 Abrar Muhammad
Abrar Muhammad Ya Fang1,2
Ya Fang1,2 Youming Hou
Youming Hou Zhanghong Shi
Zhanghong Shi- 1State Key Laboratory of Ecological Pest Control for Fujian and Taiwan Crops, Fujian Agriculture and Forestry University, Fujian, China
- 2Fujian Provincial Key Laboratory of Insect Ecology, College of Plant Protection, Fujian Agriculture and Forestry University, Fujian, China
For invasive insects, the potential roles of gut microbiota in exploiting new food resources and spreading remain elusive. Red palm weevil (RPW), Rhynchophorus ferrugineus Olivier, is an invasive destructive pest which feeds on nutrient-poor tender tissues and has caused extensive mortality of palm trees. The microbes associated with insects can improve their nutrition assimilation. However, experimental evidence on the interactions between RPW and its gut microbiota is still absent. The aim of this study is to determine the dynamics changes and the bacterial entomotype in the RPW gut and its potential physiological roles. Here, we confirmed RPW harbors a complex gut microbiota mainly constituted by bacteria in the families Enterobacteriaceae, Lactobacillaceae, Entomoplasmataceae, and Streptococcaceae. RPW gut microbiota exhibited a highly stable microbial community with low variance in abundance across different life stages and host plants. Furthermore, the abundance of Enterobacteriaceae was markedly increased but that of Acetobacteraceae was reduced significantly after administration of antibiotics. Although no significant effects were found on the body weight gain of RPW larvae, these alterations dramatically decreased the concentration of hemolymph protein and glucose while that of hemolymph triglyceride increased. In the gut of wild-caught RPW larvae, seven bacterial species in the genera Klebsiella, Serratia, Enterobacter, and Citrobacter were shown to have an ability to degrade cellulose. Together, RPW accommodate a stable gut microbiota which can degrade plant polysaccharides and confer their host optimal adaptation to its environment by modulating its metabolism.
Introduction
Rhynchophorus ferrugineus, also known as the red palm weevil (RPW), is widely considered as the most devastating insect pest of palms in the world, even in the countries where it has been accidently introduced (Ju et al., 2011; Hou et al., 2011; Wan et al., 2015). This weevil is native to southern Asia and Melanesia, but due to international exchange of infected plant materials, it has spread to the Middle East, Africa and the Mediterranean Basin. More recently, RPW has become established in China, Japan, Australia and the Caribbean (Zhang et al., 2003; Wang et al., 2015). In China, almost 20,000 coconut trees have been killed, with the area of damage by RPW covering over 10,000 km2 since its invasion, and it has seriously threatened the green ecological security of China’s coastal areas (Li et al., 2009; Ju et al., 2011; Pu and Hou, 2016; Ge et al., 2016).
The development of RPW from egg to newborn adult occurs in the palm tree trunk and the weevil larvae feed on tender tissues and sap within the apical growing point of palms (Butera et al., 2012; Shi et al., 2014). The resulting damage is often only visible long after infestation when palms are close to death (Faleiro, 2006). Both palm tissues and sap are rich in carbohydrates such as cellulose, hemicelluloses, lignin, sucrose and glucose, but poor in assimilable nitrogen sources (Nagnan et al., 1992; Behar et al., 2005; Morales-Jiménez et al., 2009). For this reason, they represent a non-easily digestible and nutrient-poor substrate for most eukaryotes.
As the most diverse biological group, insects have been found in almost every possible nutritional niche on Earth, and with a remarkable adaptation to a wide range of diets including dead wood, feathers or blood, seem incapable of sustaining basic nutritional demands. More and more evidence indicates that this capacity is not from their own unique metabolic flexibility but is conferred by their microbiota (Douglas, 2009, 2015; Storelli et al., 2011; Engel and Moran, 2013; Ben-Yosef et al., 2014; Sugio et al., 2016; Xu et al., 2016). For bark- and wood-boring species, symbioses with cellulose-degrading microorganisms appear to compensate for the inability to synthesize cellulases in many insects (Martin, 1991; Douglas, 2009, 2015; Adams et al., 2011), and these obligate or facultative associations are important factors driving the evolution of wood-feeding insects (Veivers et al., 1983). For instance, the prominent invasive pests, such as the Dendroctonus bark beetles, wood-boring emerald ash borer and tree-killing wasp, Sirex noctilio, are inhabited by gut microorganisms that contribute to host nutrition by degradation of cellulose (Vasanthakumar et al., 2008; Butera et al., 2012) and hemicelluloses (Adams et al., 2011; Butera et al., 2012) and nitrogen fixation (Morales-Jiménez et al., 2013).
Invasive insects, such as Brontispa longissima and Dendroctonus valens, have caused huge losses to economic and ecosystem services and human health (Lu et al., 2016; Wan and Yang, 2016). Unfortunately, it is less widely appreciated that these insect species often derive increased opportunities for establishment and cause additional environmental and economic loss because of their associations with symbiotic microorganisms. There are three potential threats arising from symbiotic microorganisms of invasive insects. First, symbionts may contribute to the damage caused by their insect host. The prominent example is whitefly Bemisia tabaci and its vectored begomoviruses such as tobacco curly shoot virus and tomato yellow leaf curl China virus which extensively damaged tomato and tobacco crops in southern China (Jiu et al., 2007). Second, symbionts may be released from their host and thus begin a subsequent invasion (Mota et al., 2006; Zhao et al., 2013, 2014). Finally, and probably least understood, symbionts play important roles in the survival and reproduction of their host insects during establishment in new regions (Zhou et al., 2015; Lu et al., 2016). Therefore, elucidation of the complex interactions between invasive insects and their associated microbes not only provide important information towards revealing the mechanisms underpinning their invasion success, but also drive the development of novel and more effective management approaches.
Considering the huge economic and environmental damages caused by RPW, the interest in this pest has markedly increased in recent decades. Most studies paid attention to the efficacy of different chemical and bio-control strategies (Llácer et al., 2012a,b; Mazza et al., 2014; Peng et al., 2016; Pu et al., 2017). Recently, some preliminary investigations have revealed the gut bacterial community associated with RPW larvae (Khiyami and Alyamani, 2008; Butera et al., 2012; Valzano et al., 2012; Jia et al., 2013; Tagliavia et al., 2014) and adults (Montagna et al., 2015), with some of these studies being done in the context of identifying potential insect pathogens for use in biological control (Butera et al., 2012; Valzano et al., 2012). Bacterial species with the ability to degrade polysaccharides and sucrose by hydrolase activity (e.g., Klebsiella pneumonia and Lactococcus lactis) have been identified in the gut of RPW larvae (Khiyami and Alyamani, 2008; Jia et al., 2013). Current reports on the gut microbiota of RPW have only focused on its larval and adult stage. For holometabolous insects, factors such as environmental conditions, life stages and diets have strong effects on shaping the structure and function of gut microbiota (Dethlefsen et al., 2007; Lehman et al., 2009; Sharon et al., 2010; Wang et al., 2011; Sudakaran et al., 2012; Montagna et al., 2015). Therefore, a comprehensive characterization of gut microbial dynamics across developmental stages of RPW to uncover the existence of entomotype which refers to the bacterial taxa consistently associated with the host (Montagna et al., 2016), is the prerequisite for understanding how a microbial population functions in the gut ecosystem. Unfortunately, the relationships between microbial dynamics and RPW health remain poorly understood. Given that symbionts are involved in driving the diversification and success of insects, we posit that bacteria harbored in the gut of RPW are playing pivotal roles in the host nutrition metabolism during invasion. In this study, comparisons on gut microbiota of RPW across different life stages and larvae from different host plants were performed to determine the dynamics changes and the entomotype in the RPW gut. Furthermore, the potential effects of gut microbiota on the nutrition metabolism of RPW are detected by the administration of antibiotics to disturb the inherent homeostasis of RPW gut microbiota.
Materials and Methods
Insect Sampling and Rearing
To detect the effect of developmental stages on gut microbiota, RPW larvae, pupae and adults were collected from Cocos nucifera L. in Wenchang City, Hainan Province in May 2014. Larvae from Phoenix canariensis were collected in Longyan City, Fujian Province in August 2014. The live RPW individuals were saved in plastic boxes with some soft palm tissues and then transported to Fujian Province Key Laboratory of Insect Ecology, Fujian Agriculture and Forestry University (FAFU). A laboratory population of RPW was established by adults that were trapped in Jinshan campus of FAFU (Supplementary Table S1). In our laboratory, RPW larvae were reared on sugarcane stem in the climatic chamber at 27 ± 1°C, 75% relative humidity (RH) and a photoperiod of 24 h dark, while adults were maintained under the following conditions: 27 ± 1°C, 75% RH, 12 h light/12 h dark. RPW larvae from C. nucifera, P. canariensis and our laboratory were employed to determine the influence of host plant species on its gut microbiota. All the wild-caught specimens were processed immediately in the laboratory for gut dissection and the extraction of total gut bacteria DNA upon arrival.
Gut Dissection and DNA Isolation
Before dissection, all specimens (Seventh instar larvae, pupae, and adults) were surface-sterilized in 75% ethanol for 90 s, followed by three rinses in sterilized distilled water. From RPW adults, elytra and membranous wings were removed before sterilization. Under a stereomicroscope, gut dissections were conducted in a clean Petri dish (90 mm in diameter) with sterile phosphate-buffered saline (PBS) using sterilized scissors and forceps. Three guts were pooled in 1 ml sterile PBS as a replication and each treatment had three replications.
For pyrosequencing, DNA was extracted using a DNeasy Blood & Tissue Kit (Qiagen) following a protocol modified from the manufacturer’s guidelines for Gram-positive bacteria. Briefly, samples were hand-homogenized in 20 mM Tris-HCl (pH 8.0), 1.2% Triton X-100, 2 mM sodium EDTA containing 20 mg/ml lysozyme. Gut homogenates were centrifuged for 10 min at 5000 g under room temperature and the bacterial pellets were re-suspended in 180 μl enzymatic lysis buffer. The final elution step was repeated twice with 100 μl of AE buffer for greater yield. The quality of DNA samples was checked by electrophoresis on 1% agarose gel, and quantified using Nanodrop 1000 (Thermo Scientific).
Illumina High-Throughput Sequencing of 16S rRNA Gene Sequences
The variable region 4 (V4) of the bacterial 16S rRNA gene was amplified with the general 16S rRNA primers 515F (5′-GTGYCAGCMGCCGCGGTA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′). The PCR reactions were carried out in a total volume of 30 μl which comprised 15 μl Phusion® High-Fidelity PCR Master Mix (New England Biolabs), 0.2 μM forward and reverse primers and 10 ng template DNA. The thermal conditions set for PCR were as follows: initial denaturation for 1 min at 98°C followed by 30 cycles of denaturation for 10 s at 98°C, annealing for 30 s at 50°C and elongation for 60 s at 72°C, then a final extension for 5 min at 72°C. The amplified PCR products were mixed with the same volume of 1× buffer containing SYBR green and run on a 2% agarose gel. Next, DNA from an aliquot of each PCR reaction was purified using a Qiagen Gel Extraction Kit and equal volumes of the three reaction products per sample were mixed together and the sequencing libraries were generated with TrueSeq® DNA PCR-Free Sample Preparation Kit (Illumina). The libraries were sequenced using MiSeq platform after quality assessment on the Qubit@ 2.0 fluorometer (Thermo Scientific) and Agilent Bio-analyzer 2100 system.
The paired-end sequencing reads were merged through FLASH1 (V1.2.7) (Magoč and Salzberg, 2011). The raw tags were strictly filtered (<30 Phred score) to obtain high-quality clean tags using Quantitative Insight into Microbial Ecology (QIIME) software2 (V1.7.0) (Caporaso et al., 2010; Bokulich et al., 2013) and were aligned with Gold database by running the UCHIME algorithm to remove the chimera for effective tags (Edgar et al., 2011).
Operational taxonomic units (OTUs) were clustered using Uprase3 (V7.0.1001) to group the effective tags into OTUs with ≥97% sequence identity (ID) (Edgar, 2013). The most abundant unique sequence of each OTU cluster was selected as representative, and taxonomy of the non-chimaeric sequences was assigned by RDP Classifier4 (version 2.2) (Wang et al., 2007) using GreenGene database5 with default settings (DeSantis et al., 2006). To investigate the species richness and community diversity of samples or groups, six diversity indices, Chao 1, ACE, Shannon, and Simpson’s were calculated with the software package QIIME, version 1.7.0. Beta diversity analysis of RPW gut bacterial communities was carried out through PCoA (Principal Coordinate Analysis) and PCA (Principal Component Analysis) using QIIME (Version 1.7.0). ANOSIM (Analysis of similarity) was used to estimate the differences between gut bacterial communities associated with RPW at different life stages and larvae from different host plants.
Bacterial Isolation and Identification
The larvae of R. ferrugineus were collected from the infested coconut palms C. nucifer in Hainan, China in mid-May 2014. The specimens were transported in plastic boxes along with host plant tissue to our laboratory. Upon arrival, larvae guts were dissected as above and homogenized by hand. The gut homogenates were poured on nutrient agar (NA) media in triplicate after serial dilution (10-1 to 10-6), and were incubated aerobically at 30°C for 24 h. Based on their color, size, and morphology, 200 single colonies were picked and repeatedly streaked on NA media to obtain pure cultures (Indiragandhi et al., 2007). The purified single colonies were inoculated in nutrient broth (NB) and incubated at 37°C for 24 h. Thereafter, 500 μl bacterial solutions of each colony were processed for DNA extraction with the TIANamp Stool DNA Kit DP 328 (TIANGEN) according to the manufacturer’s instructions. The bacterial species were identified as described by Butera et al. (2012) with the universal bacterial primers 27F (5′-AGAGTTTGATCATGGCTCAG-3′) and 1492 R (5′-TACGGYTACCTTGTTACGACTT-3′). The taxonomy of each 16S rRNA gene sequence was assigned with NCBI BLAST6 and Sequence Match in the Ribosomal DNA Project7 (RDP). Our representative bacterial sequences and those retrieved from the GeneBank database were aligned with ClustalW using MEGA 5.05 software.
To determine the ability of bacterial isolates to degrade cellulose, each of them was streaked on CMC (carboxymethyl-cellulose, Sigma) agar media and the plates were aerobically incubated at 37°C for 14 days. After growth, the incubated plates were covered with Congo red dye solution (Huang et al., 2012). After dye removal, cellulose degradation was indicated by the clear zone around the colonies. The cellulolytic index was calculated based on the ratio of the diameter of the clear zone to the diameter of the bacterial colony.
Effect of Gut Microbiota on the Nutrition Metabolism of RPW
To evaluate the role of gut microbiota on host nutrition metabolism, an antibiotic cocktail containing kanamycin, tetracycline, gentamicin, and erythromycin (Chu et al., 2013) with the final concentration of 600 mg/L was used to disrupt the inherent structure of RPW gut microbiota. The fourth-instar larvae from the laboratory population were randomly allocated to the following two groups: control group (CK) and antibiotic group (AT). Before treatment, the initial body weight of each individual was measured with an electric microbalance (METTLER TOLEDO AL104). The treatments were conducted for a duration of 28 days. At the end of treatment, some larvae were dissected to extract the total DNA of gut microbiota for pyrosequencing to determine the antibiotic feeding-mediated changes in the community structure of the gut microbiota. The pyrosequencing and data analysis were completed as above.
The remaining larvae were used to extract the hemolymph for the analysis of metabolic indices, containing glucose, protein, and triglyceride (TAG) concentration. Before hemolymph collection, larvae were washed under running water to remove excrements and food particles, and were then anesthetized for 3–5 min on ice to immobilize them. The larvae epidermis was pierced by a fine sharp needle and 5 μl hemolymph of each larva was collected into labeled 1.5 ml clean microcentrifuge tubes, immersed into an ice box, and containing 2 μl of 0.2% phenylthiourea (PTU) to inhibit the melanization of hemolymph. Hemolymph collections were carried out at four time points of 7, 14, 21, and 28 days after feeding. Glucose, protein, and TAG concentrations were measured with a Glucose Measurement Kit (Shanghai Rongsheng Biological Pharmaceutical Co.), a BCA Protein Assay Kit (Tiangen PA115, Tiangen Biotech) and a Triglyceride Assay Kit (Zhejiang Dongou Diagnostic Products), respectively. These assays were done with a spectrometric reader (Molecular Device®) under the manufacturer’s protocol. Ten larvae were treated in each replication and each treatment comprised three replications.
Statistical Analysis
Differences in the abundance of gut bacteria across developmental stages and host species at family and genus level, the cellulolytic ability of isolated bacterial species and that of nutrition indices were assessed by analysis of variance (ANOVA). The differences in abundance of gut bacteria and body weight between CK and AT groups were statistically detected by independent t-test. All analyses were performed using IBM SPSS Statistics (22.0). The significance level to threshold was set at 0.05 (P < 0.05).
Results
Ontogenetic Changes of RPW Gut Microbiota
A total of 733,855 raw reads from 10 RPW samples at different developmental stages (Seventh instar larvae, pupae, and adults) were characterized with the Illumina high-throughput sequencing. After quality trimming, 658,240 high-quality clean tags were obtained, which were binned into 5737 OTUs at the 3% distance level. RPW pupae had the highest species richness (Chao 1, ACE, and observed species) and the most diverse bacterial community being indicated by both Shannon and Simpson’s index, followed by larvae and adults, respectively (Table 1). The phyla Proteobacteria and Firmicutes accounted for the vast majority of reads (>93%) from all life stages. The third most abundant phylum in larvae and adults was Tenericutes at 5.0 and 1.8%, respectively. However, in pupae, Actinobacteria was the third dominant phylum at 1.11%. More than 90% of the reads were classified at the family level and 60 families were detected in the RPW gut at the abundance ≥0.01%, with Enterobacteriaceae representing 64.7% of the assemblage, followed by Lactobacillaceae (4.2%) and Streptococcaceae (1.97%). Twelve bacterial genera with the abundance ≥1% were detected and Erwinia (4.3%) is the most represented genus in the RPW gut, followed by Lactococcus (1.94%), Entomoplasma (1.66%), and Erysipelothrix (1.37%). Another 72 genera are represented at a value between 0.01 and 1%.
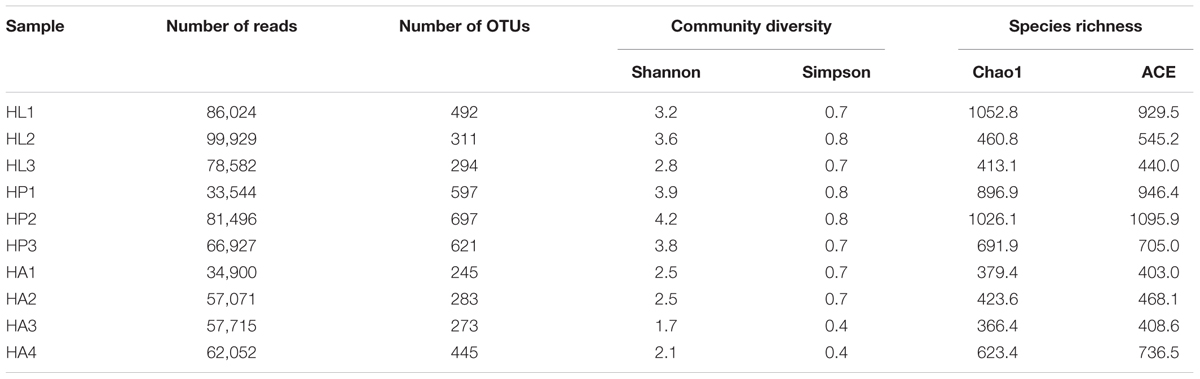
TABLE 1. Richness and diversity estimation of the gut bacterial communities associated with Rhynchophorus ferrugineus during its different life stages from the pyrosequencing analysis.
In PCA analysis, the first two components PC1 and PC2 accounted for 51.62% (PC1 = 28.4%, PC2 = 23.22%) of the variation among the gut microbiota of RPW at different life stages (Supplementary Figure S1A). While in PCoA analysis, the PC1 and PC2 coordinates explained a total variation of 41.73% (PC1 = 27.26%, PC2 = 14.47%) (Supplementary Figure S1B). The ANOSIM analysis confirmed that there were no significant differences in the bacterial communities associated with RPW at different life stages (Adult-Larvae p = 0.4, Pupae–Larvae p = 0.2, Pupae–Adult p = 0.35). The family Enterobacteriaceae prominently dominated, representing ∼60% of the assemblage, in the larvae, pupae, and adult stages. With the transition from larvae to pupae, the percentage of Lactobacillacea, Entomoplasmataceae, and Streptococcaceae decreased to 1.76, 0.40, and 0.99%, respectively. However, Enterocococcaceae, Comamonadaceae, Erysipelotrichaceae, and Moraxellaceae became more rich to 4.44, 3.85, 1.85, and 1.96%, respectively (Figure 1A). In pupal stage, the relative abundance of genera Erwinia, Acinetobacter, Pseudomonas, and Limnohabitans increased to 10.60, 1.30, 1.56, and 2.21%, respectively, and Erwinia were strongly represented, but the percentage of Entomoplasma, Lactococcus and Lactobacillus dropped to 0.40, 0.96, 0.27%, respectively. During the transition from pupae to adult, only Lactococcus in the family Streptococcaceae became more abundant (Figure 1B). These results support that the gut bacterial compositions of RPW differed in abundance across the developmental stages. Classifier analysis revealed that the 375 OTUs, common to all three developmental stages, were comprised of 56 genera in the phyla Proteobacteria, Firmicutes, Tenericutes and Actinobacteria, with Erwinia, Entomoplasma, Lactococcus, Lactobacillus, Pseudomonas, Limnohabitans, and Acinetobacter appearing the most frequent genera, constituting the “entomotype” of RPW.
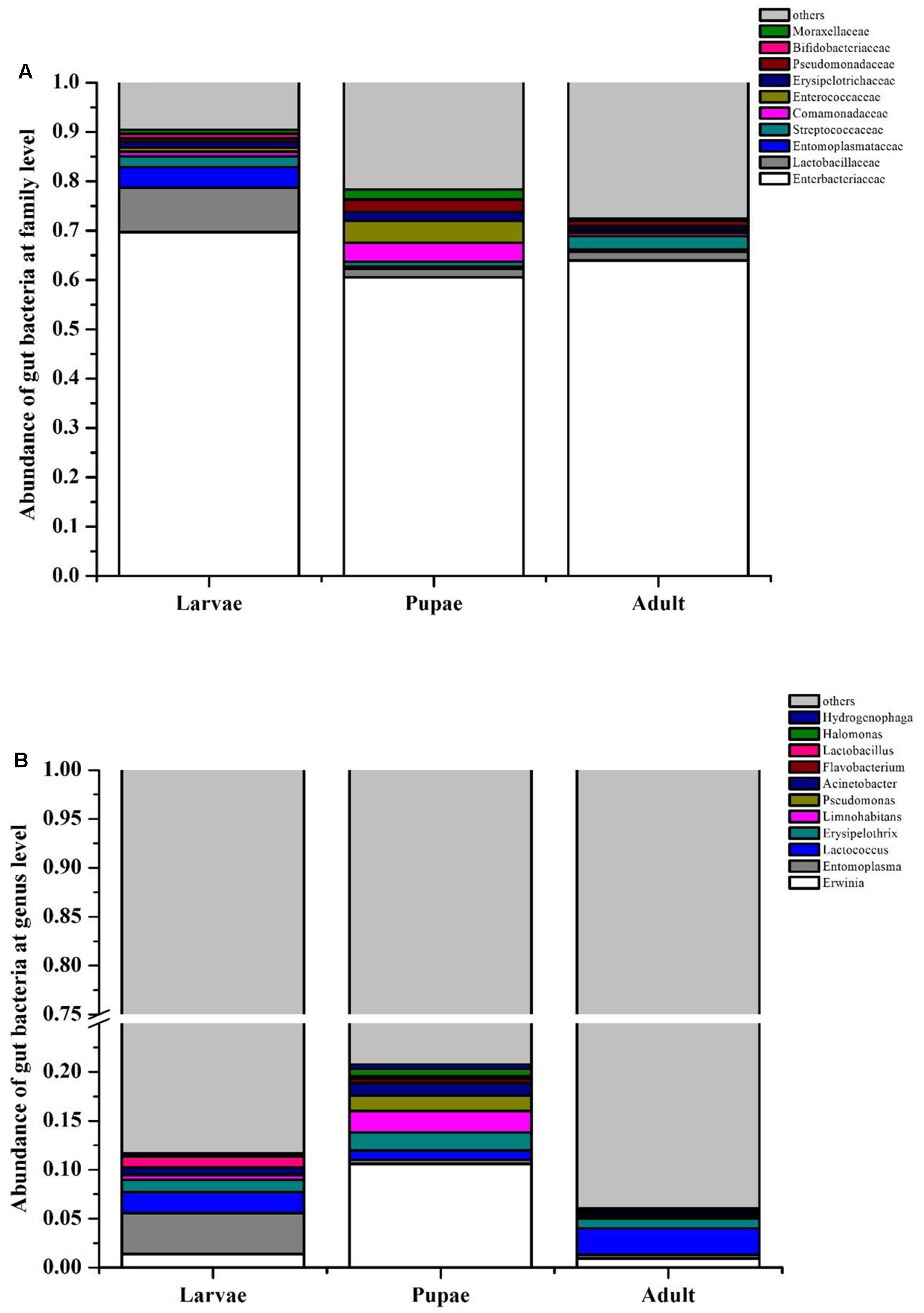
FIGURE 1. Bacterial community composition at family (A) and genus (B) level in the gut of Rhynchophorus ferrugineus during its different life stages.
The Effect of Host Plant Species on the Gut Microbiota of RPW Larvae
Principal component analysis showed that the first two components PC1 and PC2 accounted for 41.63% (PC1 = 25.28%, PC2 = 16.35%) of the variation (Supplementary Figure S2A). Meanwhile, the PCoA analysis, PC1 and PC2 explained a total variation of 44.06% (PC1 = 28.78%, PC2 = 15.28%, Supplementary Figure S2B). RPW larvae from different host plant species bore the gut microbiota containing the same compositions just with variations in their abundance (Figure 2) and no marked differences were detected in the bacterial communities associated with all three larvae groups by an ANOSIM analysis (P > 0.05). RPW larvae from three species of host plants were inhabited with a similar bacterial community which was mainly composed of Enterobacteriaceae, Lactobacillaceae, Entomoplasmataceae, Streptococcaceae, Enterocococcaceae, Erysipelotrichaceae, and Comamonadaceae. The most dominant bacterial taxa was Enterobacteriaceae, accounting for over 60% of the reads (Figure 2A). At the genus level, gut microbiota of larvae from C. nucifera mainly comprised Lactobacillus (8.26%), Entomoplasma (4.14%), and Lactococcus (2.17%). In contrast, Lactococcus and Erysipelothrix represented 14.55 and 3.51%, respectively, in the gut of RPW larvae from P. canariensis. In the larvae from laboratory populations, Lactococcus (9.13%), Dysgonomonas (3.16%), and Erysipelothrix (2.26%) comprised the major gut bacterial taxa (Figure 2B). Together, comparisons on the gut bacterial community of RPW individuals across different developmental stages and host plants revealed that the gut microbiota of RPW exhibited a highly stable microbial community with low variance in compositions.
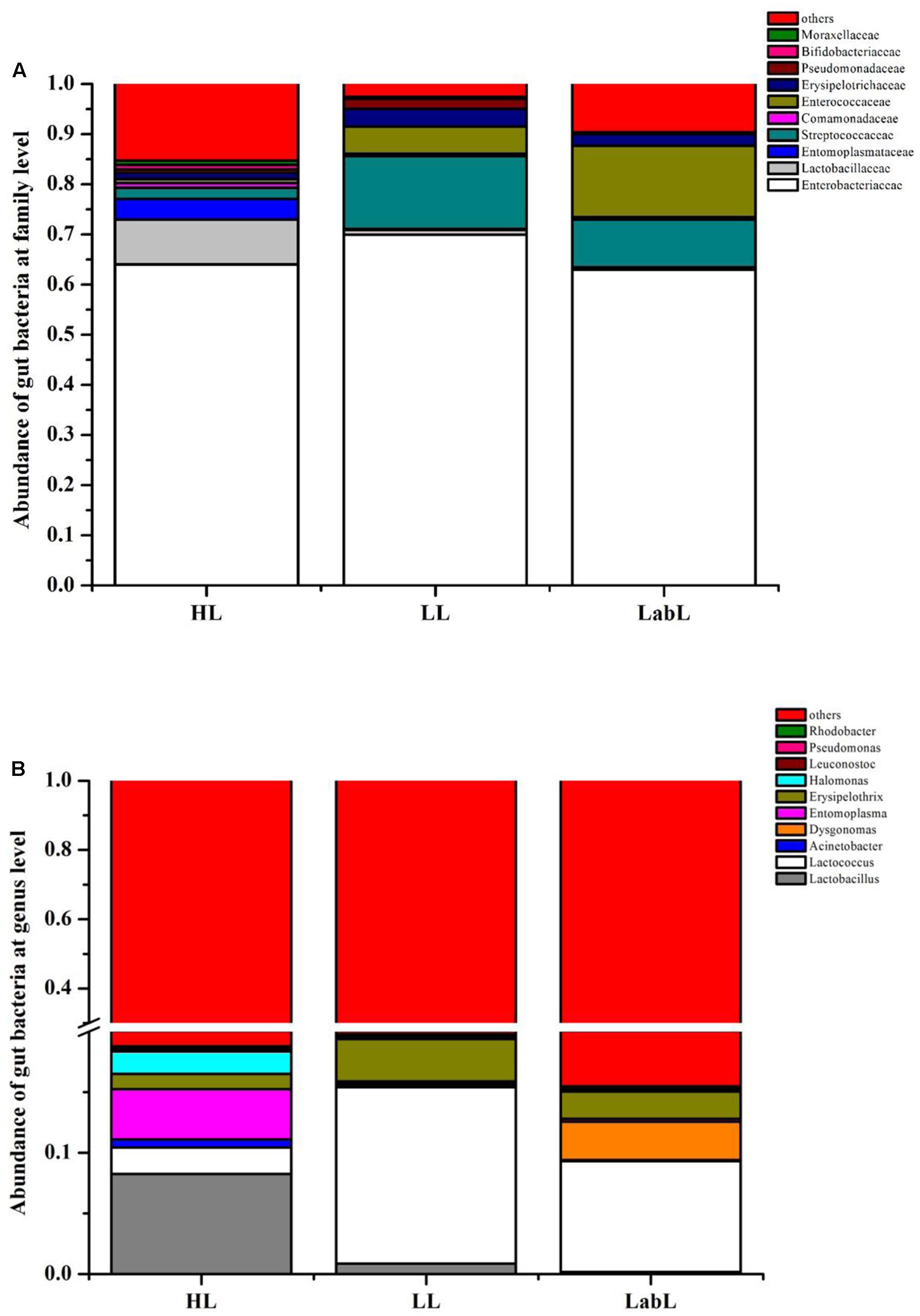
FIGURE 2. Relative abundance of gut bacterial families (A) and genera (B) in the larva of Rhynchophorus ferrugineus from different host plants. HL, LL, and LabL represent the Red palm weevil (RPW) larvae from Cocos nucifera, Phoenix canariensis and sugarcane, respectively.
Isolation and Cellulose Degradation Assay of Bacterial Isolates from RPW Larvae Gut
In total, 16 cultivable bacterial species were identified from the RPW larvae by 16S rRNA gene sequence analysis and were assigned into two phyla Proteobacteria (90.00%) and Firmicutes, (10.00%), belonging to four families, Enterobacteriaceae, Streptococcaceae, Staphylococcaceae, and Bacillaceae (Supplementary Table S2). Species of the genera Enterobacter (E. cloacae and E. aerogenes) and Klebsiella (K. oxytoca, K. variicola, and K. pneumonia) were the most dominant members, accounting for 23.00 and 20.50% in this cultivable bacteria community, respectively. The next most abundant were species of the genera Citrobacter (Citrobacter freundii and Citrobacter koseri) and Cronobacter representing 15.00 and 12.00%, respectively, of the bacterial community. The abundance of other bacterial species was less than 10.00% (Figure 3).
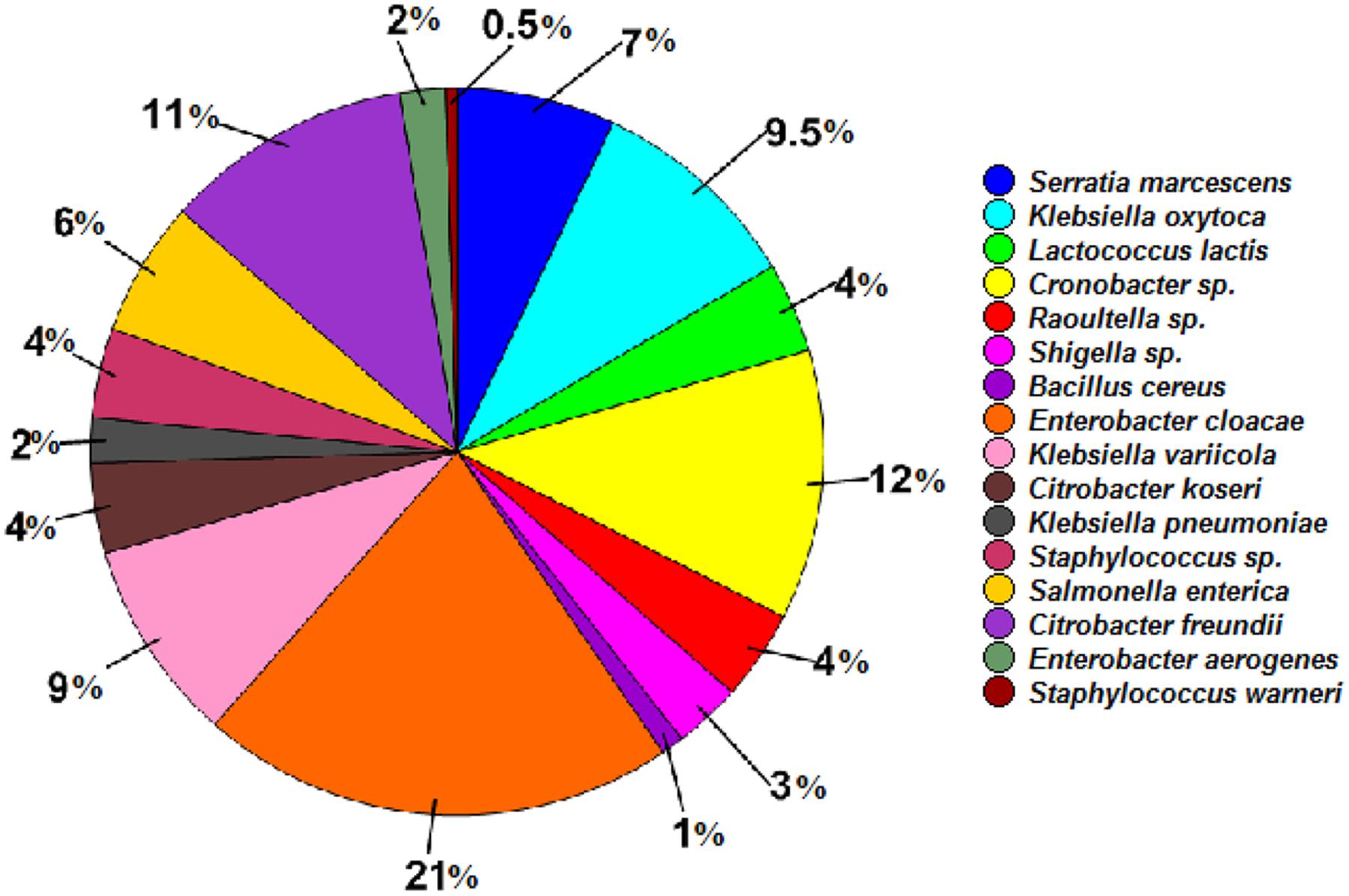
FIGURE 3. Relative abundance of cultivable bacterial species isolated from the wild-caught larvae of Rhynchophorus ferrugineus.
The CMC and Congo red assays revealed that seven bacterial species, Serratia enterica, E. cloacae, Raoultella sp., K. pneumonia, K. variicola, K. oxytoca, and Citrobacter koseri, were involved in cellulose degradation because the clearing zone was found around the colony of every bacterial species. These bacterial species performed significantly varied cellulolytic activity as they produced clear zones with different diameters ranging from 1.1 cm to 2.0 cm (ANOVA: F6,14 = 1298.04, P < 0.001). Citrobacter koseri produced the largest zones of hydrolysis, followed by E. clocae and K. oxytoca, while the species K. variicola showed the least hydrolytic activity (Figure 4). These data indicated that the gut of RPW larvae harbored bacterial species with relatively high cellulolytic activity.
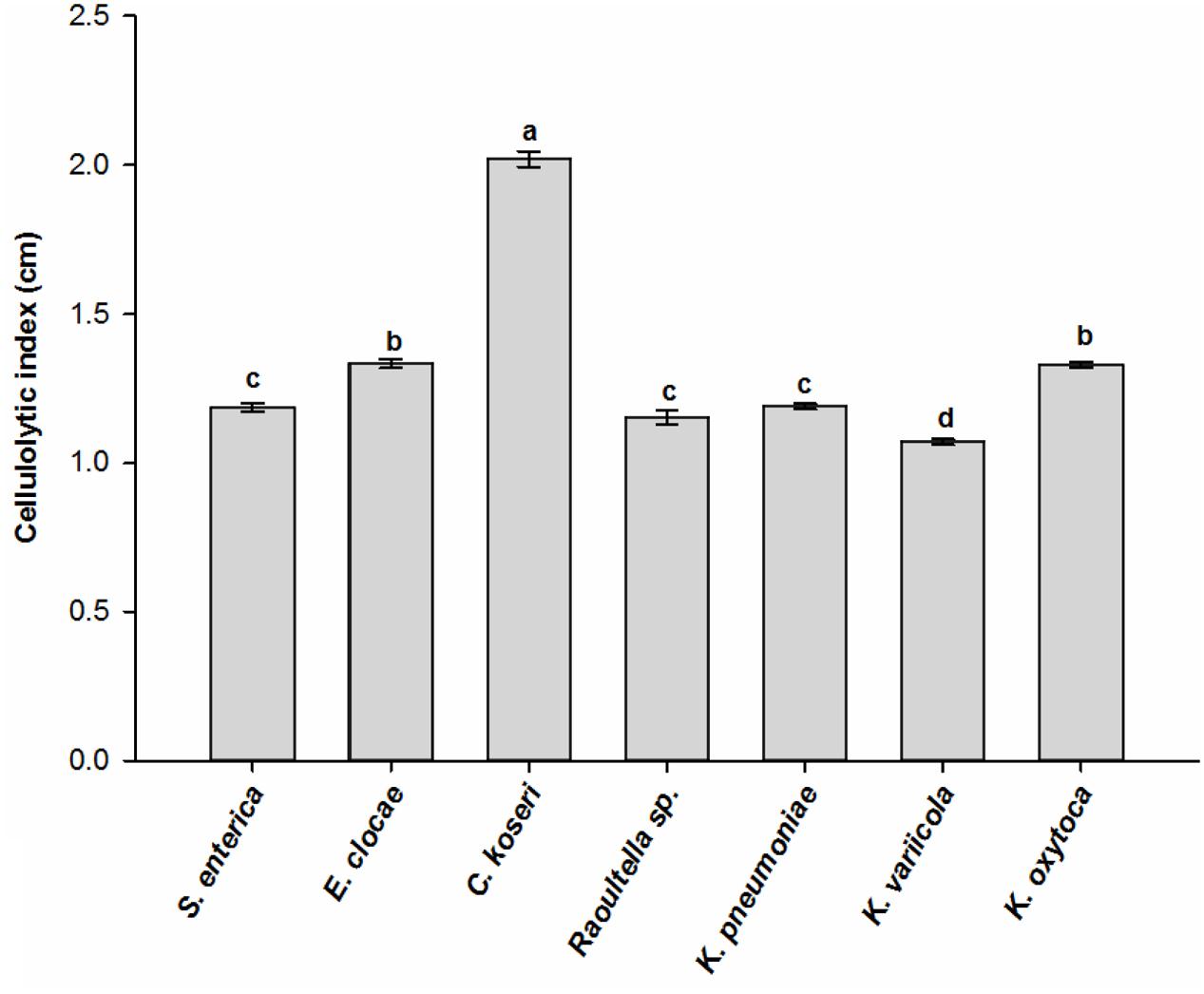
FIGURE 4. Cellulolytic activity of cultivable bacterial species isolated from the wild-caught larvae of Rhynchophorus ferrugineus. The different letters above the graph represent statistical significance (p < 0.05).
Administration of Antibiotics on the Gut Microbiota of RPW Larvae
To determine the potential physiological roles of gut microbiota in RPW, antibiotics were used to disrupt the inherent homeostasis of gut microbiota in RPW larvae. After administration of antibiotics, the species richness and community diversity of larvae gut microbiota were decreased compared with CKs (Table 2). In the CKs, the dominant bacterial taxa were Acetobacteraceae and Enterobacteriaceae which accounted for 30.45 and 16.52%, respectively. However, their abundances were dramatically changed by antibiotics administration. For instance, the relative abundance of Enterobacteriaceae was increased to 56.14% (t-test: t = 4.33, P < 0.05) while that of Acetobacteraceae dropped to 2.75% (t-test: t = -10.56, P < 0.001, Figure 5A). Klebsiella spp. (t-test: t = 5.803, P < 0.05) and Lactococcus spp. were strongly represented in the antibiotics-feeding larvae with relative abundances of 52.77 and 11.87%, respectively (Figure 5B). These data indicated that gut bacterial community structure of RPW larvae was significantly altered by antibiotics administration.
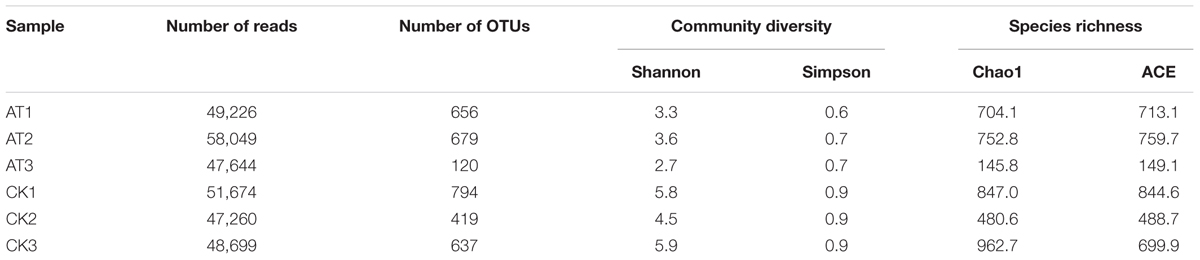
TABLE 2. Richness and diversity estimation of the gut bacterial communities associated with Rhynchophorus ferrugineus from control (CK) and antibiotics administration (AT) groups.
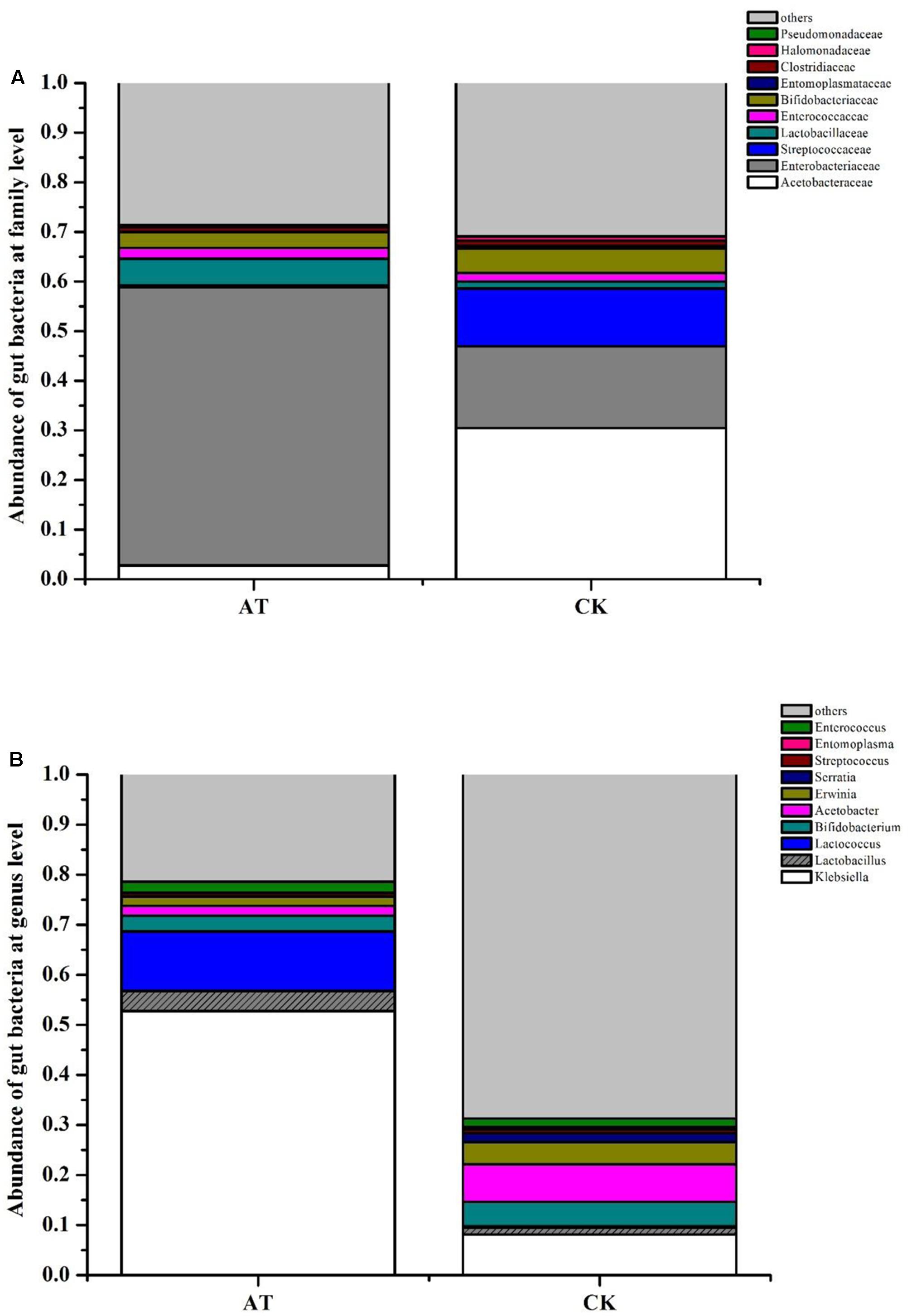
FIGURE 5. Effect of antibiotics administration on gut bacterial community composition of Rhynchophorus ferrugineus at family (A) and genus (B) level. AT and CK indicate the RPW larvae from antibiotics administration group and control group, respectively.
Perturbation of Gut Microbiota on the Nutrition Metabolism of RPW Larvae
To examine the effects of perturbation of gut microbiota on host nutrition metabolism, larva body weight, protein, glucose and TAG concentrations were measured and compared between AT and CK treatments. Although the net weight gain of antibiotics-feeding RPW larvae were higher than that of CK groups, no significant effect was determined (t-test: t = 1.68, df = 78, P > 0.05, Figure 6). Statistical analysis revealed that the protein level of RPW larvae was significantly reduced by the administration of antibiotics (ANOVA: F1,88 = 10.4, P < 0.05, Figure 7A). During the treatment period, the protein concentration of RPW increased with the development and the protein concentration of CK RPW were always higher than that of AT RPW (Figure 7B). The glucose level in AT RPW was found to be significantly lower than that in CK RPW (ANOVA: F1,88 = 5.37, P < 0.05, Figure 7C) The highest glucose level in CK and AT RPW were both recorded at the time-point of 3 weeks and then dropped 4 weeks after treatment (Figure 7D). By contrast, the concentration of TAG showed a distinct response to the treatments. The RPW larvae in the AT groups always contained more TAG than those in CK group during the treatment period (Figure 7F) and the TAG level of AT RPW was markedly higher (ANOVA: F1,88 = 12.26, P < 0.05, Figure 7E). These data highlighted that the changes in the composition of gut microbiota in RPW larvae significantly altered the host nutrition metabolism.
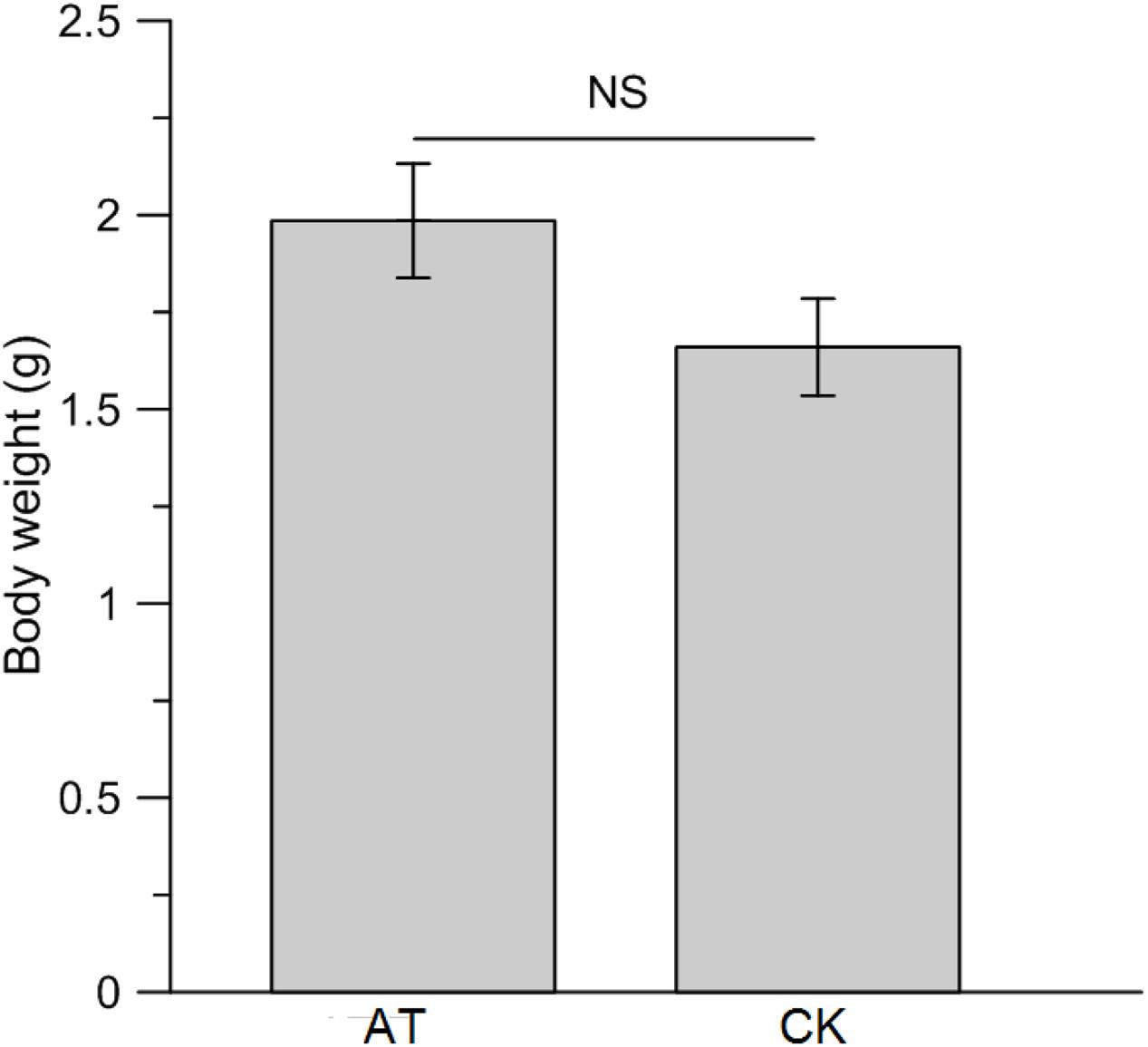
FIGURE 6. Impact of antibiotics administration-mediated alterations in gut bacterial community structure on the net body weight gain of Rhynchophorus ferrugineus larvae. AT and CK indicate the RPW larvae from antibiotics administration group and control group, respectively. NS, not significant.
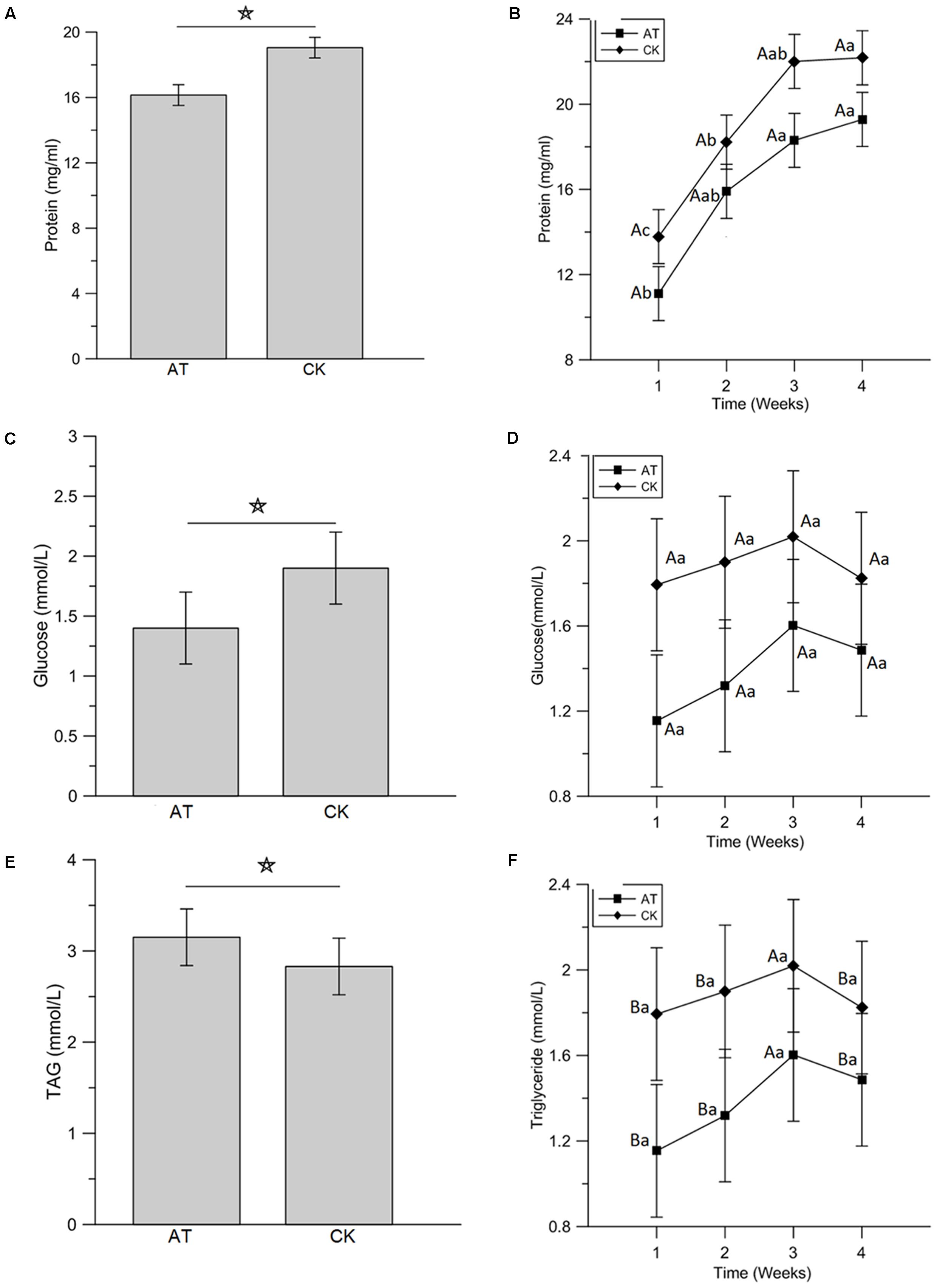
FIGURE 7. Impact of antibiotics administration-mediated alterations in gut bacterial community structure on the concentration of hemolymph protein (A,B), glucose (C,D), and triglyceride (E,F) in Rhynchophorus ferrugineus larvae. AT and CK indicate the RPW larvae from antibiotics administration group and control group, respectively. The star above the graphs indicates statistical significance was determined between two groups. The different uppercase and lowercase letters indicate the statistical significance was determined among different groups at the same time-point and among different time-points within the same group, respectively (p < 0.05).
Discussion
Previous surveys have found the associated gut microbiota of RPW larvae (Khiyami and Alyamani, 2008; Jia et al., 2013; Tagliavia et al., 2014) and adults (Montagna et al., 2015). However, the insect gut bacterial community is often dynamic in its quality and quantity, being determined by factors such as outside environmental habitat, diet, developmental stage and host phylogeny (Wong et al., 2011, 2015; Sudakaran et al., 2012; Blum et al., 2013; Yun et al., 2014; Montagna et al., 2015; Chen et al., 2016). Here, we disclosed that the RPW gut was mainly colonized by bacteria in the phyla Proteobacteria and Firmicutes. This result is congruent with the report on the gut metagenomes of RPW larvae from date palm (Jia et al., 2013) and lab-reared RPW adults on apple (Montagna et al., 2015). However, our RPW adults showed the second dominant taxon within Firmicutes that was scarcely found in the gut of Phoenix canariensis-fed RPW adults (Montagna et al., 2015). Identifying the variations of gut microbiome across various life stages and its stable indigenous gut microbiota is very important for deep elucidation of host-gut microbiota interactions. Here, we revealed that the guts of larvae, pupae and adults harbor the bacterial community primarily contained bacteria in the families Enterobacteriaceae, Lactobacillaceae, Streptococcaceae, Enterocococcaceae, and Entomoplasmataceae. Enterobacteriaceae being the most abundant family across the tested life stages is in line with previous studies (Jia et al., 2013; Tagliavia et al., 2014; Montagna et al., 2015). At the genus level, seven genera including Erwinia, Entomoplasma, Lactococcus, Lactobacillus, Pseudomonas, Limnohabitans, and Acinetobacter are stably represented in the gut of RPW through the whole life cycle, suggesting that they might have important effects on host fitness. Some minor differences in the abundance of major bacterial families or genera are detected with the transition of life stages in this beetle. The dynamics changes in gut microbial profiles of RPW might be caused by shifts in gut physiological conditions such as gut bacterial metabolism-mediated pH variations (Lowe et al., 1993; Engel and Moran, 2013; Jia et al., 2013). Comparisons on the diversity index revealed that RPW pupae host the highest species richness and community diversity, and gut microbiota of RPW larvae and adult are the subset of pupae’s. The higher species richness and community diversity in the pupae might be due to the fact that the gut is renewed during metamorphosis (Johnston and Rolff, 2015).
Comparative analysis on the gut bacterial consortia with RPW larvae from three species of host plants revealed that they share the almost same bacterial composition, just with changes in their abundance. For instance, although all the gut of larvae from different plant species was primarily colonized by the bacterial genera Lactobacillus, Entomoplasma, Lactococcus, Lactococcus, Dysgonomonas, and Erysipelothrix, they were detected in the RPW gut with various abundance. The stable existence of these bacterial genera implies that they may have functional importance in the gut of RPW. A higher number of OTUs were found in the larvae feeding on sugarcane in our laboratory (Supplementary Table S3), which might be caused by the reason that the dietary seeding was much more frequent, while in the field, larvae never come out from the galleries until pupation since they have little or no direct contact with the outside environment. These results suggest that RPW gut bacterial community is resilient to the changes of host plants, which is agreement with the view that diet has important roles in shaping the gut microbiota of RPW (Jia et al., 2013; Tagliavia et al., 2014; Montagna et al., 2015). Sixteen cultivable bacterial species were isolated from the gut of wild-caught RPW larvae by culture-dependent methods which helped in obtaining a better knowledge of RPW gut microbiota by cloning the full sequence of 16S rRNA gene. Interestingly, the majority of these species were members of the Enterobacteriaceae, Streptococcaceae, Staphylococcaceae, and Bacillaceae from the phyla Proteobacteria and Firmicutes, which supports the result from culture-independent methods. Collectively, the RPW gut entomotype is composed primarily of members of the Enterobacteriaceae, Lactobacillaceae, and Streptococcaceae, which may be critical for the health of RPW. Interestingly, some genus, including Raoltella, Serritia, Enterobacter, Citrobacter, Shigell, Cronobacter, Salmonella, and Staphylococcus, were not found by the pyrosequencing approach that relies upon a short 16S rRNA gene fragment. Failing of their detection could be due to the limited variability of the V4 region or their presence at very low levels.
Among the 16 species of isolated cultivable bacteria from wild-caught RPW larvae, Enterobacter, Klebsiella, Citrobacter and Cronobacter from the family Enterobacteriaceae are the dominant genera, representing 70.50% of this community. In the intestinal metagenomes of RPW, the open reading frames coding glycoside hydrolase have been identified in K. pneumonia, and Citrobacter koseri was also found being involved in the degradation of polysaccharides (Jia et al., 2013). In the present study, seven bacterial species, including K. pneumonia and Citrobacter koseri, were determined to have the greatest ability to degrade cellulose by in vitro CMC and Congo red assays. Isolation of cellulolytic bacteria from the gut of RPW larvae indicate that gut bacteria-mediated breakdown of plant polymers occurs inside the larvae (Butera et al., 2012). Members of the family Enterobacteriaceae are known as nitrogen fixers (Behar et al., 2005; Morales-Jiménez et al., 2009; Ben-Yosef et al., 2014), so the stable presence of this family in the RPW imply that gut bacteria of the Enterobacteriaceae can contribute to satisfy nitrogen requirements of RPW larvae feeding on nutrient-poor palm tissues, thus affecting development and fitness of RPW. Among these isolated bacteria species, Serratia marcescens was also found in RPW larvae gut with moderate abundance. Serratia plays several roles ranging from host protection against parasitoids (Costopoulos et al., 2014) to improve host fitness (Lancaster et al., 2012). The effects of S. marcescens on RPW fitness need further investigation.
The administration of antibiotics is an important alternative way to reveal the potential benefits of gut bacteria to the host by disrupting the symbiosis of gut microbiota and its host (Cho et al., 2012; Chu et al., 2013; Raymann et al., 2017). In this study, the feeding of antibiotics was found to significantly lower the community diversity and species richness of RPW larvae gut microbiota. Moreover, although no obvious variations were detected in the gut bacterial compositions, the abundance of dominant taxa was markedly changed. For example, Enterobacteriaceae became the first dominant taxon, followed by Aectobacteraceae after the administration of antibiotics. In the family Enterobacteriaceae, the abundance of the genus Klebsiella was increased significantly. In Drosophila melanogaster and honeybees, alteration of the gut microbiota community resulted in negative effects on host phenotype (Shin et al., 2011; Lee and Bray, 2013; Newell and Douglas, 2014; Wong et al., 2014; Clark et al., 2015; Raymann et al., 2017). Here, our evidence demonstrated that metabolic indices were dramatically affected by the antibiotics feeding-mediated alteration of gut microbiota community. Although no significance was detected in the weight gain of RPW larvae, the concentration of protein and glucose in hemolymph were reduced while that of hemolymph TAG was increased significantly by the perturbation of gut microbiota compared with CK groups. One possible reason for the reduced protein concentration might be that gut microbiota of conventional organisms improve the protein uptake by advancing the digestion of protein-rich These data highlighted that the changes in the composition of gut microbiota in RPW larvae, caused by the administration of antibiotics, significantly altered the host nutrition metabolism compounds or by synthesizing specific amino acids or by regulating the nutrient allocation (Blatch et al., 2010; Wong et al., 2014; Douglas, 2015). Lactobacillaceae has been uncovered to have the potential of exploiting glucose in Drosophila (Storelli et al., 2011; Wong et al., 2014). Interestingly, a higher abundance of Lactobacillaceae was detected in the AT groups with significantly lower glucose level, supporting the notion that the family Lactobacillaceae is involved in the metabolism of carbohydrates in RPW. Recent reports on the axenic mice (Bäckhed et al., 2004, 2007) and Drosophila (Ridley et al., 2012; Chaston et al., 2014) confirmed that bacteria in the family Acetobacteraceae are the main cause for the reduction of host TAG content. In the current investigation, the abundance of Acetobacteraceae obviously decreased to 2.75% which suggest that Acetobacteraceae function as vital partners in the lipid metabolism of RPW. Taken collectively, our evidence provided here suggests that gut bacteria function as obligate partners in the nutrition metabolism of RPW and might facilitate adaptation to exploiting new hosts in the phase of invasion. A recent report showed that collapse of gut symbiosis induced defective phenotypes in Nezara viridula, which present the promising notion that limitations imposed by obligate symbionts may help to counter the spread of invasive pests (Kikuchi et al., 2016; Moran, 2016). Thus, our findings provide vital implications to the development of novel pest control tactics targeting the gut bacteria of RPW. Antibiotics administartion might also have some effects on the bacteria species in hemolymph by passing through the midgut epithelial cells. This potential influence on host fitness is yet to be investigated.
Conclusion
The composition of RPW gut microbiota is stable, being comprised of bacteria in the families Enterobacteriaceae, Lactobacillaceae, and Streptococcaceae, and the entomotype is constituted by the following most frequent genera: Erwinia, Entomoplasma, Lactococcuss, Lactobacillus, Pseudomonas, Limnohabitans, and Acinetobacter. The transition of developmental stages and host plant species show no influence on the bacterial composition, just on their abundance. Furthermore, seven species of cultivable bacteria with great cellulose degradation ability were also identified. Alterations in the gut bacteria community caused by the administration of antibiotics significantly affect the nutrition metabolism of the host, suggesting that gut bacteria are involved in RPW metabolism as the obligate partners. Our results lay strong foundations to develop novel pest management strategies for RPW by disrupting gut symbiosis to retard its further spread worldwide.
Data Availability and Material
The Illumina high-throughput sequencing data are available in the NCBI database Sequence Read Archive (SRA) (BioProject PRJNA 389236). The Sanger sequencing data of the bacterial isolates can be accessed through accession numbers MF185369-MF185384 in the NCBI database.
Author Contributions
ZS and YH conceived and designed the research. AM and YF performed the experiments. AM and ZS analyzed the data and wrote the manuscript. ZS and YH revised the paper and all authors approved the final version.
Funding
This work was funded by the National Natural Science Foundation of China (31470656, ZS), the National Key Research and Development Project of China (No. 2017YFC1200600, YH) and the Program for New Century Excellent Talents in University of Fujian Province (KLa16058A, ZS).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2017.02291/full#supplementary-material
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
- ^ http://ccb.jhu.edu/software/FLASH/
- ^ http://qiime.org/index.html
- ^ http://drive5.com/uparse
- ^ http://sourceforge.net/projects/rdp-classifier/
- ^ http://greengenes.lbl.gov/
- ^ http://www.ncbi.nlm.nih.gov/blast
- ^ http://rdp.cme.msu.edu/seqmatch
References
Adams, A. S., Jordan, M. S., Adams, S. M., Suen, G., Goodwin, L. A., Davenport, K. W., et al. (2011). Cellulose-degrading bacteria associated with the invasive woodwasp Sirex noctilio. ISME J. 5, 1323–1331. doi: 10.1038/ismej.2011.14
Bäckhed, F., Ding, H., Wang, T., Hooper, L. V., Koh, G. Y., Nagy, A., et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U.S.A. 101, 15718–15723. doi: 10.1073/pnas.0407076101
Bäckhed, F., Manchester, J. K., Semenkovich, C. F., and Gordon, J. I. (2007). Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc. Natl. Acad. Sci. U.S.A. 104, 979–984. doi: 10.1073/pnas.0605374104
Behar, A., Yuval, B., and Jurkevitch, E. (2005). Enterobacteria-meidated nitrogen fixation in natural populations of the fruit fly Ceratitis capitata. Mol. Ecol. 14, 2637–2643. doi: 10.1111/j.1365-294X.2005.02615.x
Ben-Yosef, M., Pasternak, Z., Jurkevitch, E., and Yuval, B. (2014). Symbiotic bacteria enable olive flies (Bactrocera oleae) to exploit intractable sources of nitrogen. J. Evol. Biol. 27, 2695–2705. doi: 10.1111/jeb.12527
Blatch, S. A., Meyer, K. W., and Harrison, J. F. (2010). Effects of dietary folic acid level and symbiotic folate production on fitness and development in the fruit fly Drosophila melanogaster. Fly 4, 312–319. doi: 10.4161/fly.4.4.13258
Blum, J. E., Fischer, C. N., Miles, J., and Handelsman, J. (2013). Frequent replenishment sustains the beneficial microbiome of Drosophila melanogaster. mBio 4:e00860-13. doi: 10.1128/mBio.00860-13
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Butera, G., Ferraro, C., Colazza, S., Alonzo, G., and Quatrini, P. (2012). The a cultural bacterial community of frass produced by larvae of Rhynchophorus ferrugineus olivier (Coleoptera: Curculionidae) in the Canary island date palm. Lett. Appl. Microbiol. 54, 530–536. doi: 10.1111/j.1472-765X.2012.03238.x
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chaston, J. M., Newell, P. D., and Douglas, A. E. (2014). Metagenome-wide association of microbial determinants of host phenotype in Drosophila melanogaster. mBio 5:e01631-4. doi: 10.1128/mBio.01631-14
Chen, B. S., The, B., Sun, C., Hu, S. R., Lu, X. M., Boland, W., et al. (2016). Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 6:29505. doi: 10.1038/srep29505
Cho, I., Yamanishi, S., Cox, L., Methe, B. A., Zavadil, J., Li, K., et al. (2012). Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature 488, 621–626. doi: 10.1038/nature11400
Chu, C., Spencer, J. L., Curzi, M. J., Zavala, J. A., and Seufferheld, M. J. (2013). Gut bacteria facilitate adaptation to crop rotation in the western corn rootworm. Proc. Natl. Acad. Sci. U.S.A. 110, 11917–11922. doi: 10.1073/pnas.1301886110
Clark, R. I., Salazar, A., Yamada, R., Fitz-Gibbon, S., Morselli, M., Alcaraz, J., et al. (2015). Distinct shifts in microbiota composition during Drosophila aging impair intestinal function and drive mortality. Cell Rep. 12, 1656–1667. doi: 10.1016/j.celrep.2015.08.004
Costopoulos, K., Kovacs, J. L., Kamins, A., and Gerardo, N. M. (2014). Aphid facultative symbionts reduce survival of the predatory lady beetle Hippodamia convergens. BMC Ecol. 14:5. doi: 10.1186/1472-6785-14-5
DeSantis, T. Z., Hugenholtz, P., Larsen, N., Rojas, M., Brodie, E. L., Keller, K., et al. (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl. Environ. Microbiol. 72, 5069–5072. doi: 10.1128/AEM.03006-05
Dethlefsen, L., McFall-Ngai, M., and Relman, D. A. (2007). An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature 449, 811–818.
Douglas, A. E. (2009). The microbial dimension in insect nutritional ecology. Funct. Ecol. 23, 38–47. doi: 10.1111/j.1365-2435.2008.01442.x
Douglas, A. E. (2015). Multiorganismal insects: diversity and function of resident microorganisms. Annu. Rev. Entomol. 60, 17–34. doi: 10.1146/annurev-ento-010814-020822
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Engel, P., and Moran, N. A. (2013). The gut microbiota of insects - diversity in structure and function. FEMS Microbiol. Rev. 37, 699–735. doi: 10.1111/1574-6976.12025
Faleiro, J. R. (2006). A review of the issues and management of the red palm weevil Rhynchophorus ferrugineus (Coleoptera: Rhynchophoridae) in coconut and date palm during the last one hundred years. Int. J. Trop. Insect Sci. 26, 135–154. doi: 10.1079/IJT2006113
Ge, X. Z., He, S. Y., Wang, T., Yan, W., and Zong, S. X. (2016). Potential distribution predicted for Rhynchophorus ferrugineus in China under different climate warming scenarios. PLOS ONE 10:e141111. doi: 10.1371/journal.pone.0141111
Hou, Y. M., Wu, Z. J., and Wang, C. F. (2011). “The current status and damage of biological invasion in Fujian Province,” in Biological Invasions: Problems and Countermeasures, et al. Edn, eds L. H. Xie, M. S. You, and Y. M. Hou (Beijing: Science Press), 121–122.
Huang, S. W., Sheng, P., and Zhang, H. Y. (2012). Isolation and identification of cellulolytic bacteria from the gut of Holotrichia parallela larvae (Coleoptera: Scarabaeidae). Int. J. Mol. Sci. 13, 2563–2577. doi: 10.3390/ijms13032563
Indiragandhi, P., Anandham, R., Madhaiyan, M., Poonguzhali, S., Kim, G. H., Saravanan, V. S., et al. (2007). Cultivable bacteria associated with larval gut of prothiofos-resistant, prothiofos-susceptible and field-caught populations of diamondback moth, Plutella xylostella and their potential for, antagonism towards entomopathogenic fungi and host insect nutrition. J. Appl. Microbiol. 103, 2664–2675. doi: 10.1111/j.1365-2672.2007.03506.x
Jia, S. G., Zhang, X. W., Zhang, G. Y., Yin, A., Zhang, S., Li, F. S., et al. (2013). Seasonally variable intestinal metagenomes of the red palm weevil (Rhynchophorus ferrugineus). Environ. Microbiol. 15, 3020–3029. doi: 10.1111/1462-2920.12262
Jiu, M., Zhou, X. P., Tong, L., Xu, J., Yang, X., Wan, F. H., et al. (2007). Vector-virus mutualism accelerates populations increase of an invasive whitefly. PLOS ONE 2:e182. doi: 10.1371/journal.pone.0000182
Johnston, P. R., and Rolff, J. (2015). Host and symbiont jointly control gut microbiota during complete metamorphosis. PLOS Pathog. 11:e1005246. doi: 10.1371/journal.ppat.1005246
Ju, R. T., Wang, F., Wan, F. H., and Li, B. (2011). Effect of host plants on development and reproduction of Rhynchophorus ferrugineus (Olivier) (Coleoptera: Curculionidae). J. Pest Sci. 84, 33–39. doi: 10.1007/s10340-010-0323-4
Khiyami, M., and Alyamani, E. (2008). Aerobic and facultative anaerobic bacteria from gut of red palm weevil (Rhynchophorus ferrugineus). Afr. J. Biotechnol. 7, 1432–1437. doi: 10.1186/1471-2180-14-136
Kikuchi, Y., Tada, A., Musolin, D. L., Hari, N., Hosokawa, T., Fujisaki, K., et al. (2016). Collapse of insect gut symbiosis under simulated climate change. mBio 7:e01578-16. doi: 10.1128/mBio.01578-16
Lancaster, J. D., Mohammad, B., and Abebe, E. (2012). Effect of the bacterium Serratia marcescens SCBI on the longevity and reproduction of the nematode Caenorhabditis briggsae KT0001. BMC Res. Notes 5:688. doi: 10.1186/1756-0500-5-688
Lee, W., and Bray, P. T. (2013). How microbiomes influence metazoan development: insights from histroy and Drosophila modeling of gut-microbe interactions. Annu. Rev. Cell Dev. Biol. 29, 571–592. doi: 10.1146/annurev-cellbio-101512-122333
Lehman, R. M., Lundgren, J. G., and Petzke, L. M. (2009). Bacterial communities associated with the digestive tract of the predatory ground beetle, Poecilus chalcites, and their modification by laboratory rearing and antibiotic treatment. Microb. Ecol. 57, 349–358. doi: 10.1007/s00248-008-9415-6
Li, Y., Zu, Z. R., Ju, R. T., and Wang, L. S. (2009). The red palm weevil, Rhynchophorus ferrugineus (Coleoptera: Curculionidae), newly reported from Zhejiang, China and update of geographical distribution. Fla. Entomol. 92, 386–387. doi: 10.1653/024.092.0229
Llácer, E., Negre, M., and Jacas, J. A. (2012a). Evaluation of an oil dispersion formulation of imidacloprid as a drench against Rhynchophorus ferrugineus (Coleoptera, Curculionidae) in young palm trees. Pest. Manag. Sci. 68, 878–882. doi: 10.1002/ps.3245
Llácer, E., Santiago-Álvarez, C., and Jacas, J. A. (2012b). Could sterile males be used to vector a microbiological control agent? The case of Rhynchophorus ferrugineus and Beauveria bassiana. Bull. Entomol. Res. 103, 241–250. doi: 10.1017/S0007485312000582
Lowe, S. E., Jain, M. K., and Zeikus, J. G. (1993). Biology, ecology, and biotechnological applications of anaerobic bacteria adapted to environmental stresses in temperature, pH, salinity, or substrates. Microbiol. Rev. 57, 451–509.
Lu, M., Hulcr, J., and Sun, J. H. (2016). The role of symbiotic microbes in insect invasions. Annu. Rev. Ecol. Evol. Syst. 47, 487–505. doi: 10.1146/annurev-ecolsys-121415-032050
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Martin, M. M. (1991). The evolution of cellulose digestion in insects. Philos. Trans. R. Soc. Lond. B. 333, 281–288. doi: 10.1098/rstb.1991.0078
Mazza, G., Francardi, V., Simoni, S., Benvenuti, C., Cervo, R., Faleiro, J. R., et al. (2014). An overview on the natural enemies of Rhynchophorus palm weevils, with focus on R. ferrugineus. Biol. Control 77, 83–92. doi: 10.1016/j.biocontrol.2014.06.010
Montagna, M., Chouaia, B., Mazza, G., Prosdocimi, E. M., Crotti, E., Mereghetti, V., et al. (2015). Effects of the diet on the microbiota of the red palm weevil (Coleoptera: Dryophthoridae). PLOS ONE 10:e0117439. doi: 10.1371/journal.pone.0117439
Montagna, M., Mereghetti, V., Gargari, G., Guglielmetti, S., Faoro, F., Lozzia, G., et al. (2016). Evidence of a bacterial core in the stored products pest Plodia interpunctella: the influence of different diets. Environ. Microbiol. 18, 4961–4973. doi: 10.1111/1462-2920.13450
Morales-Jiménez, L., Vera-Ponce de Len, A., Garca-Domnguez, A., Martnex-Romero, E., Ziga, G., and Hernndez-Rodrguez, C. (2013). Nitrogen-fixing and uricolytic bacteria associated with the gut of Dendroctonus rhizophagus and Dendroctonus valens (Curculionidae: Scolytinae). Microbiol. Ecol. 66, 200–210. doi: 10.1007/s00248-013-0206-3
Morales-Jiménez, L., Zuniga, G., Villa-Tanaca, L., and Hernandez-Rodriguez, C. (2009). Bacterial community and nitrogen fixation in the red turpentine beetle, Dendroctonus valens LeConte (Coleoptera: Curculionidae: Scolytinae). Microb. Ecol. 58, 879–891. doi: 10.1007/s00248-009-9548-2
Mota, M. M., Takemoto, S., Takeuchi, Y., Hara, N., and Futai, K. (2006). Comparative studies between Portuguese and Japanese isolates of the pinewood nematode, Bursaphelenchus xylophilus. J. Nemotol. 38, 429–433.
Nagnan, P., Cain, A. H., and Rochat, D. (1992). Extraction et identification des composés volatils de la sève de palmier à huìle fermentée (vin de palme) attractifs potentiels pour le charancon du palmier. Oléagineux 47, 135–142.
Newell, P. D., and Douglas, A. E. (2014). Interspecies interactions determine the impact of the gut microbiota on nutrient allocation in Drosophila melanogaster. Appl. Environ. Microbiol. 80, 788–796. doi: 10.1128/AEM.02742-13
Peng, L., Miao, Y. X., and Hou, Y. M. (2016). Demographic comparison and population projection of Rhynchophorus ferrugineus (Coleoptera: Curculionidae) reared on sugarcane at different temperatures. Sci. Rep. 6:31659. doi: 10.1038/srep31659
Pu, Y. C., and Hou, Y. M. (2016). Isolation and identification of bacterial strains with insecticidal activities from Rhynchophorus ferrugineus Oliver (Coleoptera: Curculionidae). J. Appl. Entomol. 140, 617–626. doi: 10.1111/jen.12293
Pu, Y. C., Ma, T. L., Hou, Y. M., and Sun, M. (2017). An entomopathogenic bacterium strain, Bacillus thuringiensis, as a biological control agent against the red palm weevil, Rhynchophorus ferrugineus (Coleoptera: Curculionidae). Pest. Manag. Sci. 73, 1494–1502. doi: 10.1002/ps.4485
Raymann, K., Shaffer, Z., and Moran, N. A. (2017). Antibiotic exposure perturbs the gut microbiota and elevates mortality in honeybees. PLOS Biol. 15:e2001861. doi: 10.1371/journal.pbio.2001861
Ridley, E. V., Wong, A. C., Westmiller, S., and Douglas, A. E. (2012). Impact of the resident microbiota on the nutritional phenotype of Drosophila melanogaster. PLOS ONE 7:e36765. doi: 10.1371/journal.pone.0036765
Sharon, G., Segal, D., Fingo, J. M., Hefetz, A., Zilber-Rosenberg, I., and Rosenberg, E. (2010). Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 107, 20051–20056. doi: 10.1073/pnas.1009906107
Shi, Z. H., Lin, Y. T., and Hou, Y. M. (2014). Mother-derived trans-generational immune priming in the red palm weevil, Rhynchophorus ferrugineus olivier (Coleoptera, Dryophthoridae). Bull. Entomol. Res. 104, 742–750. doi: 10.1017/S0007485314000583
Shin, S. C., Kim, S. H., You, H., Kim, A. C., Lee, K. A. M., Yoon, J. H., et al. (2011). Drosophila microbiome modulates host developmental and metabolic homeostasis via insulin signaling. Science 334, 670–674. doi: 10.1126/science.1212782
Storelli, G., Defaye, A., Erkosar, B., Hols, P., Royet, J., and Leulier, F. (2011). Lactobacillus plantarum promotes Drosophila systemic growth by modulating hormonal signals through TOR-dependent nutrient sensing. Cell Metabol. 14, 403–414. doi: 10.1016/j.cmet.2011.07.012
Sudakaran, S., Salem, H., Kost, C., and Kaltenpoth, M. (2012). Geographical and ecological stability of the symbiotic mid-gut microbiota in European firebugs, Pyrrhocoris apterus (Hemiptera, Pyrrhocoridae). Mol. Ecol. 21, 6134–6151. doi: 10.1111/mec.12027
Sugio, A., Dubreuil, G., Giron, D., and Simon, J. (2016). Plant-insect interactions under bacterial influence: ecological implications and underlying mechanisms. J. Exp. Bot. 66, 467–478. doi: 10.1093/jxb/eru435
Tagliavia, M., Messina, E., Manachini, B., Cappello, S., and Quatrini, P. (2014). The gut microbiota of larvae of Rhynchophorus ferrugineus oliver (Coleoptera: Curculionidae). BMC Microbiol. 14:e136. doi: 10.1186/1471-2180-14-136
Valzano, M., Achille, G., Burzacca, F., Damiani, C., Scuppa, P., Ricci, I., et al. (2012). Deciphering microbiota associated to Rhynchophorus ferrugineus in Italian samples: a preliminary study. J. Entomol. Acarol. Res. 44:e16. doi: 10.4081/jear.2012.e16
Vasanthakumar, A., Handelsman, J., Schloss, P. D., Bauer, L. S., and Raffa, K. F. (2008). Gut microbiota of an invasive subcortical beetle, Agrilus planipennis Fairmaire, across various life stages. Environ. Entomol. 37, 1344–1353. doi: 10.1603/0046-225X/08/1344
Veivers, P. C., O’Brien, R. W., and Slaytor, M. (1983). Selective defaunation of Mastotermes darwiniensis and its effect on cellulose and starch metabolism. Insect Biochem. 12, 35–40. doi: 10.1016/0020-1790(83)90069-0
Wan, F. H., Hou, Y. M., and Jiang, M. X. (2015). “Important agriculture and forestry invasive species,” in Invasion Biology, eds F. H. Wan, Y. M. Hou, and M. X. Jiang (Beijing: Science Press), 240–244.
Wan, F. H., and Yang, N. W. (2016). Invasion and management of agricultural alien insects in China. Annu. Rev. Entomol. 61, 77–98. doi: 10.1146/annurev-ento-010715-023916
Wang, G. H., Zhang, X., Hou, Y. M., and Tang, B. Z. (2015). Analysis of the population genetic structure of Rhynchophorus ferrugineus in Fujian, China, revealed by microsatellite loci and mitochondrial COI sequences. Entomol. Exp. Appl. 155, 28–38. doi: 10.1111/eea.12282
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Wang, Y., Gilbreath, T. M., Kukutla, P., Yan, G. Y., and Xu, J. N. (2011). Dynamic gut microbiome across life history of the Malaria mosquito Anopheles gambiae in Kenya. PLOS ONE 6:e24767. doi: 10.1371/journal.pone.0024767
Wong, A. C., Dobson, A. J., and Douglas, A. E. (2014). Gut microbiota dictates the metabolic response of Drosophila to diet. J. Exp. Biol. 217, 1894–1901. doi: 10.1242/jeb.101725
Wong, A. C., Luo, Y., Jing, X., Franzenburg, S., Bost, A., and Douglas, A. E. (2015). The host as the driver of the microbiota in the gut and external environment of Drosophila melanogaster. Appl. Environ. Microbiol. 81, 6232–6240. doi: 10.1128/AEM.01442-15
Wong, A. C., Ng, P., and Douglas, A. E. (2011). Low-diversity bacterial community in the gut of the fruitfly Drosophila melanogaster. Environ. Microbiol. 13, 1889–1900. doi: 10.1111/j.1462-2920.2011.02511.x
Xu, L. T., Lu, M., and Sun, J. H. (2016). Invasive bark beetle-associated microbes degrade a host defensive monoterpene. Insect Sci. 23, 183–190. doi: 10.1111/1744-7917.12255
Yun, J., Roh, S. W., Whon, T. W., Jung, M., Kim, M., Park, D., et al. (2014). Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 80, 5254–5264. doi: 10.1128/AEM.01226-14
Zhang, R. Z., Ren, L., Sun, J. H., Wu, J., and Zeng, R. (2003). Morphological differences of the coconut pest insect, Rhynchophorus ferrugineus (Olivier) and its related species (Coleoptera: Curculionidae). Chin. For. Insect. Pest. Dis. 22, 3–6. doi: 10.1673/031.013.9101
Zhao, L. L., Lu, M., Niu, H. T., Fang, G. F., Zhang, S., and Sun, J. H. (2013). A native fungal symbionts facilitate the prevalence and development of an invasive pathogen-native vector symbiosis. Ecology 94, 2817–2826. doi: 10.1890/12-2229.1
Zhao, L. L., Mota, M., Vieira, P., Butcher, R. A., and Sun, J. H. (2014). Interspecific communication between pinewood nematode, its insect vector, and associated microbes. Trends Parasitol. 30, 299–308. doi: 10.1016/j.pt.2014.04.007
Keywords: symbiotic invasion, gut microbiota, Rhynchophorus ferrugineus, insect symbiosis, cellulose degradation
Citation: Muhammad A, Fang Y, Hou Y and Shi Z (2017) The Gut Entomotype of Red Palm Weevil Rhynchophorus ferrugineus Olivier (Coleoptera: Dryophthoridae) and Their Effect on Host Nutrition Metabolism. Front. Microbiol. 8:2291. doi: 10.3389/fmicb.2017.02291
Received: 30 August 2017; Accepted: 06 November 2017;
Published: 21 November 2017.
Edited by:
Francesca Turroni, Università degli Studi di Parma, ItalyReviewed by:
George Tsiamis, University of Patras, GreeceMatteo Montagna, Università degli Studi di Milano, Italy
Copyright © 2017 Muhammad, Fang, Hou and Shi. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Youming Hou, eW1ob3VAZmFmdS5lZHUuY24= Zhanghong Shi, c2hpemhAZmFmdS5lZHUuY24=