Abstract
Cultivation of undescribed rumen microorganisms is one of the most important tasks in rumen microbiology. In this study, we aimed to discover the potential of culturomics for characterizing the rumen microbiome and for identifying factors, specifically sample dilution and media type, which affect microbial richness on agar plates. Our cultivation experiment captured 23% of all operational taxonomic units (OTUs) found in the rumen microbiome in this study. The use of different media increased the number of cultured OTUs by up to 40%. Sample dilution had the strongest effect on increasing richness on the plates, while abundance and phylogeny were the main factors determining cultivability of rumen microbes. Our findings from phylogenetic analysis of cultured OTUs on the lower branches of the phylogenetic tree suggest that multifactorial traits govern cultivability. Interestingly, most of our cultured OTUs belonged to the rare rumen biosphere. These cultured OTUs could not be detected in the rumen microbiome, even when we surveyed it across a 38 rumen microbiome samples. These findings add another unique dimension to the complexity of the rumen microbiome and suggest that a large number of different organisms can be cultured in a single cultivation effort.
Introduction
The rumen microbiome enables ruminants to make energy stored in plant material metabolically available (Shabat et al., 2016). This achievement is the result of a complex web of interactions and energy flow among many different microorganisms, which is far from fully understood (Mizrahi, 2013). The composition of the rumen microbiome can influence the animal’s energy-harvesting ability, and the quality and quantity of specific microbial groups may tilt the scale in defining efficient vs. inefficient animals (Guan et al., 2008; Shabat et al., 2016; Sasson et al., 2017). In addition, different diets can impact the host animal by changing the rumen microbial composition and consequently, its functioning (Kittelmann et al., 2014; Friedman et al., 2017a,b). Understanding the complex interplay of the environment, microbiome, and animal host not only provides potential applicative benefits, but allows for deeper insights into microbial ecology.
This understanding implies potential answers to seemingly simple questions, such as: “Who is there?” and “What do they do?” In the past few decades, the scientific community has transformed its methodology for studying microbial communities: boosting high-throughput omics approaches, such as sequencing of metagenomes and metatranscriptomes, proteomics and large-scale metabolomics, have provided deeper insights and more available data (Yáñez-Ruiz et al., 2010; Morgavi et al., 2015; Wallace et al., 2015; Weimer et al., 2015; Huws et al., 2016). Despite these contributions, the various reference databases, which originate from microbial isolates, are still insufficient, limiting our ability to interpret and understand existing and new data, as well as our ability to experiment and recapitulate existing microbiome phenotypes. With the desire to fully understand how gut systems work synergistically and how the microbiome benefits the host, there is an urgent need to improve the reference databases and to isolate as-yet uncultured microorganisms. Therefore, in 2011, a Rumen Microbial Genomics (RGM) network was established. In addition, the “Hungate1000” project (Grigoriev et al., 2012; Seshadri et al., 2018) was initiated, aiming to increase the microbial culture collection of rumen origin. Currently, the “Hungate1000” catalog contains 410 genomes of rumen microorganisms, including different strains1. However, despite all of this effort, from the rumen environment only 3.6% of the operational taxonomic units (OTUs) found by sequencing have representative isolates (61 out of 1,698 OTUs) (Nordberg et al., 2014; Seshadri et al., 2018) in the RDP database (Cole et al., 2014), and only 117 bacterial species (not including different strains) of rumen origin are available from international culture collections: American Type Culture Collection (ATCC), Culture Collection University of Göteborg (CCUG), Leibniz Institute DSMZ-German Collection of Microorganisms and Cell Culture (DSMZ), Japan Collection of Microorganisms (JCM), the Belgian Co-Ordinated Collections of Microorganisms (BCCM/LMG), and the National Collection of Type Cultures (NCTC).
Furthermore, most of the cultures are not available in every collection, with 88 out of 117 different species being found in any one of the six international collections, and the DSMZ having the highest number of cultures available. Moreover, the composition of the available cultures does not represent the phylogenetic composition of the rumen (Figure 1A). Hence, there is still a great need to increase the number of representative isolates.
FIGURE 1
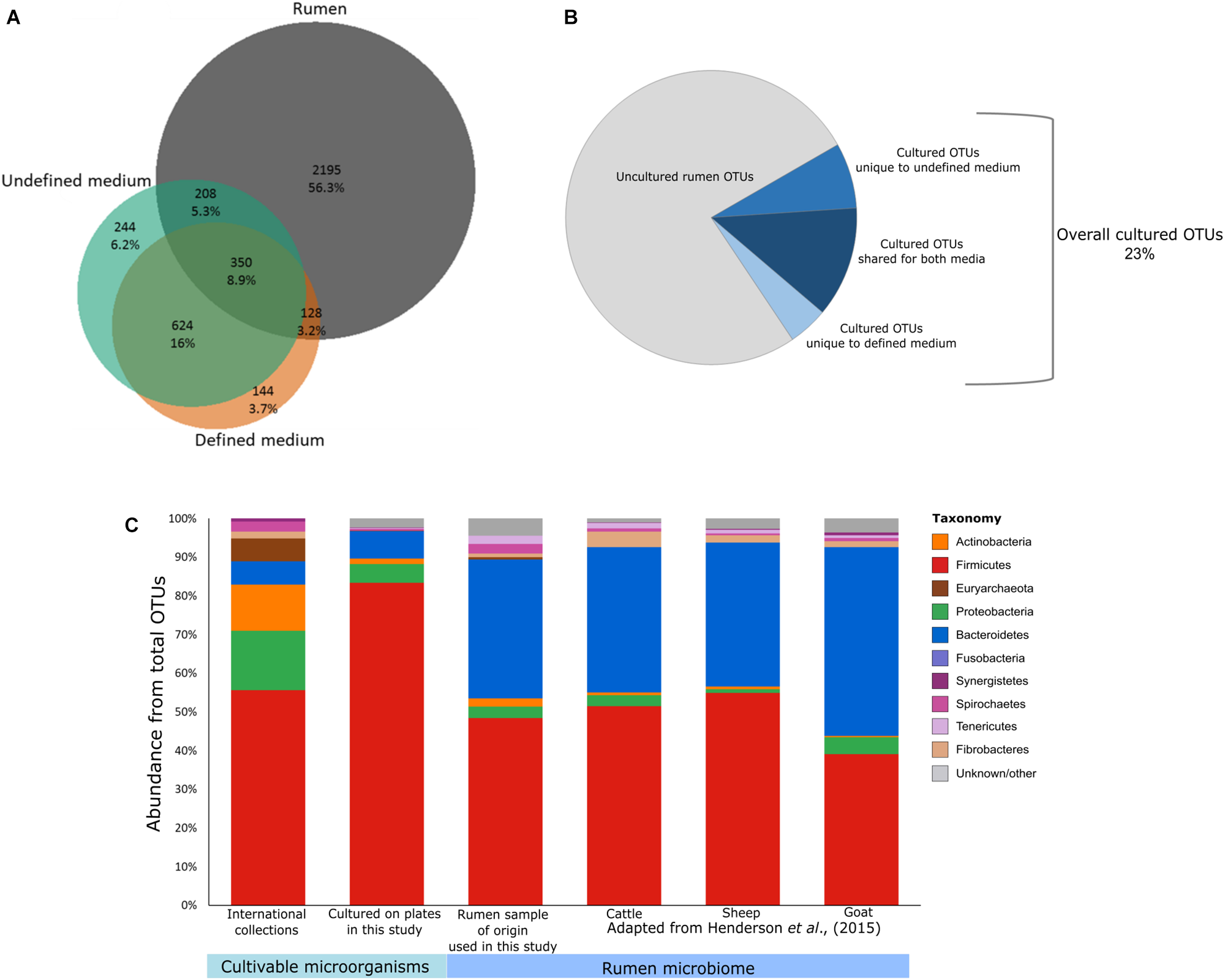
Potential of culturomics for the rumen microbiome. (A) Venn diagram illustrating the OTUs detected in the defined medium (orange), the undefined medium (green), and the original rumen sample and its dilutions (gray). Percentages were calculated from the sum of all samples. (B) Pie chart illustrating the cultivable and uncultivable fractions of the detected rumen microbiome. (C) Stacked bar plots describing the phylogenetic composition of available cultures from public collections (ATCC, CCUG, DSMZ, JCM, NCTC, and BCCM/LMG), the cultured OTUs detected on the plates, the rumen sample used for culturing in this study, and rumen microbiomes as reported previously by Henderson et al. (2015) (cattle, goat, and sheep).
The growth conditions applied in the laboratory are the most important aspect of isolating and cultivating microorganisms. With Robert E. Hungate’s development of anaerobic culturing methods (Chung and Bryant, 1997), the use of rumen fluid in growth media to isolate rumen microorganisms became the most common approach (Bryant and Burkey, 1953; Bryant and Small, 1956; Smith and Hungate, 1958; Bladen et al., 1961; Bryant and Robinson, 1961), even though “habitat-simulating” rumen fluids can vary among animals (Caldwell and Bryant, 1966) and even within the same individual following the consumption of different diets (Friedman et al., 2017b). Eventually, the transition from undefined to defined media became necessary, allowing for comparisons and reproducibility of results, and enabling different laboratories to grow the same isolates. Nonetheless, we believe that in addition to defined media, the initial use of rumen fluid for isolating as-yet uncultured rumen microorganisms can be beneficial. After isolation and characterization, the transition can be made to defined media for general availability.
Here, we aimed to (i) estimate the portion of the rumen microbiome that can be cultivated on either defined or undefined media and (ii) evaluate the effect of medium type and dilution on the overall cultivability of rumen microbes. In addition, we addressed the fraction of as-yet uncultured rumen microorganisms that can be cultured according to our results in a database-comparative manner. We hypothesized that using undefined medium would enable a higher number of different microorganisms to grow on the plates since the rumen fluid in the medium can act as a stimulating factor, making the medium compositionally more similar to the actual rumen. Diluting environmental samples prior to cultivation is a well-known method used to count cells and isolate microorganisms. Here, we sought to examine the effect of this method on our ability to culture both abundant and rare rumen microorganisms.
Materials and Methods
Animal Handling and Sampling
The experimental procedures in this study were approved by the Animal Policy and Welfare Committee of the Agricultural Research Organization Volcani Research Center and were in accordance with the guidelines of the Israel Council for Animal Care.
Israeli Holstein cows (n = 39) were fed a standard lactating cow diet ad libitum consisting of 30% roughage and 70% concentrates, and they had free access to water. The cows were sampled via the mouth using a stomach tube with a rumen vacuum sampler 1 h after the morning feeding. One rumen sample was transferred to two centrifuge tubes, one of which contained glycerol (10% final concentration). All rumen samples were kept at -20°C for 2 weeks until use.
Medium Preparation
Medium 10 (M10) (Caldwell and Bryant, 1966) was used to grow rumen microorganisms. Slight modifications were made and the final medium was composed of (per 100 mL distilled water): 0.2 g trypticase, 0.05 g yeast extract, 3.8 mL solution of K2HPO4⋅3H2O (1.57 g in 200 mL distilled water), 3.8 mL salt solution [0.32 g CaCl2⋅2H2O, 12 g KH2PO4, 2.4 g NaCl, 1.2 g (NH4)2SO4, 0.5 g MgSO4⋅7H2O in 200 mL distilled water], 100 μL Hemin solution (1 g mL-1), 100 μL Resazurin solution (1 mg mL-1), 0.4 g NaHCO3, 0.1 g L-cysteine HCl, 100 μL of 1 M complex sugar mix (arabinose, xylose, glucose, galactose, cellobiose, sucrose, lactose, mannose, maltose, and rhamnose), 1 mL vitamin mix (as described in DSMZ medium 141) and 1.5 g agar. Additionally, 22.85 μL amphotericin B (2.5 mg mL-1) was added to prevent rumen fungi from growing on the plates. Amphotericin B is not known to affect bacterial growth. For the defined medium, 310 μL volatile fatty acid mix (Caldwell and Bryant, 1966) was added. For the undefined medium, we added 30 mL of clarified rumen fluid prepared in advance as follows: 1 L of frozen rumen fluid was thawed in a vinyl anaerobic chamber (COY Lab Products, Grass Lake, MI, United States) for 24 h, incubated with 4 g yeast extract at 39°C for 24 h and subsequently centrifuged in closed anaerobic centrifuge bottles at 13,000 g, 25°C for 25 min. The supernatant was autoclaved in the anaerobic bottles for 20 min at 121°C.
Culturing of Rumen Microorganisms
A small fraction of the single rumen sample stored in glycerol was thawed on ice inside the anaerobic chamber. The anaerobic environment in the chamber was: 5% H2, 20% CO2, and 75% N2. The rumen sample was diluted in decimal format up to 10-6 using 1× anaerobic PBS which was filtered using a pore size of 0.22 μm (Merck Millipore Ltd., Tullagreen, County Cork, Ireland). A 100 μL aliquot of serially diluted rumen sample was plated in duplicate on each agar medium. The plates were incubated at 39°C inside the anaerobic chamber for 3 days, 24 plates overall. As a control, 2 plates from each media type were not plated but were incubated with the other plates. Additionally, we plated all buffers that were used for the dilutions of the rumen samples. After incubation, 1 mL of sterile anaerobic 0.85% NaCl solution was spread on each plate and used to collect all microbes from that plate by mechanically scraping and collecting the cells with a pipette.
DNA Extraction
The rumen samples were separated according to Stevenson and Weimer (2007). The rumen samples (n = 39), rumen dilutions of the selected cow as inoculum source (n = 6) and microbial consortia from the plates (n = 24) were treated as previously described for DNA extractions (Stevenson and Weimer, 2007) with some modifications according to Jami et al. (2013) to suit the needs of this experiment. Briefly, 700 μl of each sample was mechanically broken up using a bead-beater. The DNA was separated from protein twice by adding phenol. Phenol was gradually removed from the DNA sample twice using phenol:chloroform (1:1). Subsequently, the samples were washed twice with chloroform. The DNA was precipitated in NaAc and isopropanol overnight at -20°C. The pellet was washed with ethanol and finally resuspended in TE. Overall, 24 plate samples, 7 rumen samples from the cultivation experiment, and 38 rumen samples were analyzed. The extraction of DNA was performed for all samples at the same day using the same materials and equipment.
16S Ribosomal DNA Sequencing
The V4 region of 16S rDNA was amplified by PCR from 69 DNA extracts using barcoded primers 515F 5′-CCTACGGGAGGCAGCAG-3′ and 806rcbR 5′-CCGTCAATTCMTTTRAGT-3′ (Peterson et al., 2009). The barcoded samples were pooled, sequenced in a MiSeq flow cell (Illumina) for 251 cycles from one end of the fragment and analyzed with Casava 1.8. Overall 1,366,443 reads were obtained for all samples, with an average of 16,663 ± 7,561 reads per sample.
Sequence Data Analysis
Sequence data quality control and analyses were performed using the QIIME pipeline, version 1.7.0 (Caporaso et al., 2010). Briefly, the reads were demultiplexed into samples according to the different barcodes. Illumina adapters and primers were removed and the forward and reverse reads were paired. Sequences with barcode mismatches >1.5 or N bases were removed from the dataset, sequences were truncated prior to three bases with a low quality score (<3) and sequences <191 bp were excluded from further analyses. The average sequence size was 253 bp. OTUs were picked at 97% identity using UCLUST (Edgar, 2010). A de novo picking algorithm was applied in order to not exclude sequences from our dataset solely because of missing reference sequences. Taxonomic assignment of OTUs to the species level was performed by using RDP algorithm on the most abundant sequence of each OTU and the 16S rRNA reference database Greengenes (DeSantis et al., 2006). Assuming they were mainly chimeric or artifacts and in order to minimize random contaminations, singletons and doubletons were removed from the dataset. Data subsampling was performed according to the sample with the least amount of reads (8,078 sequences) and an OTU table was generated, containing the number of reads for each OTU in each sample. In addition, an OTU table containing relative abundances of non-subsampled reads was produced. Sequences were deposited to the SRA under the accession number SRP153190.
Diversity and Similarity
To assess the similarity between microbial consortia that grew on the plates to both each other and the original rumen dilution, principal coordinates analysis (PCoA) was performed on the subsampled OTU table of rumen samples (rumen sample of origin) and all the microbes collected from each plate. Distances were calculated using Euclidean distance. Eigenvalues were calculated for each axis. The significance of the dilution factor and medium type was calculated using two-way PERMANOVA on 1,000 permutations. Sum of squares was utilized to calculate the variance explained by the factors. A confidence interval of 95% was applied.
Richness Assessment
To assess the ability and reproducibility of the cultivation conditions used in the experiment to support growth of a certain number of different OTUs, the richness (observed OTUs) was examined in each sample based on two technical repetitions. The richness was then compared between different dilutions of the same rumen sample using Wilcoxon rank sum test.
Proportion of Shared OTUs
The proportion of microorganisms that are potentially cultivable using this method was estimated from the non-subsampled OTU table. The proportions of shared OTUs were calculated between the rumen samples and their matching plates per dilution, as well as the overall shared OTUs between the datasets (rumen samples, microbial consortia from defined medium and microbial consortia from undefined medium). Venn diagrams were created using the Shiny web application for Rstudio, eulerr online tool2.
Phylogenetic Distances and Tree Construction
The phylogenetic variety found on the different media and in the original rumen sample, including its plated dilutions, was compared using the average pairwise phylogenetic distance between OTUs. Therefore, a fasta file listing the sequences of all OTUs from the rumen (both cultivable and uncultivable) and a file listing all cultivable OTUs were created. A distribution of the average phylogenetic distances of randomly selected OTUs from the rumen was created (1,000 OTUs, 100 permutations). The average phylogenetic distance between cultivable OTUs was calculated and P-values were obtained using the random distribution of OTUs from the rumen.
To measure the average phylogenetic distances of OTUs within a specific taxonomic group (family level), a fasta file of all OTUs belonging to each family was generated and the permutation test was repeated. This analysis was performed for families with at least 10 OTUs in both the rumen sample of origin (including the different dilutions) and the cultured OTUs.
To create a general overview of the microorganisms detected in our dataset (Figure 2A), we searched NCBI accession numbers at the family level or higher (Sayers et al., 2009; Benson et al., 2010) and used the phyloT online tool3 to create distances between microbial families. Taxonomic phylum level and a heat map representing the log number of OTUs found in each group (unique to plates per medium, cultivable OTUs found in the microbiome per medium and uncultivable OTUs) were added. The data were visualized using the iTOL online tool (Letunic and Bork, 2016).
FIGURE 2
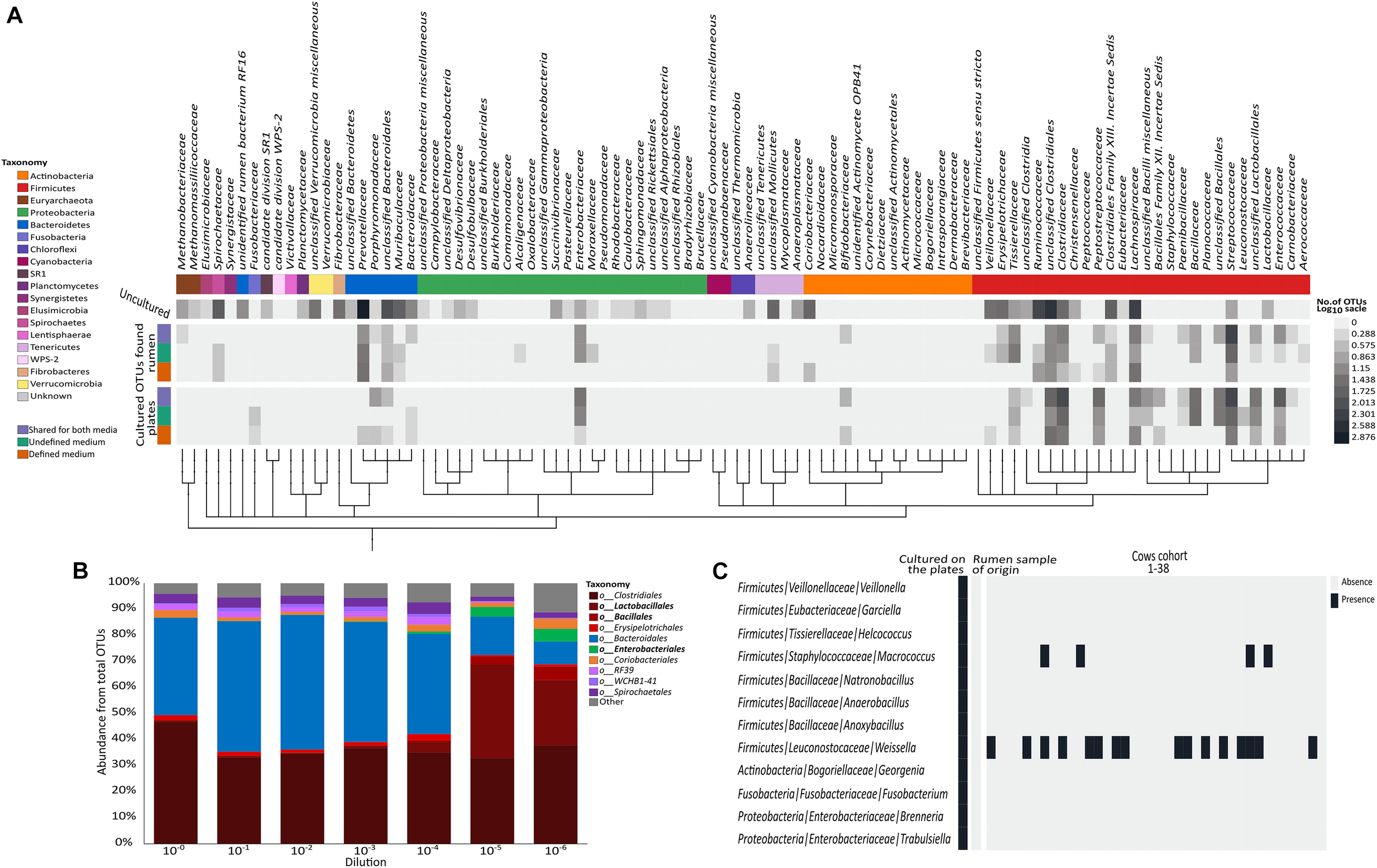
Distribution of the cultured OTUs across rumen microbiome taxonomy and structure. (A) Heat map describing the number of OTUs (in log scale) annotated to phylogenetic groups detected in each of the examined datasets. From top to bottom (color coding indicated on the left side of the figure): OTUs detected only in the original rumen sample, OTUs detected on both media and in the original rumen sample, OTUs detected on the undefined medium and in the rumen sample, OTUs detected on the defined medium and in the rumen sample, OTUs detected on both media, OTUs detected only on the undefined medium, and OTUs detected only on the defined medium. OTUs were arranged on a phylogenetic tree that was created by phyloT program using NCBI accession numbers for each phylogenetic group. The tree was visualized using iTOL online tool (Letunic and Bork, 2016). (B) Stacked bar plot describing the relative abundance at the order level of the original rumen sample and its dilutions. (C) Heat map describing the distribution of the cultured OTUs belonging to the rare biosphere across 38 rumen microbiomes. Taxonomic annotations are given as phylum/family/genus.
Comparison to “Hungate1000” Database
We used the “Hungate1000” database downloaded on June 5th 2018. The cultured OTUs from our dataset were blasted against this “Hungate1000” database (e-value = 0.005) (Seshadri et al., 2018). Results and taxonomy were tested for OTU hits of 100% similarity and 97–99.9% similarity separately. The “Hungate1000” database contains 410 genomes of rumen microorganisms (including different strains).
Results and Discussion
Most Cultivable Rumen Microbes Belong to the Rumen Rare Biosphere
For the purpose of isolating as-yet uncultured rumen microbes, it is important to identify which factors influence their cultivability. Here we focused on the effects of phylogenetic association, medium type, and sample dilution. We plated several serial dilutions (10-1–10-6) of a cow’s rumen sample on both defined medium (M10) and undefined medium (M10 supplemented with rumen fluid). We then subjected all microbes growing on the plates to 16S rDNA gene amplicon sequencing. In addition, we sequenced the original rumen sample and the serial dilutions (up to 10-6) that were used for plating, in order to capture which OTUs grew on the plates in comparison to what had been plated. Using the resulting dataset, we first examined the effect of media type on the cultivability by comparing the microbial composition growing on defined and undefined media. The undefined medium containing rumen fluid increased the cultured richness by a factor of two compared to the defined medium (Figures 1A,B). This suggests that the more natural conditions of the rumen fluid provide unidentified growth-promoting factors that enable a greater variety of OTUs to grow. Nevertheless, we found 272 specific OTUs on the defined medium as well. Therefore, utilization of both defined and undefined media can increase the number of different cultured OTUs by up to 40%. It should be noted that the sample plated had been frozen in 10% glycerol. Therefore, microbes sensitive to the freezing and thawing would not have been cultivated and thus, these numbers might increase for freshly collected samples.
Out of the total 2,881 OTUs detected in the original rumen sample and its dilutions, 686 OTUs were also found on the plates. This suggests that 23% of the rumen microbiome is potentially cultivable. These cultured OTUs belonged to at least 10 bacterial and 1 archaeal phyla (Supplementary Table S1). We found a total of 82 bacterial genera, 58 of which did not have isolated representatives in any of the international microbial collections (ATCC, CCUG, DSMZ, JCM, NCTC, or BCCM/LMG) according to our survey. Only 45 isolates in these culture collections corresponded by name to OTUs from the plates that had species level annotation in our dataset. Only 5 of these representative species had been isolated from bovine or sheep, and the rest originated from various environments (Supplementary Tables S2, S3). As the “Hungate1000” consolidates most of the rumen isolates known to date (Jun 5, 2018), we compared the OTUs found on the plates to this reference (Seshadri et al., 2018). We found a very small overlap between the “Hungate1000” database and the OTUs from our cultivation experiment (61 out of 1,698 cultivable OTUs in our study). This small overlap highlights the importance of international efforts to capture cultivable rumen diversity while it also emphasizes the enormous potential to increase the diversity of rumen culture collections, even in a single cultivation effort.
Sequencing the plated sample dilutions provided deeper insight into the complexity of the rumen microbiome as provided by sequencing the original rumen sample. With increasing dilution of the rumen sample, the percentage of reads annotated as Bacteroidetes strongly decreased, while more and more reads belonged to the phylum Firmicutes (Figure 2B and Supplementary Figure S1). At the order level, higher numbers of Lactobacilli, Bacilli, and Enterobacteria were found with increasing dilution of the rumen sample (Figure 2B), which generally fit the phylogenetic composition of microbes that grew on the plates (Figure 1C).
Interestingly, we found that most of the OTUs that grew on the plates (1,012 out of 1,698) were not detected in the original rumen sample or any of its dilutions (Figure 1A). Some of these 1,012 OTUs belonged to various lineages up to the phylum level that were not found in the original rumen samples (Figure 2C). Most of the microbes that were cultured on the plates remained undetectable, even in the extremely diluted rumen samples. With an average read number of 21,975 ± 5,847 the sequencing effort was close to saturation in individual rarefaction curves (Supplementary Figure S2). As we could exclude systematic contaminations by plating the buffers that were used for the dilutions of the rumen sample as well as by incubating the plates themselves, this finding lead us to hypothesize that the microbes cultured in our experiment might be affiliated with the rare portion of the rumen biosphere. This hypothesis was supported by the large number of OTUs which were unique to the plates. Although random contaminations can never be completely excluded, in combination with the empty control plates, it is unlikely that our finding is a result of random contaminations. Excluding contaminations as a cause for the observation, we further explored the presence of these microbes in multiple rumen ecosystems by sequencing 38 additional cow rumen microbiomes. Except for the genera Weissella and Macrococcus of the phylum Firmicutes, most of these extremely rare biosphere microbes could not be found in this large number of samples (Figure 2C). In multiple microbial ecosystems, including freshwater (Musat et al., 2008), sludge (Hernandez-Raquet et al., 2013), soil (Pester et al., 2010; Giebler et al., 2013; Philippot et al., 2013), rhizosphere (Nuccio et al., 2016), and human microbiome (Guss et al., 2011), the rare microbiome is now being acknowledged as highly influential in terms of ecosystem functions and hosts’ health. In these studies, the rare microbes are reported to affect their environment via multiple mechanisms such as enhancing functionality of abundant microbes, being more active than the abundant microbes, hampering the invasion of other species (Mallon et al., 2015) during community assembly and contributing to the metabolic potential of the community by increasing the genetic pool of their environment (Lynch and Neufeld, 2015; Jousset et al., 2017). Thus, rumen complexity seems to be even higher than previously thought, and the rare microbes of this ecosystem are not necessarily uncultivable. The abundance of these rare OTUs might vary as a function of environmental conditions. Consequently, isolating and studying these organisms becomes even more relevant given that they could have a tremendous impact on the ecosystem.
Phylogenetic Distances Between Cultured OTUs on the Lower Tree Branches Suggest That Multifactorial Traits Determine Cultivability
Current culture collections do not reflect a typical rumen composition (Figure 1C; Jami and Mizrahi, 2012; Henderson et al., 2015). Moreover, our findings show that there is a discrepancy in taxonomic composition between cultured OTUs and the rumen microbiome (Figure 1C). Nevertheless, almost all rumen microbiome phyla were represented within the cultured OTUs, and Firmicutes and Bacteroidetes made up 90% of the cultured OTUs on the plates (Figures 1C, 2A), which is comparable to the occupancy of these phyla in the original rumen sample (Figure 1C). This was surprising since the public collections ATCC, CCUG, DSMZ, JCM, NCTC, and BCCM/LMG have a lower fit to the phylogenetic distribution of a typical ruminant’s microbiome (Figure 1C) according to our survey, as well as that conducted by Creevey et al. (2014). In these collections, Proteobacteria and Actinobacteria are highly overrepresented, whereas Bacteroidetes is underrepresented (Creevey et al., 2014). Bacteroidetes, e.g., is represented by only 7 known isolates in international collections, whereas we cultured 121 OTUs belonging to six different families (Supplementary Table S1). This suggests that cultivability might be connected to the phylogenetic association of some microbial groups. If this is true, the cultured OTUs might show a phylogenetic cohesiveness that could differentiate them from the uncultured ones. With this in mind, we tested the average phylogenetic distances of OTUs cultured on the plates against randomly selected OTUs from the rumen microbiome of the original sample (Figure 3 and Supplementary Figure S3). The OTUs that were found on the plates were more closely related to each other compared to those detected in the original rumen sample (Figure 3). This supports our hypothesis that the cultured OTUs are more phylogenetically cohesive and suggests that certain genetic traits are involved in the ability to grow on plates. Indeed, the main proportion of the cultured OTUs was annotated as Firmicutes (Figure 3) which is in agreement with Creevey et al. (2014), who reported that this phylum is the most represented in cultured isolates. The 1,415 Firmicutes OTUs present in our dataset were distributed across 50 genera (Supplementary Table S1), whereas isolates from 45 genera are available in public culture collections.
FIGURE 3

Comparison of the phylogenetic distances between OTUs growing on the plates and OTUs detected in the original rumen sample. The mean phylogenetic distances between OTUs that grew on plates was calculated and compared to the distribution of phylogenetic distances between the OTUs of the original rumen sample. The distribution of distances from the rumen OTUs is illustrated as a box plot. The average phylogenetic distance between OTUs of the cultured reference group being compared to this rumen distribution is illustrated as an orange oval. The distribution of phylogenetic distances was calculated from a rumen OTU subset picked randomly in a sample size depending on the cultured OTU reference group. Permutation = 100. The following numbers of rumen OTUs were picked for each category (according to the x-axis): All = 1,012; Bacillaceae = 6, Clostridiaceae = 6, Enterococcaceae = 6, Lachnospiraceae = 6, Streptococcaceae = 6, Enterobacteriaceae = 6. Asterisks mark in which cases the average phylogenetic distance of the cultured OTUs differs from the average distance of uncultured OTUs. ∗Cultured OTUs are above/below the 10th percentile. ∗∗∗Cultured OTUs are below the 1st percentile.
We then asked whether this assumption holds true on a finer taxonomic level. We examined OTUs from the most abundant families on the plates (>10 OTUs per family) and compared them to randomly selected OTUs from the same family coming from the original rumen microbiome. The decreased phylogenetic distance was not consistent across the tested phylogenetic families. For the Firmicutes families Bacillaceae and Clostridiaceae, respectively, we detected smaller phylogenetic distances (PBacillaceae = 0.1; PClostridiaceae < 0.05) between the OTUs on the plates and those in the original rumen sample. In contrast, there was a higher phylogenetic distance between Proteobacteria OTUs annotated as Enterobacteriaceae from the plates and the Enterobacteriaceae OTUs from the original rumen sample (P < 0.01). These findings suggest that cultivability of microbes on the plates is a multifactorial trait, selecting different clades that are scattered across the branches of the phylogenetic tree rather than being confined to a specific clade.
Sample Dilutions and Different Media Increase Cultivability
To understand which factor, i.e., medium type or dilution, has a stronger effect on our ability to cultivate rumen microorganisms, PCoA based on a Euclidean distance matrix was applied (Figure 4A). Independent of their origin (plate cultivation or original rumen sample), coordinate 1 separated all consortia by the dilution factor, which explained 30% of the variance in the dataset. It should be noted that depending on the dilution, each plate contained a comparable number of OTUs (100–300). However, with increasing dilution, the compositional similarity between the plates and also between technical repetitions of the same dilution diverged (Figure 4A and Supplementary Figure S4; see also Figure 1). This phenomenon potentially stems from the random sampling of this rare biosphere during the process of diluting and plating, which resulted in a distinct microbial composition on each plate.
FIGURE 4
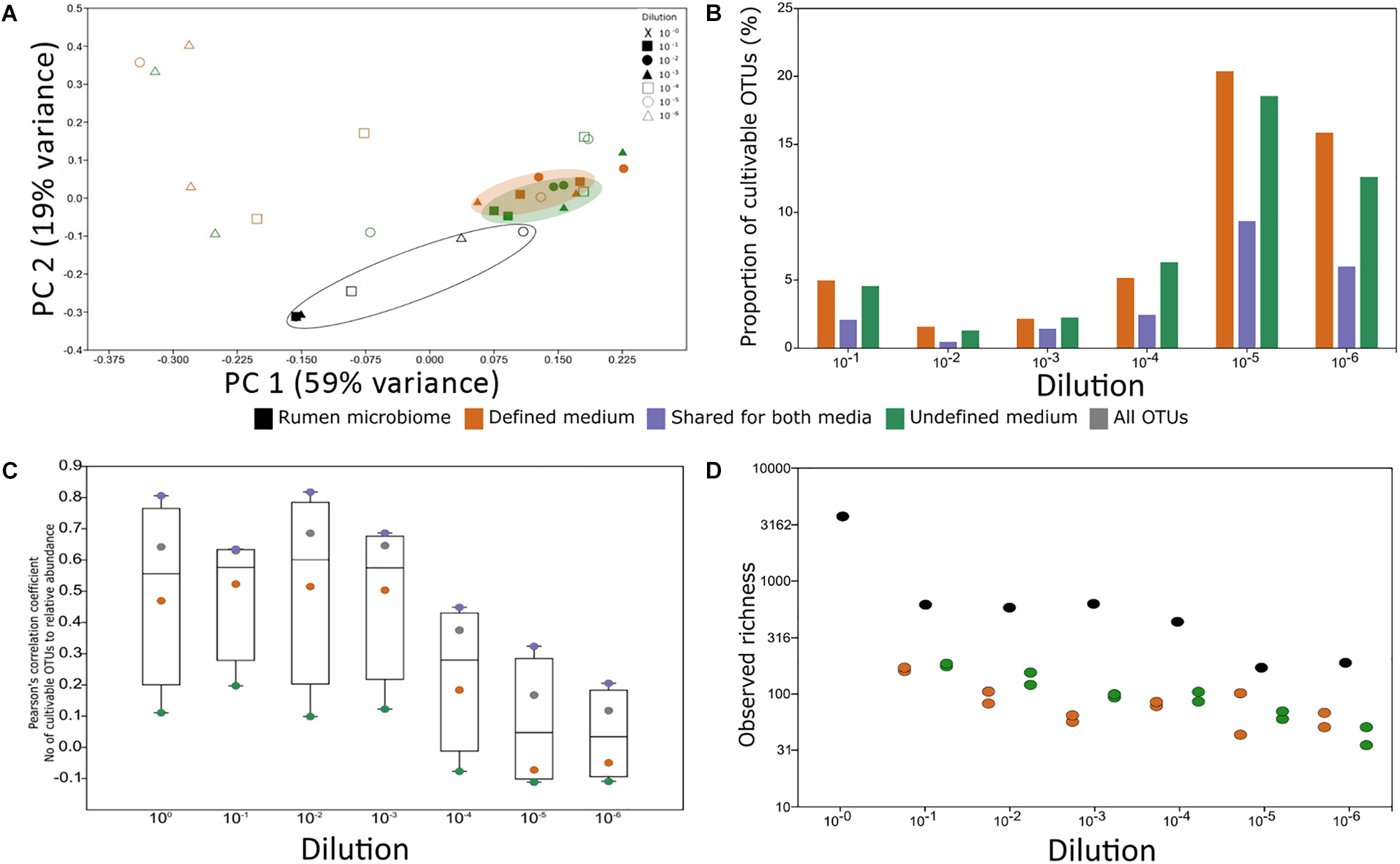
Experimental factors affecting cultivability. (A) PCoA of rumen samples and their cultivable microbial consortia. The variance explained by each component is indicated on each axis. (B) Proportion of the cultivable microbiome in each rumen dilution. The proportion was calculated as the number of OTUs found in both the rumen dilution and its matching plate. The x-axis denotes the dilution of the sample. There was no statistical difference between the proportions of OTUs growing on defined and undefined media (Wilcoxon rank sum test, P > 0.05). (C) The impact of abundance on cultivability. Pearson’s correlation coefficient of the number of cultivable OTUs in each percentile and the relative abundance in each percentile was calculated. Correlations above | r| = 0.3 and P < 0.05 after Holm–Bonferroni correction were considered significant. The y-axis represents Pearson’s correlation coefficient result and the x axis represents the dilution. The numbers of cultivable OTUs were divided according to the medium in which they were found (green – OTUs unique to undefined medium, orange – OTUs unique to defined medium, purple – OTUs found on both medium types, and gray – total number of OTUs). (D) Observed richness for rumen samples and microbial consortia from the plates in each medium type. The overall difference between groups was significant as determined by Kruskal–Wallis test (P = 0.01564). Wilcoxon rank sum paired test (P-value Holm–Bonferroni correction): rumen vs. defined medium: P = 0.03125; rumen vs. undefined medium: P = 0.03125 from each plate in each medium type. Colors represent sample source: Black – rumen samples, orange – microbial consortium that grew on defined medium, and green – microbial consortium that grew on undefined medium. X-axis denotes the dilution of the sample.
In reduced dimensionality, there was no difference in the microbial composition that grew on the defined and undefined media, independent of dilution. However, underlining the phylogenetic differences between available cultures and a typical ruminant’s microbiome, there was a clear difference between the rumen microbiome and the microbes cultured on the plates. The factor origin, i.e., rumen or medium, explained 18% of the dataset’s variance (Figure 4A). Based on these analyses, it is mainly dilutions and repetitions that allow for different microbes to grow on the plates and likely also for a higher microbial diversity to be captured by cultivation (Figures 4A,B).
OTU Abundance in the Rumen Environment Is Positively Correlated to Cultivability
We next sought to understand whether there is a connection between cultivability of an OTU and its abundance. Overall, the total number of cultured OTUs was positively and significantly correlated to their relative abundance in the less diluted rumen samples (Figure 4C). The number of cultured OTUs from highly diluted samples was less dependent on the relative abundance of OTUs in the sample (Figure 4C), suggesting that sample dilution is highly relevant to reducing the dependence of cultivability on abundance. While the number of cultured OTUs unique to the defined medium was correlated to their abundance, this dependence was practically non-existent for the undefined medium. However, OTUs which were found on both media were highly dependent on the relative abundance of the corresponding microorganisms in the sample (Figure 4C). This emphasizes the potential of using rumen fluid in the culture medium. These findings suggest that factors like sampling time and method, related to the abundance of the different rumen microbiome members, may affect the composition of cultivable microbes. For example, the rumen microbiome undergoes compositional diurnal oscillations (Shaani et al., 2018) which in turn may affect the probability of the different microbes to be sampled and subsequently cultivated.
Using sample dilutions, the culture conditions allowed growth of both the highly abundant and eminently low abundant OTUs (Figure 4C) and enabled capturing the highest proportion of cultured OTUs (Figure 4C). This is in agreement with studies on the human gut microbiome (Walker et al., 2014; Lagkouvardos et al., 2017). Thus, the isolation of rumen microorganisms should become one of the major focuses of our field, to assign better annotations and to better understand the rumen ecosystem.
Conclusion
In this study, we asked two fundamental questions: (i) What is the portion of the rumen microbiome that can potentially be cultivated? (ii) How are medium type, sample dilution and phylogeny related to cultivability? To answer these questions, we used defined and undefined (with rumen fluid) anaerobic media and plated decimal dilutions of a rumen sample. Using this methodology, 23% of the rumen microbiome is potentially cultivable. We found a positive correlation between the cultivability of an OTU and its abundance in the original rumen sample for both media. The effect of sample dilution exceeded the effect of medium type, indicating that the laborious plating effort is worth the chance of capturing rumen microbial diversity. Furthermore, technical replications in plating strongly contributed to the richness on the plates and both media had a high number of unique OTUs. Cultivation seems to select for a multifactorial set of genetic traits scattered across the phylogenetic tree. Despite the great variety of microbes cultured on the plates, the selection for genomic traits also calls for new isolation approaches based on certain sets of traits to increase cultivated diversity, as these genetic traits are not necessarily interconnected and unlikely to be fully dependent on each other.
The OTUs cultured here were mainly from the rare rumen biosphere. Taken together, the high relevance of the rare biosphere reported in the literature and its surprisingly high cultivability as found here, we conclude that the rumen’s complexity and functional repertoire are beyond our current estimation. Nonetheless part of these rare biosphere microbes can be studied in pure culture, enabling a deeper understanding of rumen ecosystem functionality. Therefore, our study presents a very promising notion: a vast variety of unknown rumen microorganisms can be grown using defined and undefined anaerobic media.
Statements
Author contributions
TZ designed the experiments, collected the samples, carried out the wet lab experiments, designed and performed the analyses, and wrote the manuscript. MP contributed to data analyses and wrote the manuscript. IM designed the experiments, designed the data analysis, and wrote the manuscript.
Funding
This research was supported by the European Research Council under the European Union’s Horizon 2020 research and innovation program, project number 640384, and Israel Science Foundation, project number 1313/13.
Acknowledgments
We thank Goor Sasson for his assistance in programming and automatization, and Matthew Metzger for English editing.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.01999/full#supplementary-material
Footnotes
1.^https://genome.jgi.doe.gov/portal/TheHunmicrobiome/TheHunmicrobiome.info.html
References
1
BensonD. A.Karsch-MizrachiI.LipmanD. J.OstellJ.SayersE. W. (2010). GenBank.Nucleic Acids Res.38D46–D51. 10.1093/nar/gkp1024
2
BladenH. A.BryantM. P.DoetschR. N. (1961). A study of bacterial species from the rumen which produce ammonia from protein hydrolyzate.Appl. Microbiol.9175–180.
3
BryantM. P.BurkeyL. A. (1953). Cultural methods and some characteristics of some of the more numerous groups of bacteria in the bovine rumen.J. Dairy Sci.36205–217. 10.3168/jds.S0022-0302(53)91482-9
4
BryantM. P.RobinsonI. M. (1961). An improved nonselective culture medium for ruminal bacteria and its use in determining diurnal variation in numbers of bacteria in the rumen.J. Dairy Sci.441446–1456. 10.3168/jds.S0022-0302(61)89906-2
5
BryantM. P.SmallN. (1956). The anaerobic monotrichous butyric acid-producing curved rod-shaped bacteria of the rumen.J. Bacteriol.7216–21.
6
CaldwellD. R.BryantM. P. (1966). Medium without rumen fluid for nonselective enumeration and isolation of rumen bacteria.Appl. Microbiol.14794–801.
7
CaporasoJ. G.KuczynskiJ.StombaughJ.BittingerK.BushmanF. D.CostelloE. K.et al (2010). QIIME allows analysis of high-throughput community sequencing data.Nat. Methods7335–336. 10.1038/nmeth.f.303
8
ChungK. T.BryantM. P. (1997). Robert E. Hungate: pioneer of anaerobic microbial ecology.Anaerobe3213–217. 10.1006/anae.1997.0109
9
ColeJ. R.WangQ.FishJ. A.ChaiB.McGarrellD. M.SunY.et al (2014). Ribosomal database project: data and tools for high throughput rRNA analysis.Nucleic Acids Res.42D633–D642. 10.1093/nar/gkt1244
10
CreeveyC. J.KellyW. J.HendersonG.LeahyS. C. (2014). Determining the culturability of the rumen bacterial microbiome.Microb. Biotechnol.7467–479. 10.1111/1751-7915.12141
11
DeSantisT. Z.HugenholtzP.LarsenN.RojasM.BrodieE. L.KellerK.et al (2006). Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB.Appl. Environ. Microbiol.725069–5072. 10.1128/AEM.03006-05
12
EdgarR. C. (2010). Search and clustering orders of magnitude faster than BLAST.Bioinformatics262460–2461. 10.1093/bioinformatics/btq461
13
FriedmanN.JamiE.MizrahiI. (2017a). Compositional and functional dynamics of the bovine rumen methanogenic community across different developmental stages.Environ. Microbiol.193365–3373. 10.1111/1462-2920.13846
14
FriedmanN.ShrikerE.GoldB. (2017b). Diet-induced changes of redox potential underlie compositional shifts in the rumen archaeal community.Environ. Microbiol.9174–184. 10.1111/1462-2920.13551
15
GieblerJ.WickL. Y.ChatzinotasA.HarmsH. (2013). Alkane-degrading bacteria at the soil–litter interface: comparing isolates with T-RFLP-based community profiles.FEMS Microbiol. Ecol.8645–58. 10.1111/1574-6941.12097
16
GrigorievI. V.NordbergH.ShabalovI.AertsA.CantorM.GoodsteinD.et al (2012). The genome portal of the department of energy joint genome institute.Nucleic Acids Res.40D26–D32. 10.1093/nar/gkr947
17
GuanL. L.NkrumahJ. D.BasarabJ. A.MooreS. S. (2008). Linkage of microbial ecology to phenotype: correlation of rumen microbial ecology to cattle’s feed efficiency.FEMS Microbiol. Lett.28885–91. 10.1111/j.1574-6968.2008.01343.x
18
GussA. M.RoeselersG.NewtonI. L.YoungC. R.Lepac-CerajV.LoryS.et al (2011). Phylogenetic and metabolic diversity of bacteria associated with cystic fibrosis.ISME J.520–29. 10.1038/ismej.2010.88
19
HendersonG.CoxF.GaneshS.JonkerA.YoungW.Global Rumenet al (2015). Rumen microbial community composition varies with diet and host, but a core microbiome is found across a wide geographical range.Sci. Rep.5:14567. 10.1038/srep14567
20
Hernandez-RaquetG.DurandE.BraunF.Cravo-LaureauC.GodonJ. J. (2013). Impact of microbial diversity depletion on xenobiotic degradation by sewage-activated sludge.Environ. Microbiol. Rep.5588–594. 10.1111/1758-2229.12053
21
HuwsS. A.EdwardsJ. E.CreeveyC. J.Rees StevensP.LinW.GirdwoodS. E.et al (2016). Temporal dynamics of the metabolically active rumen bacteria colonizing fresh perennial ryegrass.FEMS Microbiol. Ecol.92:fiv137. 10.1093/femsec/fiv137
22
JamiE.IsraeliA.KotserA.MizrahiI. (2013). Exploring the bovine rumen bacterial community from birth to adulthood.ISME J.71069–1079. 10.1038/ismej.2013.2
23
JamiE.MizrahiI. (2012). Composition and similarity of bovine rumen microbiota across individual animals.PLoS One7:e33306. 10.1371/journal.pone.0033306
24
JoussetA.BienholdC.ChatzinotasA.GallienL.GobetA.KurmV.et al (2017). where less may be more: how the rare biosphere pulls ecosystems strings.ISME J.11853–862. 10.1038/ismej.2016.174
25
KittelmannS.Pinares-PatiñoC. S.SeedorfH.KirkM. R.GaneshS.McEwanJ. C.et al (2014). Two different bacterial community types are linked with the low-methane emission trait in sheep.PLoS One9:e103171. 10.1371/journal.pone.0103171
26
LagkouvardosI.OvermannJ.ClavelT. (2017). Cultured microbes represent a substantial fraction of the human and mouse gut microbiota.Gut Microbes8493–503. 10.1080/19490976.2017.1320468
27
LetunicI.BorkP. (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees.Nucleic Acids Res.44W242–W245. 10.1093/nar/gkw290
28
LynchM. D.NeufeldJ. D. (2015). Ecology and exploration of the rare biosphere.Nat. Rev. Microbiol.13217–229. 10.1038/nrmicro3400
29
MallonC. A.PolyF.Le RouxX.MarringI.van ElsasJ. D.SallesJ. F. (2015). Resource pulses can alleviate the biodiversity–invasion relationship in soil microbial communities.Ecology96915–926. 10.1890/14-1001.1
30
MizrahiI. (2013). “Rumen Symbioses,” inThe Prokaryotes, edsRosenbergE.DeLongE. F., S. Lory, E. Stackebrandt, and F. Thompson (Berlin: Springer).
31
MorgaviD. P.Rathahao-ParisE.PopovaM.BoccardJ.NielsenK. F.BoudraH. (2015). Rumen microbial communities influence metabolic phenotypes in lambs.Front. Microbiol.6:1060. 10.3389/fmicb.2015.01060
32
MusatN.HalmH.WinterhollerB.HoppeP.PeduzziS.HillionF.et al (2008). A single-cell view on the ecophysiology of anaerobic phototrophic bacteria.Proc. Natl. Acad. Sci. U.S.A.10517861–17866. 10.1073/pnas.0809329105
33
NordbergH.CantorM.DusheykoS.HuaS.PoliakovA.ShabalovI.et al (2014). The genome portal of the department of energy joint genome institute: 2014 updates.Nucleic Acids Res.42D26–D31. 10.1093/nar/gkt1069
34
NuccioE. E.Anderson-FurgesonJ.EsteraK. Y.Pett-RidgeJ.De ValpineP.BrodieE. L.et al (2016). Climate and edaphic controllers influence rhizosphere community assembly for a wild annual grass.Ecology971307–1318. 10.1890/15-0882.1
35
PesterM.BittnerN.DeevongP.WagnerM.LoyA. (2010). A ‘Rare Biosphere’ microorganism contributes to sulfate reduction in a peatland.ISME J.41591–1602. 10.1038/ismej.2010.75
36
PetersonJ.GargesS.GiovanniM.McInnesP.WangL.SchlossJ. A.et al (2009). The NIH human microbiome project.Genome Res.192317–2323. 10.1101/gr.096651.109
37
PhilippotL.SporA.HénaultC.BruD.BizouardF.JonesC. M.et al (2013). Loss in microbial diversity affects nitrogen cycling in soil.ISME J.71609–1619. 10.1038/ismej.2013.34
38
SassonG.Kruger Ben-ShabatS.SeroussiE.Doron-FaigenboimA.ShterzerN.YaacobyS.et al (2017). Heritable bovine rumen bacteria are phylogenetically related and correlated with the cow’s capacity to harvest energy from its feed.mBio8:e00703-17. 10.1128/mBio.00703-17
39
SayersE. W.BarrettT.BensonD. A.BoltonE.BryantS. H.CaneseK.et al (2009). Database resources of the national center for biotechnology information.Nucleic Acids Res.37D5–D15. 10.1093/nar/gkp382
40
SeshadriR.LeahyS. C.AttwoodG. T.TehK. H.LambieS. C.CooksonA. (2018). Cultivation and sequencing of rumen microbiome members from the hungate1000 collection.Nat. Biotechnol.36359–367. 10.1038/nbt.4110
41
ShaaniY.ZehaviT.EyalS.MironJ.MizrahiI. (2018). Microbiome niche modification drives diurnal rumen community assembly, overpowering individual variability and diet effects.ISME J.10.1038/s41396-018-0203-0 [Epub ahead of print].
42
ShabatS. K. B.SassonG.Doron-FaigenboimA.DurmanT.YaacobyS.Berg MillerM. E.et al (2016). Specific microbiome-dependent mechanisms underlie the energy harvest efficiency of ruminants.ISME J.102958–2972. 10.1038/ismej.2016.62
43
SmithP. H.HungateR. E. (1958). Isolation and characterization of Methanobacterium ruminantium n. sp.J. Bacteriol.75713–718.
44
StevensonD. M.WeimerP. J. (2007). Dominance of Prevotella and low abundance of classical ruminal bacterial species in the bovine rumen revealed by relative quantification real-time PCR.Appl. Microbiol. Biotechnol.75165–174. 10.1007/s00253-006-0802-y
45
WalkerA. W.DuncanS. H.LouisP.FlintH. J. (2014). Phylogeny, culturing, and metagenomics of the human gut microbiota.Trends Microbiol.22267–274. 10.1016/j.tim.2014.03.001
46
WallaceR. J.RookeJ. A.McKainN.DuthieC. A.HyslopJ. J.RossD. W.et al (2015). The rumen microbial metagenome associated with high methane production in cattle.BMC Genomics16:839. 10.1186/s12864-015-2032-0
47
WeimerP. J.NerdahlM.BrandlD. J. (2015). Production of medium-chain volatile fatty acids by mixed ruminal microorganisms is enhanced by ethanol in co-culture with Clostridium kluyveri.Bioresour. Technol.17597–101. 10.1016/j.biortech.2014.10.054
48
Yáñez-RuizD. R.MacíasB.PinlocheE.NewboldC. J. (2010). The persistence of bacterial and methanogenic archaeal communities residing in the rumen of young lambs.FEMS Microbiol. Ecol.72272–278. 10.1111/j.1574-6941.2010.00852.x
Summary
Keywords
culturomics, gut microbial isolates, anaerobic microbiology, rumen microbiome, rumen microbiology, rare biosphere, cultivation
Citation
Zehavi T, Probst M and Mizrahi I (2018) Insights Into Culturomics of the Rumen Microbiome. Front. Microbiol. 9:1999. doi: 10.3389/fmicb.2018.01999
Received
14 May 2018
Accepted
08 August 2018
Published
29 August 2018
Volume
9 - 2018
Edited by
Zhongtang Yu, The Ohio State University, United States
Reviewed by
Graeme Trevor Attwood, AgResearch, New Zealand; Renee Maxine Petri, Veterinärmedizinische Universität Wien, Austria
Updates
Copyright
© 2018 Zehavi, Probst and Mizrahi.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Itzhak Mizrahi, imizrahi@bgu.ac.il
This article was submitted to Microbial Symbioses, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.