 Anastasia A. Ivanova
Anastasia A. Ivanova Daniil G. Naumoff
Daniil G. Naumoff Kirill K. Miroshnikov
Kirill K. Miroshnikov Werner Liesack
Werner Liesack Svetlana N. Dedysh
Svetlana N. Dedysh- 1Winogradsky Institute of Microbiology, Research Center of Biotechnology of the Russian Academy of Sciences, Moscow, Russia
- 2Max-Planck-Institute for Terrestrial Microbiology, Marburg, Germany
The family Isosphaeraceae accommodates stalk-free planctomycetes with spherical cells, which can be assembled in short chains, long filaments, or aggregates. These bacteria inhabit a wide variety of terrestrial environments, among those the recently described Paludisphaera borealis PX4T that was isolated from acidic boreal wetlands. Here, we analyzed its finished genome in comparison to those of three other members of the Isosphaeraceae: Isosphaera pallida IS1BT, Singulisphaera acidiphila DSM 18658T, and the uncharacterized planctomycete strain SH-PL62. The complete genome of P. borealis PX4T consists of a 7.5 Mb chromosome and two plasmids, 112 and 43 kb in size. Annotation of the genome sequence revealed 5802 potential protein-coding genes of which 2775 could be functionally assigned. The genes encoding metabolic pathways common for chemo-organotrophic bacteria, such as glycolysis, citrate cycle, pentose-phosphate pathway, and oxidative phosphorylation were identified. Several genes involved in the synthesis of peptidoglycan as well as N-methylated ornithine lipids were present in the genome of P. borealis PX4T. A total of 26 giant genes with a size >5 kb were detected. The genome encodes a wide repertoire of carbohydrate-active enzymes (CAZymes) including 44 glycoside hydrolases (GH) and 83 glycosyltransferases (GT) affiliated with 21 and 13 CAZy families, respectively. The most-represented families are GH5, GH13, GH57, GT2, GT4, and GT83. The experimentally determined carbohydrate utilization pattern agrees well with the genome-predicted capabilities. The CAZyme repertoire in P. borealis PX4T is highly similar to that in the uncharacterized planctomycete SH-PL62 and S. acidiphila DSM 18658T, but different to that in the thermophile I. pallida IS1BT. The latter strain has a strongly reduced CAZyme content. In P. borealis PX4T, many of its CAZyme genes are organized in clusters. Contrary to most other members of the order Planctomycetales, all four analyzed Isosphaeraceae planctomycetes have plasmids in numbers varying from one to four. The plasmids from P. borealis PX4T display synteny to plasmids from other family members, providing evidence for their common evolutionary origin.
Introduction
The planctomycetes is a largely unexplored bacterial phylum that accommodates microorganisms with distinctive cell morphology and a unique cellular architecture (Schlesner and Stackebrandt, 1986; Ward, 2010; Fuerst and Sagulenko, 2011). Three distinct orders of planctomycetes are currently recognized, namely the Planctomycetales, Phycisphaerales, and Candidatus Brocadiales. The order Planctomycetales accommodates chemo-organotrophs that reproduce by budding; cells of these bacteria typically possess various multifibrillar appendages described as stalks, spikes, spines, and fimbriae (Ward, 2010). Representatives of the Phycisphaerales are also chemo-organotrophs but reproduce by binary fission (Fukunaga et al., 2009). Finally, the order Candidatus Brocadiales contains chemo-lithoautotrophic planctomycetes capable of anaerobic oxidation of ammonium (anammox; Strous et al., 1999; Jetten et al., 2009).
Planctomycetes display a number of distinctive features that are highly unusual among bacteria. Cells of planctomycetes possess a well-developed endomembrane system, which is especially pronounced in some members of the Planctomycetales (Fuerst and Sagulenko, 2011; Santarella-Mellwig et al., 2013; Devos and Ward, 2014). The ability to oxidize ammonium in anammox planctomycetes is dependent on a characteristic membrane-bound cell compartment called the anammoxosome (Neumann et al., 2014). Planctomycetes are also exceptions to the otherwise dominant mode of division by binary fission, which is based on the interaction between the FtsZ protein and the peptidoglycan (PG) biosynthesis machinery (Rivas-Marín et al., 2016). They lack a recognizable homolog of FtsZ, which plays a central role in binary fission and is conserved in almost all bacteria. PG, in its turn, has historically been thought to be absent from cell walls of planctomycetes. Recently, however, it has been demonstrated that planctomycetal genomes encode the proteins required for PG synthesis and that the cells do possess a typical PG-containing cell wall (Jeske et al., 2015; van Teeseling et al., 2015). Although the alternative division mechanism as well as the proteins involved in cell morphogenesis in planctomycetes remain enigmatic (Jogler et al., 2012; Rivas-Marín et al., 2016), these bacteria are now considered as variations of, but not exceptions to, the Gram-negative cell plan (Devos and Ward, 2014).
Planctomycetes possess large genomes, 5.5–10.1 Mb in size (Guo et al., 2014). The function, however, can be predicted for only 30–55% proteins encoded in these genomes, while the remaining proteins are usually annotated as hypothetical proteins with unknown function (Jogler et al., 2012; Guo et al., 2014). One additional feature specific for all available planctomycete genomes is the presence of so-called “giant genes” (Reva and Tümmler, 2008; Kohn et al., 2016). The majority of giant genes (with a size >5 kb) were found in non-pathogenic bacteria and annotated to encode either a cell surface protein or a non-ribosomal peptide or polyketide synthase (PK) (Reva and Tümmler, 2008). Highest numbers of giant genes (up to 60 per genome) are observed in members of the order Planctomycetales (Kohn et al., 2016).
Planctomycetes are widely distributed in various environments. As revealed by molecular surveys, these bacteria are common inhabitants of boreal Sphagnum-dominated peatlands (Dedysh, 2011; Ivanova and Dedysh, 2012; Serkebaeva et al., 2013; Tveit et al., 2013; Moore et al., 2015; Ivanova et al., 2016). Although several peat-inhabiting planctomycetes have been obtained in pure cultures (Dedysh and Kulichevskaya, 2013), we know very little about their potential functions in the environment. Based on our current knowledge, these bacteria are slow-acting decomposers of plant-derived organic matter. Given that most conventional tests used for assessing hydrolytic capabilities were designed for fast-growing bacteria, the analysis of degradation capabilities of peat-inhabiting planctomycetes is complicated by their slow growth rates. One of the promising strategies to circumvent these difficulties and unveil the hidden potential of slow-growing bacteria is the comparative genomic approach. For this reason, we sequenced and analyzed the genome of the recently described planctomycete Paludisphaera borealis PX4T, which was isolated from boreal Sphagnum peat bog of northern Russia (Kulichevskaya et al., 2016). This bacterium is a chemo-organotrophic, mildly acidophilic, and psychrotolerant aerobe. Cells of this planctomycete are Gram-negative, non-motile spheres that occur singly or in short chains.
The apparent ability of hydrolyzing gellan gum (a complex heteropolysaccharide of microbial origin; PhytagelTM), as well as several other polysaccharides, makes P. borealis PX4T an attractive target for genome-based studies on its carbohydrate metabolism. Carbohydrate-active enzymes (CAZymes) are responsible for synthesis and degradation of oligo- and polysaccharides as well as their derivatives. CAZymes include glycoside hydrolases (GH), glycosyltransferases (GT), polysaccharide lyases (PL), and carbohydrate esterases (CE). Based on homology of the catalytic domains, they form about 250 protein families, including 129 families of glycoside hydrolases and transglycosidases (GH1–GH135, except for GH21, GH40, GH41, GH60, GH61, and GH69) (Lombard et al., 2014; Terrapon et al., 2017). The CAZyme repertoire of microorganisms is strongly determined by the ecological niche they occupy (Naumoff, 2011b).
Paludisphaera borealis PX4T is a member of the family Isosphaeraceae in the order Planctomycetales (Kulichevskaya et al., 2016). This family accommodates stalk-free planctomycetes with spherical cells, which can be assembled in short chains, long filaments, or shapeless aggregates. Other described genera in this family are Isosphaera, Singulisphaera, and Aquisphaera. At present, complete genome sequences are available for three members of this family, i.e., Isosphaera pallida IS1BT, S. acidiphila DSM 18658T, and the uncharacterized planctomycete strain SH-PL62 (Figure 1). Comparative genomic analysis between P. borealis PX4T and the other three members of the Isosphaeraceae revealed remarkable similarity in their genome organization. All four planctomycetes harbor plasmids which share multiple homologous regions. Many genes in P. borealis PX4T have their closest homologs in strain SH-PL62 and S. acidiphila DSM 18658T. Here, we present the genomic characteristics of P. borealis PX4T together with a detailed description of its CAZyme repertoire.
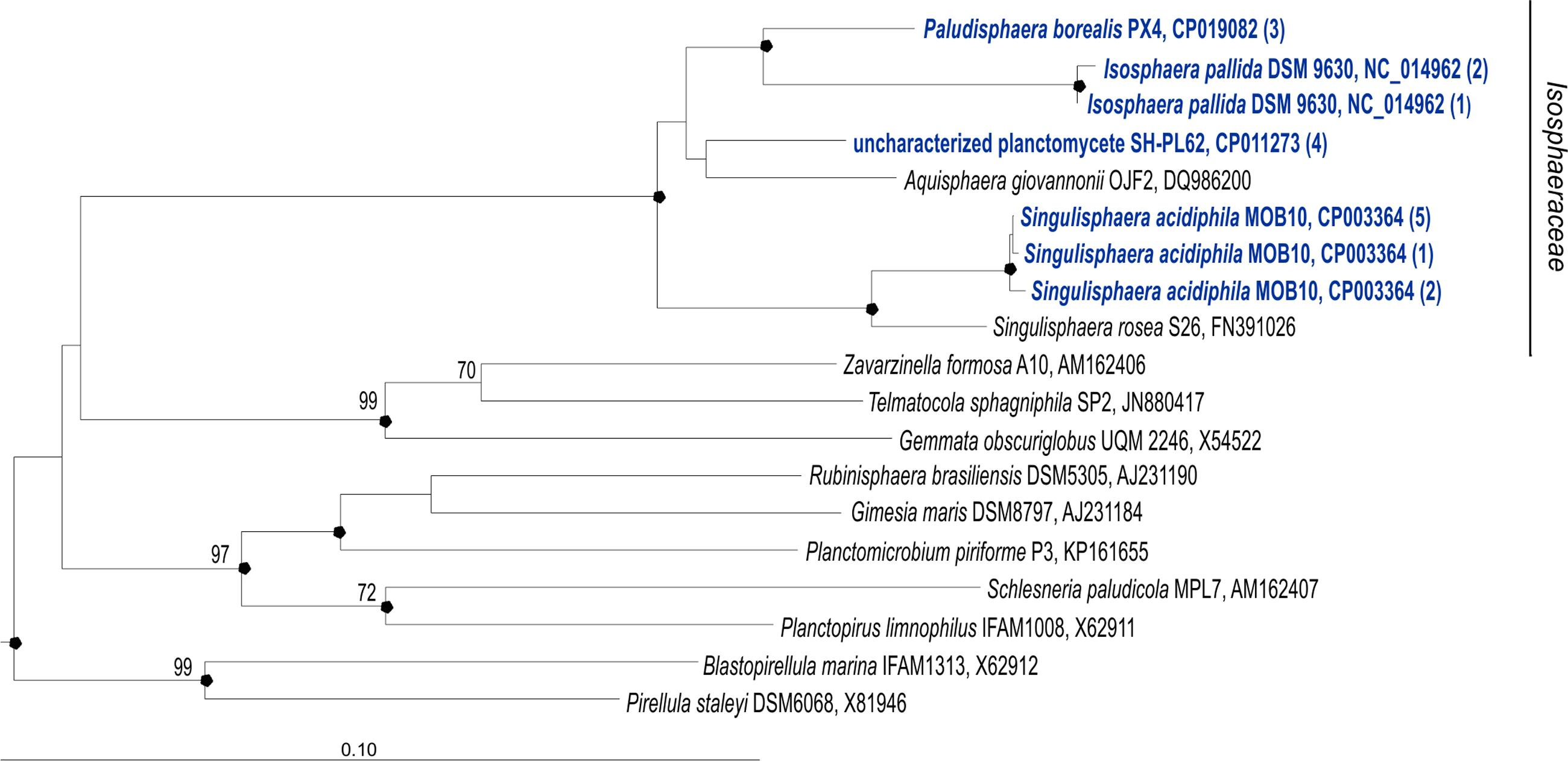
FIGURE 1. 16S rRNA gene-based neighbor-joining tree (Jukes–Cantor correction) showing the phylogenetic position of Paludisphaera borealis PX4T in relation to the three Isosphaeraceae reference organisms used in this study, i.e., Isosphaera pallida IS1BT, Singulisphaera acidiphila DSM 18658T, and undescribed planctomycete strain SH-PL62S (shown in blue). All 16S rRNA gene copies revealed in the genomes of these four planctomycetes are included in the tree. The number of identical 16S rRNA gene copies is shown in parenthesis. Different 16S rRNA gene copies of S. acidiphila DSM 18658T are grouped as follows (locus-tags are given): S. acidiphila (5)—Sinac_3310, Sinac_3707, Sinac_3773, Sinac_5935, Sinac_6238; S. acidiphila (2)—Sinac_2684, Sinac_4695; S. acidiphila (1)—Sinac_5862. Different 16S rRNA gene copies of I. pallida IS1BT are grouped as follows (locus-tags are given): I. pallida (2)— Isop_R0040, Isop_R0051; I. pallida (1)—Isop_R0061. The significance levels of interior branch points obtained in neighbor-joining analysis were determined by bootstrap analysis (1000 data re-samplings). Bootstrap values (1000 data resamplings) of ≥70% are shown. Black circles indicate that the corresponding nodes were also recovered in the maximum-likelihood and maximum-parsimony trees. The root (not shown) was composed of five 16S rRNA gene sequences from anammox planctomycetes (AF375994, AF375995, AY254883, AY257181, and AY254882). Bar, 0.1 substitutions per nucleotide position.
Materials and Methods
Cultivation Procedures
Paludisphaera borealis PX4T (= DSM 28747T = VKM B-2904T) was grown in shaking liquid cultures at 24°C in M2 medium of the following composition (g l−1 distilled water): KH2PO4, 0.1; (NH4)2SO4, 0.1; MgSO4 × 2H2O, 0.1; CaCl2 × 2H2O, 0.02; yeast extract (Difco), 0.02; glucose (Sigma), 0.5; mineral salt solution “44,” 1 ml (Staley et al., 1992); pH 5.5–5.8. After 3 weeks of incubation, the biomass was collected and transferred to the Max-Planck Genome Centre Cologne (MP-GCC, Germany) for DNA extraction, library preparation, and sequencing.
To verify the presence of several hydrolytic capabilities predicted by genome analysis, we performed additional substrate utilization tests as outlined in the original description of P. borealis (Kulichevskaya et al., 2016). For these tests, glucose was omitted from medium M2 and the latter was supplemented with 0.05 % (w/v) of the corresponding substrate. The growth was examined by measuring the rate of CO2 production in tightly closed 120 ml flasks containing 10 ml liquid medium M2 with tested substrate for 3 weeks at 24°C. Control incubations were run in parallel under the same conditions but without substrate.
Genome Sequencing
Genome sequencing of strain PX4T was performed at the MP-GCC, using the PacBio RSII platform with a single SMRT® cell (Pacific Biosciences, Menlo Park, CA, USA). A total of 56,515 sequences were obtained with a mean length of 9279.29 bp (9.28 kb) [total length = 524,419,229 bp or 524,419 kb; N50 value = 12,777 bp (12.78 kb)]. De novo assembly was done using the hierarchical genome-assembly process (HGAP2) via the SMRT Portal v.2.0 offered by Pacific Biosciences (Chin et al., 2013). The parameter settings were as follows: (i) minimum subread length: 500; (ii) minimum polymerase read quality: 0.8; (iii) minimum polymerase read length: 100; (iv) minimum seed read length: 6000; (v) overlapper error rate: 0.06; (vi) minimum overlapping length: 40; and (vii) overlapping K-mer: 14. The draft assembly was manually checked and redundant terminal sequences were removed.
Genome Annotation
Initial automated genome annotation was carried out using RAST v. 2.0 (Rapid Annotation using Subsystem Technology) with default parameters (Aziz et al., 2008; Overbeek et al., 2014; Brettin et al., 2015). Subsequent inspection was done in PROKKA package (Seemann, 2014) including all dependencies such as PRODIGAL v 2.6.2 (Hyatt et al., 2010), HMMER server (Finn et al., 2011), RNAMMER (Lagesen et al., 2007), BLAST+ (Camacho et al., 2009), and ARAGORN (Laslett and Canback, 2004). Annotation with PROKKA was performed against both the UNIPROT database (Apweiler et al., 2009) and a manually constructed database that includes all available annotated planctomycete genome sequences. Their visualization was done in BRIG program (Alikhan et al., 2011).
The analysis of P. borealis PX4T genome sequence using the Kyoto Encyclopedia of Genes and Genomes (KEGG) was performed applying GhostKOALA tool (Kanehisa et al., 2016). Screening for secondary metabolite-related genes was performed using the online web server antiSMASH3.0.5 (Antibiotics & Secondary Metabolites Analysis Shell; Medema et al., 2011; Blin et al., 2013; Weber et al., 2015).
The automated annotation of CAZymes by RAST v. 2.0 was manually checked in order to validate their affiliation to CAZy (Lombard et al., 2014) and PFAM (Finn et al., 2016) families. Additionally, all Isosphaeraceae proteins listed in the CAZy database were used as queries for blastp searches of the P. borealis PX4T proteome. Representatives of each GHL (Naumoff, 2011a, 2016; Naumoff and Stepuschenko, 2011), FURAN (Naumoff, 2012), PFAM (Naumoff, 2011b), and COG (Naumoff, 2011b) family of putative glycoside hydrolases were used as queries, as well. Identified CAZymes were analyzed for both possible alternative start-codons and domain structure. Predicted catalytic domains were used as queries for blastp searches in an iterative manner. The dbCAN server (Yin et al., 2012) was used to classify all obtained proteins into existing CAZy families. Remaining unclassified proteins were analyzed and annotated manually. Each gene encoding an incomplete catalytic domain was considered a pseudogene.
The annotated genome sequence of P. borealis PX4T has been deposited in the GenBank database under BioProject number PRJNA354242 (sequence accession numbers CP019082-CP019084).
Phylogenetic Analyses
Phylogenetic analysis of 16S rRNA gene sequences from P. borealis PX4T and other representative members of the order Planctomycetales was carried out using the ARB program (Ludwig et al., 2004). The significance levels of interior branch points obtained in the neighbor-joining analysis were determined by bootstrap analysis (based on 1000 data re-samplings). The stability of various nodes were also confirmed with PHYLIP maximum-likelihood and maximum-parsimony methods (Felsenstein, 1989) implemented in ARB package.
Comparative Genomics
In addition to P. borealis PX4T, the finished genome sequences of three other Isosphaeraceae planctomycetes were used for comparative analysis (NCBI accession number in parenthesis): I. pallida IS1BT (BioProject No PRJNA32825), S. acidiphila DSM 18658T (PRJNA52461), and the uncharacterized planctomycete strain SH-PL62 (PRJNA277747) (Figure 1). The overall similarities between the genome of P. borealis PX4T and the three reference genomes were estimated using average nucleotide identity (ANI) calculator and formula 2 of the Genome-to-Genome-Distance-Calculator (Auch et al., 2010a,b) under parameter settings proposed elsewhere (Meier-Kolthoff et al., 2013).
Results
Genome Properties
The genome of P. borealis PX4T consists of a 7.498 Mb chromosome and two plasmids, a large plasmid of 111.833 kb and a small plasmid of 42.519 kb (Tables 1, 2 and Figures 2, 3). The GC content of the chromosomal DNA is 66.3%. The corresponding values for the large and small plasmids are 65.3% and 59.3%, respectively. All three sequences (chromosome and plasmids) are finished and have no gaps. Three copies of 16S-23S-5S rRNA operon and 79 tRNA genes were identified. Annotation of the genome sequence revealed 5802 potential protein-coding genes of which 2775 could be functionally assigned. The genome contains a single Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR) locus and a set of CRISPR-associated (cas) genes.

TABLE 1. Genome statistics for planctomycetes of the family Isosphaeraceae.
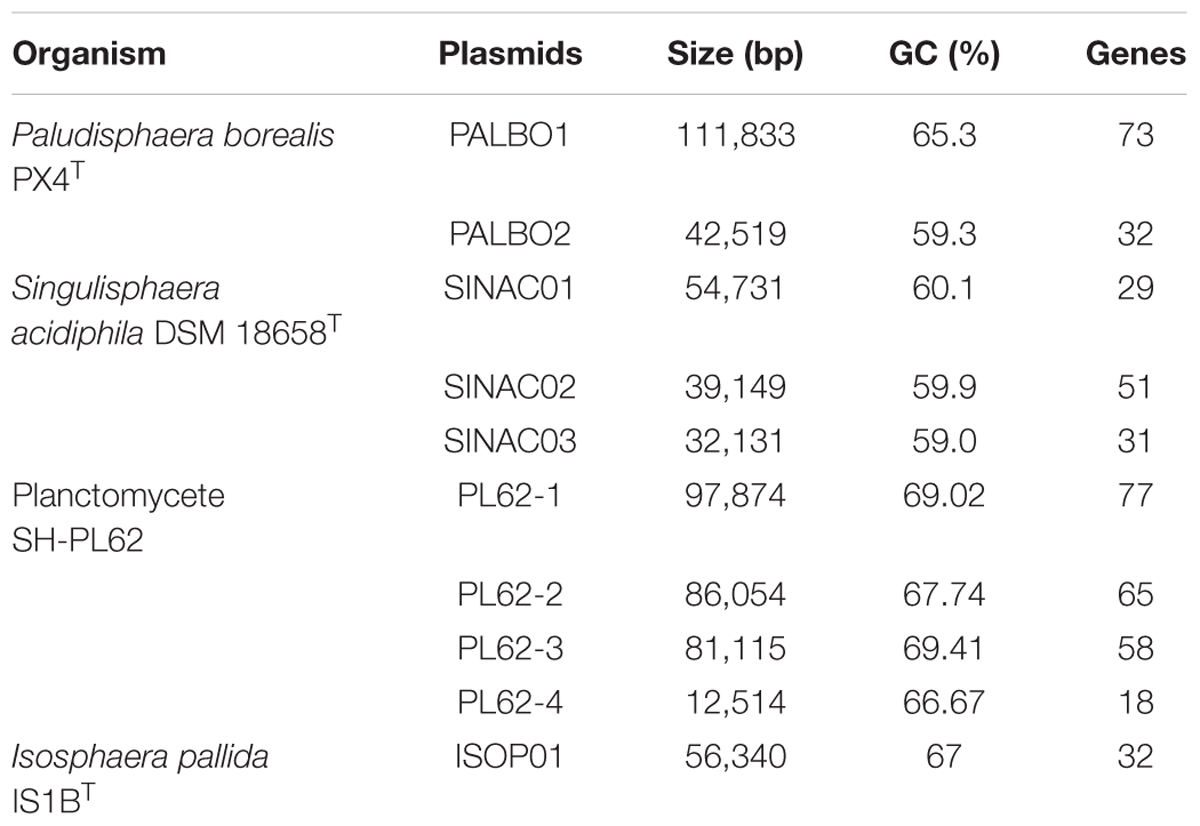
TABLE 2. Plasmid overview.
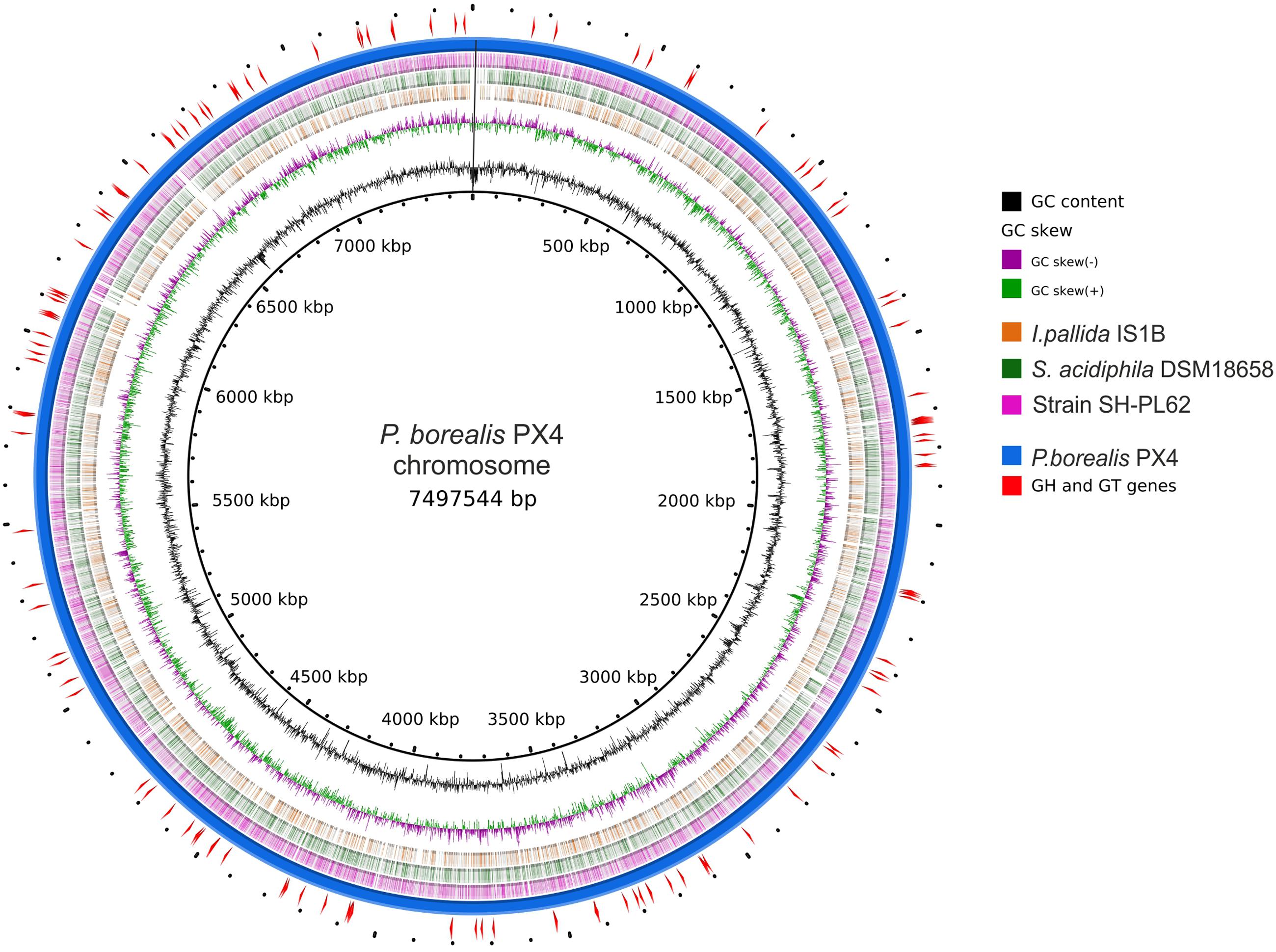
FIGURE 2. Scheme of Paludisphaera borealis PX4T genome. The innermost circle gives the genome coordinates. The next two inner rings display the GC skew and the G + C content along the genome. Next three rings indicate the chromosome regions with nucleotide sequence identity of 70–100% (based on blastn) between P. borealis PX4T and the three Isosphaeraceae reference organisms: Isosphaera pallida IS1BT, Singulisphaera acidiphila DSM 18658T, and strain SH-PL62, respectively. The outermost two rings show the chromosome of P. borealis PX4T and the positions of genes encoding glycoside hydrolases and glycosyltransferases.
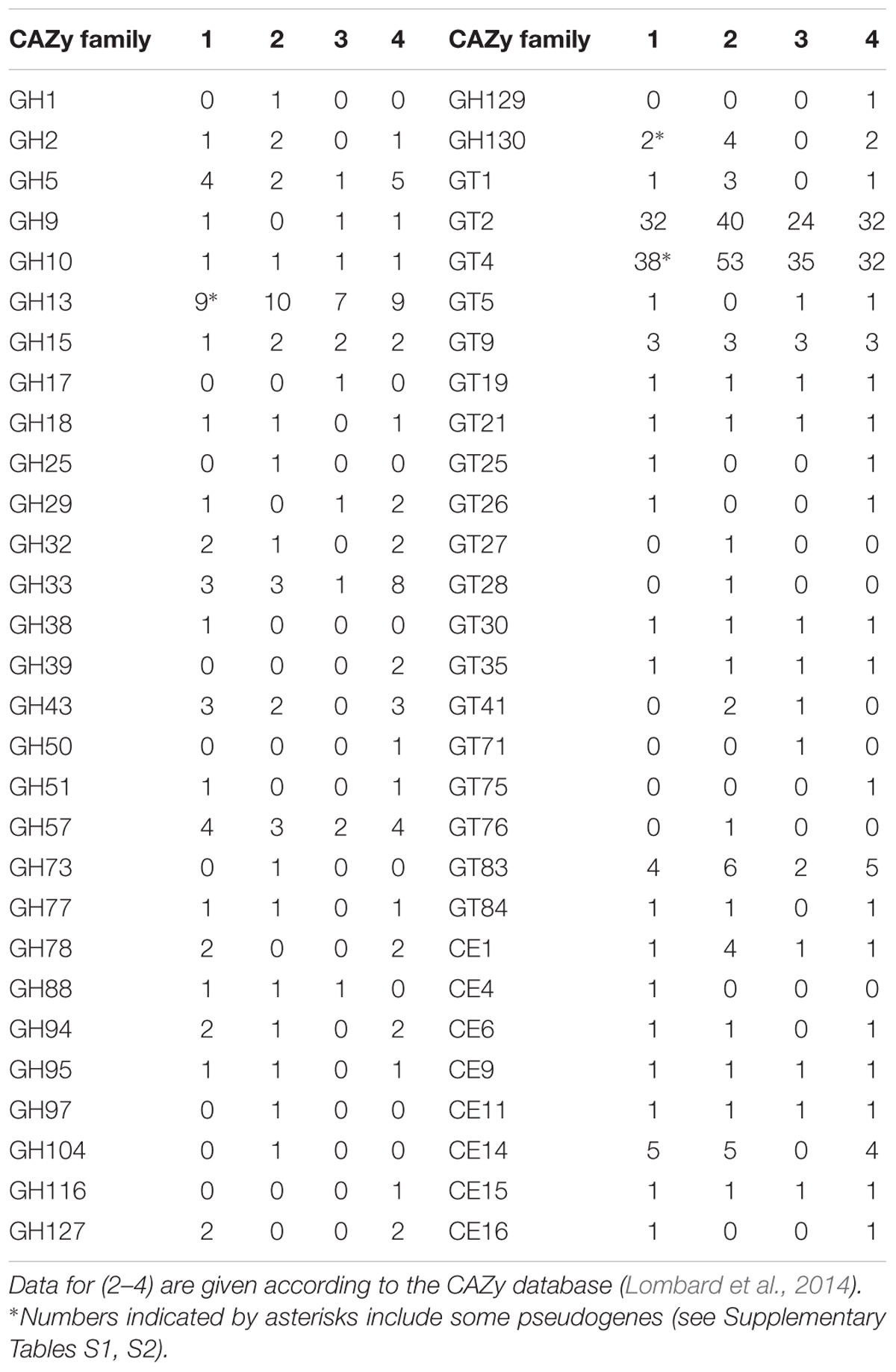
TABLE 3. Families of carbohydrate-active enzymes represented in P. borealis PX4T (1), S. acidiphila DSM18658T (2), I. pallida IS1BT (3), and planctomycete SH-PL62 (4).
The genome characteristics of P. borealis PX4T in comparison to those of I. pallida IS1BT, S. acidiphila DSM 18658T, and strain SH-PL62 are summarized in Table 1. The genome size in these bacteria varies from 5.529 Mb in thermophilic I. pallida IS1BT to 9.742 Mb in S. acidiphila DSM 18658T. The G + C content range within the Isosphaeraceae is 62.4–66.3 mol%; it is lowest in I. pallida IS1BT and highest in P. borealis PX4T. The number of ribosomal operons is highest in S. acidiphila DSM 18658T (eight), which also possesses greatest number of CRISPR repeats (five).
Phylogenetic Analyses
No sequence differences were observed between the three 16S rRNA gene copies present in the genome of P. borealis PX4T (Figure 1). The same was true for the four 16S rRNA gene copies in the genome of strain SH-PL62. Two of the three 16S rRNA gene copies in I. pallida IS1BT are also identical but display two mismatches to the third copy. Finally, eight copies of 16S rRNA gene sequences present in the genome of S. acidiphila DSM 18658T can be divided into three types, containing five, two, and one sequence(s), respectively. These three sequence types, however, differ by one to four nucleotide positions only. The use of one or another sequence type for phylogenetic inference does not affect the tree topology.
The phylogenetic position of P. borealis PX4T relative to the other Isosphaeraceae planctomycetes is shown in Figure 1. P. borealis PX4T displayed 91 and 92% 16S rRNA gene sequence similarity to I. pallida IS1BT and S. acidiphila DSM 18658T, respectively. The corresponding similarity value to the 16S rRNA gene sequence of strain SH-PL62 is 95%. This strain forms a separate phylogenetic lineage and, most likely, represents a novel genus within the Isosphaeraceae, despite its relatively high 16S rRNA gene similarity with P. borealis PX4T.
Paludisphaera borealis PX4T shares the following overall genome similarities with the three reference organisms: 20.2 ± 2.2% (S. acidiphila DSM 18658T); 19.8 ± 2.3% (I. pallida IS1BT); and 16.7 ± 2.3% (strain SH-PL62). These DNA–DNA hybridization values were estimated using formula 2 of the Genome-to-Genome-Distance-Calculator. They are in the range generally calculated for members of different genera (Scheuner et al., 2014). Accordingly, the ANI values shared between the genomes of P. borealis PX4T and the other three Isosphaeraceae are also very low: 77% (S. acidiphila DSM 18658T); 75% (I. pallida IS1BT); and 73% (strain SH-PL62).
Metabolism and Cell Biology
KEGG-based annotation of P. borealis PX4T genome sequence classified 1784 proteins into 17 major functional categories. The annotated genomes of S. acidiphila DSM 18658T, I. pallida IS1BT, and strain SH-PL62 were available via the KEGG database. The distribution of genes among the major KEGG categories was similar in all four examined planctomycete genomes, with slight variations in gene numbers in some of the categories (Supplementary Figure S1).
The genes encoding metabolic pathways common for chemo-organotrophic bacteria, such as glycolysis, the citrate cycle, the pentose-phosphate pathway, and oxidative phosphorylation were present in the genome of P. borealis PX4T. This planctomycete has the genomic potential for synthesis of all amino acids. The number of ABC-transporters in P. borealis PX4T (35) is somewhat smaller than those identified in other members of the Isosphaeraceae (40 in I. pallida IS1BT to 48 in S. acidiphila DSM 18658T). These numbers are comparable to the calculated mean of 49 ABC-transporters in free-living prokaryotes (Glöckner et al., 2003). Also, two fructose-type sugar-specific subunits of phospho-transferase system could be found in strain PX4T.
Most genes essential for chemotaxis were identified in the genome of P. borealis PX4T. These include cheA, cheB, cheR, and cheW. Similar gene arrays were present in S. acidiphila DSM 18658T and strain SH-PL62, but only cheW was identified in I. pallida IS1BT. Poor representation of genes responsible for flagellar assembly in the four examined planctomycetes agrees well with the fact that all described members of the family Isosphaeraceae do not produce motile swarmer cells as typical for planctomycetes from other families of the order Planctomycetales.
The survey for genes related to cell division revealed a situation characteristic of other planctomycetes (Rivas-Marín et al., 2016). Namely, the FtsZ-encoding gene was absent, while two copies of the gene coding for FtsK, the DNA translocase, were present in the genome of P. borealis PX4T. The cytoskeletal protein MreB whose gene has a patchy presence among the planctomycetes and is absent in S. acidiphila DSM 18658T (Rivas-Marín et al., 2016), is also missing in the other three members of the Isosphaeraceae. Several but not all genes involved in PG biosynthesis, including murB, murE, and mraY, were detected.
A few years ago, the presence of novel N-methylated ornithine membrane lipids (OLs) was reported for several peat-inhabiting planctomycetes, including S. acidiphila DSM 18658T (Moore et al., 2013). OLs are phosphorus-free membrane lipids widespread in bacteria but absent from archaea and eukaryotes. Recently, the gene encoding the key enzyme for synthesis of N-methylated OLs, N-methyltransferase (OlsG), was identified in the genome of S. acidiphila DSM 18658T (Sinac_1600; Escobedo-Hinojosa et al., 2015). We revealed homologs of Sinac_1600 in the genomes of P. borealis PX4T and the other two Isosphaeraceae members studied here as well as in the genomes of two Gemmataceae planctomycetes, Zavarzinella formosa A10T and Gemmata sp. SH-PL17. This suggests that N-methylation of OLs is a common trait among members of these planctomycete families.
Finally, P. borealis PX4T and S. acidiphila DSM 18658T possessed the greatest genetic potential for biosynthesis of secondary metabolites and antibiotics among the four Isosphaeraceae planctomycetes (Supplementary Figure S1).
CAZyme Repertoire
Homology analysis of all proteins potentially encoded in the genome of P. borealis PX4T was performed in order to reveal the complete set of CAZymes. As a result, 44 glycoside hydrolases, 83 glycosyltransferases, and 12 carbohydrate esterases belonging to, respectively, 21, 13, and 8 CAZy families were detected (Table 3 and Supplementary Tables S1–S3). The CAZyme repertoire in P. borealis PX4T was highly similar to those in strain SH-PL62 and S. acidiphila DSM 18658T, but different to that in I. pallida IS1BT (Table 3). The latter organism is thermophilic and has a strongly reduced CAZyme content, but the enzymes belong mainly to the same protein families as those from the other Isosphaeraceae planctomycetes. Additionally, we predict a significant number of proteins, which do not belong to any of the currently recognized CAZy families (Lombard et al., 2014) but display a distant relationship to some glycoside hydrolases, glycosyltransferases, or carbohydrate esterases (Supplementary Tables S4–S6). The majority of CAZymes from P. borealis PX4T was most closely related to those identified in strain SH-PL62 (Supplementary Tables S1–S6).
Many CAZyme genes of P. borealis PX4T are organized in clusters. We identified nine gene clusters, each containing at least three glycosyltransferase and/or glycoside hydrolase genes (not shown). Two largest clusters include eight (BSF38_01353–BSF38_01364) and five (BSF38_01716–BSF38_01727) glycosyltransferase genes. The third cluster is composed of three genes coding for GH13-family proteins (BSF38_02409, BSF38_02411, BSF38_02412). One of the remaining clusters is located on plasmid pPALBO1 (see below). One of two isoamylase genes (BSF38_01066 and BSF38_01066a) is partly duplicated in the form of tandem repeat (Supplementary Table S1). In addition, some other CAZyme genes are also present in the form of pseudogenes (see notes to Table 3).
Genome-Predicted versus Expressed Hydrolytic Capabilities
According to the original description (Kulichevskaya et al., 2016), P. borealis is capable of hydrolyzing aesculin, cellobiose, gellan gum, lactose, lichenin, maltose, melibiose, pectin, salicin, sucrose, trehalose, and xylan, while chondroitin sulfate and raffinose are not utilized. A large proportion of the experimentally determined substrate utilization pattern could be confirmed by the genome-derived data. A phenotypic trait that draw our attention to this planctomycete, namely the formation of visible depressions in gellan-solidified media (Kulichevskaya et al., 2016), is in agreement with the presence of genes encoding two α-L-rhamnosidases (GH78) and an unsaturated glucuronyl hydrolase (GH88). Utilization of aesculin, cellobiose, lactose, lichenin, maltose, salicin, trehalose, and xylan can be explained by the occurrence of genes encoding GH2, GH5, GH9, GH13, GH15, GH43, and GH51-domain containing proteins. The use of sucrose as growth substrate is in line with the encoded β-fructosidase (BSF38_03534). A substrate, which was not tested in the original study but suggested by the genome analysis to be utilized, is arabinan. The inability to utilize chondroitin sulfate can be explained by the absence of genes known to be responsible for its degradation: hyaluronidase (GH16, GH56, GH84), chondroitin hydrolase (GH56), hyaluronate lyase (PL8, PL16), chondroitin AC lyase (PL8), or chondroitin ABC lyase (PL8).
The earlier reported abilities of growth on melibiose and pectin, however, could not be confirmed by the genome data. The genes coding for polygalacturonase (GH28), pectate lyase (PL1, PL2, PL3, PL10), exo-pectate lyase (PL1, PL2), pectin lyase (PL1), pectin methylesterase (CE8), or pectin acetylesterase (CE12, CE13) were lacking, thereby suggesting inability of pectin utilization by P. borealis PX4T. We also did not identify genes encoding CAZymes of the GH4, GH27, GH31, GH36, GH97, or GH110 families. These are the only families known to contain bacterial α-galactosidases (Lombard et al., 2014). Inability to utilize raffinose and the absence of a secreted α-galactosidase activity (according to API ZYM test; see Kulichevskaya et al., 2016) also supported the dubiety of the earlier reported melibiose utilization by P. borealis. One additional uncertainty between the genome-predicted and experimentally determined capabilities of strain PX4T was the presence of a putative chitinase from GH18 family and the lack of experimental evidence for its growth on chitin.
These analyses prompted us to verify several genome-predicted capabilities in P. borealis PX4T, i.e., the ability to grow on arabinan and chitin. In addition, we reassessed its ability to develop on melibiose, pectin, and raffinose. With the exception that growth on chitin could not be demonstrated, the results of these experiments were in full agreement with the genome-derived data.
Plasmids
All four Isosphaeraceae planctomycetes harbor plasmids. Their numbers vary from one in I. pallida IS1BT to four in strain SH-PL62 (Table 2). P. borealis PX4T possesses two plasmids, pPALBO1 (large plasmid) and pPALBO2 (small plasmid).
pPALBO2 display synteny to plasmid pSINAC03 from S. acidiphila DSM 18658T. Their common evolutionary origin is obvious despite the fact that pPALBO2 is a bit larger (Table 2). These two plasmids have 10 regions of homology and possess 12 orthologous genes located exactly in the same order and orientation (Figure 3B). Each of the proteins encoded by these genes in P. borealis PX4T finds its counterpart in S. acidiphila DSM 18658T as the best hit in blastp search. One of them is ParB-like nuclease (BSF38_20001). The closest homologs of three other pPALBO2 genes (BSF38_20020, BSF38_20021, BSF38_20023) are located on the chromosome of S. acidiphila DSM 18658T (Figure 3B). However, BSF38_20023 homolog (Sinac_1451) is annotated as a pseudogene. The gene BSF38_20023 also has a more distant homolog located on plasmid pSINAC03 (Sinac_7675). One additional pPALBO2 gene (BSF38_20034) has a distant homolog in pSINAC03 (Sinac_7679) which is also annotated as a pseudogene. The latter two genes of pPALBO2 (BSF38_20023 and BSF38_20034) differ in their position and orientation from their counterparts in pSINAC03, suggesting an independent evolutionary history. Notably, nine genes located on pPALBO2 have close homologs on the chromosome of strain SH-PL62. All of them are located within the same 23 kb locus. Thus, it is tempting to conclude that a pPALBO2-like plasmid has been integrated into a chromosome of strain SH-PL62 ancestor.
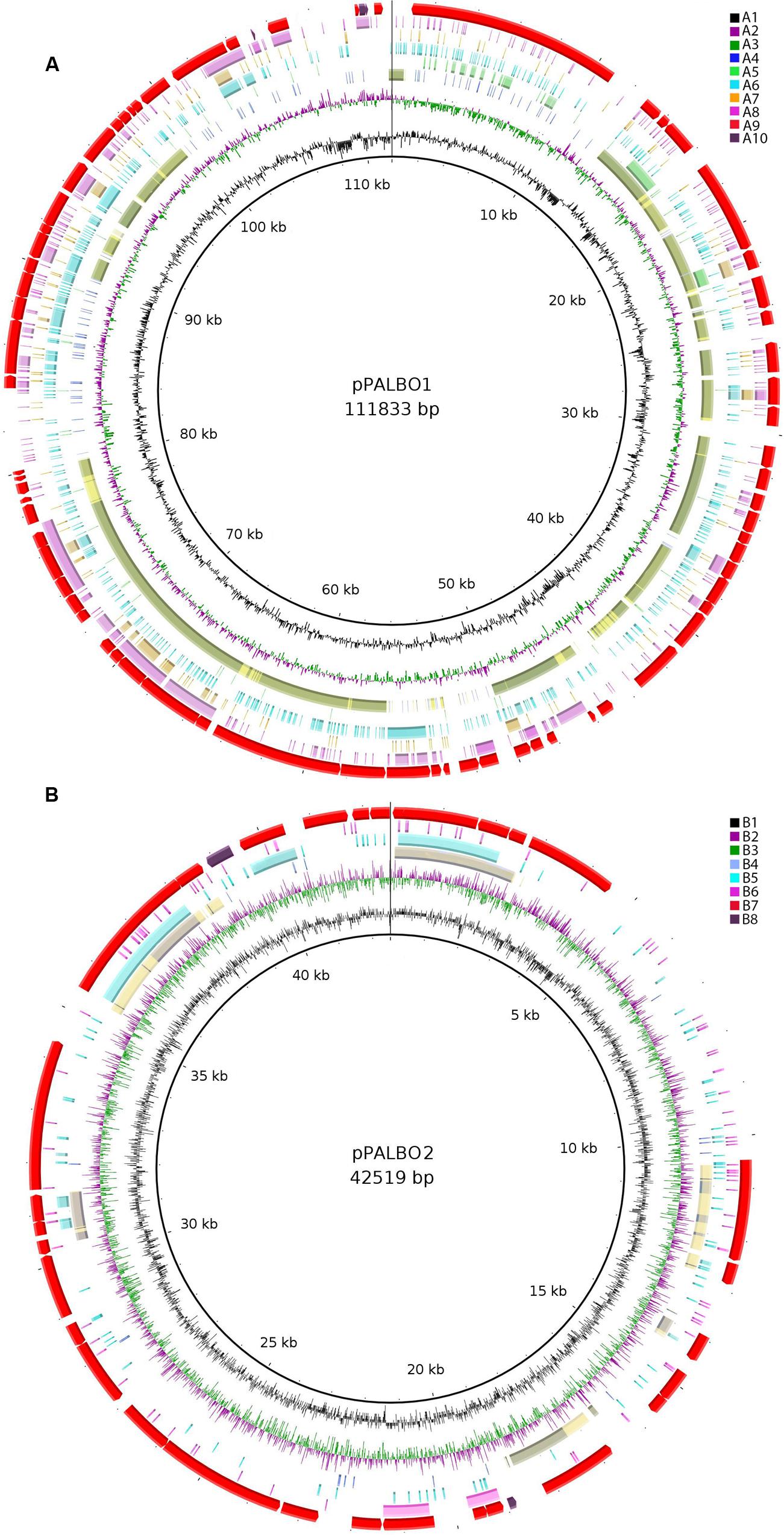
FIGURE 3. (A) Comparative genomic analysis of plasmid pPALBO1. The blastRing Image Generator (BRIG) software was used to compare pPALBO1 with nearest homologs. Different genome sequences, indicated by colors within concentric circles, are as follows: A1, GC content; A2 and A3, GC skew (+, green; −, purple); A4, plasmid pPL62-1 of strain SH-PL62 (GenBank accession CP011274.1) (blue—sequence similarity according to BRIG; yellow—manual prediction; green—both); A5, plasmid pSINAC01 (GenBank accession CP003365.1) from Singulisphaera acidiphila DSM 18658T; A6, chromosome of strain SH-PL62 (GenBank accession CP011273.1); A7, chromosome of Isosphaera pallida IS1BT (GenBank accession CP002353.1); A8, sequence similarity to the chromosome of S. acidiphila DSM 18658T (GenBank accession CP003364.1); A9, location of genes on plasmid pPALBO1; A10, pseudogenes. (B) Comparative genomic analysis of plasmid pPALBO2. The BRIG software was used to compare pPALBO2 with nearest homologs. Different genome sequences, indicated by colors within concentric circles, are as follows: B1, GC content; B2 and B3, GC skew (+, green; −, purple); B4, plasmid pSINAC03 from S. acidiphila DSM 18658T (blue—sequence similarity according to BRIG; yellow—manual prediction; green—both); B5, chromosome of strain SH-PL62; B6, chromosome of S. acidiphila DSM 18658T; B7, location of genes on plasmid pPALBO2; B8, pseudogenes.
The large plasmid pPALBO1 from P. borealis PX4T displays high similarity to plasmid pPL62-1 from strain SH-PL62. The two plasmids have 28 regions of homology and 51 orthologous genes in common (Figure 3A). Most likely, two of these genes are pseudogenes in the case of pPL62-1 (data not shown). The order of genes is essentially the same in both plasmids except for a long inversion involving 14 common genes. One of the homologous loci includes the cluster of glycosyltransferase genes mentioned above. It encodes proteins from the GT4 and GT26 families as well as two proteins from the GT2 family (Supplementary Table S2). Two other homologous loci contain a gene for phosphorylase from the GH94 family (Supplementary Table S1) and ParA ATPase (BSF38_10004). Two genes present in both plasmids, pPALBO1 and pPL62-1, also have homologs on plasmid pSINAC01 from S. acidiphila DSM 18658T. These two genes encode a replication initiator protein A (BSF38_10006) and a glucose-1-phosphate thymidylyltransferase (BSF38_10008). Plasmids pPALBO1 and pSINAC01 have an additional pair of homologous genes (BSF38_10002) coding for a putative β-propeller-type glycoside hydrolase (Supplementary Table S4), which does not belong to any of the currently recognized CAZy families (Lombard et al., 2014). Plasmids pPL62-1 and pSINAC01 also have an additional pair of homologous genes (VT85_25820) encoding a DUF1559-containing protein. Twenty-three of 51 genes shared by pPALBO1 and pPL62-1 have homologs on the chromosome of S. acidiphila DSM 18658T, but these are widely distributed over the chromosome. Thus, there may already be a long history of independent evolution between the gene homologs in pPALBO1/pPL62-1 and those in S. acidiphila DSM 18658T.
Giant Genes
The examination of P. borealis PX4T genome for the presence of giant genes (Reva and Tümmler, 2008) revealed 26 genes with a size >5 kb, among which only one gene exceeds 10 kb. Two giant genes (BSF38_10002 and BSF38_10039; Supplementary Tables S1, S4) are plasmid-borne (pPALBO1) and appear to be responsible for carbohydrate metabolism, while all the others are located on the chromosome. The genomes of S. acidiphila DSM 18658T and I. pallida IS1BT contain 36 and 16 genes, respectively, with a size >5 kb (Kohn et al., 2016). Strain SH-PL62 possesses 23 giant genes. For comparison, the highest number (60) of giant genes with a size >5 kb was detected in the genome of Z. formosa A10T, while the largest giant genes (with a size around 36 kb) were identified in “Fuerstia marisgermanica” NH11T and Gimesia maris DSM 8797T (Kohn et al., 2016).
Secondary Metabolites-Related Genes
Genome mining of gene clusters that encode biosynthetic pathways for secondary metabolites in P. borealis PX4T revealed five clusters that comprise 137 genes (Figure 4). The gene clusters 1, 2, and 4 encode PKS of type I and others (t1pks and others, respectively). Closest gene homologs to PKS-encoding gene clusters from P. borealis PX4T belong to S. acidiphila DSM 18658T, “Solibacter usitatus” Ellin6076, and Geobacter daltonii FRC-32 (Figure 4). The gene clusters 3 and 5 encode terpenes. Their closest homologs belong to S. acidiphila DSM 18658T.

FIGURE 4. Secondary metabolite-related gene clusters identified in the genome of Paludisphaera borealis PX4T with antiSMASH and displayed with homologous gene clusters from other bacteria.
Discussion
Our analysis revealed remarkable similarity in genome organization between members of the family Isosphaeraceae. All four analyzed representatives of this family have plasmids in numbers varying from one to four and sizes varying from 13 to 112 kb. Notably, the presence of plasmids has not yet been reported for any other described member of the order Planctomycetales, with one exception: a 37-kb plasmid in Planctopirus limnophila (Labutti et al., 2010). It should be noted, however, that only finished genomes without gap allow the conclusive identification of plasmids. The lack of reports on the presence of plasmids in Planctomycetales other than Isosphaeraceae has therefore to be interpreted with care. As shown in Figure 3, a number of plasmid regions in the four studied Isosphaeraceae members display synteny, providing evidence for their common evolutionary origin. The small plasmid pPALBO2 from P. borealis PX4T and the plasmid pSINAC03 from S. acidiphila DSM 18658T display 10 regions of homology and share 12 orthologous genes. Interestingly, some of these genes are also present on the chromosome of strain SH-PL62, where they are organized in a tight cluster that, most likely, originated via plasmid integration into the chromosome. The large plasmid from P. borealis PX4T, pPALBO1, carries an array of glycosyltransferase genes (families GH94, GT2, GT4, and GT26) and, in addition, two genes (BSF38_10002 and BSF38_10010) encoding CAZymes that display a distant relationship to those currently listed in the CAZy database (Lombard et al., 2014). Plasmid pPALBO1 shows high similarity to pPL62-1 from strain SH-PL62. Some of the genes common to both plasmids, pPALBO1 and pPL62-1, have homologs on plasmid pSINAC01 from S. acidiphila DSM 18658T. Apparently, the large plasmids in mesophilic Isosphaeraceae planctomycetes encode parts of the enzyme machinery required for carbohydrate biosynthesis.
One additional observation of interest is that the genes encoding ATPase ParA and nuclease ParB, both involved in DNA partitioning, are located on different plasmids in strain PX4T (pPALBO1 and pPALBO2, respectively). The activity of both proteins is required for proper distribution of each plasmid replicate to the daughter cells of P. borealis PX4T during cell division. Given this fact, the expression of ATPase ParA and nuclease ParB on the two different plasmids may thus be the reason why both plasmids, pPALBO1 and pPALBO2, are stably maintained in P. borealis PX4T, despite the increased metabolic cost.
It is well known that the CAZyme repertoire in a particular organism reflects its lifestyle and ecology and also depends on its genome size (Coutinho and Henrissat, 1999; Henrissat et al., 2002; Coutinho et al., 2003; Naumoff, 2011b). The major glycoside hydrolase families in P. borealis PX4T are GH5 (contains retaining enzymes with various β-glycopyranosidase activities), GH13 (retaining α-glycopyranosidases), and GH57 (retaining α-glucopyranosidases). Genome analysis allowed us to conclude that cells of P. borealis PX4T may possess α-L-arabinofuranosidase, β-L-arabinofuranosidase, chitinase, cyclic β-1,2-glucan synthetase, β-fructofuranosidase, α-L-fucosidase, 1,4-α-glucan branching, β-glucanase, 4-α-glucanotransferase, α-glucosidase, isoamylase, malto-oligosyltrehalose synthase, malto-oligosyltrehalose trehalohydrolase, α-mannosidase, phosphorylase, α-L-rhamnosidase, sialidase, trehalose synthase, and unsaturated glucuronyl hydrolase activities (Supplementary Table S1). In general, the genome-predicted spectrum of substrates utilized by P. borealis PX4T was in agreement with that reported in the original taxonomic description (Kulichevskaya et al., 2016). Several corrections in the list of growth substrates, however, have to be made. As suggested by the genome analysis and confirmed by cultivation experiments, arabinan should be included in the list of potential growth substrates, while melibiose and pectin should be excluded. These results clearly demonstrate the strength of genome-based predictions.
In the genome of P. borealis PX4T, we also identified several dozens of genes that encode proteins which cannot be affiliated with the currently recognized CAZy families (Lombard et al., 2014). They, however, display a distant relationship to some glycoside hydrolases, glycosyltransferases, or carbohydrate esterases (Supplementary Tables S4–S6). Particularly, we detected eighteen β-propeller-fold proteins that are homologous to an unclassified β-galactosidase from an uncultured bacterium (GenPept, AGW45552.1). This group of proteins is distantly related to the FURAN31 family of putative glycoside hydrolases (Naumoff, 2012). We also identified 20 DUF1080-containing proteins; two of these consist of two homologous domains. According to the PFAM database, the DUF1080 family is related to the GH16 family of glycoside hydrolases and belongs to the same clan (Finn et al., 2016). In addition, we detected forty one PF01408-containing proteins. In the CAZy database, many proteins of this family are classified as “glycoside hydrolases not yet assigned to a family” (also known as the GH_NC family). Given this unexpectedly large number of unclassified putative glycoside hydrolases, we conclude that P. borealis PX4T has an extremely high but partly hidden glycolytic potential.
The three mesophilic Isosphaeraceae planctomycetes examined in our study, i.e., P. borealis PX4T, S. acidiphila DSM 18658T, and strain SH-PL62, appear to possess a common CAZyme pool. Indeed, 86% of the glycoside hydrolases identified in P. borealis PX4T have their closest homologs in strain SH-PL62; one-third of these enzymes was also detected in S. acidiphila DSM 18658T (Supplementary Table S1). Similarly, 76 and 65% of the glycosyltransferases identified in P. borealis PX4T have very close homologs in strain SH-PL62 and S. acidiphila DSM 18658T, respectively (Supplementary Table S2). We made the attempt to trace the evolutionary origin of the enzymes present in P. borealis PX4T, including those shared with S. acidiphila DSM 18658T and strain SH-PL62, by examining the taxonomic source of their closest homologs (Figure 5; see organisms listed in the column “Others” in Supplementary Tables S1, S2). Apparently, a major proportion of the genes coding for glycoside hydrolases (57%) and glycosyltransferases (33%) were acquired by lateral gene transfer from other phyla, such as Proteobacteria, Chloroflexi, Cyanobacteria, Gemmatimonadetes, Verrucomicrobia, and Acidobacteria. Surprisingly, CAZyme genes from P. borealis PX4T had only a very few closest homologs with members of the Firmicutes and Bacteroidetes, the two bacterial phyla known for their high hydrolytic potential. Even more surprising, the CAZymes from P. borealis PX4T did not share any closest homolog with the Actinobacteria, the third group of hydrolytic bacteria whose genomes are overrepresented in the GenBank database. Apparently, the CAZyme repertoire in P. borealis PX4T and closely related planctomycetes significantly differs from those in well-studied hydrolytic bacteria.
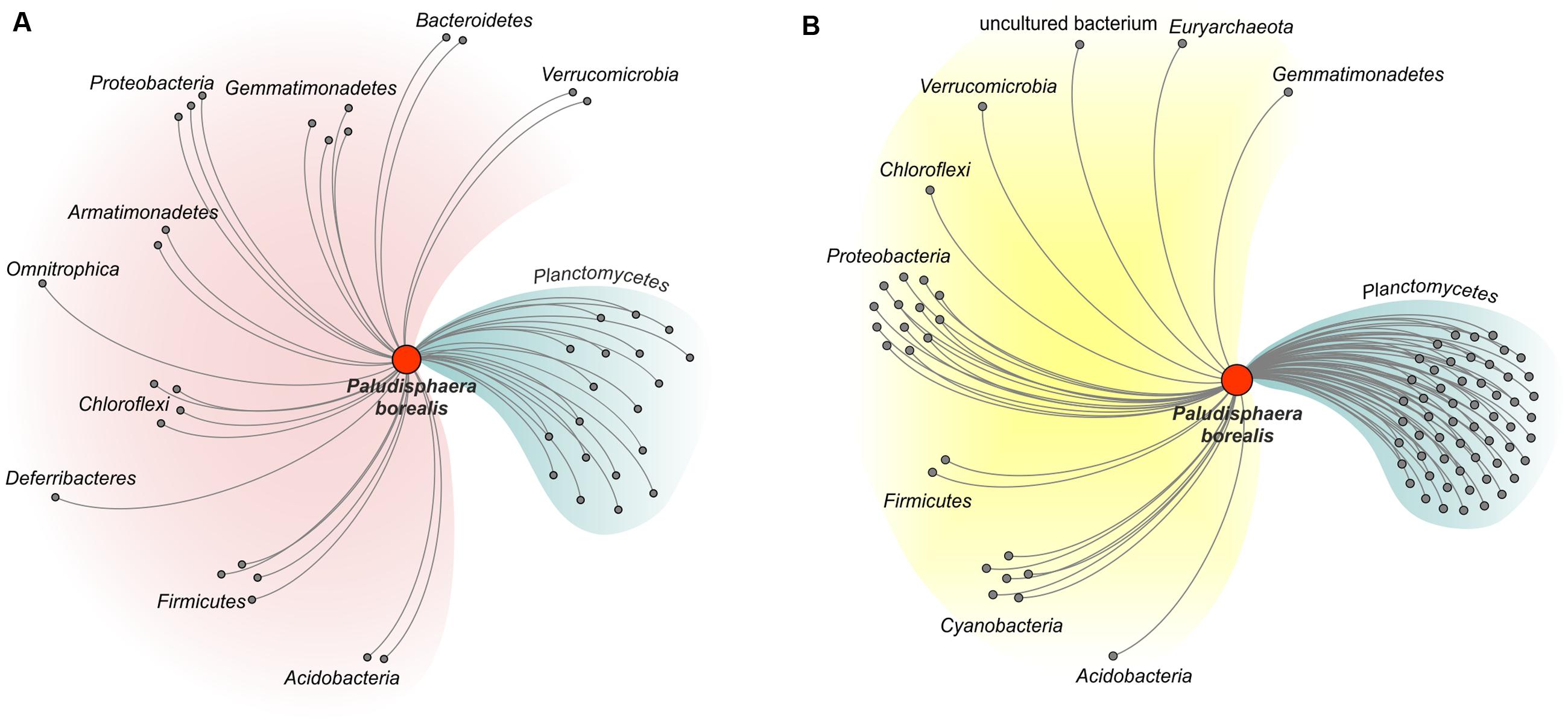
FIGURE 5. Network diagram illustrating the proposed evolutionary origin of the genes encoding glycoside hydrolases (A) and glycosyltransferases (B) in Paludisphaera borealis PX4T. The diagram was built for proteins of Paludisphaera borealis PX4T based on the list of closest homologs as shown in the column “Others” in Supplementary Tables S1, S2 (the orthologs from S. acidiphila DSM 18658T and strain SH-PL62 were ignored). CAZymes of Paludisphaera borealis PX4T with closest homologs in prokaryotic phyla other than Planctomycetes were considered as transferred horizontally. The proteins are grouped according to the host phylum.
We also analyzed the genome-encoded potential of P. borealis PX4T for secondary metabolite production. As it was shown previously, the ability to produce secondary metabolites is a “luxury” that only large-genome bacteria can afford (Donadio et al., 2007). Moreover, the bacterial genomes in general and planctomycete genomes in particular display a linear correlation between genome size and their capacity to encode putative secondary metabolites (Donadio et al., 2007; Jeske et al., 2013). Among the finished Isosphaeraceae genomes, S. acidiphila DSM 18658T has the largest one and the highest number of secondary metabolite-related gene clusters (11) (see Jeske et al., 2013). By contrast, the genome of I. pallida IS1BT is rather small (Table 1) and encodes only five secondary metabolite gene clusters (Jeske et al., 2013). The genome of strain SH-PL62 contains two terpene-encoding gene clusters, three gene clusters of PKS type I, and one gene cluster of PKS type 3. In our study, we revealed three PKS gene clusters and two terpene-encoding gene clusters in P. borealis PX4T. Terpenes are a diverse class of organic compounds that include different kinds of antibiotics, hormones, flavor components, vitamins, and pigments (Yamada et al., 2012). The planctomycete genomes are known to be extremely rich in terpenoid gene clusters (Jeske et al., 2013). Since many planctomycetes, including P. borealis PX4T, form pink-colored colonies, carotenoid synthesis might account for several planctomycetal terpenoid synthases (Jeske et al., 2013). PKS are multimodular enzymes that direct the formation of oligopeptide and polyketide secondary metabolites on a protein template. Their specific multimodular molecular assembly allows the PKS to produce numerous small bioactive compounds that can be used as antimicrobials, plant protectives, or nematicides (Fischbach and Walsh, 2006; Walsh, 2008; Jeske et al., 2013). Given that several planctomycete PKS gene clusters or genes are rather small, one of their functional roles could be in modifying the ribosomally synthesized bacteriocins and lantibiotics (Cotter et al., 2013; Jeske et al., 2013).
In summary, comparative genomics revealed high glycolytic potential in P. borealis PX4T, which remains to be explored in future studies. Many enzymes in the CAZyme pool of strain PX4T are shared with other mesophilic Isosphaeraceae planctomycetes, providing these bacteria with the ability to utilize a wide range of natural carbohydrates and glycoconjugates. The potential to produce a range of secondary metabolites is another characteristic of P. borealis PX4T that deserves additional attention.
Author Contributions
SD, AI, and WL designed the study. AI cultivated the strain. AI, DN, and KM analyzed and interpreted the sequence data. AI and DN annotated the genome sequence. DN analyzed the repertoire of carbohydrate-active enzymes. DN and KM performed the comparative analysis of plasmids. SD, AI, DN, and WL wrote the manuscript.
Funding
AI, DN, KM, and SD were supported by the Russian Science Foundation (project No. 16-14-10210), while WL was supported by the Max Planck Society.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00412/full#supplementary-material
FIGURE S1 | The heatmap summarizes the functional annotation of protein-coding genes from Paludisphaera borealis PX4T, Singulisphaera acidiphila DSM 18658T, Isosphaera pallida IS1BT, and strain SH-PL62 using KEGG database. Only the functional categories with >10 assignments for at least one organism is shown.
References
Alikhan, N. F., Petty, N. K., Ben Zakour, N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Apweiler, R., Bairoch, A., Bougueleret, L., Altairac, S., Amendolia, V., Auchincloss, A., et al. (2009). The Universal Protein resource (UniProt) 2009. Nucleic Acids Res. 37, D169–D174. doi: 10.1093/nar/gkn664
Auch, A. F., Klenk, H.-P., and Göker, M. (2010a). Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2, 142–148. doi: 10.4056/sigs.541628
Auch, A. F., von Jan, M., Klenk, H.-P., and Göker, M. (2010b). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Blin, K., Medema, M. H., Kazempour, D., Fischbach, M. A., Breitling, R., Takano, E., et al. (2013). antiSMASH 2.0-a versatile platform for genome mining of secondary metabolite producers. Nucleic Acids Res. 41, W204–W212. doi: 10.1093/nar/gkt449
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chin, C.-S., Alexander, D. H., Marks, P., Klammer, A. A., Drake, J., Heiner, C., et al. (2013). Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods 10, 563–569. doi: 10.1038/nmeth.2474
Cotter, P. D., Ross, R. P., and Hill, C. (2013). Bacteriocins - a viable alternative to antibiotics? Nat. Rev. Microbiol. 11, 95–105. doi: 10.1038/nrmicro2937
Coutinho, P. M., and Henrissat, B. (1999). Life with no sugars? J. Mol. Microbiol. Biotechnol. 1, 307–308.
Coutinho, P. M., Stam, M., Blanc, E., and Henrissat, B. (2003). Why are there so many carbohydrate-active enzyme-related genes in plants? Trends Plant Sci. 8, 563–565. doi: 10.1016/j.tplants.2003.10.002
Dedysh, S. N. (2011). Cultivating uncultured bacteria from northern wetlands: knowledge gained and remaining gaps. Front. Microbiol. 2:184. doi: 10.3389/fmicb.2011.00184
Dedysh, S. N., and Kulichevskaya, I. S. (2013). “Acidophilic Planctomycetes: expanding the horizons of new planctomycete diversity,” in Planctomycetes: Cell Structure, Origins and Biology, ed. J. A. Fuerst (New York, NY: Humana Press), 125–139. doi: 10.1007/978-1-62703-502-6
Devos, D. P., and Ward, N. L. (2014). Mind the PVCs. Environ. Microbiol. 16, 1217–1221. doi: 10.1111/1462-2920.12349
Donadio, S., Monciardini, P., and Sosio, M. (2007). Polyketide synthases and nonribosomal peptide synthetases: the emerging view from bacterial genomics. Nat. Prod. Rep. 24, 1073–1109. doi: 10.1039/b514050c
Escobedo-Hinojosa, W. I., Vences-Guzmán, M. Á., Schubotz, F., Sandoval-Calderón, M., Summons, R. E., López-Lara, I. M., et al. (2015). OlsG (Sinac_1600) is an ornithine lipid N-methyltransferase from the planctomycete Singulisphaera acidiphila. J. Biol. Chem. 290, 15102–15111. doi: 10.1074/jbc.M115.639575
Felsenstein, J. (1989). Phylip: phylogeny inference package (version 3.2). Cladistics 5, 163–166. doi: 10.1111/j.1096-0031.1989.tb00562.x
Finn, R. D., Clements, J., and Eddy, S. R. (2011). HMMER web server: interactive sequence similarity searching. Nucleic Acids Res. 39, W29–W37. doi: 10.1093/nar/gkr367
Finn, R. D., Coggill, P., Eberhardt, R. Y., Eddy, S. R., Mistry, J., Mitchell, A. L., et al. (2016). The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res. 44, D279–D285. doi: 10.1093/nar/gkv1344
Fischbach, M. A., and Walsh, C. T. (2006). Assembly-line enzymology for polyketide and nonribosomal peptide antibiotics: logic, machinery, and mechanisms. Chem. Rev. 106, 3468–3496. doi: 10.1021/cr0503097
Fuerst, J. A., and Sagulenko, E. (2011). Beyond the bacterium: planctomycetes challenge our concepts of microbial structure and function. Nat. Rev. Microbiol. 9, 403–413. doi: 10.1038/nrmicro2578
Fukunaga, Y., Kurahashi, M., Sakiyama, Y., Ohuchi, M., Yokota, A., and Harayama, S. (2009). Phycisphaera mikurensis gen. nov., sp nov., isolated from a marine alga, and proposal of Phycisphaeraceae fam. nov., Phycisphaerales ord. nov and Phycisphaerae classis nov in the phylum Planctomycetes. J. Gen. Appl. Microbiol. 55, 267–275. doi: 10.2323/jgam.55.267
Glöckner, F. O., Kube, M., Bauer, M., Teeling, H., Lombardot, T., Ludwig, W., et al. (2003). Complete genome sequence of the marine planctomycete Pirellula sp. strain 1. Proc. Natl. Acad. Sci. U.S.A. 100, 8298–8303. doi: 10.1073/pnas.1431443100
Guo, M., Zhou, Q., Zhou, Y., Yang, L., Liu, T., Yang, J., et al. (2014). Genomic evolution of 11 type strains within family Planctomycetaceae. PLoS ONE 9:e86752. doi: 10.1371/journal.pone.0086752
Henrissat, B., Deleury, E., and Coutinho, P. M. (2002). Glycogen metabolism loss: a common marker of parasitic behaviour in bacteria? Trends Genet. 18, 437–440. doi: 10.1016/S0168-9525(02)02734-8
Hyatt, D., Chen, G.-L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Ivanova, A. A., Wegner, C. E., Kim, Y., Liesack, W., and Dedysh, S. N. (2016). Identification of microbial populations driving biopolymer degradation in acidic peatlands by metatranscriptomic analysis. Mol. Ecol. 25, 4818–4835. doi: 10.1111/mec.13806
Ivanova, A. O., and Dedysh, S. N. (2012). Abundance, diversity, and depth distribution of Planctomycetes in acidic northern wetlands. Front. Microbiol. 3:5. doi: 10.3389/fmicb.2012.00005
Jeske, O., Jogler, M., Petersen, J., Sikorski, J., and Jogler, C. (2013). From genome mining to phenotypic microarrays: Planctomycetes as source for novel bioactive molecules. Antonie van Leeuwenhoek 104, 551–567. doi: 10.1007/s10482-013-0007-1
Jeske, O., Schüler, M., Schumann, P., Schneider, A., Boedeker, C., Jogler, M., et al. (2015). Planctomycetes do possess a peptidoglycan cell wall. Nat. Commun. 6:7116. doi: 10.1038/ncomms8116
Jetten, M. S. M., Niftrik, L. V., Strous, M., Kartal, B., Keltjens, J. T., and Op Den Camp, H. J. M. (2009). Biochemistry and molecular biology of anammox bacteria. Crit. Rev. Biochem. Mol. Biol. 44, 65–84. doi: 10.1080/10409230902722783
Jogler, C., Waldmann, J., Huang, X., Jogler, M., Glöckner, F. O., Mascher, T., et al. (2012). Identification of proteins likely to be involved in morphogenesis, cell division, and signal transduction in Planctomycetes by comparative genomics. J. Bacteriol. 194, 6419–6430. doi: 10.1128/JB.01325-12
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kohn, T., Heuer, A., Jogler, M., Vollmers, J., and Boedeker, C. (2016). Fuerstia marisgermanicae gen. nov., sp. nov., an unusual member of the phylum Planctomycetes from the German Wadden Sea. Front. Microbiol. 7:2079. doi: 10.3389/fmicb.2016.02079
Kulichevskaya, I. S., Ivanova, A. A., Suzina, N. E., Rijpstra, W. I. C., Damsté, J. S. S., and Dedysh, S. N. (2016). Paludisphaera borealis gen. nov., sp. nov., a hydrolytic planctomycete from northern wetlands, and the proposal of Isosphaeraceae fam. nov. Int. J. Syst. Evol. Microbiol. 66, 837–844. doi: 10.1099/ijsem.0.000799
Labutti, K., Sikorski, J., Schneider, S., Nolan, M., Lucas, S., Glavina Del Rio, T., et al. (2010). Complete genome sequence of Planctomyces limnophilus type strain (Mü 290). Stand. Genomic Sci. 3, 47–56. doi: 10.4056/sigs.1052813
Lagesen, K., Hallin, P., Rødland, E. A., Stærfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Laslett, D., and Canback, B. (2004). ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 32, 11–16. doi: 10.1093/nar/gkh152
Lombard, V., Golaconda Ramulu, H., Drula, E., Coutinho, P. M., and Henrissat, B. (2014). The carbohydrate-active enzymes database (CAZy) in 2013. Nucleic Acids Res. 42, 490–495. doi: 10.1093/nar/gkt1178
Ludwig, W., Strunk, O., Westram, R., Richter, L., Meier, H., Yadhukumar, A., et al. (2004). ARB: a software environment for sequence data. Nucleic Acids Res. 32, 1363–1371. doi: 10.1093/nar/gkh293
Medema, M. H., Blin, K., Cimermancic, P., De Jager, V., Zakrzewski, P., Fischbach, M. A., et al. (2011). AntiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res. 39, W339–W346. doi: 10.1093/nar/gkr466
Meier-Kolthoff, J. P., Göker, M., Spröer, C., and Klenk, H. P. (2013). When should a DDH experiment be mandatory in microbial taxonomy? Arch. Microbiol. 195, 413–418. doi: 10.1007/s00203-013-0888-4
Moore, E. K., Hopmans, E. C., Rijpstra, W. I. C., Villanueva, L., Dedysh, S. N., Kulichevskaya, I. S., et al. (2013). Novel mono-, di-, and trimethylornithine membrane lipids in northern wetland planctomycetes. Appl. Environ. Microbiol. 79, 6874–6884. doi: 10.1128/AEM.02169-13
Moore, E. K., Villanueva, L., Hopmans, E. C., Rijpstra, W. I. C., Mets, A., Dedysh, S. N., et al. (2015). Abundant trimethylornithine lipids and specific gene sequences are indicative of planctomycete importance at the oxic/anoxic interface in Sphagnum-dominated northern wetlands. Appl. Environ. Microbiol. 81, 6333–6344. doi: 10.1128/AEM.00324-15
Naumoff, D. G. (2011a). GHL1-GHL15: new families of the hypothetical glycoside hydrolases. Mol. Biol. 45, 983–992. doi: 10.1134/S0026893311060082
Naumoff, D. G. (2011b). Hierarchical classification of glycoside hydrolases. Biochemistry 76, 622–635. doi: 10.1134/S0006297911060022
Naumoff, D. G. (2012). Furanosidase superfamily: search of homologues. Mol. Biol. 46, 322–327. doi: 10.1134/S0026893312010153
Naumoff, D. G. (2016). GH10 family of glycoside hydrolases: structure and evolutionary connections. Mol. Biol. 50, 132–140. doi: 10.1134/S0026893315060205
Naumoff, D. G., and Stepuschenko, O. O. (2011). Endo-α-1,4-polygalactosa minidases and their homologs: structure and evolution. Mol. Biol. 45, 647–657. doi: 10.1134/S0026893311030113
Neumann, S., Wessels, H. J. C. T., Rijpstra, W. I. C., Sinninghe Damsté, J. S., Kartal, B., Jetten, M. S. M., et al. (2014). Isolation and characterization of a prokaryotic cell organelle from the anammox bacterium Kuenenia stuttgartiensis. Mol. Microbiol. 94, 794–802. doi: 10.1111/mmi.12816
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2014). The SEED and the Rapid Annotation of microbial genomes using Subsystems Technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Reva, O., and Tümmler, B. (2008). Think big - giant genes in bacteria. Environ. Microbiol. 10, 768–777. doi: 10.1111/j.1462-2920.2007.01500.x
Rivas-Marín, E., Canosa, I., and Devos, D. P. (2016). Evolutionary cell biology of division mode in the bacterial Planctomycetes-Verrucomicrobia-Chlamydiae superphylum. Front. Microbiol. 7:1964. doi: 10.3389/fmicb.2016.01964
Santarella-Mellwig, R., Pruggnaller, S., Roos, N., Mattaj, I. W., and Devos, D. P. (2013). Three-dimensional reconstruction of bacteria with a complex endomembrane system. PLoS Biol. 11:e1001565. doi: 10.1371/journal.pbio.1001565
Scheuner, C., Tindall, B. J., Lu, M., Nolan, M., Lapidus, A., Cheng, J.-F., et al. (2014). Complete genome sequence of Planctomyces brasiliensis type strain (DSM 5305T), phylogenomic analysis and reclassification of Planctomycetes including the descriptions of Gimesia gen. nov., Planctopirus gen. nov. and Rubinisphaera gen. nov. and emended descriptions of the order Planctomycetales and the family Planctomycetaceae. Stand. Genomic Sci. 9:10. doi: 10.1186/1944-3277-9-10
Schlesner, H., and Stackebrandt, E. (1986). Assignment of the genera Planctomyces and Pirella to a new family Planctomycetaceae fam. nov. and description of the order Planctomycetales ord. nov. Syst. Appl. Microbiol. 8, 174–176. doi: 10.1016/S0723-2020(86)80072-8
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Serkebaeva, Y. M., Kim, Y., Liesack, W., and Dedysh, S. N. (2013). Pyrosequencing-based assessment of the bacteria diversity in surface and subsurface peat layers of a northern wetland, with focus on poorly studied phyla and candidate divisions. PLoS ONE 8:e63994. doi: 10.1371/journal.pone.0063994
Staley, J. T., Fuerst, J. A., Giovannoni, S., and Schlesner, H. (1992). The Prokaryotes: A Handbook on the Biology of Bacteria: Ecophysiology, Isolation, Identification, Applications,” in eds A. Balows, H. G. Trüper, M. Dworkin, W. Harder, and K.-H. Schleifer (New York, NY: Springer), 3710–3731. doi: 10.1007/978-1-4757-2191-1_41
Strous, M., Fuerst, J. A., Kramer, E. H., Logemann, S., Muyzer, G., van de Pas-Schoonen, K. T., et al. (1999). Missing lithotroph identified as new planctomycete. Nature 400, 446–449. doi: 10.1038/22749
Terrapon, N., Lombard, V., Drula, E., Coutinho, P. M., and Henrissat, B. (2017). “The CAZy database/the carbohydrate-active enzyme (CAZy) database: principles and usage guidelines,” in A Practical Guide to Using Glycomics Databases, ed. K. F. Aoki-Kinoshita (Tokyo: Springer), 117–131. doi: 10.1007/978-4-431-56454-6_6
Tveit, A., Schwacke, R., Svenning, M. M., and Urich, T. (2013). Organic carbon transformations in high-Arctic peat soils: key functions and microorganisms. ISME J. 7, 299–311. doi: 10.1038/ismej.2012.99
van Teeseling, M. C. F., Mesman, R. J., Kuru, E., Espaillat, A., Cava, F., Brun, Y. V., et al. (2015). Anammox Planctomycetes have a peptidoglycan cell wall. Nat. Commun. 6:6878. doi: 10.1038/ncomms7878
Walsh, C. T. (2008). The chemical versatility of natural-product assembly lines. Acc. Chem. Res. 41, 4–10. doi: 10.1021/ar7000414
Ward, N. L. (2010). “Phylum XXV. Planctomycetes Garrity and Holt 2001, 137 emend. Ward,” in Bergey’s Manual of Systematic Bacteriology: The Bacteroidetes, Spirochaetes, Tenericutes (Mollicutes), Acidobacteria, Fibrobacteres, Fusobacteria, Dictyoglomi, Gemmatimonadetes, Lentisphaerae, Verrucomicrobia, Chlamydiae, and Planctomycetes, Vol. 4, eds N. R. Krieg, J. T. Staley, D. R. Brown, B. P. Hedlund, B. J. Paster, and N. L. Ward (New York, NY: Springer), 879–925. doi: 10.1007/978-0-387-68572-4_14
Weber, T., Blin, K., Duddela, S., Krug, D., Kim, H. U., Bruccoleri, R., et al. (2015). antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 43, W237–W243. doi: 10.1093/nar/gkv437
Yamada, Y., Cane, D. E., and Ikeda, H. (2012). “Diversity and analysis of bacterial terpene synthases,” in Methods in Enzymology, ed. D. A. Hopwood (Cambridge, MA: Academic Press), 123–162. doi: 10.1016/B978-0-12-394290-6.00007-0
Keywords: Planctomycetes, Isosphaeraceae, Paludisphaera borealis, comparative genomics, plasmids, CAZy, glycoside hydrolases, glycosyltransferases
Citation: Ivanova AA, Naumoff DG, Miroshnikov KK, Liesack W and Dedysh SN (2017) Comparative Genomics of Four Isosphaeraceae Planctomycetes: A Common Pool of Plasmids and Glycoside Hydrolase Genes Shared by Paludisphaera borealis PX4T, Isosphaera pallida IS1BT, Singulisphaera acidiphila DSM 18658T, and Strain SH-PL62. Front. Microbiol. 8:412. doi: 10.3389/fmicb.2017.00412
Received: 06 December 2016; Accepted: 27 February 2017;
Published: 16 March 2017.
Edited by:
Frank T. Robb, University of Maryland, Baltimore, USAReviewed by:
Nils-Kaare Birkeland, University of Bergen, NorwayPhillip Brumm, C5-6 Technologies, USA
Copyright © 2017 Ivanova, Naumoff, Miroshnikov, Liesack and Dedysh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Svetlana N. Dedysh, ZGVkeXNoQG1haWwucnU=
†These authors have contributed equally to this work.