 Pieter De Maayer1*
Pieter De Maayer1* Habibu Aliyu2
Habibu Aliyu2 Surendra Vikram2
Surendra Vikram2 Jochen Blom3
Jochen Blom3 Brion Duffy4
Brion Duffy4 Don A. Cowan2
Don A. Cowan2 Theo H. M. Smits4
Theo H. M. Smits4 Stephanus N. Venter5
Stephanus N. Venter5 Teresa A. Coutinho5
Teresa A. Coutinho5- 1School of Molecular and Cell Biology, University of the Witwatersrand, Johannesburg, South Africa
- 2Centre for Microbial Ecology and Genomics, Genomics Research Institute, University of Pretoria, Pretoria, South Africa
- 3Department of Bioinformatics and Systems Biology, Justus-Liebig-University Giessen, Giessen, Germany
- 4Environmental Genomics and Systems Biology Research Group, Institute for Natural Resource Sciences, Zurich University of Applied Sciences, Winterthur, Switzerland
- 5Department of Microbiology, Forestry and Agricultural Biotechnology Institute, University of Pretoria, Pretoria, South Africa
Pantoea ananatis is ubiquitously found in the environment and causes disease on a wide range of plant hosts. By contrast, its sister species, Pantoea stewartii subsp. stewartii is the host-specific causative agent of the devastating maize disease Stewart’s wilt. This pathogen has a restricted lifecycle, overwintering in an insect vector before being introduced into susceptible maize cultivars, causing disease and returning to overwinter in its vector. The other subspecies of P. stewartii subsp. indologenes, has been isolated from different plant hosts and is predicted to proliferate in different environmental niches. Here we have, by the use of comparative genomics and a comprehensive suite of bioinformatic tools, analyzed the genomes of ten P. stewartii and nineteen P. ananatis strains. Our phylogenomic analyses have revealed that there are two distinct clades within P. ananatis while far less phylogenetic diversity was observed among the P. stewartii subspecies. Pan-genome analyses revealed a large core genome comprising of 3,571 protein coding sequences is shared among the twenty-nine compared strains. Furthermore, we showed that an extensive accessory genome made up largely by a mobilome of plasmids, integrated prophages, integrative and conjugative elements and insertion elements has resulted in extensive diversification of P. stewartii and P. ananatis. While these organisms share many pathogenicity determinants, our comparative genomic analyses show that they differ in terms of the secretion systems they encode. The genomic differences identified in this study have allowed us to postulate on the divergent evolutionary histories of the analyzed P. ananatis and P. stewartii strains and on the molecular basis underlying their ecological success and host range.
Introduction
The enterobacterial genus Pantoea currently encompasses twenty-two distinct species1. A key feature of this genus is the extensive ecological and functional diversity of its members, testament to the Greek derivation of the genus name – Gr. adj. pantoios, of all sorts and sources (Gavini et al., 1989; Mergaert et al., 1993; Walterson and Stavranides, 2015). Pantoea strains have been isolated globally from diverse ecological sources, both natural and man-made, and have diverse lifestyles, including as commensals, insect symbionts and as plant and clinical pathogens (Mergaert et al., 1993; Walterson and Stavranides, 2015). There has also been increased interest in Pantoea strains from a biotechnological perspective, including in bioremediation strategies, the production of novel medical therapeutics and as biological control agent against insect pests and plant pathogens (Walterson and Stavranides, 2015; Weller-Stuart et al., 2016).
The species Pantoea ananatis (Pnan) exemplifies the ecological and functional diversity of the genus, as strains have been isolated from a wide range of environmental sources, including water, soil and man-made environments and have diverse lifestyles, including as insect commensals, clinical pathogen and as part of the epiphytic and endophytic communities on a very broad range of plant hosts (Coutinho and Venter, 2009; Weller-Stuart et al., 2016). Comparative genomic analyses have revealed a wide range of factors underlying their ecological success and ability to interact with different hosts (Weller-Stuart et al., 2016). Individual strains of Pnan have also been shown to have plant growth promoting capabilities, anti-bacterial and anti-fungal properties, and bioremediation and biofuel production capacities and as such there has been much recent interest in the biotechnological application of this species (Walterson and Stavranides, 2015; Weller-Stuart et al., 2016). Pnan is, however, best known as an emerging, and potentially opportunistic, pathogen on an extremely wide spectrum of plant hosts, including rice, maize, onions and Eucalyptus where it causes an equally vast array of disease symptoms, including stalk, fruit and bulb rot, leaf spot, blight and dieback (Coutinho and Venter, 2009; Weller-Stuart et al., 2016). To date, relatively little is known about the mechanisms of pathogenesis, epidemiology and spread, although insects such as tobacco thrips and cotton fleahoppers have been described as potential vectors of Pnan strains (Coutinho and Venter, 2009).
By contrast to Pnan, the closely related species P. stewartii (Pnst) incorporates the aggressive phytopathogen Pantoea stewartii subsp. stewartii which is host specific to maize (Zea mays), in particular sweet corn (Z. mays convar. saccharata var. rugosa) and inbred lines of maize (Roper, 2011). Stewart’s wilt of maize is a devastating disease that results in severe crop losses of susceptible varieties and is endemic to the United States, and as a result quarantine restrictions are in place to prevent spread of this pathogen (Roper, 2011). Given the agronomic importance of Pnst subsp. stewartii, its epidemiology, pathogenesis and means of disease control have been extensively researched. By contrast to Pnan, Pnst subsp. stewartii has a restricted lifecycle, overwintering in the corn flea beetle (Chaetocnema pulicularia), which subsequently introduces the pathogen into feeding wounds via its feces when it emerges in spring (Roper, 2011; Correa et al., 2012). Pnst subsp. stewartii subsequently colonizes the interstitial spaces in maize leaf tissues, where it causes the development of water-soaked lesions, and subsequently the xylem, resulting in systemic spread and wilting. The pathogen is then acquired by the last seasonal generation of the corn flea beetle in which it again overwinters (Roper, 2011). The other member of this species, Pantoea stewartii subsp. indologenes, is avirulent on maize and was long considered as non-pathogenic, although it has been linked to symptom development on pearl and foxtail millet, Eucalyptus, cluster bean and rot of pineapple fruit, suggesting that unlike the Pnst subsp. stewartii this subspecies has a broader host range (Mergaert et al., 1993; Coutinho et al., 2002). As with Pnan, little is known about the epidemiology and pathogenicity determinants of Pnst subsp. indologenes. Furthermore, two Pnst strains, which have not been characterized to the subspecies level, were isolated from a waterfall (Izzati Mohamad et al., 2015; Tan et al., 2015), suggesting that at least some isolates of this species are capable of surviving in the environment outside of a plant or insect host.
The distinct lifestyles and ecological versatility of Pnan and the Pnst subspecies raises several questions about the evolution of these bacteria. How has Pnst subsp. stewartii evolved into a host-specific pathogen on maize, while Pnan and Pnst subsp. indologenes cause disease on a broad range of plant hosts? Why is the lifecycle of Pnst subsp. stewartii largely restricted to its maize and insect hosts, while Pnan and other Pnst strains are frequently isolated from a wide range of natural sources and plant hosts? Do these pathogens make use of different pathogenicity factors for symptoms development? With the advent of cost-effective genome sequencing, comparative genomic approaches provide a viable approach to answering these questions. For example, host-specificity of Pseudomonas syringae strains isolated from different plant hosts was correlated, in part, to differences in their T3SS effector and phytotoxin production profiles by genome comparisons and genetic manipulation (Baltrus et al., 2001, 2012). Similar studies incorporating population genetics and comparative genomics have been undertaken to study genetic variation between host-adapted and broad host range Ralstonia solanacearum strains (Ailloud et al., 2015), and the distinct ecologies, physiologies and host specificities among Xanthomonas strains (Jacques et al., 2016). A previous pan-genome analysis of eight Pnan strains also revealed distinct factors which may influence their ability to interact with plant, insect, and animal hosts (De Maayer et al., 2014). Here we have employed comparative genomic analyses and a broad array of in silico tools to compare the genomes of nineteen Pnan and ten Pnst strains. Our analyses revealed a range of different evolutionary drivers, as well as strain- and species-specific factors which may explain the distinct lifestyles of Pnan and Pnst strains, and expand our understanding of these two important agronomic species.
Materials and Methods
Genomic Analyses
The genome sequences of nineteen Pnan and ten Pnst strains were obtained from the NCBI genome database2 (Table 1). The genome assemblies were refined by genome alignment using MAUVE v. 2.4.0 (Darling et al., 2010) and genome scaffolding using the MeDuSa web server (Bosi et al., 2015). General sequence manipulations, G+C % calculations and local BlastP searches were undertaken using BioEdit v. 7.2.5 (Hall, 1999). The complete and high quality draft genomes were submitted to the RAST v 2.0 server (Aziz et al., 2008) for structural and functional annotation. The genome annotations and local BlastP searches were used to identify putative plasmid (BlastP analyses with Rep and Mob proteins) sequences, ICE elements, transposases and pathogenicity determinants. For the BlastP analyses, orthologous proteins were identified as those sharing >50% amino acid identity and alignment coverage of >70%. Transposases were further categorized at the family level by identifying the best Blast hits from comparison against the Conserved Domain Database using CD-Search (Marchler-Bauer and Bryant, 2004). Similarly, integrated prophages were identified using the PHAST server (Zhou et al., 2011). Insertion Sequences were identified and analyzed using the ISSaga server (Varani et al., 2011).
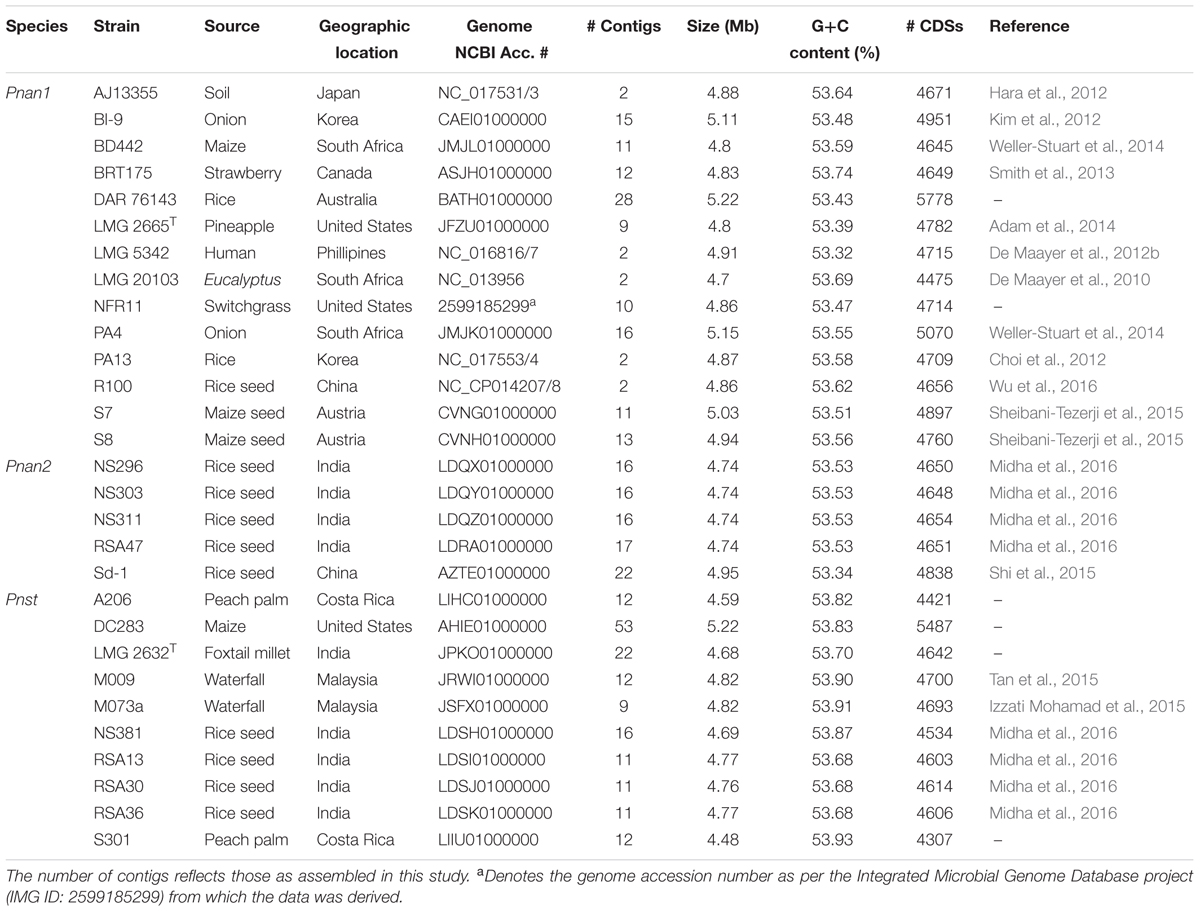
TABLE 1. General genome properties of the twenty-nine compared Pnan and Pnst strains.
Comparative Genomic Analyses
The annotated GenBank files from RAST were uploaded to the EDGAR 2.1 comparative genomic pipeline (Blom et al., 2016). Orthologous proteins among the compared genomes were identified on the basis of BLAST Score Ratio Values (SRVs) as implemented in EDGAR (Blom et al., 2009). Core proteins (shared among individual Pnan1+ Pnan2 + Pnst strains and among all twenty-nine compared strains), proteins shared among strains belonging to two groups (Pnan1 + Pnan2; Pnan1 + Pnst; Pnan2 + Pnst) and unique proteins (unique to Pnan1, Pnan2 or Pnst strains) were identified. The proteins in each of these datasets were further assigned to their Conserved Orthologous Groups supra-functional (Cellular processes; Information storage and processing; metabolism; poorly characterized) and functional categories on the basis of the HMM models for Bacteria (bactNOG 4.5) using hmmscan (Huerta-Cepas et al., 2016). Orthologous pathogenicity determinants identified from the annotated genomes were identified in EDGAR 2.1 (Blom et al., 2016) and average amino acid identity values determined using local BlastP analyses in BioEdit v 7.2.5 (Hall, 1999). Orthology was assumed for those proteins sharing >50% amino acid identity and alignment coverage of >70%.
Phylogenomic Analyses
A core genome phylogeny was constructed via the EDGAR 2.1 pipeline (Blom et al., 2016), on the basis of 2,817 core genes conserved among the compared Pnan1, Pnan2 and Pnst genomes as well as the outgroup strain P. vagans C9-1 (Smits et al., 2011). Core genes were aligned using MUSCLE (Edgar, 2004), non-matching regions in the alignments were removed using GBLOCKS (Talavera and Castresana, 2007), and the aligned sequences were concatenated and used to generate a neighbor-joining tree with Phylip (Felsenstein, 1989). The phylogeny was visualized using MEGA v 7.0.14 (Kumar et al., 2016). The two-way ANI values were calculated for the core genes for each pair of compared strains using the ani.rb script included in the Enveomics package (Rodriguez-R and Konstantinidis, 2016). Digital DNA-DNA hybridization (dDDH) values were estimated using the Genome-to-Genome Distance Calculator 2.03, with the recommended formula 2 (Auch et al., 2010; Meier-Kolthoff et al., 2013).
Results
Pantoea ananatis and Pantoea stewartii Genome Properties
The complete or high quality draft genomes of ten Pnst and nineteen Pnan strains were included in this study (Table 1). The draft genomes were assembled to between nine (Pnan LMG 2665T and Pnst M073a) and fifty-three contigs (Pnst DC283). The Pnst and Pnan genomes have an average size of 4.89 and 4.76 Mb, respectively. The genomes of three Pnan strains are substantially larger than the other sixteen compared strains (B1-9: 5.11 Mb; PA4: 5.15 Mb; and DAR76143: 5.23 Mb). Similarly, the draft genome of Pnst DC283 (5.23 Mb) is substantially larger than those of the other nine Pnst strains (average size: 4.71 Mb). Concomitantly, an additional ∼918 protein coding sequences (CDSs) are present on the Pnst DC283 genome (5,487 CDSs) compared to the other nine Pnst strains (average 4,568 CDSs). The highest number of proteins (5,778 CDSs) are encoded on the genome of Pnan DAR76143, while the strain with the smallest genome, Pnst S301 (4.48 Mb), encodes 4,307 proteins (Table 1). Slightly higher G+C contents were observed for the genomes of the Pnst strains (average G+C content: 53.80%) than those of the Pnan strains (average G+C content: 53.53%).
Phylogenomic Analyses Reveal the Existence of Two Distinct P. ananatis Clades
A core genome phylogeny was constructed on the basis of 2,810 genes conserved in all Pnan and Pnst strains used in this study, as well as the outgroup strain P. vagans C9-1 (Smits et al., 2011). This phylogeny showed distinct clustering of the Pnan and Pnst strains. Two distinct clades could be observed among the Pnan strains, with the first clade encompassing fourteen strains, including the type strain Pnan LMG 2665T, while the second clade incorporates five strains (Figure 1). dDDH values were determined using the Genome-to-Genome Distance Calculator (GGDC) tool (Supplementary Table S1) (Auch et al., 2010). On the basis of parameters for species delineation deemed valid for both wet-lab DNA-DNA hybridization and dDDH (organisms displaying DDH values < 70% are considered as distinct species) (Meier-Kolthoff et al., 2013), the nineteen Pnan strains and ten Pnst strains can be considered as distinct species (average dDDH values of 27.77 ± 0.20%; dDDH ± standard deviation). ANI threshold values of 95–96% have also been used to demarcate distinct species (Goris et al., 2007) and the average ANI value of 84.26 ± 0.14% (Average ANI value ± standard deviation) observed in this study (Supplementary Table S1) further support the delineation of Pnan and Pnst as distinct species.
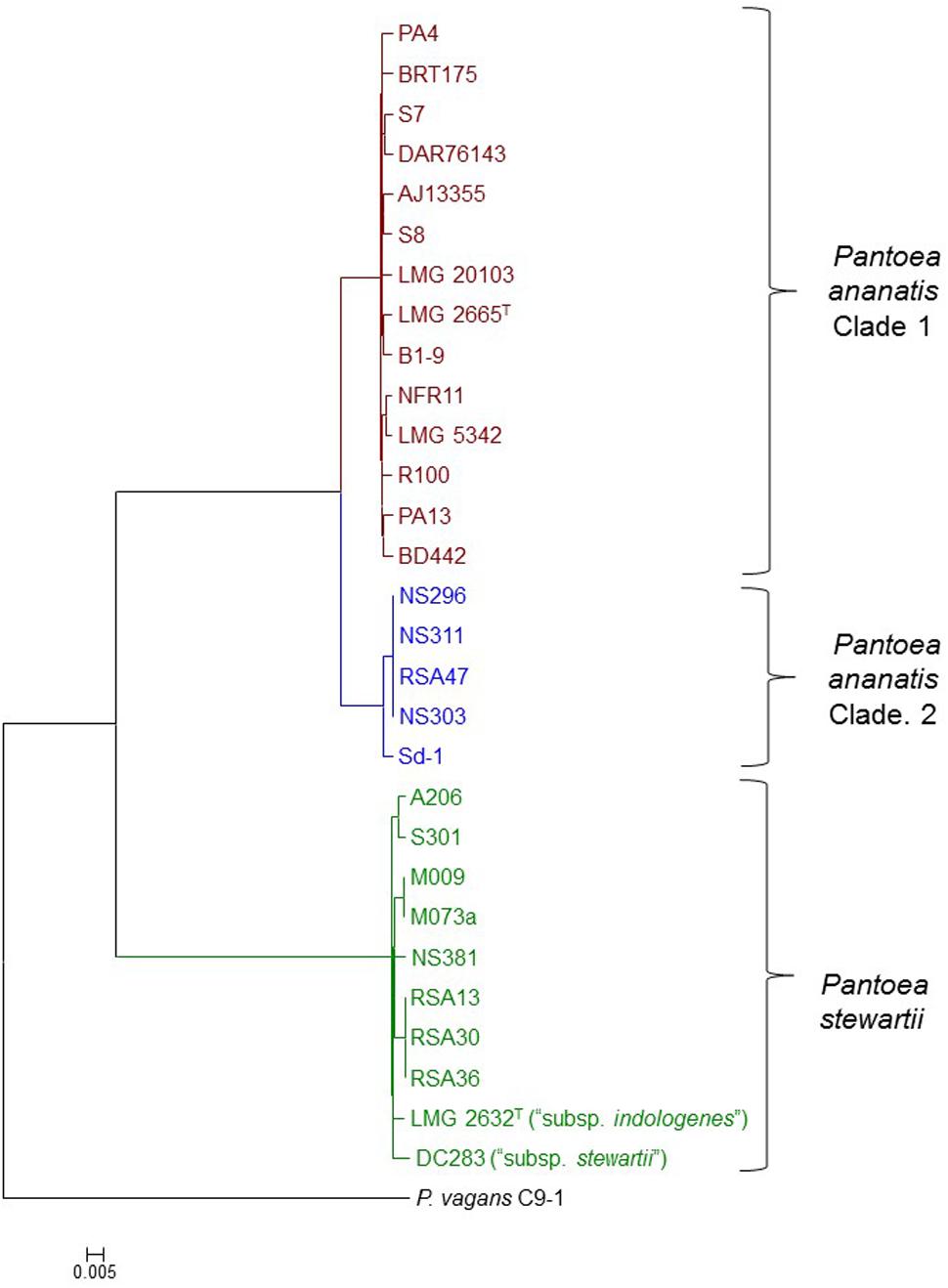
FIGURE 1. Core genome phylogeny of twenty-nine compared Pnan and Pnst strains. The core genome phylogeny was constructed on the basis of 2,817 core genes conserved among all compared strains via the EDGAR 2.1 pipeline (Blom et al., 2016). The genome of P. vagans C9-1 (Smits et al., 2011) (NCBI Acc. # CP002206.1; CP001893/4/5.1) was used as outgroup.
The five Pnan strains in the second clade of the core genome phylogeny share an average dDDH value of 69.89 ± 1.11% and average ANI value of 96.32 ± 0.10 % with the fourteen strains in the other Pnan clade (Figure 1), while the latter strains share average dDDH and ANI values of 92.04 ± 1.81% and 99.10 ± 0.13%, respectively (Supplementary Table S1). The dDDH values suggests that the strains in the second clade are on the borderline for forming a distinct species, while the lower ANI values provide support that strains in this clade are genetically distinct from strains in the other Pnan clade. Further phenotypic and biochemical characterization will need to be undertaken to validate the phylogenomic observations and to describe the potential novel species, as metadata for many of the strains are lacking. For the purpose of further comparative genomic, mobilomic and pathogenomic characterization of the Pnan and Pnst strains, we have distinguished the five Pnan strains (NS296, NS303, NS311, RSA47, and Sd-1) in the second clade as P. ananatis Clade 2 (Pnan2) while the remaining fourteen P. ananatis strains form part of P. ananatis Clade 1 (Pnan1).
Far higher average ANI (99.14 ± 0.29%) and dDDH (92.76 ± 2.53%) values were observed when comparing the ten Pnst strains (Supplementary Table S1), suggesting that less phylogenetic diversity exists between within this species and raising questions about their delineation as distinct subspecies (Meier-Kolthoff et al., 2014). This is, however, in contrast to the wet-lab DDH values observed (79%) when the genomic DNA of Pnst subsp. indologenes LMG 2632T was compared with that of the type strain of Pnst subsp. stewartii LMG 2715T and clear phenotypic differences were observed between these two subspecies in the original species description (Mergaert et al., 1993). We have therefore maintained the original subspecies nomenclature for the further comparative genomic analyses while the remaining eight Pnst strains were not delineated at the subspecies level.
Pan-genome Analyses Reveal Extensive Interspecies and Inter-subspecies Variability and Functional Diversity among Pnan Clade 1, Pnan Clade 2 and Pnst
Pan-genome analyses were undertaken using the EDGAR 2.1 pipeline, where orthologs were identified on the basis of Blast SRV (Blom et al., 2009). Individual Pnan1, Pnan2 and Pnst strains were three-way compared and average values for the core and accessory genome fractions were determined (Figure 2). An extensive core genome, consisting of an average of 3,571 CDSs (range 3,194–3,674 CDSs) was observed for the three-way comparison of the twenty-nine strains. This constitutes between 55.28 and 83.93% of the CDSs encoded on each genome, with the lower values reflecting those strains with larger genomes, including Pnan1 strains B1-9 and DAR76143 and Pnst DC283, while the higher value reflects the smallest genome of Pnst S301. It is notable that the core genome is considerably smaller when considering Pnan1+ Pnan2 + Pnst DC283 (average core CDSs = 3,241) than Pnan1+ Pnan2 + all other Pnst strains (average core CDSs = 3,612 CDSs).
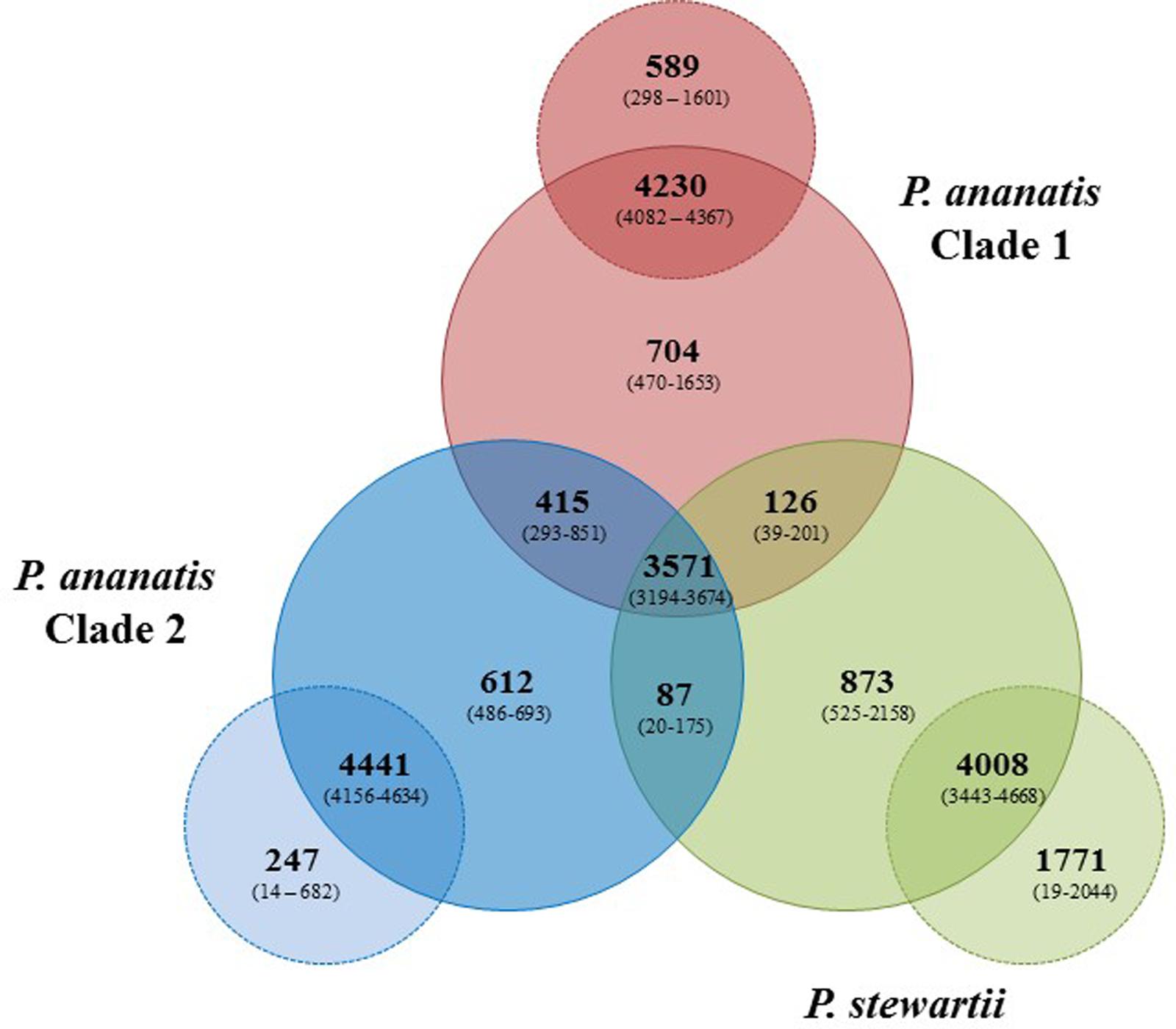
FIGURE 2. Pan-genome Venn diagram of the twenty-nine compared Pnan and Pnst strains. The average number (bold large) and the range (in brackets) of proteins comprising the core (in Pnan1+Pnan2+Pnst strains), accessory shared (between Pnan1+Pnan2; Pnan1+Pnst; Pnan2+Pnst) and accessory unique genome fractions of the three-ways compared Pnan1, Pnan2 and Pnst strains, as well as the average and range of proteins that are core and accessory within the three distinct groups are shown.
Analysis of the shared accessory fractions showed a substantial number of proteins shared only between the Pnan1 and Pnan2 strains (average: 415 CDSs; range 293–851 CDSs), which may be correlated to their closer phylogenetic relationship. By contrast, few proteins are shared between Pnan1+ Pnst and Pnan2+ Pnst strains, respectively. Similar proportions of proteins are unique to the Pnan1, Pnan2, and Pnst groups (average unique CDSs = 704, 612, and 873 CDSs, respectively) (Figure 2). Pair-wise comparisons between strains in each of the three groups were also undertaken. While large core genomes were observed for combinations of strains in the Pnan1, Pnan2 and Pnst clusters, lower intraspecific variability (average unique CDSs = 247; range 14–682 CDSs) exists among Pnan2 strains. This may be linked to the fact that four of the five Pnan2 strains analyzed were isolated from rice seeds in the same study (Midha et al., 2016). Many more unique CDSs could be observed when comparing Pnan1 strains (average unique CDSs = 589; range 298–1601 CDSs). The highest number of unique CDSs was, however, observed among the Pnst strains (average unique CDSs = 1,771 CDSs; range 525–2158 CDSs) (Figure 2). This could mainly be attributed to the maize pathogenic strain Pnst DC283. Comparisons of Pnst DC283 and other Pnst strains showed that between 1,956 and 2,044 Pnst DC283 CDSs have no orthologs in the other Pnst strains. This highlights the extensive intra-specific variability, in terms of protein content, among strains of P. ananatis and P. stewartii, particularly among the Pnan1 and Pnst strains, while the lower intra-specific variability in the Pnan2 clade may reflect clonality among four of the five studied strains that were isolated from rice seeds.
The protein datasets for the core and accessory fractions were extracted and subjected to local HMMER search against the eggNOG 4.5 BactNOG database (Huerta-Cepas et al., 2016). By this means the proteins in the distinct datasets were classified according to their Conserved Orthologous Groups functional and super-functional categories (Tatusov et al., 2000; De Maayer et al., 2014) and the relative proportions of proteins involved in each distinct functional category were determined (Figure 3). Proteins belonging to the “poorly characterized” super-functional category were removed as these make up the majority of proteins in each dataset and thus dilute the overall differences in the other super-functional categories. The core genome is predominated by proteins in the “metabolism” super-functional category. Similarly, the accessory fractions shared by Pnan1+ Pnan2 (but not Pnst) strains and Pnan1+ Pnst (but not Pnan2) are also dominated by proteins involved in metabolism. By contrast, more proteins in the “cellular processing and signaling” super-functional category are shared by the Pnan2+ Pnst strains. The proteins unique to the Pnan1 strains and Pnan2 strains are mainly involved in metabolism and information processing and storage, respectively (Figure 3). This difference is mainly due to the much higher number of proteins involved in metabolism in Pnan1 strains, in particular nucleotide transport and metabolism (3.2× as many proteins in Pnan1 than in the others) and lipid transport and metabolism (3.1× as many proteins in Pnan1 than in the others), while relatively similar numbers of proteins are involved in information storage and processing. One key observation from this analysis was the difference between the accessory fraction of Pnst DC283 and that of the other Pnst strains (Figure 3). While relatively similar numbers of proteins are involved in metabolism, much higher numbers of proteins are involved in information processing and signaling in Pnst DC283 than in the other Pnst strains. This can largely be attributed to those proteins involved in DNA replication, recombination and repair, with an average of nineteen proteins involved in this function in the other Pnst strains, while an average of 398 proteins fall in this category in Pnst DC283. This will be discussed in more detail below.
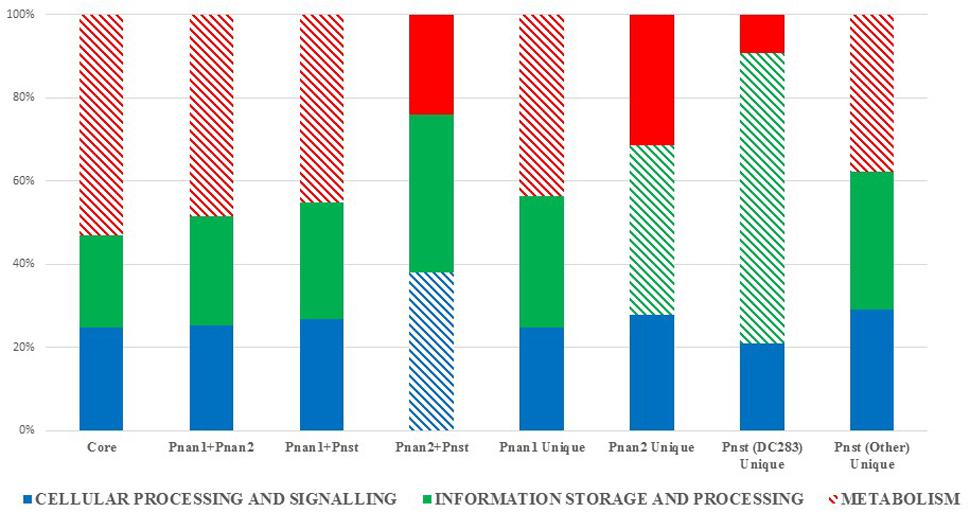
FIGURE 3. Super-functional classification of the core, accessory shared and unique proteins among the compared Pnan1, Pnan2 and Pnst strains. The relative percentages of proteins involved in the super-functional categories Cellular processing and signaling (blue), Information processing and storage (green) and Metabolism (red) were calculated. The predominant fraction is indicated with cross-hatched filling.
Plasmid Maintenance, Phage and ICE Element Integration and Transposable Elements Are Key Drivers of Evolutionary Diversification
Plasmids
A particular feature of Pnst subsp. stewartii strains is their ability to maintain large numbers of plasmids, with between eleven and thirteen plasmids, ranging in size from 4 to 320 kb, have previously been identified in 39 virulent strains by gel electrophoresis (Coplin et al., 1981). It was postulated that up to 25% of the total Pnst subsp. stewartii genomic DNA content may be plasmid-borne (Coplin et al., 1981). Using available sequences for Pnst subsp. stewartii plasmids, as well as searching for proteins involved in plasmid replication and maintenance, potential plasmids were identified in Pnst DC283. By this means, copies of four previously characterized Pnst plasmids, pSW100 (AHIE01000052.1; 4.3 kb; circular), pSW200 (AHIE01000062.1; 4.4 kb; circular), pSW800 (AHIE01000065.1; 34.4 kb; circular) and pSW1200 (AHIE01000039.1; 115.4 kb; incomplete) were identified (Fu et al., 1995, 1997, 1998; Wu et al., 2001). One further complete circular plasmid (AHIE01000057.1; 13.4 kb) was found. In addition, five further contigs containing either rep, tra/trb or mob genes were identified. We have previously analyzed a plasmid, LPP-1 (Large Pantoea Plasmid 1), which is universal in all Pantoea spp. genomes sequenced to date, and which is thought to play a major role in the ecological diversification of Pantoea spp. (De Maayer et al., 2012a). A 294.6 kb contig (AHIE01000038.1) represents an incomplete copy of this plasmid in Pnst DC283. Thus at least ten Pnst DC283 contigs, with a total size of 777.6 kb, are of plasmid origin, suggesting that plasmids contribute at least 14.90% of total genomic DNA content of Pnst DC283, with the 845 plasmid-encoded CDSs representing 15.40% of the total genomic CDSs.
On the basis of the genome annotations and local BlastP analyses with the Pnst DC283 Rep and Mob proteins, putative plasmids maintained by each of the Pnan1, Pnan2 and Pnst were characterized (Figure 4 and Supplementary Table S2). Three predicted plasmids are harbored in the genome of Pnst subsp. indologenes LMG 2632T, contributing 8.06% of the total genomic DNA content, while two plasmids are maintained by the other Pnst strains, with the exception of Pnst NS381, which only harbors a 317.5 kb LPP-1 plasmid (Figure 4 and Supplementary Table S2). Aside from the universal LPP-1 plasmid, seven Pnan1 strains harbor an additional one to four plasmids. In the case of Pnan1 PA4, its five plasmids contribute 11.51% of the total genomic DNA content and 12.25% of the total CDSs encoded on the genome. By contrast, the genomes of the Pnan2 strains generally only include LPP-1 plasmids, with the exception of Pnan2 strain Sd-1 which includes an additional 104.9 kb plasmid.
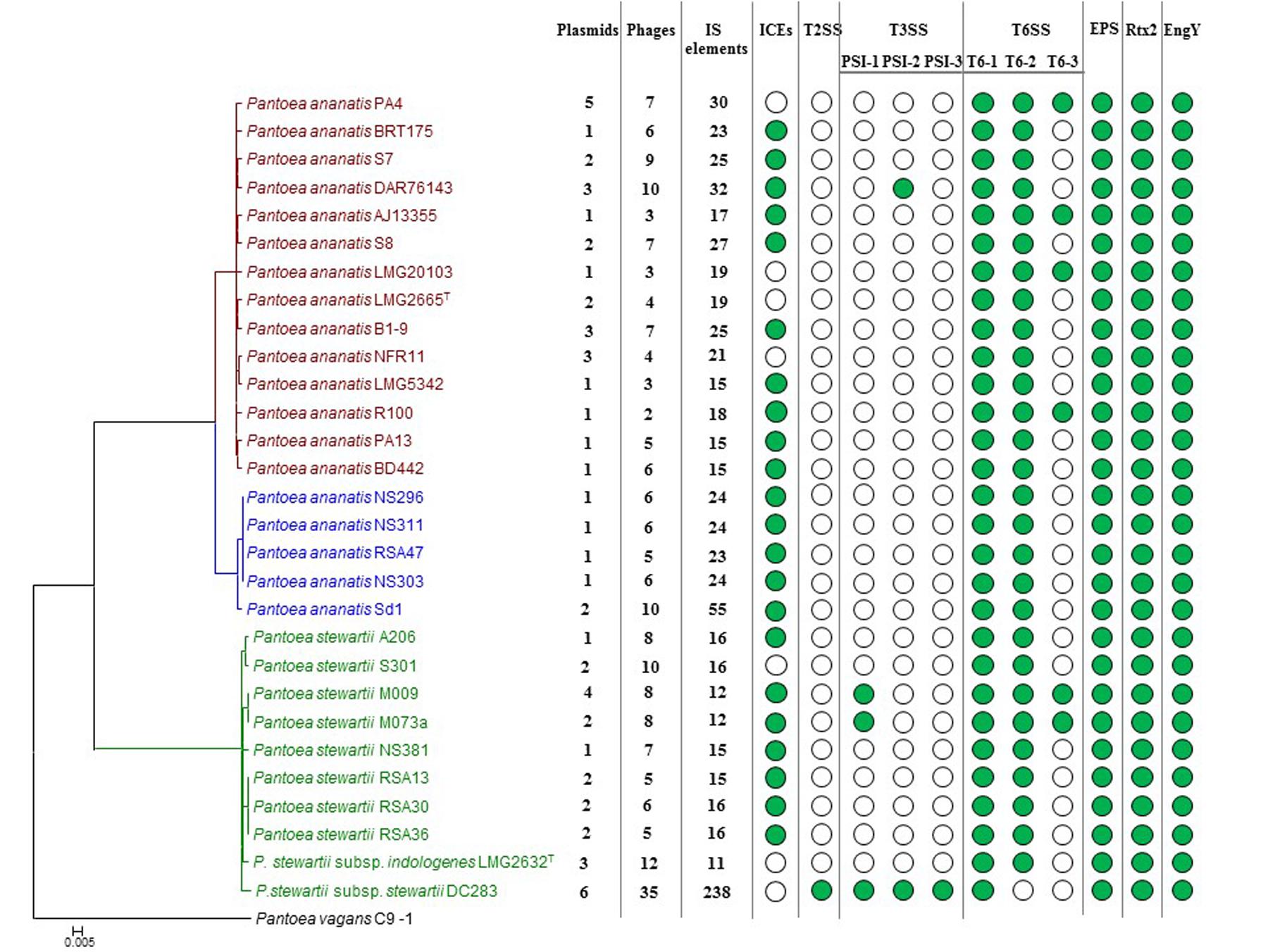
FIGURE 4. Prevalence of the mobilome and pathogenicity factors among the Pnan and Pnst strains. A core genome phylogeny is shown on the left as constructed on the basis of 2,817 core genes conserved among all compared strains. The number of predicted plasmids, prophages and insertion sequences (IS) present in each genome are indicated. Green dots indicate the presence of integrative and conjugative elements (ICEs) and pathogenicity determinants encoded on the genome.
Comparisons of the LPP-1 plasmids showed that a relatively high degree of synteny and conservation exist in this plasmid among the compared strains, with 148 CDSs conserved among all of the LPP-1 plasmids (42.17–61.67% of the total LPP-1 encoded CDSs). By contrast, the other plasmids are less well conserved. For example, only twenty, zero and eight out of the 529 CDSs encoded on the other Pnst DC283 plasmid contigs share orthology with proteins encoded on the non-LPP-1 plasmids of Pnan1 PA4 (four plasmids; 280.4 kb; 313 CDSs), Pnan2 Sd-1 (one plasmid; 104.9 kb; 103 CDSs) and Pnst LMG 2632 (two plasmids; 75.7 kb; 86 CDSs), respectively. These orthologs are largely restricted to those CDSs involved in plasmid replication, transfer and stabilization, while the plasmid cargo genes are not conserved. This would suggest that plasmids play a major role in the diversification of the Pnan and Pnst strains. Several predicted pathogenicity determinants, including a Type II secretion systems and three Type III secretion systems in Pnst DC283 and a Type VI secretion system in several Pnan1 strains are encoded by these plasmid cargo genes, further suggesting an important role of plasmids in the evolution of pathogenicity mechanisms.
Integrative and Conjugative Elements
Previously, we have shown that Integrative and Conjugative Elements (ICEs), a class of self-transmissible integrative elements, may play an important role in the ecological diversification and antibiotic capacities of P. ananatis (De Maayer et al., 2015). In the current study, ICEs were present in the genomes of 22/29 compared strains, with all Pnan2 strains carrying ICEPan elements, while the genomes of 10/14 Pnan1 and 7/10 Pnst strains incorporate ICEPan elements (Figure 4 and Supplementary Table S2). Furthermore, the genome of one strain, Pnan1 DAR76143, incorporates two distinct ICEPan elements. As was observed for the plasmids, genes coding for the excision, conjugative transfer, integration and maintenance of the ICEPan elements were conserved among the different compared strains, while variability was observed in terms of the ICEPan cargo genes. While ICEPan element proteins only constitute a small proportion of the overall genome protein content (0.71–3.51%), the versatile nature of these elements may also contribute to the diversification of the strains carrying this element.
Prophages
Phages play a major role in the evolution of bacteria. Chromosomal rearrangements and deletion events associated with prophage integration and excision have been linked to strain specific differences in a range of bacterial taxa (Canchaya et al., 2003; Brüssow et al., 2004). Prophages also carry various fitness and virulence factors that contribute toward the diversification and pathogenesis of their bacterial hosts (Canchaya et al., 2003; Brüssow et al., 2004).
Prophages were detected in the genomes of all twenty-nine strains analyzed using the PHAST server (Zhou et al., 2011). Between two and ten predicted phage elements are integrated into the genomes of Pnan1 strains (∼0.4–2 prophages integrated per Mb genomic DNA), while between six and ten prophages were identified in the Pnan2 genomes (∼2–2.7 prophages integrated per Mb) (Figure 4 and Supplementary Table S2). Similar patterns of prophage integration were observed for all of the Pnst strains compared, with the exception of Pnst DC283. In the latter strain thirty-five distinct (seven intact and twenty-eight incomplete) phage elements could be observed, with ∼7 prophages integrated per Mb of genomic DNA (Figure 4 and Supplementary Table S2). These phages encode 14.84% of the total genomic proteins indicating that, along with plasmids, bacteriophage integration probably plays an important evolutionary role in this strain.
Insertion Sequences and Transposases
In comparison to the Pnan1, Pnan2 and other Pnst genomes, a much larger proportion of the Pnst DC283 CDSs are involved in the “DNA replication, recombination and repair” COG function (560 CDSs – 10.20% of total genomic CDSs) (Figure 3). Analysis of this fraction revealed that most of the CDSs in this functional category code for up to 241 distinct transposases or their inactivated derivatives. By contrast, analysis of the annotations of the other genomes revealed that only between eleven (Pnst LMG 2632) and fifty-three (Pnan2 Sd-1) transposases are encoded on the genomes (Supplementary Table S2). Insertion Sequences (ISs) were also predicted from the genome sequences using the ISSaga server (Figure 4 and Supplementary Table S2) (Varani et al., 2011). Among the Pnan1 strains, between 15 and 32 ISs, belonging to between three and nine IS families, constitute an average 1.45% of the total genomic DNA. Between 23 and 55 ISs (1.28–3.16% of the total genomic DNA), belonging to between three and nine IS families, are incorporated in the Pnan2 genomes. By contrast, with the exception of Pnst DC283, far fewer ISs are integrated into the genomes of the other Pnst strains (between 11 and 14 ISs; 0.63–1.07% of the total genomic DNA). A total of 238 distinct IS elements were predicted in the genome of Pnst DC283, with a total predicted size of 802.8 kb, constituting 15.38% of the total genomic DNA content (Figure 4 and Supplementary Table S2). The ISs incorporated in the genome of this strain are also more diverse, belonging to sixteen distinct families, with the majority belonging to the IS66 (101) and IS630 (64) families.
The Pnan and Pnst Strains Share Several Common Pathogenicity Determinants but Vary in Terms of Their Secretion Systems
While little is known about the pathogenicity determinants of P. ananatis, the pathogenicity and virulence factors of the maize pathogenic Pnst subsp. stewartii have been extensively studied (Roper, 2011). During the early infection phase, Pnst subsp. stewartii is introduced into the plant apoplast by feeding corn flea beetles, where it subsequently causes cellular damage and cell content leakage, resulting in water soaked lesions. A recent study has attributed this activity to a large repetitive RTX-like toxin, Rtx2 (Roper et al., 2015). Analysis of the other genomes showed that this protein is well conserved among all Pnan and Pnst genomes (74.96% average amino acid identity; E-value: 0.0) (Figure 4), and hence the water-soaked lesion symptoms observed in maize, rice and Eucalyptus infected with P. ananatis (Coutinho et al., 2002; Bomfeti et al., 2008; Mondal et al., 2011) may be attributed to this pathogenicity factor. During the subsequent system infection phase of Stewart’s vascular wilt, Pnst subsp. stewartii colonizes the xylem vessels, where it produces an exopolysaccharide, stewartan that results in vascular occlusions and vascular wilt (Wang et al., 2012). Stewartan biosynthesis is dependent on a single ∼18.4 kb genetic locus (AF077292.2) which codes for fourteen proteins. Highly conserved homologous and syntenous loci (92.30% average nucleotide identity) are present in all the Pnst and Pnan genomes compared Figures 4, 5A) and may encode exopolysaccharides with similar structures and functions as stewartan. Additionally, Pnst DC283 produces an endoglucanase, EngY (CKS_4400), which has both glucanolytic and xylanolytic activities and has been shown to play a role in the systemic spread of this pathogen within its maize host (Mohammadi et al., 2012). Orthologs (94.39% average amino acid identity; E-value: 0.0) of this enzyme are encoded on the genomes of all the Pnan and Pnst strains compared (Figure 4).
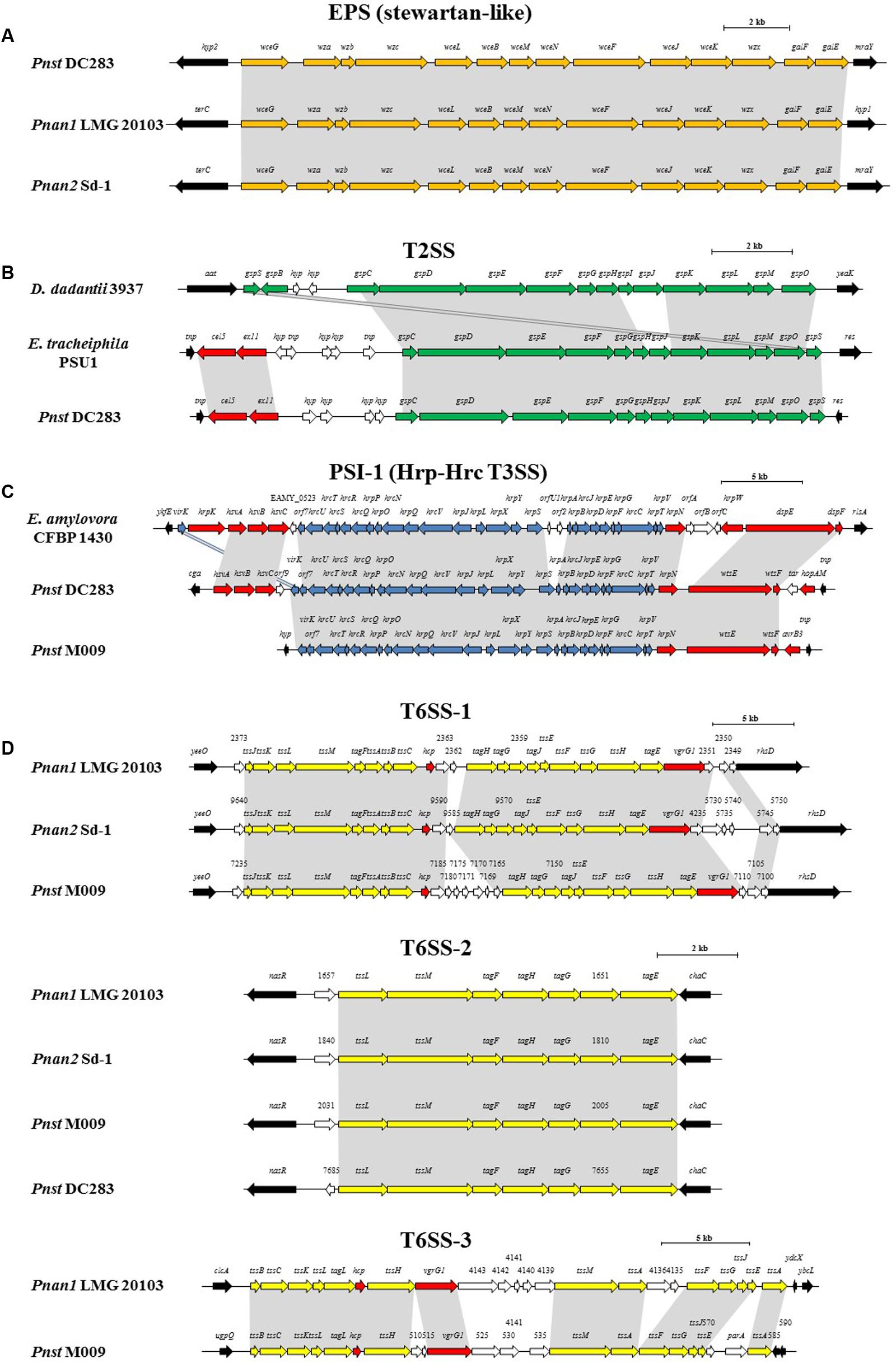
FIGURE 5. Alignments of the loci encoding for the predicted pathogenicity determinants from representative strains of each clade, Pnan1, Pnan2 and Pnst as well as representatives of other genera. (A) stewartan-like EPS (orange), (B) T2SS (green), (C) Hrp-Hrc T3SS (blue) and (D) T6SS-1, T6SS-2 and T6SS-3 loci (yellow). Red arrows indicate genes coding for predicted effectors of the T3SS and T6SSs, white arrows indicate genes coding for hypothetical proteins and black arrows denote the flanking genes. Size bars indicate the estimated sizes of the loci.
The presence of highly conserved orthologs of these Pnst subsp. stewartii pathogenicity determinants in all the Pnan and Pnst strains compared suggests they use common mechanisms to effect pathogenesis and symptom development. However, substantial differences can be observed in terms of the secretion systems encoded on their genomes, which have been shown to play an important role in the phytopathogenesis of both species. Analysis of the Pnst DC283 genome shows the presence of a locus coding for a Type II secretion system (T2SS; CKS4616-CKS4630) on the pSW1200 plasmid contig (AHIE01000039.1), unique among the strains compared (Figures 4, 5B). While the Pnst DC283 T2SS remains uncharacterized, in the related phytopathogen Dickeya dadantii it secretes a range of plant cell wall degrading enzymes, including endo-pectate lyases, esterases, a rhamnogalacturonate lyase and a cellulase (Hugouvieux-Cotte-Pattat et al., 2014; Pineau et al., 2014). The thirteen proteins encoded by the Pnst DC283 T2SS locus share highest orthology with the T2SS proteins of Erwinia tracheiphila PSU-1 (ETR_18611–18666/22561–22566; 85.51% average amino acid identity). Adjacent to the Pnst DC283 locus are two proteins which are likely to represent secreted effector proteins, including a predicted plant cell wall degrading cellulase (Cel5 – CKS4630; 63% amino acid identity to D. dadantii 3937 P07103.2; E-value: 1e-127) and an expansin-like protein (Exl1 – CKS4629; 59% amino acid identity to Pectobacterium carotovorum AHH91628.1; E-value: 5e-82) which is predicted to loosen the plant cell wall and allow the pathogen to gain access to the loosened cellulose (Chapon et al., 2001; Olarte-Lozano et al., 2014).
Type III secretion systems (T3SSs) are complex extracellular injection apparatuses that deliver effector proteins into plant and animal cells, where they play a role in colonization, modulation of host defenses and pathogenesis (Correa et al., 2012; Kirzinger et al., 2015). Three different T3SSs have been shown to be encoded in three distinct Pantoea secretion islands (PSI-1 to PSI-3) on the genome of Pnst DC283. The PSI-1 (Hrc-Hrp 1) T3SS (CKS_3241–3281), encoded on the LPP-1 plasmid contig (AHIE01000038.1), is similar to that found in other enterobacterial plant pathogens including Erwinia, Dickeya and Pectobacterium spp. and is central to Pnst DC283 pathogenesis on maize (Correa et al., 2012; Kirzinger et al., 2015). Analysis of the compared genomes showed that the PSI-1 T3SS is restricted to only four Pnst strains, Pnst M009, M073a, LMG 2632 and DC283 (Figure 4). While the PSI-1 T3SS loci are highly conserved (29 conserved CDSs; 99.54% average amino acid identity) and syntenous in terms of their structural and functional gene complements, differences could be observed in terms of the predicted effectors encoded within these loci (Figure 5C). In particular, among the PSI-1 encoding Pnst strains, only the Pnst DC283 Hrp T3SS incorporates hsvA, hsvB and hsvC genes at its 5′ end. Orthologs of the encoded proteins in E. amylovora (AAX39432-4.1; 72.25% average amino acid identity) are required for systemic virulence in apple and may be involved in the synthesis of a phytotoxin (Oh et al., 2005). The Pnst DC283 PSI-2 (CKS_4512–4544), encoded on the pSW1200 plasmid (AHIE0100039.1), is orthologous to a T3SS found in animal pathogens such as Salmonella enterica and Yersinia enterocolitica and plays a role in Pnst DC283 persistence in its corn flea beetle vector (Correa et al., 2012). Among the strains compared, PSI-2 T3SSs are restricted to Pnst DC283 and Pnan1 DAR76143 (BATH01000023.1; 25 proteins; 64.66% average amino acid identity) (Figure 4). The third T3SS, PSI-3, is likewise encoded on pSW1200 (AHIE01000039.1) and is evolutionarily related to a T3SS in Salmonella spp., but its function in Pnst DC283 remains unknown (Kirzinger et al., 2015). No orthologous PSI-3 T3SS loci are encoded on the genomes of the other twenty-eight Pnan and Pnst genomes (Figure 4).
Type VI secretion systems (T6SSs) are macromolecular injectisomes that are widely distributed among Gram-negative bacteria and have been implicated in a wide range of functions, including biofilm formation, plant and animal host cell invasion and virulence, as well as in bacterial competition (Coulthurst, 2013; Cianfanelli et al., 2016). Previously, we have shown that up to three distinct T6SSs (T6SS-1 to T6SS-3) are encoded on the genomes of different Pnan strains (Figures 4, 5D) (De Maayer et al., 2011, 2014). T6SS-1 is conserved among all Pnan1, Pnan2 and Pnst strains, with the exception of Pnst DC283 and has been implicated in both bacterial competition and virulence on onion seedlings in Pnan1 LMG 2665T (Shyntum et al., 2015). The T6SS-2 locus is conserved among all twenty-nine compared strains, including Pnst DC283 (Figure 4). This locus encodes only seven of the core proteins required for T6SS biosynthesis, and knock-out deletion does not have any effect on the virulence and antibiosis phenotypes (Shyntum et al., 2015). As such, it has been postulated that this locus encodes a non-functional T6SS and was derived through partial duplication of the T6SS-1 locus (De Maayer et al., 2011). In contrast to the other two loci, the T6SS-3 locus shows a more restricted distribution (Figure 4), being found on the LPP-1 plasmid of only four Pnan1 strains (AJ13355, LMG 20103, PA4 and R100). No orthologous loci could be observed in the genomes of any of the Pnan2 strains, but is present in two Pnst strains (M009 and M073a) where it is predicted to reside on the chromosome. Alignments of the T6SS loci shows that these loci are highly conserved and syntenous (Figure 5D). Non-conserved islands of genes are, however, present in the T6SS-1 and T6SS-3 loci adjacent to the hcp and vgrG genes, which code for predicted effector proteins of the T6SS (Durand et al., 2014). It is postulated that these non-conserved genes represent effector domains, which are tagged onto the Hcp and VgrG effector proteins and subsequently delivered into the environment or host cell (De Maayer et al., 2011; Durand et al., 2014). Orthology searches of the protein products of these non-conserved genes from eight P. ananatis strains showed that the T6SS-1 genes are restricted in distribution to plant-associated bacteria, while those from T6SS-3 are restricted to animal-associated bacteria, suggesting that these two T6SSs may be involved in plant and animal host interactions, respectively (De Maayer et al., 2014).
Discussion
Pnan strains, and some Pnst strains, are readily isolated from a wide range of environmental sources and cause disease on a broad range of plant hosts. By contrast, Pnst subsp. stewartii strains have a restricted lifecycle in that they rely on specific insect hosts for survival and transmission and have a narrow host range, causing disease only in susceptible maize varieties. The availability of a large number of genome sequences and a broad range of in silico tools has provided an enticing opportunity to determine the molecular differences underlying their distinct lifestyles.
Phylogenomic analyses indicated that there is extensive phylogenetic diversity among the Pnan strains, and that these strains form two distinct clades. Pnan Clade 1 incorporates the original type strain of the species, Pnan LMG 2665T and includes seven phytopathogenic strains (of maize, rice, pineapple, onion and Eucalyptus), three endophytic strains (from rice, maize and switchgrass), one epiphytic strain (on strawberry), one soil isolate, one plant-growth promoting strain and a clinical isolate. Pnan Clade 2 incorporates five strains, all of which were isolated as endophytes from rice seeds. Overall, less variability was observed in terms of the proteins encoded on the Pnan2 genomes than the Pnan1 strains, which may be correlated to the restricted source of isolation and clonality of most of the Pnan2 strains. As many of the genome sequences were deposited without information on its origin and ecology, additional analyses will need to be undertaken to identify cohesive and defining phenotypic differences between the Pnan1 and Pnan2 strains (Brenner et al., 2006). Far less phylogenetic diversity was observed among the Pnst strains, when considering the core genome phylogeny and phylogenomic metrics.
We propose that the different lifestyle of the Pnan and Pnst strains may largely be attributed to the distinct mobilomes observed among the compared strains. The Pnan1 strains harbor between one and five plasmids, the Pnan2 strains one or two plasmids, while the Pnst strains harbor between one and six distinct plasmids. These plasmids contribute heavily toward the total proteins encoded on the genomes (up to 15.40% in Pnst DC283) and form a substantial part of the accessory and variable fraction of the genomes. Plasmid-borne features with a more restricted distribution include the T2SS and the T3SSs, with predicted roles in pathogenicity and virulence. Pnst DC283 is also particularly prone to prophage integration, with 35 predicted prophages integrated on its chromosome, while between two and twelve phages are integrated into the genomes of the other Pnan1, Pnan2 and Pnst strains. Together, plasmids and prophages are predicted to play a major role in shaping the genomes of these distinct species and strains, contributing between 7.61 (Pnan1 R100) and 28.18% (Pnst DC283) of the total genomic DNA content and comprising largely of those proteins which form part of the accessory genome fractions of the compared strains.
Another prominent feature of the Pnst DC283 genome is the large number of insertion sequences (238 distinct IS elements) and transposons (240 distinct transposase proteins) integrated into the genome. While the direct phenotypic effects of transposon and insertion sequence integration in Pnst DC283 cannot be determined without further laboratory analyses, their integration may have contributed toward the non-deleterious “knock-out” of those phenotypes not required within the limited environmental context in which it survives (Shapiro et al., 2016), thereby restricting the metabolic and energy costs associated with these phenotypes and the genomic replication of the DNA coding for these phenotypes. Reductive genome evolution or pathoadaptation has been described in the xylem-limited plant pathogens Xylella fastidiosa and Xanthomonas albilineans and a number of obligate animal symbionts and pathogens, where initial accumulation of mobile genetic elements results in pseudogenization (gene inactivation) and ultimate gene loss (Pieretti et al., 2009; Lindeberg, 2012). Similar loss of epiphytic fitness factors and reduction in genome size, combined with acquisition of Hrp T3SS (PSI-1) and exopolysaccharide biosynthesis genes has been observed in the more closely related phytopathogens Erwinia amylovora and Erwinia tracheiphila (Kamber et al., 2012; Shapiro et al., 2016). The presence of large numbers of mobile elements in the genome of Pnst DC283 suggests that pathoadaptation into a host-specific pathogen with a restricted lifecycle is the result of recent evolutionary events or is still on-going (Shapiro et al., 2016). Genome sequencing and analyses of other Stewart’s wilt Pnst subsp. stewartii isolates may provide better insights into the distinct evolutionary history of this subspecies.
While the ten Pnst strains form a phylogenetically distinct cluster from the Pnan1 and Pnan2 strains, the nine Pnst strains (except Pnst DC283) share several genomic features with the Pnan strains that differentiate them from Pnst DC283. When comparing Pnan1+ Pnan2+ Pnst, the core genomes were consistently larger (on average 371 CDSs more) when the non-DC283 Pnst strains were considered. A broader profile of metabolic proteins was also encoded on their core and accessory genome fractions compared to Pnst DC283. This suggests that the Pnan1, Pnan2 and other Pnst strains are comparatively better adapted for survival in various environmental niches. Furthermore, as they will have to effectively outcompete other bacteria for available nutrients in these various environmental niches, they may make use of the type VI secretion systems (De Maayer et al., 2011) and the putative antimicrobial encoded on the ICEPan elements (De Maayer et al., 2015) integrated into the genomes of most of the Pnan and Pnst strains compared. A pertinent finding from the genomic analysis was that although they differ distinctly in terms of their host range and lifestyle, many predicted pathogenicity determinants are conserved among all compared Pnan and Pnst strains, regardless of whether they were isolated from diseased plant material, as part of the endophytic or epiphytic flora or from an environmental source. These putative pathogenicity determinants include an Rtx2 toxin which has been linked to the development of water-soaked lesions on the plant host (Roper et al., 2015), a stewartan-like exopolysaccharide (Wang et al., 2012), and the glucanolytic/xylanolytic endoglucanase EngY (Mohammadi et al., 2012). This shared pathogenicity factor profile, along with the extensive core genome observed among the twenty-nine compared strains emphasize the common ancestry of Pnan and Pnst, and further highlights the complex evolutionary histories and diversification of these important phytopathogens.
Availability of Data and Materials
All genome sequences incorporated in this study are publically available in the NCBI Genome database or the Joint Genome Institute – Integrated Microbial Genome Database (JGI-IMG). The accession numbers for the genome sequences are indicated in Table 1.
Author Contributions
PDM, BD, TS, SNV, and TC conceived the study. PDM, HA, SV, and JB performed data analyses. PDM wrote the original manuscript and all authors contributed to the final version. All authors have read and approved the final manuscript.
Funding
PDM was partially supported by the National Research Foundation of South Africa (NRF grant # 91447). BD and TS were supported by the Department of Life Sciences and Facility Management of ZHAW.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fmicb.2017.01755/full#supplementary-material
TABLE S1 | Digital DNA-DNA hybridization (dDDH) values and Average Nucleotide Identity (ANI) values between the twenty-nine compared Pnan1, Pnan2 and Pnst strains. ANI values are in the top half, while dDDH values are in the bottom half of the table. Values from 70 to 100% are colored in a sliding scale from light gray to black.
TABLE S2 | Mobile genetic elements among the twenty-nine compared Pnan and Pnst strains. The number of plasmids, phages, ICEs, IS elements and transposases are tabulated and their contribution to the total genomic DNA (%) and total proteins (%) encoded on the genome are described.
Footnotes
References
Adam, Z., Tambong, J. T., Lewis, C. T., Lévesque, C. A., Chen, W., Bromfield, E. S. P., et al. (2014). Draft genome sequence of Pantoea ananatis strain LMG 2665T, a bacterial pathogen of pineapple fruitlets. Genome Announc. 2:e00489-14. doi: 10.1128/genomeA.00489-14
Ailloud, F., Lowe, T., Cellier, G., Roche, D., Allen, C., and Prior, P. (2015). Comparative genomic analysis of Ralstonia solanacearum reveals candidate genes for host specificity. BMC Genomics 16:270. doi: 10.1186/s12864-015-1474-8
Auch, A. F., Klenk, H.-P., and Göker, M. (2010). Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2, 142–148. doi: 10.4056/sigs.541628
Aziz, R. K., Bartels, D., Best, A. A., DeJongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Baltrus, D. A., Nishimura, M. T., Dougherty, K. M., Biswas, S., Mukthar, M. S., Vicente, J., et al. (2012). The molecular basis of host specialization in bean pathovars of Pseudomonas syringae. Mol. Plant Microbe Interact. 25, 877–888. doi: 10.1094/MPMI-08-11-0218
Baltrus, D. A., Nishimura, M. T., Romanchuk, A., Chang, J. H., Mukthar, M. S., Cherkis, K., et al. (2001). Dynamic evolution of pathogenicity revealed by sequencing and comparative genomics of 19 Pseudomonas syringae isolates. PLOS Pathog. 7:e1002132. doi: 10.1371/journal.ppat.1002132
Blom, J., Albaum, S. P., Doppmeier, D., Pühler, A., Vorhölter, F.-J., Zakrweski, M., et al. (2009). EDGAR: a software framework for the comparative analysis of prokaryotic genomes. BMC Bioinformatics 10:154. doi: 10.1186/1471-2105-10-154
Blom, J., Kreis, J., Spänig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Bomfeti, C. A., Souza-Paccola, E. A., Massola, N. S. Jr., Marriel, I. E., Meirelles, W. F., Casela, C. R., et al. (2008). Localization of Pantoea ananatis inside lesions of maize white spot disease using transmission electron microscopy and molecular techniques. Trop. Plant Pathol. 33, 63–66. doi: 10.1590/S1982-56762008000100010
Bosi, E., Donati, B., Galardini, M., Brunetti, S., Sagot, M. F., Lió, P., et al. (2015). MeDuSa: a multi-draft based scaffolder. Bioinformatics 31, 2443–2451. doi: 10.1093/bioinformatics/btv171
Brenner, D. J., Staley, J. T., and Krieg, N. R. (2006). “Classification of procaryotic organisms and the concept of bacterial speciation,” in Bergey’s Manual® of Systematic Bacteriology, eds G. Garrity, D. J. Brenner, N. R. Krieg, and J. R. Staley (New York, NY: Springer-Verlag), 27–32.
Brüssow, H., Canchaya, C., and Hardt, W. D. (2004). Phages and the evolution of bacterial pathogens: from genomic rearrangements to lysogenic conversion. Microbiol. Mol. Biol. Rev. 68, 560–602. doi: 10.1128/MMBR.68.3.560-602.2004
Canchaya, C., Fournous, G., Chibani-Chennoufi, S., Dillmann, M. L., and Brüssow, H. (2003). Phages as agents of lateral gene transfer. Curr. Opin. Microbiol. 6, 417–442. doi: 10.1016/S1369-5274(03)00086-9
Chapon, V., Czjzek, M., El Hassouni, M., Py, B., Juy, M., and Barras, F. (2001). Type II protein secretion in gram-negative pathogenic bacteria: the study of the structure/secretion relationships of the cellulase Cel5 (formerly EGZ) from Erwinia chrysanthemi. J. Mol. Biol. 310, 1055–1066. doi: 10.1006/jmbi.2001.4787
Choi, O., Lim, J. Y., Seo, Y. S., Hwang, I., and Kim, J. (2012). Complete genome sequence of the rice pathogen Pantoea ananatis strain PA13. J. Bacteriol. 194, 531. doi: 10.1128/JB.06450-11
Cianfanelli, F. R., Monlezun, L., and Coulthurst, S. J. (2016). Aim, load, fire: the type VI secretion system, a bacterial nanoweapon. Trends Microbiol. 24, 51–62. doi: 10.1016/j.tim.2015.10.005
Coplin, D. L., Rowan, R. G., Chisholm, D. A., and Whitmoyer, R. E. (1981). Characterization of plasmids in Erwinia stewartii. Appl. Environ. Microbiol. 42, 599–604.
Correa, V. R., Majerczak, D. R., El-Ammar, D., Merighi, M., Pratt, R. C., Hougenhout, S. A., et al. (2012). The bacterium Pantoea stewartii uses two different type III secretion systems to colonize its plant host and insect vector. Appl. Environ. Microbiol. 78, 6327–6336. doi: 10.1128/AEM.00892-12
Coulthurst, S. J. (2013). The type VI secretion system - a widespread and versatile cell targeting system. Res. Microbiol. 164, 640–654. doi: 10.1016/j.resmic.2013.03.017
Coutinho, T. A., Preisig, O., Mergaert, J., Cnockaert, M. C., Riedel, K.-H., Swings, J., et al. (2002). Bacterial blight and dieback of Eucalyptus species, hybrids, and clones in South Africa. Plant Dis. 86, 20–25. doi: 10.1094/PDIS.2002.86.1.20
Coutinho, T. A., and Venter, S. N. (2009). Pantoea ananatis: an unconventional plant pathogen. Mol. Plant Pathol. 10, 325–335. doi: 10.1111/j.1364-3703.2009.00542.x
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLOS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
De Maayer, P., Chan, W. Y., Blom, J., Venter, S. N., Duffy, B., Smits, T. H. M., et al. (2012a). The large universal Pantoea plasmid LPP-1 plays a major role in biological and ecological diversification. BMC Genomics 13:625. doi: 10.1186/1471-2164-13-625
De Maayer, P., Chan, W. Y., Martin, D. A., Blom, J., Venter, S. N., Duffy, B., et al. (2015). Integrative conjugative elements of the ICEPan family play a potential role in Pantoea ananatis ecological diversification and antibiosis. Front. Microbiol. 6:576. doi: 10.3389/fmicb.2015.00576
De Maayer, P., Chan, W. Y., Rezzonico, F., Bühlmann, A., Venter, S. N., Blom, J., et al. (2012b). Complete genome sequence of clinical isolate Pantoea ananatis LMG 5342. J. Bacteriol. 194, 1615–1616. doi: 10.1128/JB.06715-11
De Maayer, P., Chan, W. Y., Rubagotti, E., Venter, S. N., Toth, I. K., Birch, P. R. J., et al. (2014). Analysis of the Pantoea ananatis pan-genome reveals factors underlying its ability to colonize and interact with plant, insect and vertebrate hosts. BMC Genomics 15:404. doi: 10.1186/1471-2164-15-404
De Maayer, P., Chan, W. Y., Venter, S. N., Toth, I. K., Birch, P. R. J., Joubert, F., et al. (2010). Genome sequence of Pantoea ananatis LMG20103, the causative agent of Eucalyptus blight and dieback. J. Bacteriol. 192, 2936–2937. doi: 10.1128/JB.00060-10
De Maayer, P., Venter, S. N., Kamber, T., Duffy, B., Coutinho, T. A., and Smits, T. H. M. (2011). Comparative genomics of the type VI secretion systems of Pantoea and Erwinia species reveals the presence of putative effector islands that may be translocated by the VgrG and Hcp proteins. BMC Genomics 12:576. doi: 10.1186/1471-2164-12-576
Durand, E., Cambillau, C., Cascales, E., and Journet, L. (2014). VgrG, Tae, Tle, and beyond: the versatile arsenal of type VI secretion effectors. Trends Microbiol. 22, 498–507. doi: 10.1016/j.tim.2014.06.004
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fu, J. F., Chang, H. C., Chen, Y. M., Chang, Y. S., and Liu, S. T. (1995). Sequence analysis of an Erwinia stewartii plasmid, pSW100. Plasmid 34, 75–84. doi: 10.1006/plas.1995.9999
Fu, J. F., Hu, J. M., Chang, Y. S., and Liu, S. T. (1998). Isolation and characterization of plasmid pSW200 from Erwinia stewartii. Plasmid 40, 100–112. doi: 10.1006/plas.1998.1350
Fu, J. F., Ying, S. W., and Liu, S. T. (1997). Cloning and characterization of the ori region of pSW1200 of Erwinia stewartii: similarity with plasmid P1. Plasmid 38, 141–147. doi: 10.1006/plas.1997.1308
Gavini, F., Mergaert, J., Beji, A., Mielcarek, C., Izard, D., Kersters, K., et al. (1989). Transfer of Enterobacter agglomerans (Beijerinck 1888) Ewing & Fife 1972 to Pantoea gen. nov. as Pantoea agglomerans comb. nov. and description of Pantoea dispersa sp. nov. Int. J. Syst. Bacteriol. 39, 337–345. doi: 10.1099/00207713-39-3-337
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Hall, T. A. (1999). BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98.
Hara, Y., Kadotani, N., Izui, H., Katashkina, J. I., Kuvaeva, T. M., Andreeva, I. G., et al. (2012). The complete genome sequence of Pantoea ananatis AJ13355, an organism of great biotechnological potential. Appl. Microbiol. Biotechnol. 93, 331–341. doi: 10.1007/s00253-011-3713-5
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Hugouvieux-Cotte-Pattat, N., Condemine, G., and Shevchik, V. E. (2014). Bacterial pectate lyases, structural and functional diversity. Environ. Microbiol. Rep. 6, 427–440. doi: 10.1111/1758-2229.12166
Izzati Mohamad, N., Tan, W. S., Keng Tee, K., Yin, F., and Chan, K. G. (2015). Analysis of quorum-sensing Pantoea stewartii strain M073A through whole-genome sequencing. Genome Announc. 3:e00022-15. doi: 10.1128/genomeA.00022-15
Jacques, M. A., Arlat, M., Boulanger, A., Boureau, T., Carrère, S., Chen, N. W., et al. (2016). Using ecology, physiology, and genomics to understand host specificity in Xanthomonas. Annu. Rev. Phytopathol. 54, 163–187. doi: 10.1146/annurev-phyto-080615-100147
Kamber, T., Smits, T. H. M., Rezzonico, F., and Duffy, B. (2012). Genomics and current genetic understanding of Erwinia amylovora and the fire blight antagonist Pantoea vagans. Trees 26, 227–238. doi: 10.1007/s00468-011-0619-x
Kim, H. J., Lee, J. H., Kang, B. R., Rong, X., McSpadden Gardener, B. B., Ji, H. J., et al. (2012). Draft genome sequence of Pantoea ananatis B1-9, a nonpathogenic plant growth-promoting bacterium. J. Bacteriol. 194, 729. doi: 10.1128/JB.06484-11
Kirzinger, M. W. B., Butz, C. J., and Stavranides, J. (2015). Inheritance of Pantoea type III secretion systems through both vertical and horizontal transfer. Mol. Genet. Genomics 290, 2075–2088. doi: 10.1007/s00438-015-1062-2
Kumar, N., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lindeberg, M. (2012). Genome-enabled perspectives on the composition, evolution and expression of virulence determinants in bacterial plant pathogens. Annu. Rev. Phytopathol. 50, 111–132. doi: 10.1146/annurev-phyto-081211-173022
Marchler-Bauer, A., and Bryant, S. H. (2004). CD-Search: protein domain annotations on the fly. Nucleic Acids Res. 32, W327–W331. doi: 10.1093/nar/gkh454
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Meier-Kolthoff, J. P., Hahnke, R. L., Petersen, J., Scheuner, C., Michael, V., Fiebig, A., et al. (2014). Complete genome sequence of DSM 30083T, the type strain (U5/41T) of Escherichia coli, and a proposal for delineating subspecies in microbial taxonomy. Stand. Genomic Sci. 9:2. doi: 10.1186/1944-3277-9-2
Mergaert, J., Verdonck, L., and Kersters, K. (1993). Transfer of Erwinia ananas (synonym, Erwinia uredovora) and Erwinia stewartii to the Genus Pantoea emend. as Pantoea ananas (Serrano 1928) comb. nov. and Pantoea stewartii (Smith 1898) comb. nov., respectively, and description of Pantoea stewartii subsp. indologenes subsp. nov. Int. J. Syst. Bacteriol. 43, 162–173. doi: 10.1099/00207713-43-1-162
Midha, S., Bansal, K., Sharma, S., Kumar, N., Patil, P. P., Chaudry, V., et al. (2016). Genomic resource of rice seed associated bacteria. Front. Microbiol. 6:1551. doi: 10.3389/fmicb.2015.01551
Mohammadi, M., Burbank, L., and Roper, M. C. (2012). Pantoea stewartii subsp. stewartii produces an endoglucanase that is required for full virulence in sweet corn. Mol. Plant Microbe Interact. 25, 463–470. doi: 10.1094/MPMI-09-11-0226
Mondal, K. K., Mani, C., Singh, J., Kim, J.-G., and Mudgett, M. B. (2011). A new leaf blight of rice caused by Pantoea ananatis in India. Plant Dis. 95, 1582–1583. doi: 10.1094/PDIS-06-11-0533
Oh, C. S., Kim, J. F., and Beer, S. V. (2005). The Hrp pathogenicity island of Erwinia amylovora and identification of three novel genes required for systemic infection. Mol. Plant Pathol. 6, 125–138. doi: 10.1111/j.1364-3703.2005.00269.x
Olarte-Lozano, M., Mendoza-Nuñez, M. A., Pastor, N., Segovia, L., Folch-Mallol, J., and Martínez-Anaya, C. (2014). PcExl1 a novel acid expansin-like protein from the plant pathogen Pectobacterium carotovorum, binds cell walls differently to BsEXLX1. PLOS ONE 9:e95638. doi: 10.1371/journal.pone.0095638
Pieretti, I., Royer, M., Barbe, V., Carrere, S., Koebnik, R., Cociancich, S., et al. (2009). The complete genome sequence of Xanthomonas albilineans provides new insights into the reductive genome evolution of the xylem-limited Xanthomonadaceae. BMC Genomics 10:616. doi: 10.1186/1471-2164-10-616
Pineau, C., Guschinskaya, N., Robert, X., Gouet, P., Ballut, L., and Shevchik, V. E. (2014). Substrate recognition by the bacterial type II secretion system: more than a simple interaction. Mol. Microbiol. 94, 126–140. doi: 10.1111/mmi.12744
Rodriguez-R, L. M., and Konstantinidis, K. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ 4, e1900v1.
Roper, M. C. (2011). Pantoea stewartii subp. stewartii: lessons learned from a xylem-dwelling pathogen of sweet corn. Mol. Plant. Pathol. 12, 628–637. doi: 10.1111/j.1364-3703.2010.00698.x
Roper, M. C., Burbank, L. P., Williams, K., Viravathana, P., Tien, H. Y., and von Bodman, S. (2015). A large repetitive RTX-like protein mediates water-soaked lesion development, leakage of plant cell content and host colonization in the Pantoea stewartii subsp. stewartii pathosystem. Mol. Plant Microbe Interact. 28, 1374–1382. doi: 10.1094/MPMI-05-15-0109-R
Shapiro, L. R., Scully, E. D., Straub, T. J., Park, J., Stephenson, A. G., Beattie, G. A., et al. (2016). Horizontal gene acquisitions, mobile element proliferation, and genome decay in the host-restricted plant pathogen Erwinia tracheiphila. Genome Biol. Evol. 8, 649–664. doi: 10.1093/gbe/evw016
Sheibani-Tezerji, R., Naveed, M., Jehl, M. A., Sessitsch, A., Rattei, T., and Mitter, B. (2015). The genomes of closely related Pantoea ananatis maize seed endophytes having different effects on the host plant differ in secretion system genes and mobile genetic elements. Front. Microbiol. 6:440. doi: 10.3389/fmicb.2015.00440
Shi, X., Liu, Q., Ma, J., Liao, H., Xiong, X., Zhang, K., et al. (2015). An acid-stable bacterial laccase identified from the endophyte Pantoea ananatis Sd-1 genome exhibiting lignin degradation and dye decolorization abilities. Biotechnol. Lett. 37, 2279–2288. doi: 10.1007/s10529-015-1914-1
Shyntum, D. Y., Theron, J., Venter, S. N., Moleleki, L. N., Toth, I. K., and Coutinho, T. A. (2015). Pantoea ananatis utilizes a type VI secretion system for pathogenesis and bacterial competition. Mol. Plant Microbe Interact. 28, 420–431. doi: 10.1094/MPMI-07-14-0219-R
Smith, D. D., Kirzinger, M. W., and Stavranides, J. (2013). Draft genome sequence of the antibiotic-producing epiphytic isolate Pantoea ananatis BRT175. Genome Announc. 1:e00902-13. doi: 10.1128/genomeA.00902-13
Smits, T. H. M., Rezzonico, F., Kamber, T., Blom, J., Goesmann, A., Ishimaru, C. A., et al. (2011). Metabolic versatility and antibacterial metabolite biosynthesis are distinguishing genomic features of the fire blight antagonist Pantoea vagans C9-1. PLOS ONE 6:e22247. doi: 10.1371/journal.pone.0022247
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Tan, W. S., Chang, C. Y., Yin, W. F., and Chan, K. G. (2015). Understanding the quorum-sensing bacterium Pantoea stewartii strain M009 with whole-genome sequence analysis. Genome Announc. 3:e01509-14. doi: 10.1128/genomeA.01509-14
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The COG database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Varani, A. M., Siquier, P., Gourbeyre, E., Charneau, V., and Chandler, M. (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12:R30. doi: 10.1186/gb-2011-12-3-r30
Walterson, A. M., and Stavranides, J. (2015). Pantoea: insights into a highly versatile and diverse genus within the Enterobacteriaceae. FEMS Microbiol. Rev. 39, 968–984. doi: 10.1093/femsre/fuv027
Wang, X., Yang, F., and von Bodman, S. B. (2012). The genetic and structural basis of two distinct terminal side branch residues in stewartan and amylovoran exopolysaccharides and their potential role in host adaptation. Mol. Microbiol. 83, 195–207. doi: 10.1111/j.1365-2958.2011.07926.x
Weller-Stuart, T., Chan, W. Y., Coutinho, T. A., Venter, S. N., Smits, T. H. M., Duffy, B., et al. (2014). The draft genome sequences of the onion centre rot pathogen Pantoea ananatis PA4 and maize brown stalk rot pathogen P. ananatis BD442. Genome Announc. 2:e00750-14. doi: 10.1128/genomeA.00750-14
Weller-Stuart, T., De Maayer, P., and Coutinho, T. A. (2016). Pantoea ananatis: genomic insights into a versatile pathogen. Mol. Plant Pathol. doi: 10.1111/mpp.12517 [Epub ahead of print].
Wu, C. Y., Fu, J. F., and Liu, S. T. (2001). The replicon of pSW800 from Pantoea stewartii. Microbiology 147, 2757–2767. doi: 10.1099/00221287-147-10-2757
Wu, L., Liu, R., Niu, Y., Lin, H., Ye, W., Guo, L., et al. (2016). Whole genome sequence of Pantoea ananatis R100, an antagonistic bacterium isolated from rice seed. J. Biotechnol. 225, 1–2. doi: 10.1016/j.jbiotec.2016.03.007
Keywords: Stewart’s wilt, mobilome, pan-genome, phylogenomics, pathogenicity factors, host range, pathoadaptation
Citation: De Maayer P, Aliyu H, Vikram S, Blom J, Duffy B, Cowan DA, Smits THM, Venter SN and Coutinho TA (2017) Phylogenomic, Pan-genomic, Pathogenomic and Evolutionary Genomic Insights into the Agronomically Relevant Enterobacteria Pantoea ananatis and Pantoea stewartii. Front. Microbiol. 8:1755. doi: 10.3389/fmicb.2017.01755
Received: 12 June 2017; Accepted: 29 August 2017;
Published: 14 September 2017.
Edited by:
Sergey M. Stolyar, University of Idaho, United StatesReviewed by:
Luis Carlos Guimarães, Federal University of Pará, BrazilNubia Seyffert, Federal University of Pará, Brazil
Copyright © 2017 De Maayer, Aliyu, Vikram, Blom, Duffy, Cowan, Smits, Venter and Coutinho. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pieter De Maayer, cGlldGVyLmRlbWFheWVyQHdpdHMuYWMuemE=