 Daniel Garrido-Sanz
Daniel Garrido-Sanz Javier Manzano
Javier Manzano Marta Martín
Marta Martín Miguel Redondo-Nieto
Miguel Redondo-Nieto Rafael Rivilla
Rafael Rivilla- Departamento de Biología, Facultad de Ciencias, Universidad Autónoma de Madrid, Madrid, Spain
Polychlorinated biphenyls (PCBs) are widespread persistent pollutants that cause several adverse health effects. Aerobic bioremediation of PCBs involves the activity of either one bacterial species or a microbial consortium. Using multiple species will enhance the range of PCB congeners co-metabolized since different PCB-degrading microorganisms exhibit different substrate specificity. We have isolated a bacterial consortium by successive enrichment culture using biphenyl (analog of PCBs) as the sole carbon and energy source. This consortium is able to grow on biphenyl, benzoate, and protocatechuate. Whole-community DNA extracted from the consortium was used to analyze biodiversity by Illumina sequencing of a 16S rRNA gene amplicon library and to determine the metagenome by whole-genome shotgun Illumina sequencing. Biodiversity analysis shows that the consortium consists of 24 operational taxonomic units (≥97% identity). The consortium is dominated by strains belonging to the genus Pseudomonas, but also contains betaproteobacteria and Rhodococcus strains. whole-genome shotgun (WGS) analysis resulted in contigs containing 78.3 Mbp of sequenced DNA, representing around 65% of the expected DNA in the consortium. Bioinformatic analysis of this metagenome has identified the genes encoding the enzymes implicated in three pathways for the conversion of biphenyl to benzoate and five pathways from benzoate to tricarboxylic acid (TCA) cycle intermediates, allowing us to model the whole biodegradation network. By genus assignment of coding sequences, we have also been able to determine that the three biphenyl to benzoate pathways are carried out by Rhodococcus strains. In turn, strains belonging to Pseudomonas and Bordetella are the main responsible of three of the benzoate to TCA pathways while the benzoate conversion into TCA cycle intermediates via benzoyl-CoA and the catechol meta-cleavage pathways are carried out by beta proteobacteria belonging to genera such as Achromobacter and Variovorax. We have isolated a Rhodococcus strain WAY2 from the consortium which contains the genes encoding the three biphenyl to benzoate pathways indicating that this strain is responsible for all the biphenyl to benzoate transformations. The presented results show that metagenomic analysis of consortia allows the identification of bacteria active in biodegradation processes and the assignment of specific reactions and pathways to specific bacterial groups.
Introduction
Biphenyl has been widely used as a mineralizable polychlorinated biphenyls (PCBs) analog in biodegradation studies (Leigh et al., 2006; Uhlik et al., 2009; Leewis et al., 2016; Vergani et al., 2017a). PCBs are a family of man-made persistent organic chemicals that consist of a biphenyl skeleton where 1–10 hydrogen atoms are substituted by chlorine giving rise to up to 209 congeners. PCBs have been widely manufactured because of their chemical and physical properties (National Research Council, 1979) and a significant amount of PCBs has been released into the environment (Pieper, 2005; Sharma et al., 2014). The relative volatility of PCBs contributes to their spread throughout the globe (Gomes et al., 2013) where they bioaccumulate and biomagnify in the food web (Turrio-Baldassarri et al., 2007). PCBs have been shown to pose a broad range of exposure-related health effects in humans (Ross, 2004; Quinete et al., 2014) and are categorized as carcinogens (Mayes et al., 1998; Lauby-Secretan et al., 2013). Because of their chemical stability, poor water solubility, and toxicity, PCBs are considered recalcitrant toxics.
Bacteria can co-metabolize PCBs anaerobically and aerobically. Anaerobic cometabolism consists of reductive dehalogenation, a process in which highly chlorinated PCBs act as electron acceptors and reduce their chlorination (Quensen et al., 1988; Fennell et al., 2004). Thus, the biphenyl skeleton is not degraded through this pathway. Aerobic biodegradation on the contrary is better suited for low chlorinated congeners (Pieper, 2005; Furukawa and Fujihara, 2008; Pieper and Seeger, 2008) and biphenyl can be aerobically mineralized either by a single microorganism or by a consortium (Hernandez-Sanchez et al., 2013). Aerobic bioremediation of PCBs has been one of the main approaches to alleviate their persistence (Harkness et al., 1993; Pieper, 2005; Sharma et al., 2017) and usually occurs through its cometabolism by enzymes of the biphenyl upper degradation pathway, encoded by the bphABCDEFG gene cluster (Furukawa and Fujihara, 2008), although gene clusters for ethylbenzene (etb) and naphthalene (nar) degradation have also been shown to contribute to biphenyl and aerobic degradation of PCBs (Kimura et al., 2006; Iwasaki et al., 2007), resulting in the formation of (chloro)benzoic acid using biphenyl as carbon and energy source (Pieper, 2005; Pieper and Seeger, 2008). The specificity toward different PCB congeners depends mainly of the particular BphA enzyme (Gibson and Parales, 2000), some of which have been shown to produce the dechlorination of certain chlorinated biphenyls (Haddock et al., 1995; Seeger et al., 2001). The genes from the biphenyl upper degradative pathway have been extensively studied in Paraburkholderia xenovorans LB400, Pseudomonas pseudoalcaligenes KF707, and Rhodococcus jostii RHA1 due to the wide range of PCB congeners that they are able to metabolize (Seeger et al., 1995; Seto et al., 1995; Mondello et al., 1997; Furukawa and Fujihara, 2008). Aerobic degradation of PCBs usually occurs via cometabolism as their chlorinated derivatives might be channeled into dead-end pathways (Brenner et al., 1994) and it has been shown that some chlorinated intermediates are toxic to bacteria (Dai et al., 2002; Camara et al., 2004). After formation of (chloro)benzoic acid, it can be further funneled through catechol, protocatechuate, or the box pathways, ending up into tricarboxylic acid (TCA) cycle intermediates (Harwood and Parales, 1996; Gescher et al., 2002), known as the lower biphenyl degradation pathways.
Strategies for bioremediation of PCBs have been mainly focused on single microorganisms, either natural or modified (Haluska et al., 1995; Abbey et al., 2003; Sierra et al., 2003; Villacieros et al., 2005; Saavedra et al., 2010), which combined with biostimulation and bioaugmentation have resulted in enhanced degradation capabilities of a wide range of congeners (Singer et al., 2000; Fava et al., 2003; Ohtsubo et al., 2004; Field and Sierra-Alvarez, 2008). On the other hand, plant–microorganism interaction also plays a major role in degradation of PCBs (Leigh et al., 2006; Gerhardt et al., 2009; Vergani et al., 2017b). The use of PCB-degrading strains together with others that are capable of degrading their metabolic products (i.e., chlorinated benzoic acids) has also shown to extend the degradation rate of PCBs and results in complete mineralization of certain chlorobiphenyls (Fava et al., 1994; Hernandez-Sanchez et al., 2013).
In this study, we report the isolation and characterization of a soil bacterial consortium that is able to grow aerobically with the PCBs analog biphenyl as the sole carbon and energy source. In order to characterize this consortium, we have followed a metagenomic approach. Previous work using stable isotope probing (SIP) has shown to be useful in order to identify the bacterial populations implicated in biphenyl and benzoate degradation in soil microcosms (Leewis et al., 2016). However, the complexity of the bacterial community and the abundance of cross-feeders limit the study. Here, we show that reducing the community complexity to a lower number of bacterial populations by means of enrichment cultures, the metagenomic analysis allows not only to identify the populations playing a role in biphenyl and benzoate degradation but also to assign specific reactions and pathways to specific populations and therefore elucidating the trophic relationships occurring within the consortium to a higher detail.
Materials and Methods
Isolation of the Biphenyl-Degrading Consortium and Growth Conditions
For the isolation of the biphenyl-degrading consortium, 2 g of rhizospheric soil collected near a petrol station (Tres Cantos, Madrid, Spain) was added to 500 ml of sterile liquid minimal salt medium (MM) (Brazil et al., 1995), supplemented with 1 ml/l of phosphate-buffered mineral medium salts (PAS) (Bedard et al., 1986) and 0.005% of yeast extract. One gram per liter of biphenyl crystals was added as the sole carbon and energy source. The culture was grown at 28°C with shaking (135 rpm) and maintained within a 9-day subculture. After five subcultures, when the culture was unable to grow without biphenyl as the sole carbon and energy source, 20 ml of the culture was centrifuged at 4,248 × g. The pellet was then resuspended in 0.75 ml of MM+PAS and mixed with 0.25 ml of glycerol (80%) and deep-frozen at -80°C. The isolated consortium was routinely grown on MM+PAS with 1 g/l of biphenyl as the sole carbon and energy source at 28°C with shaking. For solid media, 1.5% agar (w/v) was added to the media and the biphenyl crystals were placed on the Petri dish lid.
The culture growth assessment on different organic compounds was performed as above but benzoic acid, protocatechuate, benzoate, 2-chlorobenzoic acid, 3-chlorobenzoic acid, or 4-chlorobenzoic acid (1 g/l) were added as the sole carbon and energy source.
DNA Extraction, Sequencing, Processing of Reads, and Assembly
DNA extraction from the biphenyl-degrading consortium at exponential growth (OD600 = 0.6) was carried out using the Realpure Genomic DNA Extraction Kit (Durviz, Spain). The 16S rRNA gene and the complete metagenome were sequenced by means of amplification of the V3–V4 16S rRNA region (primers 16SV3-V4-CS1; 5′-ACA CTG ACG ACA TGG TTC TAC ACC TAC GGG NGG CWG CAG-3′ and 16SV3-V4-CS2; 5′-TAC GGT AGC AGA GAC TTG GTC TGA CTA CHV GGG TAT CTA ATC C-3′) prior to libraries preparation and by whole-genome shotgun sequencing, respectively. The sequencing was carried out by Parque Científico de Madrid (Spain) using Illumina MiSeq paired 300-bp reads. Reads from the 16S rRNA gene and the whole metagenome were filtered and trimmed using Trimmomatic v0.36 (Bolger et al., 2014) software. Those with less than 50 nts in the case of the 16S rRNA gene or 100 nts in the case of the whole metagenome were removed. Reads from whole-metagenome sequencing were assembled using SPAdes v.10.1 software (Bankevich et al., 2012), metaSPAdes option, and default settings. Assembly quality was assessed using QUAST v4.4 (Gurevich et al., 2013). The resulting contigs were annotated using RAST (Aziz et al., 2008).
Reconstruction of Nearly Complete Genomes from Metagenome Shotgun Sequencing
Trimmed pair-reads from the whole-metagenome shotgun sequencing (as described above) were mapped against all available and closed NCBI genomes of Achromobacter, Bordetella, Cupriavidus, Microbacterium, Pseudomonas, Rhodococcus, and Stenotrophomonas using bowtie2 v 2.3.3.1 software (Langmead and Salzberg, 2012) with an expected range of inter-mate distances between 373 and 506 nts, consecutive seed extension attempts of 20, number of mismatches allowed in a seed alignment of 0, and length of the seed substrings to align of 20. For each genus, mapping reads and those without matching alignments across all genera examined were merged, processed, and retrieved with samtools v1.6 software (Li et al., 2009) for further assembly with SPAdes. Chimeric and misassigned contigs were checked by comparing assemblies of each genus against the same databases used for reads mapping using BLAST v.2.2.28+ software (Camacho et al., 2009). Contigs without positive hits within the expected genus were removed along with those with matching hits belonging to different genera. Contigs of Cupriavidus, Microbacterium, and Rhodococcus assemblies were also removed as genomic sizes were too small for a complete or nearly complete genome. In the case of Pseudomonas, contigs were also classified as belonging to P. pseudoalcaligenes or P. putida based on best blast hits.
Diversity Analysis of the 16S rRNA Gene and Coding DNA Sequences (CDSs)
Data analysis of the 16S rRNA gene diversity was assessed with QIIME v1.9.0 (Caporaso et al., 2010) and UPARSE v9 (Edgar, 2013) following the 16S profiling data analysis pipeline specified in the Brazilian Microbiome Project1. Briefly, filtered and trimmed forward and reverse reads were assembled using the fastq-join algorithm2 and further length-filtered by a minimum of 430 nts, representing more than 99% of total reads. Singletons were also removed. These sequences were imported into UPARSE to identify operational taxonomic units (OTUs) at a 97% sequence identity. Chimeras were removed using SILVA v123 database (Quast et al., 2013) as reference, which was also used for genus assignation. QIIME was also used to perform alpha rarefaction analysis. Convergence of observed OTUs rarefaction curve was determined using R (R Core Team, 2013) and the R package iNEXT (Hsieh et al., 2016) with a bootstrapping of 1,000 and a confidence interval of 5%.
To assess the diversity of coding DNA sequences (CDSs), after whole-metagenome assembly and annotation (see above), CDSs were blasted against the NCBI nt database (on April 2017) using blastn from BLAST v2.2.28+ software (Camacho et al., 2009). For each query, the first hit was kept and further filtered by a minimum of 75% sequence identity and 50% coverage. Genus assignation of the CDSs was based on the subject entry.
Identification of CDSs Involved in Biphenyl Metabolism and Phylogenetic Analysis
Aminoacid sequences for biphenyl 2,3-dioxygenase (BphA1), BenA, benzoate-CoA ligase (BclA), CatA, CatE, PobA, protocatechuate 4,5-dioxygenase alpha subunit (LigA), and protocatechuate 3,4-dioxygenase alpha subunit (PcaG) enzymes (Supplementary File 1) were downloaded from the NCBI and used to build blast databases using makeblastdb from BLAST. These databases were used as queries for orthologs identification within the whole-metagenome proteome. Results were filtered by 75% sequence identity, 50% coverage, and 1e-10 expected value and further blasted against the nr NCBI database (on April 2017) to validate their annotation. After orthologs identification, clusters of CDSs were searched within the whole-metagenome contigs and represented using own Perl scripts. Contigs carrying bph CDSs were also compared with those reported on reference sequences of Rhodococcus strains HA99 (AB272986.1), RHA1 (AB120955.1), and SAO101 (AB110633.1) to reconstruct the gene clusters using Clustal Omega (Sievers et al., 2011). Synteny representation was based on GenBank annotations and represented as described above.
Phylogenetic Analysis
BphA1, NarA1, and EtbA1 protein sequences from the metagenome annotation of the biphenyl-degrading consortium were aligned using Clustal Omega (Sievers et al., 2011) against 15 well-known BphA1 and closely related NarA1 and EtbA1 protein sequences. Results were imported into MEGA v7 (Kumar et al., 2016) to build the phylogenetic tree using maximum-likelihood with Tamura–Nei model, 1,000 bootstrap replicates, and represented with MEGA. BenA protein sequence of Pseudomonas putida PRS200 was used as an outgroup.
Rhodococcus Isolation and Genetic Analysis
Rhodococcus sp. WAY2 was isolated by plating washed (NaCl2 0.85%) and diluted biphenyl-degrading consortium culture on MM+PAS solid medium with biphenyl (1 g/l) as the sole carbon and energy source. After 12 days of incubation at 28°C, colonies were replated under the same conditions as above. This process was repeated twice. Finally, a single colony was grown on liquid MM+PAS media supplemented with 1 g/l of biphenyl. The culture was centrifuged at 4,248 × g prior to DNA extraction using the Realpure Genomic DNA Extraction Kit (Durviz, Spain). 16S rRNA gene was amplified using the universal primer pairs 27F (5′-AGA GTT TGA TCM TGG CTC AG-3′) and 1492R (5′-CTA CGR RTA CCT TGT TAC GAC-3′) (Weisburg et al., 1991). Amplicons were cloned into pGEM®-T Easy Vector System I (Promega) and transformed into E. coli DH5α. Plasmid DNA was extracted using the kit Wizard® Plus SV Minipreps DNA Purification System (Promega). Inserts were sequenced by means of Sanger sequencing using the universal primers T7 and SP6.
The three bph gene clusters identified in the whole metagenome of the biphenyl-degrading consortium were screened by PCR on the genome of the isolated Rhodococcus sp. WAY2 using the own-designed primers BphClus1F (5′-CGC CTC ATC ACG AAT GTG ACC G-3′), BphClus1R (5′-GCG TCC TCA TGC GTA CAG GTG TCC-3′), BphClus2F (5′-CGA CTG CTC GGA CTG GAG GG-3′), BphClus2R (5′-CCC ATC GAG TTA CCG ACT ATG TGC G-3′), BphClus3F (5′-GCC CGA CCA AGC AGT ACA AAG TG-3′), and BphClus3R (5′-GTC CAG TCG GAC TTC ACG TCG-3′). Primers were designed on the genomic sequence of these clusters. Melting temperature, absence of dimerization and hairpin formation, and lack of secondary priming sites were assessed with OligoAnalyzer 3.13. PCR was carried out in a total volume of 25 μl containing 2.5 μl of 10× PCR buffer MgCl2 free, 1 μl MgCl2 50 mM, 0.5 μl dNTP mix 10 mM (2.5 μM each), 1 μl of each primer at 10 μM, 1 μl of Taq DNA polymerase 1 U/μl (Biotools), and 1 μl of DNA template 30–50 ng/μl. The cycling conditions consisted in a first denaturation step at 95°C for 5 min followed by 32 cycles of amplification (45 s denaturation at 95°C, 45 s of primer annealing at 58°C, and an elongation step at 72°C for 1.5 min) followed by a final elongation step at 72°C for 7 min. PCR products were electrophoretically separated in 0.8% (w/v) agarose gels and post-dyed with GelRed.
Sequence Deposition
Raw reads of the 16S rRNA gene amplicons and whole-metagenome shotgun sequencing of the biphenyl-degrading consortium were deposited to the NCBI Sequence Read Archive under the accession numbers SRR6076973 and SRR6076972, respectively. Assemblies of Achromobacter sp., Bordetella sp., P. pseudoalcaligenes, Pseudomonas sp., and Stenotrophomonas sp. reconstructed from the metagenome were deposited to GenBank under the accession numbers PKCB00000000, PKCD00000000, PKCC00000000, PKCE00000000, and PKCF00000000, respectively. The 16S rRNA gene sequence of the isolated Rhodococcus sp. WAY2 was submitted to GenBank and it is available under the accession number MF996860. The 16S rRNA gene sequence of the 24 identified OTUs is shown in Supplementary File 2.
Results and Discussion
Metagenomic Sequencing and Bacterial Diversity
After sequencing the 16S rRNA genes of bacteria in the biphenyl-degrading consortium, a total of 44,644 sequences were obtained and assigned to 24 OTUs (≥97% sequence identity). The rarefaction curve shows a clear and early saturation of observed OTUs, as shown in Figure 1A, which indicates that a full community coverage was achieved before 40,000 sequences and the presence of other taxa is unlikely. Furthermore, statistical analysis of the rarefaction curve (Supplementary File 3) showed that doubling the sampling would not increase the number of detected OTUs. On the other hand, the whole-genome shotgun sequencing of the metagenome resulted in 78.4 Mpb distributed in 45,046 contigs (Supplementary File 4). After annotation, 66,967 coding DNA sequences (CDSs) were obtained, from which 47,689 (71.2%) were assigned to the genus level, showing a high concordance with the identified OTUs. The relative abundance of the 16S rRNA and the CDSs (Figure 1B) shows that the biphenyl-degrading consortium is clearly dominated by Pseudomonas (28.97% 16S rRNA and 41.57% CDSs). Other genera that are present in the consortium are Bordetella (21.28% 16S rRNA and 11.75% CDSs), Achromobacter (12.67% 16S rRNA and 9.88% CDSs), Stenotrophomonas (8.57% 16S rRNA and 12.99% CDSs), Rhodococcus (2.18% 16S rRNA and 8.17% CDSs), and Cupriavidus (1.51% 16S rRNA and 7.62% CDSs). This distribution is detailed in Supplementary File 5. The main difference between the 16S rRNA and CDSs relative genus abundance lies in Pigmentiphaga, which is relatively abundant in the 16S rRNA analysis (20.54%) but is almost absent on CDSs representation (0.04%). This is probably due to lack of sequenced Pigmentiphaga genomes in the NCBI database, which makes CDSs assignation to this genus impossible and explains the higher relative abundance of the remaining genera in the CDSs diversity analysis. However, some genera, such as Bordetella and Achromobacter, have a lower relative CDSs representation than in the 16S rRNA. This could be explained by an incomplete metagenome, given that around 120 Mpb metagenome size was expected (considering an average bacterial genome size of 5 Mpb) to achieve a full genomic representation of the 24 OTUs identified in the biphenyl-degrading consortium. Furthermore, the presence of only 16 16S rRNA genes annotated in the metagenome is congruent with an incomplete one. However, it is important to indicate that the seven most represented genera represent more than 95% of the bacterial community and 96% of the identified CDS (Figure 1B), indicating a high coverage of the metagenome. This level of coverage would be impossible to achieve analyzing directly a soil sample or microcosm.
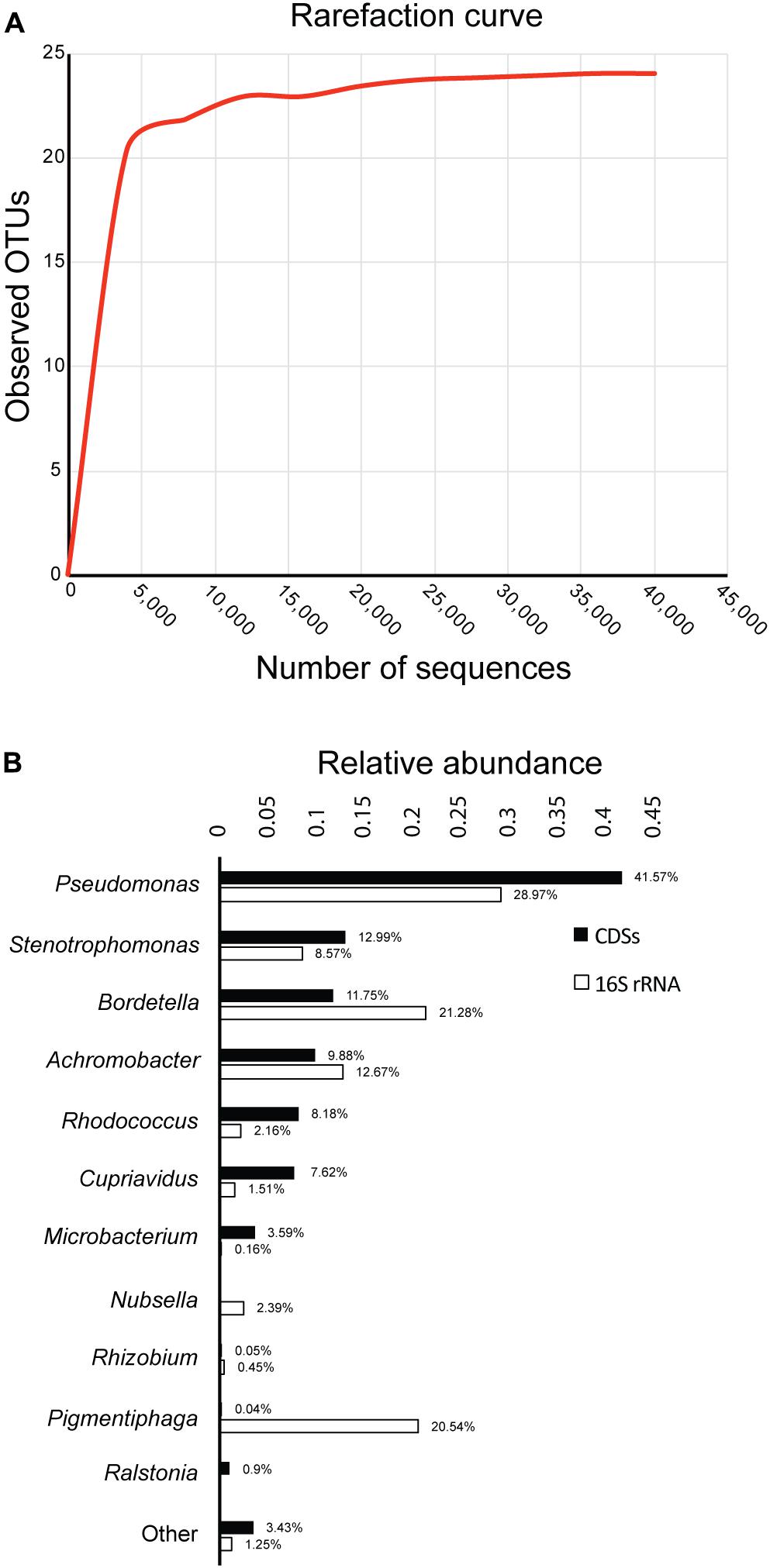
FIGURE 1. Diversity and composition of the biphenyl-degrading consortium. (A) Rarefaction curve of observed OTUs (≥97% sequence identity) over the number of 16S rRNA sequences and (B) relative abundance of genus based on 16S rRNA and CDSs taxonomic assignment. Only taxa with a minimum relative abundance of 0.15% for 16S rRNA and 0.9% for CDSs is represented.
On the other hand, we have been able to reconstruct five nearly complete genomes from the whole-metagenome sequence, which correspond with the most abundant OTUs identified in the consortium (Table 1). These include two genomes classified as P. pseudoalcaligenes and Pseudomonas sp., Achromobacter sp., Bordetella sp., and Stenotrophomonas sp. Their genomic sizes and %GC content are congruent with their closest relative genome.
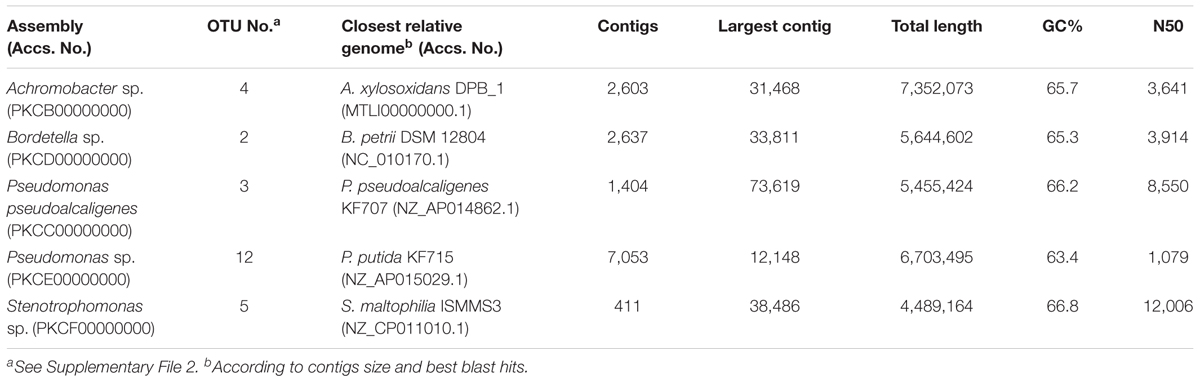
TABLE 1. Genomic statistics of the five nearly complete genomes reconstructed from the whole-metagenome sequence of the biphenyl-degrading consortium.
Identification of Biphenyl Upper Degradative Pathway Gene Clusters
In order to identify the metabolic pathways involved in the biphenyl biodegradation that are present in the whole metagenome of the biphenyl-degrading consortium, alpha subunits of the BphA1 were used as query to search for orthologous sequences. Three different BphA1 were identified (Table 2), which are present in three different contigs and are classified as belonging to the Rhodococcus genus by sequence identity (Supplementary File 6). BphA1 encodes the α subunit of biphenyl dioxygenases, and are responsible for the enzyme specificity (Gibson and Parales, 2000). As shown in Figure 2D, BphA1 proteins can be classified into three families. Typical BphA1 have been identified and characterized in many bacterial strains, including P. xenovorans LB400 (Seeger et al., 1995), P. pseudoalcaligenes KF707 (Taira et al., 1992), and R. jostii RHA1 (Seto et al., 1995). None of the BphA1 CDS identified here belongs to this family. A second family of atypical BphA1 was identified in several strains of the genus Rhodococcus, including strains HA99 and R04 (Taguchi et al., 2007; Yang et al., 2007). One of the CDS identified here is identical to these atypical BphA1. The other family is formed by proteins with proved BphA1 activity, but formerly identified as NarA1 or EtbA1. These proteins have also been identified within the genus Rhodococcus (Kimura et al., 2006; Iwasaki et al., 2007) and two of the BphA1 CDSs identified here are identical to CDSs in Rhodococcus opacus SAO101 and R. jostii RHA1, respectively. On the other hand, the comparison between these CDSs and the ones previously reported in other Rhodococcus strains sequences allowed us to reconstruct the bph gene clusters from the whole-metagenome contigs, as shown in Figure 2. The first cluster (Figure 2A) was reconstructed from four different metagenome contigs and shows high sequence identity with the bph gene clusters reported in Rhodococcus sp. HA99 (Taguchi et al., 2007). This cluster is composed by bphBCA1A2A3A4 and bphD, which are responsible for biphenyl and PCBs degradation into (chloro)benzoate and 2-hydroxypenta-2,4-dienoate (Taguchi et al., 2007). The second gene cluster (Figure 2B) was reconstructed from three different metagenome contigs and presents high sequence identity with bph and etb gene clusters which have been reported to be involved in both, biphenyl and PCBs degradation in R. jostii RHA1 (Iwasaki et al., 2006, 2007). This cluster is composed by etbA1A2C and bphDE2F2. The third gene cluster is present in a single metagenome contig (Figure 2C) and shows high sequence identity with nar gene clusters previously described in the plasmid pWK301 of R. opacus SAO101 (Kimura et al., 2006). This gene cluster is composed by narA1A2BC and two transcriptional regulators narR1R2 and it has been reported to be involved in the degradation of a wide range of substrates, including biphenyl and PCBs (Kimura and Urushigawa, 2001; Kitagawa et al., 2004; Kimura et al., 2006). These results strongly suggest that Rhodococcus is the only genus responsible for initiating the biphenyl degradation in the consortium and that initial degradation can proceed through three distinct pathways. To our knowledge, multiple pathways have only been found in R. jostii RHA1, where a bph and an etb pathways have been described (Iwasaki et al., 2006, 2007).

TABLE 2. Summary of the number and genus affiliation of the main CDSs for enzymes involved in the biphenyl and metabolic derivatives degradation identified in the biphenyl-degrading consortium.
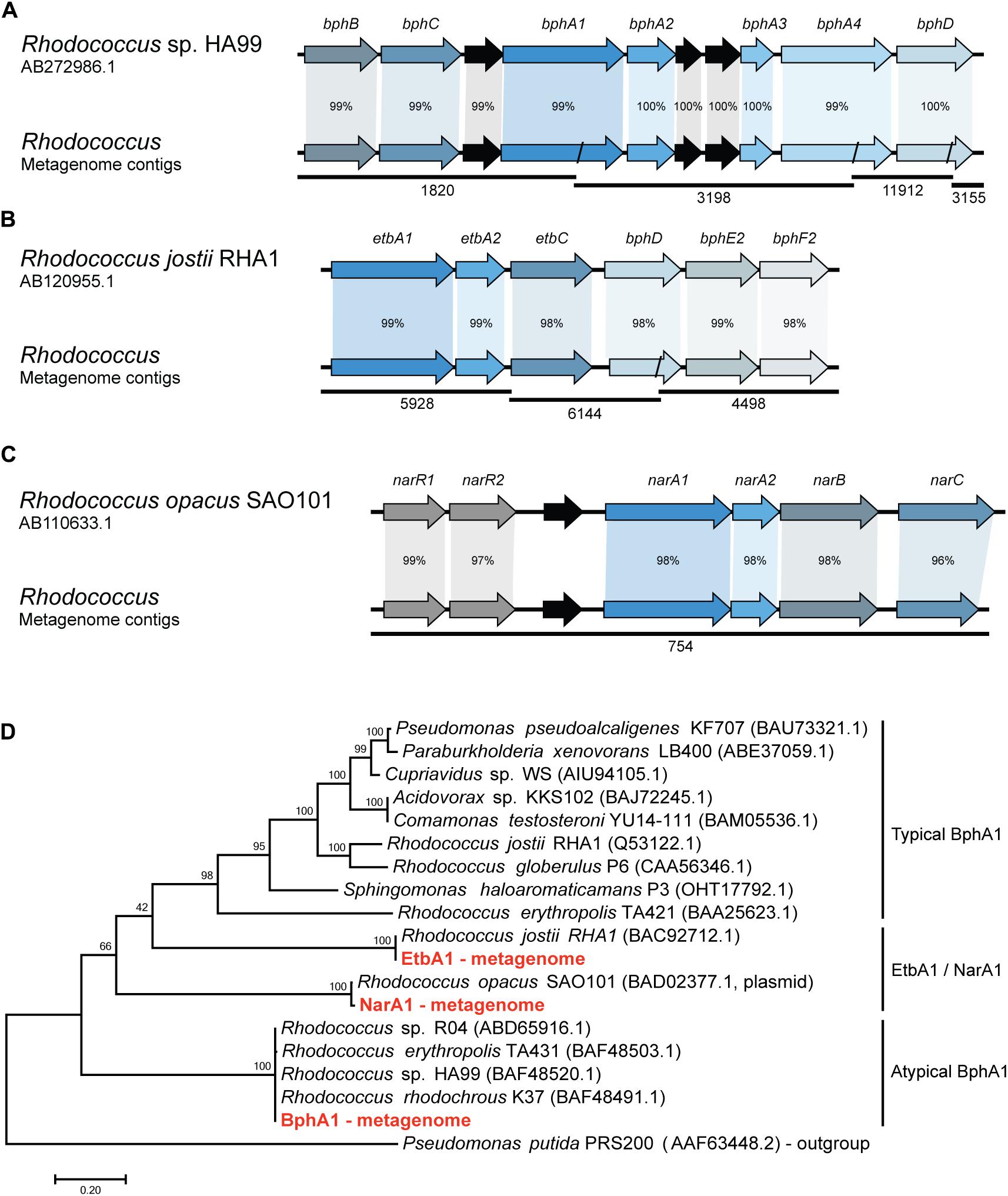
FIGURE 2. Synteny and sequence identity of gene clusters involved in biphenyl degradation compared with reference sequences. (A) Biphenyl degradative gene cluster, (B) ethylbenzene degradative gene cluster, and (C) naphthalene degradative gene cluster. Rhodococcus sp. HA99, R. jostii RHA1, and R. opacus SAO101 sequences are shown as reference. Contigs from the metagenome are represented as black lines and their ID number is shown below. Black arrows represent hypothetical genes. Percentage according to nucleotide sequence identity of the CDSs. (D) Phylogenetic tree showing the relation of the isolated BphA1 protein sequences with previously characterized proteins. A BenA protein sequence from Pseudomonas putida was used as an outgroup.
To further study if the bph, etb, and nar gene clusters identified in the metagenome belong to one or multiple Rhodococcus strains that might be present in the biphenyl-degrading consortium, we isolated a Rhodococcus strain (R. sp. WAY2) from the consortium and tested for the presence of these three gene clusters by means of PCR. The results revealed that the three clusters are present in a single Rhodococcus strain WAY2, which 16S rRNA showed a high sequence identity (>99%) with R. jostii RHA1. This might suggest that the etb gene cluster is present in the chromosome of the isolated WAY2 strain as it is in the case of RHA1, while bph and nar gene clusters could be present in plasmids, as reported in strains HA99 and SAO101, respectively (Kimura et al., 2006; Taguchi et al., 2007).
Identification of Biphenyl Lower Degradative Pathway Genes
Biphenyl is metabolized to benzoate and 2-hydroxypenta-2,4-dienoate by either the bph, etb, or nar gene clusters. Benzoate can be then further mineralized by three different aerobic pathways: catechol, protocatechuate, or benzoyl-coA ligation (Harwood and Parales, 1996; Rather et al., 2010; Fuchs et al., 2011). All the CDSs for enzymes of these aerobic benzoate degradation pathways were screened and found in the metagenome of the biphenyl-degrading consortium and are summarized in Table 2 (for details see Supplementary File 6). The benzoate degradative pathway via catechol formation is first initiated by BenABCD to form catechol. The coding sequence for benzoate 1,2-dioxygenase alpha subunit (BenA) was found 10 times in different contigs and was mainly assigned to Pseudomonas (five) and Bordetella (four). The remaining one was assigned to Rhodococcus (Table 2). After catechol formation, it can be further mineralized by ortho or meta cleavage, in which catechol 1,2-dioxygenase (CatA) or catechol 2,3-dioxygenase (CatE) is, respectively, involved. The coding sequence of CatA was found 13 times in the metagenome and was mainly assigned to Pseudomonas (five) and Rhodococcus (four). The remaining ones were assigned to Bordetella (two), Achromobacter (one), and Variovorax (one) (Table 2). On the other hand, the coding sequence for CatE was found five times in the metagenome and was assigned to Variovorax (two), Cupriavidus (two), and the remaining two could not be assigned (Table 2). These results suggest that the degradation of benzoate via catechol is mainly supported by Pseudomonas, Bordetella, and Rhodococcus, while other genera such as Achromobacter, Variovorax, and Cupriavidus have a smaller involvement in this pathway. Regarding the presence of this pathway in Rhodococcus, the isolated strain R. sp. WAY2 was unable to grow on benzoate as the sole carbon and energy source, suggesting that another Rhodococcus strain, different than the one harboring the bph, etb, and nar gene clusters, is present in the biphenyl-degrading consortium.
Benzoate can also be metabolized via protocatechuate formation, in which a benzoate 4-monooxygenase (CYP450) and a 4-hydroxybenzoate 3-monooxygenase (PobA) are involved (Fuchs et al., 2011). The coding sequence of PobA was found 10 times in different contigs in the metagenome and was assigned to Pseudomonas (four), Bordetella (two), Achromobacter (one), Ralstonia (one), Rhodococcus (one), and the remaining one could not be assigned to any genus (Table 2). After protocatechuate formation, it can also be mineralized via ortho and meta cleavage, in which protocatechuate 3,4-dioxygenase (PcaGH) and protocatechuate 4,5-dioxygenase (LigAB) are, respectively, involved. The coding sequence for PcaG was found nine times in the metagenome and was assigned to Pseudomonas (four), Achromobacter (two), Bordetella (one), Cupriavidus (one), and Ralstonia (one) (Table 2). On the other hand, the coding sequence of LigA was found four times in the metagenome and was assigned to Pseudomonas (one) and Bordetella (one). The remaining ones could not be assigned to any genus (Table 2). These results suggest that the degradation of benzoate via protocatechuate formation is also dominated by Pseudomonas and Bordetella, harboring both, the ortho and meta protocatechuate cleavage pathways, while Achromobacter, Ralstonia, and Cupriavidus only have the coding sequences for protocatechuate formation and/or its ortho-cleavage pathway.
Finally, benzoate can also be mineralized by a novel pathway in which acetyl-CoA is first ligated to benzoate by a BclA and further epoxidated by benzoyl-CoA 2,3-epoxidase (BoxAB) (Rather et al., 2010). The coding sequence for BoxA was found four times in different contigs in the metagenome and was assigned to Achromobacter (three) and Variovorax (one) (Table 2). Contigs carrying the BoxA-coding sequence were also found to contain the remaining genes for the box cluster (boxABCD and bclA), along with the transcriptional regulator boxR and several coding sequences involved in benzoate transport, as shown in Figure 3. However, two of these contigs assigned to Achromobacter lack the boxD gene, which might result in dead-end production of 3,4-didehydroadipyl-CoA semialdehyde and formate, although they could be source of carbon and energy through alternative pathways.
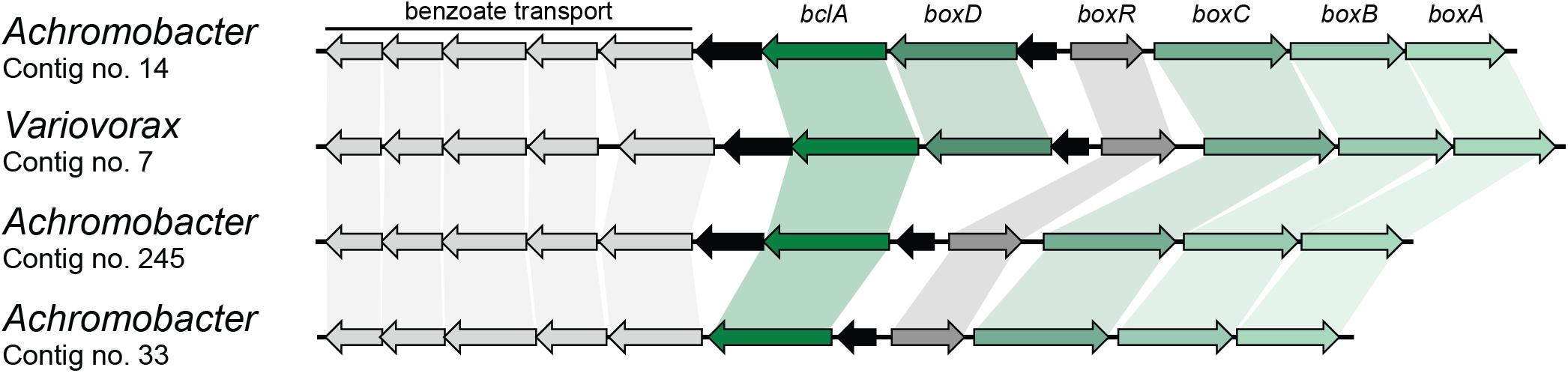
FIGURE 3. Box gene clusters identified in the metagenome of the biphenyl-degrading consortium. Black arrows represent genes with no involvement in benzoate degradation. Genus assignation of the clusters based on sequence identity of CDSs.
Population Roles in the Biphenyl-Degrading Consortium
The catabolic pathways for biphenyl and its metabolic derivatives found in the metagenome of the biphenyl-degrading consortium and the genus affiliation of the coding sequences for these pathways (Table 2 and Supplementary File 6) provide a complete understanding of the different roles of the main bacterial populations that are present in the consortium with regard of their relative abundance. It is interesting to note that the seven most represented genera in the consortium have been identified as the source of 90% of the CDSs identified in the biphenyl/benzoate degradation pathways and that these genera harbor all the enzymatic activities in the degradation pathways. These results reflect a high degree of functional redundancy, as the same reactions seem to be carried out by different taxa. These results are summarized in Table 3 and the metabolic pathways reconstructed for the biphenyl-degrading consortium is represented in Figure 4. Rhodococcus is the genus responsible for initiating the biphenyl degradation into benzoate as the three BphA1 that have been found in the metagenome have been only assigned to this genus. Furthermore, the presence of complete gene clusters for bph, etb, and nar in a single Rhodococcus strain, and the previous reports of the involvement of these clusters in both biphenyl and PCBs degradation (Kimura et al., 2006; Iwasaki et al., 2007; Taguchi et al., 2007), makes this strain suited for bioremediation of PCBs. However, although the consortium was not able to grow in any of the chlorobenzoates tested (2-, 3-, or 4-chlorobenzoic acid) as the sole carbon and energy source (Table 4), cometabolism of chlorobenzoates as well as PCB congeners should be further analyzed. After formation of benzoate as the product of biphenyl degradation, the remaining bacterial populations can thrive, either by using benzoate, catechol, or protocatechuate. Our results show that protocatechuate and catechol degradative pathways in the consortium are rather abundant (Table 2), and are dominated by Pseudomonas and Bordetella, harboring genes for both, ortho and meta cleavage of protocatechuate and ortho cleavage of catechol. The relative high abundance of this genus in the consortium can be explained by the different alternative pathways for benzoate and its metabolic derivates degradation. Other genera such as Achromobacter and Cupriavidus are likely using catechol and/or protocatechuate to grow (Table 3). In addition, the consortium was able to grow on benzoate and protocatechuate as the sole carbon and energy source (Table 4), which is in agreement with the results presented here. On the other hand, the benzoate degradative pathway via acetyl-CoA ligation was mainly assigned to Achromobacter, which explains its presence in the consortium although it could also use protocatechuate and catechol via ortho cleavage (Table 3).
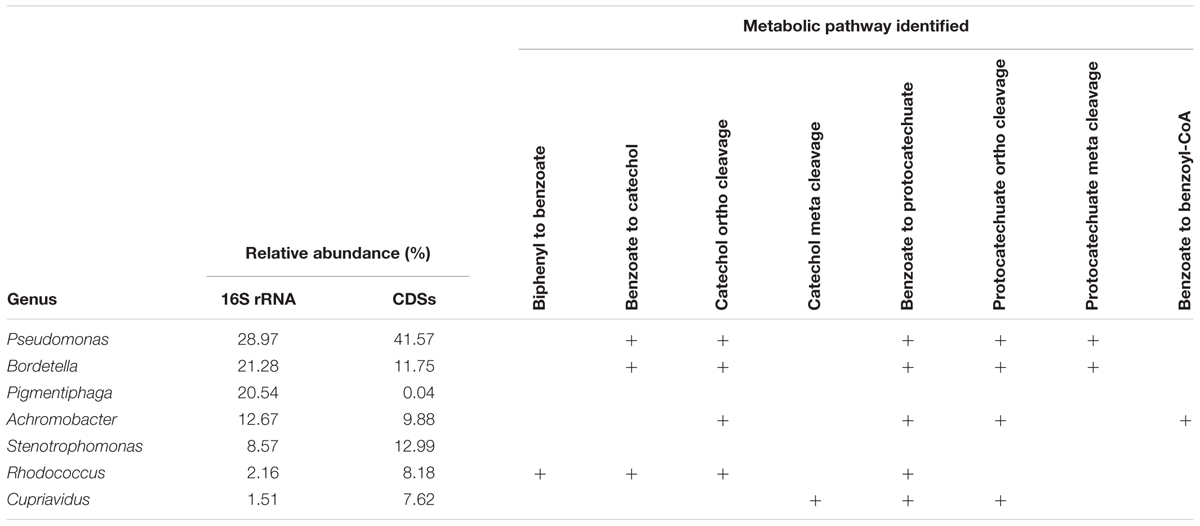
TABLE 3. Summary of the pathways assigned to the main genus present in the biphenyl-degrading consortium.
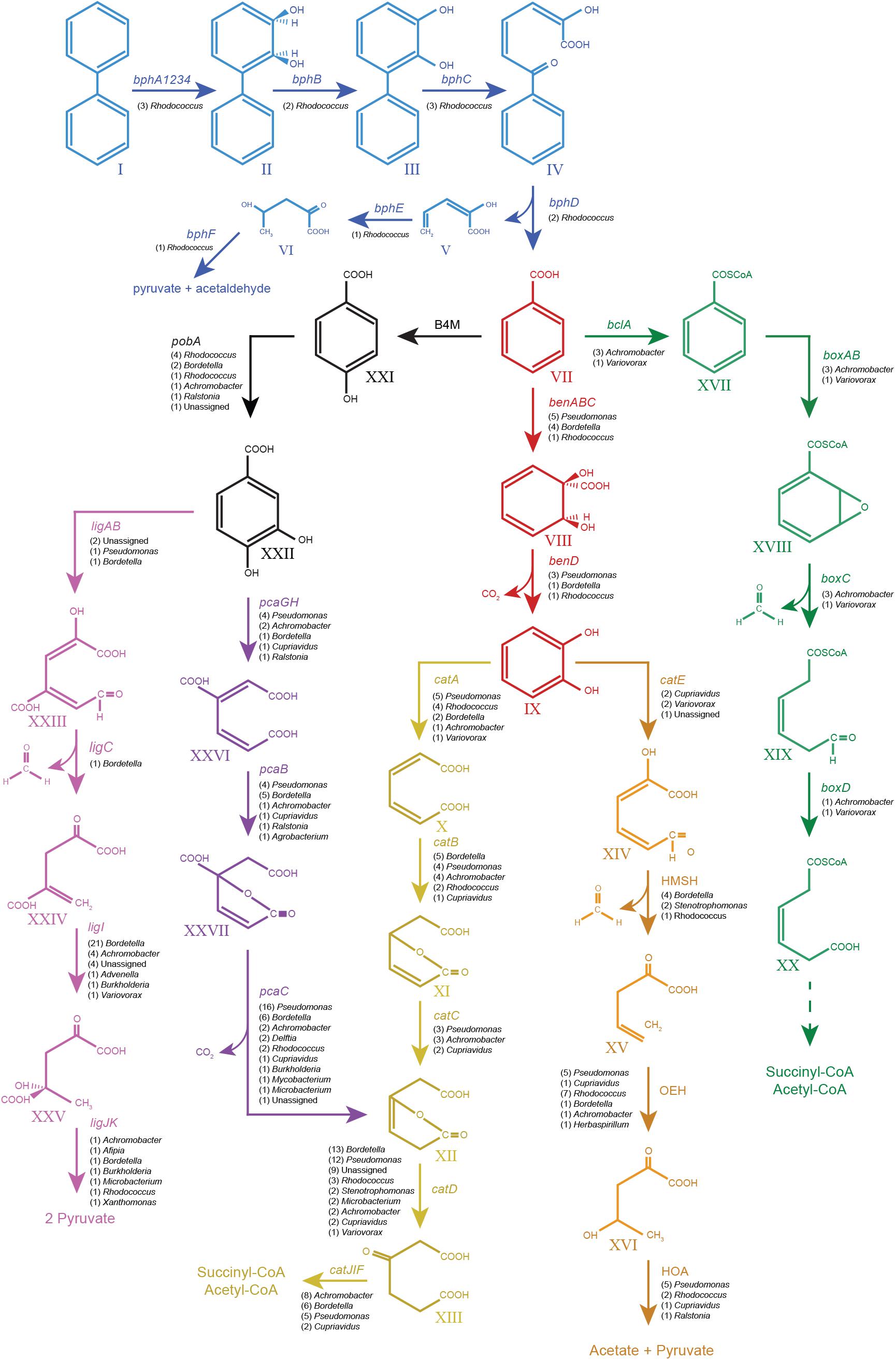
FIGURE 4. Pathways from biphenyl degradation identified in the metagenome of the biphenyl-degrading consortium. Blue, biphenyl degradation; red, benzoate degradation via catechol; black, benzoate degradation via protocatechuate; violet, protocatechuate degradation via meta cleavage; purple, protocatechuate degradation via ortho cleavage; yellow, catechol degradation via ortho cleavage; orange, catechol degradation via meta cleavage; green, benzoate degradation via benzoyl-CoA formation. All the genes shown in the graph have been found in the metagenome of the biphenyl-degrading consortium. Their number of CDSs and genus assignation are specified under the gene names. Compounds: I, biphenyl; II, 2,3-dihydroxy-4-phenylhexa-4,6-diene; III, 2,3-dihydroxybiphenyl; IV, 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate; V, 2-hydroxypenta-2,4-dienoate; VI, 4-hydroxy-2-oxopenta; VII, benzoate; VIII, 2-hydro-1,2-dihydroxybenzoate; IX, catechol; X, cis,cis-muconate; XI, mucolactone; XII, 3-oxooadipate enol-lactone; XIII, 3-oxoadipate; XIV, 2-hydroxy-muconate-6-semialdehyde; XV, 2-oxo-penta-4-enoate; XVI, 4-hydroxy-2-oxovalerate; XVII, benzoyl-CoA; XVIII, 2,3-epoxy-benzoyl-CoA; XIX, 3,4-dehydroadipyl-CoA semialdehyde; XX, 3,4-dehydroadipyl-CoA; XXI, hydroxybenzoate; XXII, protocatechuate; XXIII, 2-hydroxy-4-carboxymuconic semialdehyde; XXIV, 2-keto-4-carboxypenta-enoate; XXV, 4-hydroxy-4-carboxy-2-ketovalerate; XXVI, 3-carboxy-cis,cis-muconate; and XXVII, 4-carbxymucolactone. Genes: bphA1A2A3A4, biphenyl 2,3-dioxygenase; bphB, cis-2,3-dihydrobiphenyl-2,3-diol dehydrogenase; bphC, biphenyl-2,3-diol 1,2-dioxygenase; bphD, 2,6-dioxo-6-phenylhexa-3-enoate hydrolase; bphE, 2-hydroxypenta-2,4-dienoate hydratase; bphF, 4-hydroxy-2-oxovalerate aldolase; benABC, benzoate 1,2-dioxygenase; benD, 1,6-dihydroxycyclohexa-2,4-diene-1-carboxylate dehydrogenase; catA, catechol 1,2-dioxygenase; catB, muconate cycloisomerase; catC, muconolactone delta-isomerase; catD, 3-oxoadipate enol-lactonase; catIJ, 3-oxoadipate CoA-transferase; catF, 3-oxoadipyl-CoA thiolase; catE, catechol 2,3-dioxygenase; 2HM H, 2-hydroxymuconate semialdehyde hydrolase; 2OE H, 2-oxopent-4-enoate hydratase, 4HO A, 4-hydroxy-2-oxovalerate aldolase; B4M, benzoate 4-monooxygenase; pobA, 4-hydroxybenzoate 3-monooxygenase; ligAB, protocatechuate 4,5-dioxygenase; ligC, 2-hydroxy-4-carboxymuconate semialdehyde hemiacetal dehydrogenase; ligI, 2-pyrone-4,6-dicarboxylate lactonase; ligJ, 4-oxalomesaconate hydratase; ligK, 4-hydroxy-4-methy-2-oxoglutarate aldolase; pcaGH, protocatechuate 3,4-dioxygenase; pcaB, 3-carboxy-cis,cis-muconate cycloisomerase; pcaC, 4-carboxymuconolactone decarboxylase; blcA, benzoate CoA-ligase; boxAB, benzoyl-CoA 2,3-epoxidase; boxC, 2,3-epoxybenzoyl-CoA dihydrolase; and boxD, 3,4-dehydroadipyl-CoA semialdehyde dehydrogenase (NADP(+)).

TABLE 4. Consortium growth on different organic compounds as the sole carbon and energy source.
Interestingly, two of the most abundant genera within the consortium, Pigmentiphaga and Stenotrophomonas (20.54 and 8.57% 16S rRNA relative abundance, respectively) do not have any of the coding sequences for enzymes screened in the metagenome (Table 2 and Supplementary File 6). In the case of Pigmentiphaga, it is clear that the lack of sequenced genomes available on the NCBI database (on April 2017) prevented the affiliation of CDSs to this genus. However, it is unclear if any of the coding sequences for enzymes of these pathways that could not been assigned to any genus (Table 2) might belong to Pigmentiphaga or if other metabolic abilities are involved. Regarding Stenotrophomonas, it is a common member of biphenyl, PCBs, and other aromatics-degrading communities (Leigh et al., 2007; Uhlik et al., 2013; Wald et al., 2015) and exhibits high metabolic versatility (Hauben et al., 1999). Its presence in the biphenyl-degrading consortium might be explained by cross-feeding on secondary metabolites produced by the rest of the consortium members, as it has been previously suggested (Wald et al., 2015). These results show that the metagenomic analysis of this consortium allows the determination of the biodegradation network involved in biphenyl degradation, being able to determine the specific role of different bacterial populations in the biodegradation process. The combination of these data with transcriptomic/proteomic and metabolomic approaches could result in robust models of biodegradation processes, explaining the metabolic fluxes. This approach is also a proof of concept of the possibility of generating rationally designed inoculants for environmental restoration. Consortia, as this described here, can be thoroughly characterized and could be used as an inoculant, as a source of novel bioremediation strains or as a background for bioaugmentation with previously isolated strains.
The results presented here show that metagenomic analysis is a powerful tool for the functional characterization of consortia designed for bioremediation of complex contaminants. The analysis of consortia rather than soil microcosms has obvious advantages. First of all, while a typical soil microcosm usually contains thousands of genotypes, a consortium such as the one shown here contains less than a hundred genotypes, and therefore the depth of sequencing is much higher. Furthermore, while most of the genotypes detected in the consortium play a role in the biodegradation process, as shown here, most of the populations in a microcosm are irrelevant for the process. Furthermore, metagenomic analysis has proven to be advantageous over SIP in analyzing the biodegrading populations. While SIP was able to identify the bacterial populations involved in biphenyl and benzoate degradation in a soil microcosm and to determine that biphenyl and benzoate were mostly degraded by different populations (Leewis et al., 2016), here we have been able to determine not only the biodegrading populations, but also to assign specific functions and reactions to specific populations, identifying all the biodegradation pathways and therefore providing a deeper insight in the biodegradation process.
Author Contributions
DG-S and JM performed the experiments and bioinformatic analysis. MM, MR-N, and RR designed the study and supervised the work. DG-S and RR drafted the manuscript.
Funding
This research was funded by grant BIO2015-64480-R from MINECO/FEDER EU. DG-S was granted by FPU fellowship program (FPU14/03965) from Ministerio de Educación, Cultura y Deporte, Spain.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00232/full#supplementary-material
Footnotes
- ^ http://www.brmicrobiome.org/
- ^ https://github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqJoin.md
- ^ https://eu.idtdna.com/calc/analyzer
References
Abbey, A. M., Beaudette, L. A., Lee, H., and Trevors, J. T. (2003). Polychlorinated biphenyl (PCB) degradation and persistence of a gfp-marked Ralstonia eutropha H850 in PCB-contaminated soil. Appl. Microbiol. Biotechnol. 63, 222–230. doi: 10.1007/s00253-003-1380-x
Aziz, R. K., Bartels, D., Best, A. A., Dejongh, M., Disz, T., Edwards, R. A., et al. (2008). The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. doi: 10.1186/1471-2164-9-75
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Bedard, D. L., Unterman, R., Bopp, L. H., Brennan, M. J., Haberl, M. L., and Johnson, C. (1986). Rapid assay for screening and characterizing microorganisms for the ability to degrade polychlorinated biphenyls. Appl. Environ. Microbiol. 51, 761–768.
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brazil, G. M., Kenefick, L., Callanan, M., Haro, A., De Lorenzo, V., Dowling, D. N., et al. (1995). Construction of a rhizosphere pseudomonad with potential to degrade polychlorinated biphenyls and detection of bph gene expression in the rhizosphere. Appl. Environ. Microbiol. 61, 1946–1952.
Brenner, V., Arensdorf, J. J., and Focht, D. D. (1994). Genetic construction of PCB degraders. Biodegradation 5, 359–377. doi: 10.1007/BF00696470
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Camara, B., Herrera, C., Gonzalez, M., Couve, E., Hofer, B., and Seeger, M. (2004). From PCBs to highly toxic metabolites by the biphenyl pathway. Environ. Microbiol. 6, 842–850. doi: 10.1111/j.1462-2920.2004.00630.x
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Dai, S., Vaillancourt, F. H., Maaroufi, H., Drouin, N. M., Neau, D. B., Snieckus, V., et al. (2002). Identification and analysis of a bottleneck in PCB biodegradation. Nat. Struct. Biol. 9, 934–939. doi: 10.1038/nsb866
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Fava, F., Bertin, L., Fedi, S., and Zannoni, D. (2003). Methyl-beta-cyclodextrin-enhanced solubilization and aerobic biodegradation of polychlorinated biphenyls in two aged-contaminated soils. Biotechnol. Bioeng. 81, 381–390. doi: 10.1002/bit.10579
Fava, F., Di Gioia, D., Cinti, S., Marchetti, L., and Quattroni, G. (1994). Degradation and dechlorination of low-chlorinated biphenyls by a three-membered bacterial co-culture. Appl. Microbiol. Biotechnol. 41, 117–123. doi: 10.1007/BF00166092
Fennell, D. E., Nijenhuis, I., Wilson, S. F., Zinder, S. H., and Haggblom, M. M. (2004). Dehalococcoides ethenogenes strain 195 reductively dechlorinates diverse chlorinated aromatic pollutants. Environ. Sci. Technol. 38, 2075–2081. doi: 10.1021/es034989b
Field, J. A., and Sierra-Alvarez, R. (2008). Microbial transformation and degradation of polychlorinated biphenyls. Environ. Pollut. 155, 1–12. doi: 10.1016/j.envpol.2007.10.016
Fuchs, G., Boll, M., and Heider, J. (2011). Microbial degradation of aromatic compounds - from one strategy to four. Nat. Rev. Microbiol. 9, 803–816. doi: 10.1038/nrmicro2652
Furukawa, K., and Fujihara, H. (2008). Microbial degradation of polychlorinated biphenyls: biochemical and molecular features. J. Biosci. Bioeng. 105, 433–449. doi: 10.1263/jbb.105.433
Gerhardt, K. E., Huang, X.-D., Glick, B. R., and Greenberg, B. M. (2009). Phytoremediation and rhizoremediation of organic soil contaminants: potential and challenges. Plant Sci. 176, 20–30. doi: 10.1016/j.plantsci.2008.09.014
Gescher, J., Zaar, A., Mohamed, M., Schagger, H., and Fuchs, G. (2002). Genes coding for a new pathway of aerobic benzoate metabolism in Azoarcus evansii. J. Bacteriol. 184, 6301–6315. doi: 10.1128/JB.184.22.6301-6315.2002
Gibson, D. T., and Parales, R. E. (2000). Aromatic hydrocarbon dioxygenases in environmental biotechnology. Curr. Opin. Biotechnol. 11, 236–243. doi: 10.1016/S0958-1669(00)00090-2
Gomes, H. I., Dias-Ferreira, C., and Ribeiro, A. B. (2013). Overview of in situ and ex situ remediation technologies for PCB-contaminated soils and sediments and obstacles for full-scale application. Sci. Total Environ. 445–446, 237–260. doi: 10.1016/j.scitotenv.2012.11.098
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Haddock, J. D., Horton, J. R., and Gibson, D. T. (1995). Dihydroxylation and dechlorination of chlorinated biphenyls by purified biphenyl 2,3-dioxygenase from Pseudomonas sp. strain LB400. J. Bacteriol. 177, 20–26. doi: 10.1128/jb.177.1.20-26.1995
Haluska, L., Barancikova, G., Balaz, S., Dercova, K., Vrana, B., Paz-Weisshaar, M., et al. (1995). Degradation of PCB in different soils by inoculated Alcaligenes xylosoxidans. Sci. Total Environ. 175, 275–285. doi: 10.1016/0048-9697(95)04927-4
Harkness, M. R., Mcdermott, J. B., Abramowicz, D. A., Salvo, J. J., Flanagan, W. P., Stephens, M. L., et al. (1993). In situ stimulation of aerobic PCB biodegradation in Hudson River sediments. Science 259, 503–507. doi: 10.1126/science.8424172
Harwood, C. S., and Parales, R. E. (1996). The beta-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 50, 553–590. doi: 10.1146/annurev.micro.50.1.553
Hauben, L., Vauterin, L., Moore, E. R., Hoste, B., and Swings, J. (1999). Genomic diversity of the genus Stenotrophomonas. Int. J. Syst. Bacteriol. 49(Pt 4), 1749–1760. doi: 10.1099/00207713-49-4-1749
Hernandez-Sanchez, V., Lang, E., and Wittich, R. M. (2013). The three-species consortium of genetically improved strains Cupriavidus necator RW112, Burkholderia xenovorans RW118, and Pseudomonas pseudoalcaligenes RW120 Grows with Technical Polychlorobiphenyl, Aroclor 1242. Front. Microbiol. 4:90. doi: 10.3389/fmicb.2013.00090
Hsieh, T. C., Ma, K. H., and Chao, A. (2016). iNEXT: an R package for rarefaction and extrapolation of species diversity (Hill numbers). Methods Ecol. Evol. 7, 1451–1456. doi: 10.1111/2041-210X.12613
Iwasaki, T., Miyauchi, K., Masai, E., and Fukuda, M. (2006). Multiple-subunit genes of the aromatic-ring-hydroxylating dioxygenase play an active role in biphenyl and polychlorinated biphenyl degradation in Rhodococcus sp. strain RHA1. Appl. Environ. Microbiol. 72, 5396–5402. doi: 10.1128/AEM.00298-06
Iwasaki, T., Takeda, H., Miyauchi, K., Yamada, T., Masai, E., and Fukuda, M. (2007). Characterization of two biphenyl dioxygenases for biphenyl/PCB degradation in A PCB degrader, Rhodococcus sp. strain RHA1. Biosci. Biotechnol. Biochem. 71, 993–1002. doi: 10.1271/bbb.60663
Kimura, N., Kitagawa, W., Mori, T., Nakashima, N., Tamura, T., and Kamagata, Y. (2006). Genetic and biochemical characterization of the dioxygenase involved in lateral dioxygenation of dibenzofuran from Rhodococcus opacus strain SAO101. Appl. Microbiol. Biotechnol. 73, 474–484. doi: 10.1007/s00253-006-0481-8
Kimura, N., and Urushigawa, Y. (2001). Metabolism of dibenzo-p-dioxin and chlorinated dibenzo-p-dioxin by a gram-positive bacterium, Rhodococcus opacus SAO101. J. Biosci. Bioeng. 92, 138–143. doi: 10.1016/S1389-1723(01)80214-0
Kitagawa, W., Kimura, N., and Kamagata, Y. (2004). A novel p-nitrophenol degradation gene cluster from a gram-positive bacterium, Rhodococcus opacus SAO101. J. Bacteriol. 186, 4894–4902. doi: 10.1128/JB.186.15.4894-4902.2004
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lauby-Secretan, B., Loomis, D., Grosse, Y., El Ghissassi, F., Bouvard, V., Benbrahim-Tallaa, L., et al. (2013). Carcinogenicity of polychlorinated biphenyls and polybrominated biphenyls. Lancet Oncol. 14, 287–288. doi: 10.1016/S1470-2045(13)70104-9
Leewis, M. C., Uhlik, O., and Leigh, M. B. (2016). Synergistic processing of Biphenyl and Benzoate: carbon flow through the bacterial community in polychlorinated-biphenyl-contaminated soil. Sci. Rep. 6:22145. doi: 10.1038/srep22145
Leigh, M. B., Pellizari, V. H., Uhlik, O., Sutka, R., Rodrigues, J., Ostrom, N. E., et al. (2007). Biphenyl-utilizing bacteria and their functional genes in a pine root zone contaminated with polychlorinated biphenyls (PCBs). ISME J. 1, 134–148. doi: 10.1038/ismej.2007.26
Leigh, M. B., Prouzova, P., Mackova, M., Macek, T., Nagle, D. P., and Fletcher, J. S. (2006). Polychlorinated biphenyl (PCB)-degrading bacteria associated with trees in a PCB-contaminated site. Appl. Environ. Microbiol. 72, 2331–2342. doi: 10.1128/AEM.72.4.2331-2342.2006
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Mayes, B. A., Mcconnell, E. E., Neal, B. H., Brunner, M. J., Hamilton, S. B., Sullivan, T. M., et al. (1998). Comparative carcinogenicity in Sprague-Dawley rats of the polychlorinated biphenyl mixtures Aroclors 1016, 1242, 1254, and 1260. Toxicol. Sci. 41, 62–76.
Mondello, F. J., Turcich, M. P., Lobos, J. H., and Erickson, B. D. (1997). Identification and modification of biphenyl dioxygenase sequences that determine the specificity of polychlorinated biphenyl degradation. Appl. Environ. Microbiol. 63, 3096–3103.
National Research Council (1979). Polychlorinated Biphenyls. Washington, DC: The National Academies Press.
Ohtsubo, Y., Kudo, T., Tsuda, M., and Nagata, Y. (2004). Strategies for bioremediation of polychlorinated biphenyls. Appl. Microbiol. Biotechnol. 65, 250–258. doi: 10.1007/s00253-004-1654-y
Pieper, D. H. (2005). Aerobic degradation of polychlorinated biphenyls. Appl. Microbiol. Biotechnol. 67, 170–191. doi: 10.1007/s00253-004-1810-4
Pieper, D. H., and Seeger, M. (2008). Bacterial metabolism of polychlorinated biphenyls. J. Mol. Microbiol. Biotechnol. 15, 121–138. doi: 10.1159/000121325
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Quensen, J. F. III, Tiedje, J. M., and Boyd, S. A. (1988). Reductive dechlorination of polychlorinated biphenyls by anaerobic microorganisms from sediments. Science 242, 752–754. doi: 10.1126/science.242.4879.752
Quinete, N., Schettgen, T., Bertram, J., and Kraus, T. (2014). Occurrence and distribution of PCB metabolites in blood and their potential health effects in humans: a review. Environ. Sci. Pollut. Res. Int. 21, 11951–11972. doi: 10.1007/s11356-014-3136-9
R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rather, L. J., Knapp, B., Haehnel, W., and Fuchs, G. (2010). Coenzyme A-dependent aerobic metabolism of benzoate via epoxide formation. J. Biol. Chem. 285, 20615–20624. doi: 10.1074/jbc.M110.124156
Ross, G. (2004). The public health implications of polychlorinated biphenyls (PCBs) in the environment. Ecotoxicol. Environ. Saf. 59, 275–291. doi: 10.1016/j.ecoenv.2004.06.003
Saavedra, J. M., Acevedo, F., Gonzalez, M., and Seeger, M. (2010). Mineralization of PCBs by the genetically modified strain Cupriavidus necator JMS34 and its application for bioremediation of PCBs in soil. Appl. Microbiol. Biotechnol. 87, 1543–1554. doi: 10.1007/s00253-010-2575-6
Seeger, M., Camara, B., and Hofer, B. (2001). Dehalogenation, denitration, dehydroxylation, and angular attack on substituted biphenyls and related compounds by a biphenyl dioxygenase. J. Bacteriol. 183, 3548–3555. doi: 10.1128/JB.183.12.3548-3555.2001
Seeger, M., Timmis, K. N., and Hofer, B. (1995). Conversion of chlorobiphenyls into phenylhexadienoates and benzoates by the enzymes of the upper pathway for polychlorobiphenyl degradation encoded by the bph locus of Pseudomonas sp. strain LB400. Appl. Environ. Microbiol. 61, 2654–2658.
Seto, M., Kimbara, K., Shimura, M., Hatta, T., Fukuda, M., and Yano, K. (1995). A novel transformation of polychlorinated biphenyls by Rhodococcus sp. Strain RHA1. Appl. Environ. Microbiol. 61, 3353–3358.
Sharma, J. K., Gautam, R. K., Misra, R. R., Kashyap, S. M., Singh, S. K., and Juwarkar, A. A. (2014). Degradation of Di- through hepta-chlorobiphenyls in clophen oil using microorganisms isolated from long term PCBs contaminated soil. Indian J. Microbiol. 54, 337–342. doi: 10.1007/s12088-014-0459-7
Sharma, J. K., Gautam, R. K., Nanekar, S. V., Weber, R., Singh, B. K., Singh, S. K., et al. (2017). Advances and perspective in bioremediation of polychlorinated biphenyl-contaminated soils. Environ. Sci. Pollut. Res. Int. doi: 10.1007/s11356-017-8995-4 [Epub ahead of print].
Sierra, I., Valera, J. L., Marina, M. L., and Laborda, F. (2003). Study of the biodegradation process of polychlorinated biphenyls in liquid medium and soil by a new isolated aerobic bacterium (Janibacter sp.). Chemosphere 53, 609–618. doi: 10.1016/S0045-6535(03)00418-1
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Singer, A. C., Gilbert, E. S., Luepromchai, E., and Crowley, D. E. (2000). Bioremediation of polychlorinated biphenyl-contaminated soil using carvone and surfactant-grown bacteria. Appl. Microbiol. Biotechnol. 54, 838–843. doi: 10.1007/s002530000472
Taguchi, K., Motoyama, M., Iida, T., and Kudo, T. (2007). Polychlorinated biphenyl/biphenyl degrading gene clusters in Rhodococcus sp. K37, HA99, and TA431 are different from well-known bph gene clusters of Rhodococci. Biosci. Biotechnol. Biochem. 71, 1136–1144. doi: 10.1271/bbb.60551
Taira, K., Hirose, J., Hayashida, S., and Furukawa, K. (1992). Analysis of bph operon from the polychlorinated biphenyl-degrading strain of Pseudomonas pseudoalcaligenes KF707. J. Biol. Chem. 267, 4844–4853.
Turrio-Baldassarri, L., Abate, V., Alivernini, S., Battistelli, C. L., Carasi, S., Casella, M., et al. (2007). A study on PCB, PCDD/PCDF industrial contamination in a mixed urban-agricultural area significantly affecting the food chain and the human exposure. Part I: soil and feed. Chemosphere 67, 1822–1830. doi: 10.1016/j.chemosphere.2006.05.124
Uhlik, O., Jecna, K., Mackova, M., Vlcek, C., Hroudova, M., Demnerova, K., et al. (2009). Biphenyl-metabolizing bacteria in the rhizosphere of horseradish and bulk soil contaminated by polychlorinated biphenyls as revealed by stable isotope probing. Appl. Environ. Microbiol. 75, 6471–6477. doi: 10.1128/AEM.00466-09
Uhlik, O., Musilova, L., Ridl, J., Hroudova, M., Vlcek, C., Koubek, J., et al. (2013). Plant secondary metabolite-induced shifts in bacterial community structure and degradative ability in contaminated soil. Appl. Microbiol. Biotechnol. 97, 9245–9256. doi: 10.1007/s00253-012-4627-6
Vergani, L., Mapelli, F., Marasco, R., Crotti, E., Fusi, M., Di Guardo, A., et al. (2017a). Bacteria associated to plants naturally selected in a historical PCB polluted soil show potential to sustain natural attenuation. Front. Microbiol. 8:1385. doi: 10.3389/fmicb.2017.01385
Vergani, L., Mapelli, F., Zanardini, E., Terzaghi, E., Di Guardo, A., Morosini, C., et al. (2017b). Phyto-rhizoremediation of polychlorinated biphenyl contaminated soils: an outlook on plant-microbe beneficial interactions. Sci. Total Environ. 575, 1395–1406. doi: 10.1016/j.scitotenv.2016.09.218
Villacieros, M., Whelan, C., Mackova, M., Molgaard, J., Sanchez-Contreras, M., Lloret, J., et al. (2005). Polychlorinated biphenyl rhizoremediation by Pseudomonas fluorescens F113 derivatives, using a Sinorhizobium meliloti nod system to drive bph gene expression. Appl. Environ. Microbiol. 71, 2687–2694. doi: 10.1128/AEM.71.5.2687-2694.2005
Wald, J., Hroudova, M., Jansa, J., Vrchotova, B., Macek, T., and Uhlik, O. (2015). Pseudomonads rule degradation of polyaromatic hydrocarbons in aerated sediment. Front. Microbiol. 6:1268. doi: 10.3389/fmicb.2015.01268
Weisburg, W. G., Barns, S. M., Pelletier, D. A., and Lane, D. J. (1991). 16S ribosomal DNA amplification for phylogenetic study. J. Bacteriol. 173, 697–703. doi: 10.1128/jb.173.2.697-703.1991
Keywords: biphenyl, PCBs, bacterial consortium, metagenomics, Rhodococcus
Citation: Garrido-Sanz D, Manzano J, Martín M, Redondo-Nieto M and Rivilla R (2018) Metagenomic Analysis of a Biphenyl-Degrading Soil Bacterial Consortium Reveals the Metabolic Roles of Specific Populations. Front. Microbiol. 9:232. doi: 10.3389/fmicb.2018.00232
Received: 28 September 2017; Accepted: 30 January 2018;
Published: 15 February 2018.
Edited by:
Diana Elizabeth Marco, National Scientific and Technical Research Council (CONICET), ArgentinaReviewed by:
Marc Viñas, Institut de Recerca i Tecnologia Agroalimentàries (IRTA), SpainSonja Kristine Fagervold, Université Pierre et Marie Curie (UPMC), France
Copyright © 2018 Garrido-Sanz, Manzano, Martín, Redondo-Nieto and Rivilla. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rafael Rivilla, cmFmYWVsLnJpdmlsbGFAdWFtLmVz