictive nature of the host has led to many bacterial pathogens coordinating the production of virulence factors to the detection of low iron availability. H. pylori encodes two important virulence factors, the VacA, and the cytotoxin-associated gene A (CagA) toxin (Cover and Peek, 2013). VacA is secreted via the Type V autotransporter pathway (Fischer et al., 2001; Letley et al., 2006). VacA causes numerous changes in host cells including vacuolation, depolarization of the membrane potential, permeabilization of epithelial monolayers, disruption of lysosomes, inhibition of T-cell activation and proliferation, and ultimately leads to cell death via programmed necrosis (Satin et al., 1997; Szabò et al., 1999; Sundrud et al., 2004; Torres et al., 2007; Radin et al., 2011). CagA is secreted via a type IV secretion system (T4SS), which is encoded within the cag pathogenicity island (Tummuru et al., 1995). Upon translocation into host cells, CagA is phosphorylated, and induces a cascade of changes within the host cell ultimately leading to changes in immune signaling and cell morphology (Odenbreit et al., 2000). Interestingly, one effect of CagA translocation into host cells is the marked alteration in host cell polarity, which results in apical release of transferrin (Tan et al., 2011). VacA is also implicated in perturbing transferrin trafficking in epithelial monolayers to the apical cell surface (Tan et al., 2011). This apically localized transferrin is believed to be used as an iron source by H. pylori, thus allowing this bacterium to modify the host environment and create a more hospitable replicative niche. Given its role in immune modulation and iron acquisition, it is not surprising that Fur has been implicated as a potential regulator of cagA expression (Pich et al., 2012; Vannini et al., 2014). Along with these observations, our work has shown that conditions of low nutrient iron availability induce expression of the Cag-T4SS pili at the host-pathogen interface, and also enhance the activity of the Cag-T4SS (Noto et al., 2013, 2014; Haley et al., 2014). Additionally, transcription of cagA has shown to be upregulated in iron-replete conditions suggesting Fe-Fur activation of cagA (Pich et al., 2012). These seemingly contradictory findings suggest an additional, as yet unknown, environmental signal modifies the Fur-mediated regulation of cagA activity. Thus, these observations reveal that H. pylori toxin secretion is regulated by iron and plays a role in iron homeostasis as well as suggests a role for additional signals to influence iron-mediated regulation of cagA.
Importance of fur in vivo
The importance of sensing environmental iron levels, and responding with global transcriptional changes, is underscored by the colonization defect of the H. pylori Δfur strain within a Mongolian gerbil animal model (Gancz et al., 2006; Miles et al., 2010). Interestingly, the colonization defect of the Δfur strain as compared to the wild-type strain was most severe early in infection with a >50 fold decrease in the number of CFU/g stomach tissue recoverable at day 3 post infection (Miles et al., 2010). Importantly, this initial decrease in bacterial burden dissipates over time and by day 14 no discernable differences between the wild-type and Δfur strain could be detected, indicating that Fur-mediated transcriptional regulation is most critical early in the infection process (Miles et al., 2010). Furthermore, while H. pylori has been shown to localize to both the corpus and antrum of the stomach the highest level of bacterial burden is typically associated with the antral region. This localization pattern is maintained within a gerbil infection model where it has been shown that in the absence of Fur, H. pylori preferentially colonizes the corpus as opposed to the antrum (Miles et al., 2010). The precise mechanism for this aberrant distribution within the Δfur strain may be due to transcriptional changes of cheV2, a Fur-regulated gene shown to be involved in chemotaxis (Miles et al., 2010).
Nickel
The transition metal nickel plays an important role in H. pylori physiology and pathogenesis (Figure 2). Nickel is a cofactor for two critical metalloenzymes, urease and [NiFe]-hydrogenase (de Reuse et al., 2013; Sydor et al., 2014). The former catalyzes the generation of ammonia and bicarbonate from urea, a process that provides a protective increase in pH, enabling the bacterium to withstand the acidic environment of the stomach (Evans et al., 1991; Hawtin et al., 1991; Maier, 2005; Benoit and Maier, 2008). The latter, enables the bacterium to utilize hydrogen gas as an energy source within the gastric niche. Because nickel is required for the activity of these important enzymes, nickel acquisition is a nutritional requirement for H. pylori (de Reuse et al., 2013). However, accumulation of high concentrations of intracellular nickel can be toxic to the cells (Benoit and Maier, 2008). Thus, nickel acquisition and distribution are tightly controlled by multiple features including transport, storage, and efflux.
Figure 2
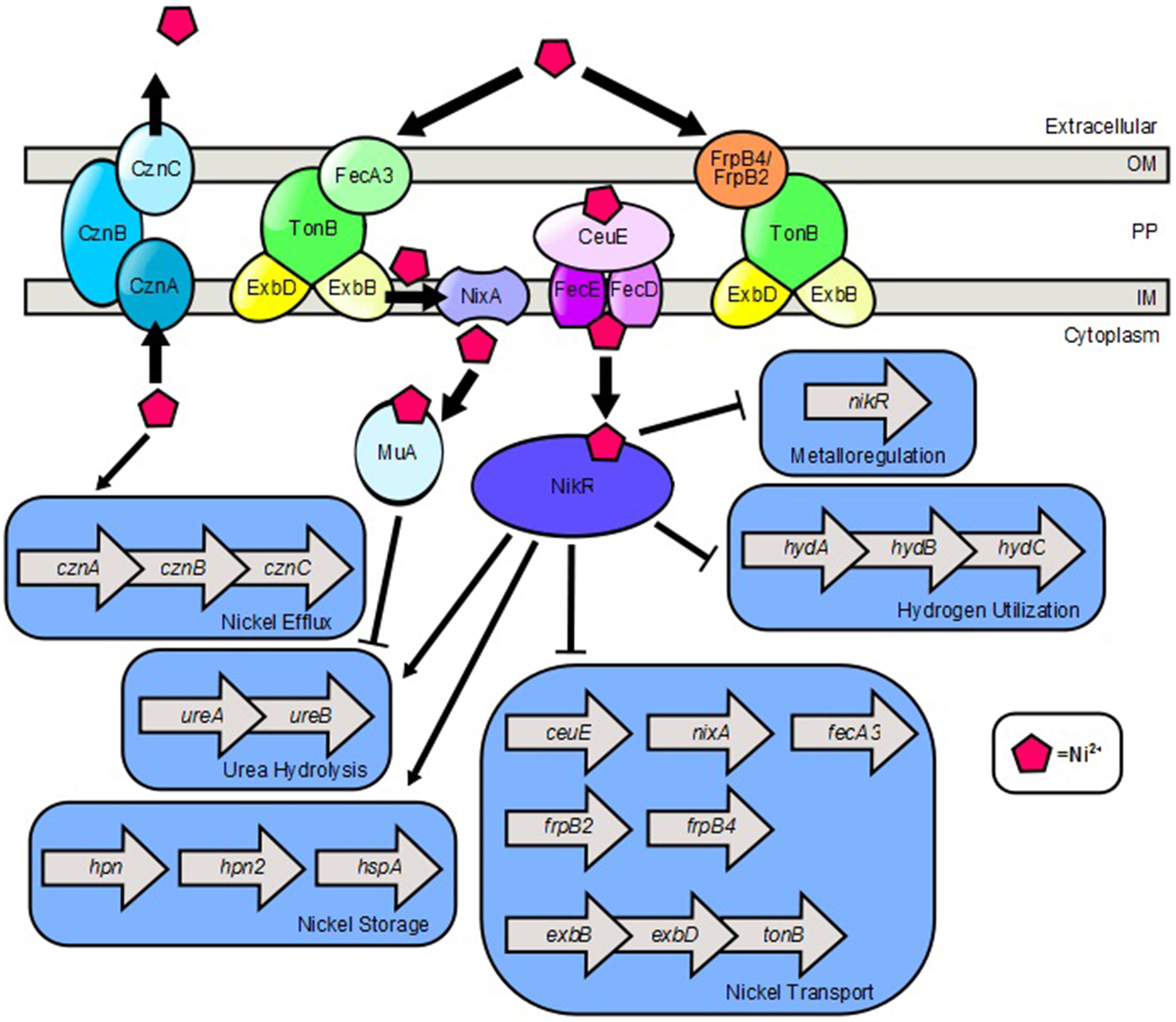
Model of nickel transport and nickel-dependent gene regulation in H. pylori. FecA3, FrpB4, and FrpB2 are implicated as outer membrane nickel receptors which transport nickel through the TonB-ExbB-ExbD complex. NixA is a nickel permease which shuttles nickel from the periplasm, across the inner membrane to the cytoplasm. CeuE is a periplasmic nickel binding protein that is believed to shuttle nickel through the FecD/FecE system to the cytoplasm. Once in the cytoplasm, nickel promotes expression of the cznABC operon which encodes a nickel efflux pump. Nickel binds to NikR which promotes expression of genes involved in nickel storage and urea hydrolysis. Ni2+-NikR represses its own coding, as well as nickel transport genes and hydrogen utilization genes. Nickel also binds to the Mua protein which represses transcription of genes encoding urease subunits.
The master regulator for nickel homeostasis is a nickel-responsive TR referred to as, NikR (Dosanjh and Michel, 2006). H. pylori NikR can act as either a repressor or an activator, although the precise mechanism by which nickel-sensing affects the DNA-binding capacity of NikR remains largely unknown, however a putative NikR- DNA-binding consensus sequence has been identified (TATWATT-N11-AATWATA) (Ernst et al., 2006). The NikR-regulon includes multiple genes involved in metal homeostasis, hydrogen utilization, and acid response with chronological hierarchy (Jones et al., 2014). NikR represses transcription of HPG27_1499 (ceuE) and HP1077 (nixA), genes which encode a periplasmic nickel transporter and an inner membrane nickel permease, respectively, HPG27_866, a gene encoding FrpB2, (Muller et al., 2011) which contributes to the accumulation of cell-associated nickel, HP1339, the first gene in the operon which encodes ExbB/ExbD/TonB energy transport system, and the hydABC locus which encodes proteins involved in hydrogen utilization (Contreras et al., 2003). NikR also represses fecA3 and frpB4, which encode a putative outer membrane receptors for nickel (Ernst et al., 2006; Romagnoli et al., 2011). Furthermore, NikR is an autoregulator that binds to its own intergenic region and represses expression of the nikR locus in conditions of excess nickel (Contreras et al., 2003). Interestingly, Ni2+-NikR also induces transcription of numerous genes including hpn, hpn2, and hspA which are involved in nickel storage, and the ureA locus (HP0073) in which the first gene in the operon encodes the subunits of the urease enzyme (Contreras et al., 2003). Urease is an important nickel-containing dodecameric enzyme that buffers the cytoplasm and the periplasm of H. pylori during colonization of the acidic gastric niche (Khan et al., 2009). Urease is the most abundant enzyme in H. pylori cells, accounting for almost 10% of the total cellular protein content (Benoit and Maier, 2011). Urease expression is critical for early colonization and virulence in the vertebrate host (Eaton et al., 1991). The urease complex is comprised of subunits organized into two transcriptional units, ureAB and ureIEFGH, which produces three transcripts, ureAB, ureIEFGH, and ureABIEFGH (Akada et al., 2000). These transcripts are induced, as part of the acid stress response through the phosphorylation of the ArsRS TCS (Pflock et al., 2005). Conversely, these transcripts are repressed at neutral pH by the unphosphorylated ArsRS system via a cis-encoded antisense small RNA to ureB (Wen et al., 2011). In addition to the canonical NikR-mediated activation of urease expression in response to nickel availability, recent evidence suggests that the HP0868 (Mua) protein can repress expression of ureA under conditions where nickel is abundant in the intracellular compartment (Benoit and Maier, 2011).
Nickel transport into the cell is facilitated through FrpB4, FrpB2, or FecA3, outer membrane receptors involved in nickel uptake work in tandem with ExbB/ExbD/TonB machinery to import nickel across the outer membrane (Schauer et al., 2007). NixA, an inner membrane protein then facilitates nickel transport from the periplasm to the cytoplasm. Alternatively, nickel can be transported by the periplasmic transporter CeuE, which likely works cooperatively with the inner membrane FecD/E ABC transporter, (Figure 2) which has been implicated in H. mustelae nickel and cobalt acquisition (Stoof et al., 2010). As nickel concentration increases in the cytoplasm, H. pylori induces expression of the cznABC loci, which encode components of a putative cobalt, zinc, and nickel efflux pump. This efflux system is critical for resistance to nickel stress and colonization of the vertebrate host (Stähler et al., 2006). Although H. pylori does not seem to have a strict nutritional requirement for nickel to survive (Testerman et al., 2006), it is clear that nickel sensing and nickel homeostasis are important for H. pylori persistence in the human gastric niche as underscored by the significant colonization defect of H. pylori strains lacking a functional nikR gene as compared to wildtype H. pylori strains (Bury-Mone et al., 2004).
Copper
In the gastric environment, dietary copper intake can exceed 1 mg, indicating copper is present in micromolar concentration at the lumen of the stomach (Barceloux, 1999). Copper can also be sequestered by S100A12 (Haley et al., 2015). Copper is gaining increasing recognition as an important component of macrophage-mediated antimicrobial activity (Wolschendorf et al., 2011; Johnson et al., 2015; Neyrolles et al., 2015). Macrophages exploit copper toxicity to poison bacteria within the phagosome presumably by inducing Fenton-like reactions which produce hydroxyl radicals (Pham et al., 2013; Neyrolles et al., 2015). Conversely, bacteria also utilize copper as a cofactor for oxidases, electron transport proteins, and hydroxylases. H. pylori encodes both menaquinone-6 and a cbb3-type cytochrome-c oxidase, which harbors a heme-copper binuclear center similar to the cytochrome aa3-type oxidase (Nagata et al., 1996). This is likely the terminal oxidase in the H. pylori respiratory chain, and thus, copper is a critical cofactor for respiration. Copper promotes H. pylori colonization of mucosal surfaces and also acts as a chemotactic repellant for bacterial cell motility (Montefusco et al., 2013; Sanders et al., 2013). Thus, H. pylori must balance the need for copper as a respiration cofactor and the importance of protection against copper toxicity (Figure 3).
Figure 3
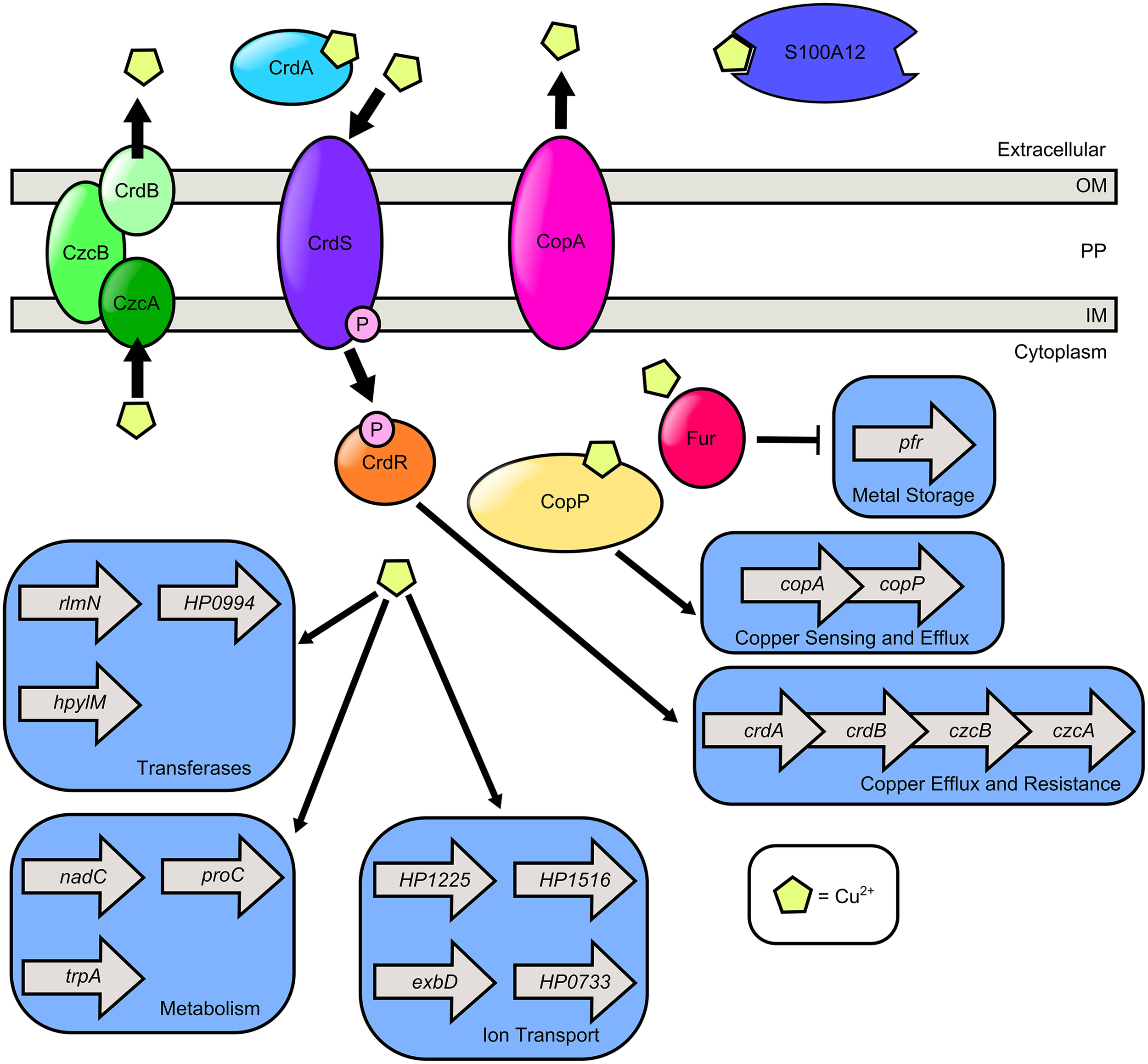
Model of copper transport and copper-dependent gene regulation in H. pylori. H. pylori secretes CrdA, a copper-binding protein that facilitates copper resistance. Extracellular copper is sensed by the CrdS sensor kinase which phosphorylates the cognate response regulator CrdR, an activator of copper efflux and resistance expression. S100A12 is a host protein that can bind copper as well as zinc. It is hypothesized that cytoplasmic copper levels are sensed by Cu2+-CopP which upregulates the copAP operon encoding the CopA copper resistance protein and the CopP regulator. In the presence of copper, cytoplasmic levels of ferritin (Pfr) are reduced in a Fur dependent manner. Copper exposure promotes expression of the methyltransferase (rlmN), the nucleotidyl transferase (HP0994), and the DNA-methyltransferase (hpylM). Copper induces transcription of nicotinate-nucleotide pyrophosphorylase (nadC), pyrroline-5-carboxylate reductase (proC), and tryptophan synthase (trpA). Copper promotes transcription of genes involved in ion transport including exbD, crcB which encodes a camphor resistance protein involved in fluoride ion transport (HP1225), a putative ABC-transporter (HP1516), and a GTPase with putative ABC-transporter activity (HP0733). H. pylori encodes two Cu2+ efflux systems, the CrdB-CzcAB complex and the CopA system.
Exposure to copper alters the H. pylori transcriptional profile in numerous ways. In the presence of copper and other metals, the cytoplasmic levels of Pfr are reduced in a Fur dependent manner, suggesting Fur-mediated repression of pfr transcription in an undefined mechanism (Bereswill et al., 2000). This is likely to facilitate survival under conditions of metal stress. Copper is also associated with upregulation of numerous genes involved in a variety of cellular responses including motility, fliS- encoding a putative flagellin chaperone, ion transport, exbD which encodes an energy transport protein involved in iron and nickel acquisition, crcB, which encodes camphor resistance protein involved in fluoride ion transport (HP1225), a putative ABC-transporter (HP1516), and a GTPase with putative ABC-transporter activity (HP0733); transferase activity (rlmN- encoding methyltransferase), the nucleotidyl transferase (HP0994), and the DNA-methyltransferase (hpylM) (Waidner et al., 2002). Copper upregulates transcription of genes involved in several metabolic pathways including nicotinate-nucleotide pyrophosphorylase (nadC), pyrroline-5-carboxylate reductase (proC), and tryptophan synthase (trpA). H. pylori also upregulates numerous genes involved in copper homeostasis such as crdAB, czcAB, and the copAP operon in response to copper stress (Waidner et al., 2002).
To evade copper toxicity, H. pylori has evolved elegant efflux strategies governed by copper sensing mechanisms. H. pylori employs a TCS crdRS encoding a sensor kinase (CrdS) which phosphorylates a cognate response regulator (CrdR) in the presence of copper (Waidner et al., 2005). CrdR induces expression of crdAB and czcAB, which encode a secreted copper resistance protein (CrdA) and a copper efflux complex comprised of CrdB, CzcB, and CzcA. H. pylori also utilizes the copAP operon, which encodes a cytoplasmic copper-binding regulator homologous to CopZ from E. coli (CopP) that promotes expression of CopA, a protein which promotes resistance to copper (Beier et al., 1997). Taken together, these results reveal that tight molecular mechanisms of gene regulation have evolved in the human pathogen H. pylori in response to changes in copper availability, and that these regulatory events are critical for adaptation to the gastric environment.
Zinc
Zinc is a micronutrient required for all forms of life, including H. pylori (Testerman et al., 2006; Kehl-Fie and Skaar, 2010). Zinc availability is controlled tightly at the host-pathogen interface by host S100A-family proteins including calprotectin, which participate in nutritional immunity via transition metal sequestration (Kehl-Fie and Skaar, 2010). Coinciding with this, increased severity of H. pylori-induced gastric inflammation is inversely correlated with zinc concentrations within the gastric mucosa (Sempértegui et al., 2007). Conversely, humans consume large quantities of transition metals such as zinc, so it is likely that H. pylori is exposed to micromolar concentrations of zinc at the lumen of the stomach (Stähler et al., 2006). To persist in the gastric environment, H. pylori must quickly adapt to variations in zinc availability (Figure 4). One way that H. pylori senses and responds to metal signals in the gastric niche is through chemotaxis, the sensing of and response to a chemical signal. Zinc is a chemotactic attractant for H. pylori, whereas nickel is a repellent (Sanders et al., 2013). H. pylori possesses four chemotactic receptors, TlpABCD, which participate in environmental sensing (Lertsethtakarn et al., 2011; Rader et al., 2011). TlpD is required for colonization of the antrum of the stomach and is important for bacterial motility (Rolig et al., 2012). In H. pylori, chemotaxis has been shown to be important for the elicitation of an inflammation response during infection (McGee et al., 2005; Williams et al., 2007). Interestingly, TlpD has a zinc-binding domain which could participate in its chemotactic activity and zinc-sensing (Draper et al., 2011). A tlpD mutant elicited more IL-8 from epithelial cells and produced more CagA compared to the WT parental strain, although the mechanism of this regulation remains obscure (Behrens et al., 2013). Recent work indicates that zinc sequestration by host antimicrobial proteins such as calprotectin (S100A8/A9 heterodimer) or calgranulin C (S100A12 homodimer) represses the elaboration of the extracellular pilus associated with the H. pylori cag-type IV secretion system (Cag-T4SS) and its associated activity (Gaddy et al., 2014; Haley et al., 2015). The Cag-T4SS is a macromolecular machine that is responsible for translocation of the oncogenic effector cytotoxin CagA. Strains of H. pylori that possess CagA are associated with increased risk of disease outcomes including gastritis and gastric cancer (Blaser et al., 1995). Zinc is required for Cag-T4SS pilus formation, CagA translocation, activation of nuclear factor kappa-β, and IL-8 secretion by host cells (Gaddy et al., 2014). Together, these results reveal that zinc is an important global signal that regulates virulence.
Figure 4
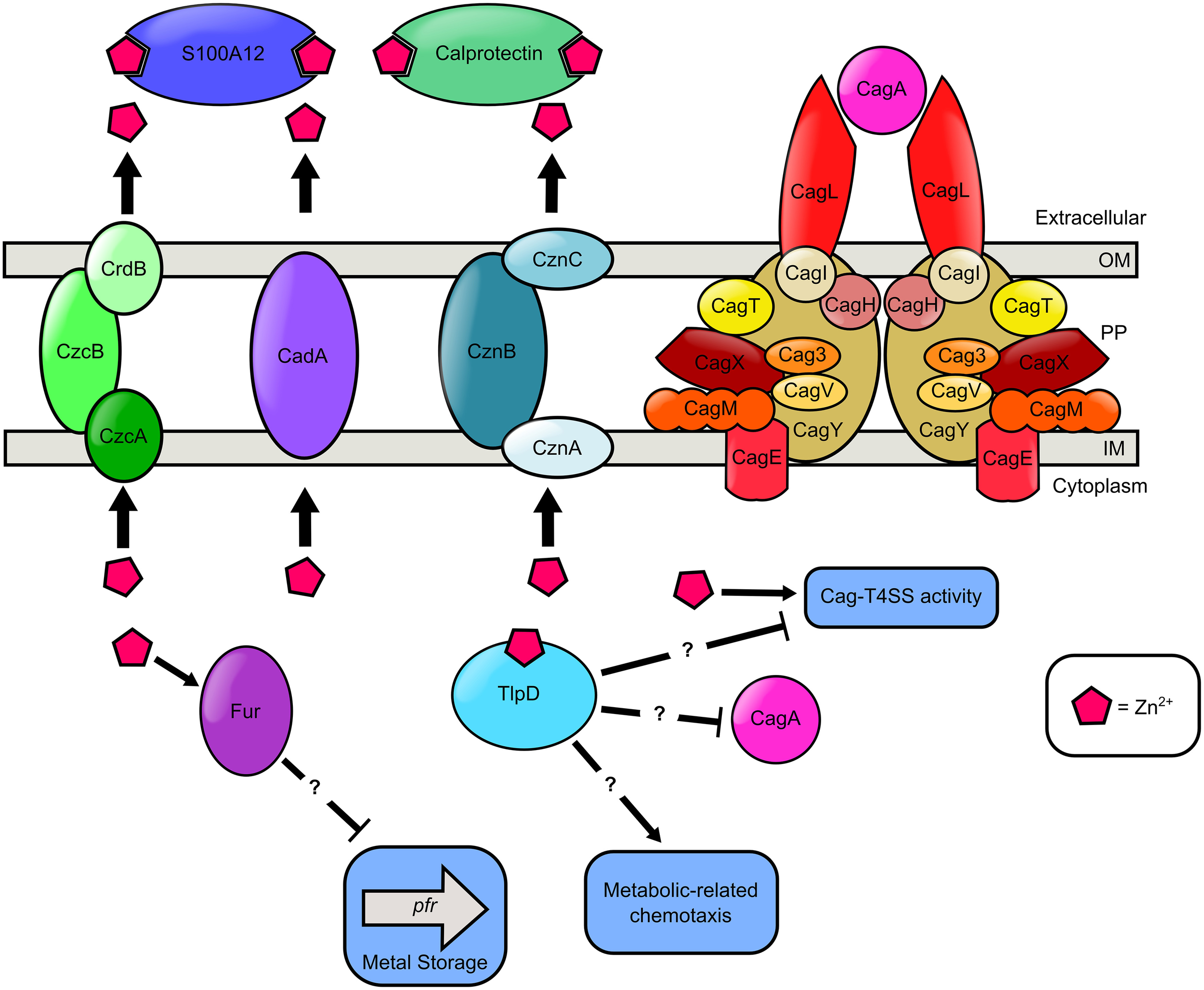
Model of zinc transport and zinc-dependent regulation of chemotaxis, metal storage, and virulence in H. pylori. Extracellular zinc is bound by the S100A-family host antimicrobial proteins, calprotectin and S100A12, produced by innate immune cells. Cytoplasmic zinc binds the cytoplasmic chemotactic receptor TlpD. High cytoplasmic levels of zinc results in a reduction in Pfr expression in a Fur dependent manner. TlpD putatively participates in metabolic-related chemotaxis, CagA repression, and repression of the Cag-T4SS activity. Nutrient zinc is required for Cag-T4SS pilus deployment and activity. In the presence of excess zinc, H. pylori utilizes zinc efflux pumps as a detoxification strategy. H. pylori encodes three Zn2+ efflux systems, the CrdB-CzcAB complex, the CadA transporter, and the CznABC system. Question marks “?” indicate proteins involved in zinc-responsive changes in cell biology are influencing regulation in an undefined manner.
In addition to being an important environmental signal, high levels of zinc are toxic to bacterial cells (Braymer and Giedroc, 2014). There is growing evidence that the host innate immune system exploits zinc and copper to poison bacteria trapped within the macrophage phagosome (Neyrolles et al., 2013). To evade zinc toxicity, bacterial pathogens have evolved efflux strategies to transport zinc to the extracellular space (Braymer and Giedroc, 2014). H. pylori has three zinc transport systems that participate in zinc resistance including CadA, the CrdB-CzcAB complex, and the CznABC system (Waidner et al., 2002, 2005; Stähler et al., 2006). H. pylori mutants harboring inactivation in the coding region of the CadA transition metal ATPase exhibit increased sensitivity to both cadmium and zinc (Herrmann et al., 1999). H. pylori cznA, cznB, and cznC mutants have increased sensitivity to zinc toxicity, and also exhibit elevated cytoplasmic levels of zinc, indicating zinc ions are trapped within the cell (Stähler et al., 2006). These mutants are defective for colonization of rodent models, implicating the CznABC efflux pump as an important virulence determinant (Stähler et al., 2006). Furthermore, the H. pylori genome encodes a redundant CrdB-CzcAB complex purported to be involved in zinc efflux (Stähler et al., 2006). Interestingly, while it has been unequivocally demonstrated that zinc is a vital nutrient for H. pylori survival no zinc import system has been identified. One possible explanation for the absence of a zinc uptake system is that many metal import systems are promiscuous and facilitate the acquisition of multiple metals. It is therefore feasible that the nickel import systems also facilitate zinc acquisition. Additionally, the promiscuity of metal influx structures may hinder traditional genetic screening methods used to identify putative import proteins as the ablation of any one single gene will not result in a significant growth defect. These results indicate that H. pylori must maintain zinc homeostasis as a prerequisite for colonization of the host and persistence within the host to establish a chronic infection.
Magnesium, manganese, and cobalt
While it is clear that magnesium, manganese, and cobalt are important cofactors for various bacterial proteins, the current depth of understanding regarding the role, regulation, and acquisition of these metals is limited relative to iron, nickel, zinc, and copper. Magnesium is required for the activity of several biochemical pathways in central metabolism, which are essential for bacterial growth and viability (Smith and Maguire, 1998). For example, H. pylori heavily relies on Mg2+ for phosphate metabolism, including the catabolism of phosphonates, which can function as both a vital source of phosphorous and as phosphorous storage (Ford et al., 2010). H. pylori is capable of degrading the phosphonate, phenylphosphonate, to use as a sole phosphate source. This catabolism was shown to be enhanced by the presence of exogenous MgCl2+ and inhibited upon the addition of the chelator EDTA (Ford et al., 2010). Mg2+ also plays a critical role in H. pylori phosphate metabolism as a metal cofactor within pyrophosphatase. Pyrophosphatases are ubiquitious enzymes that catalyze the interconversion of inorganic pyrophosphate and orthophosphate, and function as a catalyst within cellular bioenergetics, making this group of enzymes essential for life. The pyrophosphatases within H. pylori requires Mg2+ for enzymatic activity (Lee et al., 2007). In addition to its role as a cofactor within H. pylori phosphate metabolism, Mg2+ may also play an important role in H. pylori transcriptional regulation through the binding of a lower affinity site within the transcription factor NikR. Although NikR binds nickel and has been shown to be nickel responsive, a second binding site has been shown to require either Mg2+ or calcium for NikR promoter binding (Dosanjh et al., 2007). The importance of Mg2+ is underscored by the strict requirement of magnesium for H. pylori cell viability (Testerman et al., 2006). To satisfy this Mg2+ requirement H. pylori has evolved systems to transport this important metal. CorA, a distant homolog (15–20% sequence identity) of the Salmonella enterica serovar Typhimurium ZntB Zn2+ efflux transporter, is involved in manganese uptake in H. pylori (Wan et al., 2011). Isogenic corA mutants have diminished growth in the absence of an exogenous source of nutrient magnesium compared to the parental strain, and expression of the H. pylori corA locus in trans in an E. coli corA mutant enhanced magnesium toxicity compared to the E. coli corA mutant, demonstrating its role in magnesium transport (Pfeiffer et al., 2002). CorA is also implicated in cobalt and nickel transport, however, magnesium is the dominant substrate for this protein (Pfeiffer et al., 2002; Wan et al., 2011).
Manganese is an important nutrient for H. pylori due to its use as a cofactor within the pyrimidine metabolic pathway. Uridine monophosphate (UMP) kinase is a necessary enzyme for the synthesis of pyrimidine nucleotides. The UMP kinase in H. pylori is unique in its use of manganese as a cofactor as opposed to the canonical magnesium (Lee et al., 2010). Manganese also functions as a critical cofactor within the H. pylori phosphatidylserine synthase (PSS), an enzyme necessary for the biosynthesis of phospholipids. Specifically, PSS is responsible for the catalysis of the first committed step in the formation of the phospholipid phosphatidylethanolamine, a major component of the phospholipid membrane (Ge and Taylor, 1997). The importance of PSS and therefore manganese is highlighted by the inability to ablate the gene encoding PSS as it is essential for cell viability (Ge and Taylor, 1997).
Cobalt is gaining appreciation as an important micronutrient. Cobalt, among several other transition metals, can serve as a cofactor for HpyAV, a type II restriction-modification system in H. pylori. Type II restriction-modification endonucleases are highly conserved in prokaryotic organisms, but H. pylori is particularly rich with these, likely due to acquisition through natural competence. Type II restriction-modification endonucleases are implicated in phage resistance and transcriptional regulation of gene expression (Chan et al., 2010). Additionally, cobalt is utilized as a cofactor within the H. pylori arginase. Arginases are a group of enzymes that are responsible for catalyzing the conversion of L-arginine to L-ornithine. The ubiquitous use of arginases across multiple kingdoms underscores the importance of this enzyme in maintaining arginine homeostasis. The H. pylori arginase has been shown to be critical for acid protection in vitro and to play an important role during colonization. Many arginases utilize Mn2+ as a metal cofactor; however, the H. pylori arginase is unique in its use of Co2+ as a cofactor (Srivastava et al., 2011). To fulfill its need for cobalt H. pylori transports cobalt into the cell using the metal transporter CorA (Pfeiffer et al., 2002). Although, cobalt is an invaluable micronutrient at high quantities cobalt can exert cellular toxicity. To prevent metal toxicity H. pylori utilizes the metal ion efflux pump CadA which has specificity for Co (II), Cd (II), and Zn (II) (Herrmann et al., 1999).
Conclusions
The human stomach is an incredibly dynamic and seemingly inhospitable environment for an invading prokaryote. Yet, it is within this environment that H. pylori establishes its replicative niche. To thrive in an environment of oscillating extremes including pH, immune assault and nutrient availability H. pylori alters its transcriptional profile through the intricate interplay of multiple transcriptional regulators. Transcriptional changes mediated through the sensing of metals enables H. pylori to detect changes in nutrient availability and deploy an impressive array of metal acquisition systems, store the corresponding influx of metals, avoid metal toxicity and circumnavigate the host immune assault. Many bacteria use metal sensing as a way to precisely coordinate virulence factor expression as changes in metal abundance can be associated with important bacterial life cycle events such as host entry or the arrival of recruited immune cells, and H. pylori is no exception. It is no surprise that several genes are regulated by more than one metal condition and concomitant regulators (Table 1). While two major regulators including Fur and NikR participate in H. pylori metal response and have been characterized in some depth, it is very likely that additional, as yet unknown, regulators exist and additional roles for the two major regulators have yet to be defined.
Statements
Author contributions
KH and JG conceived, designed, wrote, reviewed, and edited this manuscript for critical content. Both authors approve this manuscript for integrity and accuracy.
Acknowledgments
This work has been funded primarily by a Career Development Award IK2BX001701 (to JG) from the Office of Medical Research, Department of Veterans Affairs and Childhood Infections Research Program T32-AI095202 (to KH) and by Vanderbilt University Medical Center's Digestive Disease Research Center supported by NIH grant P30DK058404 and Vanderbilt Institute for Clinical and Translational Research program supported by the National Center for Research Resources, Grant UL1 RR024975-01, and the National Center for Advancing Translational Sciences, Grant 2 UL1 TR000445-06. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
1
AguirreJ. D.CulottaV. C. (2012). Battles with iron: manganese in oxidative stress protection. J. Biol. Chem.287, 13541–13548. 10.1074/jbc.R111.312181
2
AkadaJ. K.ShiraiM.TakeuchiH.TsudaM.NakazawaT. (2000). Identification of the urease operon in Helicobacter pylori and its control by mRNA decay in response to pH. Mol. Microbiol.36, 1071–1084. 10.1046/j.1365-2958.2000.01918.x
3
AlgoodH. M. S.CoverT. (2006). Helicobacter pylori persistence: an overview of interactions between H. pylori and host immune defenses. Clin. Microbiol. Rev.19, 597–613. 10.1128/CMR.00006-06
4
AthertonJ. C.BlaserM. J. (2009). Coadaptation of Helicobacter pylori and humans: ancient history, modern implications. J. Clin. Invest.119, 2475–2487. 10.1172/JCI38605
5
BarcelouxD. G. (1999). Copper. J. Toxicol. Clin. Toxicol.37, 217–230. 10.1081/CLT-100102421
6
BeckerK. W.SkaarE. P. (2014). Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol. Rev.38, 1235–1249. 10.1111/1574-6976.12087
7
BehrensW.SchweinitzerT.BalJ.DorschM.BleichA.KopsF.et al. (2013). Role of energy sensor TlpD of Helicobacter pylori in gerbil colonization and genome analyses after adaptation in the gerbil. Infect. Immun.81, 3534–3551. 10.1128/IAI.00750-13
8
BeierD.SpohnG.RappuoliR.ScarlatoV. (1997). Identification and characterization of an operon of Helicobacter pylori that is involved in motility and stress adaptation. J. Bacteriol.179, 4676–4683.
9
BelzerC.StoofJ.van VlietA. H. (2007). Metal-responsive gene regulation and metal transport in Helicobacter species. Biometals20, 417–429. 10.1007/s10534-006-9028-9
10
BenoitS. L.MaierR. J. (2008). Hydrogen and nickel metabolism in Helicobacter species. Ann. N.Y. Acad. Sci.1125, 242–251. 10.1196/annals.1419.014
11
BenoitS. L.MaierR. J. (2011). Mua (HP0868) is a nickel-binding protein that modulates urease activity in Helicobacter pylori. MBio2, e00039–e00011. 10.1128/mBio.00114-11
12
BereswillS.GreinerS.van VlietA. H.WaidnerB.FassbinderF.SchiltzE.et al. (2000). Regulation of ferritin-mediated cytoplasmic iron storage by the ferric uptake regulator homolog (Fur) of Helicobacter pylori. J. Bacteriol.182, 5948–5953. 10.1128/JB.182.21.5948-5953.2000
13
BikE. M.EckburgP. B.GillS. R.NelsonK. E.PurdomE. A.FrancoisF.et al. (2006). Molecular analysis of the bacterial microbiota in the human stomach. Proc. Natl. Acad. Sci. U.S.A.103, 732–737. 10.1073/pnas.0506655103
14
BlaserM. J.Perez-PerezG. I.KleanthousH.CoverT. L.PeekR. M.ChyouP. H.et al. (1995). Infection with Helicobacter pylori strains possessing cagA is associated with increased risk of developing adenocarcinoma of the stomach. Cancer Res.55, 2111–2115.
15
BraymerJ. J.GiedrocD. P. (2014). Recent developments in copper and zinc homeostasis in bacterial pathogens. Curr. Opin. Chem. Biol.19, 59–66. 10.1016/j.cbpa.2013.12.021
16
Bury-MoneS.ThibergeJ.ContrerasM.MaitournamA.LabigneA.De ReuseH. (2004). Responsiveness to acidity via metal ion regulators mediates virulence in the gastric pathogen Helicobacter pylori. Mol. Microbiol.53, 623–638. 10.1111/j.1365-2958.2004.04137.x
17
CarpenterB. M.GilbreathJ. J.PichO. Q.McKelveyA. M.MaynardE. L.LiZ. Z.et al. (2013). Identification and characterization of novel Helicobacter pylori apo-Fur-regulated target genes. J. Bacteriol.195, 5526–5539. 10.1128/JB.01026-13
18
CassatJ. E.SkaarE. P. (2013). Iron in infection and immunity. Cell Host Microbe. 13, 509–519. 10.1016/j.chom.2013.04.010
19
ChanS. H.OpitzL.HigginsL.O'LoaneD.XuS. Y. (2010). Cofactor requirement of HpyAV restriction endonuclease. PLoS ONE5:e9071. 10.1371/journal.pone.0009071
20
ContrerasM.ThibergeJ. M.Mandrand-BerthelotM. A.LabigneA. (2003). Characterization of the roles of NikR, a nickel-responsive pleiotropic autoregulator of Helicobacter pylori. Mol. Microbiol.49, 947–963. 10.1046/j.1365-2958.2003.03621.x
21
CoverT. L.BlaserM. J. (2009). Helicobacter pylori in health and disease. Gastroenterology136, 1863–1873. 10.1053/j.gastro.2009.01.073
22
CoverT. L.PeekR. M.Jr. (2013). Diet, microbial virulence, and Helicobacter pylori-induced gastric cancer. Gut Microbes4, 482–493. 10.4161/gmic.26262
23
DanielliA.RomagnoliS.RoncaratiD.CostantinoL.DelanyI.ScarlatoV. (2009). Growth phase and metal-dependent transcriptional regulation of the fecA genes in Helicobacter pylori. J. Bacteriol.191, 3717–3725. 10.1128/JB.01741-08
24
DanielliA.RoncaratiD.DelanyI.ChiariniV.RappuoliR.ScarlatoV. (2006). In vivo dissection of the Helicobacter pylori Fur regulatory circuit by genome-wide location analysis. J. Bacteriol.188, 4654–4662. 10.1128/JB.00120-06
25
DanielliA.ScarlatoV. (2010). Regulatory circuits in Helicobacter pylori: network motifs and regulators involved in metal-dependent responses. FEMS Microbiol. Rev.34, 738–752. 10.1111/j.1574-6976.2010.00233.x
26
de BernardM.JosenhansC. (2014). Pathogenesis of Helicobacter pylori infection. Helicobacter19(Suppl. 1), 11–18. 10.1111/hel.12160
27
de ReuseH.VinellaD.CavazzaC. (2013). Common themes and unique proteins for the uptake and trafficking of nickel, a metal essential for the virulence of Helicobacter pylori. Front. Cell. Infect. Microbiol.3:94. 10.3389/fcimb.2013.00094
28
DelanyI.PachecoA. B.SpohnG.RappuoliR.ScarlatoV. (2001). Iron-dependent transcription of the frpB gene of Helicobacter pylori is controlled by the Fur repressor protein. J. Bacteriol.183, 4932–4937. 10.1128/JB.183.16.4932-4937.2001
29
DhaenensL.SzczebaraF.Van NieuwenhuyseS.HussonM. O. (1999). Comparison of iron uptake in different Helicobacter species. Res. Microbiol.150, 475–481. 10.1016/S0923-2508(99)00109-6
30
Diaz-OchoaV. E.JellbauerS.KlausS.RaffatelluM. (2014). Transition metal ions at the crossroads of mucosal immunity and microbial pathogenesis. Front. Cell. Infect. Microbiol.4:2. 10.3389/fcimb.2014.00002
31
DosanjhN. S.HammerbacherN. A.MichelS. L. J. (2007). Characterization of the Helicobacter pylori NikR-PureA DNA interaction:? Metal ion requirements and sequence specificity. Biochemistry46, 2520–2529. 10.1021/bi062092w
32
DosanjhN. S.MichelS. L. (2006). Microbial nickel metalloregulation: NikRs for nickel ions. Curr. Opin. Chem. Biol.10, 123–130. 10.1016/j.cbpa.2006.02.011
33
DraperJ.KarplusK.OttemannK. M. (2011). Identification of a chemoreceptor zinc-binding domain common to cytoplasmic bacterial chemoreceptors. J. Bacteriol.193, 4338–4345. 10.1128/JB.05140-11
34
EatonK. A.BrooksC. L.MorganD. R.KrakowkaS. (1991). Essential role of urease in pathogenesis of gastritis induced by Helicobacter pylori in gnotobiotic piglets. Infect. Immun. 59, 2470–2475.
35
EguchiY.UtsumiR. (2008). Introduction to bacterial signal transduction networks. Adv. Exp. Med. Biol.631, 1–6. 10.1007/978-0-387-78885-2_1
36
ErnstF. D.BereswillS.WaidnerB.StoofJ.MäderU.KustersJ. G.et al. (2005a). Transcriptional profiling of Helicobacter pylori Fur- and iron-regulated gene expression. Microbiology151(Pt 2), 533–546. 10.1099/mic.0.27404-0
37
ErnstF. D.HomuthG.StoofJ.MäderU.WaidnerB.KuipersE. J.et al. (2005b). Iron-responsive regulation of the Helicobacter pylori iron-cofactored superoxide dismutase SodB is mediated by Fur. J. Bacteriol.187, 3687–3692. 10.1128/JB.187.11.3687-3692.2005
38
ErnstF. D.StoofJ.HorrevoetsW. M.KuipersE. J.KustersJ. G.van VlietA. H. (2006). NikR mediates nickel-responsive transcriptional repression of the Helicobacter pylori outer membrane proteins FecA3 (HP1400) and FrpB4 (HP1512). Infect. Immun.74, 6821–6828. 10.1128/IAI.01196-06
39
EvansD. J.Jr.EvansD. G.KirkpatrickS. S.GrahamD. Y. (1991). Characterization of the Helicobacter pylori urease and purification of its subunits. Microb. Pathog.10, 15–26. 10.1016/0882-4010(91)90062-F
40
FassbinderF.van VlietA. H.GimmelV.KustersJ. G.KistM.BereswillS. (2000). Identification of iron-regulated genes of Helicobacter pylori by a modified fur titration assay (FURTA-Hp). FEMS Microbiol. Lett.184, 225–229. 10.1111/j.1574-6968.2000.tb09018.x
41
FischerW.BuhrdorfR.GerlandE.HaasR. (2001). Outer membrane targeting of passenger proteins by the vacuolating cytotoxin autotransporter of Helicobacter pylori. Infect. Immun.69, 6769–6775. 10.1128/IAI.69.11.6769-6775.2001
42
FordJ. L.KaakoushN. O.MendzG. L. (2010). Phosphonate metabolism in Helicobacter pylori. Antonie Leeuwenhoek97, 51–60. 10.1007/s10482-009-9387-7
43
FosterA. W.OsmanD.RobinsonN. J. (2014). Metal preferences and metalation. J. Biol. Chem.289, 28095–28103. 10.1074/jbc.R114.588145
44
GaddyJ. A.RadinJ. N.LohJ. T.PiazueloM. B.Kehl-FieT. E.DelgadoA. G.et al. (2014). The host protein calprotectin modulates the Helicobacter pylori cag type IV secretion system via zinc sequestration. PLoS Pathog.10:e1004450. 10.1371/journal.ppat.1004450
45
GanczH.CensiniS.MerrellD. S. (2006). Iron and pH homeostasis intersect at the level of Fur regulation in the gastric pathogen Helicobacter pylori. Infect. Immun.74, 602–614. 10.1128/IAI.74.1.602-614.2006
46
GeR. G.WangD. X.HaoM. C.SunX. S. (2013). Nickel trafficking system responsible for urease maturation in Helicobacter pylori. World J. Gastroenterol.19, 8211–8218. 10.3748/wjg.v19.i45.8211
47
GeZ.TaylorD. E. (1997). The Helicobacter pylori gene encoding phosphatidylserine synthase: sequence, expression, and insertional mutagenesis. J. Bacteriol.179, 4970–4976.
48
GilbreathJ. J.WestA. L.PichO. Q.CarpenterB. M.MichelS.MerrellS. (2012). Fur activates expression of the 2-oxoglutarate oxidoreductase genes (oorDABC) in Helicobacter pylori. J. Bacteriol.194, 6490–6497. 10.1128/JB.01226-12
49
GroismanE. A. (2001). The pleiotropic two-component regulatory system PhoP-PhoQ. J. Bacteriol.183, 1835–1842. 10.1128/JB.183.6.1835-1842.2001
50
HaleyK. P.BlanzE. J.GaddyJ. A. (2014). High resolution electron microscopy of the Helicobacter pylori Cag type IV secretion system pili produced in varying conditions of iron availability. J. Vis. Exp.93:e52122. 10.3791/52122
51
HaleyK. P.DelgadoA. G.PiazueloM. B.MortensenB. L.CorreaP.DamoS. M.et al. (2015). The human antimicrobial protein calgranulin C participates in control of Helicobacter pylori growth and regulation of virulence. Infect. Immun.83, 2944–2956. 10.1128/IAI.00544-15
52
HaleyK. P.GaddyJ. A. (2015). Helicobacter pylori: Genomic insight into the host-pathogen interaction. Int. J. Genomics2015:386905. 10.1155/2015/386905
53
HaleyK. P.SkaarE. P. (2012). A battle for iron: host sequestration and Staphylococcus aureus acquisition. Microbes Infect.14, 217–227. 10.1016/j.micinf.2011.11.001
54
HawtinP. R.DelvesH. T.NewellD. G. (1991). The demonstration of nickel in the urease of Helicobacter pylori by atomic absorption spectroscopy. FEMS Microbiol. Lett.61, 51–54. 10.1111/j.1574-6968.1991.tb04320.x
55
HerrmannL.SchwanD.GarnerR.MobleyH. L.HaasR.SchäferK. P.et al. (1999). Helicobacter pylori cadA encodes an essential Cd(II)-Zn(II)-Co(II) resistance factor influencing urease activity. Mol. Microbiol.33, 524–536. 10.1046/j.1365-2958.1999.01496.x
56
HoodM. I.SkaarE. P. (2012). Nutritional immunity: transition metals at the pathogen-host interface. Nat. Rev. Microbiol.10, 525–537. 10.1038/nrmicro2836
57
JohnsonM. D.Kehl-FieT. E.KleinR.KellyJ.BurnhamC.MannB.et al. (2015). Role of copper efflux in pneumococcal pathogenesis and resistance to macrophage-mediated immune clearance. Infect. Immun.83, 1684–1694. 10.1128/IAI.03015-14
58
JonesM. D.AdemiI.YinX.GongY.ZambleD. B. (2014). Nickel-responsive regulation of two novel Helicobacter pylori NikR-targeted genes. Metallomics7, 662–673. 10.1039/C4MT00210E
59
Kehl-FieT. E.SkaarE. P. (2010). Nutritional immunity beyond iron: a role for manganese and zinc. Curr. Opin. Chem. Biol.14, 218–224. 10.1016/j.cbpa.2009.11.008
60
KhanS.KarimA.IqbalS. (2009). Helicobacter urease: niche construction at the single molecule level. J. Biosci.34, 503–511. 10.1007/s12038-009-0069-4
61
LeeM. J.Chien-LiangL.TsaiJ. Y.SueW. T.HsiaW. S.HuangH. (2010). Identification and biochemical characterization of a unique Mn2+-dependent UMP kinase from Helicobacter pylori. Arch. Microbiol.192, 739–746. 10.1007/s00203-010-0600-x
62
LeeM. J.HuangH.LinW.YangR. R.LiuC. L.HuangC. Y. (2007). Activation of Helicobacter pylori inorganic pyrophosphatase and the importance of Cys16 in thermostability, enzyme activation and quaternary structure. Arch. Microbiol.188, 473–482. 10.1007/s00203-007-0267-0
63
LertsethtakarnP.OttemannK. M.HendrixsonD. R. (2011). Motility and chemotaxis in Campylobacter and Helicobacter. Annu. Rev. Microbiol.65, 389–410. 10.1146/annurev-micro-090110-102908
64
LetleyD. P.RheadJ. L.BishopK.AthertonJ. C. (2006). Paired cysteine residues are required for high levels of the Helicobacter pylori autotransporter VacA. Microbiology152(Pt 5), 1319–1325. 10.1099/mic.0.28548-0
65
MaierR. J. (2005). Use of molecular hydrogen as an energy substrate by human pathogenic bacteria. Biochem. Soc. Trans.33(Pt 1), 83–85. 10.1042/BST0330083
66
McGeeD. J.LangfordM. L.WatsonE. L.CarterJ. E.ChenY. T.OttemannK. M. (2005). Colonization and inflammation deficiencies in Mongolian gerbils infected by Helicobacter pylori chemotaxis mutants. Infect. Immun.73, 1820–1827. 10.1128/IAI.73.3.1820-1827.2005
67
MilesS.PiazueloM. B.Semino-MoraC.WashingtonM. K.DuboisA.PeekR. M.Jr.et al. (2010). Detailed in vivo analysis of the role of Helicobacter pylori Fur in colonization and disease. Infect. Immun.78, 3073–3082. 10.1128/IAI.00190-10
68
MontefuscoS.EspositoR.D'AndreaL.MontiM. C.DunneC.DolanB.ToscoA.et al. (2013). Copper promotes TFF1-mediated Helicobacter pylori colonization. PLoS ONE8:e79455. 10.1371/journal.pone.0079455
69
MullerC.BahlawaneC.AubertS.DelayC. M.SchauerK.Michaud-SoretI.et al. (2011). Hierarchical regulation of the NikR-mediated nickel response in Helicobacter pylori. Nucleic Acids Res.39, 7564–7575. 10.1093/nar/gkr460
70
NagataK.TsukitaS.TamuraT.SoneN. (1996). A cb-type cytochrome-c oxidase terminates the respiratory chain in Helicobacter pylori. Microbiology142(Pt 7), 1757–1763. 10.1099/13500872-142-7-1757
71
NeyrollesO.MintzE.CattyP. (2013). Zinc and copper toxicity in host defense against pathogens: Mycobacterium tuberculosis as a model example of an emerging paradigm. Front. Cell. Infect. Microbiol.3:89. 10.3389/fcimb.2013.00089
72
NeyrollesO.WolschendorfF.MitraA.NiederweisM. (2015). Mycobacteria, metals, and the macrophage. Immunol. Rev.264, 249–263. 10.1111/imr.12265
73
NotoJ. M.GaddyJ. A.LeeJ. Y.PiazueloM. B.FriedmanD. B.ColvinD. C.et al. (2013). Iron deficiency accelerates Helicobacter pylori-induced carcinogenesis in rodents and humans. J. Clin. Invest.123, 479–492. 10.1172/JCI64373
74
NotoJ. M.LeeJ. Y.GaddyJ. A.CoverT. L.AmievaM. R.PeekR. M.et al. (2014). Regulation of Helicobacter pylori virulence within the context of iron deficiency. J. Infect. Dis.211, 1790–1794. 10.1093/infdis/jiu805
75
OdenbreitS.PülsJ.SedlmaierB.GerlandE.FischerW.HaasR. (2000). Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science287, 1497–1500. 10.1126/science.287.5457.1497
76
PanthelK.DietzP.HaasR.BeierD. (2003). Two-component systems of Helicobacter pylori contribute to virulence in a mouse infection model. Infect. Immun.71, 5381–5385. 10.1128/IAI.71.9.5381-5385.2003
77
PfeifferJ.GuhlJ.WaidnerB.KistM.BereswillS. (2002). Magnesium uptake by CorA is essential for viability of the gastric pathogen Helicobacter pylori. Infect. Immun.70, 3930–3934. 10.1128/IAI.70.7.3930-3934.2002
78
PflockM.KennardS.DelanyI.ScarlatoV.BeierD. (2005). Acid-induced activation of the urease promoters is mediated directly by the ArsRS two-component system of Helicobacter pylori. Infect. Immun. 73, 6437–6445. 10.1128/IAI.73.10.6437-6445.2005
79
PhamA. N.XingG.MillerC. J.WaiteD. (2013). Fenton-like copper redox chemistry revisited: hydrogen peroxide and superoxide mediation of copper-catalyzed oxidant production. J. Catal.301, 54–64. 10.1016/j.jcat.2013.01.025
80
PichO. Q.CarpenterB. M.GilbreathJ. J.MerrellS. D. (2012). Detailed analysis of Helicobacter pylori Fur-regulated promoters reveals a Fur box core sequence and novel Fur-regulated genes. Mol. Microbiol.84, 921–941. 10.1111/j.1365-2958.2012.08066.x
81
RaderB. A.WredenC.HicksK. G.SweeneyE. G.OttemannK. M.GuilleminK. (2011). Helicobacter pylori perceives the quorum-sensing molecule AI-2 as a chemorepellent via the chemoreceptor TlpB. Microbiology157(Pt 9), 2445–2455. 10.1099/mic.0.049353-0
82
RadinJ. N.González-RiveraC.IvieS. E.McClainM. S.CoverT. L. (2011). Helicobacter pylori VacA induces programmed necrosis in gastric epithelial cells. Infect. Immun.79, 2535–2543. 10.1128/IAI.01370-10
83
RoligA. S.ShanksJ.CarterJ. E.OttemannK. M. (2012). Helicobacter pylori requires TlpD-driven chemotaxis to proliferate in the antrum. Infect. Immun.80, 3713–3720. 10.1128/IAI.00407-12
84
RomagnoliS.AgriestiF.ScarlatoV. (2011). In vivo recognition of the fecA3 target promoter by Helicobacter pylori NikR. J. Bacteriol.193, 1131–1141. 10.1128/JB.01153-10
85
SalamaN. R.OttoG.TompkinsL.FalkowS. (2000). Vacuolating cytotoxin of Helicobacter pylori plays a role during colonization in a mouse model of infection. Infect. Immun.69, 730–736. 10.1128/IAI.69.2.730-736.2001
86
SandersL.AndermannT. M.OttemannK. M. (2013). A supplemented soft agar chemotaxis assay demonstrates the Helicobacter pylori chemotactic response to zinc and nickel. Microbiology159(Pt 1), 46–57. 10.1099/mic.0.062877-0
87
SatinB.NoraisN.TelfordJ.RappuoliR.MurgiaM.MontecuccoC.et al. (1997). Effect of Helicobacter pylori vacuolating toxin on maturation and extracellular release of procathepsin D and on epidermal growth factor degradation. J. Biol. Chem.272, 25022–25028. 10.1074/jbc.272.40.25022
88
SchauerK.GougetB.CarrièreM.LabigneA.de ReuseH. (2007). Novel nickel transport mechanism across the bacterial outer membrane energized by the TonB/ExbB/ExbD machinery. Mol. Microbiol.63, 1054–1068. 10.1111/j.1365-2958.2006.05578.x
89
SempérteguiF.DíazM.MejíaR.Rodríguez-MoraO. G.RenteríaE.GuarderasC.et al. (2007). Low concentrations of zinc in gastric mucosa are associated with increased severity of Helicobacter pylori-induced inflammation. Helicobacter12, 43–48. 10.1111/j.1523-5378.2007.00476.x
90
SenkovichO.CeaserS.McGeeD. J.TestermanT. L. (2010). Unique host iron utilization mechanisms of Helicobacter pylori revealed with iron-deficient chemically defined media. Infect. Immun.78, 1841–1849. 10.1128/IAI.01258-09
91
SharmaC. M.HoffmannS.DarfeuilleF.ReignierJ.FindeissS.SittkaA.et al. (2010). The primary transcriptome of the major human pathogen Helicobacter pylori. Nature464, 250–255. 10.1038/nature08756
92
SilverS.WalderhaugM. (1992). Gene regulation of plasmid- and chromosome-determined inorganic ion transport in bacteria. Microbiol. Rev.56, 195–228.
93
SmithR. L.MaguireM. E. (1998). Microbial magnesium transport: unusual transporters searching for identity. Mol. Microbiol.28, 217–226. 10.1046/j.1365-2958.1998.00810.x
94
SrivastavaA.DwivediN.SamantaU.SauA. K. (2011). Insight into the role of a unique SSEHA motif in the activity and stability of Helicobacter pylori arginase. IUBMB Life63, 1027–1036. 10.1002/iub.552
95
StählerF. N.OdenbreitS.HaasR.WilrichJ.Van VlietA. H.KustersJ. G.et al. (2006). The novel Helicobacter pylori CznABC metal efflux pump is required for cadmium, zinc, and nickel resistance, urease modulation, and gastric colonization. Infect. Immun.74, 3845–3852. 10.1128/IAI.02025-05
96
StoofJ.KuipersE. J.KlaverG.van VlietA. H. (2010). An ABC transporter and a TonB ortholog contribute to Helicobacter mustelae nickel and cobalt acquisition. Infect. Immun.78, 4261–4267. 10.1128/IAI.00365-10
97
SundrudM. S.TorresV. J.UnutmazD.CoverT. L. (2004). Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. U.S.A.101, 7727–7732. 10.1073/pnas.0401528101
98
SydorA. M.LebretteH.AriyakumaranR.CavazzaC.ZambleD. B. (2014). Relationship between Ni(II) and Zn(II) coordination and nucleotide binding by the Helicobacter pylori [NiFe]-hydrogenase and urease maturation factor HypB. J. Biol. Chem.289, 3828–3841. 10.1074/jbc.M113.502781
99
SzabòI.BrutscheS.TombolaF.MoschioniM.SatinB.TelfordJ. L.et al. (1999). Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J.18, 5517–5527. 10.1093/emboj/18.20.5517
100
TanS.NotoJ. M.Romero-GalloJ.PeekR. M.Jr.AmievaM. R. (2011). Helicobacter pylori perturbs iron trafficking in the epithelium to grow on the cell surface. PLoS Pathog.7:e1002050. 10.1371/journal.ppat.1002050
101
TestermanT. L.ConnP. B.MobleyH. L.McGeeD. J. (2006). Nutritional requirements and antibiotic resistance patterns of Helicobacter species in chemically defined media. J. Clin. Microbiol.44, 1650–1658. 10.1128/JCM.44.5.1650-1658.2006
102
TorresV. J.Van CompernolleS. E.SundrudM. S.UnutmazD.CoverT. L. (2007). Helicobacter pylori vacuolating cytotoxin inhibits activation-induced proliferation of human T and B lymphocyte subsets. J. Immunol.179, 5433–5440. 10.4049/jimmunol.179.8.5433
103
TroxellB.HassanH. M. (2013). Transcriptional regulation by Ferric Uptake Regulator (Fur) in pathogenic bacteria. Front. Cell. Infect. Microbiol.3:59. 10.3389/fcimb.2013.00059
104
TummuruM. K.SharmaS. A.BlaserM. J. (1995). Helicobacter pylori picB, a homologue of the Bordetella pertussis toxin secretion protein, is required for induction of IL-8 in gastric epithelial cells. Mol. Microbiol.18, 867–876. 10.1111/j.1365-2958.1995.18050867.x
105
VanniniA.RoncaratiD.SpinsantiM.ScarlatoV.DanielliA. (2014). In depth analysis of the Helicobacter pylori cag pathogenicity island transcriptional responses. PLoS ONE9:e98416. 10.1371/journal.pone.0098416
106
WaidnerB.MelchersK.IvanovI.LofererH.BenschK. W.KistM.et al. (2002). Identification by RNA profiling and mutational analysis of the novel copper resistance determinants CrdA (HP1326), CrdB (HP1327), and CzcB (HP1328) in Helicobacter pylori. J. Bacteriol.184, 6700–6708. 10.1128/JB.184.23.6700-6708.2002
107
WaidnerB.MelchersK.StählerF. N.KistM.BereswillS. (2005). The Helicobacter pylori CrdRS two-component regulation system (HP1364/HP1365) is required for copper-mediated induction of the copper resistance determinant CrdA. J. Bacteriol.187, 4683–4688. 10.1128/JB.187.13.4683-4688.2005
108
WanQ.AhmadM. F.FairmanJ.GorzelleB.de la FuenteM.DealwisC.et al. (2011). X-ray crystallography and isothermal titration calorimetry studies of the Salmonella zinc transporter ZntB. Structure19, 700–710. 10.1016/j.str.2011.02.011
109
WenY.FengJ.ScottD. R.MarcusE. A.SachsG. (2011). A cis-encoded antisense small RNA regulated by the HP0165-HP0166 two-component system controls expression of ureB in Helicobacter pylori. J. Bacteriol.193, 40–51. 10.1128/JB.00800-10
110
WilliamsS. M.ChenY. T.AndermannT. M.CarterJ. E.McGeeD. J.OttemannK. M. (2007). Helicobacter pylori chemotaxis modulates inflammation and bacterium-gastric epithelium interactions in infected mice. Infect. Immun.75, 3747–3757. 10.1128/IAI.00082-07
111
WolschendorfF.AckartD.ShresthaT. B.Hascall-DoveL.NolanS.LamichhaneG.et al. (2011). Copper resistance is essential for virulence of Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A.108, 1621–1626. 10.1073/pnas.1009261108
112
WorstD. J.GerritsM. M.Vandenbroucke-GraulsC. M.KustersJ. G. (1998). Helicobacter pylori ribBA-mediated riboflavin production is involved in iron acquisition. J. Bacteriol.180, 1473–1479.
113
YenC. C.ShenC. J.HsuW. H.ChangY. H.LinH. T.ChenH. L.et al. (2011). Lactoferrin: an iron-binding antimicrobial protein against Escherichia coli infection. Biometals24, 585–594. 10.1007/s10534-011-9423-8
114
ZhaoX.DrlicaK. (2014). Reactive oxygen species and the bacterial response to lethal stress. Curr. Opin. Chem. Biol.21, 1–6. 10.1016/j.mib.2014.06.008
Summary
Keywords
Helicobacter pylori, gene, regulation, metal, virulence
Citation
Haley KP and Gaddy JA (2015) Metalloregulation of Helicobacter pylori physiology and pathogenesis. Front. Microbiol. 6:911. doi: 10.3389/fmicb.2015.00911
Received
29 May 2015
Accepted
19 August 2015
Published
02 September 2015
Volume
6 - 2015
Edited by
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, China
Reviewed by
Dominic Chih-Cheng Voon, Cancer Science Institute of Singapore, Singapore; D. Scott Merrell, Uniformed Services University, USA; Mathieu F. Cellier, Institut National de la Recherche Scientifique, Canada; Flavia Vischi Winck, Brazilian Center for Research in Energy and Materials, Brazil
Updates
Copyright
© 2015 Haley and Gaddy.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jennifer A. Gaddy, Division of Infectious Diseases, Vanderbilt University School of Medicine, A2200 Medical Center North, 1161 21st Avenue South, Nashville, TN 37232, USA jennifer.a.gaddy@vanderbilt.edu
This article was submitted to Food Microbiology, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.