 Benjamin R. LaFrentz1*
Benjamin R. LaFrentz1* Julio C. García1
Julio C. García1 Geoffrey C. Waldbieser2
Geoffrey C. Waldbieser2 Jason P. Evenhuis3
Jason P. Evenhuis3 Thomas P. Loch4
Thomas P. Loch4 Mark R. Liles5
Mark R. Liles5 Fong S. Wong6Siow F. Chang6
Fong S. Wong6Siow F. Chang6- 1Aquatic Animal Health Research Unit, United States Department of Agriculture – Agricultural Research Service, Auburn, AL, United States
- 2Warmwater Aquaculture Research Unit, Thad Cochran National Warmwater Aquaculture Center, United States Department of Agriculture – Agricultural Research Service, Stoneville, MS, United States
- 3National Center for Cool and Cold Water Aquaculture, United States Department of Agriculture – Agricultural Research Service, Kearneysville, WV, United States
- 4Department of Pathobiology and Diagnostic Investigation, College of Veterinary Medicine, Michigan State University, East Lansing, MI, United States
- 5Department of Biological Sciences, Auburn University, Auburn, AL, United States
- 6MSD Animal Health Innovation Pte. Ltd., Singapore, Singapore
Columnaris disease, caused by the Gram-negative bacterium Flavobacterium columnare, is one of the most prevalent fish diseases worldwide. An exceptionally high level of genetic diversity among isolates of F. columnare has long been recognized, whereby six established genomovars have been described to date. However, little has been done to quantify or characterize this diversity further in a systematic fashion. The objective of this research was to perform phylogenetic analyses of 16S rRNA and housekeeping gene sequences to decipher the genetic diversity of F. columnare. Fifty isolates and/or genomes of F. columnare, originating from diverse years, geographic locations, fish hosts, and representative of the six genomovars were analyzed in this study. A multilocus phylogenetic analysis (MLPA) of the 16S rRNA and six housekeeping genes supported four distinct F. columnare genetic groups. There were associations between genomovar and genetic group, but these relationships were imperfect indicating that genomovar assignment does not accurately reflect F. columnare genetic diversity. To expand the dataset, an additional 90 16S rRNA gene sequences were retrieved from GenBank and a phylogenetic analysis of this larger dataset also supported the establishment of four genetic groups. Examination of isolate historical data indicated biological relevance to the identified genetic diversity, with some genetic groups isolated preferentially from specific fish species or families. It is proposed that F. columnare isolates be assigned to the four genetic groups defined in this study rather than genomovar in order to facilitate a standard nomenclature across the scientific community. An increased understanding of which genetic groups are most prevalent in different regions and/or aquaculture industries may allow for the development of improved targeted control and treatment measures for columnaris disease.
Introduction
Columnaris disease, caused by the Gram-negative bacterium Flavobacterium columnare, is a prevalent disease of fish due to an ability to infect most freshwater species in a range of environmental temperatures. The disease was first described in the early 1900s and clinical signs include lethargy, loss of appetite, necrotic gills, depigmented and necrotic lesions of the skin, and necrotic or frayed fins. Upon the discovery of this disease, it was suggested to be a significant pathogen due to an ability to infect numerous fish species (Davis, 1922). Currently, columnaris disease continues to impact wild (Morris et al., 2006; Scott and Bollinger, 2014; Faisal et al., 2017) and cultured species of fish worldwide such as salmonids in Finland (Suomalainen et al., 2005), channel catfish and rainbow trout in the United States (Wagner et al., 2002; Evenhuis et al., 2014), salmonids in Chile (Avendaño-Herrera et al., 2011), tilapia in Thailand (Dong et al., 2015), various species in Brazil (Barony et al., 2015), as well as other species and locations.
Although globally important, a thorough understanding of this bacterium and the disease it causes is lacking. Recent research has closed gaps in our knowledge, including host–pathogen–environment interactions (Kinnula et al., 2017), identification of functional virulence factors (Li et al., 2017), selective breeding for columnaris disease resistance (Evenhuis et al., 2015) and other important research areas (Declercq et al., 2013). It is anticipated that an increased understanding of these research areas will lead to improved methods to prevent and control columnaris disease. Currently, columnaris disease is best controlled by antibiotic or chemical use and preventative measures include management strategies and use of autogenous and licensed vaccines. While there has been some success in controlling and preventing columnaris using these measures, losses in aquaculture operations due to this disease remain substantial.
The genetic diversity among F. columnare isolates has been studied for several decades. Song et al. (1988) performed DNA hybridization experiments on isolates from geographically distant regions which indicated that three genetic groups existed based upon DNA homology. Toyama et al. (1996) demonstrated intraspecific nucleotide diversity in the 16S rRNA gene of isolates, and this diversity was exploited to develop a PCR-based assay for typing F. columnare (Triyanto and Wakabayashi, 1999). In this restriction fragment length polymorphism (RFLP) assay, a portion of the 16S rRNA gene is amplified, digested with a single restriction enzyme, and resultant DNA fragments are resolved by electrophoresis. Use of this assay on a small number of isolates categorized them into three genomovars (i.e., genetic groups), and the assay results were supported by phylogenetic analysis and DNA hybridization (Triyanto and Wakabayashi, 1999).
Following these original reports of genetic diversity, numerous studies have analyzed isolates from various locations using different molecular approaches such as random amplified polymorphic DNA (RAPD), pulsed-field gel electrophoresis (PFGE), amplified fragment length polymorphism (AFLP), single-strand conformation polymorphism (SSCP), repetitive extragenic palindromic PCR (REP-PCR), multilocus sequence analysis (MLSA), and sequencing of the 16S rRNA gene and 16S–23S intergenic spacer region. The results from these studies are strikingly similar in that most indicate at least two different groups among F. columnare isolates (Table 1). The first whole genome sequence of F. columnare was published in 2012 (Tekedar et al., 2012) and subsequently several other genomes have been published (Bartelme et al., 2016; Kumru et al., 2016; Zhang et al., 2016; Evenhuis et al., 2017; Kayansamruaj et al., 2017) allowing for genome based analyses. Kumru et al. (2017) performed a thorough comparative analysis of a genomovar I and II genome, representative of two genetic types of F. columnare (described further below). Their research suggested that the core genomes were mostly conserved, but the isolates were genetically distinct as defined by in silico DNA–DNA hybridization methods. Kayansamruaj et al. (2017) performed a comparative genome analysis of several genomes and their research indicated three or four groups depending on the locus used for analysis. These studies highlight the genetic diversity among F. columnare isolates and that phylogenetic resolution is affected by the methods used to compare isolates.
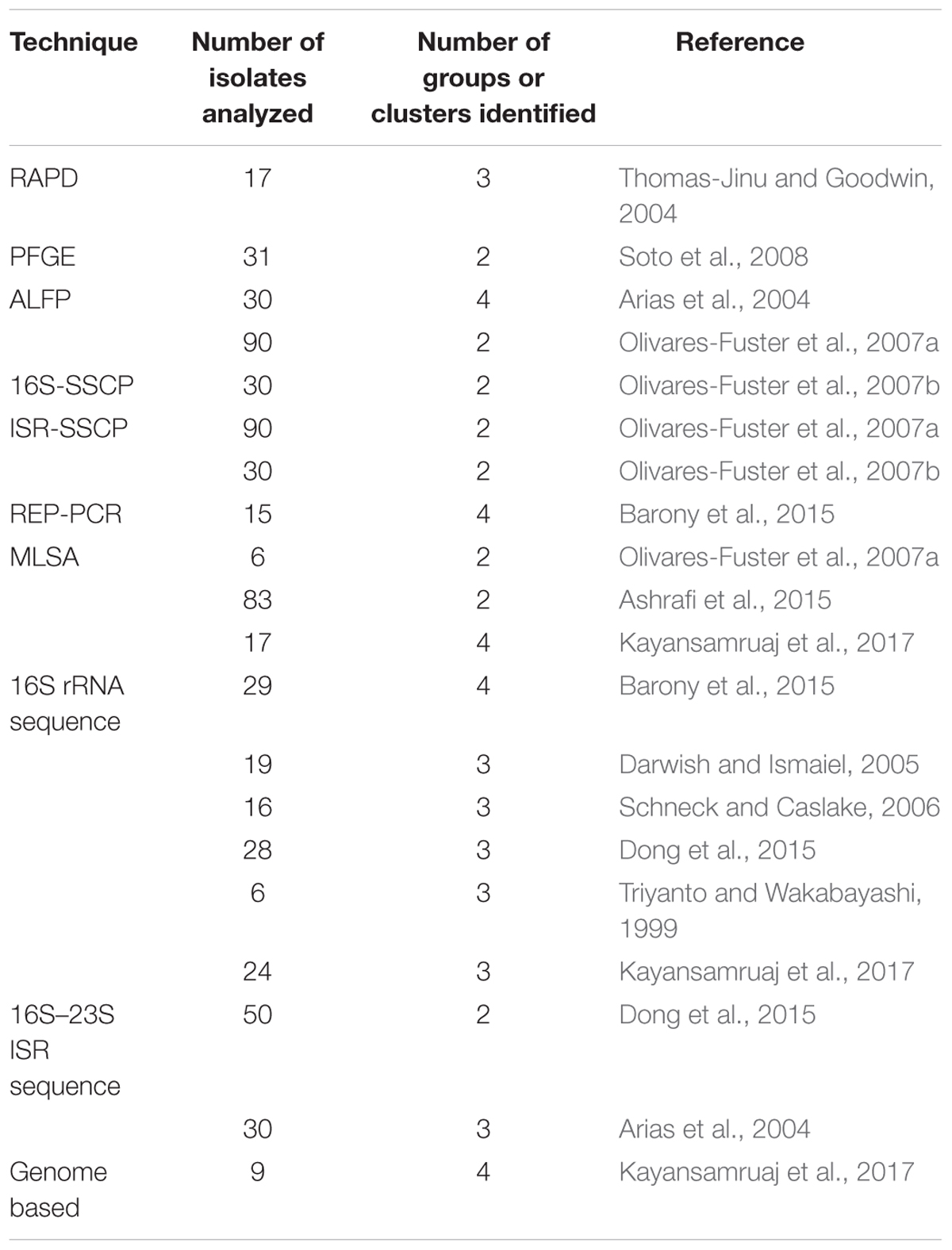
TABLE 1. Summary of publications that used molecular approaches to define the genetic diversity of Flavobacterium columnare.
It is well known that development of control methods and vaccines can be complicated by genetic diversity of bacterial pathogens. In regards to vaccines, genetic diversity can result in antigenic variation that may render vaccines ineffective against heterologous isolates (Telford, 2008). The difficulty in preventing and controlling columnaris disease may reflect an inadequate understanding of F. columnare genetic diversity. To begin to understand the genetic diversity of F. columnare, we first standardized the 16S-RFLP technique (LaFrentz et al., 2014) developed by Triyanto and Wakabayashi (1999). In that study, five genomovars were described (I, II, II-B, III, and I/II) and subsequently one primer was optimized to overcome difficulties in amplifying 16S rRNA genes from all F. columnare isolates (LaFrentz et al., 2017). More recently, a new genomovar, II-A, was described (García et al., 2018). With this established genotyping system, the objective of the present study was to decipher F. columnare genetic diversity using higher resolution methods and to compare the phylogenies derived from a multilocus phylogenetic analysis (MLPA) with previously assigned genomovars.
Materials and Methods
Bacterial and Culture Conditions
Fifty isolates and/or genomes of F. columnare, originating from diverse years, geographic locations, and fish hosts, were analyzed in this study (Tables 2, 3). Of these, 26 (Table 2) were cultured from archive laboratory stocks, and extracted DNA was used to sequence genes of interest as described below. Eleven publicly available genomes and thirteen additional genomes (LaFrentz et al., unpublished; Table 3) were used to obtain DNA sequences from annotated genomes. The identity of isolates was confirmed by PCR using species specific primers as described by Welker et al. (2005). Frozen material from archived glycerol stocks were plated onto modified Shieh agar (LaFrentz and Klesius, 2009) plates and incubated at 28°C for 48–72 h. Single isolated colonies were then cultured in 25 mL of modified Shieh broth and incubated at 28°C for 24–36 h with shaking at 175 rpm. These cultures were used for DNA extraction.
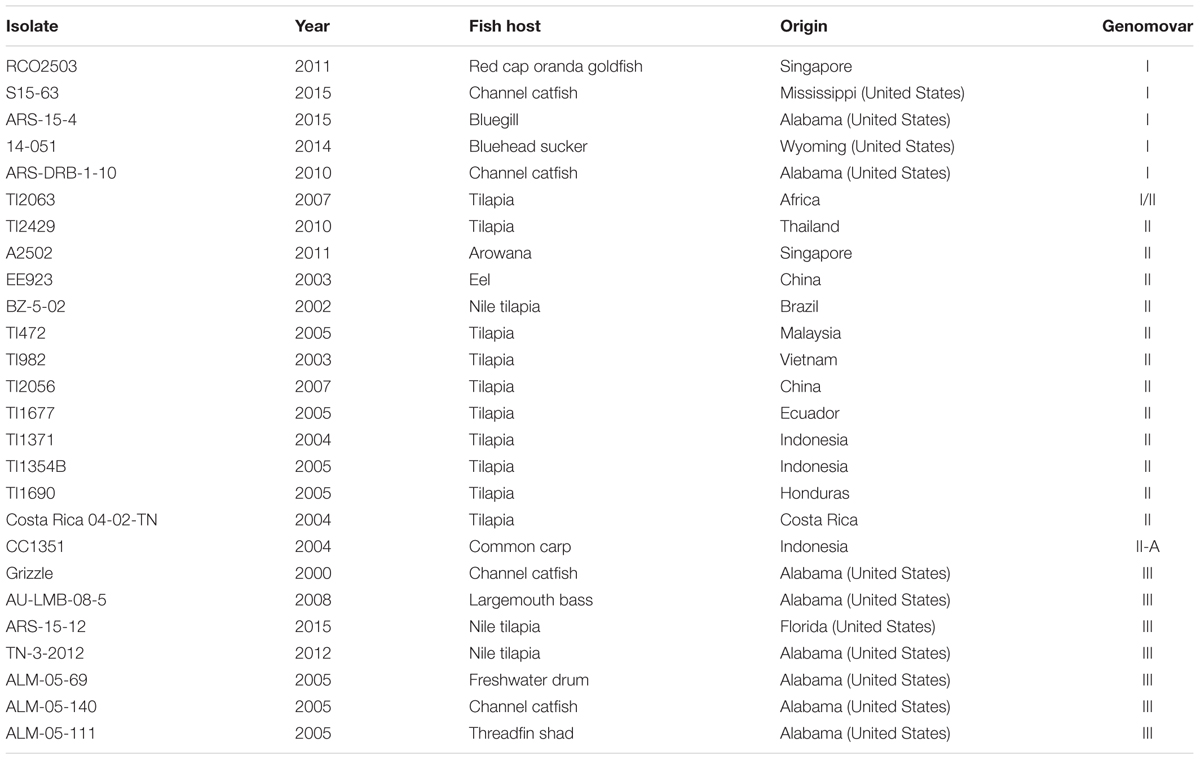
TABLE 2. Description of Flavobacterium columnare isolates used in this study, including the year and fish host of isolation, geographic origin, and genomovar assignment.
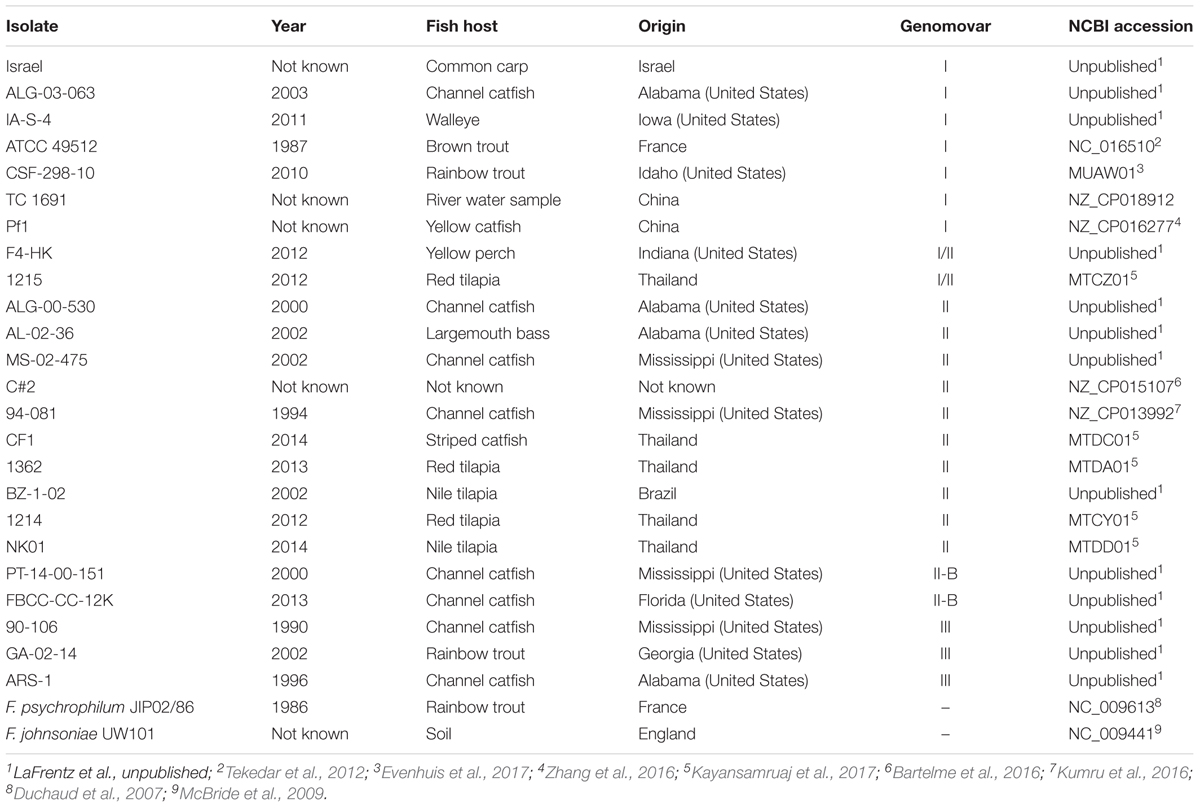
TABLE 3. Description of Flavobacterium columnare, F. psychrophilum, and F. johnsoniae genomes analyzed in this study, including the year and fish host of isolation, geographic origin, genomovar assignment, and NCBI accession number.
DNA Extraction and RFLP
Bacterial genomic DNA (gDNA) was extracted from isolates using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s protocol for Gram-negative bacteria and quantified using a NanoDrop ND-1000 spectrophotometer (NanoDrop). Each isolate was assigned to genomovar as described by LaFrentz et al. (2014) using an optimized reverse primer (1500R-1; LaFrentz et al., 2017).
Primer Design
Ashrafi et al. (2015) previously published a MLSA scheme for F. columnare, in which the primers were designed from the DNA sequence of a genomovar I isolate (ATCC 49512). In the present study, the same six housekeeping genes were used for a MLPA, including trpB, gyrB, dnaK, tuf, atpA, and rpoD. To ensure primer specificity for isolates assigned to different genomovars, these gene sequences were extracted from 13 draft genomes (Table 3; LaFrentz et al., unpublished) and analyzed. For each gene, a multiple sequence alignment was performed using CLC Genomics Workbench (version 9.5.2) and the primer binding sites designed by Ashrafi et al. (2015) were examined. Nucleotide heterogeneity was observed at the binding site of most primers (i.e., up to eight positions with differing nucleotides; data not shown); thus, new forward and reverse degenerate primers for each gene were designed using the consensus sequences at the primer binding sites used by Ashrafi et al. (2015) (Table 4).

TABLE 4. Sequences of primers designed for the amplification of Flavobacterium columnare housekeeping genes used for the MLSA and corresponding PCR cycling parameters.
PCR Amplification and Sequencing
The 16S rRNA and each of the six housekeeping genes were amplified from each F. columnare isolate (Table 2) by PCR using the primers designed in this study or previously published (Table 4). PCR was performed with AccuPrime Pfx DNA polymerase (Invitrogen), and the final concentrations of each component in the 50 μL reaction mixture were as follows: 5 μL 10X AccuPrime Pfx reaction mix, 0.4 μM forward and reverse primer, 1 unit AccuPrime Pfx DNA polymerase and 25 ng total gDNA. PCR amplification was performed with a MyCycler thermal cycler (Bio-Rad) using a cycling protocol as follows: 1 cycle of 5 min at 95°C; 30–40 cycles of 30 s at 95°C, 20 s at 45–62°C and 45 s at 68°C; and a final cycle of 10 min at 68°C. Different numbers of PCR cycles and annealing temperatures were used for each primer pair (Table 4). Following amplification, PCR products were detected by subjecting 5 μL of the PCR reaction to 1.5% (w/v) agarose gel electrophoresis in Tris-acetate-EDTA (TAE buffer). Gels were precast with 1X SYBR Safe DNA gel stain (Invitrogen), and the products were visualized using ultraviolet transillumination. PCR products were purified using the QIAquick PCR Purification Kit (Qiagen) and were sequenced commercially (Eurofins Genomics) using the same primers used for PCR. Sequence reads were assembled into contigs using BioNumerics (version 6.6, Applied Maths), and sequences were verified by manually examining chromatograms. The sequences for the gyrB, tuf, dnaK, rpoD, atpA, and trpB genes were deposited into GenBank under accession numbers MG516221–MG516454 and the 16S rRNA gene sequences were deposited under accession numbers MG516944–MG516975.
Phylogenetic Analyses
The 16S rRNA gene sequences from the 26 isolates of F. columnare (Table 2), sequences extracted from F. columnare genomes (Table 3), and sequences from F. psychrophilum and F. johnsoniae were aligned and trimmed using the Molecular Evolutionary Genetics Analysis (MEGA6) software (Tamura et al., 2013). The best nucleotide substitution model was tested in MEGA6 and the model with the lowest Bayesian Information Criterion (BIC) scores was used for the phylogenic analysis. The evolutionary relatedness of the 50 16S rRNA gene sequences was inferred using the maximum likelihood method based upon the Kimura 2-parameter model (K2+G; Kimura, 1980). Initial trees for the heuristic search were obtained by applying the neighbor-joining method to a matrix of pairwise distances estimated using the maximum composite likelihood (MCL) approach. All positions containing gaps and missing data were eliminated, leaving a total of 1,338 positions in the final dataset. The final tree was constructed from 1,000 bootstrap replicates and was rooted with F. psychrophilum and F. johnsoniae.
For MLPA, the six housekeeping gene sequences from the 26 isolates of F. columnare (Table 2), sequences extracted from annotated F. columnare genomes (Table 3), and sequences from F. psychrophilum and F. johnsoniae were aligned and trimmed using MEGA6. The six gene sequences from each individual isolate were then concatenated and used to infer the evolutionary relatedness by using the maximum likelihood method based upon the general time reversible model (GTR+G+I; Nei and Kumar, 2000). There were a total of 3,633 positions in the final dataset, the final tree was constructed from 1,000 bootstrap replicates, and was rooted with F. psychrophilum and F. johnsoniae. Additionally, a phylogenetic analysis was performed on each individual gene as indicated above.
An additional 16S rRNA gene phylogeny was performed. All available F. columnare 16S rRNA gene sequences were downloaded from GenBank (performed on 9/26/2017). From these, multiple sequences were removed because Basic Local Alignment Search Tool (BLASTn; Johnson et al., 2008) at the National Center for Biotechnology Information (NCBI1) analysis indicated they were not related to F. columnare. Sequences less than 1,000 nucleotides were omitted as were duplicate sequences of the same isolate. A total of 90 sequences were obtained that met these criteria and were added to the 50 sequences described above for phylogenetic analysis (Supplementary Table 1). The 140 F. columnare 16S rRNA gene sequences and 16S rRNA gene sequences from F. psychrophilum and F. johnsoniae were aligned and trimmed in MEGA6, whereby the evolutionary relatedness was inferred using the maximum likelihood method based upon the Kimura 2-parameter model (K2+G) as described above. There were a total of 1,061 positions in the final dataset, the final tree was constructed from 1,000 bootstrap replicates, and rooted with F. psychrophilum and F. johnsoniae.
Results and Discussion
The objective of the present study was to decipher F. columnare genetic diversity using phylogenetic analyses. In order to be inclusive, a diverse panel of isolates was collected from broad geographical regions, different fish species, and across a range of years. Additionally, it was important to include isolates assigned to the six established genomovars of F. columnare in order to use assigned genomovars as the basis to compare phylogenies derived from DNA sequences. Therefore, 12 genomovar I, 3 genomovar I/II, 22 genomovar II, 1 genomovar II-A, 2 genomovar II-B, and 10 genomovar III isolates were included and encompassed a large degree of diversity as indicated above (Tables 2, 3).
The 16S rRNA gene sequences from these isolates were obtained by sequencing or were downloaded from publicly available and unpublished genomes. Phylogenetic analysis of these sequences resulted in the establishment of four distinct genetic groups as evidenced by bootstrap values >70 (Figure 1). There was an association between genomovar and genetic group, but this association was imperfect. Genetic group 1 was comprised of all genomovar I isolates and the genomovar I/II isolate, F4-HK. Genetic group 2 was comprised of a portion of the genomovar II isolates, the only genomovar II-A isolate (CC1351), and the two genomovar II-B isolates. Genetic group 3 was comprised of all genomovar III isolates and two genomovar I/II isolates. Within genetic group 3, the two genomovar I/II isolates, TI2063 and 1215, formed a separate supported group with strong bootstrap support (98). Genetic group 4 was comprised of all the remaining genomovar II isolates. Within genetic group 4, two isolates, BZ-1-02 and BZ-5-02, formed a separate group with strong bootstrap support (100). Although there were four distinct genetic groups, the presence of the two supported sub-groups within genetic groups 3 and 4, may indicate the existence of additional genetic groups. Three of the genetic groups described above contained members from more than one genomovar as assigned by 16S-RFLP. These results indicate the 16S-RFLP method may not accurately reflect the genetic diversity in the 16S rRNA gene sequences of F. columnare. This may be explained by the nature of this technique which only interrogates sequence variation at restriction sites. Similar results were suggested by Kayansamruaj et al. (2017). For example, an isolate assigned to genomovar I/II by RFLP could be either a genetic group 1 or 3 isolate. Additionally, the RFLP technique was unable to differentiate between isolates contained in genetic groups 2 and 4. Interestingly, all of the genomovar II isolates contained in genetic group 4 were isolates that could not be assigned to a genomovar using the standardized RFLP method as described by LaFrentz et al. (2014). The 16S rRNA genes of these isolates could not be amplified by PCR using the original typing primers (20F/1500R) due to nucleotide heterogeneity in the primer binding site in these isolates (Dong et al., 2015; LaFrentz et al., 2017). Thus, a new degenerate reverse primer was designed (1500R-1) that allowed for amplification of the 16S rRNA genes and subsequent typing to genomovar II (LaFrentz et al., 2017). This finding indicates that the nucleotide heterogeneity observed in these isolates was not likely a random occurrence in some isolates, but is an accurate reflection of their phylogeny.
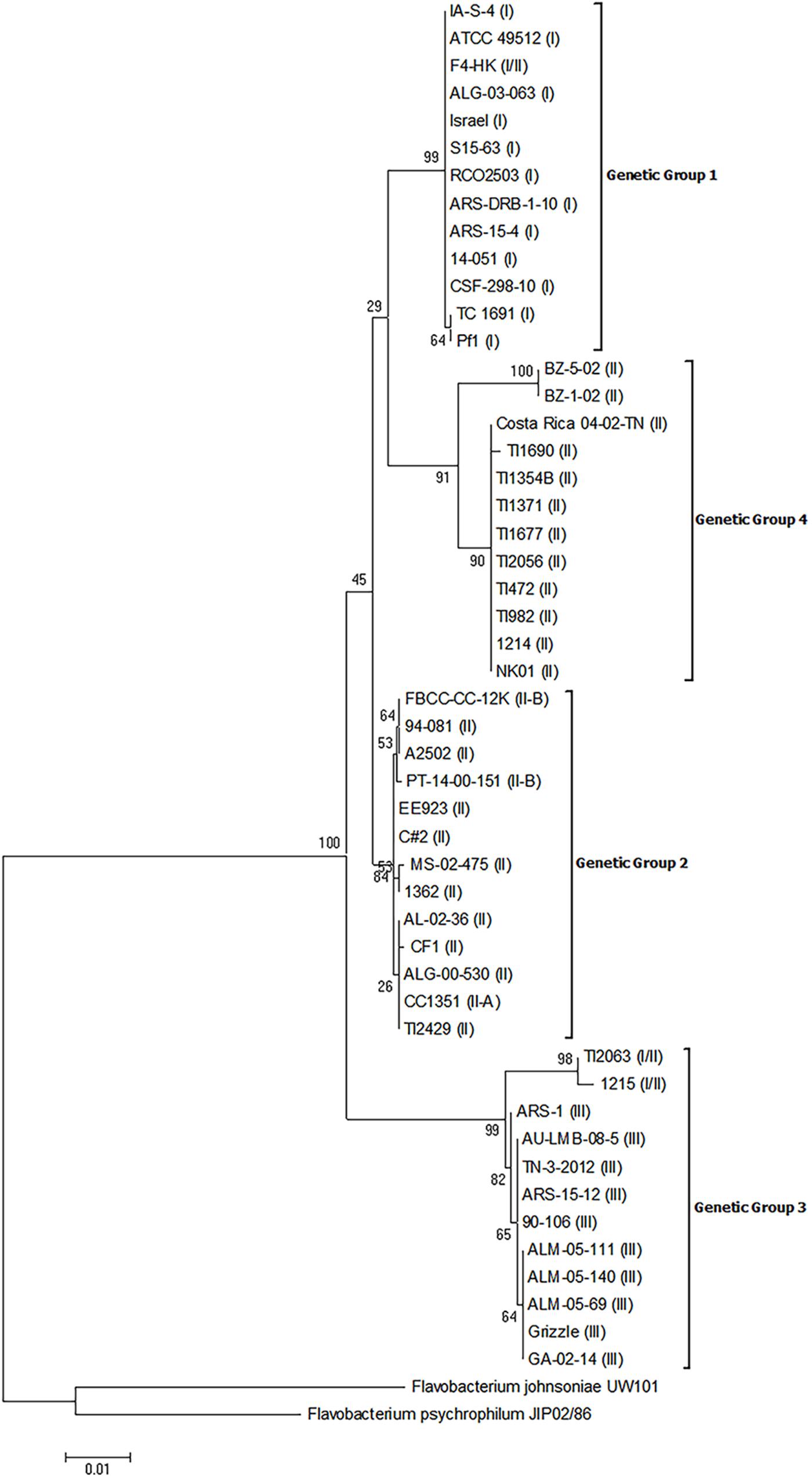
FIGURE 1. Phylogenetic relationships based on 16S rRNA gene sequences of 50 isolates of Flavobacterium columnare. Relatedness was inferred using the maximum likelihood method based upon the Kimura 2-parameter model (K2+G) and rooted with F. johnsoniae and F. psychrophilum. The percentage of trees in which the associated sequences clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The analysis involved 52 nucleotide sequences, all positions containing gaps and missing data were eliminated, and there were a total of 1,338 positions in the final dataset. The assigned genomovar of the isolate is in parentheses adjacent to the isolate designation.
The MLPA analysis of the concatenated housekeeping gene sequences also resulted in the establishment of four distinct genetic groups (Figure 2) and there was 100% agreement in the placement of isolates into each genetic group between the analysis of the 16S rRNA genes and the MLPA. Moreover, each genetic group was robustly supported as distinct from all other genetic groups as evidenced by bootstrap values >70. Additionally, there was a greater level of sequence heterogeneity in the MLPA analysis compared to the 16S rRNA phylogeny, as expected. However, the supported subgroups within genetic groups 3 and 4 that were present in the 16S rRNA gene phylogeny were not present in the MLPA phylogeny, which suggests these subgroups are not unique genetic groups.
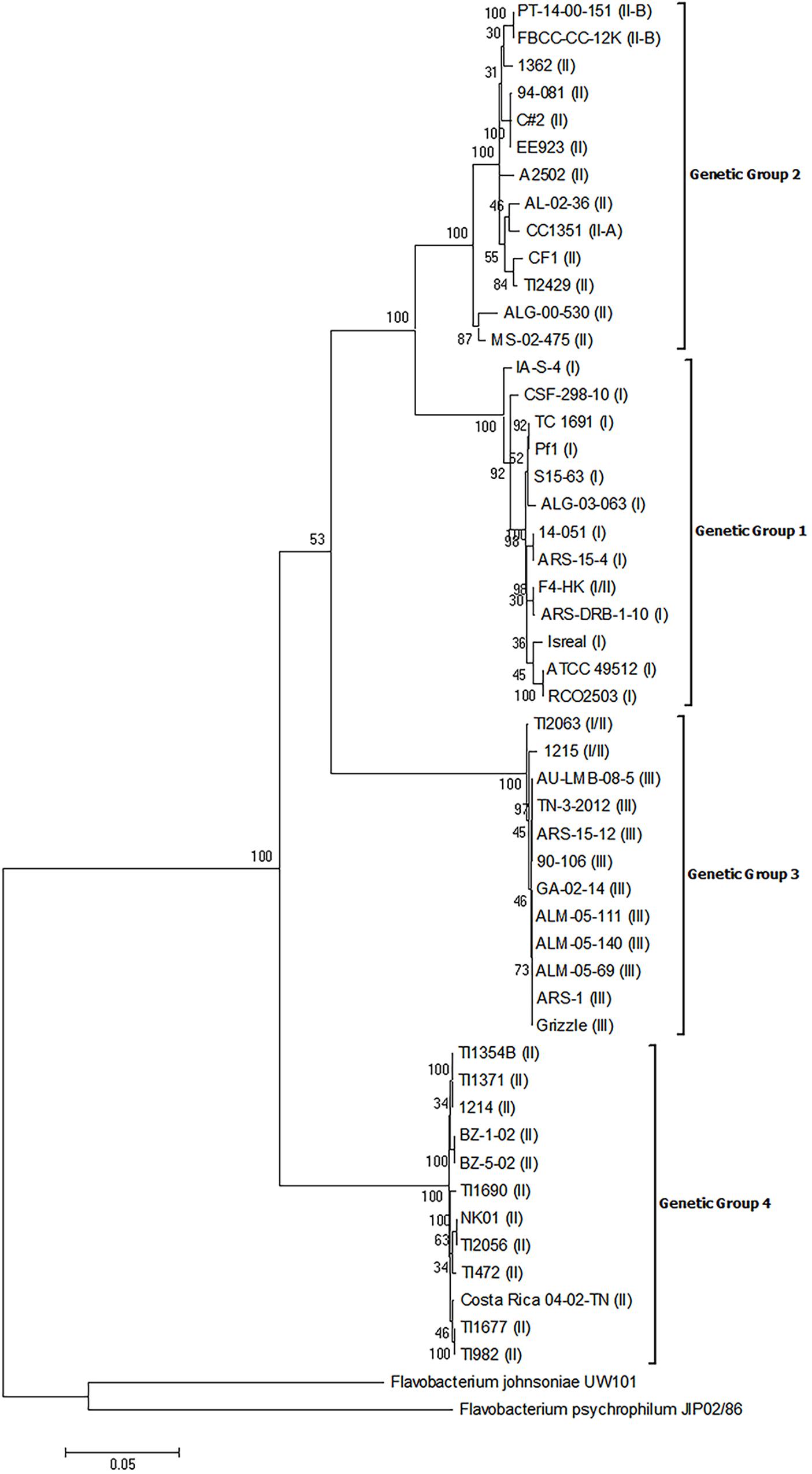
FIGURE 2. Phylogenetic relationships based on six concatenated housekeeping gene sequences of 50 isolates of Flavobacterium columnare. Relatedness was inferred using the maximum likelihood method based upon the general time reversible model (GTR+G+I) and rooted with F. johnsoniae and F. psychrophilum. The percentage of trees in which the associated sequences clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The analysis involved 52 nucleotide sequences, all positions containing gaps and missing data were eliminated, and there were a total of 3,633 positions in the final dataset. The assigned genomovar of the isolate is in parentheses adjacent to the isolate designation.
Phylogenetic analyses were also performed using each of the six individual gene sequences used in the MLPA. Analysis of the gyrB, tuf, and dnaK gene sequences also resulted in the formation of four distinct genetic groups with bootstrap values >70 (Supplementary Figures 1–3). Analysis of the dnaK gene resulted in the most robustly supported tree delineating the four genetic groups (bootstrap values of 99–100), and all basal nodes within the dendrogram were also highly supported (bootstrap values of 99–100). Thus the dnaK gene may represent an optimal locus to assign F. columnare isolates to a genetic group. Analysis of the rpoD gene sequences resulted in robust support for each genetic group; however, three genetic group 1 isolates (CSF-298-10, ALG-03-063, and IA-S-4) could not be ascribed to this genetic group via this locus (Supplementary Figure 4). Phylogenetic analysis of the atpA gene sequences resulted in robust support for each genetic group; however, two genetic group 2 isolates (MS-02-475 and ALG-00-530) formed a separate clade (Supplementary Figure 5). Phylogenetic analysis of the trpB gene sequences resulted in robust support for genetic groups 3 and 4; however, the topology was mostly unresolved for genetic groups 1 and 2 (Supplementary Figure 6). This is likely due to the high allelic diversity previously observed in this gene following an analysis of genomovar I (i.e., genetic group 1) isolates (Ashrafi et al., 2015).
The phylogenetic analyses provided strong support for the presence of four genetic groups among the F. columnare isolates described in this study. To expand the dataset, all 16S rRNA gene sequences deposited into GenBank meeting the criteria defined in the materials and methods (n = 90) were included with the 50 sequences of the present study for phylogenetic analysis. Similar to the previous phylogenetic analyses, analysis of these sequences resulted in the establishment of four distinct genetic groups as evidenced by bootstrap values >70. (Figure 3 and Supplementary Table 1). All 16S rRNA sequences retrieved from GenBank fell into the four genetic groups with the exception of one isolate, CUVET1216. However, it is unknown whether the sequence of this isolate is divergent from the four genetic groups or if there may have been sequencing errors. This isolate was recovered from red tilapia in Thailand (Dong et al., 2015), and the majority of isolates from this source were assigned to genetic group 4. It is possible that there is additional genetic diversity in F. columnare; however, these results suggest that the isolates selected for analysis in the present study are representative of the currently known genetic diversity in F. columnare.
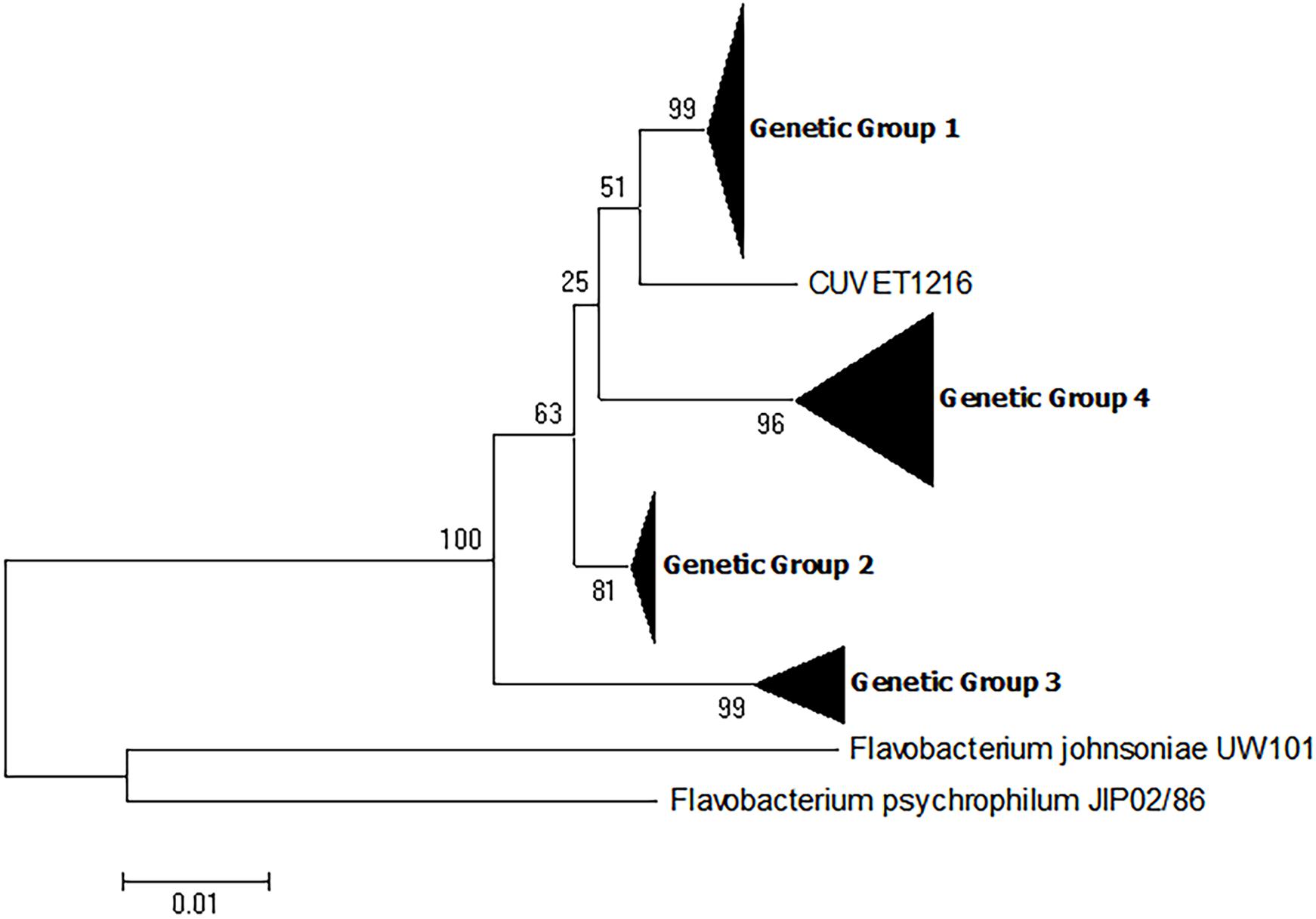
FIGURE 3. Phylogenetic relationships based on 16S rRNA gene sequences of 140 isolates of Flavobacterium columnare. Relatedness was inferred using the maximum likelihood method based upon the Kimura 2-parameter model (K2+G) and rooted with F. johnsoniae and F. psychrophilum. The percentage of trees in which the associated sequences clustered together in the bootstrap test (1,000 replicates) is shown next to the branches. The analysis involved 142 nucleotide sequences, all positions containing gaps and missing data were eliminated, and there were a total of 1,061 positions in the final dataset. See Supplementary Table 1 for individual isolates falling into each genetic group.
Although the phylogenetic analyses identified four genetic groups, there were differences in the topologies depending upon the marker used (i.e., 16S rRNA, MLPA, or individual housekeeping genes). For example the phylogenetic analysis of the 16S rRNA gene sequences placed genetic group 3 isolates as a sister group to all other genetic groups, while the analysis of the concatenated housekeeping genes placed genetic group 4 as the sister group (Figures 1, 2). Kayansamruaj et al. (2017) also observed a similar difference in branching order when the 16S rRNA and the same concatenated housekeeping genes were used for phylogenetic analyses of F. columnare. These authors also generated a whole genome phylogenetic tree based on concatenated single nucleotide polymorphisms (SNPs) of the core genomes and the resultant tree topology was different from both the 16S rRNA and multilocus sequence phylogenies. Additionally, there were variable topologies upon phylogenetic analyses of the individual housekeeping genes in the present study (Supplementary Figures 1–6). Ashrafi et al. (2015) also observed variable topologies for F. columnare between phylogenetic analyses of the individual six housekeeping genes. One potential reason for these differences may be the existence of multiple copies of the 16S rRNA gene in F. columnare, and/or polymorphisms among the multiple rrn copies. LaFrentz et al. (2014) demonstrated that in the 1,254 bp region of the 16S rRNA gene used for the 16S-RFLP of F. columnare, some isolates exhibited up to 10 SNPs between the multiple copies of this gene. Since most 16S rRNA sequences are obtained from direct sequencing of PCR products, the resultant sequence is a consensus from the multiple copies and use of these may impact phylogenetic analyses. Another potential reason for these differences may be due to different evolutionary histories of the housekeeping genes compared the 16S rRNA gene. For instance comparison of the 16S rRNA genes between representative isolates from each genetic group show a percent identity >96%, while for the sequences of the housekeeping genes, the percent identities were as low as 85% (data not shown). The use of low variation and higher variation genes likely contribute to the topology inconsistencies observed between markers. Due to the observation of variable topologies following phylogenetic analyses of different markers in the present and previous studies, future research should critically analyze whole genome sequences and construct genome based phylogenies to gain a better understanding of how these genetic groups were established as was recently published with F. psychrophilum (Duchaud et al., 2018).
Study findings highlight the existence of four phylogenetically distinct genetic groups within the species F. columnare. Since the results demonstrate that 16S-RFLP and genomovar assignment does not accurately reflect the genetic diversity in F. columnare, we propose that instead of genomovars, isolates be assigned to the genetic groups defined in this study. This can be achieved by sequencing the 16S rRNA gene or the dnaK gene (Table 4) and subsequent phylogenetic analysis that includes both the isolate(s) of interest and sequences belonging to each of the four genetic groups. This can be laborious and time consuming, so our laboratory is currently developing a specific multiplex PCR that can assign an unknown F. columnare isolate to genetic group in a single PCR. This assay will facilitate standard nomenclature for the genetically disparate groups within F. columnare across the scientific community, a matter of importance given the worldwide distribution of this important fish pathogen and the need for universally comparable genetic typing schemes. Recent analyses of F. columnare genomes further illustrate the importance of this. For example, Kumru et al. (2016, 2017) determined the average nucleotide identity (ANI) between a genetic group 1 isolate (ATCC 49512) and a genetic group 2 isolate (94-081) to be 90.71% and suggested that the two isolates may be classified as different species based upon the <95% ANI cut-off for species delineation (Goris et al., 2007). Kayansamruaj et al. (2017) recently published and analyzed five F. columnare genomes and suggested that species designation of F. columnare may need to be amended. Their phylogenetic analyses are consistent with those of the present study and identified four distinct clusters. Additionally, they determined the digital DNA–DNA hybridization (dDDH) values between the genomes of isolates they sequenced as well as publicly available F. columnare genomes. In the context of this study, comparison of the genomes between isolates corresponding to the different genetic groups 1, 2, 3, and 4, resulted in dDDH values of less than 43% (Kayansamruaj et al., 2017). These dDDH values were well below the <70% cut-off for species delineation (Auch et al., 2010). Therefore, based solely upon analysis of DNA sequences, the genetic groups identified in this study may represent four different species of bacteria; F. columnare (genetic group 1) and three additional species (genetic groups 2, 3, and 4). Additional research is needed to determine whether these are unique species of bacteria or different genetic types of the same species of bacteria.
It is imperative to determine whether there is any biological relevance to the genetic diversity revealed from MLPA analyses. To address this, historical data (species and family of fish, and country of origin) for each of the 140 isolates included in the 16S rRNA phylogeny were retrieved from GenBank and/or publications (Supplementary Table 1). Although this dataset may be limited and biased based upon publication of F. columnare 16S rRNA gene sequences, some interesting associations were found between genetic groups, the family of fish from which the isolate was recovered, and country of origin. For genetic group 1, the majority of isolates were recovered from the families Salmonidae (44.4%), Cyprinidae (16.7%), and Ictaluridae (9.3%; Figure 4) in North and South America, Asia, and Europe. The association between genomovar I isolates (i.e., genetic group 1) and salmonids was previously noted (LaFrentz et al., 2012), and in this study all but one isolate recovered from salmonids were genetic group 1 (Supplementary Figure 7). Evenhuis and LaFrentz (2016) determined the virulence of genetic group 1, 2, and 3 isolates in rainbow trout (Oncorhynchus mykiss) and the results clearly demonstrated genetic group 1 isolates were more virulent than isolates in genetic groups 2 and 3. Salmonids are cold water fish species, and early research demonstrated that genomovar I isolates (i.e., genetic group 1) were the only isolates capable of growth at 15°C (Triyanto and Wakabayashi, 1999), which may explain this association. Evenhuis and LaFrentz (2016) also showed that challenge of rainbow trout with some genomovar III (i.e., genetic group 3) isolates resulted in moderate mortality (up to 30%). The one isolate recovered from a salmonid that did not belong to genetic group 1 belonged to genetic group 3 (Supplementary Figure 7). The virulence studies of Evenhuis and LaFrentz (2016) may provide some basis for the isolation and virulence of a genetic group 3 isolate from infected rainbow trout.
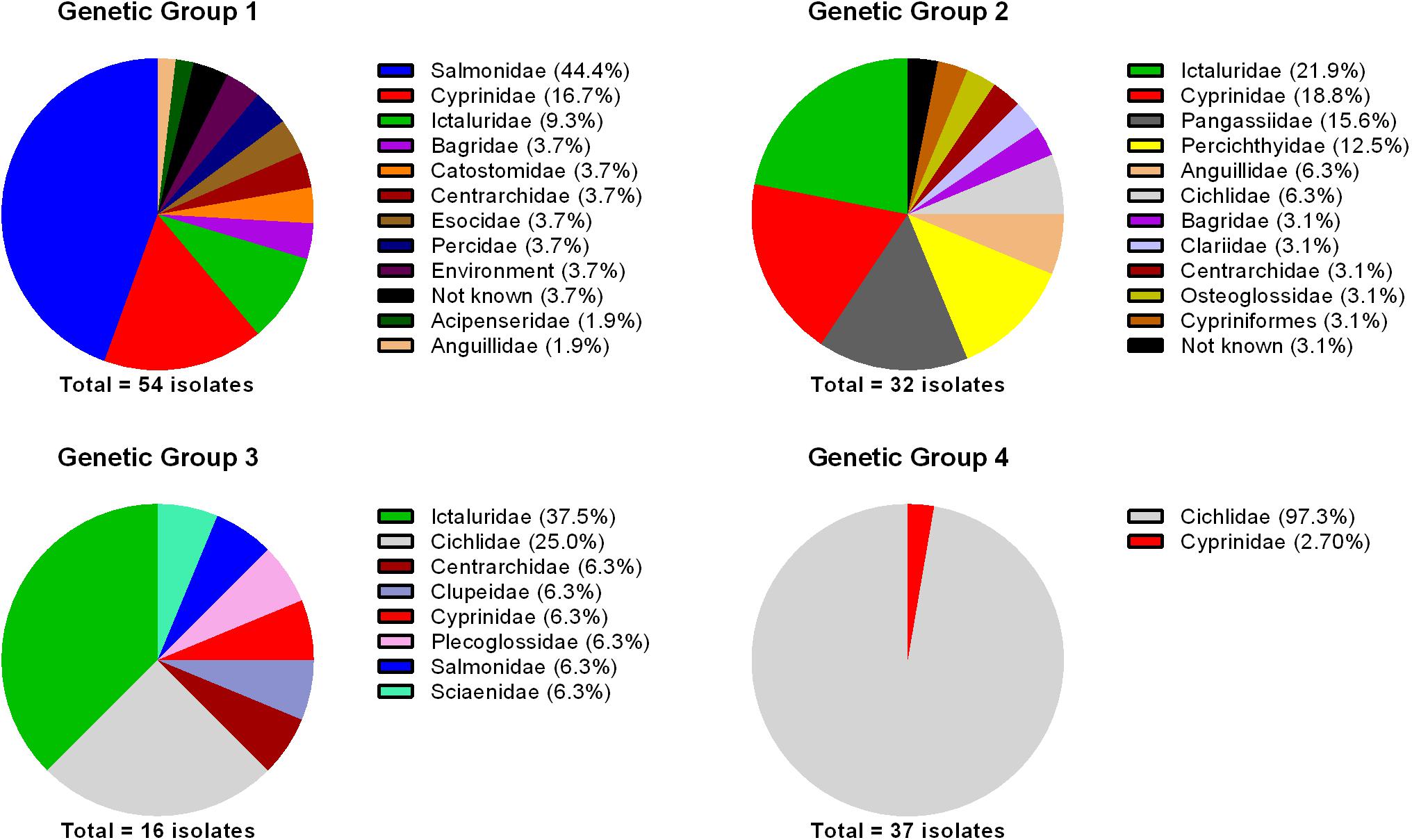
FIGURE 4. Percentage of Flavobacterium columnare isolates in each genetic group recovered from fish species in the families indicated. The number of isolates included in the analysis is indicated below the pie charts.
For genetic group 2, the majority of isolates were recovered from the families Ictaluridae (21.9%), Cyprinidae (18.8%), Pangasiidae (15.6%), and Percichthyidae (12.5%; Figure 4) from North America and Asia. For genetic group 3, which contained the lowest number of isolates (n = 16), the majority of isolates were recovered from the families Ictaluridae (37.5%) and Cichlidae (25%; Figure 4) from North America, Asia, and Africa. Shoemaker et al. (2008) tested the virulence of several genomovar I (i.e., genetic group 1) and genomovar II (i.e., genetic group 2) isolates in channel catfish (Ictalurus punctatus). The research demonstrated that genetic group 2 isolates were extremely virulent, with mortality greater than 60% for all isolates tested and the genetic group 1 isolates were less virulent. However, LaFrentz et al. (2014) discovered that some of the genetic group 1 isolates tested were misclassified and were actually genetic group 3. Therefore, in that study, only two genetic group 1 isolates (ATCC 23463 and MS-02-463) were tested and these did not cause any mortality. The genetic group 3 isolates tested were virulent and resulted in up to 46% mortality (Shoemaker et al., 2008). This virulence data tends to agree with the finding that the majority of isolates recovered from fish in the family Ictaluridae were genetic groups 2 and 3 (Figure 4 and Supplementary Figure 7). Additional studies are needed to determine the virulence of genetic group 1 isolates in fish of the family Ictaluridae because they have been recovered from epizootics caused by these isolates (Supplementary Figure 7).
For genetic group 4, all isolates, with the exception of one were recovered from tilapia (Oreochromis sp.) in the family Cichlidae (97.3%; Figure 4). These isolates were recovered from infected fish in South America, Central America, and Asia. Shoemaker and LaFrentz (2015) determined the virulence of isolates from genetic groups 1, 2, and 3 in hybrid tilapia and demonstrated that there was no association between virulence and genetic group; some isolates from each genetic group were capable of causing moderate to high mortality. Unfortunately, the virulence of genetic group 4 isolates were not tested in this research. Since genetic groups 2, 3, and 4 have been recovered from infected tilapia (Figure 4 and Supplementary Figure 7) and Shoemaker and LaFrentz (2015) demonstrated genetic group 1 isolates are virulent in this species, it would be of interest to determine if genetic group 4 isolates exhibit higher virulence in tilapia. Enhanced knowledge on this may provide support for the apparent association between genetic group 4 isolates and tilapia. Also of interest in regards to genetic group 4 isolates was the finding that they have only been recovered from Asia and South/Central America. If these isolates do have an association with tilapia, it may be possible that these isolates were spread to South and Central America from Asia with the movement of tilapia for aquaculture production. Our laboratory has analyzed over 200 F. columnare isolates, mostly of United States origin, and thus far none have been assigned to genetic group 4.
The results from this research establish the existence of four phylogenetically distinct genetic groups within the species F. columnare and demonstrate that 16S-RFLP and genomovar assignment does not accurately reflect this genetic diversity. Therefore, we propose that isolates be assigned to the genetic groups defined in this study rather than genomovar to facilitate a standard nomenclature across the scientific community. Analysis of the historical data of the isolates utilized indicates there is biological relevance to the genetic diversity identified. Thus, increased knowledge on the genetic groups of F. columnare that are most prevalent in different regions and/or aquaculture industries may allow for the development of better targeted control and treatment measures for columnaris disease.
Author Contributions
BL conceived the research, analyzed the DNA sequence data, performed the phylogenetic analyses, interpreted the data, and prepared the manuscript. JG managed the bacterial isolates, performed DNA extraction, PCR, and RFLP. GW and JE assisted with sequencing and data analysis. TL and ML assisted with the interpretation of the phylogenetic data. FW and SC assisted with bacterial isolate management. All authors contributed to manuscript revision, and read and approved the submitted version.
Funding
This research was supported by USDA-ARS CRIS Project No. 6010-32000-026-00D, Pathogen Characterization, Host Immune Response and Development of Strategies to Reduce Losses to Disease in Aquaculture.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors thank Kirsten Forster and Jordan Lehman, interns through the USDA Wallace-Carver Fellowship Program, for technical support. Flavobacterium columnare isolates RCO2503, TI2063, TI2429, A2502, EE923, TI472, TI982, TI2056, TI1677, TI1371, TI1354B, TI1690, and CC1351 were transferred to USDA-ARS through Material Transfer Research Agreement Number 58-6010-6-006-F. Mention of trade names or commercial products in this publication is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the United States Department of Agriculture.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00452/full#supplementary-material
Footnotes
References
Arias, C. R., Welker, T. L., Shoemaker, C. A., Abernathy, J. W., and Klesius, P. H. (2004). Genetic fingerprinting of Flavobacterium columnare isolates from cultured fish. J. Appl. Microbiol. 97, 421–428. doi: 10.1111/j.1365-2672.2004.02314.x
Ashrafi, R., Pulkkinen, K., Sundberg, L. R., Pekkala, N., and Ketola, T. (2015). A multilocus sequence analysis scheme for characterization of Flavobacterium columnare isolates. BMC Microbiol. 15:243. doi: 10.1186/s12866-015-0576-4
Auch, A. F., von Jan, M., Klenk, H. P., and Goker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Avendaño-Herrera, R., Gherardelli, V., Olmos, P., Godoy, M. G., Heisinger, A., and Fernandez, J. (2011). Flavobacterium columnare associated with mortality of salmonids farmed in Chile: a case report of two outbreaks. Bull. Eur. Assoc. Fish Pathol. 31, 36–44.
Barony, G. M., Tavares, G. C., Assis, G. B. N., Luz, R. K., Figueiredo, H. C. P., and Leal, C. A. G. (2015). New hosts and genetic diversity of Flavobacterium columnare isolated from Brazilian native species and Nile tilapia. Dis. Aquat. Organ. 117, 1–11. doi: 10.3354/dao02931
Bartelme, R. P., Newton, R. J., Zhu, Y., Li, N., LaFrentz, B. R., and McBride, M. J. (2016). Complete genome sequence of the fish pathogen Flavobacterium columnare strain C#2. Genome Announc. 4:e00624-16. doi: 10.1128/genomeA.00624-16
Darwish, A. M., and Ismaiel, A. A. (2005). Genetic diversity of Flavobacterium columnare examined by restriction fragment length polymorphism and sequencing of the 16S ribosomal RNA gene and the 16S-23S rDNA spacer. Mol. Cell. Probes 19, 267–274. doi: 10.1016/j.mcp.2005.04.003
Davis, H. S. (1922). A new bacterial disease of fresh-water fishes. Bull. U. S. Bur. Fish. 38, 261–280. doi: 10.5962/bhl.title.49773
Declercq, A. M., Haesebrouck, F., Van den Broeck, W., Bossier, P., and Decostere, A. (2013). Columnaris disease in fish: a review with emphasis on bacterium-host interactions. Vet. Res. 44:27. doi: 10.1186/1297-9716-44-27
Dong, H. T., LaFrentz, B., Pirarat, N., and Rodkhum, C. (2015). Phenotypic characterization and genetic diversity of Flavobacterium columnare isolated from red tilapia, Oreochromis sp., in Thailand. J. Fish Dis. 38, 901–913. doi: 10.1111/jfd.12304
Duchaud, E., Boussaha, M., Loux, V., Bernardet, J. F., Michel, C., Kerouault, B., et al. (2007). Complete genome sequence of the fish pathogen Flavobacterium psychrophilum. Nat. Biotechnol. 25, 763–769. doi: 10.1038/nbt1313
Duchaud, E., Rochat, T., Habib, C., Barbier, P., Loux, V., Guérin, C., et al. (2018). Genomic diversity and evolution of the fish pathogen Flavobacterium psychrophilum. Front. Microbiol. 9:138. doi: 10.3389/fmicb.2018.00138
Evenhuis, J. P., and LaFrentz, B. R. (2016). Virulence of Flavobacterium columnare genomovars in rainbow trout Oncorhynchus mykiss. Dis. Aquat. Organ. 120, 217–224. doi: 10.3354/dao03027
Evenhuis, J. P., LaPatra, S. E., and Graf, J. (2017). Draft genome sequence of the fish pathogen Flavobacterium columnare strain CSF-298-10. Genome Announc. 5:e00173-17. doi: 10.1128/genomeA.00173-17
Evenhuis, J. P., LaPatra, S. E., and Marancik, D. (2014). Early life stage rainbow trout (Oncorhynchus mykiss) mortalities due to Flavobacterium columnare in Idaho, USA. Aquaculture 418-419, 126–131. doi: 10.1016/j.aquaculture.2013.09.044
Evenhuis, J. P., Leeds, T. D., Marancik, D. P., LaPatra, S. E., and Wiens, G. D. (2015). Rainbow trout (Oncorhynchus mykiss) resistance to columnaris disease is heritable and favorably correlated with bacterial cold water disease resistance. J. Anim. Sci. 93, 1546–1554. doi: 10.2527/jas.2014-8566
Faisal, M., Diamanka, A., Loch, T. P., LaFrentz, B. R., Winters, A. D., García, J. C., et al. (2017). Isolation and characterization of Flavobacterium columnare strains infecting fishes inhabiting the Laurentian Great Lakes basin. J. Fish Dis. 40, 637–648. doi: 10.1111/jfd.12548
García, J. C., LaFrentz, B. R., Waldbieser, G. C., Wong, F. S., and Chang, S. F. (2018). Characterization of atypical Flavobacterium columnare and identification of a new genomovar. J. Fish Dis. (in press). doi: 10.1111/jfd.12778
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Johnson, M., Zaretskaya, I., Raytselis, Y., Merezhuk, Y., McGinnis, S., and Madden, T. L. (2008). NCBI BLAST: a better web interface. Nucleic Acids Res. 36, W5–W9. doi: 10.1093/nar/gkn201
Kayansamruaj, P., Dong, H. T., Hirono, I., Kondo, H., Senapin, S., and Rodkhum, C. (2017). Comparative genome analysis of fish pathogen Flavobacterium columnare reveals extensive sequence diversity within the species. Infect. Genet. Evol. 54(Suppl. C), 7–17. doi: 10.1016/j.meegid.2017.06.012
Kimura, M. (1980). A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120. doi: 10.1007/BF01731581
Kinnula, H., Mappes, J., Valkonen, J. K., Pulkkinen, K., and Sundberg, L.-R. (2017). Higher resource level promotes virulence in an environmentally transmitted bacterial fish pathogen. Evol. Appl. 10, 462–470. doi: 10.1111/eva.12466
Kumru, S., Tekedar, H. C., Gulsoy, N., Waldbieser, G. C., Lawrence, M. L., and Karsi, A. (2017). Comparative analysis of the Flavobacterium columnare genomovar I and II genomes. Front. Microbiol. 8:1375. doi: 10.3389/fmicb.2017.01375
Kumru, S., Tekedar, H. C., Waldbieser, G. C., Karsi, A., and Lawrence, M. L. (2016). Genome sequence of the fish pathogen Flavobacterium columnare genomovar II strain 94-081. Genome Announc. 4:e00430-16. doi: 10.1128/genomeA.00430-16
LaFrentz, B. R., García, J. C., Dong, H. T., Waldbieser, G. C., Rodkhum, C., Wong, F. S., et al. (2017). Optimized reverse primer for 16S-RFLP analysis and genomovar assignment of Flavobacterium columnare. J. Fish Dis. 40, 1103–1108. doi: 10.1111/jfd.12583
LaFrentz, B. R., and Klesius, P. H. (2009). Development of a culture independent method to characterize the chemotactic response of Flavobacterium columnare to fish mucus. J. Microbiol. Methods 77, 37–40. doi: 10.1016/j.mimet.2008.12.011
LaFrentz, B. R., LaPatra, S. E., Shoemaker, C. A., and Klesius, P. H. (2012). Reproducible challenge model to investigate the virulence of Flavobacterium columnare genomovars in rainbow trout Oncorhynchus mykiss. Dis. Aquat. Organ. 101, 115–122. doi: 10.3354/dao02522
LaFrentz, B. R., Waldbieser, G. C., Welch, T. J., and Shoemaker, C. A. (2014). Intragenomic heterogeneity in the 16S rRNA genes of Flavobacterium columnare and standard protocol for genomovar assignment. J. Fish Dis. 37, 657–669. doi: 10.1111/jfd.12166
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (New York, NY: John Wiley & Sons, Inc), 115–176.
Li, N., Zhu, Y., LaFrentz, B. R., Evenhuis, J. P., Hunnicutt, D. W., Conrad, R. A., et al. (2017). The type IX secretion system is required for virulence of the fish pathogen Flavobacterium columnare. Appl. Environ. Microbiol. 83:e01769-17. doi: 10.1128/aem.01769-17
McBride, M. J., Xie, G., Martens, E. C., Lapidus, A., Henrissat, B., Rhodes, R. G., et al. (2009). Novel features of the polysaccharide-digesting gliding vacterium Flavobacterium johnsoniae as revealed by genome sequence analysis. Appl. Environ. Microbiol. 75, 6864–6875. doi: 10.1128/aem.01495-09
Morris, J., Snyder-Conn, E., Foott, J., Holt, R., Suedkamp, M., Lease, H., et al. (2006). Survival of Lost River suckers (Deltistes luxatus) challenged with Flavobacterium columnare during exposure to sublethal ammonia concentrations at pH 9.5. Arch. Environ. Contam. Toxicol. 50, 256–263. doi: 10.1007/s00244-004-0194-x
Nei, M., and Kumar, S. (2000). Molecular Evolution and Phylogenetics. Oxford: Oxford University Press.
Olivares-Fuster, O., Baker, J. L., Terhune, J. S., Shoemaker, C. A., Klesius, P. H., and Arias, C. R. (2007a). Host-specific association between Flavobacterium columnare genomovars and fish species. Syst. Appl. Microbiol. 30, 624–633.
Olivares-Fuster, O., Shoemaker, C. A., Klesius, P. H., and Arias, C. R. (2007b). Molecular typing of isolates of the fish pathogen, Flavobacterium columnare, by single-strand conformation polymorphism analysis. FEMS Microbiol. Lett. 269, 63–69. doi: 10.1111/j.1574-6968.2006.00605.x
Schneck, J. L., and Caslake, L. F. (2006). Genetic diversity of Flavobacterium columnare isolated from fish collected from warm and cold water. J. Fish Dis. 29, 245–248. doi: 10.1111/j.1365-2761.2006.00698.x
Scott, S. J., and Bollinger, T. K. (2014). Flavobacterium columnare: an important contributing factor to fish die-offs in southern lakes of Saskatchewan, Canada. J. Vet. Diagn. Invest. 26, 832–836. doi: 10.1177/1040638714553591
Shoemaker, C. A., and LaFrentz, B. R. (2015). Lack of association between Flavobacterium columnare genomovar and virulence in hybrid tilapia Oreochromis niloticus (L.) × Oreochromis aureus (Steindachner). J. Fish Dis. 38, 491–498. doi: 10.1111/jfd.12262
Shoemaker, C. A., Olivares-Fuster, O., Arias, C. R., and Klesius, P. H. (2008). Flavobacterium columnare genomovar influences mortality in channel catfish (Ictalurus punctatus). Vet. Microbiol. 127, 353–359. doi: 10.1016/j.vetmic.2007.09.003
Song, Y.-L., Fryer, J. L., and Rohovec, J. S. (1988). Comparison of gliding bacteria isolated from fish in North America and other areas of the pacific rim. Fish Pathol. 23, 197–202. doi: 10.3147/jsfp.23.197
Soto, E., Mauel, M. J., Karsi, A., and Lawrence, M. L. (2008). Genetic and virulence characterization of Flavobacterium columnare from channel catfish (Ictalurus punctatus). J. Appl. Microbiol. 104, 1302–1310. doi: 10.1111/j.1365-2672.2007.03632.x
Suomalainen, L.-R., Tiirola, M. A., and Valtonen, E. T. (2005). Influence of rearing conditions on Flavobacterium columnare infection of rainbow trout, Oncorhynchus mykiss (Walbaum). J. Fish Dis. 28, 271–277. doi: 10.1111/j.1365-2761.2005.00631.x
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tekedar, H. C., Karsi, A., Gillaspy, A. F., Dyer, D. W., Benton, N. R., Zaitshik, J., et al. (2012). Genome sequence of the fish pathogen Flavobacterium columnare ATCC 49512. J. Bacteriol. 194, 2763–2764. doi: 10.1128/jb.00281-12
Telford, J. L. (2008). Bacterial genome variability and its impact on vaccine design. Cell Host Microbe 3, 408–416. doi: 10.1016/j.chom.2008.05.004
Thomas-Jinu, S., and Goodwin, A. E. (2004). Morphological and genetic characteristics of Flavobacterium columnare isolates: correlations with virulence in fish. J. Fish Dis. 27, 29–35. doi: 10.1046/j.1365-2761.2003.00507.x
Toyama, T., Tsukamoto, K. K., and Wakabayashi, H. (1996). Identification of Flexibacter maritimus, Flavobacterium branchiophilum and Cytophaga columnaris by PCR targeted 16S ribosomal DNA. Fish Pathol. 31, 25–31. doi: 10.3147/jsfp.31.25
Triyanto, A. K., and Wakabayashi, H. (1999). Genotypic diversity of strains of Flavobacterium columnare from diseased fishes. Fish Pathol. 34, 65–71. doi: 10.3147/jsfp.34.65
Wagner, B. A., Wise, D. J., Khoo, L. H., and Terhune, J. S. (2002). The epidemiology of bacterial diseases in food-size channel catfish. J. Aquat. Anim. Health 14, 263–272. doi: 10.1577/1548-86672002014
Welker, T. L., Shoemaker, C. A., Arias, C. R., and Klesius, P. H. (2005). Transmission and detection of Flavobacterium columnare in channel catfish Ictalurus punctatus. Dis. Aquat. Organ. 63, 129–138. doi: 10.3354/dao063129
Keywords: Flavobacterium columnare, columnaris disease, 16S rRNA, genomovar, multilocus phylogenetic analysis, genetic diversity
Citation: LaFrentz BR, García JC, Waldbieser GC, Evenhuis JP, Loch TP, Liles MR, Wong FS and Chang SF (2018) Identification of Four Distinct Phylogenetic Groups in Flavobacterium columnare With Fish Host Associations. Front. Microbiol. 9:452. doi: 10.3389/fmicb.2018.00452
Received: 28 November 2017; Accepted: 27 February 2018;
Published: 13 March 2018.
Edited by:
Catherine Ayn Brissette, University of North Dakota, United StatesReviewed by:
Jason Sahl, Northern Arizona University, United StatesDeng Liu, Washington University in St. Louis, United States
Copyright © 2018 LaFrentz, García, Waldbieser, Evenhuis, Loch, Liles, Wong and Chang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Benjamin R. LaFrentz, YmVuamFtaW4ubGFmcmVudHpAYXJzLnVzZGEuZ292