 Tao Wang
Tao Wang Jiangfan Xiu1
Jiangfan Xiu1 Yu Wang
Yu Wang- 1School of Basic Medical Sciences, Guizhou Medical University, Guiyang, China
- 2Guizhou Provincial Center for Disease Control and Prevention, Guiyang, China
- 3School of Biology and Engineering, Guizhou Medical University, Guiyang, China
Candida albicans is a major fungal pathogen in humans. Novel antifungal agents are urgent demanded due to the challenges of the resistance. Antimicrobial peptides (AMPs) are critical components of the innate immune system against pathogenic microorganism infection. MAF-1A is a novel cationic AMP that comes from Musca domestica and is effective against C. albicans, but the antifungal mechanism remains unclear. In this study, we performed a transcriptomics analysis in C. albicans using RNA-seq technique under the treatment of MAF-1A. A total of 5654 genes were identified. Among these, 1032 were differentially expressed genes (DEGs), including 575 up-regulated genes and 457 down-regulated genes. In these DEGs, genes encoding ergosterol metabolism and fatty acid biosynthesis were identified to be significantly down-regulated, while genes associated with oxidative stress response and cell wall were identified to be significantly up-regulated. Using pathway enrichment analysis, 12 significant metabolic pathways were identified, and ribosome, oxidative phosphorylation, citrate cycle were mainly involved. The results revealed that MAF-1A induces complex responses in C. albicans. This study provides evidence that MAF-1A may inhibit the growth through affect multi-targets in C. albicans cells.
Introduction
Candida albicans is an opportunistic fungal pathogen of humans that can cause human mycoses, ranging from superficial mucosal and skin to invasive candidiasis, especially in immunocompromised patients (Eggimann et al., 2003; Linares et al., 2013; Guilhelmelli et al., 2016). In the past two decades, infections caused by C. albicans have increased significantly (Lo et al., 2015). Invasive candidiasis has an estimated mortality about 40%, even with the use of antifungal drugs (Horn et al., 2009; Lu et al., 2014). Currently, only a limited number of antifungal agents are being used to treat these infections, including azoles, polyene, and echinocandins drugs. The persistent use of these drugs have caused the emergence of drug-resistant strains. The resistance and shortage of antifungal agents have potentially limited the management of infections. Besides being pathogenic, C. albicans also provides an excellent eukaryotic model system to explore the antifungal mechanisms of potent drugs (Khan et al., 2015).
Recently, the therapeutic application of antimicrobial peptides (AMPs) or their analogs have received a great deal of attention (Cruz et al., 2014; Lakshmaiah Narayana and Chen, 2015; Mahlapuu et al., 2016). AMPs are relatively small (less than 100 amino acid residues), cationic, amphipathic, variable in length, sequence or structure peptides which have been isolated from animals, plants, bacteria, or fungi (Pushpanathan et al., 2013; Malmsten, 2014; Silva et al., 2014). AMPs are important components of the innate immunity system against invading pathogens. Many AMPs are effective against multi-drug resistant (MDR) microorganism and less developing resistance due to their distinguished mode of action (Seo et al., 2012). Thus, AMPs could be promising candidates for developing novel therapeutic agents against fungi.
Musca domestica antifungal peptide-1 (MAF-1), isolated from the instar larvae of housefly, is a kind of a novel cationic AMP with excellent antimicrobial effect (Fu et al., 2009). In previous study, full-length of MAF-1 gene was cloned, and bioinformatics analysis was used to explore structure domain and its potential physiological function (Luo et al., 2013). MAF-1A peptide was derived from the structure domain of MAF-1 and contained 26 amino acid residues (KKFKETADKLIESAKQQLESLAKEMK). MAF-1A showed its remarkable antifungal effect (Luo et al., 2013; Zhou et al., 2016), but the detailed antifungal actions of MAF-1A remain unclear.
The classic action mechanism of AMPs is cause microbial cell membrane damage. So far, most research has been focused on the use of model membrane systems such as lipid vesicles, to determine the mode of action of AMPs. Even though this knowledge is essential in our understanding of the mode of action of AMPs, it does not fully explain their interaction with microbial cell membrane or the response of microbes to the AMPs (Omardien et al., 2016). In addition, it has been proposed that some AMPs can interact with intracellular specific targets inducing cell damages, such as the inhibition of DNA, RNA, protein and cell wall synthesis (Guilhelmelli et al., 2013; van der Weerden et al., 2013; Scocchi et al., 2016; Shah et al., 2016). The analysis of microbial transcriptome may contribute to the understanding of their interaction with novel AMPs (Tavares et al., 2013). The next generation sequencing technologies for transcriptome analysis have opened the opportunity to understand a wide variety of physiological response of microorganism affected by drugs or environmental conditions treatment. RNA-Seq (deep-sequencing of cDNA) has been used successfully to identify and quantify gene expression at a genome scale level. RNA-Seq shows significant advantages such as sensitive, resolution and comprehensive, and is becoming more popular for various gene expression studies (Nagalakshmi et al., 2008; Wang et al., 2009; Nookaew et al., 2012). RNA-Seq enables genome-wide expression studies on the cellular responses and pathways of microbe affected by drug treatment via differential gene expression profiling (Heo et al., 2014; Qin et al., 2014; Le et al., 2016). This approach has already been used in the cellular responses of C. albicans under several different environmental conditions (Bruno et al., 2010; Cottier et al., 2015). In the present work, we investigated the differentially expressed genes and the pathways involved using high-throughput RNA-Seq technique to explore the mechanisms of MAF-1A on C. albicans.
Materials and Methods
Strain and Growth Conditions
The C. albicans ATCC 10231 was used throughout this study. Strain was preserved in nutrient broth supplemented with 30% glycerol stocked and stored at -80°C and streaked on Sabouraud Dextrose agar (SDA) plates (Sangon, Shanghai, China) at 37°C when used.
Peptide Synthesis
The synthetic MAF-1A peptide is a linear peptide consisting of a 26 amino acid sequences as follows: KKFKETADKLIESAKQ QLESLAKEMK. Peptides were synthesized by Sangon Biotech Co., Ltd. (Shanghai, China). The purity of peptides was confirmed as higher than 95% using analytical reverse-phase high-performance liquid chromatography (RP-HPLC). Peptides were dissolved in sterilized de-ion water to 10 mg/mL and stored -20°C.
MIC Determinations
Antifungal activity of MAF-1A was monitored according to Clinical and Laboratory Standards Institute (CLSI) M27-A2. Briefly, the colonies from 24 hour cultures of C. albicans were picked and resuspended in 0.9% sterile saline and adjusted to 0.5 Mc Farland standard (1–5 × 106 CFU/mL). The yeast stock suspension was then diluted to obtain a starting inoculum of 0.5 × 103 to 2.5 × 103 CFU/mL. Peptides were then serially diluted in RPMI 1640 medium (Sangon, Shanghai, China) in volume of 100 μL per well, giving final concentrations ranging from 5 mg/mL to 0.1 mg/mL in sterile round bottom 96-well polypropylene microplates. Hundred microliter of standardized yeast suspension was then added to each well. After 24 h of incubation at 37°C, the minimum inhibitory concentration (MIC) was defined as the lowest drug concentration that caused 90% growth inhibition compared to drug-free growth control well by visual evaluation. Amphotericin B (Sigma, United States) and fluconazole (Sigma, United States) were used as controls. The MICs were determined in triplicate.
Total RNA Extraction
The cells of C. albicans were inoculated into Sabouraud dextrose broth (SDB) medium (Sangon, Shanghai, China) and cultured at 37°C for 24 h. Before C. albicans were harvested for RNA extraction, the cells were treated with MAF-1A at MIC for 2 h (CA_DT). The untreated cultures were used as the control (CA_D). Total RNA was isolated with RNAiso Plus (Takara, Dalian, China) following the manufacturer’s instructions. Each RNA sample had an A260: A280 ratio between 1.8 and 2.0. RNA purity was checked using the NanoPhotometer® spectrophotometer (Implen, CA, United States). RNA concentration was measured using Qubit® RNA Assay Kit in Qubit® 2.0 Flurometer (Life Technologies, CA, United States). RNA integrity was assessed using the RNA Nano 6000 Assay Kit of the Bioanalyzer 2100 system (Agilent Technologies, CA, United States).
cDNA Library Construction and Illumina RNA-Seq
Libraries construction and RNA-Seq were performed by the Novogene Corporation (Beijing, China). A total amount of 3 μg RNA per sample was used as input material for the RNA sample preparations. Sequencing libraries were generated using NEBNext® UltraTM RNA Library Prep Kit for Illumina® (NEB, United States) following manufacturer’s recommendations and index codes were added to attribute sequences to each sample. Briefly, mRNA was purified from total RNA using poly-T oligo-attached magnetic beads. Fragmentation was carried out using divalent cations under elevated temperature in NEB Next First Strand Synthesis Reaction Buffer. First strand cDNA was synthesized using random hexamer primer and M-MuLV Reverse Transcriptase (RNase H-). Second strand cDNA synthesis was subsequently performed using DNA Polymerase I and RNase H. Remaining over hangs were converted into blunt ends via exonuclease/polymerase activities. After adenylation of 3′ ends of DNA fragments, NEBNext Adaptor with hairpin loop structure were ligated to prepare for hybridization. In order to select cDNA fragments of preferentially 150∼200 bp in length, the library fragments were purified with AMPure XP system (Beckman Coulter, Beverly, United States). Sequencing library quality was assessed on the Agilent Bioanalyzer 2100 system. Paired-end sequencing of cDNA was carried out with Illumina HiseqTM 2000. Raw data was filtered by removing reads with adaptor sequences, as well as low quality reads. Then, clean reads were aligned to the reference genome using TopHat v2.0.12. (Trapnell et al., 2009).
Reads Mapping to the Reference Genome
Reference genome and gene model annotation files of C. albicans SC5314 were downloaded from GenBank (NW_139421). Index of the reference genome was built using Bowtie v2.2.3 and paired-end clean reads were aligned to the reference genome using TopHat v2.0.12. (Trapnell et al., 2009).
Analysis of Differential Expressed Genes
The expression level for each gene is determined by the numbers of reads uniquely mapped to the specific gene and the total number of uniquely mapped reads in the sample. HTSeq v0.6.1 was used to count the reads numbers mapped to each gene. And then expected number of Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced (FPKM) of each gene was calculated based on the length of the gene and reads count mapped to this gene (Trapnell et al., 2010).
Differential expression analysis of two conditions was performed using the DEGSeq R package (1.20.0). The P-values were adjusted using the Benjamini and Hochberg method. Corrected P-value < 0.005 and log2(Fold change) > 1 were set as the threshold for significantly differential expression (Wang et al., 2010).
Enrichment Analysis of Gene Ontology (GO) and KEGG Pathways
Gene Ontology (GO) enrichment analysis of differentially expressed genes (DEGs) was implemented by the GO seq (Young et al., 2010). GO terms with corrected P-value < 0.05 were considered significantly enriched by differential expressed genes. KOBAS 2.0 software was used to test the statistical enrichment of differential expression genes in KEGG pathway (Mao et al., 2005). False discovery rate (FDR) of pathways was calculated. The threshold of significance of pathways was set as FDR < 0.05.
Validation of RNA-Seq by quantitative RT-PCR (qRT-PCR)
To validate the results of RNA-Seq, eight DEGs (five down-regulated and three up-regulated) were selected for qRT-PCR. According to the SYBR Premix Ex TaqTM Kit (Takara, Dalian, China) protocol, the reactions were run on an ABI7300 real-time PCR system using a 20 μL reaction system with reaction procedures of 40 cycles of 95°C for 5 s and 60°C for 30 s and 72°C for 30 s. Gene expression levels were calculated using the 2-ΔΔCt method (Livak and Schmittgen, 2001) and normalized to the abundance of a house-keeping gene 18S rRNA. All samples were examined in triplicate. Target genes using primers listed in Supplementary Table S1.
Results
MIC Assay
The MIC value of the MAF-1A against C. albicans was 0.6 mg/mL.
RNA Sequencing and Gene Prediction
Approximately 50,000,000 raw reads were obtained from each sample. After filtering by quality, 80.30 and 80.01% clean reads from the two groups were mapped to C. albicans genome. The profile of transcriptome sequence data was shown in Table 1. Sequence reads have been deposited in the NCBI Sequence Read Archive (SRA) under accession number PRJNA3751091.
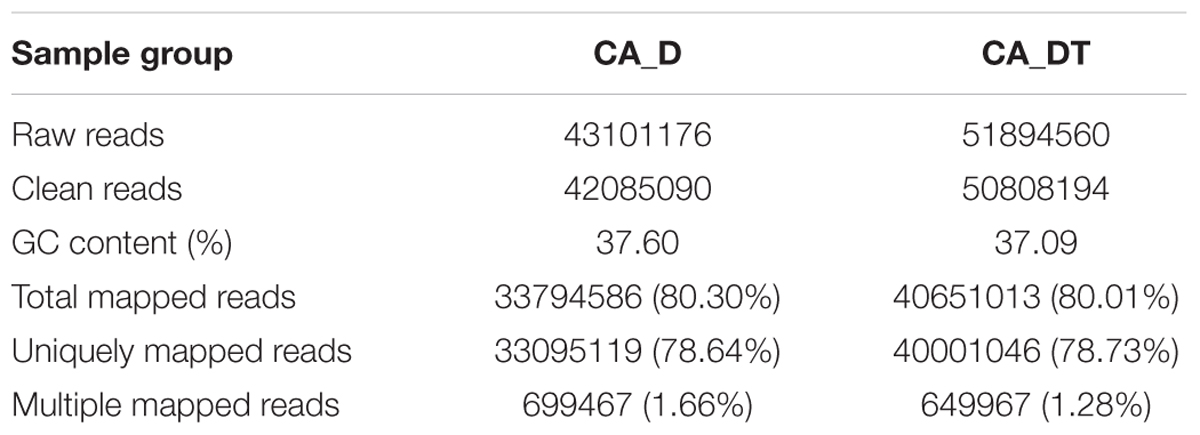
TABLE 1. Profile of the transcriptome sequence data.
Transcriptional Stress Response of C. albicans to MAF-1A
The RNA sequencing results revealed the differences in distribution and density distribution of gene expression between CA_DT and CA_D (Figure 1). CA_DT was detected 5612 genes expression, CA_D was detected 5546 genes expression. There are 5504 genes co-expression in these two samples, 108 genes were specificity expressed in CA_DT and 42 genes were specificity expressed in CA_D (Figure 2). The volcano plots of the differentially expressed genes demonstrated that 1032 genes in C. albicans were differentially expressed after MAF-1A treatment, with either an increase or decrease of more than twofold (the spots marked in red or green), including 575 up-regulated genes and 457 down-regulated genes (Figure 3).
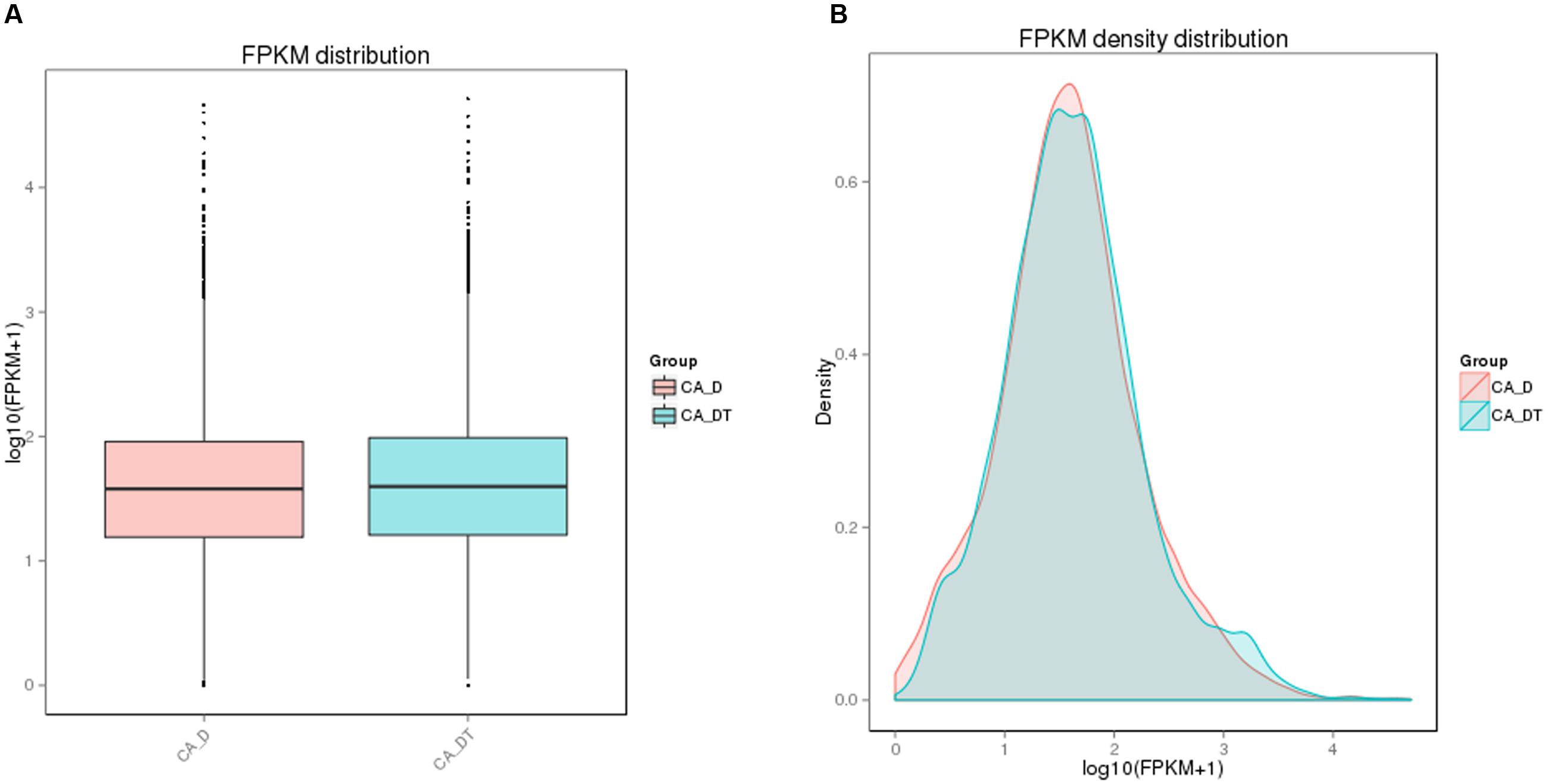
FIGURE 1. The comparison diagram of gene expression level. (A) FPKM (Fragments Per Kilobase of transcript sequence per Millions base pairs sequenced) distribution, (B) FPKM density distribution.
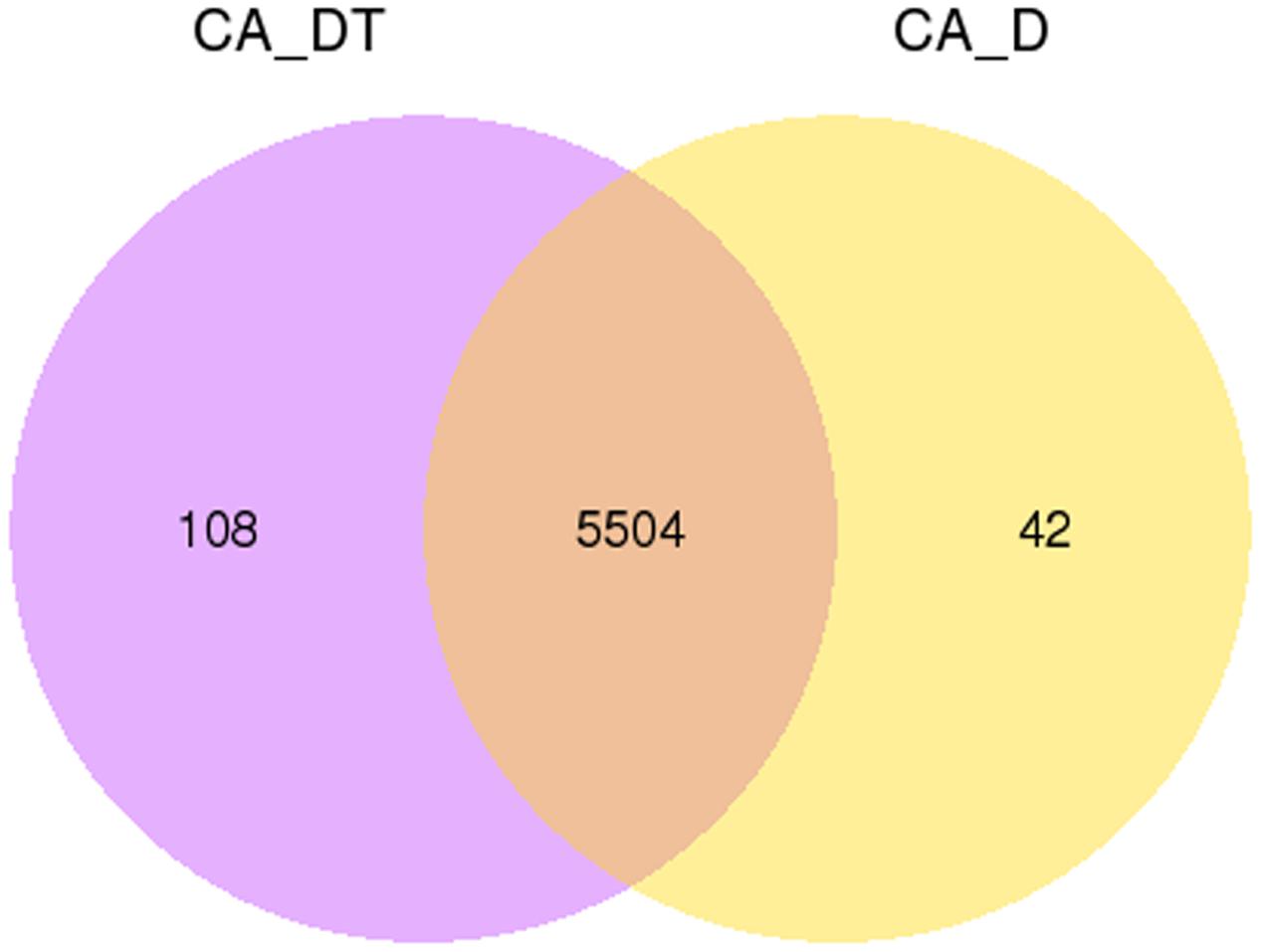
FIGURE 2. Gene expression Venn diagram. The number in each circle represents the total number of genes that are expressed in each sample, and the overlapping part of circles indicates that the gene is co-expressed in both samples.
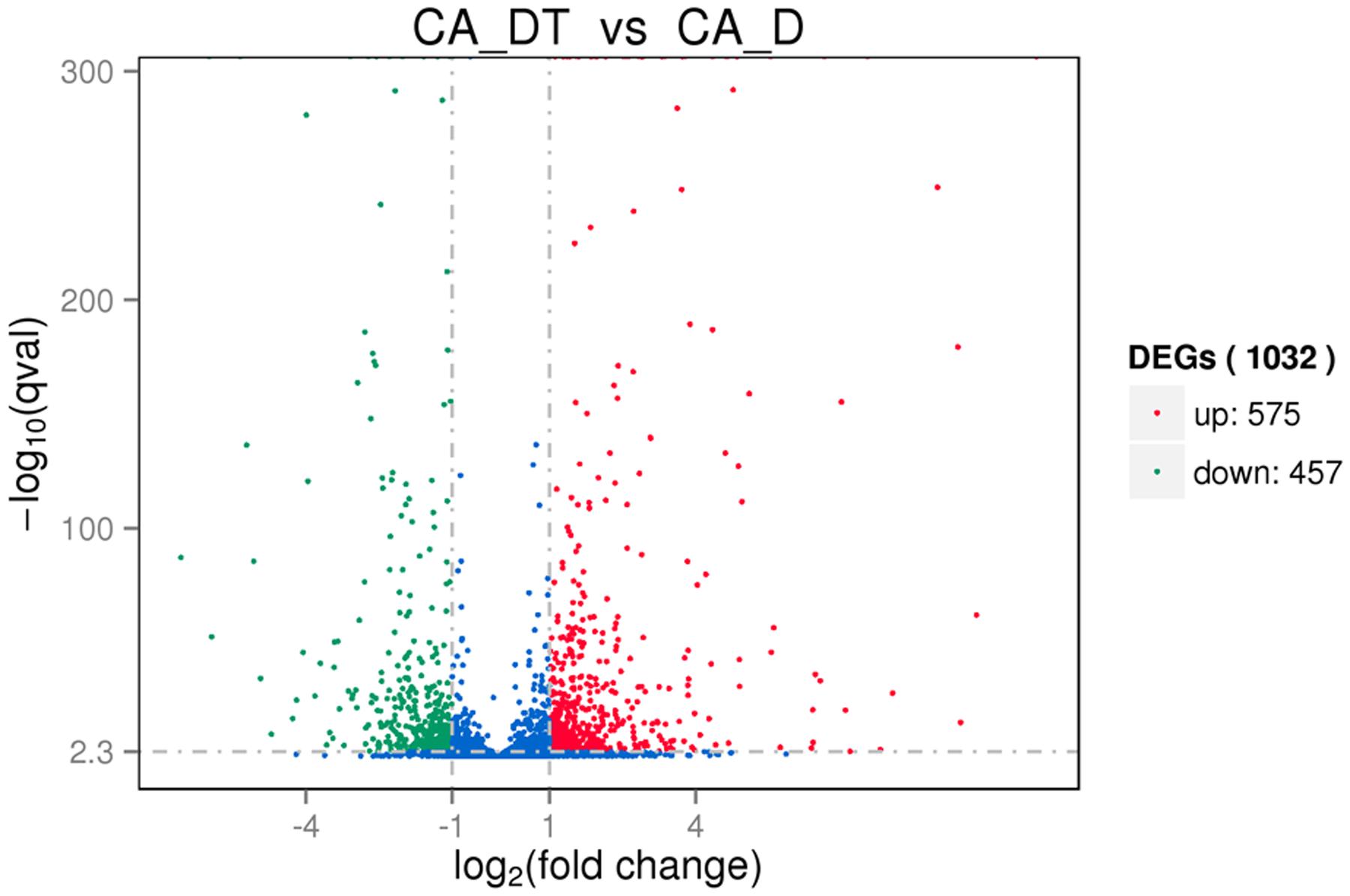
FIGURE 3. The volcano plots of the differentially expressed genes. Significantly differentially expressed genes were treated with red dots (up-regulated) or green dots (down-regulated), others indicated with blue dots. The abscissa represents fold change, and the ordinate represents statistical significance.
Verification of Differentially Expressed Genes
To validate the RNA-Seq results, a total of eight genes were selected including five up-regulated and three down-regulated from DGE libraries for qRT-PCR analysis. The results indicated the expression levels have a consistent change for both RNA-Seq and qRT-PCR. Hence, the qRT-PCR results confirmed the reliability of our RNA-Seq data (Figure 4).
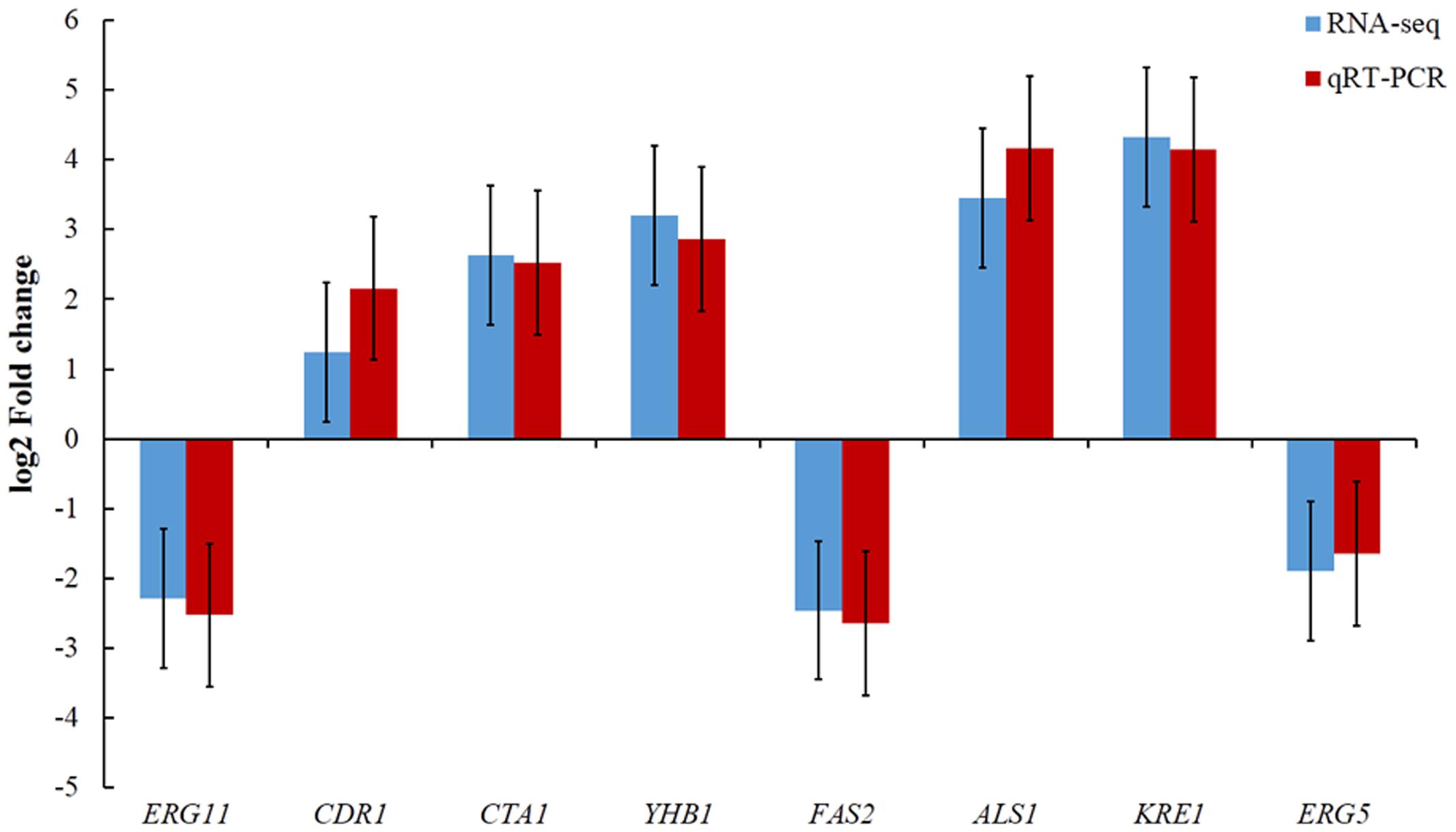
FIGURE 4. Comparison of eight gene expression levels between RNA-Seq and qRT-PCR.
Analysis of Differential Gene Expression
The Cell Wall Synthesis Related Genes
In this study, two genes involved in the biosynthesis of β-glucan (KRE1, KRE6) were up-regulated 4.32-fold, 1.31-fold, respectively (Table 2); one gene involved in the biosynthesis of mannoprotein (MP65) was up-regulated 5.73-fold (Table 2).
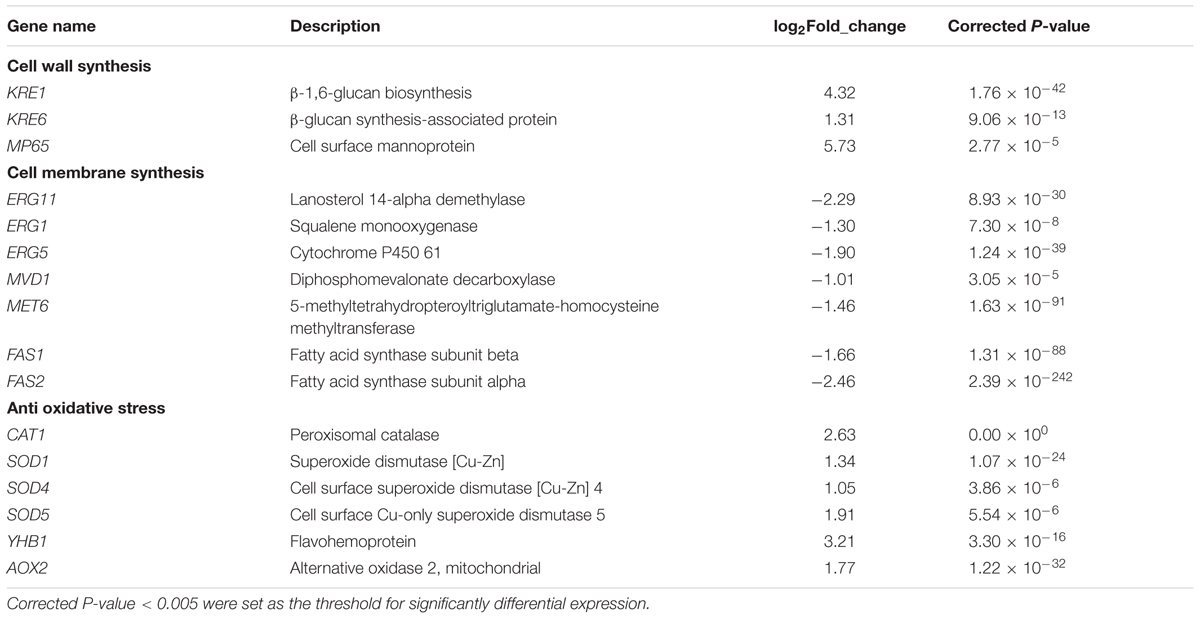
TABLE 2. The genes up-regulated or down-regulated response to MAF-1A.
The Cell Membrane Synthesis and Stability Related Genes
Several genes involved in ergosterol biosynthesis pathway were down-regulated about twofold (ERG11, ERG1, ERG5, MET6, MVD1). Among these genes, ERG11expression decreased mostly (Table 2).
FAS1 and FAS2 are fatty acid synthesis genes, which are involved in the membrane biosynthesis. After MAF-1A treatment, FAS1 and FAS2 were down-regulated 1.66-fold and 2.46-fold, respectively (Table 2).
Anti Oxidative Stress Genes
In this work, six genes (CAT1, SOD1, SOD4, SOD5, YHB1, AOX2) were identified to be up-regulated (1.03-fold to 3.21-fold) in C. albicans cells after MAF-1A treatment (Table 2).
Enrichment Analysis of GO and KEGG Pathways
Gene Ontology and KEGG analysis were used to classify the DEGs in the response of C. albicans to MAF-1A. The GO categories significantly enriched (corrected P-value < 0.05) among those DEGs are shown in Tables 3, 4. In the DEGs, a total of 483 up-regulated and 386 down-regulated genes were classified into the following three functional categories: cellular component, molecular function and biological process, respectively. It is noteworthy that within the biological process category of up-regulated DEGs, ribosome, mitochondrion, mitochondrial part, mitochondrial inner membrane, mitochondrial envelope, mitochondrial membrane, mitochondrial membrane part were significantly enriched. The structural constituent of ribosome, oxidoreductase activity terms were enriched in the molecular function category. Oxidation-reduction process, translation terms were enriched in the cellular component category.
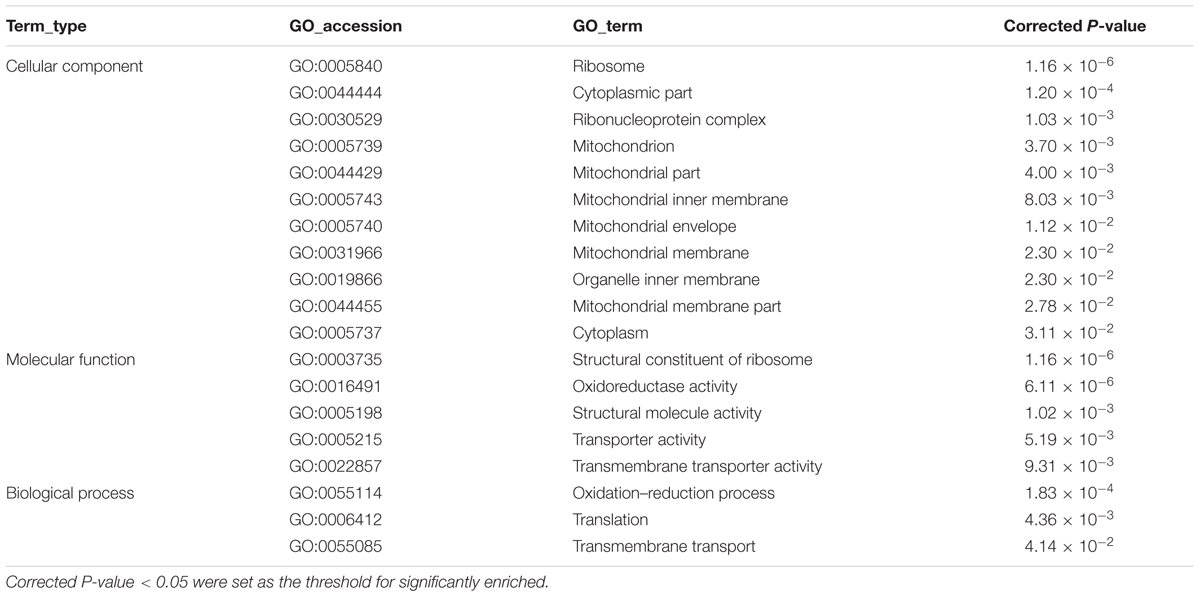
TABLE 3. The significant enrich Gene Ontology (GO) terms of up-regulated differentially expressed genes (DEGs).

TABLE 4. The significant enrich GO terms of down-regulated DEGs.
By enrichment analysis, up-regulated genes were involved in 11 KEGG metabolic pathways, and down-regulated genes were involved in 1 KEGG metabolic pathway (Table 5). In the mapped pathways of the up-regulated genes, the abundant genes mapped onto ribosome, oxidative phosphorylation, peroxisome, carbon metabolism and citrate cycle (TCA cycle). While, the down-regulated genes were primarily related to proteasome.
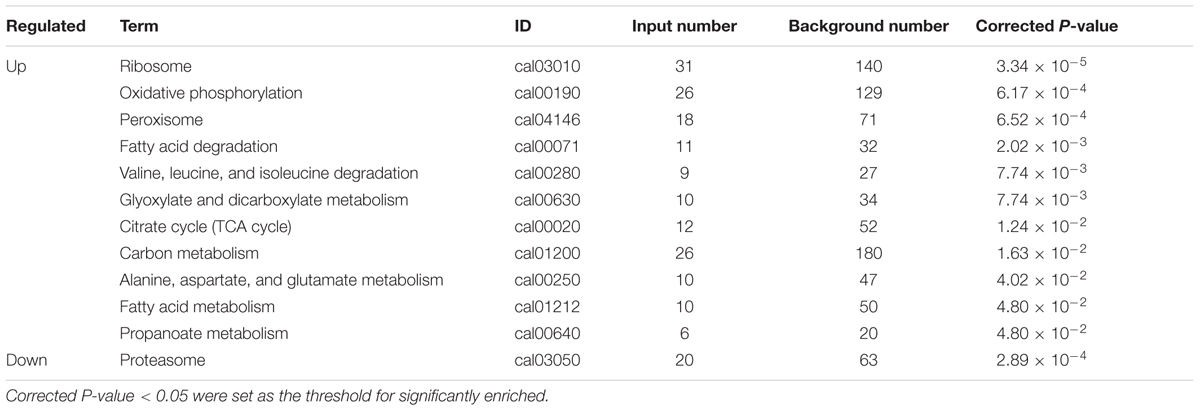
TABLE 5. Differentially expressed genes were involved in significant KEGG metabolic pathways.
Discussion
Antimicrobial peptides are important components of natural defenses against pathogens. The mechanism of antimicrobial activity for these peptides is generally more complex. AMPs were originally proposed to act via plasma membrane permeabilization, leading to membrane rupture and rapid lysis of microbial cells. Recently, it has been proposed that AMP driven microbial death in addition to membrane disruption (Nguyen et al., 2011; Pasupuleti et al., 2012). In our preliminary work, MAF-1A was shown its remarkable activity anti-C. albicans. To further investigate the mechanism of actions of MAF-1A, we used RNA-Seq to study the transcriptomic profile of C. albicans treated with MAF-1A. The RNA-Seq results showed that the gene expression of C. albicans was extensively altered by MAF-1A treatment. These genes that related to cell wall, plasma membrane and anti-oxidative stress had differentially expressed comparing to control.
The fungal cell wall is essential for sustaining cell morphology and for protection against life threatening environmental conditions (Bowman and Free, 2006; Ruiz-Herrera et al., 2006). The cell wall of C. albicans contains three major macromolecules, including mannoproteins, β-glucan and Chitin (Klis et al., 2001). Composition of the cell wall can be quite dynamic, as it changes during cell response to environmental stresses. C. albicans constantly remodels the cell wall by breaking and reforming chemical bonds within and between polysaccharides to maintain integrity of the cell wall structure (Nather and Munro, 2008). C. albicans could change its expression of cell wall related genes to reduce the toxic effect of caspofungin (Liu et al., 2005; Nailis et al., 2010). In this work, three genes involved in the biosynthesis of cell wall were obviously up-regulated (KRE1, KRE6, MP65) after exposure to MAF-1A. The KRE1, KRE6 encoding proteins are involved in β-glucan biosynthesis, and MP65 encodes cell surface mannoprotein, which plays a significant role in maintaining cell wall integrity, morphogenesis and pathogenicity (Sandini et al., 2011). Therefore, the results indicated that these genes may play a role in the mitigation of cell wall damage.
The fungal plasma membrane is similar to those of other eukaryotic cells, composed of a lipid bilayer with proteins embedded within it. Sterols are major components of fungal plasma membranes. The sterol present in animal plasma membranes is cholesterol, while fungi plasma membranes contain ergosterol (Thevissen et al., 2003). This difference in sterol content is exploited in the mechanisms of antifungal agents (Spampinato and Leonardi, 2013). Itraconazole, widely used antifungal drugs, belongs to the azoles and interferes with ergosterol biosynthesis. Up-regulation of ERG genes were detected in C. albicans cells after exposure to Itraconazole in vitro (De Backer et al., 2001). On the contrary, in vitro exposure of C. albicans to amphotericin B (AMB) is correlated with under-expression of ERG genes (Liu et al., 2005). Binding to ergosterol is sufficient for antifungal activity of AMB and therefore considered the primary mode of fungicidal action (Gray et al., 2012). Besides, AMPs could kill the target cells by disrupting the integrity of fungal membranes (Bahar and Ren, 2013). In the current study, several genes (ERG11, ERG1, ERG5, MET6, MVD1) involved in ergosterol biosynthesis pathway were under-expression. The results indicated that MAF-1A could interfere with ergosterol biosynthesis. We speculate that down-regulation of certain ergosterol biosynthesis genes may reduce the binding of MAF-1A to ergosterol. In addition, FAS1 and FAS2 involved in fatty acid biosynthesis were down-regulated, indicating the composition in cell membrane was also affected by the presence of MAF-1A.
ROS, such as hydrogen peroxide and hydroxyl radicals, cause damage to proteins, lipids and nucleic acids, resulting in irreversible damage and loss of viability. A well-characterized response of eukaryotic microbes to ROS is the rapid induction of mRNAs that encode oxidative stress detoxification and repair proteins. These include catalase (CAT1), glutathione peroxidase (GPX) and superoxide dismutase (SOD) antioxidant-encoding genes (Dantas Ada et al., 2015). Various antifungal agents have been confirmed to cause oxidative damage to C. albicans (Delattin et al., 2014). For example, a number of genes involved in oxidative stress response (YHB1, CTA1, AOX1, and SOD2) were found to be up-regulated upon AMB treatment (Liu et al., 2005). In this research, such oxidative stress-responsive genes in C. albicans are also up-regulated following exposure to MAF-1A. These genes included CAT1, SOD1, SOD4, SOD5, AOX2, and YHB1, encodes catalase, superoxide dismutase, alternative oxidases and flavohemoprotein, respectively.
The significantly enriched GO term in the cellular component category suggest that nearly all cellular components were affected including the ribosomes, mitochondrion, cytoplasm, and organelle inner membrane. The significant enrichment of GO terms associated with molecular function indicates that the transcription levels of structural constituent of ribosome, oxidoreductase, structural molecule activity, transmembrane transporter genes were significantly changed. In addition, KEGG analysis showed that twelve metabolic pathways were affected in C. albicans upon MAF-1A treatment. Three metabolic pathways, including “ribosome”, “oxidative phosphorylation”, and “citrate cycle”, are the metabolic pathways of protein biosynthesis, cellular respiration and ATP generation.
It is widely accepted that membrane interaction is a key factor for antimicrobial activity of AMPs. Furthermore, AMPs can also act up on multiple cell targets, involving a mixed multi-hit mechanism (Guilhelmelli et al., 2013; van der Weerden et al., 2013; Silva et al., 2014; Shah et al., 2016). Considering the changes of expression pattern, it is strongly suggested that C. albicans inhibition caused by MAF-1A should be a result of multiple and complementary actions, involving in cell wall, plasma membrane, as well as protein synthesis and energy metabolism (Figure 5). It should be noted that this study has examined only on transcriptional level and further experiments are required to determine the anti-C. albicans key mechanisms.
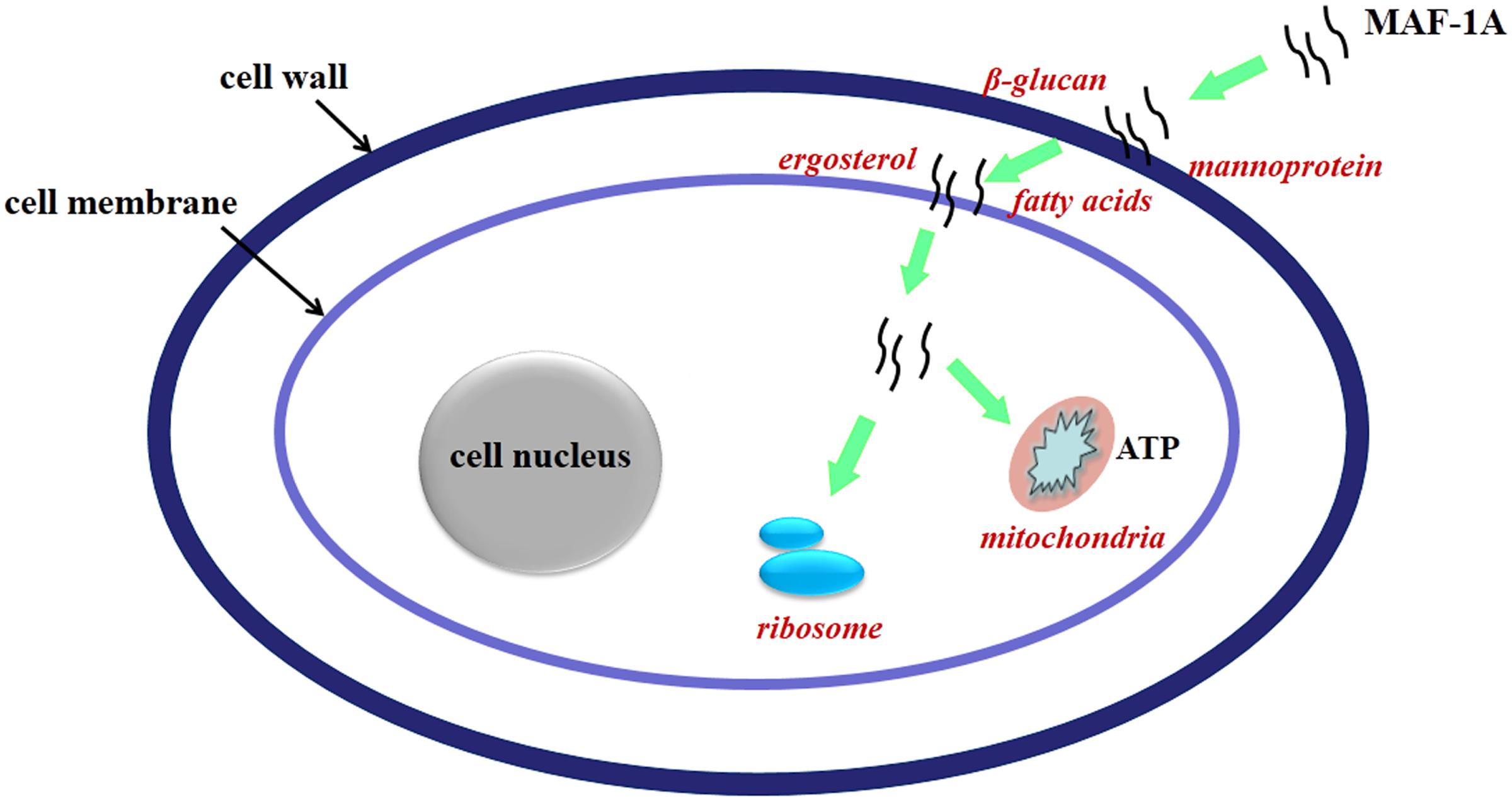
FIGURE 5. Schematic overview of the proposed mechanisms of action of MAF-1A.
Conclusion
We analyzed the gene expression changes for C. albicans under treatment of MAF-1A using RNA-seq. The results from this study suggest that MAF-1A induces complex responses in C. albicans and MAF-1A has the potential to inhibit C. albicans growth through affect multi-targets of C. albicans cells. This study provides important insights into the mechanisms of action of MAF-1A and the responds of C. albicans under AMPs stress. However, further studies are needed to determine the key mechanisms that C. albicans responses to MAF-1A.
Author Contributions
TW and JW designed the project, analyzed data and wrote the manuscript. TW, JX, YZ, and XM carried out the experiments. YW, GG, and XS reviewed the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (81360254), the Guiyang Science and Technology Bureau and Guizhou Medical University United Foundation (GY2015-23), the Science and Technology Program of Guizhou Province Agricultural research Project [NY (2014)3054].
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.00894/full#supplementary-material
Footnotes
References
Bahar, A. A., and Ren, D. (2013). Antimicrobial peptides. Pharmaceuticals 6, 1543–1575. doi: 10.3390/ph6121543
Bowman, S. M., and Free, S. J. (2006). The structure and synthesis of the fungal cell wall. Bioessays 28, 799–808. doi: 10.1002/bies.20441
Bruno, V. M., Wang, Z., Marjani, S. L., Euskirchen, G. M., Martin, J., Sherlock, G., et al. (2010). Comprehensive annotation of the transcriptome of the human fungal pathogen Candida albicans using RNA-seq. Genome Res. 20, 1451–1458. doi: 10.1101/gr.109553.110
Cottier, F., Tan, A. S., Chen, J., Lum, J., Zolezzi, F., Poidinger, M., et al. (2015). The transcriptional stress response of Candida albicans to weak organic acids. G3 5, 497–505. doi: 10.1534/g3.114.015941
Cruz, J., Ortiz, C., Guzmán, F., Fernández-Lafuente, R., and Torres, R. (2014). Antimicrobial peptides: promising compounds against pathogenic microorganisms. Curr. Med. Chem. 21, 2299–2321. doi: 10.2174/0929867321666140217110155
Dantas Ada, S., Day, A., Ikeh, M., Kos, I., Achan, B., and Quinn, J. (2015). Oxidative stress responses in the human fungal pathogen, Candida albicans. Biomolecules 5, 142–165. doi: 10.3390/biom5010142
De Backer, M. D., Ilyina, T., Ma, X. J., Vandoninck, S., Luyten, W. H., and Vanden Bossche, H. (2001). Genomic profiling of the response of Candida albicans to itraconazole treatment using a DNA microarray. Antimicrob. Agents Chemother. 45, 1660–1670. doi: 10.1128/AAC.45.6.1660-1670.2001
Delattin, N., Cammue, B. P., and Thevissen, K. (2014). Reactive oxygen species-inducing antifungal agents and their activity against fungal biofilms. Future Med. Chem. 6, 77–90. doi: 10.4155/fmc.13.189
Eggimann, P., Garbino, J., and Pittet, D. (2003). Epidemiology of Candida species infections in critically ill non-immunosuppressed patients. Lancet Infect. Dis. 3, 685–702. doi: 10.1016/S1473-3099(03)00801-6
Fu, P., Wu, J., and Guo, G. (2009). Purification and molecular identificationn of an antifungal peptide from the hemolymph of Musca domestica (housefly). Cell Mol. Immunol. 6, 245–251. doi: 10.1038/cmi.2009.33
Gray, K. C., Palacios, D. S., Dailey, I., Endo, M. M., Uno, B. E., Wilcock, B. C., et al. (2012). Amphotericin primarily kills yeast by simply binding ergosterol. Proc. Natl. Acad. Sci. U.S.A. 109, 2234–2239. doi: 10.1073/pnas.1117280109
Guilhelmelli, F., Vilela, N., Albuquerque, P., Derengowski Lda, S., Silva-Pereira, I., and Kyaw, C. M. (2013). Antibiotic development challenges: the various mechanisms of action of antimicrobial peptides and of bacterial resistance. Front. Microbiol. 4:353. doi: 10.3389/fmicb.2013.00353
Guilhelmelli, F., Vilela, N., Smidt, K. S., de Oliveira, M. A., da Cunha Morales Álvares, A., Rigonatto, M. C., et al. (2016). Activity of scorpion venom-derived antifungal peptides against planktonic cells of Candida spp. and Cryptococcus neoformans and Candida albicans biofilms. Front. Microbiol. 7:1844. doi: 10.3389/fmicb.2016.01844
Heo, A., Jang, H. J., Sung, J. S., and Park, W. (2014). Global transcriptome and physiological responses of Acinetobacter oleivorans DR1 exposed to distinct classes of antibiotics. PLoS ONE 9:e110215. doi: 10.1371/journal.pone.0110215
Horn, D. L., Neofytos, D., Anaissie, E. J., Fishman, J. A., Steinbach, W. J., Olyaei, A. J., et al. (2009). Epidemiology and outcomes of candidemia in 2019 patients: data from the prospective antifungal therapy alliance registry. Clin. Infect. Dis. 48, 1695–1703. doi: 10.1086/599039
Khan, A., Ahmad, A., Ahmad Khan, L., Padoa, C. J., van Vuuren, S., and Manzoor, N. (2015). Effect of two monoterpene phenols on antioxidant defense system in Candida albicans. Microb. Pathog. 80, 50–56. doi: 10.1016/j.micpath.2015.02.004
Klis, F. M., de Groot, P., and Hellingwerf, K. (2001). Molecular organization of the cell wall of Candida albicans. Med. Mycol. 39(Suppl. 1), 1–8. doi: 10.1080/mmy.39.1.1.8-0
Lakshmaiah Narayana, J., and Chen, J. Y. (2015). Antimicrobial peptides: possible anti-infective agents. Peptides 72, 88–94. doi: 10.1016/j.peptides.2015.05.012
Le, C. F., Gudimella, R., Razali, R., Manikam, R., and Sekaran, S. D. (2016). Transcriptome analysis of Streptococcus pneumoniae treated with the designed antimicrobial peptides, DM3. Sci. Rep. 6:26828. doi: 10.1038/srep26828
Linares, C. E., Giacomelli, S. R., Altenhofen, D., Alves, S. H., Morsch, V. M., and Schetinger, M. R. (2013). Fluconazole and amphotericin-B resistance are associated with increased catalase and superoxide dismutase activity in Candida albicans and Candida dubliniensis. Rev. Soc. Bras. Med. Trop. 46, 752–758. doi: 10.1590/0037-8682-0190-2013
Liu, T. T., Lee, R. E., Barker, K. S., Lee, R. E., Wei, L., Homayouni, R., et al. (2005). Genome-wide expression profiling of the response to azole, polyene, echinocandin, and pyrimidine antifungal agents in Candida albicans. Antimicrob. Agents Chemother. 49, 2226–2236. doi: 10.1128/AAC.49.6.2226-2236.2005
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Lo, H. J., Tseng, K. Y., Kao, Y. Y., Tsao, M. Y., Lo, H. L., and Yang, Y. L. (2015). Cph1p negatively regulates MDR1 involved in drug resistance in Candida albicans. Int. J. Antimicrob. Agents 45, 617–621. doi: 10.1016/j.ijantimicag.2015.01.017
Lu, Y., Su, C., and Liu, H. (2014). Candida albicans hyphal initiation and elongation. Trends Microbiol. 22, 707–714. doi: 10.1016/j.tim.2014.09.001
Luo, Z. H., Wu, J. W., Fu, P., Guo, G., and Jiang, S. (2013). Effect of the synthetic Musca domestica antifungal peptide-1a (MAF-1A) on the pathogenicity of Candida albicans. J. Third Military Med. Univ. 35, 2203–2207.
Mahlapuu, M., Håkansson, J., Ringstad, L., and Björn, C. (2016). Antimicrobial peptides: an emerging category of therapeutic agents. Front. Cell Infect. Microbiol. 6:194. doi: 10.3389/fcimb.2016.00194
Malmsten, M. (2014). Antimicrobial peptides. UPS J. Med. Sci. 119, 199–204. doi: 10.3109/03009734.2014.899278
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Nagalakshmi, U., Wang, Z., Waern, K., Shou, C., Raha, D., Gerstein, M., et al. (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344–1349. doi: 10.1126/science.1158441
Nailis, H., Vandenbosch, D., Deforce, D., Nelis, H. J., and Coenye, T. (2010). Transcriptional response to fluconazole and amphotericin B in Candida albicans biofilms. Res. Microbiol. 161, 284–292. doi: 10.1016/j.resmic.2010.02.004
Nather, K., and Munro, C. A. (2008). Generating cell surface diversity in Candida albicans and other fungal pathogens. FEMS Microbiol. Lett. 285, 137–145. doi: 10.1111/j.1574-6968.2008.01263.x
Nguyen, L. T., Haney, E. F., and Vogel, H. J. (2011). The expanding scope of antimicrobial peptide structures and their modes of action. Trends Biotechnol. 29, 464–472. doi: 10.1016/j.tibtech.2011.05.001
Nookaew, I., Papini, M., Pornputtapong, N., Scalcinati, G., Fagerberg, L., Uhlén, M., et al. (2012). A comprehensive comparison of RNA-Seq-based transcriptome analysis from reads to differential gene expression and cross-comparison with microarrays: a case study in Saccharomyces cerevisiae. Nucleic Acids Res. 40, 10084–10097. doi: 10.1093/nar/gks804
Omardien, S., Brul, S., and Zaat, S. A. (2016). Antimicrobial activity of cationic antimicrobial peptides against gram-positives: current progress made in understanding the mode of action and the response of bacteria. Front. Cell Dev. Biol. 4:111. doi: 10.3389/fcell.2016.00111
Pasupuleti, M., Schmidtchen, A., and Malmsten, M. (2012). Antimicrobial peptides: key components of the innate immune system. Crit. Rev. Biotechnol. 32, 143–171. doi: 10.3109/07388551.2011.594423
Pushpanathan, M., Gunasekaran, P., and Rajendhran, J. (2013). Antimicrobial peptides: versatile biological properties. Int. J. Pept. 2013:675391. doi: 10.1155/2013/675391
Qin, N., Tan, X., Jiao, Y., Liu, L., Zhao, W., Yang, S., et al. (2014). RNA-Seq-based transcriptome analysis of methicillin-resistant Staphylococcus aureus bioflm inhibition by ursolic acid and resveratrol. Sci. Rep. 4:5467. doi: 10.1038/srep05467
Ruiz-Herrera, J., Elorza, M. V., Valentín, E., and Sentandreu, R. (2006). Molecular organization of the cell wall of Candida albicans and its relation to pathogenicity. FEMS Yeast Res. 6, 14–29. doi: 10.1111/j.1567-1364.2005.00017.x
Sandini, S., Stringaro, A., Arancia, S., Colone, M., Mondello, F., Murtas, S., et al. (2011). The MP65 gene is required for cell wall integrity, adherence to epithelial cells and biofilm formation in Candida albicans. BMC Microbiol. 11:106. doi: 10.1186/1471-2180-11-106
Scocchi, M., Mardirossian, M., Runti, G., and Benincasa, M. (2016). Non-membrane permeabilizing modes of action of antimicrobial peptides on bacteria. Curr. Top. Med. Chem. 16, 76–88. doi: 10.2174/1568026615666150703121009
Seo, M. D., Won, H. S., Kim, J. H., Mishig-Ochir, T., and Lee, B. J. (2012). Antimicrobial peptides for therapeutic applications: a review. Molecules 17, 12276–12286. doi: 10.3390/molecules171012276
Shah, P., Hsiao, F. S., Ho, Y. H., and Chen, C. S. (2016). The proteome targets of intracellular targeting antimicrobial peptides. Proteomics 16, 1225–1237. doi: 10.1002/pmic.201500380
Silva, P. M., Gonçalves, S., and Santos, N. C. (2014). Defensins: antifungal lessons from eukaryotes. Front. Microbiol. 5:97. doi: 10.3389/fmicb.2014.00097
Spampinato, C., and Leonardi, D. (2013). Candida infections, causes, targets, and resistance mechanisms: traditional and alternative antifungal agents. Biomed. Res. Int. 2013:204237. doi: 10.1155/2013/204237
Tavares, L. S., Silva, C. S., de Souza, V. C., da Silva, V. L., Diniz, C. G., and Santos, M. O. (2013). Strategies and molecular tools to fight antimicrobial resistance: resistome, transcriptome, and antimicrobial peptides. Front. Microbiol. 4:412. doi: 10.3389/fmicb.2013.00412
Thevissen, K., Ferket, K. K., François, I. E., and Cammue, B. P. (2003). Interactions of antifungal plant defensins with fungal membrane components. Peptides 24, 1705–1712. doi: 10.1016/j.peptides.2003.09.014
Trapnell, C., Pachter, L., and Salzberg, S. L. (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. doi: 10.1093/bioinformatics/btp120
Trapnell, C., Williams, B. A., Pertea, G., Mortazavi, A., Kwan, G., van Baren, M. J., et al. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. doi: 10.1038/nbt.1621
van der Weerden, N. L., Bleackley, M. R., and Anderson, M. A. (2013). Properties and mechanisms of action of naturally occurring antifungal peptides. Cell Mol. Life Sci. 70, 3545–3570. doi: 10.1007/s00018-013-1260-1
Wang, L., Feng, Z., Wang, X., Wang, X., and Zhang, X. (2010). DEGseq: an R package for identifying differentially expressed genes from RNA-seq data. Bioinformatics 26, 136–138. doi: 10.1093/bioinformatics/btp612
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Young, M. D., Wakefield, M. J., Smyth, G. K., and Oshlack, A. (2010). Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol. 11:R14. doi: 10.1186/gb-2010-11-2-r14
Keywords: Candida albicans, transcriptional responses, antimicrobial peptides, MAF-1A, RNA-Seq
Citation: Wang T, Xiu J, Zhang Y, Wu J, Ma X, Wang Y, Guo G and Shang X (2017) Transcriptional Responses of Candida albicans to Antimicrobial Peptide MAF-1A. Front. Microbiol. 8:894. doi: 10.3389/fmicb.2017.00894
Received: 05 March 2017; Accepted: 03 May 2017;
Published: 17 May 2017.
Edited by:
Octavio Luiz Franco, Universidade Católica de Brasília, BrazilReviewed by:
Humberto Lanz-Mendoza, Instituto Nacional de Salud Pública, MexicoCamila Guimarães Freitas, Federal Institute of Brasilia, Brazil
Copyright © 2017 Wang, Xiu, Zhang, Wu, Ma, Wang, Guo and Shang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jianwei Wu, d3VqdzkxNUBzaW5hLmNvbQ==