 Feng Liao1†
Feng Liao1† Zhishuo Mo
Zhishuo Mo Bo Pang
Bo Pang Huaiqi Jing
Huaiqi Jing Biao Kan
Biao Kan Wenpeng Gu
Wenpeng Gu- 1Department of Respiratory Medicine, First People’s Hospital of Yunnan Province, Kunming, China
- 2The Third Affiliated Hospital of Sun Yat-sen University, Guangzhou, China
- 3National Institute for Communicable Disease Control and Prevention, Chinese Center for Disease Control and Prevention, State Key Laboratory of Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases, Beijing, China
- 4Department of Acute Infectious Diseases Control and Prevention, Yunnan Provincial Centre for Disease Control and Prevention, Kunming, China
- 5Institute of Medical Biology, Chinese Academy of Medical Science and Peking Union Medical School, Kunming, China
Vibrio cholerae O1 strains taken from the repository of Yunnan province, southwest China, were abundant and special. We selected 70 typical toxigenic V. cholerae (69 O1 and one O139 serogroup strains) isolated from Yunnan province, performed the pulsed field gel electrophoresis (PFGE), multilocus sequence typing (MLST), and MLST of virulence gene (V-MLST) methods, and evaluated the resolution abilities for typing methods. The ctxB subunit sequence analysis for all strains have shown that cholera between 1986 and 1995 was associated with mixed infections with El Tor and El Tor variants, while infections after 1996 were all caused by El Tor variant strains. Seventy V. cholerae obtained 50 PFGE patterns, with a high resolution. The strains could be divided into three groups with predominance of strains isolated during 1980s, 1990s, and 2000s, respectively, showing a good consistency with the epidemiological investigation. We also evaluated two MLST method for V. cholerae, one was used seven housekeeping genes (adk, gyrB, metE, pntA, mdh, purM, and pyrC), and all the isolates belonged to ST69; another was used nine housekeeping genes (cat, chi, dnaE, gyrB, lap, pgm, recA, rstA, and gmd). A total of seven sequence types (STs) were found by using this method for all the strains; among them, rstA gene had five alleles, recA and gmd have two alleles, and others had only one allele. The virulence gene sequence typing method (ctxAB, tcpA, and toxR) showed that 70 strains were divided into nine STs; among them, tcpA gene had six alleles, toxR had five alleles, while ctxAB was identical for all the strains. The latter two sequences based typing methods also had consistency with epidemiology of the strains. PFGE had a higher resolution ability compared with the sequence based typing method, and MLST used seven housekeeping genes showed the lower resolution power than nine housekeeping genes and virulence genes methods. These two sequence typing methods could distinguish some epidemiological special strains in local area.
Introduction
Vibrio cholerae is a Gram-negative intestinal pathogen, causing serious human diarrhea, mainly distributed in southern Asia, parts of Africa, Latin America, and other regions (Heidelberg et al., 2000; Morris, 2011). Toxigenic V. cholerae is the strain carrying cholera toxin (CT), and mainly refers to O1 and O139 serogroup (Faruque et al., 1998; Nair et al., 2006). However, non-O1/non-O139 V. cholerae is not carrying CT, and can only cause mild diarrhea diseases (Singh et al., 2001). Therefore, the prevention and control of toxic strains are more important for humans. In China, cholera was considered to be one of the most serious infectious diseases, although the incidence rate has been maintained at a relatively low level in recent years, the epidemic or outbreak still existed in few areas (Gu et al., 2014). It is very important to perform the molecular typing research for toxigenic V. cholerae and clarify the variation and changes of bacteria.
At present, the majority molecular typing methods of V. cholerae comprised of pulsed field gel electrophoresis (PFGE), multilocus sequence typing (MLST), MLVA (multiple-locus variable number tandem repeat analysis), or genome sequencing (Karaolis et al., 2001; O’Shea et al., 2004a,b; Danin-Poleg et al., 2007; Grim et al., 2010; Taviani et al., 2010; Okada et al., 2012; Sealfon et al., 2012; Tran et al., 2012). PFGE is considered to have highly discrimination efficiency, and commonly used in the epidemiological or outbreak investigation. Two MLST typing methods have been reported, one was used seven housekeeping genes for adk, gyrB, metE, pntA, mdh, purM, and pyrC, established by Octavia1 (Octavia et al., 2013). This method has established the database, and researchers in different countries could submit and compare their results. Another was used nine housekeeping genes for cat, chi, dnaE, gyrB, lap, pgm, recA, rstA, and gmd. This method was developed by Garg et al. (2003), several studies have used this method to perform their researches, showing a good discriminatory power (Bhattacharya et al., 2006; Ang et al., 2010). However, this method has not yet established a public database. Researchers from different regions were unable to exchange and share their data. In addition, some studies performed the molecular typing researches by using virulence genes; the results also had effective resolving abilities (Rivera et al., 2001). Up to present, there was no systemic evaluation for molecular typing methods of toxigenic V. cholerae, especially for two MLST methods mentioned above. The applicability of different typing methods was still unknown.
Yunnan located in southwest China, bordering Myanmar, Vietnam, and Laos, has an extended frontier. V. cholerae resources here were abundant and special, indicated that the cholera was endemic in these regions. Although cholera cases were seldom found in recent years, the imported strains from neighboring countries still existed (Liao et al., 2016). It was very important to find the epidemic consistency of cholera by molecular typing methods. In this study, we selected 70 typical toxigenic V. cholerae isolated from different areas and years in Yunnan province, performed the PFGE, two MLST typing, and MLST of virulence gene (V-MLST) methods, and compared the distinguish ability for different molecular typing methods in local epidemic area.
Materials and Methods
Strains
Seventy V. cholerae strains (already-existing collections) were isolated from different regions, years, and sources in Yunnan province between 1986 and 2012. Sixty-nine strains were O1 serogroup, included 43 Ogawa and 26 Inaba serotype, and one O139 serogroup isolates (we only have three O139 serogroup strains, and selected one as the representative for the study purpose). Fifty-four strains were isolated from the feces samples of patients, 11 from water samples, and five from the external environment (surface of objects), as shown in Table 1.
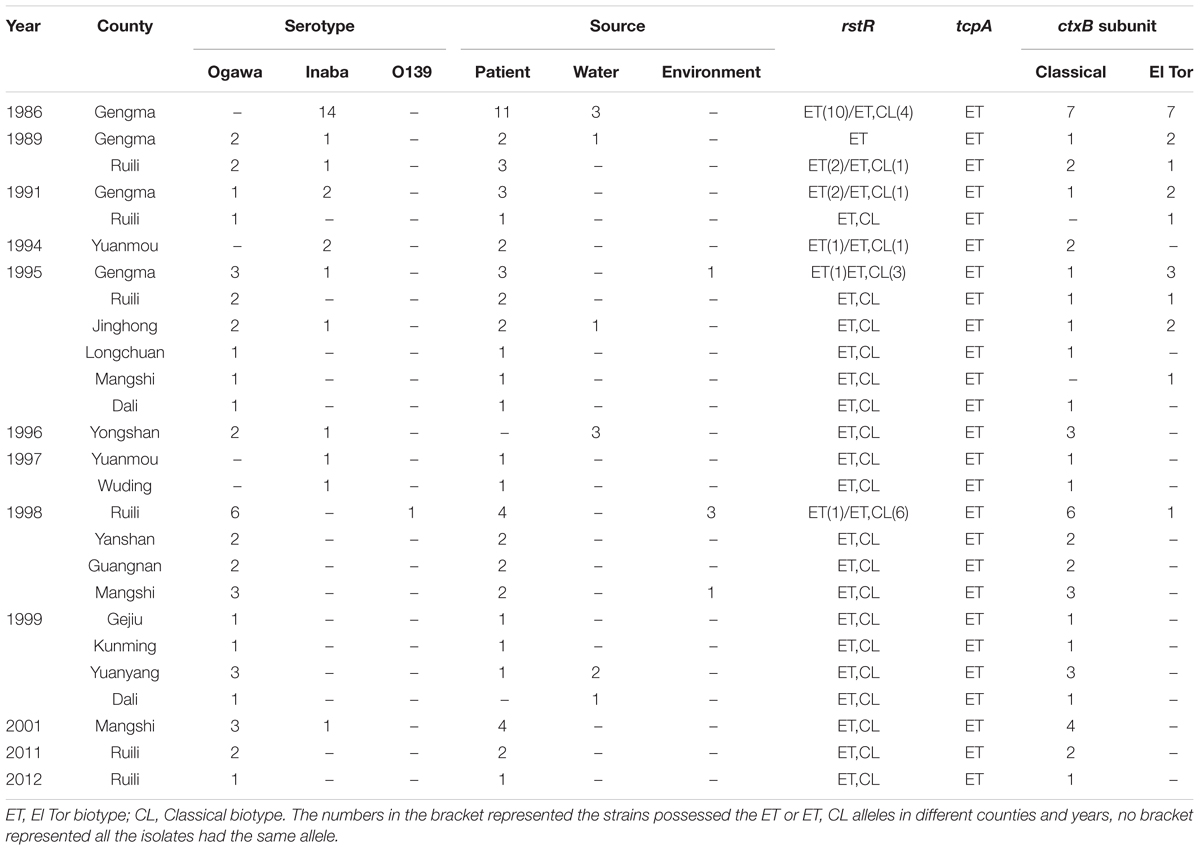
TABLE 1. The 70 V. cholerae strains used in this study.
PCR Detection of Virulence Genes and ctxB Sequencing
Genomic DNA was extracted from each isolate using a DNA extraction kit (Tiangen, Beijing) according to the manufacturers’ instructions. The virulence genes for ctxAB, ompU, ace, zot, toxR, rtxC, and CTX phage rstR (Classical/El Tor) and tcpA (Classical/El Tor) were amplified using Taq premix (TaKaRa, Japan), the primers and amplification procedures were as described previously (Chow et al., 2001; Singh et al., 2001, 2002; O’Shea et al., 2004b). All of the strains were sequenced for ctxB gene subunit to further identify the characters of the CTX phage, Taq premix (TaKaRa, Japan) was used as described above, and amplification processes were performed as previously described (Goel et al., 2010). The amplification products were sent for bidirectional sequencing (TaKaRa, Japan), and the results were analyzed using DNAStar (DNASTAR, Inc., United States) and MEGA 4 software (Tamura et al., 2007). The ctxB sequences of N16961 of El Tor V. cholerae (GenBank: NC-002505) and O395 Classical strain (GenBank: NC-012582) were used as the standards for comparison.
Pulsed Field Gel Electrophoresis
Pulsed field gel electrophoresis was performed based on the PulseNet protocol for V. cholerae and procedures described previously (Gu et al., 2014). The enzyme digestion for each plug was NotI 40U at 37°C for 4 h. The CHEF-Mapper (Bio-Rad) was used for electrophoresis, and the pulse time ranged from 1 to 20 s for 13 h, and 20 to 25 s for 6 h. The gel was stained using Gel Red (Biotium) and visualized using the gel imaging system (Bio-Rad, Gel Doc XR). PFGE patterns were analyzed with BioNumerics version 6.6 (Applied Maths, Belgium), and a dendrogram was produced using the Dice coefficient and un-weighted pair group method with arithmetic mean algorithm (UPGMA). A pairwise distance matrix was also created.
MLST and V-MLST
Seven Housekeeping Genes
PCR amplification was performed according to the public database (see text footnote 1) for adk, gyrB, metE, pntA, mdh, purM, and pyrC, and a list of primers were shown in Table 2. A 100 μl reaction system was used, including 50 μl Taq premix (TaKaRa, Japan), 40 μl water, upstream and downstream of primers 2.5 μl, respectively, and template 5 μl. Amplification procedure was: 94°C 5 min; 94°C 15 s, 50°C 30 s, 72°C 30 s, 35 cycles; the last 72°C 10 min. Amplified products were sent to bidirectional sequencing (TaKaRa, Japan).
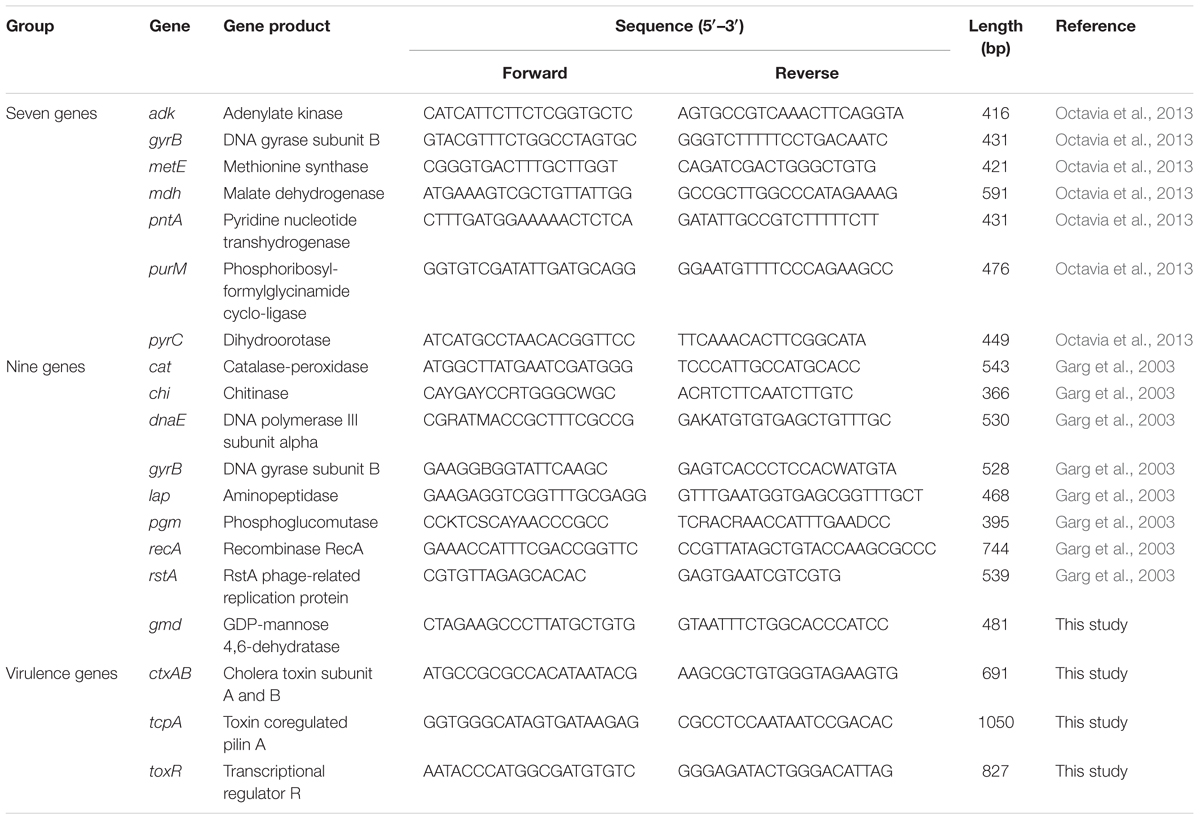
TABLE 2. The primers used in this study.
Nine Housekeeping Genes
PCR amplification was made following the published work (Garg et al., 2003) targeting the genes cat, chi, dnaE, gyrB, lap, pgm, recA, rstA, and gmd. The primers were shown in Table 2, gmd gene could not amplified by reference primer, so we designed the new primers by using Clone Manager Professional 8.0 software (Scientific & Educational), and the gmd gene of V. cholerae reference strain N16961 was used. The reaction system was identical as mentioned above. Amplification procedure was: 94°C 5 min; 94°C 15 s, 55°C 30 s, 72°C 30 s, 35 cycles; the last 72°C 10 min. Amplified products were sent to bidirectional sequencing (TaKaRa, Japan).
Virulent Genes of MLST
We designed the ctxAB, tcpA, and toxR genes primers (Table 2) by using Clone Manager Professional 8.0 software (Scientific & Educational) as well, V. cholerae reference strain N16961 was also used. The reaction system and amplification procedure were identical as mentioned above. Amplified products were sent to bidirectional sequencing (TaKaRa, Japan).
Data Analysis
All the sequencing results were assembled by DNAStar 6.0 software (DNASTAR, Inc., United States), compared and aligned by MEGA 4.0 (Tamura et al., 2007). The seven housekeeping genes sequences were submitted to the public database (see text footnote 1), the alleles of different genes and sequence types (STs) were obtained. The sequence alignments were performed for nine housekeeping and virulence genes, when a new sequence appeared; we gave a new allele for each gene, and finally got the STs by permutation and combination of the nine genes or virulent genes. The minimum spanning tree was constructed by using BioNumerics 6.6 software (Applied Maths, Belgium) for sequences based typing methods.
Nucleotide Sequence Accession Numbers
All the genes of different sequences were deposited in the GenBank with the accession numbers: KX960341 to KX960367.
Ethics Approval Statement
The human sample collection and detection protocols were carried out in accordance with relevant guidelines and regulations approved by Ethical Committee of Yunnan Provincial Centre for Disease Control and Prevention. All experimental procedures were approved by the Ethics Review Committee [Institutional Review Board (IRB)] of Yunnan Provincial Centre for Disease Control and Prevention. All adult subjects provided informed consent, and a parent or guardian of any child participant provided informed consent on their behalf. The informed consents were oral for all the participants, because the samples were too large; we could not get all the written ones. All samples collections and experimental procedures were approved by the Ethics Review Committee, according to Chinese ethics laws and regulations. The anonymization strategy was used for the human sample collection and detection protocols used in this study. The details of patients, such as name, address, age, and sex were anonymous, and we just defined the numbers of patients or samples.
Results
PCR Test for Virulence Genes and ctxB Sequencing
The ctxAB, ompU, ace, zot, toxR, and rtxC for all of the isolates were positive; tcpAElTor was positive for all of the isolates as well, while tcpAClassical was negative. For the rstR, most of the strains carried rstRElTor and rstRClassical; however, some of the strains possessed only rstRElTor. The ctxB subunit showed mixed infection with El Tor type and El Tor variant strains before 1995; after 1996 all of the isolates harbored the ctxB Classical except one O139 V. cholerae that possessed ctxB El Tor (Table 1).
PFGE Results
Seventy toxigenic V. cholerae obtained 50 PFGE patterns, with a high resolution. The clusters could be divided into three groups, named as A-1, A-2, and B with predominance of strains isolated during 1980s, 1990s, and 2000s, respectively (Figure 1). A total of 88.00% similarity of PFGE pattern was found between all the isolates and the pattern similarity scale was 88.96% for group A-1, 89.47% for group A-2, and 94.58% for group B. The green areas of group A-1 and A-2 mainly referred to native epidemic strains, while the yellow areas of group B were mostly imported strains from Myanmar for epidemiological investigation, except one O139 strain. Some V. cholerae isolated in different years and areas had identical PFGE patterns, such as Gengma in 1986 and Gejiu in 1999 (KZGN11O1.CN1241); Mangshi in 2001, Dali and Yuanyang in 1999 (KZGN11O1.CN0322); and Gengma in 1989 and 1991, Yuanmou in 1994 and 1997, Ruili in 1995 and 1998, Yongshan in 1996, Yanshan, Guangnan, and Mangshi in 1998 (KZGN11O1.CN0736). Compared the PFGE result with our previous study (Liao et al., 2016), we found that PFGE had highly discrimination power with whole genomic sequencing method, since the imported strain YN2011QXL (YN2011004) was separated from other three V. cholerae in our previous work used genomic sequencing. And in this study, YN2011QXL was also clustered to different groups with other V. cholerae.
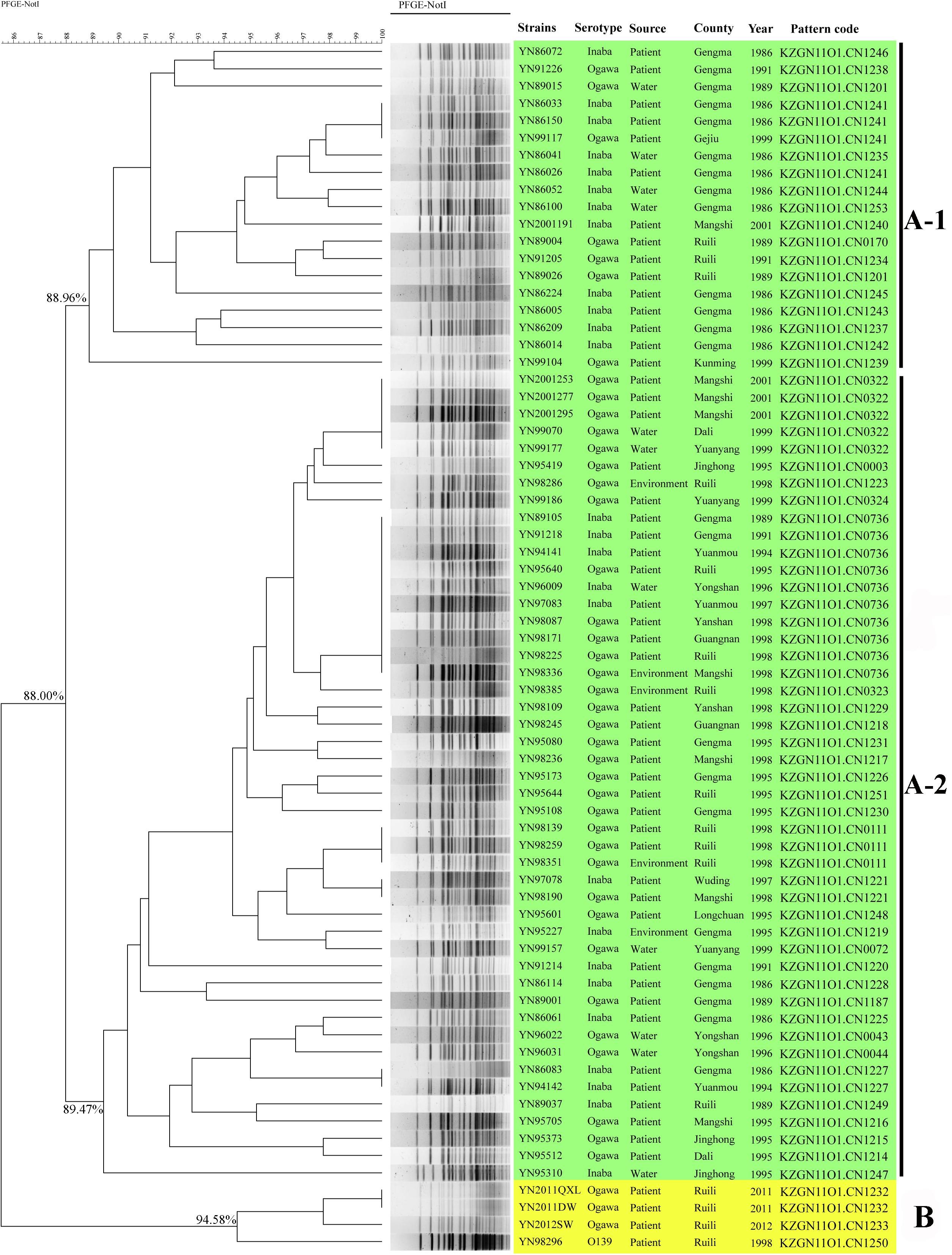
FIGURE 1. PFGE-NotI dendrogram for 70 V. cholerae in this study. The green areas of group A mainly referred to native epidemic strains, while the yellow areas of group B were mostly imported strains from Myanmar.
MLST and V-MLST Results
The MLST results used seven housekeeping genes showed that all the strains belonged to ST69. adk allele was 7, gyrB was 11, metE 37, pntA 12, mdh 4, purM 1, and pyrC 20. The results had no relations with isolated areas or years of the strains (Figure 2A). Nine housekeeping genes were arranged and combined to produce seven different STs, named as ST1–ST7, as shown in Figure 2B. Three imported strains from Myanmar after 2011 formed their own ST (Figure 2B, blue area). Three virulence genes were analyzed and produced nine STs, named as ST1–ST9, as shown in Figure 2C. The imported strains also formed their own STs, while YN2011QXL and other two strains were divided into different types. The latter two sequences based typing methods had consistency with epidemiology of the strains.
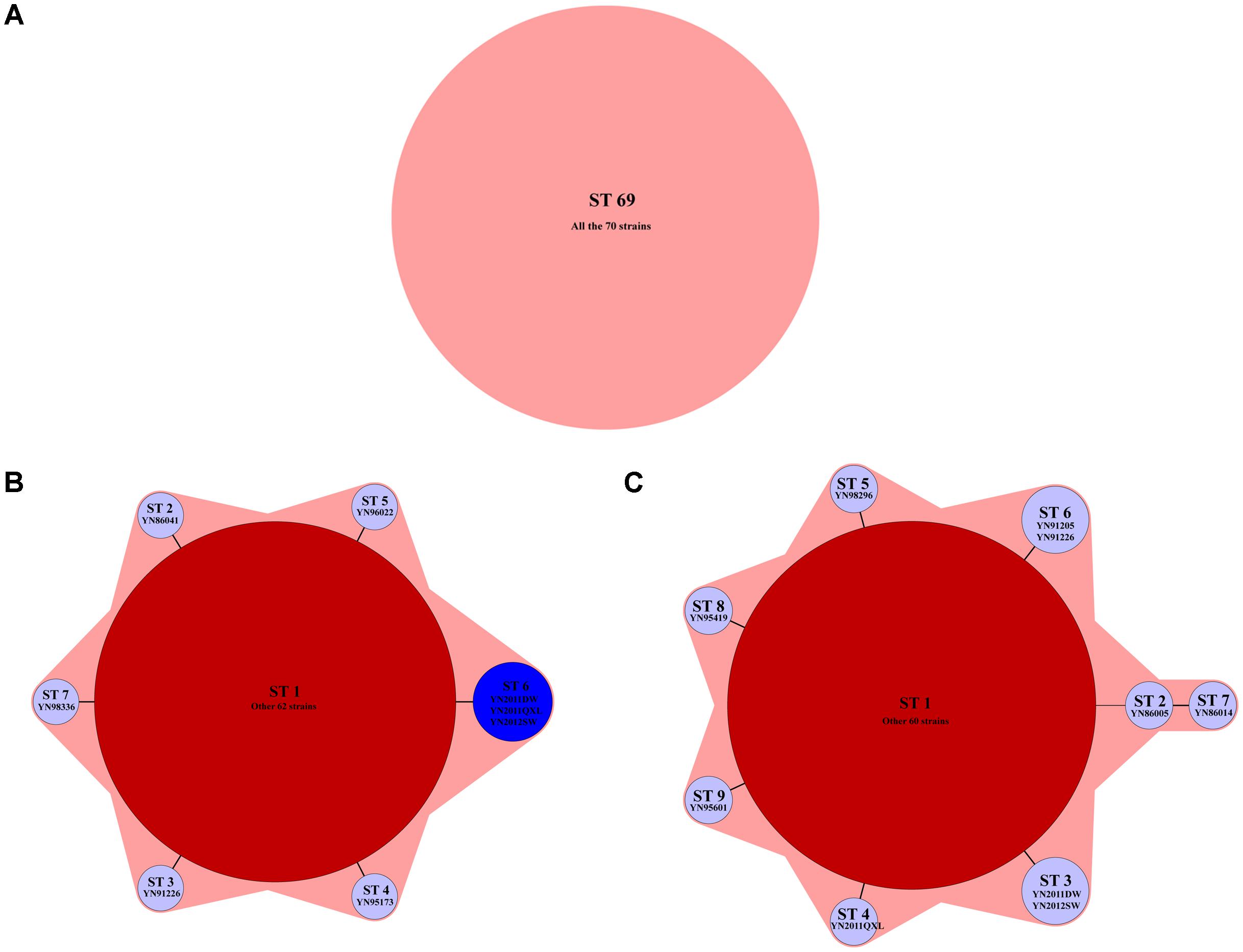
FIGURE 2. The minimum spanning tree of three sequence based genotyping methods. (A) Seven housekeeping genes: all the strains belonged to ST69; (B) nine housekeeping genes: seven STs were found by using this method, named as ST1–ST7. The typical isolates were shown below each STs; and (C) virulence genes: nine STs were found by using this method, named as ST1–ST9. The typical isolates were shown below each STs.
Seven STs were found for all the isolates used nine housekeeping genes method, rstA gene had five alleles (YN2011DW, YN2011QXL, and YN2012SW; YN91226; YN95173; YN96022; and other strains), recA had two alleles (YN86041 and other strains), gmd had two alleles (YN98336 and other strains). Other six housekeeping genes had only one allele, respectively. For rstA gene, YN91226 mutated at position 505 nt; YN2011DW mutated at 453, 459, and 468 nt; YN95173 mutated at 453 nt; and YN96022 mutated at 468 nt, as Figure 3A shown. For recA gene, YN86041 inserted a “T” at position 105 nt (Figure 3B). For gmd gene, YN98336 mutated at 11, 17, 20, 22, 34, 36, 42, 43, 56, and 104 nt position (Figure 3C).
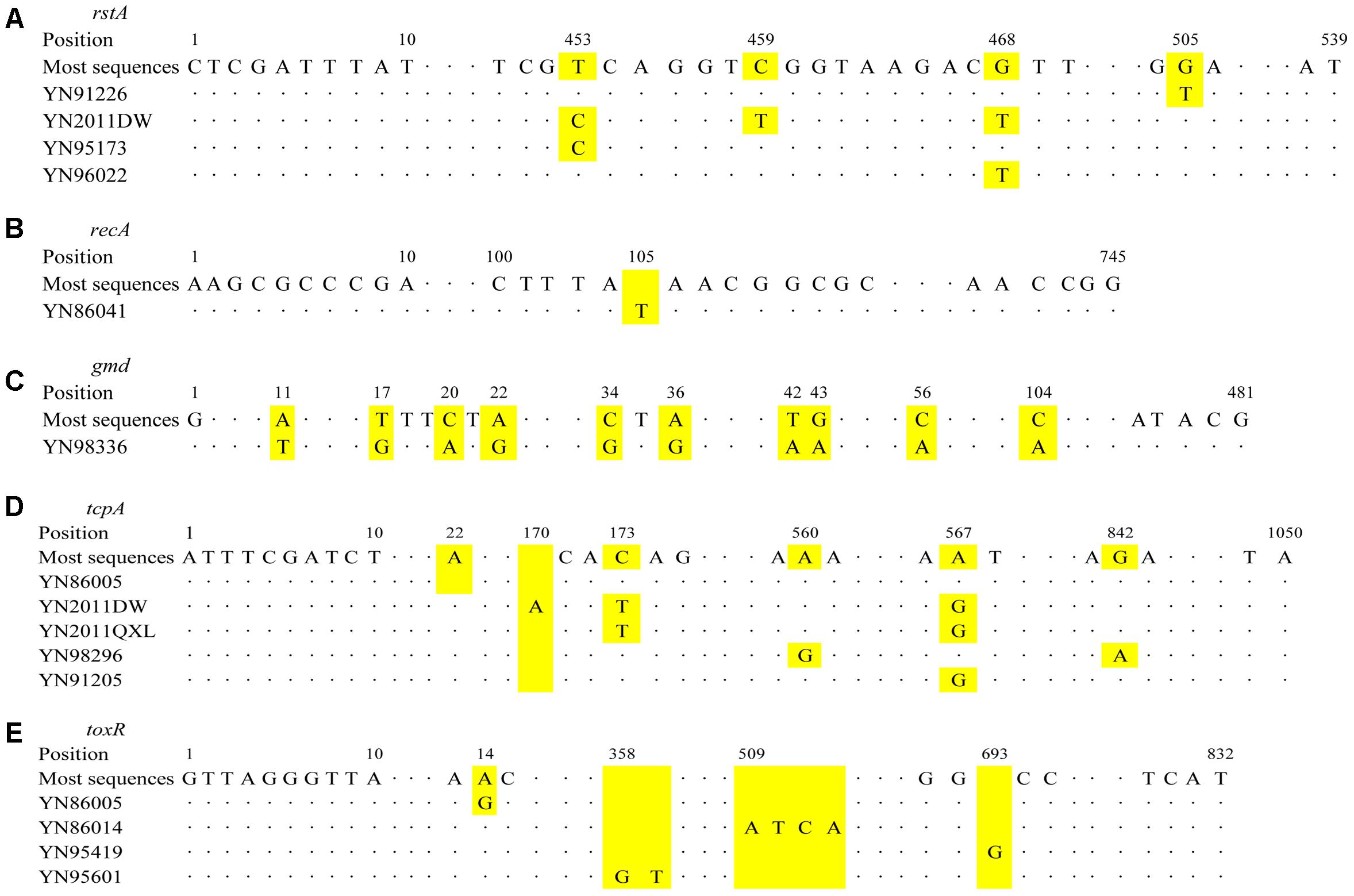
FIGURE 3. Schematic diagram of base position changes for different genes. (A) rstA; (B) recA; (C) gmd; (D) tcpA; and (E) toxR. The yellow areas for each gene represent the changes of base positions.
The virulence genes sequences results showed ctxAB gene was identical for all the strains, tcpA gene had six alleles (YN86005 and YN86014; YN2012SW and YN2011DW; YN2011QXL; YN98296; YN91205 and YN91226; and other strains), toxR gene had five alleles (YN86005; YN95601; YN86014; YN95419; and other strains). For tcpA, YN86005 deleted an “A” at position 22 nt; YN2011DW mutated at 173 and 567 nt, and inserted an “A” at 170 nt; YN2011QXL mutated at 173 and 567 nt; YN98296 mutated at 560 and 842 nt; YN91205 mutated at 567 nt, as Figure 3D shown. For toxR gene, YN86005 mutated at 14 nt; YN86014 inserted “ATCA” at 509 nt; YN95419 inserted a “G” at 693 nt; and YN95601 inserted “GT” at 358 nt (Figure 3E).
All the sequence alignments results for genotyping methods were shown in Supplementary Material.
Comparison the Molecular Typing Methods
Compared the sequence based typing methods in this study, genotyping of seven housekeeping genes was unable to distinguish between strains from different epidemiological resources; genotyping of nine housekeeping genes divided 70 strains into seven STs, and the different epidemiological resources of isolates were distinguished by this method; genotyping of three virulence genes had the similar discriminatory power with nine housekeeping genes. However, the discriminatory ability based on sequence typing methods was lower than PFGE in the local epidemic areas. For example, the cholera epidemic happened in 1986 of Gengma County (A-1 group); 10 patterns were found among 11 strains, while only two STs were found used nine housekeeping genes method (ST1 and ST2); and three STs were identified used virulence genes method (ST1, ST2, and ST7).
Discussion
Pulsed field gel electrophoresis is considered to be the “golden standard” for pathogen molecular typing techniques, showing the highly resolution power, frequently used for outbreak investigation and traceability analysis. MLST is often used for analysis the long-term variability and changes of strains. The purpose of our study was evaluated different molecular typing methods for toxigenic V. cholerae in Yunnan province. From our previous works (Gu et al., 2014; Liao et al., 2016), V. cholerae in Yunnan province, southwest China had similar homology with strains from other part of China, or even some southeast Asia countries. Therefore, the isolates used in this study could reflect the general V. cholerae distributions characteristics in China or southeast Asia, and have enough representatives of the bacteria.
At present, PFGE have the standard experimental procedure, the data could be exchanged and analyzed between different laboratories. Two MLST methods have been reported, Garg et al. (2003) analyzed 96 O139 strains used nine housekeeping genes in 2003, and they found 64 new alleles in 51 STs. Several studies have used this method to perform their researches, for example, Nguyen et al. (2009) performed the MLST method for cholera outbreak in Vietnam in 2009 by using nine housekeeping genes, and all the strains had the same ST with N16961. Lee et al. (2006) analyzed the Mozambique V. cholerae by using nine genetic loci showed that the Mozambique isolates have the same ST as O1 El Tor N16961. Kotetishvili et al. (2003) used 22 V. cholerae isolates to perform the PFGE and MLST by using three housekeeping genes, gyrB, pgm, and recA; sequence data were also obtained for the virulence-associated genes tcpA, ctxA, and ctxB. Their results showed that MLST had better discriminatory ability than PFGE; On MLST analysis, there was clear clustering of epidemic serogroups; much greater diversity was seen among tcpA and ctxAB positive V. cholerae strains from others, non-epidemic serogroups, with a number of tcpA and ctxAB alleles identified. However, this method has not established public database, and its application was limited. Octavia et al. (2013) developed a new MLST method for V. cholerae in 2013; they found a total of 77 isolates were divided into 66 STs, including 55 non-O1/non-O139 strains. While, in this study, 70 toxigenic strains had only one ST, no correction with epidemiological information could be found with the typing results, we considered this method was more suitable for genomic diversity of non-O1/non-O139 V. cholerae. In fact, in our study, PFGE had higher discriminatory power than all sequence based typing method. The cholera epidemic happened in 1986 of Gengma County (A-1 group); 10 patterns were found among 11 strains, while only two STs were found used nine housekeeping genes method (ST1 and ST2); and three STs were identified used virulence genes method (ST1, ST2, and ST7). Therefore, we considered that PFGE was more suitable for molecular typing in cholera epidemic local area.
In Boyd and Waldor (2002) study, genetic variation at the tcpA locus in toxigenic isolates of V. cholerae was investigated; the results showed tcpA sequences were far more diverse than other loci. This diversity was a reflection of diversifying selection in adaptation to the host immune response. Therefore, we selected three major virulence genes of V. cholerae to perform the molecular typing analysis. Its discriminatory ability was similar with nine housekeeping genes method, and the tcpA gene discriminatory effect was the best compared with ctxAB and toxR.
Compared the sequence based typing methods in this study, genotyping of seven housekeeping genes was unable to distinguish between strains from different epidemiological resources; genotyping of nine housekeeping genes divided 70 strains into seven STs, and the different epidemiological resources of isolates were distinguished by this method; genotyping of three virulence genes had the similar discriminatory power with nine housekeeping genes. However, the discriminatory ability based on sequence typing methods was lower than PFGE in the local epidemic areas.
Author Contributions
BK, HJ, and WG designed the work. FL, MC, BP, XF, and WX did the experiments. ZM and WG analyzed the data. ZM drafted the work. BK and HJ revised it critically for important intellectual content.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sent our manuscript to American Journal Experts (www.aje.com) for English language revisions.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.00905/full#supplementary-material
Footnotes
References
Ang, G. Y., Yu, C. Y., Balqis, K., Elina, H. T., Azura, H., Hani, M. H., et al. (2010). Molecular evidence of cholera outbreak caused by a toxigenic Vibrio cholerae O1 El tor variant strain in Kelantan, Malaysia. J. Clin. Microbiol. 48, 3963–3969. doi: 10.1128/JCM.01086-10
Bhattacharya, T., Chatterjee, S., Maiti, D., Bhadra, R. K., Takeda, Y., Nair, G. B., et al. (2006). Molecular analysis of the rstR and orfU genes of the CTX prophages integrated in the small chromosomes of environmental Vibrio cholerae non-O1, non-O139 strains. Environ. Microbiol. 8, 526–634. doi: 10.1111/j.1462-2920.2005.00932.x
Boyd, E. F., and Waldor, M. K. (2002). Evolutionary and functional analyses of variants of the toxin-coregulated pilus protein TcpA from toxigenic Vibrio cholerae non-O1/non-O139 serogroup isolates. Microbiology 148, 1655–1666. doi: 10.1099/00221287-148-6-1655
Chow, K. H., Ng, T. K., Yuen, K. Y., and Yam, W. C. (2001). Detection of RTX toxin gene in Vibrio cholerae by PCR. J. Clin. Microbiol. 39, 2594–2597. doi: 10.1128/JCM.39.7.2594-2597.2001
Danin-Poleg, Y., Cohen, L. A., Gancz, H., Broza, Y. Y., Goldshmidt, H., Malul, E., et al. (2007). Vibrio cholerae strain typing and phylogeny study based on simple sequence repeats. J. Clin. Microbiol. 45, 736–746. doi: 10.1128/JCM.01895-06
Faruque, S. M., Albert, M. J., and Mekalanos, J. J. (1998). Epidemiology, genetics, and ecology of toxigenic Vibrio cholerae. Microbiol. Mol. Biol. Rev. 62, 1301–1314.
Garg, P., Aydanian, A., Smith, D. J., Glenn, M. J., Nair, G. B., and Stine, O. C. (2003). Molecular epidemiology of O139 Vibrio cholerae: mutation, lateral gene transfer, and founder flush. Emerg. Infect. Dis. 9, 810–814. doi: 10.3201/eid0907.020760
Goel, A. K., Jain, M., Kumar, P., and Jiang, S. C. (2010). Molecular characterization of Vibrio cholerae outbreak strains with altered El Tor biotype from southern India. World J. Microbiol. Biotechnol. 26, 281–287. doi: 10.1007/s11274-009-0171-7
Grim, C. J., Hasan, N. A., Taviani, E., Haley, B., Chun, J., Brettin, T. S., et al. (2010). Genome sequence of hybrid Vibrio cholerae O1 MJ-1236, B-33, and CIRS101 and comparative genomics with V. cholerae. J. Bacteriol. 192, 3524–3533. doi: 10.1128/JB.00040-10
Gu, W., Yin, J., Yang, J., Li, C., Chen, Y., Xu, W., et al. (2014). Characterization of Vibrio cholerae from 1986 to 2012 in Yunnan Province, southwest China bordering Myanmar. Infect. Genet. Evol. 21, 1–7. doi: 10.1016/j.meegid.2013.10.015
Heidelberg, J. F., Eisen, J. A., Nelson, W. C., Clayton, R. A., Gwinn, M. L., Dodson, R. J., et al. (2000). DNA sequence of both chromosomes of the cholera pathogen Vibrio cholerae. Nature 406, 477–483. doi: 10.1038/35020000
Karaolis, D. K., Lan, R., Kaper, J. B., and Reeves, P. R. (2001). Comparison of Vibrio cholerae pathogenicity islands in sixth and seventh pandemic strains. Infect. Immun. 69, 1947–1952. doi: 10.1128/IAI.69.3.1947-1952.2001
Kotetishvili, M., Stine, O. C., Chen, Y., Kreger, A., Sulakvelidze, A., Sozhamannan, S., et al. (2003). Multilocus sequence typing has better discriminatory ability for typing Vibrio cholerae than does pulsed-field gel electrophoresis and provides a measure of phylogenetic relatedness. J. Clin. Microbiol. 41, 2191–2196. doi: 10.1128/JCM.41.5.2191-2196.2003
Lee, J. H., Han, K. H., Choi, S. Y., Lucas, M. E., Mondlane, C., Ansaruzzaman, M., et al. (2006). Multilocus sequence typing (MLST) analysis of Vibrio cholerae O1 El Tor isolates from Mozambique that harbour the classical CTX prophage. J. Med. Microbiol. 55, 165–170. doi: 10.1099/jmm.0.46287-0
Liao, F., Pang, B., Fu, X., Xu, W., Kan, B., Jing, H., et al. (2016). The complete genomic analysis of an imported Vibrio cholerae from Myanmar in southwest China. Infect. Genet. Evol. 44, 272–277. doi: 10.1016/j.meegid.2016.07.023
Morris, J. G. Jr. (2011). Cholera–modern pandemic disease of ancient lineage. Emerg. Infect. Dis. 17, 2099–2104. doi: 10.3201/eid1711.111109
Nair, G. B., Qadri, F., Holmgren, J., Svennerholm, A. M., Safa, A., Bhuiyan, N. A., et al. (2006). Cholera due to altered El Tor strains of Vibrio cholerae O1 in Bangladesh. J. Clin. Microbiol. 44, 4211–4213. doi: 10.1128/JCM.01304-06
Nguyen, B. M., Lee, J. H., Cuong, N. T., Choi, S. Y., Hien, N. T., Anh, D. D., et al. (2009). Cholera outbreaks caused by an altered Vibrio cholerae O1 El Tor biotype strain producing classical cholera toxin B in Vietnam in 2007 to 2008. J. Clin. Microbiol. 47, 1568–1571. doi: 10.1128/JCM.02040-08
Octavia, S., Salim, A., Kurniawan, J., Lam, C., Leung, Q., Ahsan, S., et al. (2013). Population structure and evolution of non-O1/non-O139 Vibrio cholerae by multilocus sequence typing. PLoS One 8:e65342. doi: 10.1371/journal.pone.0065342
Okada, K., Roobthaisong, A., Nakagawa, I., Hamada, S., and Chantaroj, S. (2012). Genotypic and PFGE/MLVA analyses of Vibrio cholerae O1: geographical spread and temporal changes during the 2007-2010 cholera outbreaks in Thailand. PLoS One 7:e30863. doi: 10.1371/journal.pone.0030863
O’Shea, Y. A., Finnan, S., Reen, F. J., Morrissey, J. P., O’gara, F., and Boyd, E. F. (2004a). The Vibrio seventh pandemic island-II is a 26.9 kb genomic island present in Vibrio cholerae El Tor and O139 serogroup isolates that shows homology to a 43.4 kb genomic island in V. vulnificus. Microbiology 150, 4053–4063.
O’Shea, Y. A., Reen, F. J., Quirke, A. M., and Boyd, E. F. (2004b). Evolutionary genetic analysis of the emergence of epidemic Vibrio cholerae isolates on the basis of comparative nucleotide sequence analysis and multilocus virulence gene profiles. J. Clin. Microbiol. 42, 4657–4671.
Rivera, I. N., Chun, J., Huq, A., Sack, R. B., and Colwell, R. R. (2001). Genotypes associated with virulence in environmental isolates of Vibrio cholerae. Appl. Environ. Microbiol. 67, 2421–2429. doi: 10.1128/AEM.67.6.2421-2429.2001
Sealfon, R., Gire, S., Ellis, C., Calderwood, S., Qadri, F., Hensley, L., et al. (2012). High depth, whole-genome sequencing of cholera isolates from Haiti and the Dominican Republic. BMC Genomics 13:468. doi: 10.1186/1471-2164-13-468
Singh, D. V., Isac, S. R., and Colwell, R. R. (2002). Development of a Hexaplex PCR assay for rapid detection of virulence and regulatory genes in Vibrio cholerae and Vibrio mimicus. J. Clin. Microbiol. 40, 4321–4324. doi: 10.1128/JCM.40.11.4321-4324.2002
Singh, D. V., Matte, M. H., Matte, G. R., Jiang, S., Sabeena, F., Shukla, B. N., et al. (2001). Molecular analysis of Vibrio cholerae O1, O139, non-O1, and non-O139 strains: clonal relationships between clinical and environmental isolates. Appl. Environ. Microbiol. 67, 910–921. doi: 10.1128/AEM.67.2.910-921.2001
Tamura, K., Dudley, J., Nei, M., and Kumar, S. (2007). MEGA4: molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol. 24, 1596–1599. doi: 10.1093/molbev/msm092
Taviani, E., Grim, C. J., Choi, J., Chun, J., Haley, B., Hasan, N. A., et al. (2010). Discovery of novel Vibrio cholerae VSP-II genomic islands using comparative genomic analysis. FEMS Microbiol. Lett. 308, 130–137. doi: 10.1111/j.1574-6968.2010.02008.x
Keywords: Vibrio cholerae, molecular typing methods, pulsed field gel electrophoresis, multilocus sequence typing, southwest China
Citation: Liao F, Mo Z, Chen M, Pang B, Fu X, Xu W, Jing H, Kan B and Gu W (2018) Comparison and Evaluation of the Molecular Typing Methods for Toxigenic Vibrio cholerae in Southwest China. Front. Microbiol. 9:905. doi: 10.3389/fmicb.2018.00905
Received: 19 December 2017; Accepted: 18 April 2018;
Published: 08 May 2018.
Edited by:
Dongsheng Zhou, Beijing Institute of Microbiology and Epidemiology, ChinaReviewed by:
Thandavarayan Ramamurthy, Translational Health Science and Technology Institute, IndiaXiaohui Zhou, University of Connecticut, United States
Copyright © 2018 Liao, Mo, Chen, Pang, Fu, Xu, Jing, Kan and Gu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenpeng Gu, Z3VfMDI3ODhAMTYzLmNvbQ==
†These authors have contributed equally to this work.