 Koo Nagasawa1,2†Yuki Matsushima3†Takumi Motoya4
Koo Nagasawa1,2†Yuki Matsushima3†Takumi Motoya4 Fuminori Mizukoshi5Yo Ueki6
Fuminori Mizukoshi5Yo Ueki6 Naomi Sakon7
Naomi Sakon7 Koichi Murakami1Tomomi Shimizu3Nobuhiko Okabe3Noriko Nagata4
Koichi Murakami1Tomomi Shimizu3Nobuhiko Okabe3Noriko Nagata4 Komei Shirabe8
Komei Shirabe8 Hiroto Shinomiya9Wataru Suzuki10
Hiroto Shinomiya9Wataru Suzuki10 Makoto Kuroda11
Makoto Kuroda11 Tsuyoshi Sekizuka11
Tsuyoshi Sekizuka11 Yoshiyuki Suzuki12
Yoshiyuki Suzuki12 Akihide Ryo13Kiyotaka Fujita14Kazunori Oishi1
Akihide Ryo13Kiyotaka Fujita14Kazunori Oishi1 Kazuhiko Katayama15*
Kazuhiko Katayama15* Hirokazu Kimura1,13,14*
Hirokazu Kimura1,13,14*- 1Infectious Disease Surveillance Center, National Institute of Infectious Diseases, Musashimurayama, Japan
- 2Department of Pediatrics, Graduate School of Medicine, Chiba University, Chiba, Japan
- 3Division of Virology, Kawasaki City Institute for Public Health, Kawasaki, Japan
- 4Division of Virology, Ibaraki Prefectural Institute of Public Health, Mito, Japan
- 5Department of Microbiology, Tochigi Prefectural Institute of Public Health and Environmental Science, Utsunomiya, Japan
- 6Division of Virology, Department of Microbiology, Miyagi Prefectural Institute of Public Health and Environment, Sendai, Japan
- 7Department of Microbiology, Osaka Institute of Public Health, Osaka, Japan
- 8Yamaguchi Prefectural Institute of Public Health and Environment, Yamaguchi, Japan
- 9Department of Microbiology, Ehime Prefectural Institute of Public Health and Environmental Science, Matsuyama, Japan
- 10Eiken Chemical Co., Ltd., Biochemical Research Laboratory I Department-I, Shimotsuga, Japan
- 11Pathogen Genomics Center, National Institute of Infectious Diseases, Shinjuku, Japan
- 12Division of Biological Science, Department of Information and Basic Science, Graduate School of Natural Sciences, Nagoya City University, Nagoya, Japan
- 13Department of Microbiology, Yokohama City University School of Medicine, Yokohama, Japan
- 14School of Medical Technology, Faculty of Health Science, Gunma Paz University, Takasaki, Japan
- 15Laboratory of Viral Infection I, Kitasato Institute for Life Sciences, Graduate School of Infection Control Sciences, Kitasato University, Minato, Japan
In the 2016/2017 winter season in Japan, HuNoV GII.P16-GII.2 strains (2016 strains) emerged and caused large outbreaks of acute gastroenteritis. To better understand the outbreaks, we examined the molecular evolution of the VP1 gene and RdRp region in 2016 strains from patients by studying their time-scale evolutionary phylogeny, positive/negative selection, conformational epitopes, and phylodynamics. The time-scale phylogeny suggested that the common ancestors of the 2016 strains VP1 gene and RdRp region diverged in 2006 and 1999, respectively, and that the 2016 strain was the progeny of a pre-2016 GII.2. The evolutionary rates of the VP1 gene and RdRp region were around 10-3 substitutions/site/year. Amino acid substitutions (position 341) in an epitope in the P2 domain of 2016 strains were not found in pre-2016 GII.2 strains. Bayesian skyline plot analyses showed that the effective population size of the VP1 gene in GII.2 strains was almost constant for those 50 years, although the number of patients with NoV GII.2 increased in 2016. The 2016 strain may be involved in future outbreaks in Japan and elsewhere.
Introduction
Human norovirus (HuNoV) is a common cause of acute gastroenteritis in humans (Matson, 2004). HuNoV is classified into two genogroups (GI and GII), including many genotypes (Vinjé, 2015). Of GII’s confirmed 22 genotypes (GII.1-GII.22) (Vinjé, 2015), GII.4 was a major cause of pandemics in 2006–2012 (Siebenga et al., 2007; Eden et al., 2014). Furthermore, other some genotypes, including GII.2, GII.3, GII.6, and GII.17, were detected worldwide, including Japan (Thongprachum et al., 2017).
During the 2016–2017 winter season in Japan, GII.P16-GII.2 strains (2016 strains) suddenly appeared and caused large outbreaks of gastroenteritis in Japanese children (Thongprachum et al., 2017). The Japanese national surveillance system reported in Infectious Diseases Weekly Report (IDWR) that the outbreaks of acute gastroenteritis in children in this season were the second largest number of patients in the past 11 seasons [National Institute of Infectious Diseases, Japan, Infectious Gastroenteritis (cited 2017 February 16, in Japanese)1]. Similar findings were reported from Germany, France, China, and United States (Bidalot et al., 2017; Lu et al., 2017; Niendorf et al., 2017; Tohma et al., 2017).
To better understand the genetics of the 2016 strains, we completed a detailed analysis of the full length of the RNA-dependent RNA polymerase (RdRp) region and capsid (VP1) gene of the GII.P16-GII.2 strains detected in Japan during 2016–2017 winter season.
Materials and Methods
Samples and Patients
In the present study, we analyzed 19 strains of the 2016 strains. Samples were collected from patients (children and adults) with acute gastroenteritis (9.2 years ± 12.4, mean ± standard deviation) from October to December in 2016. The samples were obtained from clinics or hospitals, such as the sentinel surveillance medical institutions in Ibaraki prefecture and Kawasaki city in Japan. Written informed consent was obtained from the patients or their guardian for the donation of samples. The study protocols were approved by National Institute of Infectious Diseases for Public Health Ethics Committees (No. 576).
RNA Extraction, Reverse Transcription Polymerase Chain Reaction, and Sequencing
Viral RNA was extracted from samples with a QIAamp Viral RNA Mini kit (Qiagen). Reverse transcription–polymerase chain reaction (RT-PCR) was performed using a PrimeScript II High Fidelity One Step RT-PCR Kit (TaKaRa). Using primer walking methods, we analyzed the complete RdRp region and VP1 gene. The primers were designed by the PrimaClade server (Supplementary Table S1) (Gadberry et al., 2005). After the PCR products were purified using a MinElute PCR Purification Kit (Qiagen), they were sequenced using a BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems). We used the Norovirus Genotyping Tool version 1.0 to genotype the present virus (Kroneman et al., 2011). The accession numbers of these strains are shown in Supplementary Table S2.
Phylogenetic Analyses and Estimating the Evolutionary Rate by a Bayesian Markov Chain Monte Carlo (MCMC) Method
To examine the evolution of the strains, we collected complete sequences of the VP1 gene and RdRp region of HuNoV from GenBank and constructed phylogenetic trees of the genes by the MCMC method. The full-length VP1 gene sequences of the HuNoV GII.2 strains with the detection year were collected in December 2016. A total of 186 strains were obtained, including our present strains. Furthermore, the full-length RdRp region sequences of the HuNoV GII.P16 strains and HuNoV GII.2 strains with the detection year were also collected in December 2016. A total of 107 strains were obtained, including the present strains. At that time, new GII.2 variants’ sequence data detected from other areas were not disclosed in GenBank. Thus, the evolutionary analyses were performed on our strains alone. The best substitution models were selected using the BIC method by MEGA6.0 (Tamura et al., 2013). Appropriate clock and tree models were determined by the pass sampling method using the BEAST2 packages (Bouckaert et al., 2014). To generate the posterior set of trees, we used BEAST v2.4.5 (Bouckaert et al., 2014) (Supplementary Table S3). After the first 10% of the chain was omitted, effective sample sizes greater than 200 were accepted, as described in the manual. Maximum clade credibility trees were constructed using Tree Annotator v2.4.5, and the MCC phylogenetic trees were visualized using FigTree v1.4.0. Moreover, we estimated the evolutionary rates of the VP1 gene and RdRp region using BEAST v2.4.5. Appropriate models were selected as described above.
Calculation of Pairwise Distances of the VP1 Gene and RdRp Region in HuNoV Strains
To evaluate the genetic divergence of the VP1 gene and RdRp region, the pairwise distances were calculated using MEGA 6.0, as described (Tamura et al., 2013). In addition, to recognize genetic diversity among the GII.P16-GII.2 strains, we constructed phylogeny trees of the VP1 gene and RdRp region using neighbor-joining trees. In these phylogenic trees, we added six strains detected in other countries in the 2016/2017 winter season as reference strains (VP1: KY421044, KY485115, KY485122, VP1, and RdRp: KY771081, KY865306, KY865307).
Construction of the Capsid Protein Structural Models and Prediction of the B-Cell Conformational Epitopes in the Capsid Protein
To examine the relationship between amino acid substitutions and B-cell epitopes, we constructed structural models of the capsid protein and their predicted B-cell conformational epitopes. We made two models with the strains Hu/GII/JP/2010/GII.2/Ehime43 (one of the strains with highest identity score to the 2016 strains in the past GII.2 strains) and Hu/GII/JP/2016/GII.P16-GII.2/Kawasaki129 (2016 strain). The homology modeling was based on the crystal structures of the 1IHM and 4RPB (Protein Data Bank accession numbers). After aligning these data using MAFFTash (Katoh et al., 2002), the models were constructed using MODELER v9.15 (Webb and Sali, 2014) and adjusted using Swiss PDB Viewer v4.1 (Guex and Peitsch, 1997). Then, they were confirmed by Ramachandran plot analysis using the RAMPAGE server (Lovell et al., 2003), and the residues of putative B-cell epitopes and amino acid substitutions were mapped on the predicted structure using Chimera v1.10.2 (Pettersen et al., 2004).
Next, we predicted B-cell conformational epitopes in the capsid protein in the structural models by DiscoTope2.0 (Kringelum et al., 2012), BEPro (Sweredoski and Baldi, 2008), EPCES (Liang et al., 2007), and EPSVR (Liang et al., 2010). We used these tools with cut-offs of -3.7 (DiscoTope2.0), 1.3 (BEPro), and 70 (EPCES, EPSVR). We accepted sites as B-cell conformational epitopes when they were inferred by three or more methods. Amino acid substitutions of the strain Hu/GII/JP/2016/GII.P16-GII.2/Kawasaki129 that corresponded to the strain Hu/GII/JP/2010/GII.2/Ehime43 and predicted epitopes were mapped onto the model. Moreover, to estimate whether the amino acid substitutions in the predicted epitopes were characteristic for the 2016 strains, we examined the number of strains with these amino acid substitutions among all present GII.2 strains (186 strains), compared to Snow mountain strain.
Selective Pressure Analysis
To estimate sites under positive or negative selection in the capsid protein of the 2016 strains, we calculated the rates of non-synonymous (dN) and synonymous (dS) substitutions at every codon position with the Datamonkey package (Delport et al., 2010). In the present study, we analyzed only the 2016 strains. We used the fixed effects likelihood (FEL) method, the internal fixed effects likelihood (IFEL), the single likelihood ancestor (SLAC), and the random effects likelihood (REL) methods.
Bayesian Skyline Plot Analyses
To examine the changes in the effective population size of the HuNoV GII.2 strains, Bayesian skyline plot (BSP) analyses were performed using all collected strains (VP1: 186 strains; RdRp: 88 strains), including 100% identical sequences by BEAST v2.4.5, as reported (Drummond et al., 2005; Bouckaert et al., 2014). The appropriate models for these analyses were selected as described above. The detailed conditions of each analysis are shown in Supplementary Table S4.
Results
Phylogenetic and Evolutionary Rate Analyses of the VP1 Gene in HuNoV GII.2
Time-scale evolutional phylogenetic trees were constructed by an MCMC method, based on the full-length nucleotide sequences of VP1 (Figure 1). To perform this analysis, we used 19 2016 strains that were obtained by December 2016, and 167 reference strains from GenBank. The MCC tree of the VP1 genes shows that the VP1 gene was divided into many clusters. Interestingly, the VP1 sequences clustered according to their RdRp sequence. The most recent common ancestor of the GII.2 strains was estimated to have circulated in 1966 [95% highest posterior density (HPD) interval, 1953–1975]. Furthermore, the most recent common ancestor of the GII.P16-GII.2 strains was in 2002 (95% HPD interval, 2000–2003). Subsequently, the common ancestors of the GII.P16-GII.2 cluster, detected in 2010–2012, and the 2016 strains diverged in 2006 (95% HPD interval, 2004–2007). The evolutionary rate of the VP1 gene in HuNoV Gll.2 strains was estimated as 3.26 × 10-3 substitutions/site/year (95% HPD interval, 2.74–3.78 × 10-3 substitutions/site/year).
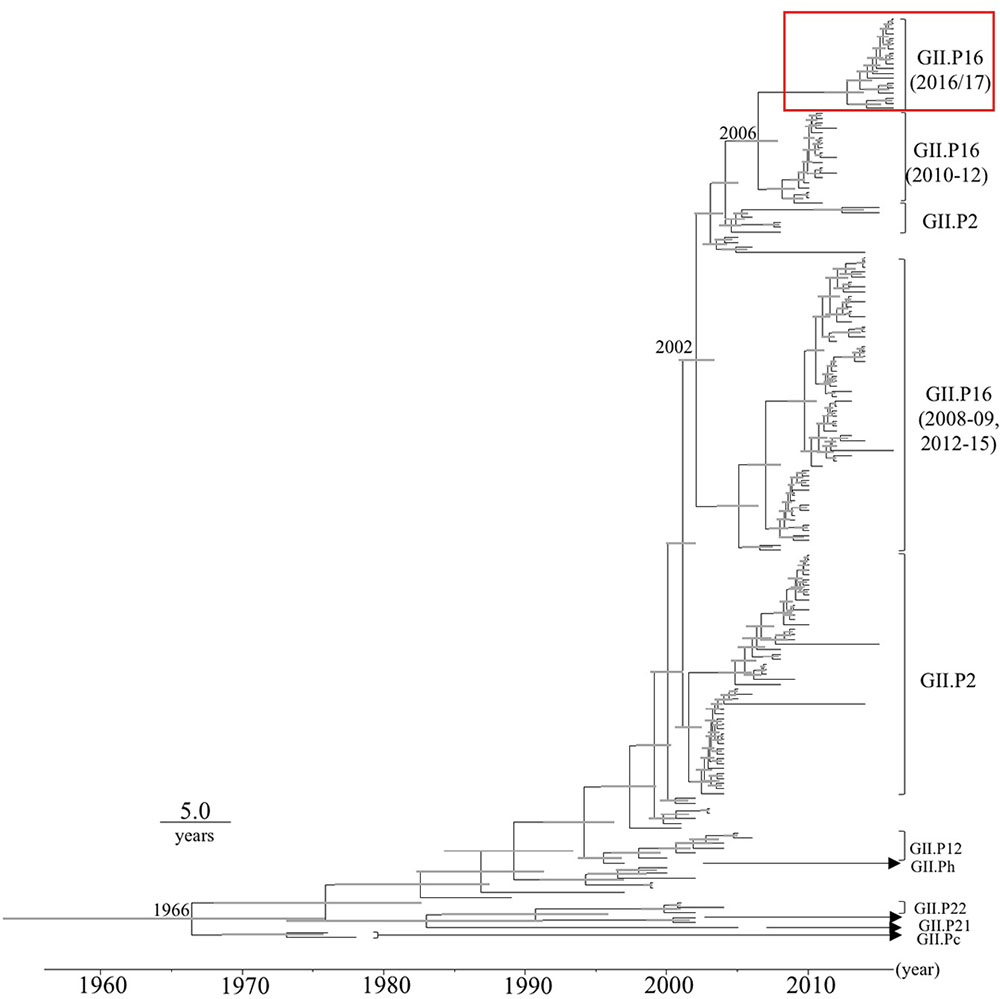
FIGURE 1. Phylogenetic tree for the VP1 gene in the HuNoV GII strains was constructed using the Bayesian Markov chain Monte Carlo method. The scale bar represents time (years). Gray bars indicate the 95% highest probability density (HPD) for the branched year. The genotypes of the RdRp region of the strains are shown with the RdRp region sequences. A red square shows 2016 strains.
Phylogenetic and Evolutionary Rate Analyses of the RdRp Region in HuNoV
Next, we determined the time-scale of the evolutionary phylogenetic tree of the various genotypes of RdRp region, based on their full-length nucleotide sequences (Figure 2). We used 19 2016 strains, including 100% identical sequences. The common ancestors of the 2016 strains, GII.P16-GII.4, GII.P16-GII.3, GII.P16-GII.13, and GII.P16-GII.2 strains detected in 2010–2012 diverged in 1972 (95% HPD interval, 1954–1987), and the common ancestors of the 2016 strains and GII.P16-GII.4 strains diverged in 1999 (95% HPD interval, 1991–2005). The evolutionary rate of the RdRp region in HuNoV Gll.P16 strains was estimated as 2.03 × 10-3 substitutions/site/year (95% HPD interval, 1.36–2.75 × 10-3 substitutions/site/year). These results suggest that the RdRp region and the VP1 gene may have evolved independently.
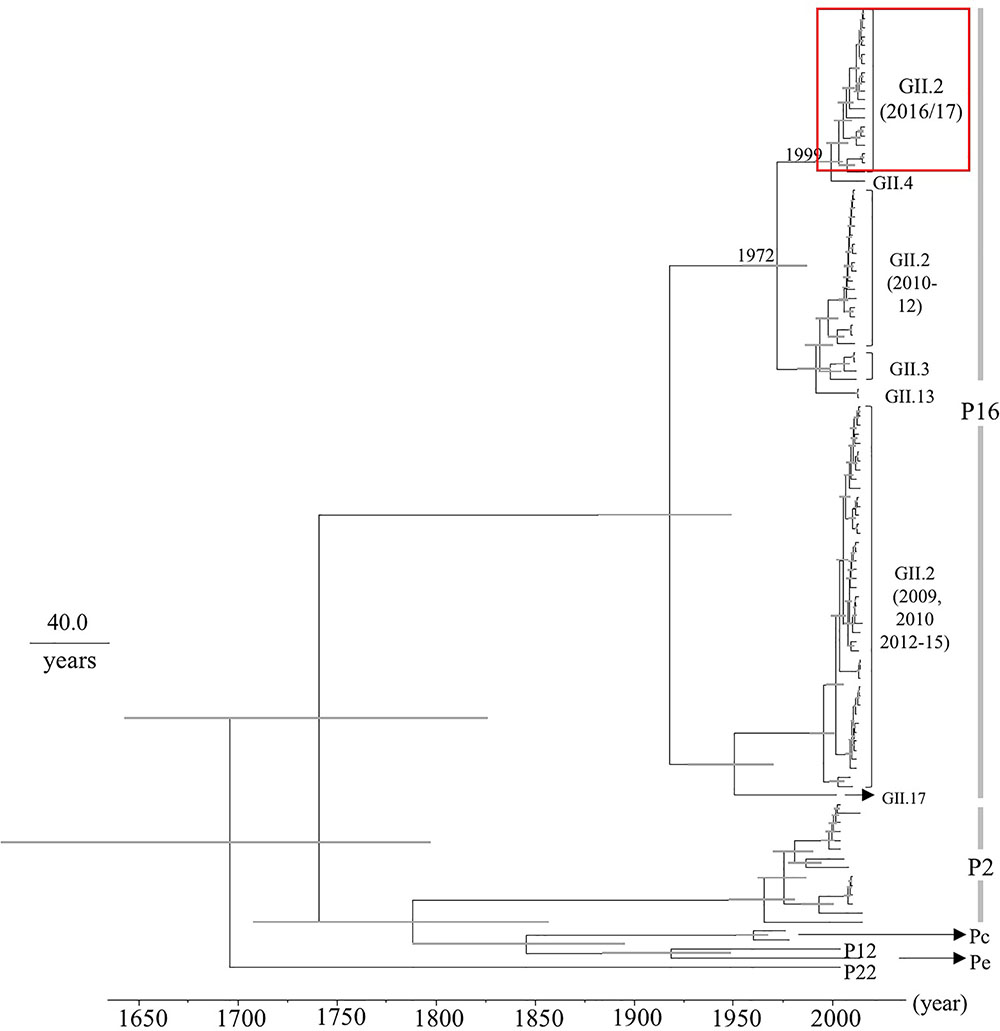
FIGURE 2. Phylogenetic tree for the RdRp region in the HuNoV strains was constructed using the Bayesian Markov chain Monte Carlo method. The scale bar represents time (years). Gray bars indicate the 95% HPD for the branched year. The genotypes of the VP1 gene of the strains are shown with the VP1 gene sequences. A red square shows 2016 strains.
Pairwise Distances of the VP1 Gene and RdRp Region Sequences
To evaluate the genetic divergence between the 2016 strains and the other GII.2 strains, we calculated pairwise distances as the genetic distances among the present strains (Figures 3A–D and Supplementary Figures S1a,b). The pairwise distances values of the VP1 gene among all GII.2 strains were 0.065 ± 0.033 (mean ± SD), and the histogram was bimodal (Figure 3A). Furthermore, the pairwise distances of the VP1 genes in the GII.P16–GII.2 strains were 0.042 ± 0.0024, and the histogram was also bimodal (Figure 3B). The pairwise distances between the 2016 strains and the other GII.P16–GII.2 strains were relatively long (>0.043), and those of the RdRp region among all GII.P16 strains were 0.064 ± 0.043 (Figure 3C). Moreover, the pairwise distances between the 2016 strains and the other GII.P16 strains were also relatively long (>0.041). In contrast, pairwise distances between the 2016 strains and GII.P16-GII.2 strains detected in America and China in the 2016/2017 winter season were very short (Supplementary Figures S1a,b). These results suggested that the 2016 strains evolved further from pre-2016 GII.P16-GII.2 strains genetically.
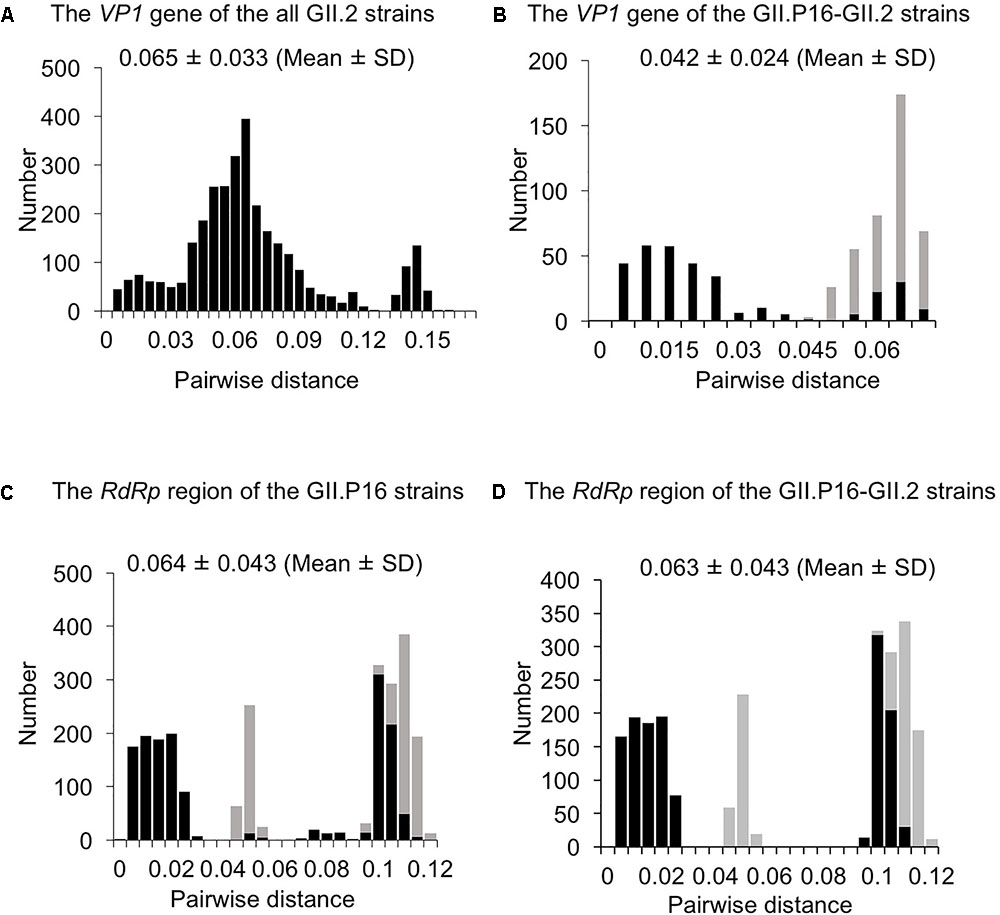
FIGURE 3. Distribution of pairwise distances between HuNoV strains for (A) the VP1 gene of the all GII.2 strains, (B) the VP1 gene of the GII.P16-GII.2 strains, (C) the RdRp region of the GII.P16 strains, and (D) the RdRp region of the GII.P16-GII.2 strains, based on the nucleotide sequences. The pairwise distances between the 2016 strains and the other strains are shown by gray in (B–D).
Structural Models of the Capsid Protein and the Predicted B-Cell Conformational Epitopes
We constructed two structural models of the capsid protein (Figures 4A,B). Ramachandran plots showed that over 94% of the residues in each model were evaluated as geometrically favored regions. Using these models, we predicted the B-cell conformational epitopes in the capsid protein, and the predicted epitopes were mapped on the models. The prediction of the B-cell epitopes from two predicted structural models resulted in similar results (Figure 4). As shown in Figure 4, Hu/GII/JP/GII.P16-GII.2/Kawasaki129 strains had five amino acid substitutions on the surface of the structural model that corresponded to the strain Hu/GII/JP/2010/GII.2/Ehime43 (Figure 4B). Moreover, three amino acid substitutions (Lys341Arg, Gly344Ser, and Gln347His) were detected in the epitope on the protruding 2 (P2) domain. Among them, 14 2016 strains have an amino acid substitution at position 341 (Lys341Arg: 12 strains, Lys341Thr: two strains), but no pre-2016 GII.P16-GII.2 strains had this substitution. These results suggested that the VP1 protein of the 2016 strains has multiple amino acid substitutions in these epitopes, compared with the pre-2016 GII.P16-GII.2 strains.
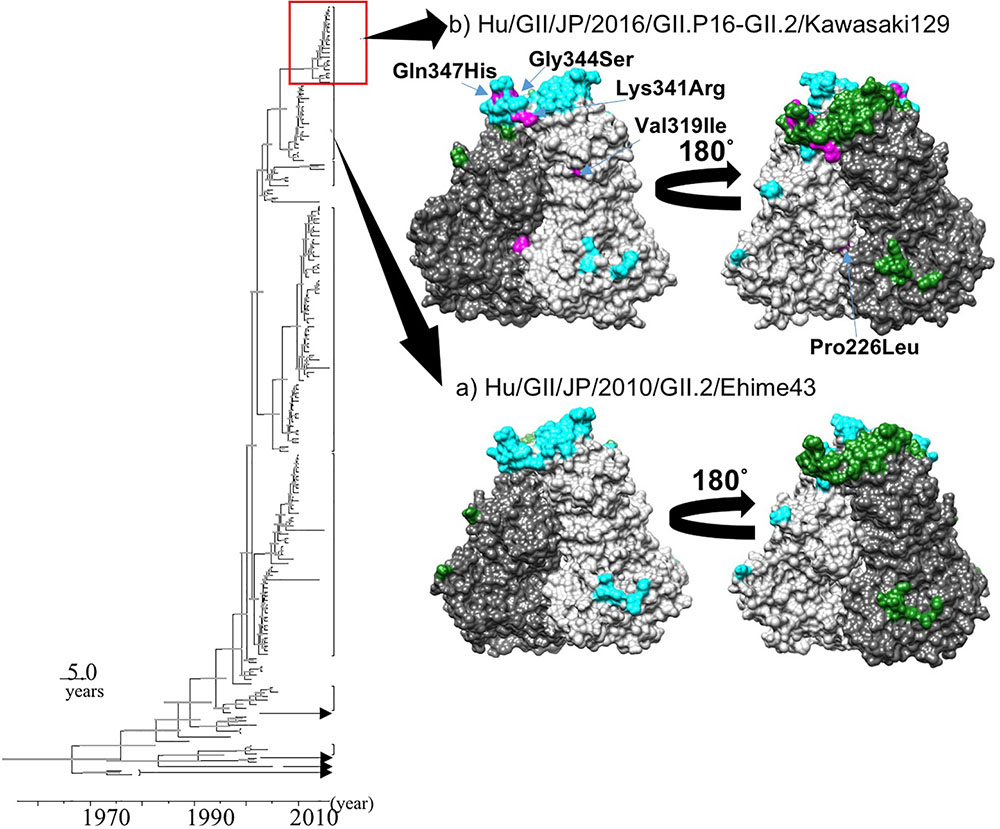
FIGURE 4. Structural models of the capsid protein in strains (A) Hu/GII/JP/2010/GII.2/Ehime43, and (B) Hu/GII/JP/2016/GII.P16-GII.2/Kawasaki129. Chains (A) and (B) are colored by gray and dim gray, respectively. The predicted conformational epitopes are shown in cyan (chain A) and green (chain B). Amino acid substitutions in the protein corresponding to the strain Hu/GII/JP/2010/GII.2/Ehime43 are indicated in magenta.
Positive and Negative Selection Site Analyses
Positive and negative selection sites in the capsid protein of the 2016 strains were inferred using FEL, IFEL, SLAC, and REL models. As a result, no positive selection sites and three negative selection sites (aa 234, 429, and 439) were estimated by FEL, but no positive or negative selection site was estimated by the SLAC, IFEL, and REL methods. These results suggested that the sequence variation of the capsid gene of 2016 strains was influenced by natural evolution rather than by selective pressure from the host immune system.
Bayesian Skyline Plot Analyses
We used BSP analyses to estimate the fluctuations of the effective population sizes for the HuNoV (Figure 5). The effective population size of the VP1 gene in the HuNoV GII.2 strains was nearly constant until the year 2005. After that, the population size dropped sharply (Figure 5A). The fluctuation of the effective population size of RdRp region in the GII.P16 strains was almost constant since the year 2000 (Figure 5B). We could not perform BSP analyses of the 2016 strains due to their small number. These results suggest that GII.2 strains have been constantly distributed to humans since 1960, although the numbers of GII.2 detected increased in 2016 with the emergence of the 2016 strains.
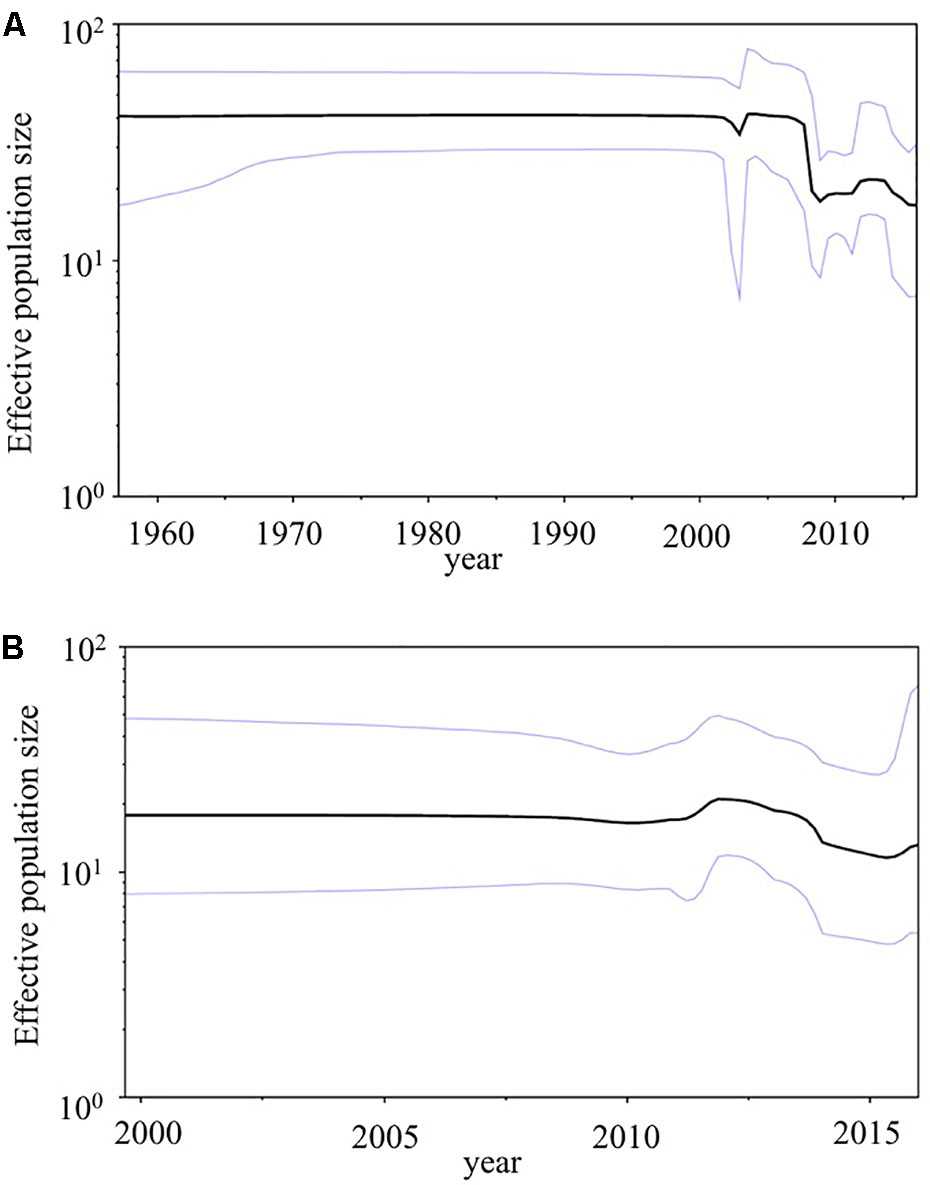
FIGURE 5. Phylodynamics of the HuNoV. (A) The VP1 gene in the present GII.2 strains, and (B) the RdRp region in the present GII.P16 strains using Bayesian skyline plot analyses. The black lines indicate median effective population sizes, and the blue lines indicate 95% highest probability density.
Discussion
Here we describe the evolution of the VP1 gene and RdRp region in the HuNoV GII.P16-GII.2 strains detected in the 2016/2017 winter season (2016 strains) that suddenly emerged in Japan. Our main findings are as follows. (1) A common ancestor of the VP1 gene of the 2016 strains diverged from another GII.P16-GII.2 strain around 10 years ago with an evolutionary rate as described (Mizukoshi et al., 2017) (Figure 1). (2) A common ancestor of the RdRp region of the 2016 strains diverged from the RdRp region of the GII.P16-GII.4 rather than the previous GII.P16-GII.2 strains in 1999 with a rapid evolutionary rate (around 10-3 substitutions/site/year) (Figure 2). Some reports estimated that the GII.P16-GII.2 strains detected in 2016 were a new recombinant strain, and both the RdRp region and VP1 gene sequences of our strains and the new strains were very similar (identity: about 98–99%, data not shown), although the analyzed sequence lengths were different (Bidalot et al., 2017; Lu et al., 2017). Based on these reports, the 2016 strains might be a new chimeric virus. For example, it might be a recombination of GII.P16-GII.4 and the previous GII.P16-GII.2 (2010–2012 type) viruses, although only a limited number of strains were used in the present MCC tree.
We found that the rate of the HuNoV VP1 gene in GII.2 strains, including 2016 strains was 3.26 × 10-3 substitutions/site/year. However, previous studies showed different evolutionary rates (Eden et al., 2014; Kobayashi et al., 2016; Mizukoshi et al., 2017). For example, Kobayashi et al. (2016) suggested it was around 3.76 × 10-3 substitutions/site/year for the VP1 gene of all HuNoV GII. Another report showed that the GII.4 variant strains (GII.4v Sydney 2012 strains, GII4v New Orleans 2009 strains, and Apeldoorn 2007 strains) had an evolutionary rate of 7.20 × 10-3 substitutions/site/year (Eden et al., 2014). A very recent report showed that the rate of pre-2016 GII.2 VP1 gene was 2.987 × 10-3 substitutions/site/year (Mizukoshi et al., 2017). Thus, the VP1 gene of the present 2016 strains may evolve as well as the previous GII.2 strains (Mizukoshi et al., 2017).
Next, the current structural models of the HuNoV capsid protein suggest that amino acid substitutions occurred in the 2016 strains. In particular, an amino acid substitution at position 341, which was involved in a predicted B-cell epitope in the P2 domain, was detected in almost all 2016 strains, but was not detected in pre-2016 GII.P16-GII.2 strains. The amino acid sequences of the P2 domain are reportedly closely related to the antigenicity of the HuNoV capsid protein (Lindesmith et al., 2012). In contrast, Swanstrom et al. (2014) suggested that the GII.2 strains have evolved very little in the blockade epitopes for 35 years. In addition, a very recent report suggested that the reactivity to the immunochromatography kits showed no differences between 2016 strains and pre-2016 strains (Nagasawa et al., 2018). From these reports, it is unclear whether the amino acid substitution in the epitope causes antigenic changes or not. To the best of our knowledge, no report has investigated HuNoV neutralization assay using infectious HuNoV particles, HuNoV susceptible cells, and human antibodies, such as the secreted IgA-related gut mucosal immune response. Further studies may be needed to clarify the cause of GII.2 outbreaks.
We also estimated the positive/negative selection sites in the VP1 gene of 2016 strains. No positive selection site was found, but there were three negative selection sites by the FEL method. In general, positive selection sites may be responsible for the immune pressure leading to an escape mutation, and negative selection sites may prevent deterioration of antigenic function and structures (Domingo, 2006). Previous reports suggested that HuNoV GII capsid proteins may not receive strong host immune pressure, resulting in no or only small numbers of positive selection sites in these VP1 proteins (Kobayashi et al., 2016; Parra et al., 2017). Thus, the present results seem to be compatible with earlier reports, although the strains we analyzed were limited in number (Kobayashi et al., 2016; Parra et al., 2017).
In addition, we calculated genetic distances of the VP1 gene and RdRp region. The present 2016 strains had relatively longer pairwise distance values than the previously detected GII.P16-GII.2 strains, and relatively short pairwise distance values were found among 2016 strains (Figures 3B–D). Thus, the 2016 strains had larger genetic divergences than the previously detected GII.P16-GII.2 strains. In contrast, GII.P16-GII.2 strains detected in 2016/2017 winter seasons in various areas were located in a same cluster of the phylogenetic trees (Supplementary Figures S1a,b). In the analyzed regions, the sequence identities of these strains detected in various countries, including the United States, China, France, Germany and Japan were also very high (identity: about 98–99%, data not shown). These results suggested that genetically similar GII.P16-GII.2 strains cause the prevalence of acute gastroenteritis in many counties (Nagasawa et al., 2018). Next, we examined the phylodynamics of the HuNoV GII.2 (Figure 5). Notably, the effective population sizes of the VP1 gene seem to be almost constant or to decrease slightly, but the number of acute gastroenteritis patients with HuNoV GII.2 strains increased in 2016. From these results, the symptoms of the pre-2016 GII.2 strains might be less severe than the symptoms of the 2016 strains, although these analyses are limited by the small number of old strains. Moreover, the GII.P16 RdRp region was also constant. To test our hypothesis, further studies on the molecular epidemiology are needed.
Molecular epidemiological studies suggested that the HuNoV genotype GII.4 caused multiple pandemics of acute gastroenteritis in people of all ages. For example, Den Haag 2006b suddenly emerged and caused a pandemic worldwide in 2006 (Siebenga et al., 2007), and Sydney 2012 caused a pandemic in 2012 (Eden et al., 2014). Detailed evolutionary analyses of these strains showed that significant amino acid substitutions occurred in the P2 domain of the GII.4 virus responsible for that pandemic, compared with the previous GII.4 strains (Siebenga et al., 2007; Eden et al., 2014). In the 2016 strains, multiple amino acid substitutions were found in the P2 domain. Indeed, this 2016 strain caused large outbreaks of acute gastroenteritis in Japanese children. In addition, the 2016 strains caused outbreaks of acute gastroenteritis in many countries (Bidalot et al., 2017; Lu et al., 2017; Niendorf et al., 2017; Tohma et al., 2017). Therefore, these strains may cause further outbreaks in Japan and elsewhere.
Author Contributions
HK and KK designed this study. KN, YM, TM, FM, WS, and TSe analyzed the data. YM, TM, FM, TSh, NO, and NN collected the samples. YU, NS, KM, KS, HS, MK, YS, AR, KF, and KO contributed analyses tools. KN, YM, KK, and HK wrote this paper. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Funding
This work was supported by a commissioned project for Research on Emerging and Re-emerging Infectious Diseases from Japan Agency for Medical Research and Development, AMED (grant number J17JK00050).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Dr. Gary C. Howard for critically proofreading/reviewing the draft and revised manuscripts.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00001/full#supplementary-material
FIGURE S1 | Phylogenetic trees for (a) the VP1 gene and (b) RdRp region in the HuNoV GII.P16-GII.2 strains were constructed using the neighbor-joining method. The scale bars represent substitution per site. Bootstrap values of more than 70% were shown. Circles indicate strains detected in the United States in the 2016/2017 winter season, and the square indicates the strain detected in China in the 2016/2017 winter season.
TABLE S1 | Primers used in this study.
TABLE S2 | Strains used in this study.
TABLE S3 | Conditions of Bayesian Markov chain Monte Carlo method for the phylogenic trees.
TABLE S4 | Conditions of Bayesian skyline plot analyses.
Footnotes
References
Bidalot, M., Théry, L., Kaplon, J., De Rougemont, A., and Ambert-Balay, K. (2017). Emergence of new recombinant noroviruses GII.p16-GII.4 and GII.p16-GII.2, France, winter 2016 to 2017. Euro Surveill. 22:30508. doi: 10.2807/1560-7917.ES.2017.22.15.30508
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu,C-H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLOS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Delport, W., Poon, A. F., Frost, S. D., and Kosakovsky Pond, S. L. (2010). Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457. doi: 10.1093/bioinformatics/btq429
Domingo, E. (2006). “Virus evolution,” in Fields Virology, 5th Edn, eds D. M. Knipe, P. M. Howley, and P. A. Philadelphia (New York, NY: Lippincott Williams & Wilkins), 389–421.
Drummond, A. J., Rambaut, A., Shapiro, B., and Pybus, O. G. (2005). Bayesian coalescent inference of past population dynamics from molecular sequences. Mol. Biol. Evol. 22, 1185–1192. doi: 10.1093/molbev/msi103
Eden, J. S., Hewitt, J., Lim, K. L., Boni, M. F., Merif, J., Greening, G., et al. (2014). The emergence and evolution of the novel epidemic norovirus GII.4 variant Sydney 2012. Virology 450–451, 106–113. doi: 10.1016/j.virol.2013.12.005
Gadberry, M. D., Malcomber, S. T., Doust, A. N., and Kellogg, E. A. (2005). Primaclade–a flexible tool to find conserved PCR primers across multiple species. Bioinformatics 21, 1263–1264. doi: 10.1093/bioinformatics/bti134
Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723. doi: 10.1002/elps.1150181505
Katoh, K., Misawa, K., Kuma, K., and Miyata, T. (2002). MAFFT: a novel method for rapid multiple sequence alignment based on fast fourier transform. Nucleic Acids Res. 30, 3059–3066. doi: 10.1093/nar/gkf436
Kobayashi, M., Matsushima, Y., Motoya, T., Sakon, N., Shigemoto, N., Okamoto-Nakagawa, R., et al. (2016). Molecular evolution of the capsid gene in human norovirus genogroup II. Sci. Rep. 6:29400. doi: 10.1038/srep29400
Kringelum, J. V., Lundegaard, C., Lund, O., and Nielsen, M. (2012). Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLOS Comput. Biol. 8:e1002829. doi: 10.1371/journal.pcbi.1002829
Kroneman, A., Vennema, H., Deforche, K., v d Avoort, H., Peñaranda, S., Oberste, M. S., et al. (2011). An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 51, 121–125. doi: 10.1016/j.jcv.2011.03.006
Liang, S., Liu, S., Zhang, C., and Zhou, Y. (2007). A simple reference state makes a significant improvement in near-native selections from structurally refined docking decoys. Proteins 69, 244–253. doi: 10.1002/prot.21498
Liang, S., Zheng, D., Standley, D. M., Yao, B., Zacharias, M., and Zhang, C. (2010). EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results. BMC Bioinformatics 11:381. doi: 10.1186/1471-2105-11-381
Lindesmith, L. C., Beltramello, M., Donaldson, E. F., Corti, D., Swanstrom, J., Debbink, K., et al. (2012). Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLOS Pathog. 8:e1002705. doi: 10.1371/journal.ppat.1002705
Lovell, S. C., Davis, I. W., Arendall, W. B. III, de Bakker, P. I., Word, J. M., Prisant, M. G., et al. (2003). Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450. doi: 10.1002/prot.10286
Lu, J., Fang, L., Sun, L., Zeng, H., Li, Y., Zheng, H., et al. (2017). Association of GII.P16-GII.2 recombinant norovirus strain with increased norovirus outbreaks, Guangdong, China, 2016. Emerg. Infect. Dis. 23, 1188–1190. doi: 10.3201/eid2307.170333
Matson, D. O. (2004). “Caliciviruses and hepatitis E virus,” in Textbook of Pediatric Infectious Diseases, 5th Edn, eds R. D. Feigin, J. D. Cherry, G. J. Demmler, and S. L. Kaplan (Philadelphia, PA: Saunders Company), 2087–2101.
Mizukoshi, F., Nagasawa, K., Doan, Y. H., Haga, K., Yoshizumi, S., Ueki, Y., et al. (2017). Molecular evolution of the RNA-Dependent RNA polymerase and capsid genes of human norovirus genotype GII.2 in Japan during 2004–2015. Front. Microbiol. 8:705. doi: 10.3389/fmicb.2017.00705
Nagasawa, K., Matsuhima, Y., Motoya, T., Mizukoshi, F., Ueki, Y., Sakon, N., et al. (2018). Phylogeny and immunoreactivity of norovirus GII.P16-GII.2, Japan, winter 2016–17. Emerg. Infect. Dis. 24, 144–148. doi: 10.3201/eid2401.170284
Niendorf, S., Jacobsen, S., Faber, M., Eis-Hübinger, A. M., Hofmann, J., Zimmermann, O., et al. (2017). Steep rise in norovirus cases and emergence of a new recombinant strain GII.P16-GII.2, Germany, winter 2016. Euro Surveill. 22:30447. doi: 10.2807/1560-7917.ES.2017.22.4.30447
Parra, G. I., Squires, R. B., Karangwa, C. K., Johnson, J. A., Lepore, C. J., Sosnovtsev, S. V., et al. (2017). Static and evolving norovirus genotypes: implications for epidemiology and immunity. PLOS Pathog. 13:e1006136. doi: 10.1371/journal.ppat.1006136
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Siebenga, J. J., Vennema, H., Renckens, B., de Bruin, E., van der Veer, B., Siezen, R. J., et al. (2007). Epochal evolution of GGII.4 norovirus capsid proteins from 1995 to 2006. J. Virol. 81, 9932–9941. doi: 10.1128/JVI.00674-07
Swanstrom, J., Lindesmith, L. C., Donaldson, E. F., Yount, B., and Baric, R. S. (2014). Characterization of blockade antibody responses in GII.2.1976 snow mountain virus-infected subjects. J. Virol. 88, 829–837. doi: 10.1128/JVI.02793-13
Sweredoski, M. J., and Baldi, P. (2008). PEPITO: improved discontinuous B-cell epitope prediction using multiple distance thresholds and half sphere exposure. Bioinformatics 24, 1459–1460. doi: 10.1093/bioinformatics/btn199
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Thongprachum, A., Okitsu, S., Khamrin, P., Maneekarn, N., Hayakawa, S., and Ushijima, H. (2017). Emergence of norovirus GII.2 and its novel recombination during the gastroenteritis outbreak in Japanese children in mid-2016. Infect. Genet. Evol. 51, 86–88. doi: 10.1016/j.meegid.2017.03.020
Tohma, K., Lepore, C. J., Ford-Siltz, L. A., and Parra, G. I. (2017). Phylogenetic analyses suggest that factors other than the capsid protein play a role in the epidemic potential of GII.2 norovirus. mSphere 2:e00187-17. doi: 10.1128/mSphereDirect.00187-17
Vinjé, J. (2015). Advances in laboratory methods for detection and typing of norovirus. J. Clin. Microbiol. 53, 373–381. doi: 10.1128/JCM.01535-14
Keywords: norovirus, capsid, RNA-dependent RNA polymerase, phylogeny, molecular evolution, epitope mapping
Citation: Nagasawa K, Matsushima Y, Motoya T, Mizukoshi F, Ueki Y, Sakon N, Murakami K, Shimizu T, Okabe N, Nagata N, Shirabe K, Shinomiya H, Suzuki W, Kuroda M, Sekizuka T, Suzuki Y, Ryo A, Fujita K, Oishi K, Katayama K and Kimura H (2018) Genetic Analysis of Human Norovirus Strains in Japan in 2016–2017. Front. Microbiol. 9:1. doi: 10.3389/fmicb.2018.00001
Received: 07 November 2017; Accepted: 03 January 2018;
Published: 18 January 2018.
Edited by:
José A. Melero, Instituto de Salud Carlos III, SpainReviewed by:
Hirotaka Ode, Nagoya Medical Center (NHO), JapanHiroshi Ushijima, Nihon University School of Medicine, Japan
Copyright © 2018 Nagasawa, Matsushima, Motoya, Mizukoshi, Ueki, Sakon, Murakami, Shimizu, Okabe, Nagata, Shirabe, Shinomiya, Suzuki, Kuroda, Sekizuka, Suzuki, Ryo, Fujita, Oishi, Katayama and Kimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hirokazu Kimura, a2ltaGlyb0BuaWguZ28uanA= Kazuhiko Katayama, a2F0YXlhbWFAbGlzY2kua2l0YXNhdG8tdS5hYy5qcA==
†These authors have contributed equally to this work.