 Jeffrey T. Foster1,2*
Jeffrey T. Foster1,2* Faith M. Walker2Brandy D. Rannals2
Faith M. Walker2Brandy D. Rannals2 M. Hammad Hussain3,4Kevin P. Drees1,2Rebekah V. Tiller5Alex R. Hoffmaster5
M. Hammad Hussain3,4Kevin P. Drees1,2Rebekah V. Tiller5Alex R. Hoffmaster5 Abdulmajeed Al-Rawahi4Paul Keim2
Abdulmajeed Al-Rawahi4Paul Keim2 Muhammad Saqib3*
Muhammad Saqib3*- 1Department of Molecular, Cellular, and Biomedical Sciences, University of New Hampshire, Durham, NH, United States
- 2Pathogen and Microbiome Institute, Northern Arizona University, Flagstaff, AZ, United States
- 3Department of Clinical Medicine and Surgery, University of Agriculture, Faisalabad, Pakistan
- 4Animal Health Research Center, Ministry of Agriculture and Fisheries, Muscat, Oman
- 5National Center for Emerging and Zoonotic Infectious Diseases, Centers for Disease Control, Atlanta, GA, United States
Brucellosis is a common livestock disease in the Middle East and North Africa, but remains poorly described in the region both genetically and epidemiologically. Traditionally found in goats and sheep, Brucella melitensis is increasingly recognized as infecting camels. Most studies of brucellosis in camels to date have focused on serological surveys, providing only limited understanding of the molecular epidemiology of circulating strains. We genotyped B. melitensis isolates from Omani camels using whole genome SNP assays and VNTRs to provide context for regional brucellosis cases. We identified a lineage of B. melitensis circulating in camels as well as in goats, sheep, and cattle in Oman. This lineage is genetically distinct from most genotypes from the Arabian Peninsula and from isolates from much of the rest of the Middle East. We then developed diagnostic assays that rapidly identify strains from this lineage. In analyses of genotypes from throughout the region, Omani isolates were genetically most closely related to strains from brucellosis cases in humans and livestock in North Africa. Our findings suggest an African origin for B. melitensis in Oman that has likely occurred through the trade of infected livestock. Moreover, African lineages of B. melitensis appear to be undersampled and consequently are underrepresented in genetic databases for Brucella. As we begin to more fully understand global genomic diversity of B. melitensis, finding and characterizing these unique but widespread lineages is essential. We predict that increased sampling of humans and livestock in Africa will reveal little known diversity in this important zoonotic pathogen.
Introduction
Brucella melitensis is a ubiquitous and common pathogen of goats and sheep worldwide (Seleem et al., 2010; Moreno, 2014). This pathogen was first identified in Malta by David Bruce in 1887, with subsequent discovery of the role of contaminated goat's milk for brucellosis infections in humans by Themistocles Zammit in 1905 (Vassallo, 1996; Wyatt, 2005). Despite apparent host specificity of B. melitensis to caprines, this bacterium also infects camels (Abbas and Agab, 2002; Gwida et al., 2012; Sprague et al., 2012; Wernery, 2014). Arabian camels (Camelus dromedarius) occur throughout the deserts of North Africa and across the Middle East to North India, a region where they are critical for meat, milk, leather, wool and transport (Wilson, 1984). Camel brucellosis was first reported in 1931 and has since been found in all camel-keeping countries in this region but are particularly well documented for infected herds from Africa and the Arabian Peninsula (Gwida et al., 2012).
Camels are not a primary host for Brucella spp. but infections with B. melitensis occur due to the co-mingling of camels and ruminant livestock (Sprague et al., 2012). In fact, among the highest prevalence rates in camels have been documented when camel herds are intermixed with ruminants (Musa et al., 2008). Despite this cross-species transmission, epidemiological links between brucellosis in camels and other livestock are poorly understood. Prevalence rates of brucellosis in camels vary widely based on several factors, especially animal husbandry practices (Gwida et al., 2012). Camels can also be infected with B. abortus, likely due to the commingling of camel herds with infected cattle (Sprague et al., 2012). Thus, brucellosis prevalence in camels is complex and the role of infections in the primary caprine and bovine hosts must be considered. The pathology of brucellosis infection in camels is poorly known as well. Consistent with findings from other livestock, the bacteria appear to localize in reproductive tissues, lymph nodes, and spleen, causing inflammation, edema, and necrosis (Wernery, 2014). Infection of pregnant camels can result in placental and fetal pathologies resulting in abortion (Narnaware et al., 2017). As with brucellosis in other animals, these abortion events likely disseminate the bacteria broadly and allow for transmission to other livestock and to animal handlers. Not surprisingly, the disease is prevalent in Bedouin in Oman (Scrimgeour et al., 1999).
Several serological tests, such as Rose Bengal, tube and serum agglutination tests and ELISAs that have been optimized for testing cattle are used to determine Brucella seroprevalence in camels (Gwida et al., 2012), but epidemiological investigations often stop at this point. Furthermore, the lack of validated serological tests that detect Brucella infection in camels pose a challenge to definitive diagnosis. A combination of real-time PCR and serological tests provides a solution to many of the diagnostic challenges (Gwida et al., 2011). To determine the causative species, bacterial culturing from milk, blood or tissues of infected animals is performed to recover bacterial isolates. For B. melitensis, subsequent testing is required to distinguish the three biovars that are traditionally assessed in characterizing this species.
Brucellosis is a public health concern throughout the Greater Middle East (Pappas and Memish, 2007) and has been considered “hyperendemic” in Saudi Arabia with ~8,000 reported cases per annum (Memish and Mah, 2001). Comprehensive reporting of the disease throughout the region has been elusive due to limited public health infrastructure in many countries. Refai (2002) documented that brucellosis was ubiquitous throughout the Near East, with highest human incidence in Saudi Arabia, Iran, Syria, Jordan, and Oman. In fact, Western Asia contains among the highest incidence of brucellosis globally (Pappas et al., 2006). Human exposure to brucellosis from camels occurs primarily from contaminated milk (Shimol et al., 2012; Garcell et al., 2016). More broadly across other animal hosts, brucellosis infections most often occur in people working in close contact with animal tissues such as slaughterhouse workers, veterinarians, and farmers (Kaufmann et al., 1980; Whatmore, 2009). Determining the genetic diversity of B. melitensis in camels would provide valuable information about disease dispersal and transmission among camels, goats, and sheep in endemic areas, and particularly help better understand the role of camels in human infections.
As part of a brucellosis control program, we collected animal samples from routine surveillance activities. Tissues or bodily fluids were collected and isolates of B. melitensis were recovered from camels, goats, sheep, and cattle in the Dhofar governorate of Oman, an area with the highest brucellosis prevalence in the country (El Tahir and Nair, 2011). In 1996, a brucellosis control program that implemented both vaccination and culling of infected animals was initiated in the Dhofar region. As a part of an animal disease surveillance system, the regional Brucellosis Diagnostic Lab in Salalah, Dhofar identified all Brucella species that were involved in animal brucellosis cases. Because B. melitensis isolates from this region are not well characterized, we took two approaches to assess genetic relationships. First, we placed these isolates into a global phylogeny using single nucleotide polymorphism (SNP)-based genotyping assays specific to major evolutionary lineages of B. melitensis. We then genotyped a subset of these samples using multilocus variable number tandem repeats analysis (MLVA), following Huynh et al. (2008), to confirm their placement into well characterized clades. Our results suggest the genetic lineage of B. melitensis isolates in camels in Oman extends from North Africa into the Arabian Peninsula.
Materials and Methods
Aborted fetuses, placental membranes and vaginal swabs/discharge from aborted animals, and milk secretions and blood from suspected animal brucellosis cases were collected in the Dhofar governorate of southwestern Oman. Samples were handled under BSL-3 containment at the Animal Health Research Center, Ministry of Agriculture and Fisheries, Sultanate of Oman. Putative Brucella samples were inoculated on sheep blood agar plates together with Brucella selective supplement (SR 0083; Oxoid, Hampshire, UK) and 2.5% glucose. The plates were incubated at 37°C with (10%) or without CO2 for 7 days. Colonies were presumptively identified as Brucella by morphology and Gram staining and further biotyped using standard microbiological lab procedures (Alton et al., 1988; OIE, 2012). Brucella genus and species identification was confirmed by PCR (Hinic et al., 2008). Thirty-four isolates of B. melitensis were recovered from four animal species: camels (n = 15), goats (n = 8), cattle (n = 7), and sheep (n = 4) from 1997 to 2010 (Table 1). These samples are stored in the Brucella repository of the Animal Health Research Center. Twenty-hour individual broth cultures (~2 × 109/ml) were pelleted by centrifugation (7,500 rpm) for 10 min and genomic DNA was extracted and purified using Qiagen DNeasy Blood and Tissue Kits (Hilden, Germany) following the manufacturer's protocol for Gram-negative bacteria.
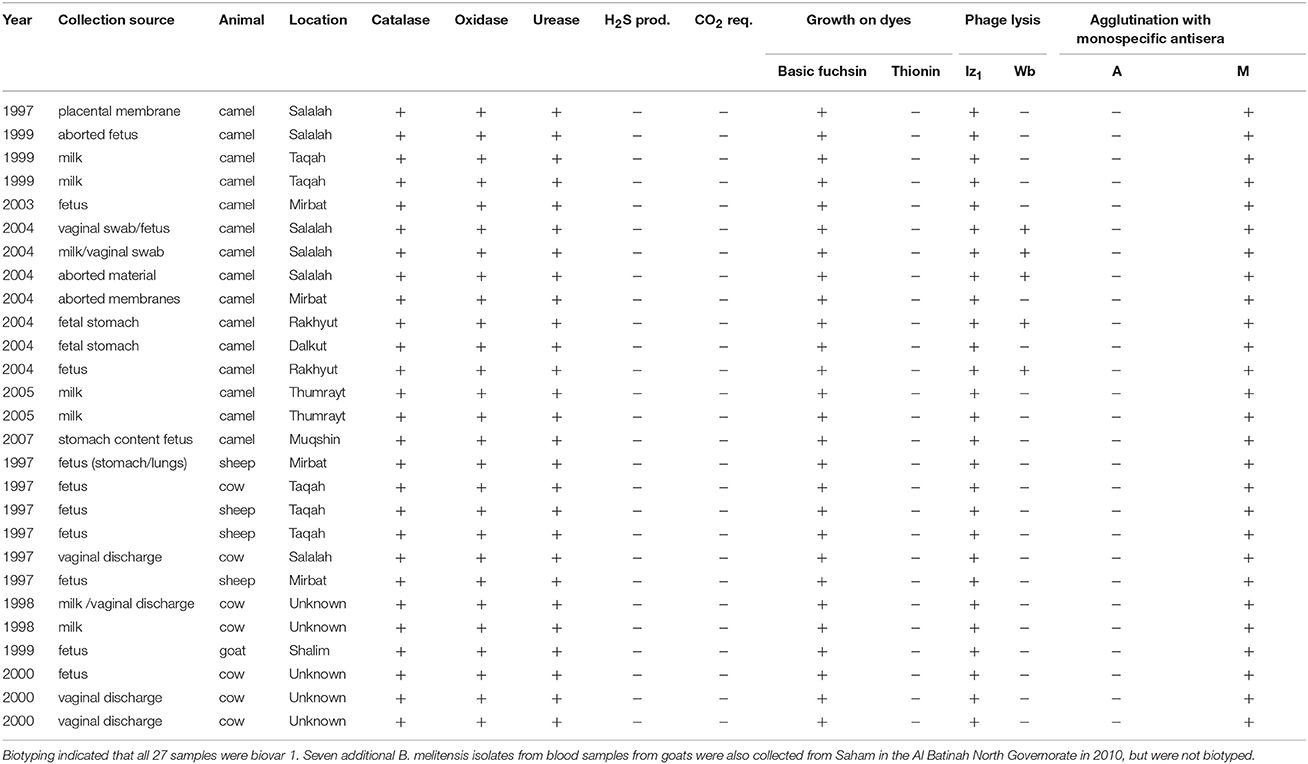
Table 1. Characteristics of 27 Brucella melitensis isolates collected from livestock in Dhofar governorate, Oman.
A sub-set of MLVA profiles derived from geographically diverse B. melitensis isolates causing human infection was generated at the U.S. Centers for Disease Control and Prevention (CDC) and used in the analysis to understand the distribution of B. melitensis genotypes in the region. Samples originating from Egypt with the naming designation starting with “E” were collected from an acute febrile illness surveillance study in Egypt as described by Afifi et al. (2005) and a MLVA study of this sub-set of isolates is described by Tiller et al. (2009). The remainder of the patient samples came from reference diagnostic specimens or were recovered from outbreak or support testing by the CDC. Samples were de-identified prior to analyses. Isolation and DNA extraction methods followed those detailed in Tiller et al. (2009). Three reference strains (16 M, 63/9, Rev-1) came from the CDC strain collection but their complete culture history is not known. These strains were MLVA genotyped using the actual isolates rather than using the genome sequence. All strains were minimally passaged after collection from human or animal sources. All culture and manipulation of Brucella isolates were conducted in BSL-3 conditions.
To develop a B. melitensis SNP genotyping assay, we conducted an in silico analysis of 29 B. melitensis genomes that were then available in GenBank and generated a phylogenetic tree using Northern Arizona SNP Pipeline (NASP) (Sahl et al., 2016; Figure 1). We used default parameters for SNP calling for each genome, which included a minimum of 10X coverage at a locus, at least 90% consensus for a SNP allele, and the requirement that the SNP locus was present in all genomes (i.e., no missing data).
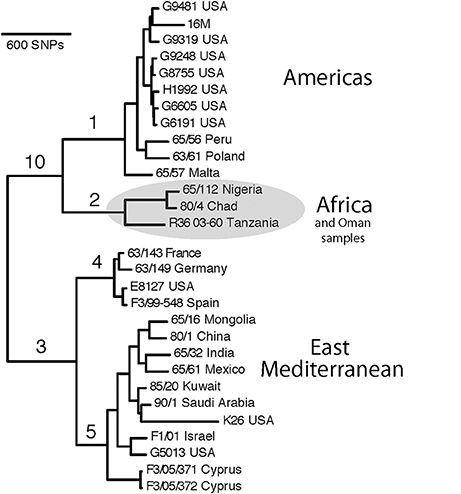
Figure 1. SNP-based phylogenetic tree of Brucella melitensis using 29 genomes available at time of assay development. Numbered branches correspond to SNP-based assays for each of the corresponding lineages. Note that the B. melitensis lineage known as the W. Mediterranean group (includes the Ether strain), is a basal group and is not shown in this tree. Country names indicate where the isolate was identified, and due to travel and sharing of strains among labs, this is not necessarily the country of origin.
Using these genome sequences, flanking regions were aligned using Sequencher 5.0, and Melt-MAMA (Mismatch Amplification Mutation Assays) were designed (Birdsell et al., 2012) targeting at least two randomly selected SNPs on each major branch (Table 2). Changes to primer base composition are detectable in melt-curve analysis, allowing differential fluorescence to determine the allele. Using DNA from the 34 isolates from Omani livestock, we performed Real-Time PCR with SYBR Green incorporation on an Applied Biosystems 7900HT Fast Real-Time PCR system. We ran 5 μL reactions with 1 μL DNA standardized to 2 ng/μL, when possible, and 4 μL of PCR reagents. Final concentration per reaction was 1X ABI SYBR Green Universal PCR Master Mix, and 0.15 μM each of the derived MAMA primer, ancestral MAMA primer, and reverse consensus primer. In practice this converts to 1.28 μL molecular grade water, 2.5 μL of 2X Universal PCR Master mix, and 0.08 μL each of the three primers at 10 μM each. In melt-MAMA reactions, the derived or ancestral primer containing the SNP allele more effectively amplifies in a competing reaction. Thermo-cycling conditions were as follows: initial UNG activation of 2 min at 50°C, a hot start of 10 min at 95°C, followed by 40 PCR cycles of 15 s at 95°C for denaturation and 1 min at 60°C for annealing, and a final stage of 15 s at 95°C, 15 s at 60°C, and 15 s at 95°C. Alleles were readily distinguished by the melting step and were confirmed with positive controls for each allele state (Birdsell et al., 2012). The SNP genotyping assays were run on all 34 Omani livestock B. melitensis samples.
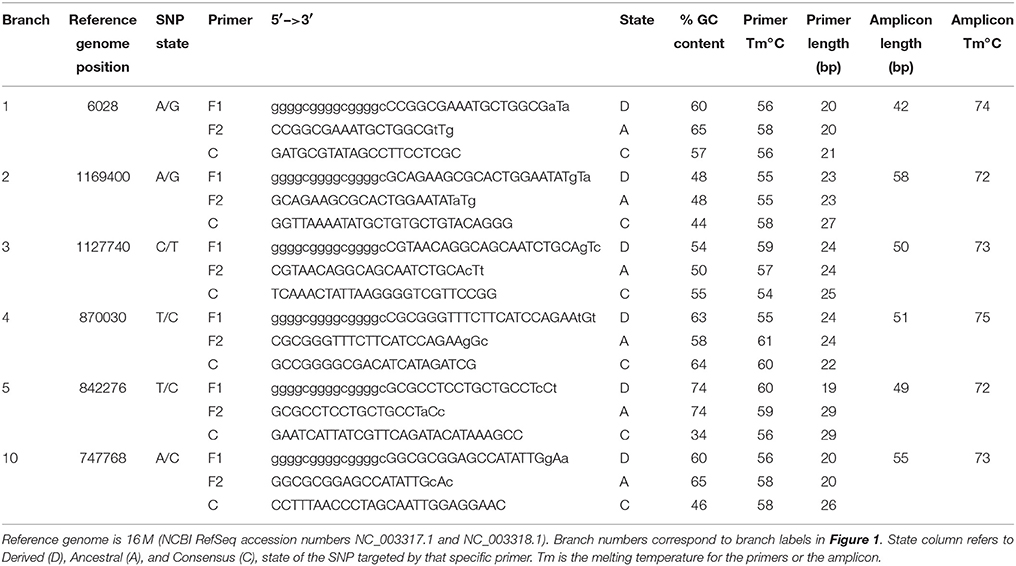
Table 2. Assay design parameters for primer sets targeting 6 major branches of Brucella melitensis.
We also performed MLVA on the 15 camel isolates following the methods of Huynh et al. (2008). Briefly, this is a 15 locus VNTR panel that uses four multiplex PCRs with fluorescently labeled forward primers (6-FAM, NED, PET, or VIC) and unlabeled reverse primers. Fragment sizes for each locus were visualized by capillary electrophoresis on an ABI 3130 Genetic Analyzer and converted to a repeat number corresponding to each fragment size using the LIZ 1200 size standard in GeneMapper version 4.0. As detailed by Tiller et al. (2009), the MLVA approach we used compares favorably to the more widely used MLVA approach (Le Fleche et al., 2006), although the latter has an expansive database of MLVA genotypes (http://mlva.u-psud.fr/brucella/).
Results
In this work, we used SNPs discovered in whole genome comparisons to develop multiple SNP assays that distinguish six major branches in B. melitensis. We present details on the assays that provided the greatest peak separation for SNPs found on each branch (Table 2). Of particular interest to this study was the phylogenetic placement of isolates from Omani livestock; all 34 of these Omani isolates are part of the clade on the assay branch 2 (Figure 1). The finding that Omani livestock samples all came from a distinct branch in the B. melitensis phylogeny indicates that these animals all contain relatively closely related isolates from a single lineage.
SNP analyses indicate that all of our B. melitensis isolates from Omani camels belong to a distinct clade that also contains isolates originating in Africa (Nigeria, Chad, Tanzania). This key finding was unexpected, as one would predict that isolates from the Arabian Peninsula would be part of the E. Mediterranean lineage that predominates the region (Gyuranecz et al., 2016). Thus, SNP analysis indicates that Omani livestock isolates are part of a group of B. melitensis that is related to isolates from Africa and distantly related to most other isolates from the Middle East. Moreover, it suggests that the lineages of B. melitensis in Egypt and probably throughout much of N. Africa are distinct from most strains from the rest of the Middle East. Our SNP assays also identify two major groups within the E. Mediterranean clade, branches 4 and 5, suggesting substructure within this group. Interestingly, the African clade isolates are more closely related to isolates from the Americas clade than the E. Mediterranean clade in this rooted tree, a pattern not seen in MLVA (Gyuranecz et al., 2016), likely due to the higher resolution of whole genome sequencing.
MLVA results were compared to our database of MLVA genotypes from the Middle East (Huynh et al., 2008; Tiller et al., 2009). We utilized all MLVA genotypes from the African clade and a representative sample of genotypes from the E. Mediterranean and Americas clades (Table 3). A fourth lineage, W. Mediterranean, is not part of our collection, nor was it detected in our sampling so was not included in our phylogeny. The W. Mediterranean clade is largely limited to Italy (Garofolo et al., 2013) but does occur in other countries, including along the Mediterranean (Lounes et al., 2014). Low DNA quality prevented us from running MLVA on the 12 other animal isolates from Oman but this DNA was still of sufficient quality to SNP-based analysis on Real-Time PCR due to the smaller amplicons sizes and higher sensitivity of these Real-Time assays (Birdsell et al., 2012). The 10 most stable of the 15 Variable Number Tandem Repeat (VNTR) loci were used in our analyses (VNTRs: 1, 3, 7, 14, 16, 20, 21, 25 27, 28). Limiting VNTR loci to the most stable markers, or placing greater weighting on these markers, is a common practice in Brucella MLVA (e.g., Al Dahouk et al., 2007a). Such a practice reduces homoplasy and allows for understanding deeper phylogenetic relationships but potentially sacrifices more recent epidemiological connections (Keim et al., 2004); this potential loss of resolution was not a concern for our study because we were focused on these deeper connections.
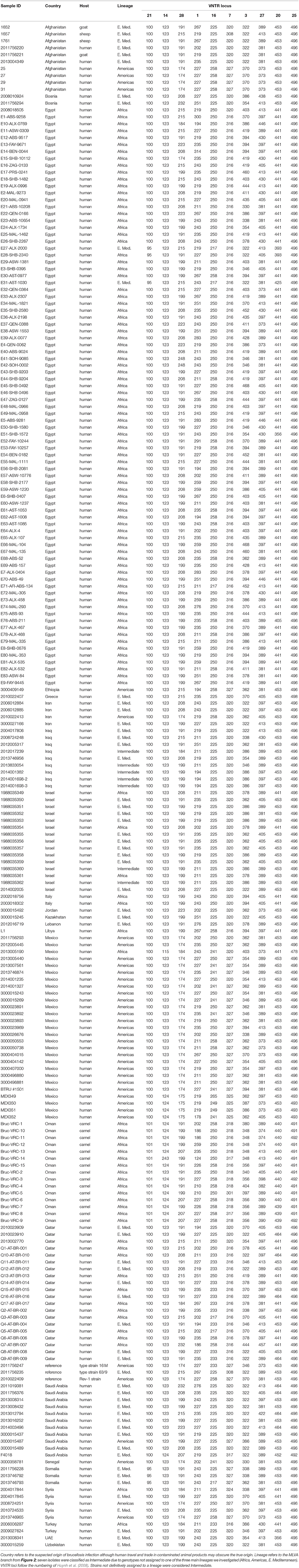
Table 3. MLVA-10 genotypes for 221 isolates of Brucella melitensis from the Middle East. Isolates from additional regions added for context.
Three distinct lineages of B. melitensis were found in our sampling, consistent with geographic groupings of Americas, E. Mediterranean, and Africa clades. Al Dahouk et al. (2007a) first identified the Americas and E. Mediterranean clades but used a different MLVA genotyping scheme (Le Fleche et al., 2006). Subsequent work by Gyuranecz et al. (2016), identified another lineage, the Africa clade. This consistency between the two MLVA schemes, as well as support from whole genome analyses, suggests that either scheme is capable of identifying these major lineages although differentiation of the Africa clade is more pronounced with the Huynh et al. (2008) MLVA scheme. VNTR genotypes were compared using minimum spanning trees in Phyloviz 2.0 (Nascimento et al., 2017).
Using MLVA genotyping, the VNTR diversity we observed in these 15 camel isolates divide into 3–6 distinct lineages, depending on clustering criteria (Figure 2). Use of all 15 MLVA loci gave similar overall patterns (data not shown). All camel isolates from Oman, except for one highly similar to a human isolate from Qatar (Q7), are closely related to samples from human patients from Egypt, typically differing from each other by only 1–3 VNTR mutations; indicating that highly related strains circulating across the region in both livestock and humans. Our MLVA results also support the SNP analyses that the Omani camel isolates have greatest similarity to B. melitensis isolates from North Africa. Omani camel isolates are related to isolates originating from humans in Egypt, sometimes separated by only one VNTR repeat. Epidemiological trace back may allow for identification of these potential animal to human transmission events. We also note the presence of African lineage isolates in other countries in the region, including Libya, Israel, Syria, Turkey, and Qatar.
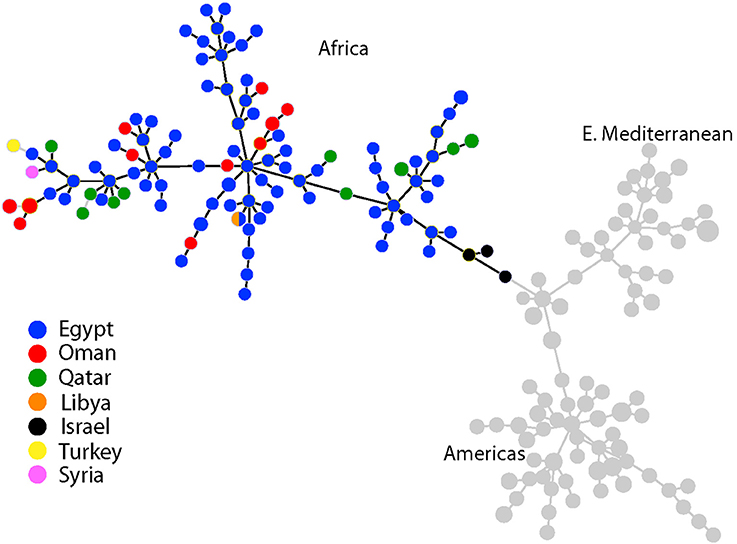
Figure 2. Minimum spanning tree of 221 Brucella melitensis isolates based on MLVA-10 genotyping. Country of origin for isolates from the Middle East is represented by the colored circles. Additional isolate details are found in Table 3.
Discussion
Our study highlights the presence of a unique lineage of B. melitensis in the Middle East, provides assays to quickly identify the strain types circulating and causing animal and human brucellosis in the region, and suggests that substantial diversity remains to be uncovered in Africa. The SNP assays we designed, particularly those for branch 2, allow for rapid identification of African lineage strains and thus can be used to focus on finding new genetic variation among this poorly sampled group. When samples are identified as part of the African lineage, MLVA can then be utilized for higher resolution analyses, such as understanding the relationships among African isolates and for epidemiological investigations.
SNP Genotyping
The SNP-based results from the whole genome analyses and SNP genotyping support the finding that an African lineage of B. melitensis exists in the Arabian Peninsula (e.g., Oman and Qatar), and other nearby countries such as Israel, Turkey, and Syria. Although the African lineage appears to be uncommon elsewhere, it nonetheless has been introduced into many Middle Eastern countries. The two SNP assays specific for branch 2 can quickly identify isolates that are part of this unique African lineage, allowing for a determination of the spread of this clade.
We emphasize that the African clade is a phylogenetic assignment and not a geographic one, and that not all isolates from Africa will be a part of this clade. Accordingly, isolates from Algeria in western North Africa were predominantly from the W. Mediterranean clade (Lounes et al., 2014), suggesting a connection to Europe (especially Italy) rather than the rest of North Africa or other parts of the Middle East. Our SNP findings, and those of Georgi et al. (2017), highlight the power of whole genome analyses and this genomic approach is able to clearly distinguish isolates from the African and E. Mediterranean clades as evolutionarily distinct lineages.
MLVA Genotyping
The MLVA groupings show broad and consistent geographic representations of MLVA types from the Americas, E. Mediterranean clade (consisting primarily of West/Central Asia and Middle Eastern isolates), and Africa. Although this method uses different loci than SNP approaches, these groupings match those from the SNP-based genotyping. Using the MLVA method of Le Fleche et al. (2006), B. melitensis isolates from the United Arab Emirates from camels, cattle, and a goat had genotypes from the E. Mediterranean clade but also contained isolates from four camels and a gazelle from the African clade (Gyuranecz et al., 2016). Indeed, the expectation for B. melitensis isolates collected from humans or livestock from the Arabian Peninsula and most of the Middle East is membership in the E. Mediterranean clade (Al Dahouk et al., 2007a; Kattar et al., 2008; Kilic et al., 2011). MLVA and whole genome analyses indicate that the E. Mediterranean clade is the principal lineage in the region, continuing from the Middle East along the Mediterranean and into and throughout Asia (Jiang et al., 2011; Tan et al., 2015; Tay et al., 2015). Whole genome comparisons from brucellosis patients infected in the Middle East support this predominance of the E. Mediterranean clade but also identify three isolates of Somali origin from an Africa clade (Georgi et al., 2017). Despite its broad sampling and thousands of genotypes for B. melitensis, the MLVA database (http://mlva.u-psud.fr/brucella/) contains few representatives from North Africa so it appears to be currently missing much of this genetic diversity. We encourage researchers using the Le Fleche MLVA scheme to genotype samples from this region for a better global understanding of this lineage, after first using our SNP-based assays to identify this clade.
Resolving Apparent Inconsistencies
Our findings challenge common approaches used in Brucella epidemiology. While we detail our results from traditional phenotyping approaches, these data are typically not informative for understanding the genetic relationships of isolates within B. melitensis. For example, distinguishing the three biovars of B. melitensis provides limited resolution when attempting to establish epidemiological links between outbreaks; likely due to the limited association between genotype and phenotype (biovar) in this species (Whatmore et al., 2016). In fact, isolates from biovars 1, 2, and 3 can be closely related genetically (e.g., Jiang et al., 2011) and isolates from the same biovar can be distantly related and from evolutionarily distinct clades (e.g., De Massis et al., 2015). Apparent discrepancies also occur for the country of origin for some isolates; a handful of SNP-based and MLVA genotypes appear to not match the expected clade corresponding to the region. For example, in the SNP phylogeny an isolate from Poland is in the Americas clade, and samples from the USA are in the E. Mediterranean clade. Such discordance is expected. First, as mentioned previously these groupings are phylogenetic assignments and should not be misconstrued as geographic origins. In addition, epidemiological histories of various strains are not always precisely known and the country names may indicate where the isolate was identified and not necessarily the country of origin. Finally, international travel of patients infected with brucellosis or movement of infected animals may potentially obscure the country of origin unless detailed epidemiological data are available (Al Dahouk et al., 2007b; Garofolo et al., 2013; De Massis et al., 2015; Georgi et al., 2017).
Brucellosis Management
Disease management approaches involving vaccination, treatment with antibiotics, and test-and-slaughter programs for camels can lower disease incidence and even eliminate brucellosis from some farms (Radwan et al., 1995; Abbas and Agab, 2002; Wernery, 2014). Nonetheless, comprehensive elimination of B. melitensis in camels from larger regions will require brucellosis management in goats and sheep. Despite calls for the elimination of brucellosis from the Arabian Peninsula for over two decades (Tabbara, 1993), the disease persists throughout the region and indeed continues to be a successful pathogen throughout much of world. High prevalence rates in sheep, goats and cattle, and the hundreds of millions of these animals in the region make brucellosis elimination difficult (Refai, 2002). Although the economic cost of brucellosis to livestock production in Oman is not well-known, globally the economic burden on both animal and human health is significant and far reaching (Seleem et al., 2010). Understanding historic and contemporary movement of animals infected with brucellosis is an important step in improving disease management strategies.
Brucellosis Movement
The presence of an African lineage from North Africa in the Arabian Peninsula indicates the interconnectedness of livestock in the Greater Middle East, likely due to historical trade and movement between the two regions, although the E. Mediterranean lineage still predominates. Contemporary introductions of B. melitensis also appear to occur—the four camels with the African clade genotypes in the United Arab Emirates came from Sudan (Gyuranecz et al., 2016)—connectedness previously suggested by Wernery (2014). Our results also indicate that not all B. melitensis isolates from Africa are part of this “Africa” lineage. For example, some samples from Somalia, Kenya, and Egypt appear to be part of the E. Mediterranean clade and Ethiopian and some Egyptian samples are part of the Americas clade. Moreover, Lounes et al. (2014) identified isolates from the W. Mediterranean clade in the Maghreb region of Algeria. Even less is known from sub-Saharan Africa, where brucellosis is widespread among livestock (Mcdermott and Arimi, 2002). Clearly, there is tremendous diversity of B. melitensis in Africa, potentially due to many introductions of infected livestock from many regions as well as widespread trade. Additional sampling throughout the African continent is needed to better understand the evolutionary history regarding the origins and spread of B. melitensis in the region. Although the African lineage may be more localized than the three widely distributed B. melitensis clades, substantial undiscovered diversity likely exists, and as we have discovered in Oman, extends beyond the African continent into new lands that have been connected by trade in camels, goats, and sheep.
Author Contributions
JF, RT, AH, PK, and MS designed the study. MH, AA-R, RT, and MS collected and cultured the samples. JF, FW, BR, KD, and RT analyzed the data. All authors contributed to writing and editing the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
Funding was provided by the U.S. Department of Homeland Security to PK and JF (HSHQDC-10-C-00139) and by the Agriculture and Fisheries Development Fund (AFDF) of Ministry of Agriculture and Fisheries, Oman. The findings and conclusions in this report are those of the authors and do not necessarily represent the official position of the Centers for Disease Control and Prevention.
References
Abbas, B., and Agab, H. (2002). A review of camel brucellosis. Prev. Vet. Med. 55, 47–56. doi: 10.1016/S0167-5877(02)00055-7
Afifi, S., Earhart, K., Azab, M. A., Youssef, F. G., El Sakka, H., Wasfy, M., et al. (2005). Hospital-based surveillance for acute febrile illness in Egypt: a focus on community-acquired bloodstream infections. Am. J. Trop. Med. Hygiene 73, 392–399. doi: 10.4269/ajtmh.2005.73.392
Al Dahouk, S., Le Fleche, P., Nockler, K., Jacques, I., Grayon, M., Scholz, H. C., et al. (2007a). Evaluation of Brucella MLVA typing for human brucellosis. J. Microbiol. Methods 69, 137–145. doi: 10.1016/j.mimet.2006.12.015
Al Dahouk, S., Neubauer, H., Hensel, A., Schoneberg, I., Nockler, K., Alpers, K., et al. (2007b). Changing epidemiology of human brucellosis, Germany, 1962–2005. Emerg. Infect. Dis. 13, 1895–1900. doi: 10.3201/eid1312.070527
Alton, G. G., Jones, L. M., Angus, R. D., and Verger, J. M. (1988). “Bacteriological methods,” in Techniques for the Brucellosis Laboratory (Paris: Institut National de la Recherche Agronomique), 13–61.
Birdsell, D. N., Pearson, T., Price, E. P., Hornstra, H. M., Nera, R. D., Stone, N., et al. (2012). Melt analysis of mismatch amplification mutation assays (Melt-MAMA): a functional study of a cost-effective SNP genotyping assay in bacterial models. PLoS ONE 7:e32866. doi: 10.1371/journal.pone.0032866
De Massis, F., Ancora, M., Atzeni, M., Rolesu, S., Bandino, E., Danzetta, M. L., et al. (2015). MLVA as an epidemiological tool to trace back Brucella melitensis biovar 1 re-emergence in Italy. Transbound. Emerg. Dis. 62, 463–469. doi: 10.1111/tbed.12397
El Tahir, Y. E. H., and Nair, R. R. (2011). Prevalence of brucellosis in the sultanate of Oman with reference to some Middle-East countries. Vet. Res. 4, 71–76. doi: 10.3923/vr.2011.71.76
Garcell, H. G., Garcia, E. G., Pueyo, P. V., Martín, I. R., Arias, A. V., and Alfonso Serrano, R. N. (2016). Outbreaks of brucellosis related to the consumption of unpasteurized camel milk. J. Infect. Public Health 9, 523–527. doi: 10.1016/j.jiph.2015.12.006
Garofolo, G., Di Giannatale, E., De Massis, F., Zilli, K., Ancora, M., Camma, C., et al. (2013). Investigating genetic diversity of Brucella abortus and Brucella melitensis in Italy with MLVA-16. Infect. Genetics. Evol. 19, 59–70. doi: 10.1016/j.meegid.2013.06.021
Georgi, E., Walter, M. C., Pfalzgraf, M.-T., Northoff, B. H., Holdt, L. M., Scholz, H. C., et al. (2017). Whole genome sequencing of Brucella melitensis isolated from 57 patients in Germany reveals high diversity in strains from Middle East. PLoS ONE 12:e0175425. doi: 10.1371/journal.pone.0175425
Gwida, M., El-Gohary, A., Melzer, F., Khan, I., Rosler, U., and Neubauer, H. (2012). Brucellosis in camels. Res. Vet. Sci. 92, 351–355. doi: 10.1016/j.rvsc.2011.05.002
Gwida, M. M., El-Gohary, A. H., Melzer, F., Tomaso, H., Rosler, U., Wernery, U., et al. (2011). Comparison of diagnostic tests for the detection of Brucella spp. in camel sera. BMC Res. Notes 4:525. doi: 10.1186/1756-0500-4-525
Gyuranecz, M., Wernery, U., Kreizinger, Z., Juhász, J., Felde, O., and Nagy, P. (2016). Genotyping of Brucella melitensis strains from dromedary camels (Camelus dromedarius) from the United Arab Emirates with multiple-locus variable-number tandem repeat analysis. Vet. Microbiol. 186, 8–12. doi: 10.1016/j.vetmic.2016.02.009
Hinic, V., Brodard, I., Thomann, A., Cvetnic, Z., Makaya, P. V., Frey, J., et al. (2008). Novel identification and differentiation of Brucella melitensis, B. abortus, B. suis, B. ovis, B. canis, and B. neotomae suitable for both conventional and real-time PCR systems. J. Microbiol. Methods 75, 375–378. doi: 10.1016/j.mimet.2008.07.002
Huynh, L. Y., Van Ert, M. N., Hadfield, T., Probert, W. S., Bellaire, B. H., Dobson, M., et al. (2008). “Multiple locus variable number tandem repeat (VNTR) analysis (MLVA) of Brucella spp. identifies species-specific markers and provides insights into phylogenetic relationships,” in NIH: Frontiers in Research, ed V. St. Georgiev (Totowa, NJ: Humana Press), 47–54.
Jiang, H., Fan, M., Chen, J., Mi, J., Yu, R., Zhao, H., et al. (2011). MLVA genotyping of Chinese human Brucella melitensis biovar 1, 2 and 3 isolates. BMC Microbiol. 11:256. doi: 10.1186/1471-2180-11-256
Kattar, M. M., Jaafar, R. F., Araj, G. F., Le Fleche, P., Matar, M. G., Rached, R. A., et al. (2008). Evaluation of a multilocus variable-number tandem-repeat analysis scheme for typing human Brucella isolates in a region of brucellosis endemicity. J. Clin. Microbiol. 45, 3935–3940. doi: 10.1128/JCM.00464-08
Kaufmann, A. F., Fox, M. D., Boyce, J. M., Anderson, D. C., Potter, M. E., Martone, W. J., et al. (1980). Airborne spread of brucellosis. An. N.Y. Acad. Sci. 353, 105–114. doi: 10.1111/j.1749-6632.1980.tb18912.x
Keim, P., Van Ert, M. N., Pearson, T., Vogler, A. J., Huynh, L. Y., and Wagner, D. M. (2004). Anthrax molecular epidemiology and forensics: using the appropriate marker for different evolutionary scales. Infect. Genetics Evol. 4, 205–213. doi: 10.1016/j.meegid.2004.02.005
Kilic, S., Ivanov, I. N., Durmaz, R., Bayraktar, M. R., Ayaslioglu, E., Uyanik, M. H., et al. (2011). Multiple-locus variable-number tandem-repeat analysis genotyping of human Brucella isolates from Turkey. J. Clin. Microbiol. 49, 3276–3283. doi: 10.1128/JCM.02538-10
Le Fleche, P., Jacques, I., Grayon, M., Al Dahouk, S., Bouchon, P., Denoeud, F., et al. (2006). Evaluation and selection of tandem repeat loci for a Brucella MLVA typing assay. BMC Microbiol. 6:9. doi: 10.1186/1471-2180-6-9
Lounes, N., Cherfa, M.-A., Le Carrou, G., Bouyoucef, A., Jay, M., Garin-Bastuji, B., et al. (2014). Human brucellosis in Maghreb: existence of a lineage related to socio-historical connections with Europe. PLoS ONE 9:e115319. doi: 10.1371/journal.pone.0115319
Mcdermott, J. J., and Arimi, S. M. (2002). Brucellosis in sub-Saharan Africa: epidemiology, control and impact. Vet. Microbiol. 90, 111–134. doi: 10.1016/S0378-1135(02)00249-3
Memish, Z. A., and Mah, M. W. (2001). Brucellosis in laboratory workers at a Saudi Arabian hospital. Am. J. Infect. Control 29, 48–52. doi: 10.1067/mic.2001.111374
Moreno, E. (2014). Retrospective and prospective perspectives on zoonotic brucellosis. Front. Microbiol. 5:213. doi: 10.3389/fmicb.2014.00213
Musa, M. T., Eisa, M. Z., El Sanousi, E. M., Abdel Wahab, M. B., and Perrett, L. (2008). Brucellosis in camels (Camelus dromedarius) in Darfur, Western Sudan. J. Comp. Pathol. 138, 151–155. doi: 10.1016/j.jcpa.2007.10.005
Narnaware, S. D., Dahiya, S. S., Kumar, S., Tuteja, F. C., Nath, K., and Patil, N. V. (2017). Pathological and diagnostic investigations of abortions and neonatal mortality associated with natural infection of Brucella abortus in dromedary camels. Comp. Clin. Path. 26, 79–85. doi: 10.1007/s00580-016-2348-4
Nascimento, M., Sousa, A., Ramirez, M., Francisco, A. P., Carriço, J. A., and Vaz, C. (2017). PHYLOViZ 2.0: Providing scalable data integration and visualization for multiple phylogenetic inference methods. Bioinformatics 33, 128–129. doi: 10.1093/bioinformatics/btw582
OIE, A. H. S. (2012). Manual of Diagnostic Tests and Vaccines for Terrestrial Animals, Chapter 2.4.3 Bovine brucellosis. Paris: Office International des Epizooties.
Pappas, G., and Memish, Z. A. (2007). Brucellosis in the Middle East: a persistent medical, socioeconomic and political issue. J. Chemother. 19, 243–248. doi: 10.1179/joc.2007.19.3.243
Pappas, G., Papadimitriou, P., Akritidis, N., Christou, L., and Tsianos, E. V. (2006). The new global map of human brucellosis. Lancet Infect. Dis. 6, 91–99. doi: 10.1016/S1473-3099(06)70382-6
Radwan, A. I., Bekairi, S. I., Mukayel, A. A., Albokmy, A. M., Prasad, P. V. S., Azar, F. N., et al. (1995). Control of Brucella melitensis infection in a large camel herd in Saudi Arabia using antibiotherapy and vaccination with Rev. 1 vaccine. Rev. Sci. Tech. 14, 719–732. doi: 10.20506/rst.14.3.860
Refai, M. (2002). Incidence and control of brucellosis in the Near East region. Vet. Microbiol. 90, 81–110. doi: 10.1016/S0378-1135(02)00248-1
Sahl, J. W., Lemmer, D., Travis, J., Schupp, J., Gillece, J., Aziz, M., et al. (2016). The Northern Arizona SNP Pipeline (NASP): accurate, flexible, and rapid identification of SNPs in WGS datasets. bioRxiv. doi: 10.1101/037267
Scrimgeour, E. M., Mehta, F. R., and Suleiman, A. J. (1999). Infectious and tropical diseases in Oman: a review. Am. J. Trop. Med. Hyg. 61, 920–925. doi: 10.4269/ajtmh.1999.61.920
Seleem, M. N., Boyle, S. M., and Sriranganathan, N. (2010). Brucellosis: a re-emerging zoonosis. Vet. Microbiol. 140, 392–398. doi: 10.1016/j.vetmic.2009.06.021
Shimol, S. B., Dukhan, L., Belmaker, I., Bardenstein, S., Sibirsky, D., Barrett, C., et al. (2012). Human brucellosis outbreak acquired through camel milk ingestion in southern Israel. Isr. Med. Assoc. J. 14, 475–478.
Sprague, L. D., Al-Dahouk, S., and Neubauer, H. (2012). A review on camel brucellosis: a zoonosis sustained by ignorance and indifference. Pathog. Glob. Health 106, 144–149. doi: 10.1179/2047773212Y.0000000020
Tabbara, K. F. (1993). Brucellosis: a model for eradication of endemic diseases from the Arabian peninsula. Ann. Saudi Med. 13, 1–2. doi: 10.5144/0256-4947.1993.1
Tan, K.-K., Tan, Y.-C., Chang, L.-Y., Lee, K. W., Nore, S. S., Yee, W. -Y., et al. (2015). Full genome SNP-based phylogenetic analysis reveals the origin and global spread of Brucella melitensis. BMC Genomics 16:93. doi: 10.1186/s12864-015-1294-x
Tay, B. Y., Ahmad, N., Hashim, R., Mohamed Zahidi, J. A., Thong, K. L., Koh, X. P., et al. (2015). Multiple-locus variable-number tandem-repeat analysis (MLVA) genotyping of human Brucella isolates in Malaysia. BMC Infect. Dis. 15:220. doi: 10.1186/s12879-015-0958-0
Tiller, R. V., De, B. K., Boshra, M., Huynh, L. Y., Van Ert, M. N., Wagner, D. M., et al. (2009). Comparison of two multiple-locus variable-number tandem-repeat analysis methods for molecular strain typing of human Brucella melitensis isolates from the Middle East. J. Clin. Microbiol. 47, 2226–2231. doi: 10.1128/JCM.02362-08
Vassallo, D. J. (1996). The saga of brucellosis: controversy over credit for linking Malta fever with goats' milk. Lancet 348, 804–808. doi: 10.1016/S0140-6736(96)05470-0
Wernery, U. (2014). Camelid brucellosis: a review. Rev. Sci. Tech. 33, 839–857. doi: 10.20506/rst.33.3.2322
Whatmore, A. M. (2009). Current understanding of the genetic diversity of Brucella, an expanding genus of zoonotic pathogens. Infect. Genet. Evol. 9, 1168–1184. doi: 10.1016/j.meegid.2009.07.001
Whatmore, A. M., Koylass, M. S., Muchowski, J., Edwards-Smallbone, J., Gopaul, K. K., and Perrett, L. L. (2016). Extended multilocus sequence analysis to describe the global population structure of the genus Brucella: phylogeography and relationship to biovars. Front. Microbiol. 7:2049. doi: 10.3389/fmicb.2016.02049
Keywords: brucellosis, Brucella melitensis, camels, Oman, MLVA, SNP genotyping
Citation: Foster JT, Walker FM, Rannals BD, Hussain MH, Drees KP, Tiller RV, Hoffmaster AR, Al-Rawahi A, Keim P and Saqib M (2018) African Lineage Brucella melitensis Isolates from Omani Livestock. Front. Microbiol. 8:2702. doi: 10.3389/fmicb.2017.02702
Received: 22 June 2017; Accepted: 29 December 2017;
Published: 15 January 2018.
Edited by:
Axel Cloeckaert, Institut National de la Recherche Agronomique (INRA), FranceReviewed by:
Thomas A. Ficht, Texas A&M University College Station, United StatesMenachem Banai, Kimron Veterinary Institute, Israel
Copyright © 2018 Foster, Walker, Rannals, Hussain, Drees, Tiller, Hoffmaster, Al-Rawahi, Keim and Saqib. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey T. Foster, amVmZi5mb3N0ZXJAbmF1LmVkdQ==
Muhammad Saqib, ZHJzYXFpYl92ZXRAaG90bWFpbC5jb20=