 Karel Schoonvaere
Karel Schoonvaere Guy Smagghe
Guy Smagghe Frédéric Francis
Frédéric Francis Dirk C. de Graaf
Dirk C. de Graaf- 1Laboratory of Molecular Entomology and Bee Pathology, Department of Biochemistry and Microbiology, Faculty of Sciences, Ghent University, Ghent, Belgium
- 2Functional and Evolutionary Entomology, Gembloux Agro-Bio Tech, University of Liege, Gembloux, Belgium
- 3Laboratory of Agrozoology, Department of Crop Protection, Faculty of Bioscience Engineering, Ghent University, Ghent, Belgium
Bees are associated with a remarkable diversity of microorganisms, including unicellular parasites, bacteria, fungi, and viruses. The application of next-generation sequencing approaches enables the identification of this rich species composition as well as the discovery of previously unknown associations. Using high-throughput polyadenylated ribonucleic acid (RNA) sequencing, we investigated the metatranscriptome of eight wild bee species (Andrena cineraria, Andrena fulva, Andrena haemorrhoa, Bombus terrestris, Bombus cryptarum, Bombus pascuorum, Osmia bicornis, and Osmia cornuta) sampled from four different localities in Belgium. Across the RNA sequencing libraries, 88–99% of the taxonomically informative reads were of the host transcriptome. Four viruses with homology to insect pathogens were found including two RNA viruses (belonging to the families Iflaviridae and Tymoviridae that harbor already viruses of honey bees), a double stranded DNA virus (family Nudiviridae) and a single stranded DNA virus (family Parvoviridae). In addition, we found genomic sequences of 11 unclassified arthropod viruses (related to negeviruses, sobemoviruses, totiviruses, rhabdoviruses, and mononegaviruses), seven plant pathogenic viruses, and one fungal virus. Interestingly, nege-like viruses appear to be widespread, host-specific, and capable of attaining high copy numbers inside bees. Next to viruses, three novel parasite associations were discovered in wild bees, including Crithidia pragensis and a tubulinosematid and a neogregarine parasite. Yeasts of the genus Metschnikowia were identified in solitary bees. This study gives a glimpse of the microorganisms and viruses associated with social and solitary wild bees and demonstrates that their diversity exceeds by far the subset of species first discovered in honey bees.
Introduction
Transcriptomics is the study of all ribonucleic acid (RNA) molecules in a biological sample (Wang et al., 2009). In bees (Hymenoptera; Anthophila clade), transcriptomics has been used to study evolutionary relationships (Peters et al., 2017), eusociality (Woodard et al., 2011), developmental stage and caste differentiation (Harrison et al., 2015), tissue functionality (Yin et al., 2013), and genetic responses to external stimuli (Aufauvre et al., 2014; Riddell et al., 2014). Herein, next-generation sequencing (NGS) followed by bioinformatics analysis of short sequence reads is the most cost-effective method and supplanted previous approaches like expressed sequence tag sequencing or microarrays (Oppenheim et al., 2015). To date, public repositories hold transcriptomics data of at least 62 bee species, the majority of which originate from evolutionary biology initiatives such as the 1K Insect Transcriptome Evolution project (Peters et al., 2017).
Depending on the extraction source, NGS data often comprise a mixture of reads from different species, that is, the host and its co-inhabiting (micro)organisms and viruses (Whitacre et al., 2015). This is notable when the source includes the digestive system (Douglas, 2015) or a specific tissue wherein symbionts reside (DeLay et al., 2012). In case of RNA sequencing, the collection of multi-specific RNA transcripts is then referred to as a metatranscriptome. Where a transcriptomics workflow includes a data preprocessing step to filter out undesired species, such as mapping reads to a database of known co-inhabitants, a metatranscriptomics workflow intentionally retains those reads to simultaneously study species composition and their function. So far, metatranscriptomics studies in bees are limited in number and most are dedicated to the domesticated honey bee, Apis mellifera. These studies include investigations of the gut microbiome function (Lee et al., 2015), host-parasite interactions, and viral metagenomics (Runckel et al., 2011; Schoonvaere et al., 2016; Remnant et al., 2017) or both (Cox-Foster et al., 2007; Tozkar et al., 2015).
Bees interact with a remarkable diversity of microorganisms and viruses (Evans and Schwarz, 2011). These interactions constitute either a form of symbiosis with respect to the host (Engel et al., 2016), ranging from beneficial to harmful, or temporarily hitchhiking with respect to a broader plant-pollinator ecology (Lachance, 2016). Bees fall victim to three major groups of unicellular parasites being trypanosomatids (Schmid-Hempel and Tognazzo, 2010), microsporidians (Brown, 2017), and apicomplexans (Lipa and Triggiani, 1996). Also metazoan organisms are common parasites including nematodes (Pionar and Van der Laan, 1972), tracheal mites (Husband and Shina, 1970), and conopid flies (Smith, 1969). Further, honey bees are associated with a variety of viruses, the majority of which bear a single-stranded RNA genome (de Miranda et al., 2013). Despite studies have demonstrated the broad host range of some of those viruses (McMahon et al., 2015; Dolezal et al., 2016) and their capability of inter-taxa replication (Peng et al., 2011), the full impact of bee viruses in a pollinator community is not well understood (Singh et al., 2010).
In this study, we aimed at exploring novel viruses and parasites associated with wild bees using high-throughput RNA sequencing. We adopted a polyadenylation enrichment strategy for two reasons: (1) to selectively enrich for RNA viruses with a polyadenylated genome (which are common in bees) or DNA virus transcripts and (2) to detect as well a significant fraction of the transcriptomes of eukaryote bee parasites and host. We increased the likelihood to encounter infected bees by pooling five individuals per sequencing library and sampling populations from different localities. Despite of the modest selection of only 8 of the nearly 400 wild bee species that occur in Belgium, the diversity of viruses and parasites associated with those species was high and comprised numerous novel associations to bees.
Materials and Methods
Sample Collection
Four sampling locations were selected in correspondence to historical collection sites in Belgium (Figure 1B); see also Maebe et al. (2016). At each sampling location, female individuals of the commonest bee species of genera Bombus, Osmia, and Andrena were collected during the spring of 2015. In case of Bombus, only queens that fly early in the bumble bee life cycle could be sampled. Sampling authorization was obtained for protected areas in the southern provinces of the country. We only sampled on days with favorable weather conditions to ensure high bee activity. Bees were individually captured using a sweep net and directly euthanized by placing them on dry ice. At the end of the day, whole bees were stored in the lab at -80°C until further processing. The species determination was confirmed by sequencing of the COI barcode gene from a DNA extract of a single hind leg (see Supplementary File S1). Per locality, five individual bees of the same species were selected for RNA extraction. The species included were (Figure 1C): Andrena cineraria (Acin), Andrena fulva (Aful), Andrena haemorrhoa (Ahae), Bombus cryptarum (Bcry), Bombus pascuorum (Bpas), Bombus terrestris (Bter), Osmia bicornis (Obic), and Osmia cornuta (Ocor). The species name abbreviation is extended by a number that refers to one of the four sampling localities, for example, Bter1 refers to B. terrestris sampled from locality 1 (Figure 1B).
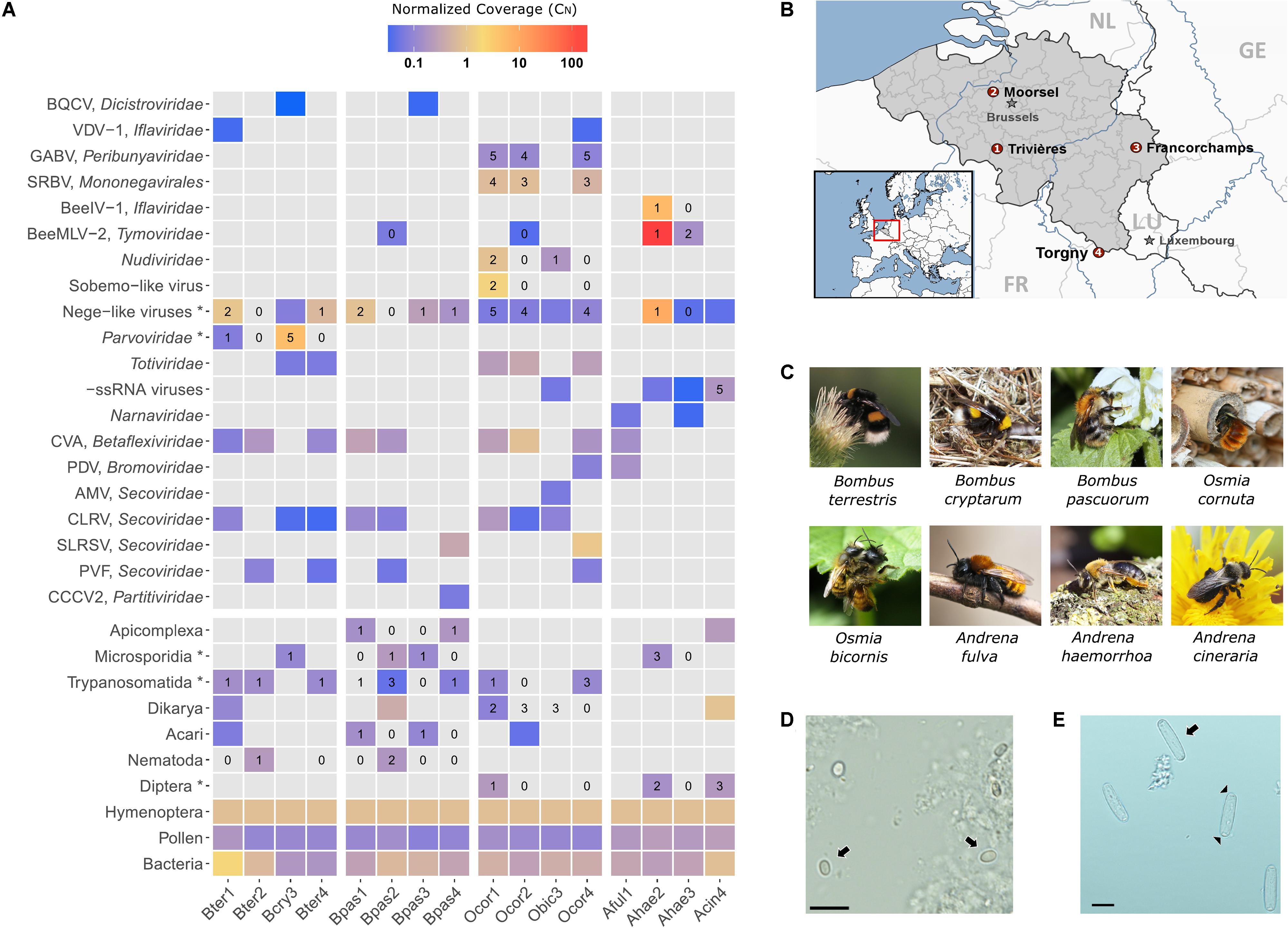
FIGURE 1. Metagenomics sequencing in eight wild bee species in Belgium. (A) Heatmap depicting the relative abundance of the identified bee viruses and other well-supported taxa across the 16 sequencing libraries. Gray-colored tiles indicate that the taxon was not detected by RNA-Seq. Numerical values represent the number of individuals per library (out of five) that were positive by RT-PCR analysis for the corresponding virus or parasite. Taxa marked with an asterisk (∗) include more than one species. For example, the taxon “Microsporidia” includes Nosema bombi, Nosema thomsoni, and Tubulinosema sp. (B) Geographical map of Belgium illustrating the four sampling locations (numbers 1–4 correspond to the sequencing libraries). The map was generated using StepMap. (C) Natural photographs of the eight different bee species investigated in this study. (D) Light microscopic image of spores (arrows) of the parasite Tubulinosema sp., bar: 10 μm. (E) Light microscopic image of sporocysts (arrows) of the parasite Apicystis sp. Note the navicular shape of the sporocysts. Black arrow heads indicate the dense polar plugs at both ends of the sporocyst, bar: 10 μm.
RNA Extraction and Sequencing
Prior to all tissue handlings, areas and pipettes were wiped with RNase Away® Reagent (Ambion®, Life Technologies). We assumed that the head and abdomen tagmata consist of the predominant target tissues for pathogenic viruses and parasites including the alimentary tract, fat bodies, and hypopharyngeal glands. Hence, the thorax tagma was entirely (including wings and legs) left out in order to reduce sample complexity and host fraction. Accordingly, a single bee at a time was dissected on a -80°C surface and tagmata were directly submerged in QIAzol Lysis Reagent (Qiagen). In case of non-corbiculate Osmia and Andrena bees, external pollen was removed from the abdominal scopa using a sterile knife. Tissues were disrupted using a tissue homogenizer (Precellys) for 90 s at 5000 Hz in the presence of 250 mg 0.1 mm Ø zirconium beads and five 2.3 mm Ø stainless steel beads. RNA was extracted using the RNeasy Lipid Tissue Mini kit (Qiagen) following manufacturer’s instructions including a 15 min on-column DNase I (Qiagen) digestion step. RNA concentration was determined using an ND-1000 UV–Vis Spectrophotometer (Nanodrop Technologies). At this stage, the total RNA was pooled per 5 individuals (10 μg RNA each) per species group and per locality, resulting in 16 pooled total RNA samples. cDNA library construction and sequencing were performed at Omega Bioservices, Norcross, GA, United States (Supplementary File S1). The cDNA library preparation method included a polyA+ RNA selective enrichment step and preserved stranded information of the original RNA fragments. An average of 32.7 M (27.2–40.5 M) paired-end 100 bp reads per library was obtained.
Data Analysis
A detailed report of all in silico protocols is given in Supplementary File S1. Briefly, after quality assessment, reads were trimmed and de novo assembled into contiguous sequences (contigs) using two independent algorithms, CLC Genomics Workbench 9 (Qiagen) and Trinity v2.2.0 (Grabherr et al., 2011). CLC de novo contigs were used for homology-based annotation while Trinity contigs were used for validation or refinement purposes. Contigs were queried against the latest versions (August, 2016) of the non-redundant nucleotide and protein sequence databases using BLAST+ v2.2.31 (Camacho et al., 2009) and DIAMOND v0.8.17 (Buchfink et al., 2015), respectively. Blast queries were split over multiple threads using GNU Parallel (Tange, 2011). Annotated contigs were binned based on the lowest common ancestor method implemented in MEGAN6 (Huson et al., 2007) and their fasta sequences extracted for each of the following taxa: Bacteria, Alveolata, Euglenozoa, Dikarya, Microsporidia, Nematoda, Chelicerata, Diptera, Hymenoptera, Viridiplantae, and Viruses. The taxon Viruses was manually refined to the family or species level. Subsequently, nucleotide and protein bins were merged using a custom bash script. Contigs of the taxon Hymenoptera were analyzed by the Benchmarking Universal Single-Copy Orthologs (BUSCO) v2 completeness assessment (Simao et al., 2015). All relevant hits to known or unknown viruses and organisms (except from plants and bacteria) are summarized in Supplementary Tables S1, S2, respectively. Further, the multi-fasta files per taxon were used as a reference for abundance analysis by aligning forward reads using HISAT2 v2.0.4 (Kim et al., 2015). Count data were normalized to a representative coverage value (scaled by reference length and host average coverage) in R v3.3.0 (R Development Core Team, 2011) and the normalized coverage (CN) was visualized as a heatmap using ggplot2 v2.2.1 (Wickham, 2009). The CN for each taxonomic group provides a rough abundance estimate where a value of 1 represents an equal transcript coverage relative to the host.
RT-PCR Confirmation and Cloning
All primer sequences and corresponding polymerase chain reaction (PCR) parameters used in this study are included in Supplementary File S1. As a general note, PCRs for screening purpose were carried out using HotStartTaq DNA polymerase (DNApol; QIAgen), PCRs for sanger sequencing and GenBank submissions using proof-reading Platinum Pfx DNApol (Thermo Scientific), or in case of difficult templates using expand long template PCR system (Roche). Amplicons were cloned using the TOPO-TA cloning system (Life Technologies) and plasmids were extracted using the GeneJET Plasmid Miniprep Kit (Thermo Scientific). Purified plasmids were sequenced using M13 and custom primers at GATC Biotech (Constance, Germany).
Virus Genome Organization and Phylogenetic Analyses
Open reading frames (ORFs) were predicted using the online Expasy Translate tool. Conserved domains were predicted from amino acid (aa) sequences using the web-based Interproscan portal. Coverage graphs were exported from CLC Genomics Workbench v8.5 (Qiagen) after mapping paired-end reads to the contigs. Viral taxonomy is based on the International Committee on Taxonomy of Viruses website with the inclusion of the genus type species where possible. All virus abbreviations and accession numbers are summarized in Supplementary File S2. The viral replicase protein was used as a phylogenetic marker for RNA viruses, the NS1 protein for ssDNA viruses, and the DNApol, per os infectivity factor 2 (pif-2), and late expression factor 8 proteins for dsDNA viruses. Multiple sequence analyses were carried out using MAFFT v7 (Katoh and Standley, 2013) and the L-INS-i (if single domain) or E-INS-i (if multiple domains) iterative refinement methods. The alignment was evaluated (TCS score higher than 5) (Chang et al., 2014) and trimmed. Using the best-fit protein evolution model as determined by ProtTest3 (Darriba et al., 2011), a maximum likelihood tree was estimated using PhyML v3.1 (Guindon et al., 2010) embedded in SeaView (Gouy et al., 2010).
Data Accessibility
Raw NGS data have been submitted to the SRA repository and can be accessed via the BioProject PRJNA411946. Nucleotide sequences that were cloned and confirmed by Sanger sequencing have been submitted to GenBank under the following name and accessions: Tubulinosema sp. isolate Bpas2 (MF998087) and Apicystis sp. isolate Bpas1 (MF998086). Other GenBank submissions based on in silico assembled contigs are: A. haemorrhoa nege-like virus isolate Ahae2 (MF998082), Bee iflavirus 1 isolate Ahae2 (MF998083), Bee macula-like virus 2 isolate Ahae2 (MF998084), and B. cryptarum densovirus isolate bcry3 (MF998085). CLC de novo and Trinity contigs were stored at a freely accessible Figshare collection (Schoonvaere, 2017), along with taxon-binned multi-fasta sequences, custom bash and R scripts that were generated in this study.
Results and Discussion
Host Transcripts Occupied the Highest Proportion of the Metatranscriptome
The host transcriptome fraction, referred to as Hymenoptera, ranged from 88 to 99% of the taxonomically informative reads across the 16 sequencing libraries. To measure the transcriptome completeness of the host, we performed a BUSCO assessment of contigs that were binned to Hymenoptera by MEGAN6 analysis. The different libraries produced a similar profile (Supplementary Figure S1) and the average BUSCO notation was C:66.4% [S:66.3%, D:0.1%], F:15.3%, M:18.3%, n:4415. In full, out of the 4415 single-copy orthologs in the Hymenoptera lineage, 2930 (66.3%) were complete and single, 5 (0.1%) were complete and duplicated, 667 (15.1%) were fragmented, and 807 (18.3%) were missing. The transcriptome completeness was similar to an independent RNA-Seq study of B. terrestris queens (Harrison et al., 2015) indicating that around 70% transcriptome completeness is generally achieved for this caste and sequencing method.
Insect-Specific Viruses with an RNA Genome
Two honey bee RNA viruses were detected in wild bees. Black queen cell virus (BQCV) was found in sympatric B. cryptarum and B. pascuorum of a single locality. BQCV was not found elsewhere. In both occurrences, the BQCV read count was very low (Figure 1A) and nucleotide (nt) similarity to honey bee BQCV strains was low. Varroa destructor virus-1 (VDV-1) was detected in B. terrestris and O. cornuta. Read count was very low (Figure 1A) but nt similarity to honey bee isolates was high. Intuitively, both BQCV and VDV-1 were present in very low copy number. Either the viruses were at an early infection stage in the bee host or they were associated with pollen (Singh et al., 2010). Two solitary bee viruses that we discovered earlier (Schoonvaere et al., 2016) were also detected here in O. cornuta: Scaldis River bee virus (SRBV) and Ganda bee virus (GABV). RT-PCR analysis confirmed SRBV in 66% individuals and GABV in 93% individuals (Figure 1A). GABV is related to Apis bunyavirus 2 that was recently discovered in honey bees (Remnant et al., 2017).
A novel iflavirus (Picornavirales), referred to as bee iflavirus 1 (BeeIV-1), was present in A. haemorrhoa. The virus is phylogenetically related to honey bee pathogenic viruses deformed wing virus, sacbrood virus, and slow bee paralysis virus (Figure 2B). However, the partial polyprotein of BeeIV-1 had highest aa identity to Dinocampus coccinelae paralysis virus, a virus that induces behavioral changes in its parasitoid host (Dheilly et al., 2015). We should note that the presence of BeeIV-1 in A. haemorrhoa was confounded by the co-infection of a conopid fly larva and a microsporidian. The possibility exists that BeeIV-1 was present as an infection of the fly larva rather than the bee host. However, BeeIV-1 was absent from another conopid-infected A. haemorrhoa individual suggesting no obligate relationship between BeeIV-1 and parasitic flies.

FIGURE 2. Genome organizations and phylogenetic analyses of wild bee viruses and insect-specific viruses distantly related to plant viruses. (A) The genome structure with in silico predicted ORF annotations (gray arrow boxes) on the assembled viral transcripts (black lines). In case a genome was not fully recovered by RNA-Seq, the probable complete genome sequence is represented by a gray dashed line. The sequence length in nucleotides is given by the numbers below the lines. Protein domains were predicted using interproscan and are depicted by yellow boxes on top of the ORFs. In blue is the coverage by re-alignment of RNA-Seq reads to the contigs. Below left each map is the sequencing library name wherein the virus was detected and it is underlined in case the depicted transcript originated from that library. ∗In case of O. cornuta nege-like virus, the contig was obtained by the combined de novo assembly of all Ocor datasets. Below right is the normalized coverage corresponding to the heatmap depicted in Figure 1A. (B) Maximum likelihood trees based on the partial replicase protein (RNA viruses) or non-structural protein 1 (densoviruses). Red abbreviations correspond to the new virus(es) for which a genome organization is given in (A). Blue abbreviations correspond to viruses also detected in the current study but not illustrated in (A). Green abbreviations correspond to known bee pathogenic viruses (not detected in this study). Virus full names and protein accession numbers are included in Supplementary File S2.
A novel macula-like virus (Tymovirales), referred to as bee macula-like virus 2 (BeeMLV-2), was detected in multiple hosts B. pascuorum, O. cornuta, and A. haemorrhoa but was only present in high copy number in library Ahae2 (Figure 1A). The recovered genome sequence of BeeMLV-2 was nearly complete and a similar organization was observed compared to related macula-like viruses (Figure 2A). Based on RdRp protein phylogeny, BeeMLV-2 clustered close to members of Maculavirus in the family Tymoviridae and its closest relative was Bombyx mori macula-like virus (Figure 2B). This virus was originally identified in cell cultures (Katsuma et al., 2005). BeeMLV-2 was more distantly related to bee macula-like virus (BeeMLV) that is currently unassigned in Tymoviridae awaiting genus designation. BeeMLV is a pathogen of honey bees and the parasitic mite Varroa destructor (de Miranda et al., 2015).
Two contigs in O. cornuta library Ocor2 showed homology to arthropod-infecting +ssRNA viruses that are distantly related to sobemoviruses. Although the primary hosts of sobemoviruses are plants, sobemo-like viruses have been reported in fruit flies (Webster et al., 2016), mosquitoes (Shi et al., 2016), ants, and mites (Tokarz et al., 2014). Phylogenetic analysis based on the replicase protein placed the sobemo-like virus together with other arthropod viruses in a clade close to Luteoviridae (Figure 2B). The longest transcript had a similar genome organization to that of Ixodes scapularis associated virus-2 where the viral serine protease ORF1 overlaps the RdRp protein ORF2, a feature that is shared among sobemoviruses.
Contigs related to insect-specific negeviruses and plant viruses of the family Virgaviridae were identified in all libraries except Aful1, Bter2, and Bpas2. Each bee species was associated with a particular nege-like virus. The genome sequence of viruses from different localities shared less than 1% sequence dissimilarity, which can be attributed to the natural variation in RNA viruses. The genome organization of four nege-like viruses are depicted in Figure 2A. Comparison of nege-like viruses from different bee species revealed strong analogy in genome organization and evolutionary relationship (Figure 2B). Based on RdRp protein phylogeny, the A. haemorrhoa nege-like virus was related to the Sandewavirus phylogenetic group of negeviruses. The RdRp protein was most similar (49% overall aa identity) to Tanay virus, a nege-like virus of mosquitoes (Nabeshima et al., 2014). The O. cornuta nege-like virus was phylogenetically related to nege-like viruses of the moth Epirrita autumnata (de Miranda et al., 2017), dipterans (Shi et al., 2016; Webster et al., 2016), and the plant bug Adelphocoris suturalis (Li et al., 2017). Bombus terrestris and B. pascuorum nege-like viruses were most related to viruses associated with nematodes (Shi et al., 2016). The viruses in B. terrestris and B. pascuorum were highly similar to each other (ORF1-3 predicted viral proteins had 92, 86, and 91% protein identity, respectively). It is worth noting that the nege-viral sequences were not detected in nematode-associated B. terrestris and B. pascuorum libraries (locality 2), despite of their relatively abundant presence in nematode-free libraries.
In all O. cornuta libraries, one or two sets of contigs had homology to totiviruses. The most representative genome sequence was obtained in library Ocor2 with a length of 6.6 Kb (Figure 2A). Genome organization is similar to that of Leptopilina boulardi toti-like virus (LbTV) where ORF1 overlaps ORF2 and translation of ORF2 is mediated by a -1 ribosomal frameshift. Sequences of libraries Ocor1 and Ocor2 are very similar (99% nt identity), but both are more dissimilar to the totivirus in library Ocor4 (93% nt identity) suggesting that different strains exist. Members of the family Totiviridae have a linear dsRNA genome of 4.6–6.7 Kb in size and are originally described from fungal hosts. However, several insect totiviruses have been discovered that currently fall apart in two groups, the one infecting Hymenoptera including parasitic wasps (Martinez et al., 2016) and ants (Koyama et al., 2015) and the other infecting Diptera including mosquitoes (Zhai et al., 2010) and fruit flies (Wu et al., 2010). The RdRp and capsid proteins predicted from the toti-like viral sequences in Ocor2 showed highest homology to a member of the first group (Figure 2B). In fact, Martinez et al. (Martinez et al., 2016) already showed the presence of fragmentary transcripts in O. cornuta transcriptome shotgun assembly data related to LbTV. Experimental studies with LbTV in a parasitoid wasp system showed that it is biparentally transmitted (through both eggs and sperm) and that infection positively correlates with developmental success (Martinez et al., 2016).
Sequences of –ssRNA viruses were too fragmented in order to discuss their genome organization and phylogeny. Two contigs of A. haemorrhoa had 68–70% aa identity to viruses of the genus Quaranjavirus, family Orthomyxoviridae. Eleven contigs (summed length 8837 nt) of O. bicornis consistently had around 80% aa identity to SRBV RdRp, nucleoprotein, and glycoprotein. Nine contigs (summed length 6394 nt) of A. cineraria had 36–42% aa identity to RdRp proteins of rhabdoviruses (Mononegavirales) including Wuhan ant virus and Apis rhabdovirus 1 and 2. Both rhabdoviruses were shown to elicit an anti-viral immune response in honey bees (Remnant et al., 2017). The rhabdo-like virus was confirmed to be present in all five A. cineraria individuals. Together, these reports add to the growing evidence that –ssRNA viruses are widespread in arthropods (Li et al., 2015) including bees (Schoonvaere et al., 2016; Remnant et al., 2017).
Insect-Specific Viruses with a DNA Genome
A dsDNA virus belonging to the Nudiviridae family was present in allopatric O. cornuta and O. bicornis females. The same virus was encountered in a previous metagenomic survey (Schoonvaere et al., 2016). We confirmed that both reports concern the same virus, which we tentatively name O. cornuta nudivirus (OcNV). OcNV is phylogenetically related to pathogenic nudiviruses in beetles (Huger, 2005), crickets (Huger, 1985), and dipterans (Webster et al., 2015) that belong to the genus Alphanudivirus (Figure 3B). Because it is not possible to obtain the complete genome sequence of a DNA virus by RNA sequencing, we mapped the OcNV transcriptome to the complete genome of a related nudivirus (Figure 3A) to estimate its completeness. Strikingly, OcNV homologs to all of the 20 core baculoviral genes shared among nudiviruses and baculoviruses (Wang et al., 2012) were expressed. The summed transcript length of the 74 nudivirus orthologs identified in library Ocor1 was 94.4 Kbp which is nearly the genome length of Kallithea virus (96.9 Kbp). We expect that the genome of OcNV exceeds 100 Kbp because there are likely more OcNV-specific proteins that were not detected here. The abundant presence of viral RNA transcripts that originate from a common DNA template at least indicates that OcNV is actively replicating inside bees. Further, the relatively low number of OcNV positive bees in spatially separated populations of two closely related bee species suggests a specialized relationship, though further studies need to confirm this hypothesis. Since O. cornuta and O. bicornis are commercially distributed pollinators in Europe, caution should be taken in the trade of bees that carry OcNV before more is known about possible pathogenicity and reproductive success effects of this virus.
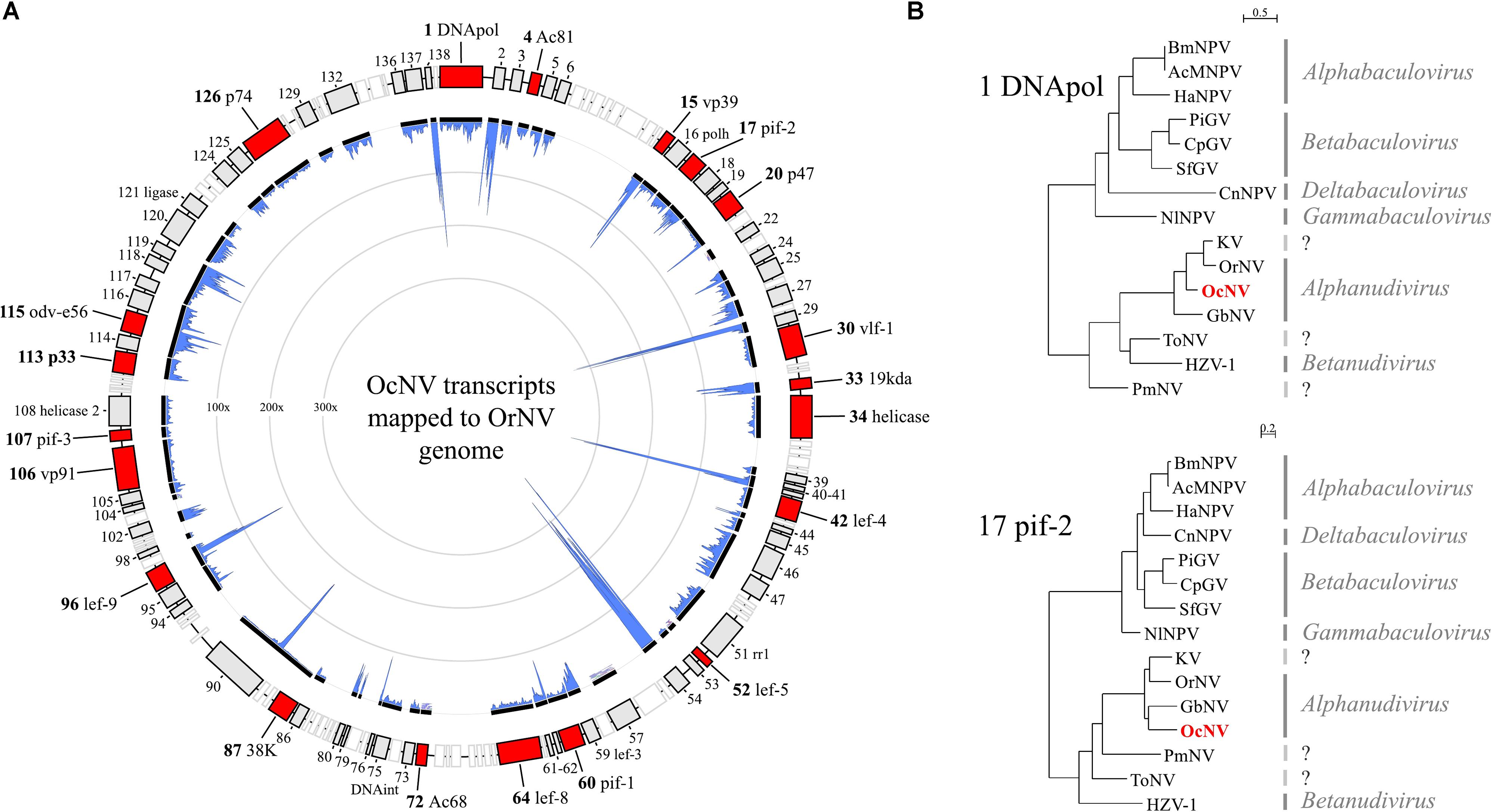
FIGURE 3. The transcriptome and phylogenetic analysis of Osmia cornuta nudivirus (OcNV). (A) Nudiviral transcripts (black lines) were mapped to the genome map of Oryctes rhinoceros nudivirus (OrNV). Note that the order of the transcripts is not representative for OcNV since genomic reorganizations are common in dsDNA viruses. In total, 74 orthologs with respect to OrNV genes (boxes) were identified. Red boxes represent the 20 core baculoviral genes that are shared among Baculoviridae and Nudiviridae. Empty (white) boxes represent OcNV genes that were not found in the OrNV transcriptome. In blue is the coverage (100–300x) by realignment of RNA-Seq reads to the contigs which correlates to the abundance of the viral transcript in the library Ocor2. (B) Maximum likelihood trees of OcNV based on DNApol and pif-2 conserved domains pfam03104 and pfam04631, respectively. The phylogenetic analyses included viruses of the families Baculoviridae (four genera) and Nudiviridae (two genera). A third analysis with late expression factor 8 protein gave a tree with a similar topology to that of DNApol and was considered redundant. Both trees depicted here placed OcNV close to OrNV and Gryllus bimaculatus nudivirus of the genus Alphanudivirus. Virus full names and protein accession numbers are included in Supplementary File S2.
Bombus cryptarum (Bcry3) and one B. terrestris library (Bter1) were associated with viral sequences of ssDNA viruses of the family Parvoviridae. The closest relative (36% protein identity) was the ambidensovirus Diaphorina citri densovirus (DcDNV), although phylogenetic analysis of the replicase protein placed the B. cryptarum densovirus together with Lone star tick densovirus 1 (LstDNV-1) in a separate clade (Figure 2B). A characteristic feature of ambidensoviruses is that structural (VP) and non-structural (NS) cassettes are transcribed from opposite DNA strands. In case of DcDNV, the VP and NS cassettes are separated by 21 nt (Nigg et al., 2016). Since we initially identified full-length VP and NS densoviral ORFs in two Bcry3 transcripts that could not be linked by paired-end RNA-Seq read data (Figure 2A), we anticipated a non-overlapping, ambisense genome organization similar to that of DcDNV. Indeed, gap-spanning PCR on a genomic DNA extract of an infected bee and Sanger sequencing of the amplicon confirmed that both transcripts originate from the same DNA template but are in opposite orientation and separated by 30 nt (the actual number depends on the true size of the viral transcripts). Densoviral contigs were also found in library Bter1 with 70–75 and 88% nucleotide identity to the densoviral NS and VP transcripts in Bcry3, respectively, although the coverage was much lower. RT-PCR revealed that all (5) B. cryptarum queens and one B. terrestris queen were associated with a densovirus (Figure 1A). Despite of the broad host range of densoviruses in insects, none so far have been reported in bees. The relation of densoviruses of the subfamily Densovirinae to their arthropod host ranges from mutualistic (Xu et al., 2014) to severe pathology (Dhar et al., 2014), which is especially problematic in large insect rearing facilities (Szelei et al., 2011). In this respect, it is worth to investigate the biological relationship between the densoviruses that were discovered here and the commercial pollinator species B. terrestris.
Plant and Fungal Viruses
Seven well-characterized plant viruses were detected: cherry virus A (CVA), Prunus dwarf virus (PDV), cherry leaf roll virus (CLRV), Prunus virus F (PVF), strawberry latent ringspot virus (SLRSV), arabis mosaic virus (AMV), and crimson clover cryptic virus 2 (CCCV-2). Except from CCCV-2, all are +ssRNA viruses and some are serious pathogens of crops (Cooper, 1979). Only the genome of PDV is not polyadenylated. The normalized coverage of the plant viruses was low to moderate in comparison to most insect-specific viruses; however, CVA and SLRSV were, respectively, more abundant in Ocor2 and Ocor4 libraries (Figure 1A). Because the primary source of plant viruses in bees is likely to be dietary pollen, we anticipated a correlation between the abundance of plant virus and pollen-derived reads. Surprisingly, no plant viruses were detected in A. haemorrhoa and A. cineraria despite of the larger proportion of pollen-derived reads in these hosts. Four of the plant viruses detected in this study belong to the family Secoviridae (order Picornavirales) of which AMV and CLRV are nepoviruses. Recently, a plant pathogenic nepovirus (Secoviridae) was demonstrated to replicate in and negatively affect honey bees (Li et al., 2014). We could not infer from our data whether these viruses replicated in bees. However, CLRV was detected in each locality and associated with all species except Andrena spp.
In two Andrena libraries, low-abundant sequences were detected with up to 40% protein homology to narna-like viruses isolated from yeasts and crustaceans. Narnaviruses (Narnaviridae) have a monopartite, linear +ssRNA genome of 2.5–2.9 Kb in size and naturally infect fungi. Two fragmentary, narna-like viral contigs in A. fulva had a summed length of 2077 nt and a candidate partial genome sequence of 2.6 Kb was manually refined.
Typical Bee Parasites: Trypanosomes, Microsporidia, and Neogregarines
The trypanosome Crithidia bombi was commonly detected in B. terrestris, B. pascuorum, and O. cornuta (Figure 1A), and typically occupied less than 0.6% of sequencing libraries. A trypanosome nearly identical to Crithidia pragensis was identified in O. cornuta (Table 1). Crithidia pragensis is originally described from the fly Cordilura albipes in Central Europe (Yurchenko et al., 2014). The library fraction that was occupied by C. pragensis was similar to that of C. bombi indicating that it was present as an internal parasite. There was no evidence for a co-infection of a parasitic fly species although the same individual carried the viruses OcNV, GABV, SRBV, O. cornuta nege-like virus, and a sobemo-like virus. The microsporidian Nosema bombi was found in sympatric B. cryptarum and B. pascuorum but not elsewhere (Figure 1A). Either the microsporidium Nosema thomsoni or a very related species was associated with A. haemorrhoa (Figure 1A). It was most identical to the Nosema associated with Andrena vaga (Ravoet et al., 2014). A third microsporidian transcriptome was detected in library Bpas2 that was clearly different from Nosema. The partial ribosomal DNA cassette of the microsporidium was cloned and its SSU rRNA sequence was equally similar to that of Tubulinosema pampeana and Tubulinosema loxostegi (Table 2), which, respectively, are low-virulent parasites recently described from Bombus atratus in S-America (Plischuk et al., 2015) and the moth Loxostege sticticalis in W-Siberia (Malysh et al., 2013). The Tubulinosema infected bumble bee was co-infected with C. bombi and the nematode Sphaerularia bombi. Ovoid morphologies that strongly resemble the spores of T. pampeana were observed in the thorax of the infected bumble bee (Figure 1D). An apicomplexan parasite equally similar to Apicystis bombi as Mattesia geminata (Table 3) was associated with two B. pascuorum libraries. However, microscopical examination of the infected thoraces revealed morphisms similar in shape as the navicular sporocysts of A. bombi (Figure 1E).
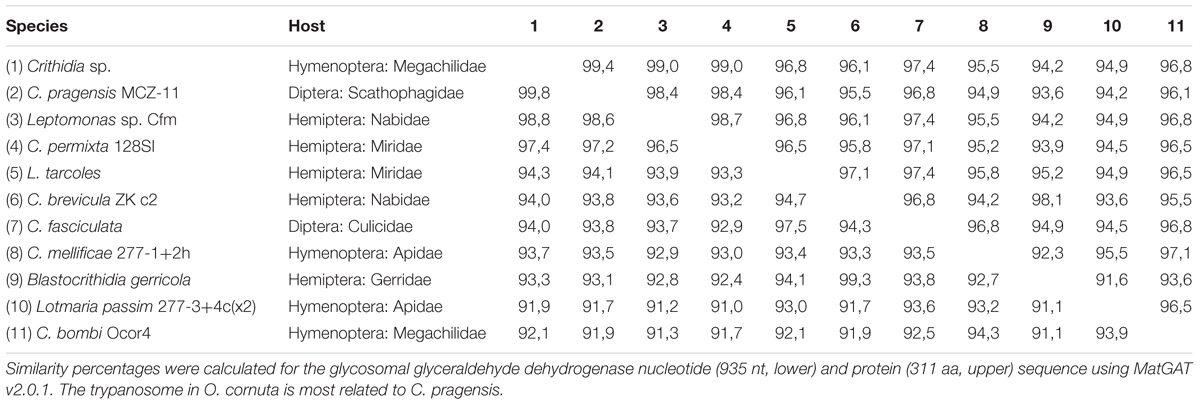
TABLE 1. Sequence similarity matrix of C. pragensis and related trypanosomes.
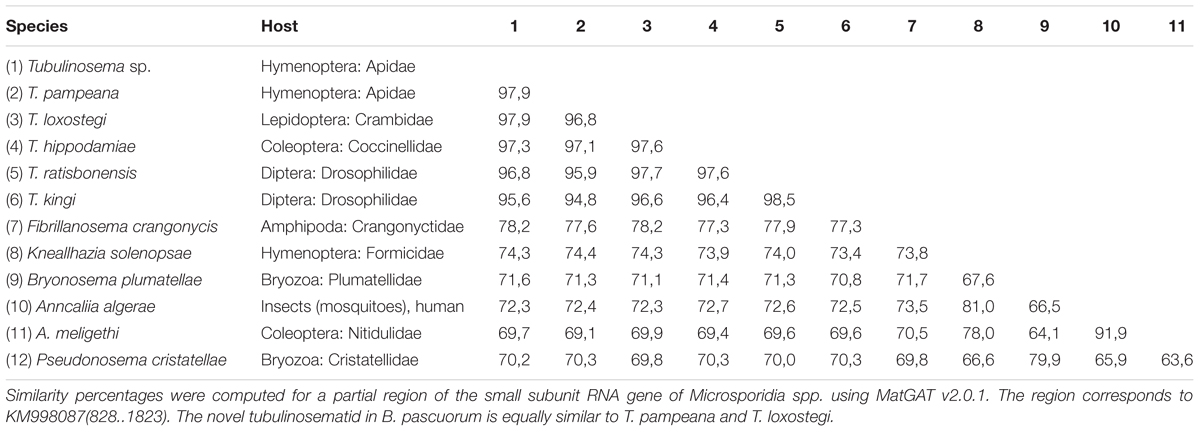
TABLE 2. Sequence similarity matrix of Tubulinosema sp. and related apicomplexans.
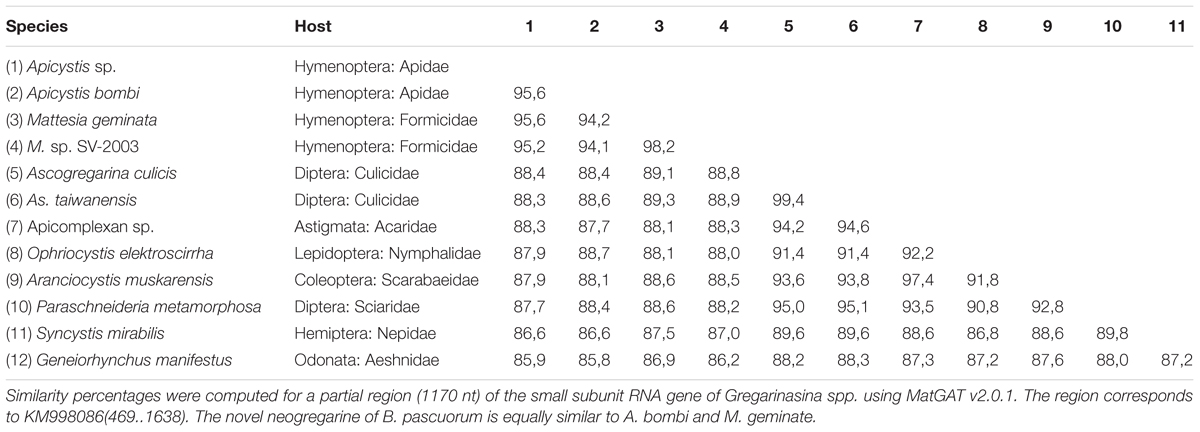
TABLE 3. Sequence similarity matrix of Apicystis sp. and related neogregarines.
Ascomycetous Yeasts and Basidiomycete Fungi
The summit of an ascomycetous yeast transcriptome was observed. About 100 protein-coding transcripts were annotated. The four most abundant transcripts were cytochrome C oxidase subunits I and II, elongation factor 1 alpha, and a mitochondrial carrier protein. Based on sequence similarity of the 5′ partial large subunit RNA and small subunit RNA, the closest related species were Metschnikowia pulcherima, Metschnikowia chrysoperlae, and Metschnikowia (Candida) pimensis. All are members of the M. pulcherima clade which is primarily associated with nectar and fruit-feeding insects (Lachance, 2016). Some members of this clade produce pulcherrimin, an iron-chelating molecule that induces antagonistic activity against fungi and bacteria (Sipiczki, 2006). Other clades are pathogens of marine arthropods or are strictly associated with beetles (Lachance, 2016). Yeasts of the genus Metschnikowia are not new to bees. In fact, it was long assumed that a mutualistic relation exists between both, wherein the yeast hibernates inside its bee host, although recent experimental evidence contradicts this hypothesis (Brysch-Herzberg, 2004). The size of the yeast transcriptome detected here is similar to that of internal bee parasites such as Crithidia and Nosema spp. and at least indicates that the yeasts cells were abundantly present. Metschnikowia contigs were also found in library Acin4 but only originated from ribosomal RNA genes. Further, several contigs were found in libraries Bter1 and Bpas2 that had annotations to basidiomycete genes including Hypsizygus marmoreus, Serpula himantioides, Pycnoporus cocci, and Pleurotus sp. The significance of these associations is likely very low and probably results from external contaminants.
Metazoan Bee Parasites
The parasitic nematode S. bombi was present in one B. terrestris queen and two B. pascuorum queens of a single locality (Bter2 and Bpas2), but was absent elsewhere (Figure 1A). The nematode transcriptome fraction occupied 5.5 and 2.3% of the sequencing library, respectively. Sequence comparison of conserved house-keeping transcripts revealed that S. bombi of interspecific hosts was genetically distinct, which suggests a host-specific haplotype structuring of this parasite. Further, the tracheal mite Locustacarus buchneri was molecularly detected in B. pascuorum queens (Figure 1A). Finally, the larvae of two conopid fly species, Myopa testacea and Myopa tesselatipennis, were detected as abdominal parasites. Myopa testacea was present in one O. cornuta female and three A. cineraria females whereas M. tesselatipennis was present in two A. haemorrhoa females (Figure 1A). The fly transcriptome typically occupied less than 3% of the sequencing library.
Conclusion
In this study, we combined a polyadenylated RNA enrichment with high-throughput RNA sequencing to explore the metatranscriptome of eight social and solitary wild bee species, of which the transcriptomes of five species (A. cineraria, A. haemorrhoa, A. fulva, B. cryptarum, and B. pascuorum) were not sequenced earlier. We identified over 20 associated viruses, 10 different species of bee parasites, and associated fungi. Two unknown RNA viruses in A. haemorrhoa (bee iflavirus 1 and bee macula-like virus 2) and two unknown DNA viruses in Osmia spp. and Bombus spp. (OcNV and a densovirus, respectively) were related to other pathogenic viruses of insects. These viruses may be of pathological importance to the studied bees and other bee species, or in case of the DNA viruses to the rearing of the commercial pollinator species O. cornuta, O. bicornis, and B. terrestris. However, the potential pathological association of the viruses has to be validated by follow-up studies that include isolation of the virus particles and experimental inoculation in the bee host. We also discovered the widespread occurrence of nege-like viruses, negative-stranded RNA viruses, and other insect-specific viruses, which is in congruence with recent large-scale, viral metagenomics studies in arthropods. However, the role of these viruses and their significance with respect to insects in general is unknown. Further, next to typical bee parasites, we detected for the first time in Europe a Tubulinosema parasite in a bumble bee host, a fly trypanosome parasite that infected O. cornuta, and a neogregarine parasite that is related but molecularly dissimilar to A. bombi. We encourage future prevalence studies to include the novel associations of viruses and parasites reported here to learn more about their geographical distribution and impact on pollinator health.
Author Contributions
DdG, GS, and KS conceived the study. KS performed the sampling and practical work. DdG, FF, and KS wrote the manuscript. DdG, GS, FF, and KS revised the manuscript.
Funding
Belgian Science Policy (BELSPO; BR/132/A1/BELBEES; Multi-disciplinary assessment of BELgian wild BEE decline to adapt mitigation management policy).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Prof. Dr. Bart Braeckman and Andy Vierstraete for kindly providing access and support to a Linux platform. We also thank entomologist and field expert Chantal Deschepper who provided high-quality natural photographs of the wild bee species studied in this work. The photograph of Andrena fulva was taken by S. Robinson and purchased from Shutterstock.com (ID 102069679). The computational resources (Stevin Supercomputer Infrastructure) and services used in this work were provided by the VSC (Flemish Supercomputer Center), funded by Ghent University, the Hercules Foundation, and the Flemish Government Department EWI.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00177/full#supplementary-material
FIGURE S1 | BUSCO completeness assessment.
TABLE S1 | Description of viruses associated with wild bees.
TABLE S2 | Description of protozoan and metazoan parasites associated with wild bees.
FILE S1 | Supplementary information for Materials and Methods.
FILE S2 | Virus abbreviations and protein accessions used in phylogenetics analyses.
References
Aufauvre, J., Misme-Aucouturier, B., Vigues, B., Texier, C., Delbac, F., and Blot, N. (2014). Transcriptome analyses of the honeybee response to Nosema ceranae and insecticides. PLOS ONE 9:e91686. doi: 10.1371/journal.pone.0091686
Brown, M. J. F. (2017). Microsporidia: an emerging threat to bumblebees? Trends Parasitol. 33, 754–762. doi: 10.1016/j.pt.2017.06.001
Brysch-Herzberg, M. (2004). Ecology of yeasts in plant-bumblebee mutualism in Central Europe. FEMS Microbiol. Ecol. 50, 87–100. doi: 10.1016/j.femsec.2004.06.003
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Chang, J. M., Di Tommaso, P., and Notredame, C. (2014). TCS: a new multiple sequence alignment reliability measure to estimate alignment accuracy and improve phylogenetic tree reconstruction. Mol. Biol. Evol. 31, 1625–1637. doi: 10.1093/molbev/msu117
Cooper, J. I. (1979). Virus Diseases of Trees and Shrubs. Cambridge: Institute of Terrestrial Ecology.
Cox-Foster, D. L., Conlan, S., Holmes, E. C., Palacios, G., Evans, J. D., Moran, N. A., et al. (2007). A metagenomic survey of microbes in honey bee colony collapse disorder. Science 318, 283–287. doi: 10.1126/science.1146498
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). ProtTest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
de Miranda, J. R., Bailey, L., Ball, B. V., Blanchard, P., Budge, G. E., Chejanovsky, N., et al. (2013). Standard methods for virus research in Apis mellifera. J. Apic. Res. 52, 1–56. doi: 10.3896/IBRA.1.52.4.22
de Miranda, J. R., Cornman, R. S., Evans, J. D., Semberg, E., Haddad, N., Neumann, P., et al. (2015). Genome characterization, prevalence and distribution of a macula-like virus from Apis mellifera and Varroa destructor. Viruses 7, 3586–3602. doi: 10.3390/v7072789
de Miranda, J. R., Hedman, H., Onorati, P., Stephan, J., Karlberg, O., Bylund, H., et al. (2017). Characterization of a novel RNA virus discovered in the autumnal moth Epirrita autumnata in Sweden. Viruses 9, 214. doi: 10.3390/v9080214
DeLay, B., Mamidala, P., Wijeratne, A., Wijeratne, S., Mittapalli, O., Wang, J., et al. (2012). Transcriptome analysis of the salivary glands of potato leafhopper, Empoasca fabae. J. Insect Physiol. 58, 1626–1634. doi: 10.1016/j.jinsphys.2012.10.002
Dhar, A. K., Robles-Sikisaka, R., Saksmerprome, V., and Lakshman, D. K. (2014). Biology, genome organization, and evolution of parvoviruses in marine shrimp. Adv. Virus Res. 89, 85–139. doi: 10.1016/B978-0-12-800172-1.00003-3
Dheilly, N. M., Maure, F., Ravallec, M., Galinier, R., Doyon, J., Duval, D., et al. (2015). Who is the puppet master? Replication of a parasitic wasp-associated virus correlates with host behaviour manipulation. Proc. Biol. Sci. 282:20142773. doi: 10.1098/rspb.2014.2773
Dolezal, A. G., Hendrix, S. D., Scavo, N. A., Carrillo-Tripp, J., Harris, M. A., Wheelock, M. J., et al. (2016). Honey bee viruses in wild bees: viral prevalence, loads, and experimental inoculation. PLOS ONE 11:e0166190. doi: 10.1371/journal.pone.0166190
Douglas, A. E. (2015). Multiorganismal insects: diversity and function of resident microorganisms. Annu. Rev. Entomol. 60, 17–34. doi: 10.1146/annurev-ento-010814-020822
Engel, P., Kwong, W. K., McFrederick, Q., Anderson, K. E., Barribeau, S. M., Chandler, J. A., et al. (2016). The Bee microbiome: impact on bee health and model for evolution and ecology of host-microbe interactions. mBio 7:e2164-15. doi: 10.1128/mBio.02164-15
Evans, J. D., and Schwarz, R. S. (2011). Bees brought to their knees: microbes affecting honey bee health. Trends Microbiol. 19, 614–620. doi: 10.1016/j.tim.2011.09.003
Gouy, M., Guindon, S., and Gascuel, O. (2010). SeaView version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Harrison, M. C., Hammond, R. L., and Mallon, E. B. (2015). Reproductive workers show queenlike gene expression in an intermediately eusocial insect, the buff-tailed bumble bee Bombus terrestris. Mol. Ecol. 24, 3043–3063. doi: 10.1111/mec.13215
Huger, A. M. (1985). A new virus disease of crickets (Orthoptera: Gryllidae) causing macronucleosis of fatbody. J. Invertebr. Pathol. 45, 108–111. doi: 10.1016/0022-2011(85)90055-2
Huger, A. M. (2005). The Oryctes virus: its detection, identification, and implementation in biological control of the coconut palm rhinoceros beetle, Oryctes rhinoceros (Coleoptera: Scarabaeidae). J. Invertebr. Pathol. 89, 78–84. doi: 10.1016/j.jip.2005.02.010
Husband, R. W., and Shina, R. N. (1970). A revision of the genus Locustacarus with a key to the genera of the family Podapolipidae (Acarina). Ann. Entomol. Soc. Am. 63, 1152–1162. doi: 10.1093/aesa/63.4.1152
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Katsuma, S., Tanaka, S., Omuro, N., Takabuchi, L., Daimon, T., Imanishi, S., et al. (2005). Novel macula-like virus identified in Bombyx mori cultured cells. J. Virol. 79, 5577–5584. doi: 10.1128/JVI.79.9.5577-5584.2005
Kim, D., Langmead, B., and Salzberg, S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12, 357–360. doi: 10.1038/nmeth.3317
Koyama, S., Urayama, S., Ohmatsu, T., Sassa, Y., Sakai, C., Takata, M., et al. (2015). Identification, characterization and full-length sequence analysis of a novel dsRNA virus isolated from the arboreal ant Camponotus yamaokai. J. Gen. Virol. 96(Pt 7), 1930–1937. doi: 10.1099/vir.0.000126
Lachance, M. A. (2016). Metschnikowia: half tetrads, a regicide and the fountain of youth. Yeast 33, 563–574. doi: 10.1002/yea.3208
Lee, F. J., Rusch, D. B., Stewart, F. J., Mattila, H. R., and Newton, I. L. (2015). Saccharide breakdown and fermentation by the honey bee gut microbiome. Environ. Microbiol. 17, 796–815. doi: 10.1111/1462-2920.12526
Li, C. X., Shi, M., Tian, J. H., Lin, X. D., Kang, Y. J., Chen, L. J., et al. (2015). Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife 4:e05378. doi: 10.7554/eLife.05378
Li, J. L., Cornman, R. S., Evans, J. D., Pettis, J. S., Zhao, Y., Murphy, C., et al. (2014). Systemic spread and propagation of a plant-pathogenic virus in European Honeybees, Apis mellifera. mBio 5:e898-13. doi: 10.1128/mBio.00898-13
Li, X., Xu, P., Yang, X., Yuan, H., Chen, L., and Lu, Y. (2017). The genome sequence of a novel RNA virus in Adelphocoris suturalis. Arch. Virol. 162, 1397–1401. doi: 10.1007/s00705-016-3211-2
Lipa, J. J., and Triggiani, O. (1996). Apicystis gen nov and Apicystis bombi (Liu, Macfarlane & Pengelly) comb nov (Protozoa: Neogregarinida), a cosmopolitan parasite of Bombus and Apis (Hymenoptera: Apidae). Apidologie 27, 29–34. doi: 10.1051/apido:19960104
Maebe, K., Meeus, I., Vray, S., Claeys, T., Dekoninck, W., Boeve, J. L., et al. (2016). A century of temporal stability of genetic diversity in wild bumblebees. Sci. Rep. 6:38289. doi: 10.1038/srep38289
Malysh, J. M., Tokarev, Y. S., Sitnicova, N. V., Martemyanov, V. V., Frolov, A. N., and Issi, I. V. (2013). Tubulinosema loxostegi sp. n. (Microsporidia: Tubulinosematidae) from the Beet Webworm Loxostege sticticalis L. (Lepidoptera: Crambidae) in Western Siberia. Acta Protozool. 52, 299–309. doi: 10.4467/16890027AP.13.028.1319
Martinez, J., Lepetit, D., Ravallec, M., Fleury, F., and Varaldi, J. (2016). Additional heritable virus in the parasitic wasp Leptopilina boulardi: prevalence, transmission and phenotypic effects. J. Gen. Virol. 97, 523–535. doi: 10.1099/jgv.0.000360
McMahon, D. P., Furst, M. A., Caspar, J., Theodorou, P., Brown, M. J., and Paxton, R. J. (2015). A sting in the spit: widespread cross-infection of multiple RNA viruses across wild and managed bees. J. Anim. Ecol. 84, 615–624. doi: 10.1111/1365-2656.12345
Nabeshima, T., Inoue, S., Okamoto, K., Posadas-Herrera, G., Yu, F., Uchida, L., et al. (2014). Tanay virus, a new species of virus isolated from mosquitoes in the Philippines. J. Gen. Virol. 95(Pt 6), 1390–1395. doi: 10.1099/vir.0.061887-0
Nigg, J. C., Nouri, S., and Falk, B. W. (2016). Complete genome sequence of a putative densovirus of the Asian citrus psyllid, Diaphorina citri. Genome Announc. 4:e589-16. doi: 10.1128/genomeA.00589-16
Oppenheim, S. J., Baker, R. H., Simon, S., and DeSalle, R. (2015). We can’t all be supermodels: the value of comparative transcriptomics to the study of non-model insects. Insect Mol. Biol. 24, 139–154. doi: 10.1111/imb.12154
Peng, W. J., Li, J. L., Boncristiani, H., Strange, J. P., Hamilton, M., and Chen, Y. P. (2011). Host range expansion of honey bee black queen cell virus in the bumble bee, Bombus huntii. Apidologie 42, 650–658. doi: 10.1007/s13592-011-0061-5
Peters, R. S., Krogmann, L., Mayer, C., Donath, A., Gunkel, S., Meusemann, K., et al. (2017). Evolutionary history of the hymenoptera. Curr. Biol. 27, 1013–1018. doi: 10.1016/j.cub.2017.01.027
Pionar, G. O., and Van der Laan, P. A. (1972). Morphology and life history of Sphaerularia bombi. Nematologica 18, 239–252. doi: 10.1163/187529272X00476
Plischuk, S., Sanscrainte, N. D., Becnel, J. J., Estep, A. S., and Lange, C. E. (2015). Tubulinosema pampeana sp. n. (Microsporidia, Tubulinosematidae), a pathogen of the South American bumble bee Bombus atratus. J. Invertebr. Pathol. 126, 31–42. doi: 10.1016/j.jip.2015.01.006
R Development Core Team (2011). R: A Language and Environment for Statistical Computing. Available at: http://www.R-project.org/
Ravoet, J., De Smet, L., Meeus, I., Smagghe, G., Wenseleers, T., and de Graaf, D. C. (2014). Widespread occurrence of honey bee pathogens in solitary bees. J. Invertebr. Pathol. 122, 55–58. doi: 10.1016/j.jip.2014.08.007
Remnant, E. J., Shi, M., Buchmann, G., Blacquiere, T., Holmes, E. C., Beekman, M., et al. (2017). A diverse range of novel RNA viruses in geographically distinct honey bee populations. J. Virol. 91:e158-17. doi: 10.1128/JVI.00158-17
Riddell, C. E., Lobaton Garces, J. D., Adams, S., Barribeau, S. M., Twell, D., and Mallon, E. B. (2014). Differential gene expression and alternative splicing in insect immune specificity. BMC Genomics 15:1031. doi: 10.1186/1471-2164-15-1031
Runckel, C., Flenniken, M. L., Engel, J. C., Ruby, J. G., Ganem, D., Andino, R., et al. (2011). Temporal analysis of the honey bee microbiome reveals four novel viruses and seasonal prevalence of known viruses, Nosema, and Crithidia. PLOS ONE 6:e20656. doi: 10.1371/journal.pone.0020656
Schmid-Hempel, R., and Tognazzo, M. (2010). Molecular divergence defines two distinct lineages of Crithidia bombi (Trypanosomatidae), parasites of bumblebees. J. Eukaryot. Microbiol. 57, 337–345. doi: 10.1111/j.1550-7408.2010.00480.x
Schoonvaere, K. (2017). Study of the virome of 8 wild bee species in Belgium. doi: 10.6084/m9.figshare.c.3902704.v1
Schoonvaere, K., De Smet, L., Smagghe, G., Vierstraete, A., Braeckman, B. P., and de Graaf, D. C. (2016). Unbiased RNA shotgun metagenomics in social and solitary wild bees detects associations with Eukaryote Parasites and New Viruses. PLOS ONE 11:e0168456. doi: 10.1371/journal.pone.0168456
Shi, M., Lin, X. D., Tian, J. H., Chen, L. J., Chen, X., Li, C. X., et al. (2016). Redefining the invertebrate RNA virosphere. Nature 540, 539–543. doi: 10.1038/nature20167
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Singh, R., Levitt, A. L., Rajotte, E. G., Holmes, E. C., Ostiguy, N., Vanengelsdorp, D., et al. (2010). RNA Viruses in hymenopteran pollinators: evidence of inter-taxa virus transmission via pollen and potential impact on non-Apis hymenopteran species. PLOS ONE 5:e14357. doi: 10.1371/journal.pone.0014357
Sipiczki, M. (2006). Metschnikowia strains isolated from botrytized grapes antagonize fungal and bacterial growth by iron depletion. Appl. Environ. Microbiol. 72, 6716–6724. doi: 10.1128/AEM.01275-06
Smith, K. G. V. (1969). Diptera, Conopidae, Handbooks for The Identification of British Insects, Vol. 10. London: Royal Entomological Society.
Szelei, J., Woodring, J., Goettel, M. S., Duke, G., Jousset, F. X., Liu, K. Y., et al. (2011). Susceptibility of North-American and European crickets to Acheta domesticus densovirus (AdDNV) and associated epizootics. J. Invertebr. Pathol. 106, 394–399. doi: 10.1016/j.jip.2010.12.009
Tange, O. (2011). GNU parallel - the command-line power tool. USENIX Mag. 36, 42–47. doi: 10.5281/zenodo.16303
Tokarz, R., Williams, S. H., Sameroff, S., Sanchez Leon, M., Jain, K., and Lipkin, W. I. (2014). Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J. Virol. 88, 11480–11492. doi: 10.1128/JVI.01858-14
Tozkar, C. O., Kence, M., Kence, A., Huang, Q., and Evans, J. D. (2015). Metatranscriptomic analyses of honey bee colonies. Front. Genet. 6:100. doi: 10.3389/fgene.2015.00100
Wang, Y., Bininda-Emonds, O. R., and Jehle, J. A. (2012). “Nudivirus genomics and phylogeny,” in Viral Genomes - Molecular Structure, Diversity, Gene Expression Mechanisms and Host-Virus Interactions, ed. M. Garcia (Rijeka: InTech).
Wang, Z., Gerstein, M., and Snyder, M. (2009). RNA-Seq: a revolutionary tool for transcriptomics. Nat. Rev. Genet. 10, 57–63. doi: 10.1038/nrg2484
Webster, C. L., Longdon, B., Lewis, S. H., and Obbard, D. J. (2016). Twenty-five new viruses associated with the Drosophilidae (Diptera). Evol. Bioinform. Online 12(Suppl. 2), 13–25. doi: 10.4137/EBO.S39454
Webster, C. L., Waldron, F. M., Robertson, S., Crowson, D., Ferrari, G., Quintana, J. F., et al. (2015). The discovery, distribution, and evolution of viruses associated with Drosophila melanogaster. PLOS Biol. 13:e1002210. doi: 10.1371/journal.pbio.1002210
Whitacre, L. K., Tizioto, P. C., Kim, J., Sonstegard, T. S., Schroeder, S. G., Alexander, L. J., et al. (2015). What’s in your next-generation sequence data? An exploration of unmapped DNA and RNA sequence reads from the bovine reference individual. BMC Genomics 16:1114. doi: 10.1186/s12864-015-2313-7
Woodard, S. H., Fischman, B. J., Venkat, A., Hudson, M. E., Varala, K., Cameron, S. A., et al. (2011). Genes involved in convergent evolution of eusociality in bees. Proc. Natl. Acad. Sci. U.S.A. 108, 7472–7477. doi: 10.1073/pnas.1103457108
Wu, Q., Luo, Y., Lu, R., Lau, N., Lai, E. C., Li, W. X., et al. (2010). Virus discovery by deep sequencing and assembly of virus-derived small silencing RNAs. Proc. Natl. Acad. Sci. U.S.A. 107, 1606–1611. doi: 10.1073/pnas.0911353107
Xu, P., Liu, Y., Graham, R. I., Wilson, K., and Wu, K. (2014). Densovirus is a mutualistic symbiont of a global crop pest (Helicoverpa armigera) and protects against a baculovirus and Bt biopesticide. PLOS Pathog. 10:e1004490. doi: 10.1371/journal.ppat.1004490
Yin, X. W., Iovinella, I., Marangoni, R., Cattonaro, F., Flamini, G., Sagona, S., et al. (2013). Odorant-binding proteins and olfactory coding in the solitary bee Osmia cornuta. Cell. Mol. Life Sci. 70, 3029–3039. doi: 10.1007/s00018-013-1308-2
Yurchenko, V., Votypka, J., Tesarova, M., Klepetkova, H., Kraeva, N., Jirku, M., et al. (2014). Ultrastructure and molecular phylogeny of four new species of monoxenous trypanosomatids from flies (Diptera: Brachycera) with redefinition of the genus Wallaceina. Folia Parasitol. 61, 97–112.
Zhai, Y., Attoui, H., Mohd Jaafar, F., Wang, H. Q., Cao, Y. X., Fan, S. P., et al. (2010). Isolation and full-length sequence analysis of Armigeres subalbatus totivirus, the first totivirus isolate from mosquitoes representing a proposed novel genus (Artivirus) of the family Totiviridae. J. Gen. Virol. 91(Pt 11), 2836–2845. doi: 10.1099/vir.0.024794-0
Keywords: Bombus, Osmia, Andrena, viruses, bee parasites, metatranscriptomics
Citation: Schoonvaere K, Smagghe G, Francis F and de Graaf DC (2018) Study of the Metatranscriptome of Eight Social and Solitary Wild Bee Species Reveals Novel Viruses and Bee Parasites. Front. Microbiol. 9:177. doi: 10.3389/fmicb.2018.00177
Received: 15 October 2017; Accepted: 25 January 2018;
Published: 14 February 2018.
Edited by:
Steven M. Short, University of Toronto Mississauga, CanadaReviewed by:
Declan C. Schroeder, Marine Biological Association of the United Kingdom, United KingdomPeter John Walker, The University of Queensland, Australia
Copyright © 2018 Schoonvaere, Smagghe, Francis and de Graaf. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Karel Schoonvaere, a2FyZWwuc2Nob29udmFlcmVAZ21haWwuY29t