 Xingang Zhou
Xingang Zhou Zhilin Wang1
Zhilin Wang1- 1Department of Horticulture, Northeast Agricultural University, Harbin, China
- 2Key Laboratory of Biology and Genetic Improvement of Horticultural Crops, Northeast Region, Ministry of Agriculture, Harbin, China
- 3Institute of Horticulture, Qinghai Academy of Agriculture and Forestry Sciences, Xining, China
Soil microbial communities have profound effects on the growth, nutrition and health of plants in agroecosystems. Understanding soil microbial dynamics in cropping systems can assist in determining how agricultural practices influence soil processes mediated by microorganisms. In this study, soil bacterial communities were monitored in a continuously monocropped Jerusalem artichoke (JA) system, in which JA was successively monocropped for 3 years in a wheat field. Soil bacterial community compositions were estimated by amplicon sequencing of the 16S rRNA gene. Abundances of ammonia-oxidizing and denitrifying bacteria were estimated by quantitative PCR analysis of the amoA, nirS, and nirK genes. Results showed that 1–2 years of monocropping of JA did not significantly impact the microbial alpha diversity, and the third cropping of JA decreased the microbial alpha diversity (P < 0.05). Principal coordinates analysis and permutational multivariate analysis of variance analyses revealed that continuous monocropping of JA changed soil bacterial community structure and function profile (P < 0.001). At the phylum level, the wheat field was characterized with higher relative abundances of Latescibacteria, Planctomycetes, and Cyanobacteria, the first cropping of JA with Actinobacteria, the second cropping of JA with Acidobacteria, Armatimonadetes, Gemmatimonadetes, and Proteobacteria. At the genus level, the first cropping of JA was enriched with bacterial species with pathogen-antagonistic and/or plant growth promoting potentials, while members of genera that included potential denitrifiers increased in the second and third cropping of JA. The first cropping of JA had higher relative abundances of KO terms related to lignocellulose degradation and phosphorus cycling, the second cropping of JA had higher relative abundances of KO terms nitrous-oxide reductase and nitric-oxide reductase, and the third cropping of JA had higher relative abundances of KO terms nitrate reductase and nitrite reductase. The abundances of amoA genes decreased while nirK increased in the third cropping of JA, nirS continuously increased in the second and third cropping of JA (P < 0.05). Redundancy analysis and Mantel test found that soil organic carbon and Olsen phosphorus contents played important roles in shaping soil bacterial communities. Overall, our results revealed that continuous monocropping of JA changed soil bacterial community composition and its functional potentials.
Introduction
The rapidly increasing global food demand poses a huge challenge for the sustainability of agricultural production (Tilman et al., 2011). Modern agricultural practices are often characterized by monocropping, which leads to the simplification of the components of agricultural systems (Cook, 2006). The continuous monocropping system, in which the same crop is repeatedly monocropped on the same land, is not long-term sustainable, because it usually results in reduction of crop yield and quality, a phenomenon which has been described as ‘soil sickness’ (Cook, 2006; van der Putten et al., 2013).
Plants can change soil biology, chemistry, and structure in ways that alter subsequent plant growth, and this process is referred as plant–soil feedback (van der Putten et al., 2013). As a kind of negative plant–soil feedback in agricultural ecosystems, soil sickness has been reported for several crops, such as corn (Zea mays L.) (Gentry et al., 2013), pea (Pisum sativum L.) (Nayyar et al., 2009) and cucumber (Cucumis sativus L.) (Zhou and Wu, 2012). Possible factors that contribute to soil sickness include accumulation of phytotoxic compounds, build-up of soil-borne pathogens, deterioration of soil physico-chemical characteristics, and changes in nutrient availability (Zhou and Wu, 2012; Huang et al., 2013). Recently, changes in soil biological properties have also been proposed to account for the yield decline in continuous monocropping systems (Nayyar et al., 2009; Huang et al., 2013; Zhou et al., 2017a).
Soil microorganisms are responsible for key processes associated with soil fertility and plant health, and are an important driver of the functioning of terrestrial ecosystems (Bever et al., 2012; Bhattacharyya and Jha, 2012). Changes in soil microbial communities may lead to alterations in the functions performed by the community and thus have feedbacks on plant health and fitness (Bever et al., 2012; Zhou et al., 2017a). Soil microbial communities are driven by a myriad of factors, including soil physical and chemical properties, aboveground plant species abundance and diversity, and agricultural practices (such as monocropping, crop rotation, intercropping, fertilization, irrigation, and tillage) (Acosta-Martínez et al., 2008; Nayyar et al., 2009; Rosenzweig et al., 2012; Sun et al., 2014; Zhou et al., 2017a). For example, it has been shown that diversified cropping systems (such as intercropping, crop rotation, and cover crop) usually have higher diversities and abundances of soil microbial communities than monocropping systems (Zhou et al., 2011, 2017a; Tiemann et al., 2015). Knowledge about how continuous monocropping influences soil microbial communities is helpful for the development of practices to relieve soil sickness in agriculture production.
Jerusalem artichoke (JA) (Helianthus tuberosus L.) is an economically important crop, which can be used as a food for direct human consumption and livestock feed after silage (Kaur and Gupta, 2002). Market forces have encouraged farmers to cultivate JA in monoculture and a reduction in tuber yield and quality was usually observed (Chi et al., 2009; Zhou et al., 2016). Previously, we found that continuous monocropping of JA changed the structure and decreased the diversity of soil bacterial communities as estimated by PCR-denaturing gradient gel electrophoresis analysis (Zhou et al., 2016). High-throughput sequencing techniques, such as 454 pyrosequencing and Illumina sequencing, can provide a higher resolution and a better understanding of environmental microbial communities than the PCR-based fingerprinting techniques (Schöler et al., 2017). High-throughput sequencing techniques also greatly facilitated the diversity and the composition analyses of microbial communities in agricultural soils (Acosta-Martínez et al., 2008; Sun et al., 2014; Bainard et al., 2016; Chavez-Romero et al., 2016; Zhou et al., 2017a). Therefore, it is necessary to deepen our understanding about the dynamic changes in soil microbial communities during continuous monocropping of crops with high-throughput sequencing techniques.
In this study, we evaluated the responses of soil bacterial communities to continuous monocropping of JA with high-throughput sequencing. JA was grown in a long-term cultivated wheat field for three successive years. Bulk soil bacterial communities were assessed by amplicon sequencing of the V3-V4 region of the 16S rRNA gene on an Illumina MiSeq platform. In addition, bacterial community potential functions were inferred from the amplicon data using Phylogenetic Investigation of Communities by Reconstruction of Unobserved States (PICRUSt) (Langille et al., 2013). Abundances of ammonia-oxidizing and denitrifying bacteria were estimated by quantitative PCR analysis of the amoA, nirS, and nirK genes. Plant litter and root exudates are important carbon resources for soil microorganisms, and the chemistries of these organic matters differ among plant species (Fierer et al., 2007; Eilers et al., 2010; Birouste et al., 2012). Therefore, we first hypothesized that soil bacterial communities differed between wheat- and JA-cultivated soils. Since it is usually observed that continuous monocropping had an adverse effect on soil bacterial community diversity, while crop rotation were able to increase soil bacterial community diversity (Zhou et al., 2011, 2017a; Tiemann et al., 2015). The first cropping of JA can be viewed as a wheat-JA rotation system. Therefore, our second hypothesis was that soils from the wheat field and the third cropping of JA would have lower soil bacterial community diversities than the first cropping of JA.
Materials and Methods
Field Experiment
The experimental site was located in field of Mojiaquanwan village, Chengbei District, Xining, China (36°42’N, 101°45’E), which has been continuously cultivated with wheat (Triticum aestivum L.) for more than 20 years. Wheat was grown from early March to early September and thereafter left fallow till next wheat crop. The soil was castanozem (FAO/UNESCO System of Soil Classification), containing organic matter: 2.03%, available N (NH4+ and NO3-): 69 mg kg-1, Olsen P: 65 mg kg-1, available K: 229 mg kg-1, EC (1:2.5, w/v) and pH (1:2.5, w/v), 8.12.
The field experiment was conducted from April 2010 to October 2012. The annual precipitations in these 3 years were 405.0, 390.4, and 446.1 mm, respectively, and the mean annual temperatures were 6.4, 5.7, and 5.2°C, respectively. There are four treatments in the experiment, namely, W, F, S, and T (Table 1). W was the long-term cultivated wheat field. F, S, and T were designed to be planted with JA for 1, 2, and 3 years, respectively. Briefly, in 2010, treatment T was planted with JA, the other three treatments were planted with wheat. In 2011, treatments T and S were planted with JA, the other two treatments were planted with wheat. In 2012, treatments T, S, and F were planted with JA, the treatment W was planted with wheat. The experiment was set up in a randomized block design, with three replicate plots for each treatment. Each plot measured 120 m long and 80 m wide.

TABLE 1. Experiment setup of the field experiment.
Jerusalem artichoke tubers (cv. Qingyu 2), provided by Institute of Horticulture, Qinghai Academy of Agriculture and Forestry Sciences, China, were planted on April 5 each year and harvested on October 25 each year. Within-row spacing was 40 cm and the row width was 60 cm. Wheat was broadcast seeded in early March and harvested in early September. There was one crop (wheat or JA) per year. After the harvest of JA and wheat, the fields were left fallow until to plant the next crop. Both diammonium hydrogen phosphate and urea were applied at the rate of 300 kg ha-1 as basal fertilizer. Flooding irrigation with groundwater was performed when necessary. Weeds were manually removed once a month in May and June.
Soil Sampling and DNA Extraction
Bulk soil samples were collected on November 25, 2012, 1 month after JA harvest. Eight soil cores (5 cm diameter, 15 cm deep) were randomly collected between rows of crops from each plot to make a composite sample. Large stones and root debris were removed by sieving (2 mm), then fresh soils were transported to laboratory and stored at -70°C. There were triplicate soil samples for each treatment and there were 12 soil samples in total.
Total soil DNA was extracted with the PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, United States) as per the manufacturer’s instructions. Each composite soil sample was extracted in triplicate and the extracted DNA solutions were pooled.
High-Throughput Amplicon Sequencing and Data Processing
Soil bacterial community compositions were analyzed with high-throughput sequencing on an Illumina MiSeq platform. Primers of F338/R806 were used to amplify V3-V4 region of the bacterial 16S rRNA gene as described before (Derakhshani et al., 2016; Zhou et al., 2017a). Both the forward and reverse primers also had a 6-bp barcode unique to each sample, which were used to permit multiplexing of samples. Each composite soil sample was independently amplified in triplicate, the products of the triplicate PCR reactions were pooled and purified using the Agarose Gel DNA purification kit (TaKaRa, China). Then, purified amplicons were quantified by a TBS-380 micro fluorometer with Picogreen reagent (Invitrogen, United States), and pooled in equal amounts. The mixture was then paired-end sequenced (2 × 300) on an Illumina Miseq platform at Majorbio Bio-Pharm Technology Co., Ltd., Shanghai, China.
Raw sequence reads were de-multiplexed, quality-filtered, and processed using FLASH (Magoc and Salzberg, 2011) as described before (Zhou et al., 2017a). Operational taxonomic units (OTUs) were delineated at 97% sequence similarity with USEARCH using an agglomerative clustering algorithm (Edgar, 2010). Then, a representative sequence of each OTU was taxonomically classified through BLAST against the SILVA (Quast et al., 2013). Chimeric sequences were identified and removed using USEARCH 6.1 in QIIME 1.9.1 (Caporaso et al., 2010). Functions of soil bacterial communities were predicted by PICRUSt from the 16S rRNA marker gene sequences on the Galaxy platform1 (Langille et al., 2013), and the biological functions were annotated in the KEGG database (Kanehisa et al., 2012). Specifically, we focused on functions associated with carbon, nitrogen, phosphorus, and sulfur cycling. The data set was deposited in the NCBI-Sequence Read Archive with the submission Accession Number SRP115368.
Quantitative PCR Analysis
Abundances of ammonia-oxidizing and denitrifier communities were estimated by quantitative PCR assays with an IQ5 real-time PCR system (Bio-Rad Lab, Los Angeles, CA, United States). For the ammonia-oxidizing community, the gene encoding ammonia monooxygenase catalytic subunit A (amoA) was amplified using the primer set of amoA1F/amoA2R (Rotthauwe et al., 1997) according to the methods described by Glaser et al. (2010). For the denitrifier community, the cytochrome cd1-containing nitrite reductase gene (nirS) and the Cu-dependent nitrite reductase gene (nirK) were amplified using the primer sets of nirSCd3aF/nirSR3cd (Kandeler et al., 2006) and nirK1F/nirK5R (Braker et al., 1998), respectively, according to the methods described before (Hai et al., 2009; Braker et al., 2015). A 20 μl PCR reaction mixture contained 10 μl of 2× Real SYBR Mixture (Tiangen Biotech, Beijing, China), 0.2 μM of each primer, 2.5 ng of soil DNA. Standard curves were created with 10-fold dilution series of plasmids containing the ITS regions from soil samples. The specificity of the products was confirmed by melting curve analysis and agarose gel electrophoresis. The threshold cycle (Ct) values obtained for each sample were compared with the standard curve to determine the initial copy number of the target gene. Sterile water was used as a negative control to replace the template. All amplifications were performed in triplicate.
Statistical Analysis
Read counts from high-throughput amplicon sequencing were not rarefied to equal sampling depths because this unnecessarily discards data (McMurdie and Holmes, 2014). For alpha diversity analysis, square root transformed read counts (Balint et al., 2015) were used to calculated Hill’s series of diversity. The series consists of three numbers: N0 is the number of species in a sample; N1 is the antilogarithm of the Shannon diversity (representing the abundant species in a sample); and N2 is the inverse Simpson diversity (representing the very abundant species in a sample) (Hill, 1973). To compare with alpha diversity indices from the unrarefied data, alpha diversity indices were also calculated from a randomly selected subset of 22,503 16S rRNA gene sequences per sample.
For beta diversity analysis, read counts were centered log-ratio (CLR) transformed (Fernandes et al., 2014). Bacterial community structure and function profile were analyzed using principal coordinates analysis (PCoA) based on a Euclidean distance matrix. Permutational multivariate analysis of variance (PerMANOVA) was used to test the differences in microbial communities with the Euclidean distance and 999 permutations. The PCoA and PerMANOVA analyses were performed with the pcoa and adonis functions in ‘vegan’ package in ‘R’ (Version 3.3.1), respectively.
Linear discriminant effect size (LEfSe) analysis was used to identify biomarkers that were significantly associated with each treatment with an alpha value of 0.05 for the Kruskal–Wallis test and a threshold of 2.0 for logarithmic linear discriminant analysis (LDA) scores (Segata et al., 2011).
Previously, we found that continuously monocropped JA did not change soil pH and inorganic N content, the first cropping of JA had the highest soil organic carbon (SOC) content while the third cropping of JA had the lowest soil Olsen P (Zhou et al., 2017b). Redundancy analysis (RDA) was used to identify soil properties that predict the variation of bacterial communities. Mantel test with a Monte Carlo simulation with 999 randomizations was used to assess the relationships between the Euclidean distance of bacterial community and soil chemical properties. RDA and Mantel test analyses were performed with the rda function in the ‘vegan’ package and the mantel.rtest function in the ‘ade4’ package in ‘R’ (version 2.1.3), respectively. Spearman’s rank correlations between soil properties and relative abundances of bacterial classes and genus, and predicted functions were calculated in ‘psych’ package in ‘R’ (Version 3.3.1).
Differences in Hill’s series of diversity, relative abundances of microbial taxa and abundances of amoA, nirS, and nirK genes among treatments were analyzed using one-way analysis of variance (ANOVA) followed by Tukey’s honestly significant difference (HSD) test at the 0.05 probability level.
Results
Amplicon Sequencing Data
After filtering reads by basal quality control and removing singletons, Illumina Miseq sequencing of bacterial 16S rRNA gene fragments generated 322,976 quality bacterial sequences with an average read length of 397 bp, and 22,503–30,340 sequences were obtained per sample (mean = 26,915). The Good’s coverage, which reflects the captured diversity, was larger than 98% for all samples (data not shown). Rarefaction curves of OTUs at 97% sequence similarity and Shannon’s diversity indices of all samples tended to approach the saturation plateau (Supplementary Figure S1), which indicates that the majority of the bacterial diversity was recovered by the surveying effort.
Bacterial Community Composition
In total, 32 phyla were detected across all samples and 0.99% bacterial sequences were unclassified at the phylum level (Unclassified Bacteria). The dominant phyla (relative abundance > 5%) across all soil samples were Proteobacteria, Actinobacteria, Acidobacteria, Bacteroidetes, Planctomycetes, and Chloroflexi, which accounted for more than 92% of the bacterial sequences (Figure 1A). The top three phyla were Proteobacteria, Actinobacteria, and Acidobacteria, which had relative abundances ranging from 26.36 to 32.37%, 21.17 to 39.64%, and 9.31 to 17.00%, respectively. Gemmatimonadetes and Firmicutes were less abundant phyla (relative abundance < 5% but > 1%) with relative abundances ranging from 1.76 to 3.25% and 1.13 to 2.00%, respectively. Groups of Nitrospirae, Verrucomicrobia, Cyanobacteria, Latescibacteria, Armatimonadetes, and JL-ETNP-Z39 were also detected at relatively low abundances in all samples (relative abundance > 0.1%).
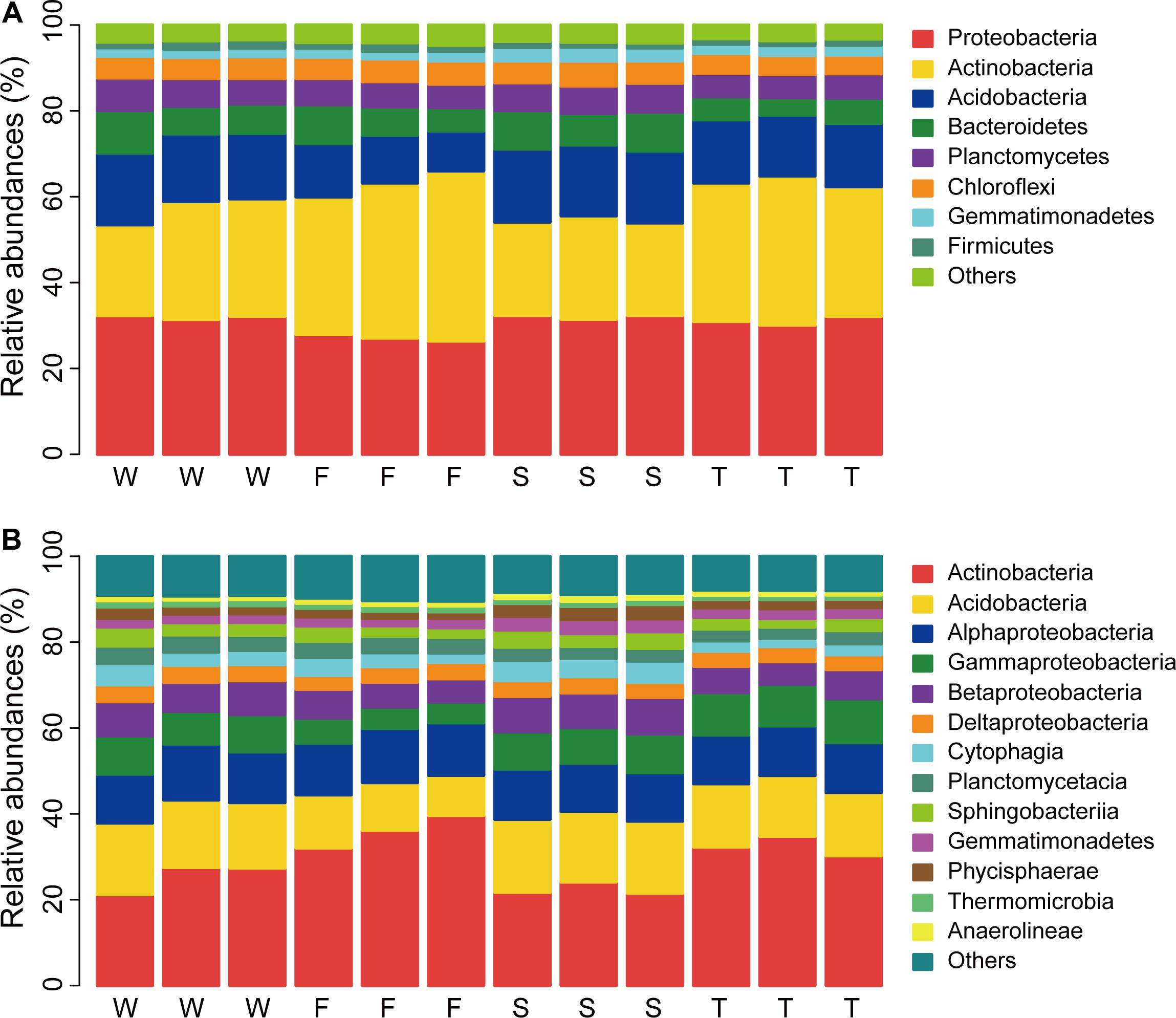
FIGURE 1. Relative abundances of main soil bacterial phyla (A) and classes (B). W represents the wheat field; F, S, and T represent the first, second, and third cropping of Jerusalem artichoke (JA), respectively.
Linear discriminant effect size analysis identified 107 differentially abundant taxa from the phylum to the genus level (Figure 2 and Supplementary Figure S2). The first cropping of JA had the most (41) and the third cropping of JA has the least (13) number of differentially abundant taxa. Among these differentially abundant taxa, nine were found to be genus-level biomarkers. At the phylum level, the wheat field was enriched with Latescibacteria, Planctomycetes, and Cyanobacteria; the first cropping of JA with Actinobacteria and Unclassified Bacteria; the second cropping of JA with Acidobacteria, Armatimonadetes, Gemmatimonadetes, and Proteobacteria (P < 0.05).
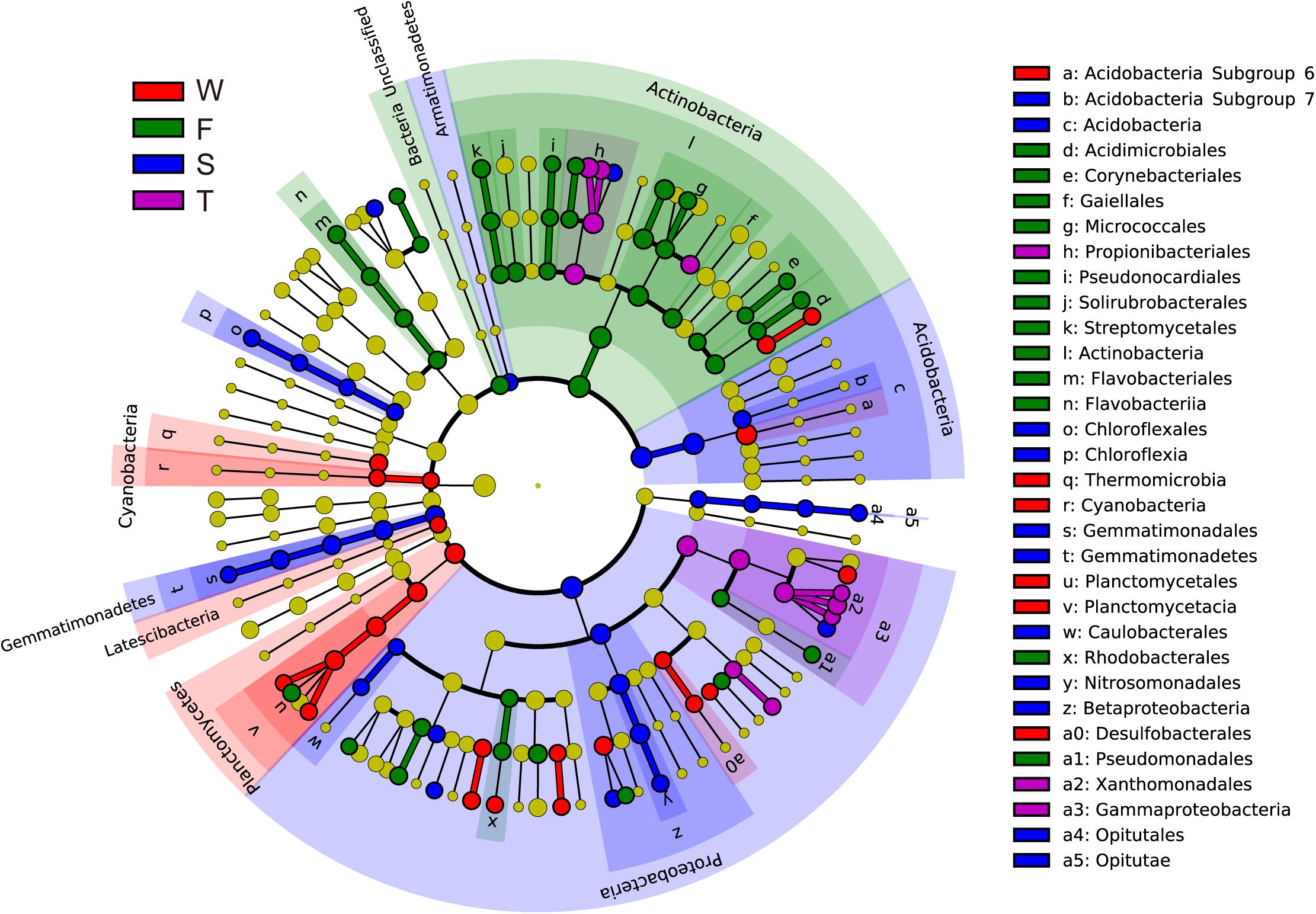
FIGURE 2. Cladograms, generated from LEfSe analysis, represent the polygenetic distribution of soil bacterial taxa. Biomarkers that are significantly associated with each treatment with LDA scores larger than 2 are shown. Significantly discriminant taxon nodes are colored: red for the wheat field (W), green, blue and purple for the first (F), second (S), and third (T) cropping of Jerusalem artichoke (JA), respectively. Yellow circles represent non-significant differences in abundance between treatment groups for that particular taxon. Each circle’s diameter is proportional to the taxon’s abundance. Labels are shown of the phylum, class and order levels. The LDA scores of each identified biomarker from the phylum to genus levels are shown in Supplementary Figure S1.
At the class level, more than 70 bacterial taxa were detected. All samples were dominated by Actinobacteria, Acidobacteria, Alphaproteobacteria, Gammaproteobacteria, and Betaproteobacteria (Figure 1B). LEfSe analysis identified 11 biomarkers at the class level (Figure 2 and Supplementary Figure S2). The wheat field was characterized with higher relative abundances of Thermomicrobia, Cyanobacteria, and Planctomycetacia; the first cropping of JA with Actinobacteria and Flavobacteriia; the second cropping of JA with Acidobacteria, Chloroflexia, Gemmatimonadetes, Betaproteobacteria, and Opitutae; the third cropping of JA with Gammaproteobacteria (P < 0.05).
At the genus level, more than 500 bacterial taxa were detected. Arthrobacter, Blastococcus, Chryseolinea, Gaiella, Lysobacter, Marmoricola, Nocardioides, Skermanella, and Streptomyces spp. were dominant classified genera (relative abundance > 1%) (Table 2). The relative abundances of Variibacter, Illumatobacter, Altererythrobacter, and Gemmata spp. were higher in the wheat field than in other treatments (P < 0.05) (Table 2 and Supplementary Figure S2). The relative abundances of Microbacterium, Mycobacterium, Pseudonocardia, Algoriphagus, Flavobacterium, Bosea, Microvirga, and Pseudomonas spp. were higher in the first cropping of JA than in other treatments (P < 0.05). The relative abundances of Adhaeribacter, Roseiflexus, Gemmatimonas, Rhizobium, Caenimonas, and Nitrosospira spp. were higher in the second cropping of JA than in other treatments (P < 0.05). The relative abundances of Nitrospira, Marmoricola, Nocardioides, Haliangium, and Lysobacter were higher in the third cropping of JA than in other treatments (P < 0.05).
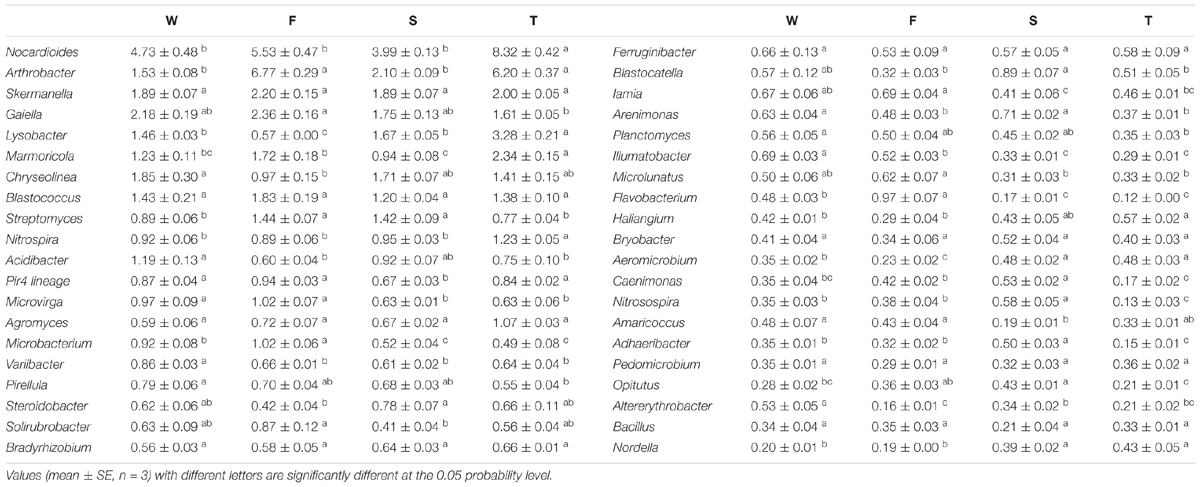
TABLE 2. The relative abundances of main classified bacterial genera (average relative abundance > 0.3%) in the wheat field (W), the first (F), second (S), and third (T) cropping of Jerusalem artichoke (JA).
A total of 2,395 OTUs were identified at 97% similarity. Most dominated OTUs, with relative abundances greater than 0.5% of the total sequences, were mainly assigned to the Acidobacteria, Actinobacteria, and Gammaproteobacteria at the class level (Supplementary Table S1). The relative abundances of one OTUs assigned to Acidibacter, Chryseolinea, Lysobacter, Acidobacteria Subgroup 6 norank, uncultured Nitrosomonadaceae and unclassified Xanthomonadaceae were higher in the wheat field than in the first cropping of JA; while the relative abundances of OTUs assigned to Arthrobacter and Streptomyces spp. was higher in the first cropping of JA than in the wheat field (P < 0.05). The third cropping of JA had the highest relative abundances of two OTUs assigned to Nocardioides, Marmoricola and Lysobacter spp. and the lowest Comamonadaceae unclassified among all treatments (P < 0.05).
Bacterial Community Diversity and Structure
For alpha diversities calculated from unrarefied data (Figure 3A) and rarefied data (Supplementary Figure S3), the number of OTUs (Hill’s N0) was lower in the third cropping of JA than in the first cropping of JA (ANOVA, P < 0.05). Hill’s N1 and N2 were significantly lower in third cropping of JA than in other treatments (ANOVA, P < 0.05).
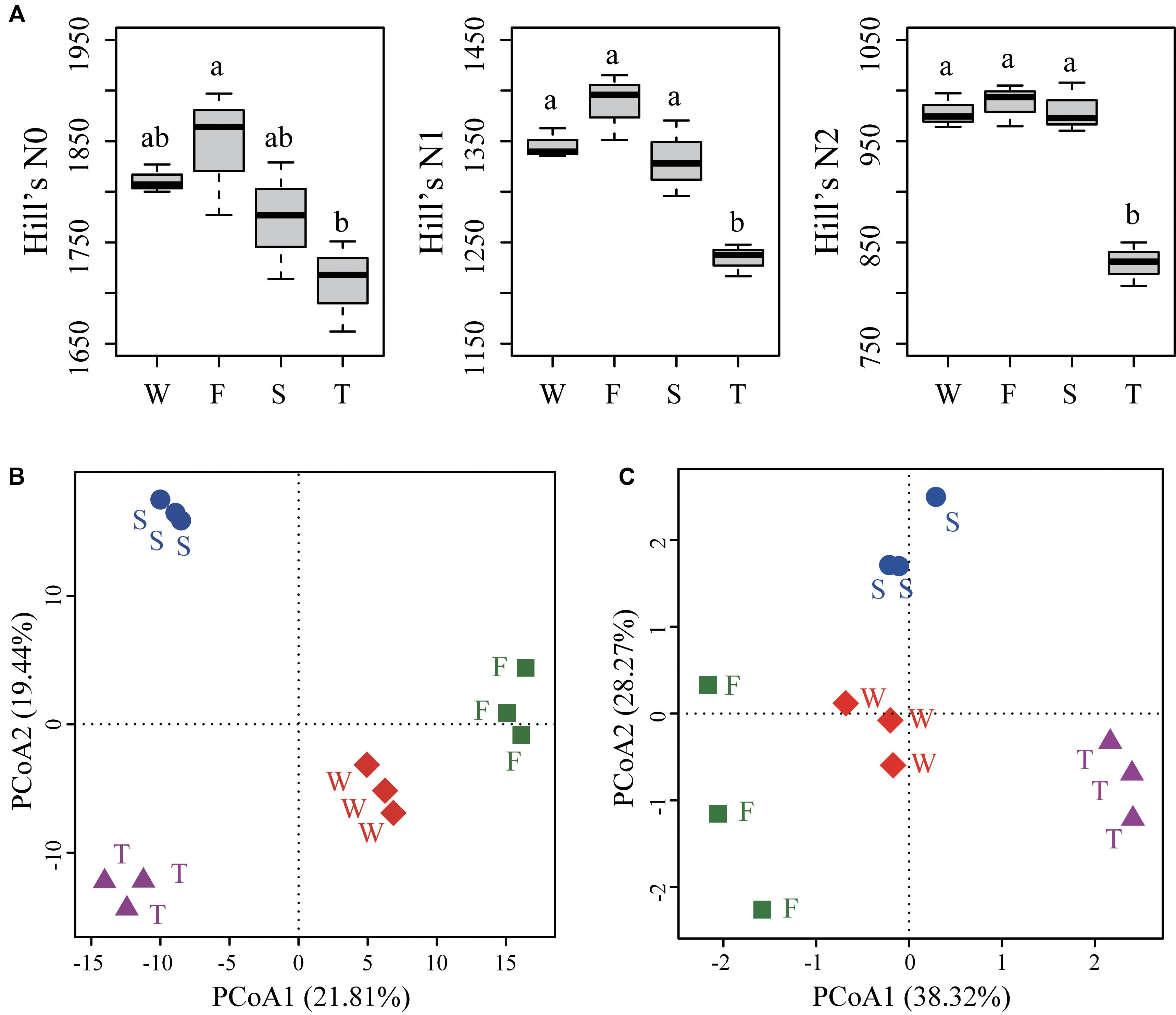
FIGURE 3. Alpha diversities (A) and PCoA analyses of soil bacterial community structure (B) and function profile (C). (A) Hill’s series of diversity was calculated from unrarefied data. Different letters indicate significant difference based on Tukey’s HSD test (P < 0.05). (B) The PCoA analysis of soil bacterial community structure was based on the Euclidean distance of the centered log-ratio (CLR) transformed read counts at the OTU level. (C) The PCoA analysis of soil bacterial community function profile was based on the Euclidean distance of the CLR transformed pathway numbers at KEGG level 3. OTUs were delineated at 97% sequence similarity. W represents the wheat field; F, S, and T represent the first, second, and third cropping of Jerusalem artichoke (JA), respectively.
The PCoA analysis at the OTU level showed a clear separation among samples from the wheat field, the first, second, and third cropping of JA (Figure 3B). PerMANOVA analysis demonstrated that continuous cropping of JA significantly changed soil bacterial community structure (F = 3.034, R2 = 0.532, P < 0.001).
Predicted Functions of Bacterial Communities
The majority of the predicted functional gene categories were related to metabolism (52.09%), followed by genetic information (15.76%), environmental information processing (13.27%), and unclassified (12.91%). LEfSe analysis identified five differentially abundant KEGG pathways at KEGG level 1, 21 differentially abundant KEGG pathways at KEGG level 2 (Figure 4), and 89 differentially abundant KEGG pathways at KEGG level 3 (Supplementary Figure S4). The first cropping of JA was characterized by enrichment of functions related to metabolism at KEGG level 1, and amino acid metabolism and carbohydrate metabolism at KEGG level 2; the second cropping of JA was characterized by enrichment of functions related to genetic information processing and cellular processes at KEGG level 1; the third cropping of JA was characterized by enrichment of functions related to organismal systems at KEGG level 1 (Figure 4).
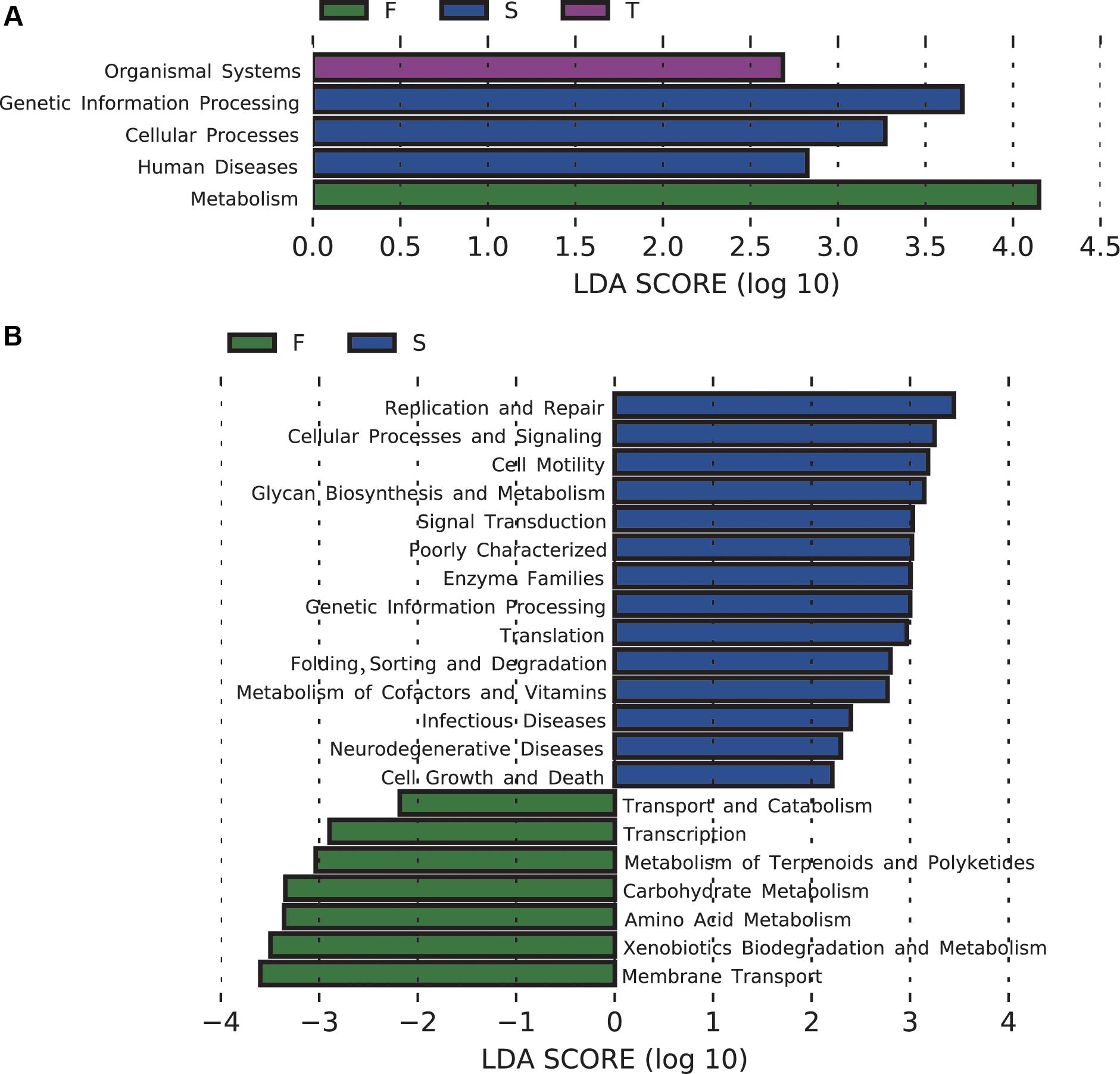
FIGURE 4. Changes in soil bacterial community functional profiles. Histograms (A,B) showed the LDA scores calculated for the differentially abundant biomarkers at KEGG level 1 and 2, respectively (P < 0.05). W represents the wheat field; F, S, and T represent the first, second, and third cropping of Jerusalem artichoke (JA), respectively.
For pathways at level 3 that are involved in carbohydrate metabolism, pentose and glucuronate interconversions, galactose metabolism, inositol phosphate metabolism, glyoxylate and dicarboxylate metabolism, fructose and mannose metabolism, glycolysis/gluconeogenesis, pyruvate metabolism, propanoate metabolism, butanoate metabolism, were enriched in the first cropping of JA; TCA cycle was enriched in the third cropping of JA. At KEGG level 3, the second cropping of JA was also enriched with sulfur metabolism.
Several predicted KEGG Ortholog (KO) terms related to carbon, nitrogen, phosphorus and sulfur cycling were also differed among treatments (Figure 5). For example, the relative abundances of KO terms related to lignocellulose degradation, such as 6-phospho-beta-glucosidase, neutral alpha-glucosidase C and catalase, were higher in the first cropping of JA than in other treatments (P < 0.05). The relative abundances of KO terms ammonia monooxygenase subunit C and hydroxylamine oxidase, which are involved in ammonification, were higher in the second cropping of JA than in the wheat field and third cropping of JA (P < 0.05). Among all treatments, the third cropping of JA had the highest relative abundances of KO terms nitrate reductase and nitrite reductase, while the second cropping of JA had the highest relative abundances of KO terms nitrous-oxide reductase and nitric-oxide reductase (P < 0.05). The relative abundance of KO term inosose dehydratase, which participates in phosphorus cycling was higher in the first cropping of JA than in other treatments (P < 0.05). For KO terms related to sulfur cycling, the relative abundance of sulfate adenylyltransferase and adenylylsulfate reductase subunit B were higher in the second cropping of JA than in the wheat field and the first cropping of JA (P < 0.05).
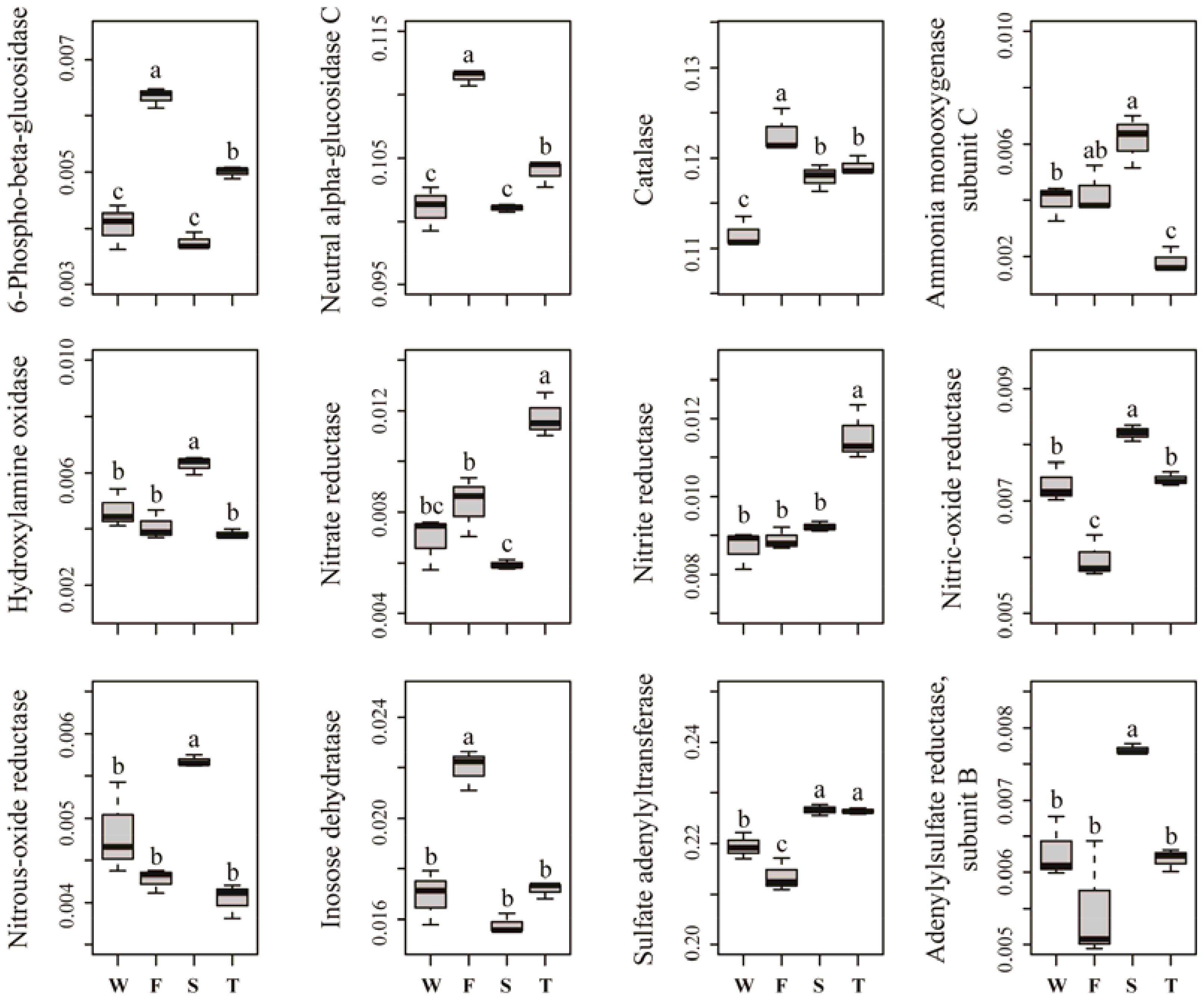
FIGURE 5. Relative abundances of significantly changed predicted KEGG Orthologs related to carbon, nitrogen, phosphorus, and sulfur cycling. Different letters indicate significant difference based on Kruskal–Wallis test (P < 0.05). W represents the wheat field; F, S, and T represent the first, second, and third cropping of Jerusalem artichoke (JA), respectively.
Principal coordinates analysis showed that soil bacterial function profiles at KEGG level 3 differed among samples from the wheat field, the first, second, and third cropping of JA (Figure 3C). PerMANOVA analysis also demonstrated that continuous cropping of JA significantly changed the function of soil bacterial communities (F = 6.478, R2 = 0.708, P < 0.001).
Relationships Between Soil Bacterial Communities and Soil Chemical Properties
The RDA analysis and Mantel test were conducted to identify the key drivers of soil bacterial community structure and function profile. In the RDA plots of both bacterial community structure and function, soil SOC, inorganic N, and Olsen P had longer arrows than the soil pH (Figure 6). Mantel test demonstrated that soil bacterial community structure was significantly correlated to SOC (r = 0.339, P = 0.002) and Olsen P (r = 0.395, P = 0.004) but not to inorganic N (r = 0.395, P = 0.105) and soil pH (r = 0.075, P = 0.238); soil bacterial function profile was significantly correlated to SOC (r = 0.406, P = 0.003) and Olsen P (r = 0.426, P = 0.004) but not to inorganic N (r = 0.126, P = 0.220) and soil pH (r = -0.085, P = 0.692). Spearman’s rank correlation test showed that the relative abundances of bacterial class Gammaproteobacteria was negatively correlated with SOC (r = -0.85, P < 0.05), and genus Caenimonas spp. was positively correlated with SOC (r = 0.82, P < 0.05) and Olsen P (r = 0.83, P < 0.05). The relative abundance of the relative abundances of KO terms neutral alpha-glucosidase C and inosose dehydratase were positively correlated with SOC (r = 0.75, P < 0.05; r = 0.75, P < 0.05, respectively). The relative abundance of KO term ammonia monooxygenase subunit C was positively correlated with Olsen P (r = 0.71, P < 0.05).
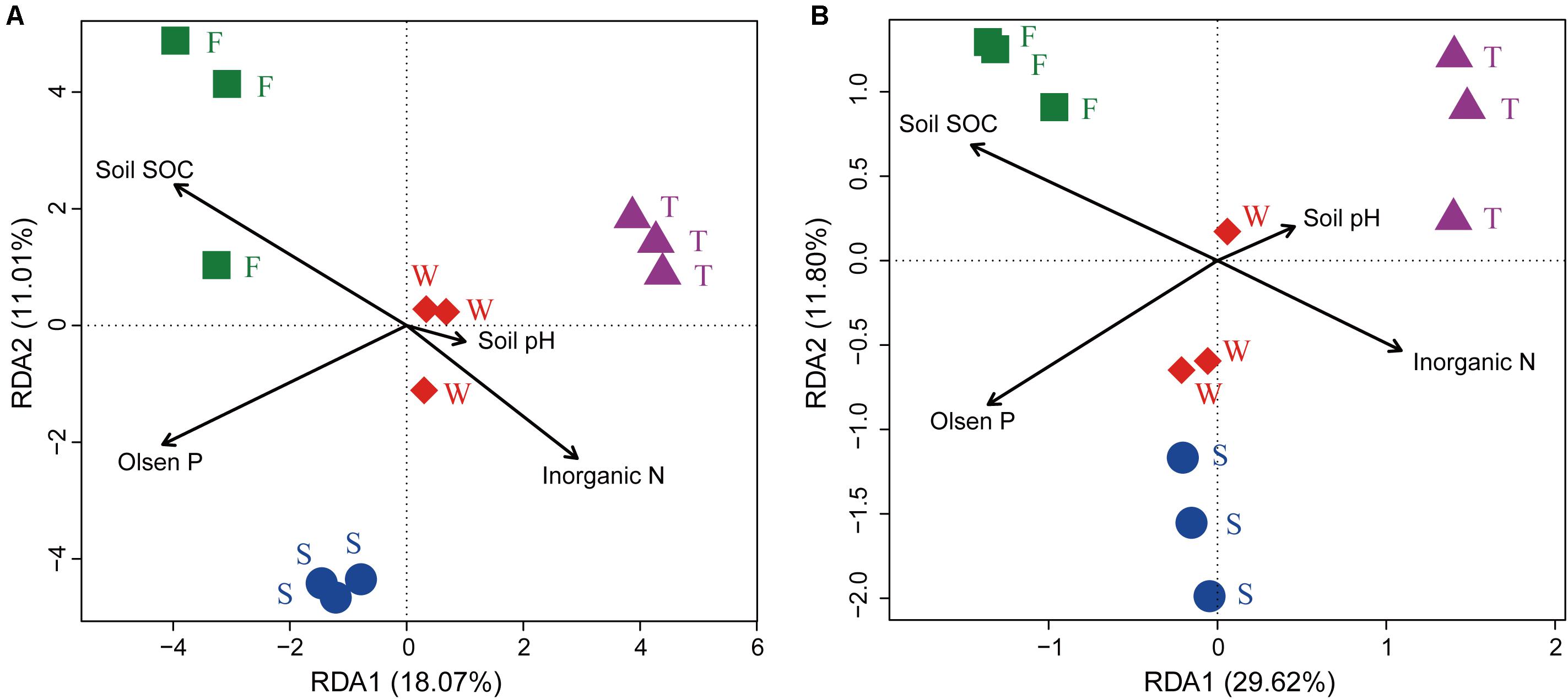
FIGURE 6. Ordination plots of the results from the redundancy analysis. (A) Relationship between soil bacterial community structures at the OTU level (97% sequence similarity) with soil chemical properties. (B) Relationship between soil bacterial function profiles at KEGG level 3 with soil chemical properties. W represents the wheat field; F, S, and T represent the first, second, and third cropping of Jerusalem artichoke (JA), respectively.
Soil Ammonia-Oxidizing and Denitrifier Community Abundances
Quantitative PCR analysis showed that the ammonia-oxidizing abundance, expressed as amoA gene copy number, was significantly lower in the third cropping of JA than in other treatments (P < 0.05) (Figure 7). However, the denitrifier community abundance, expressed as nirS and nirK gene copy numbers, was significantly higher in the third cropping of JA than in other treatments (P < 0.05). Meanwhile, nirS gene copy number was higher in the second cropping of JA than in the wheat field and the first cropping of JA (P < 0.05).
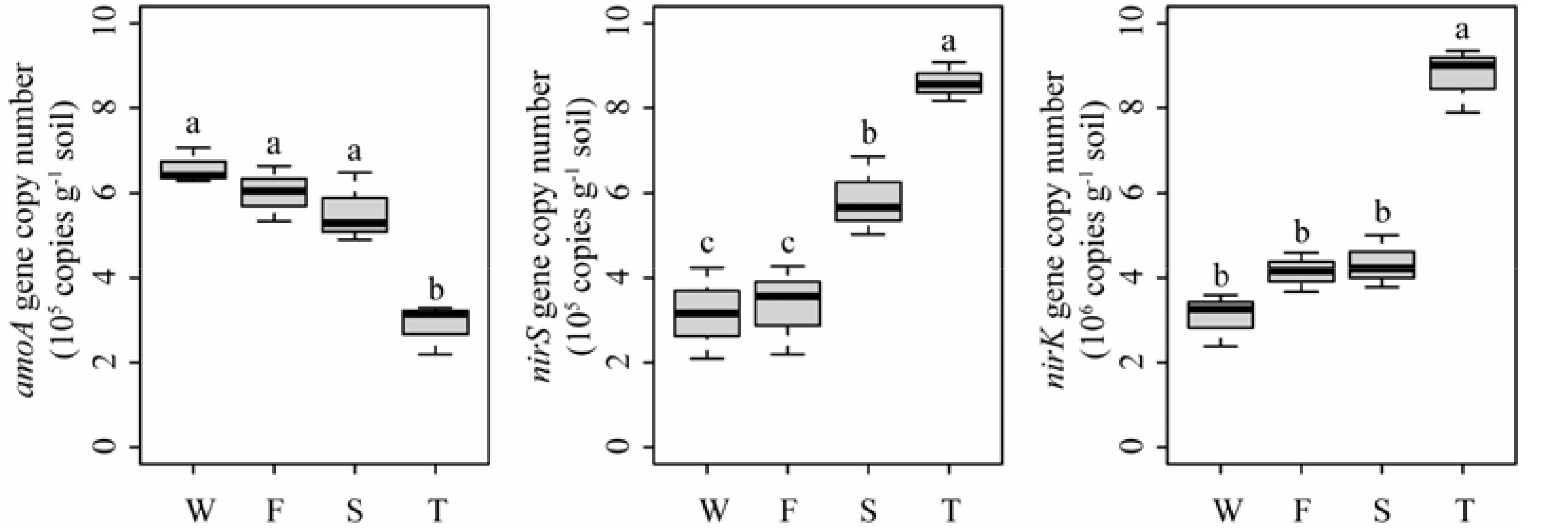
FIGURE 7. Abundances of amoA, nirS, and nirK genes in the wheat field (W), the first (F), second (S), and third (T) cropping of Jerusalem artichoke (JA) as determined by quantitative PCR. Different letters indicate significant difference based on Tukey’s HSD test (P < 0.05).
Discussion
The productivity and sustainability of agricultural system depend greatly on the functional processes carried out by soil microorganisms (Bever et al., 2012). Mounting evidences demonstrated that agricultural practices, such as fertilization, tillage, crop rotation, and intercropping, could alter soil microbial community (Garbeva et al., 2008; Zhou et al., 2011, 2017a; Bever et al., 2012). The present study stressed the influences of continuous monocropping on soil bacterial communities by amplicon sequencing of the 16S rRNA marker gene. PCoA and PerMANOVA analyses revealed that soil bacterial community composition and function profile changed during continuous monocropping of JA, which supported our first hypothesis.
It has been observed that soil microbial community can become compositionally adapted to utilize certain plant litter type (Ayres et al., 2009). Some bacterial taxa [such as Gemmata spp. (Bastian et al., 2009)], that were involved in degrading wheat residues, were enriched in the wheat field. These results indicate that long-term wheat cultivation selected for specific microorganisms that can degrade wheat residues. Several other bacterial taxa that associated with decomposing plant-derived organic matters were also enriched in other treatments. For example, Bosea and Pseudonocardia spp. were enriched in the first cropping of JA; Adhaeribacter and Gemmatimonas spp. were enriched in the second cropping of JA; Lysobacter and Nocardioides spp. were enriched in the third cropping of JA. Previous studies demonstrated that Adhaeribacter (Bastian et al., 2009), Gemmatimonas (Bastian et al., 2009), Pseudonocardia (Espana et al., 2011), Lysobacter (Chavez-Romero et al., 2016) were involved in decomposition of crop residues, such as potato (Solanum tuberosum L.), rice, maize and wheat; Species in Bosea (Houfani et al., 2017) have cellulolytic activities. Moreover, continuous monocropping of JA altered the predicted bacterial functions related to lignocellulose degradation. This would be possibly explained by the species-specific effects of plants on soil microbial communities through varying quantities and qualities of plant-derived organic matters, which can be used as substrates by soil microorganisms, as the chemistries of plant-derived organic matters were shown to differ among plant species (Fierer et al., 2007; Meier and Bowman, 2008; Eilers et al., 2010; Bever et al., 2012; Birouste et al., 2012).
Linear discriminant effect size analysis revealed that the first cropping of JA were enriched with bacterial species with pathogen-antagonistic and/or plant growth promoting potentials, including Bosea (Cavalca et al., 2010), Microbacterium (Bhattacharyya and Jha, 2012), Mycobacterium (Hayat et al., 2010), and Pseudonocardia spp. (Nimnoi et al., 2010). In our experiment, soils from the wheat field mainly contained wheat debris while soils from the JA-cultivated fields contained crop debris from both wheat and JA. It was found that most wheat residues (more that 80%) was decomposed within 320 days after wheat residues incorporated into the soil (Cookson et al., 1998). Thus, the diversity of plant-derived organic matters may be higher in the first cropping of JA, which also had higher soil SOC (Zhou et al., 2017b). Therefore, our results were in line with previous studies showing that increasing resource quantity and quality through increasing the temporal and spatial plant diversity can enhance the function of soil microbial communities (Rosenzweig et al., 2012; Tiemann et al., 2015). In our experiment, the first cropping of JA can be viewed as a wheat-JA rotation system. Thus, wheat-JA rotation may be used in production to stimulate soil bacteria beneficial to plants.
Quantitative PCR showed that the third cropping of JA has the lowest amoA gene copy number but had the highest nirS and nirK gene copy number. The second cropping of JA had higher nirS gene copy number than the wheat field and the first cropping of JA. This may be attributed to the lower available P in the third cropping of JA since soil P availability play an important in modulating soil N cycle. For example, it has been reported that nitrification was dependent on P availability (Sierra et al., 2003) and poor P availability can promote denitrification at higher N fertilizer inputs (Baral et al., 2014). PICRUSt revealed that continuous monocropping of JA altered the predicted bacterial functions related to nitrogen cycling. Specifically, the relative abundances of one KO term related to ammonification (ammonia monooxygenase subunit C) was lower in the third cropping of JA than in other treatments. Meanwhile, the relative abundances of KO terms of nitrate reductase and nitrite reductase were higher in the third cropping of JA. The relative abundances of KO terms of nitrous-oxide reductase and nitric oxide reductase were higher in the second cropping of JA. Caenimonas, Gemmatimonas, and Rhodopirellula spp. were enriched in the second cropping of JA. Haliangium, Marmoricola, and Nocardioides spp. were enriched in the third cropping of JA. Members of these taxa were reported to be involved in denitrification. For example, Gemmatimonas (Coyotzi et al., 2016) and Rhodopirellula spp. (Coyotzi et al., 2016) were shown to possess nitrite reductase gene and nitrous-oxide reductase gene, while Arenimonas spp. (Remmas et al., 2016) harbors nitrite reductase gene. Denitrifying strains have been described in Caenimonas (Ryu et al., 2008), Haliangium (McIlroy et al., 2016), Marmoricola (Dastager et al., 2008), and Nocardioides spp. (Woo et al., 2012). These indicated that soil nitrogen cycling may be changed by continuously monocropped JA.
Several studies have reported that soil edaphic properties, especially soil pH, were important determinants of soil bacterial community structures (Fierer and Jackson, 2006; Liu et al., 2014). However, the present study found that soil bacterial community structure was not correlated to soil pH. This may be due to the fact that soil pH was relatively stable in our cropping system (Zhou et al., 2017b). The first cropping of JA had higher soil SOC (Zhou et al., 2017b) and was characterized with higher relative abundance of Actinobacteria and lower relative abundance of Acidobacteria, which was in agreement with others’ finding that Actinobacteria responded positively while Acidobacteria responded negatively to exogenously applied labile carbon resources (Fierer et al., 2007; Eilers et al., 2010). The third cropping of JA had lower soil Olsen P (Zhou et al., 2017b) and higher relative abundance of Marmoricola spp., which was consistent with previous studies reporting that these bacterial taxa had negative relationship with soil P (Sun et al., 2014; Bainard et al., 2016). Our RDA analysis and Mantel test also confirmed that soil SOC played an important role in shaping soil bacterial communities, which was in accordance with the observation that soil carbon and P status are important factors in structuring soil bacterial communities (Griffiths et al., 2011; Liu et al., 2014).
Generally, it is suggested that intensive agricultural practices, such as continuous monocropping, had adverse effects on soil microbial community diversity (Zhou et al., 2011, 2017a; Tiemann et al., 2015; Tsiafouli et al., 2015). Our results showed that the third cropping of JA had lower bacterial community diversity indices than the first cropping of JA. However, the wheat field and the first cropping of JA had similar bacterial community diversity indices. Therefore, our second hypothesis was only partially validated. Accumulating evidence suggests that increasing soil microbial diversity can have positive effects on pathogen suppression, nutrient cycling, and plant growth (Bever et al., 2012). Therefore, the declined bacterial community diversity in the third cropping of JA maybe associated with the soil sickness in JA production. Long-term monocropping of several crops can induce soil suppressiveness against soil-borne diseases (Berendsen et al., 2012). For example, the decline of take-all of wheat, caused by Gaeumannomyces graminis var. tritici, has been observed during wheat monocropping (de Souza et al., 2003). The induction of soil suppressiveness was associated with the build-up of antagonistic microorganisms, such as fluorescent Pseudomonas spp. and increased bacterial community diversity (de Souza et al., 2003; Rosenzweig et al., 2012). However, it was not known whether suppressive soil was induced in our wheat field and its relationship with soil bacterial diversities, which should be stressed in future studies.
One shortcoming of this experiment was that soil samples in only one time point were analyzed. Environmental variables, which change across seasons, are main governors of soil microbial communities (Bell et al., 2009). It has been demonstrated that there were seasonal variations in the effects of agricultural practices on soil microbial communities (Spedding et al., 2004; Wolsing and Prieme, 2004). Therefore, seasonal changes in soil bacterial communities in our continuously monocropped JA system should be investigated in more detail. Agricultural weeds were shown to affect soil microbial functional group abundance and community composition (Wortman et al., 2013). However, weeds were only manually removed in the early growth season of JA and the total amount of weeds on the field was not measured in this study. Therefore, there was possibility that the total of weeds differed among treatments and contributed to the changes in soil bacterial communities observed in our cropping system.
Conclusion
In summary, our results demonstrated that continuous monocropping of JA changed soil bacterial community composition and function profile, and soil bacterial community diversity was lower the third cropping of JA. Soil SOC and Olsen P were the important predictors of soil bacterial community in our cropping system. Our results also suggested that wheat rotated with JA can stimulate potentially beneficial bacteria. Soil microbial community composition and function are tightly linked (Bever et al., 2012). However, we only predicted bacteria function from a taxonomy assignment in this study (Langille et al., 2013). Further researches should focus on getting direct evidence of changes in soil microbial functions in our continuously monocropped JA system through approaches such as metagenomic or metatranscriptomic sequencing (Choi et al., 2017; Ofaim et al., 2017).
Author Contributions
XZ, LL, and FW conceived and designed the study. XZ, ZW, and HJ performed the experiments. XZ analyzed the data and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (31772361), ‘Academic Backbone’ Project of Northeast Agricultural University (17XG05), University Nursing Program for Young Scholars with Creative Talents in Heilongjiang Province (UNPYSCT-2015002), and China Agricultural Research System (CARS-23-C-10).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00705/full#supplementary-material
Footnotes
References
Acosta-Martínez, V., Dowd, S., Sun, Y., and Allen, V. (2008). Tag-encoded pyrosequencing analysis of bacterial diversity in a single soil type as affected by management and land use. Soil Biol. Biochem. 40, 2762–2770. doi: 10.1016/j.soilbio.2008.07.022
Ayres, E., Steltzer, H., Simmons, B. L., Simpson, R. T., Steinweg, J. M., Wallenstein, M. D., et al. (2009). Home-field advantage accelerates leaf litter decomposition in forests. Soil Biol. Biochem. 41, 606–610. doi: 10.1016/j.soilbio.2008.12.022
Bainard, L. D., Hamel, C., and Gan, Y. T. (2016). Edaphic properties override the influence of crops on the composition of the soil bacterial community in a semiarid agroecosystem. Appl. Soil Ecol. 105, 160–168. doi: 10.1016/j.apsoil.2016.03.013
Balint, M., Bartha, L., O’hara, R. B., Olson, M. S., Otte, J., Pfenninger, M., et al. (2015). Relocation, high-latitude warming and host genetic identity shape the foliar fungal microbiome of poplars. Mol. Ecol. 24, 235–248. doi: 10.1111/mec.13018
Baral, B. R., Kuyper, T. W., and Van Groenigen, J. W. (2014). Liebig’s law of the minimum applied to a greenhouse gas: alleviation of P-limitation reduces soil N2O emission. Plant Soil 374, 539–548. doi: 10.1007/s11104-013-1913-8
Bastian, F., Bouziri, L., Nicolardot, B., and Ranjard, L. (2009). Impact of wheat straw decomposition on successional patterns of soil microbial community structure. Soil Biol. Biochem. 41, 262–275. doi: 10.1016/j.soilbio.2008.10.024
Bell, C. W., Acosta-Martínez, V., Mcintyre, N. E., Cox, S., Tissue, D. T., and Zak, J. C. (2009). Linking microbial community structure and function to seasonal differences in soil moisture and temperature in a Chihuahuan desert grassland. Microb. Ecol. 58, 827–842. doi: 10.1007/s00248-009-9529-5
Berendsen, R. L., Pieterse, C. M., and Bakker, P. A. (2012). The rhizosphere microbiome and plant health. Trends Plant Sci. 17, 478–486. doi: 10.1016/j.tplants.2012.04.001
Bever, J. D., Platt, T. G., and Morton, E. R. (2012). Microbial population and community dynamics on plant roots and their feedbacks on plant communities. Annu. Rev. Microbiol. 66, 265–283. doi: 10.1146/annurev-micro-092611-150107
Bhattacharyya, P. N., and Jha, D. K. (2012). Plant growth-promoting rhizobacteria (PGPR): emergence in agriculture. World J. Microbiol. Biotechnol. 28, 1327–1350. doi: 10.1007/s11274-011-0979-9
Birouste, M., Kazakou, E., Blanchard, A., and Roumet, C. (2012). Plant traits and decomposition: are the relationships for roots comparable to those for leaves? Ann. Bot. 109, 463–472. doi: 10.1093/aob/mcr297
Braker, G., Fesefeldt, A., and Witzel, K. P. (1998). Development of PCR primer systems for amplification of nitrite reductase genes (nirK and nirS) to detect denitrifying bacteria in environmental samples. Appl. Environ. Microbiol. 64, 3769–3775.
Braker, G., Matthies, D., Hannig, M., Brandt, F. B., Brenzinger, K., and Gröngröft, A. (2015). Impact of land use management and soil properties on denitrifier communities of Namibian savannas. Microb. Ecol. 70, 981–992. doi: 10.1007/s00248-015-0623-6
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cavalca, L., Zanchi, R., Corsini, A., Colombo, M., Romagnoli, C., Canzi, E., et al. (2010). Arsenic-resistant bacteria associated with roots of the wild Cirsium arvense (L.) plant from an arsenic polluted soil, and screening of potential plant growth-promoting characteristics. Syst. Appl. Microbiol. 33, 154–164. doi: 10.1016/j.syapm.2010.02.004
Chavez-Romero, Y., Navarro-Noya, Y. E., Reynoso-Martinez, S. C., Sarria-Guzman, Y., Govaerts, B., Verhulst, N., et al. (2016). 16S metagenomics reveals changes in the soil bacterial community driven by soil organic C, N-fertilizer and tillage -crop residue management. Soil Tillage Res. 159, 1–8. doi: 10.1016/j.still.2016.01.007
Chi, J., Long, X., and Liu, Z. (2009). Effects of continuous cropping on yield, quality of Jerusalem artichoke and soil enzyme activities. Jiangsu J. Agric. Sci. 25, 775–780.
Choi, S., Song, H., Tripathi, B. M., Kerfahi, D., Kim, H., and Adams, J. M. (2017). Effect of experimental soil disturbance and recovery on structure and function of soil community: a metagenomic and metagenetic approach. Sci. Rep. 7:2260. doi: 10.1038/s41598-017-02262-6
Cook, R. J. (2006). Toward cropping systems that enhance productivity and sustainability. Proc. Nat. Acad. Sci. U.S.A. 103, 18389–18394. doi: 10.1073/pnas.0605946103
Cookson, W. R., Beare, M. H., and Wilson, P. E. (1998). Effects of prior crop residue management on microbial properties and crop residue decomposition. Appl. Soil Ecol. 7, 179–188. doi: 10.1016/S0929-1393(97)00032-2
Coyotzi, S., Doxey, A. C., Clark, I. D., Lapen, D. R., Van Cappellen, P., and Neufeld, J. D. (2016). Agricultural soil denitrifiers possess extensive nitrite reductase gene diversity. Environ. Microbiol. 19, 1189–1208. doi: 10.1111/1462-2920.13643
Dastager, S. G., Lee, J.-C., Ju, Y.-J., Park, D.-J., and Kim, C.-J. (2008). Marmoricola bigeumensis sp. nov., a member of the family Nocardioidaceae. Int. J. Syst. Evol. Microbiol. 58, 1060–1063. doi: 10.1099/ijs.0.65576-0
de Souza, J. T., Weller, D. M., and Raaijmakers, J. M. (2003). Frequency, diversity, and activity of 2,4-diacetylphloroglucinol-producing fluorescent Pseudomonas spp. in Dutch take-all decline soils. Phytopathology 93, 54–63. doi: 10.1094/PHYTO.2003.93.1.54
Derakhshani, H., Tun, H. M., and Khafipour, E. (2016). An extended single-index multiplexed 16S rRNA sequencing for microbial community analysis on MiSeq illumina platforms. J. Basic Microb. 56, 321–326. doi: 10.1002/jobm.201500420
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Eilers, K. G., Lauber, C. L., Knight, R., and Fierer, N. (2010). Shifts in bacterial community structure associated with inputs of low molecular weight carbon compounds to soil. Soil Biol. Biochem. 42, 896–903. doi: 10.1016/j.soilbio.2010.02.003
Espana, M., Rasche, F., Kandeler, E., Brune, T., Rodriguez, B., Bending, G. D., et al. (2011). Identification of active bacteria involved in decomposition of complex maize and soybean residues in a tropical Vertisol using 15N-DNA stable isotope probing. Pedobiologia 54, 187–193. doi: 10.1016/j.pedobi.2011.03.001
Fernandes, A. D., Reid, J. N., Macklaim, J. M., Mcmurrough, T. A., Edgell, D. R., and Gloor, G. B. (2014). Unifying the analysis of high-throughput sequencing datasets: characterizing RNA-seq, 16S rRNA gene sequencing and selective growth experiments by compositional data analysis. Microbiome 2:15. doi: 10.1186/2049-2618-2-15
Fierer, N., Bradford, M. A., and Jackson, R. B. (2007). Toward an ecological classification of soil bacteria. Ecology 88, 1354–1364. doi: 10.1890/05-1839
Fierer, N., and Jackson, R. B. (2006). The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. U.S.A. 103, 626–631. doi: 10.1073/pnas.0507535103
Garbeva, P., van Elsas, J. D., and van Veen, J. A. (2008). Rhizosphere microbial community and its response to plant species and soil history. Plant Soil 302, 19–32. doi: 10.1038/srep45318
Gentry, L. F., Ruffo, M. L., and Below, F. E. (2013). Identifying factors controlling the continuous corn yield penalty. Agron. J. 105, 295–303. doi: 10.2134/agronj2012.0246
Glaser, K., Hackl, E., Inselsbacher, E., Strauss, J., Wanek, W., Zechmeister-Boltenstern, S., et al. (2010). Dynamics of ammonia-oxidizing communities in barley-planted bulk soil and rhizosphere following nitrate and ammonium fertilizer amendment. FEMS Microbiol. Ecol. 74, 575–591. doi: 10.1111/j.1574-6941.2010.00970.x
Griffiths, R. I., Thomson, B. C., James, P., Bell, T., Bailey, M., and Whiteley, A. S. (2011). The bacterial biogeography of British soils. Environ. Microbiol. 13, 1642–1654. doi: 10.1111/j.1462-2920.2011.02480.x
Hai, B., Diallo, N. H., Sall, S., Haesler, F., Schauss, K., Bonzi, M., et al. (2009). Quantification of key genes steering the microbial nitrogen cycle in the rhizosphere of sorghum cultivars in tropical agroecosystems. Appl. Environ. Microbiol. 75, 4993–5000. doi: 10.1128/AEM.02917-08
Hayat, R., Ali, S., Amara, U., Khalid, R., and Ahmed, I. (2010). Soil beneficial bacteria and their role in plant growth promotion: a review. Ann. Microbiol. 60, 579–598. doi: 10.1007/s13213-010-0117-1
Hill, M. O. (1973). Diversity and Evenness: a unifying notation and its consequences. Ecology 54, 427–432. doi: 10.2307/1934352
Houfani, A. A., Vetrovsky, T., Baldrian, P., and Benallaoua, S. (2017). Efficient screening of potential cellulases and hemicellulases produced by Bosea sp. FBZP-16 using the combination of enzyme assays and genome analysis. World J. Microbiol. Biotechnol. 33:29. doi: 10.1007/s11274-016-2198-x
Huang, L., Song, L., Xia, X., Mao, W., Shi, K., Zhou, Y., et al. (2013). Plant-soil feedbacks and soil sickness: from mechanisms to application in agriculture. J. Chem. Ecol. 39, 232–242. doi: 10.1007/s10886-013-0244-9
Kandeler, E., Deiglmayr, K., Tscherko, D., Bru, D., and Philippot, L. (2006). Abundance of narG, nirS, nirK, and nosZ genes of denitrifying bacteria during primary successions of a glacier foreland. Appl. Environ. Microbiol. 72, 5957–5962. doi: 10.1128/AEM.00439-06
Kanehisa, M., Goto, S., Sato, Y., Furumichi, M., and Tanabe, M. (2012). KEGG for integration and interpretation of large-scale molecular data sets. Nucleic Acids Res. 40, D109–D114. doi: 10.1093/nar/gkr988
Kaur, N., and Gupta, A. K. (2002). Applications of inulin and oligofructose in health and nutrition. J. Biosci. 27, 703–714. doi: 10.1007/BF02708379
Langille, M. G. I., Zaneveld, J., Caporaso, J. G., Mcdonald, D., Knights, D., Reyes, J. A., et al. (2013). Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 31, 814–821. doi: 10.1038/nbt.2676
Liu, J., Sui, Y., Yu, Z., Shi, Y., Chu, H., Jin, J., et al. (2014). High throughput sequencing analysis of biogeographical distribution of bacterial communities in the black soils of northeast China. Soil Biol. Biochem. 70, 113–122. doi: 10.1016/j.soilbio.2013.12.014
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
McIlroy, S. J., Starnawska, A., Starnawski, P., Saunders, A. M., Nierychlo, M., Nielsen, P. H., et al. (2016). Identification of active denitrifiers in full-scale nutrient removal wastewater treatment systems. Environ. Microbiol. 18, 50–64. doi: 10.1111/1462-2920.12614
McMurdie, P. J., and Holmes, S. (2014). Waste not, want not: why rarefying microbiome data is inadmissible. PLoS Comput. Biol. 10:e1003531. doi: 10.1371/journal.pcbi.1003531
Meier, C. L., and Bowman, W. D. (2008). Links between plant litter chemistry, species diversity, and below-ground ecosystem function. Proc. Nat. Acad. Sci. U.S.A. 105, 19780–19785. doi: 10.1073/pnas.0805600105
Nayyar, A., Hamel, C., Lafond, G., Gossen, B. D., Hanson, K., and Germida, J. (2009). Soil microbial quality associated with yield reduction in continuous-pea. Appl. Soil Ecol. 43, 115–121. doi: 10.1111/j.1574-6941.2012.01312.x
Nimnoi, P., Pongsilp, N., and Lumyong, S. (2010). Endophytic actinomycetes isolated from Aquilaria crassna Pierre ex Lec and screening of plant growth promoters production. World J. Microbiol. Biotechnol. 26, 193–203. doi: 10.1007/s11274-009-0159-3
Ofaim, S., Ofek-Lalzar, M., Sela, N., Jinag, J., Kashi, Y., Minz, D., et al. (2017). Analysis of microbial functions in the rhizosphere using a metabolic-network based framework for metagenomics interpretation. Front. Microbiol. 8:1606. doi: 10.3389/fmicb.2017.01606
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Remmas, N., Melidis, P., Katsioupi, E., and Ntougias, S. (2016). Effects of high organic load on amoA and nirS gene diversity of an intermittently aerated and fed membrane bioreactor treating landfill leachate. Bioresour. Technol. 220, 557–565. doi: 10.1016/j.biortech.2016.09.009
Rosenzweig, N., Tiedje, J. M., Quensen, J. F., Meng, Q. X., and Hao, J. J. J. (2012). Microbial communities associated with potato common scab-suppressive soil determined by pyrosequencing analyses. Plant Dis. 96, 718–725. doi: 10.1094/PDIS-07-11-0571
Rotthauwe, J. H., Witzel, K. P., and Liesack, W. (1997). The ammonia monooxygenase structural gene amoA as a functional marker: molecular fine-scale analysis of natural ammonia-oxidizing populations. Appl. Environ. Microbiol. 63, 4704–4712.
Ryu, S. H., Lee, D. S., Park, M., Wang, Q., Jang, H. H., Park, W., et al. (2008). Caenimonas koreensis gen. nov., sp. nov., isolated from activated sludge. Int. J. Syst. Evol. Microbiol. 58, 1064–1068. doi: 10.1099/ijs.0.65416-0
Schöler, A., Jacquiod, S., Vestergaard, G., Schulz, S., and Schloter, M. (2017). Analysis of soil microbial communities based on amplicon sequencing of marker genes. Biol. Fertil. Soils 53, 485–489. doi: 10.1007/s00374-017-1205-1
Segata, N., Izard, J., Waldron, L., Gevers, D., Miropolsky, L., Garrett, W. S., et al. (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12:R60. doi: 10.1186/gb-2011-12-6-r60
Sierra, J., Brisson, N., Ripoche, D., and Noël, C. (2003). Application of the STICS crop model to predict nitrogen availability and nitrate transport in a tropical soil cropped with maize. Plant Soil 256, 333–345. doi: 10.1023/A:1026106208320
Spedding, T. A., Hamel, C., Mehuys, G. R., and Madramootoo, C. A. (2004). Soil microbial dynamics in maize-growing soil under different tillage and residue management systems. Soil Biol. Biochem. 36, 499–512. doi: 10.1016/j.soilbio.2003.10.026
Sun, J., Zhang, Q., Zhou, J., and Wei, Q. (2014). Pyrosequencing technology reveals the impact of different manure doses on the bacterial community in apple rhizosphere soil. Appl. Soil Ecol. 78, 28–36. doi: 10.1016/j.apsoil.2014.02.004
Tiemann, L. K., Grandy, A. S., Atkinson, E. E., Marin-Spiotta, E., and Mcdaniel, M. D. (2015). Crop rotational diversity enhances belowground communities and functions in an agroecosystem. Ecol. Lett. 18, 761–771. doi: 10.1111/ele.12453
Tilman, D., Balzer, C., Hill, J., and Befort, B. L. (2011). Global food demand and the sustainable intensification of agriculture. Proc. Nat. Acad. Sci. U.S.A. 108, 20260–20264. doi: 10.1073/pnas.1116437108
Tsiafouli, M. A., Thebault, E., Sgardelis, S. P., de Ruiter, P. C., van der Putten, W. H., Birkhofer, K., et al. (2015). Intensive agriculture reduces soil biodiversity across Europe. Glob. Change Biol. 21, 973–985. doi: 10.1111/gcb.12752
van der Putten, W. H., Bardgett, R. D., Bever, J. D., Bezemer, T. M., Casper, B. B., Fukami, T., et al. (2013). Plant–soil feedbacks: the past, the present and future challenges. J. Ecol. 101, 265–276. doi: 10.1111/1365-2745.12054
Wolsing, M., and Prieme, A. (2004). Observation of high seasonal variation in community structure of denitrifying bacteria in arable soil receiving artificial fertilizer and cattle manure by determining T-RFLP of nir gene fragments. FEMS Microbiol. Ecol. 48, 261–271. doi: 10.1016/j.femsec.2004.02.002
Woo, S. G., Srinivasan, S., Yang, J., Jung, Y. A., Kim, M. K., and Lee, M. (2012). Nocardioides daejeonensis sp. nov., a denitrifying bacterium isolated from sludge in a sewage-disposal plant. Int. J. Syst. Evol. Microbiol. 62, 1199–1203. doi: 10.1099/ijs.0.033308-0
Wortman, S. E., Drijber, R. A., Francis, C. A., and Lindquist, J. L. (2013). Arable weeds, cover crops, and tillage drive soil microbial community composition in organic cropping systems. Appl. Soil Ecol. 72, 232–241. doi: 10.1016/j.apsoil.2013.07.014
Zhou, X., Gao, D., Zhao, M., Zhang, J., Li, L., and Wu, F. (2016). Dynamics of soil bacterial communities in Jerusalem artichoke monocropping system. Allelopathy J. 39, 167–178.
Zhou, X., Liu, J., and Wu, F. (2017a). Soil microbial communities in cucumber monoculture and rotation systems and their feedback effects on cucumber seedling growth. Plant Soil 415, 507–520. doi: 10.1007/s11104-017-3181-5
Zhou, X., and Wu, F. (2012). Dynamics of the diversity of fungal and Fusarium communities during continuous cropping of cucumber in the greenhouse. FEMS Microbiol. Ecol. 80, 469–478. doi: 10.1111/j.1574-6941.2012.01312.x
Zhou, X., Yu, G., and Wu, F. (2011). Effects of intercropping cucumber with onion or garlic on soil enzyme activities, microbial communities and cucumber yield. Eur. J. Soil Biol. 47, 279–287. doi: 10.1016/j.ejsobi.2011.07.001
Keywords: Helianthus tuberosus L., soil bacterial community, bacterial diversity, monocropping, nitrogen cycling
Citation: Zhou X, Wang Z, Jia H, Li L and Wu F (2018) Continuously Monocropped Jerusalem Artichoke Changed Soil Bacterial Community Composition and Ammonia-Oxidizing and Denitrifying Bacteria Abundances. Front. Microbiol. 9:705. doi: 10.3389/fmicb.2018.00705
Received: 16 January 2018; Accepted: 27 March 2018;
Published: 10 April 2018.
Edited by:
Marcus A. Horn, Leibniz University of Hanover, GermanyReviewed by:
Annika Vaksmaa, Radboud University Nijmegen, NetherlandsXuesong Luo, Huazhong Agricultural University, China
Copyright © 2018 Zhou, Wang, Jia, Li and Wu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Fengzhi Wu, Znp3dW5lYXVAeWFob28uY29t