 Juanping Wang1,2†
Juanping Wang1,2† Chang Wang1,2†
Chang Wang1,2† Jionghui Li1,2
Jionghui Li1,2 Peng Bai1,2
Peng Bai1,2 Qi Li1
Qi Li1 Mengyuan Shen1,2
Mengyuan Shen1,2 Renhui Li1*
Renhui Li1* Tao Li1*
Tao Li1* Jindong Zhao1,3
Jindong Zhao1,3- 1State Key Laboratory of Freshwater Ecology and Biotechnology, Institute of Hydrobiology, Chinese Academy of Sciences, Wuhan, China
- 2University of Chinese Academy of Sciences, Beijing, China
- 3State Key Laboratory of Protein and Plant Genetic Engineering, College of Life Sciences, Peking University, Beijing, China
Bacteria in genus Novosphingobium associated with biodegradation of substrates are prevalent in environments such as lakes, soil, sea, wood and sediments. To better understand the characteristics linked to their wide distribution and metabolic versatility, we report the whole genome sequence of Novosphingobium sp. THN1, a microcystin-degrading strain previously isolated by Jiang et al. (2011) from cyanobacteria-blooming water samples from Lake Taihu, China. We performed a genomic comparison analysis of Novosphingobium sp. THN1 with 21 other degradative Novosphingobium strains downloaded from GenBank. Phylogenetic trees were constructed using 16S rRNA genes, core genes, protein-coding sequences, and average nucleotide identity of whole genomes. Orthologous protein analysis showed that the 22 genomes contained 674 core genes and each strain contained a high proportion of distributed genes that are shared by a subset of strains. Inspection of their genomic plasticity revealed a high number of insertion sequence elements and genomic islands that were distributed on both chromosomes and plasmids. We also compared the predicted functional profiles of the Novosphingobium protein-coding genes. The flexible genes and all protein-coding genes produced the same heatmap clusters. The COG annotations were used to generate a dendrogram correlated with the compounds degraded. Furthermore, the metabolic profiles predicted from KEGG pathways showed that the majority of genes involved in central carbon metabolism, nitrogen, phosphate, sulfate metabolism, energy metabolism and cell mobility (above 62.5%) are located on chromosomes. Whereas, a great many of genes involved in degradation pathways (21–50%) are located on plasmids. The abundance and distribution of aromatics-degradative mono- and dioxygenases varied among 22 Novosphingoibum strains. Comparative analysis of the microcystin-degrading mlr gene cluster provided evidence for horizontal acquisition of this cluster. The Novosphingobium sp. THN1 genome sequence contained all the functional genes crucial for microcystin degradation and the mlr gene cluster shared high sequence similarity (≥85%) with the sequences of other microcystin-degrading genera isolated from cyanobacteria-blooming water. Our results indicate that Novosphingobium species have high genomic and functional plasticity, rearranging their genomes according to environment variations and shaping their metabolic profiles by the substrates they are exposed to, to better adapt to their environments.
Introduction
Hazardous compounds are produced in large quantities by natural and anthropogenic activities. Their release into the environment causes public health concerns. Polycyclic and heterocyclic aromatic compounds have potential toxic, mutagenic, and carcinogenic effects on humans (Dean, 1985; Yoshikawa et al., 1985; Hu et al., 2014; Nguyen et al., 2014; Zhao et al., 2017). For example, organophosphorus insecticide inhibits acetylcholinesterase, which is crucial for the transmission of normal nerve impulses (Jariyal et al., 2018). Microcystins produced by some genera of cyanobacteria inhibit protein phosphatases 1 and 2A and promote tumor formation (Mackintosh et al., 1990; Campos and Vasconcelos, 2010), which causes severe health risks to plants, animals, and humans. Therefore, the elimination of these substances from the environment is of great importance. Microorganisms have evolved efficient degradative pathways for most of these compounds, including bacterial isolates in genera Rhodococcus, Mycobacterium, Pseudomonas, Sphingomonas, and Novosphingobium (Stolz, 2009).
Novosphingobium bacteria are widely distributed and can degrade a wide range of xenobiotic compounds (Tiirola et al., 2005; Hegedus et al., 2017; Kampfer et al., 2018; Sheu et al., 2018). They have been detected in rhizosphere soil (Krishnan et al., 2017), wood (Ohta et al., 2015), contaminated environments (Saxena et al., 2013; Hyeon et al., 2017; Singh et al., 2017), and marine and freshwater environments (Chen et al., 2017; Sheu et al., 2017; Zhang et al., 2017). Novosphingobium isolates display a wide variety of metabolic features. For example, Novosphingobium pokkalii L3E4T, which was isolated from soil of saline-tolerant Pokkali rice, promoted plant growth; Novosphingobium sp. MBES04 and Novosphingobium sp. B-7 isolated from wood degraded lignin-related compounds; and other Novosphingobium strains from water or solid supports were found to degrade a wide range of xenobiotic aromatic compounds (Tiirola et al., 2002; Hashimoto et al., 2010; Notomista et al., 2011; Choi et al., 2015). A Novosphingobium strain isolated from a water sample of Lake Taihu was classified as Novosphingobium sp. THN1 (hereafter referred to as THN1) by searching its 16S rRNA sequence against 16S rRNA sequences in GenBank using the BLAST service (Jiang et al., 2011). The isolated strain effectively degraded microcystin-LR (MC-LR), eliminating 91.2% of the MC-LR in the THN1 culture during the first 12 h and completely removing it after 60 h (Jiang et al., 2011).
Although many species in family Sphingomonadaceae, including those in genera Sphingomonas, Sphingobium, and Sphingopyxis, have been reported to be capable of degrading microcystins (MCs), to our knowledge, THN1 is the only Novosphingobium strain found so far that can degrade MCs. Typically, a cluster of four genes (mlrA, mlrB, mlrC, and mlrD) has been characterized as being responsible for MC degradation (Bourne et al., 1996, 2001). The mlrA, mlrB, and mlrC genes encode proteins involved in the degradation of MCs, whereas mlrD probably encodes a transporter protein. In the first step of the degradation pathway, microcystinase (MlrA) hydrolyzes cyclic MC-LR to form a linear intermediate. Then, MlrB hydrolyzes the linear MC-LR to form a tetrapeptide that is subsequently degraded by MlrC. Recently, two other genes, mlrE and mlrF, were identified in the genome of Sphingopyxis sp. strain C-1 and may be involved in MC degradation in addition to the four genes in the mlr cluster (Okano et al., 2015). The mlrE and mlrF genes were also found in the THN1 genome sequence reported in this study. Comparing the mlr gene cluster in different species and strains may help to elucidate how their MC-degrading capacity was acquired.
The genomes of Novosphingobium strains isolated from various habitats have been sequenced and characterized, including strains that degrade substrates they have been exposed to. The availability of the genome sequences has facilitated further analysis of the wide distribution and metabolic versatility of Novosphingobium strains. In a previous study, the genomes of six marine Novosphingobium strains were compared and genes involved in marine adaptation and cell–cell signaling were identified (Gan et al., 2013). Recently, Kumar et al. (2017) identified habitat-specific genes from 27 Novosphingobium strains from diverse habitats. Both these studies focused on habitat-specific traits. How these strains evolved their efficient degradative capabilities for the compounds they were exposed to remains to be revealed. Comparative analysis of the genomic contents and functional profiles of Novosphingobium strains may throw some light on this issue.
In this study, we sequenced the Novosphingobium sp. THN1 genome using a third-generation PacBio RSII system and assembled and annotated the genome sequence. Our aim was to identify potentially important functional genomic characteristics that may facilitate the adaptation and degradation of compounds they were exposed to in the environment.
Materials and Methods
Genome Sequencing, Assembly, and Annotation
Single-molecule real-time (SMRT) genome sequencing (Gupta, 2008; McCarthy, 2010) of the THN1 genome was performed on a Pacific Biosciences (PacBio) RSII platform (Pacific Biosciences, Menlo Park, CA, USA) by the Annoroad Gene Technology Co. Ltd. (Beijing, China). The Novosphingobium strain THN1 previously isolated by Jiang et al. (2011) from a water sample of Lake Taihu was spread onto fresh R2A plates. All plates were incubated at 37°C. After 48-h cultivation, cells were collected and used for genomic DNA extraction. Genomic DNA was extracted using an EZNA Bacterial DNA kit (Omega) according to the manufacturer's instructions. The genomic DNA was sheared using the G-tubes protocol (Covaris, Inc., Woburn, MA, USA). A 20-Kb library was constructed using a PacBio Template Prep kit and sequenced on a PacBio SMRT platform. A total of 916.83 Mb raw data comprising 70,424 reads was generated. De novo assembly was performed using the SMRT Analysis pipeline v2.3.0 in conjunction with the HGAP assembler (Chien et al., 2016). Additional assemblies were performed using minimus2 (Treangen et al., 2011). The assembly was validated by aligning the raw reads onto the finished contigs using the Burrows-Wheeler Aligner v0.7.9a (Li and Durbin, 2009). Genes in the assembled sequence were predicted using Prodigal (Hyatt et al., 2010). tRNA, rRNA, and ncRNA were identified by Infernal (Nawrocki et al., 2009) and RNAmmer (Lagesen et al., 2007). Protein-coding sequences were annotated based on BLASTP searches against the downloaded Rfam (Griffiths-Jones et al., 2005), NCBI NR, COG, KEGG (Kanehisa et al., 2008), and Swiss-Prot (Watanabe and Harayama, 2001) databases with an E-value cut-off of 1e−20 and subsequent filtering for the best hits.
Phylogenetic Analysis
For comparative analysis, we downloaded 21 Novosphingobium genome sequences and their annotations (including 5 complete genomes) from the NCBI GenBank database as shown in Table 1. These 21 strains were isolated from different types of ecological niches and can degrade a wide range of natural or xenobiotic compounds, including aromatic hydrocarbons, hexachlorocyclohexane, lignin and thiocyanate. The average chromosome length of the 22 Novosphingobium genomes (including THN1) was 5.19 Mb with a range of 3.71–6.92 Mb. Accession numbers of the genomes included in this study and their genomic features are listed in Table 1.
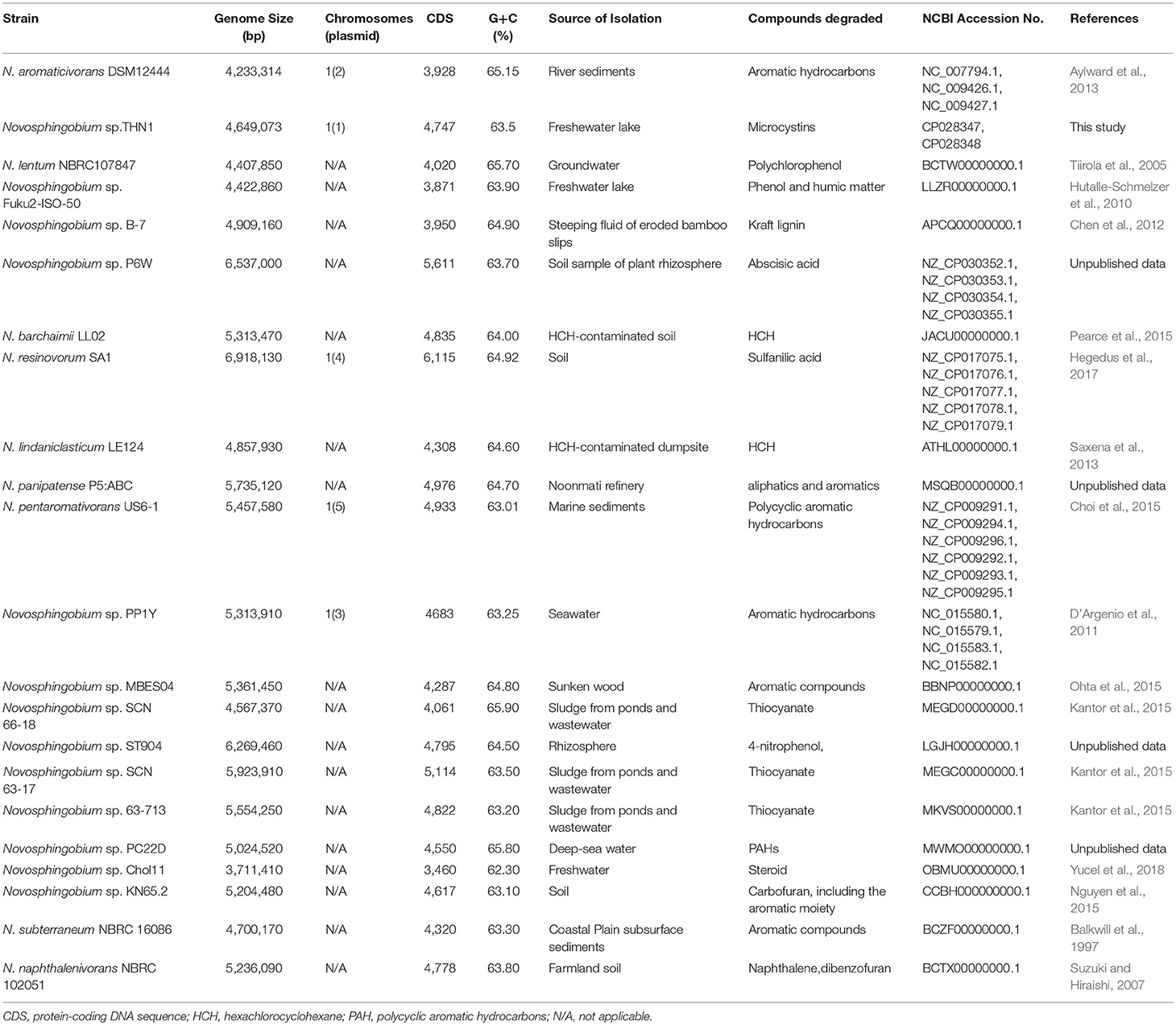
Table 1. General features of the 22 genome-sequenced degradative Novosphingobium strains used in this study.
We used two different datasets of representative markers, 16S rRNA sequences and core genes, to construct phylogenetic trees. We performed a multiple sequence alignment of the 16S rRNA sequences from the 22 genomes using Muscle (Edgar, 2004). Unaligned sequences were trimmed from the ends. We used the aligned 16S rRNA sequences to construct a phylogenetic tree using MEGA v7.0.26 (Kumar et al., 2016) with the neighbor-joining method. The robustness of clustering was evaluated by 1000 bootstrap replicates. Phylogeny based on only one common gene may lead to bias; therefore, we used the core genes of the 22 Novosphingobium strains to construct another phylogenetic tree with Sphingobium sp. YBL2 as an outgroup. The core genes were computed by OrthoMCL v2.0.9 (Li et al., 2003) and retrieved using inhouse scripts. We performed a multiple sequence alignment of the core genes from the 22 genomes using Clustal Omega (Sievers et al., 2011). The end-trimmed core sequences were joined and a phylogenetic tree was constructed using MEGA v7.0.26 with the neighbor-joining method.
We also estimated the phylogenetic relationships based on the whole genomes of 22 Novosphingobium strains. Average nucleotide identity (ANI) values were calculated using the MUMmer algorithm of JSpecies v1.2.1 (Kurtz et al., 2004; Richter and Rossello-Mora, 2009; Chan et al., 2012) and visualized as a heatmap using the Morpheus software (https://software.broadinstitute.org/morpheus/). We used CVTree (v3.9.6) (Qi et al., 2004) with a K value of 6 to compute the whole-genome composition vector of the 22 Novosphingobium strains. PHYLIP (Retief, 2000) was used to construct a neighbor-joining phylogenetic tree, which was visualized using MEGA v7.0.26 (Kumar et al., 2016).
Identification of Orthologous Proteins
Orthologous proteins were detected based on the classification of all encoded proteins in protein families, excluding transposable elements. Groups of orthologous sequences (orthogroups, hereafter referred to as protein families) in all 22 Novosphingobium strains were classified by clustering with OrthoMCL v2.0.9 (Li et al., 2003) using a Markov cluster algorithm. Protein families were constructed using the cut-off of 60% percentage identity in the alignments (Snipen and Ussery, 2010) and each protein was assigned to one protein family. The protein families were classified as core, distributed, or unique according to their distribution across the genomes. The core families comprised predicted proteins shared by all 22 strains, the distributed families comprised proteins assigned to a subset of strains, and the unique families comprised proteins assigned to a single strain. The core, distributed, and unique subgroups made up the pan-genome (i.e., the union of all the protein-coding genes present in the 22 Novosphingobium genomes; Tettelin et al., 2005). The number and percentage of each family in each strain were calculated for plotting.
Insertion Sequence Elements, Genomic Islands, and Crispr Detection
Insertion sequences (ISs) were detected by BLAST comparisons (E-value ≤ 1e−5) against the ISFinder database (Siguier et al., 2006). IslandViewer 4 (Bertelli et al., 2017), which integrates IslandPick (Langille et al., 2008), IslandPathDIMOB (Hsiao et al., 2003), and SIGI-HMM (Waack et al., 2006), were used to predict genomic islands (GIs), as described previously (Zhang et al., 2016). CRISPR arrays were detected using the CRISPRFinder (Grissa et al., 2007) online server to perform BLAST searches against dbCRISPR (CRISPR database). The identified CRISPR arrays were validated as true or false based on whether they were associated with CRISPR associated genes (Cas). Only candidate CRISPRs with Cas genes in the vicinity were designated as true CRISPRs and selected.
Functional Annotation and Metabolic Pathway Reconstruction
Functional predictions using the COG (Clusters of Orthologous Groups of proteins) database are believed to provide a fast way of describing the functional characteristics of one microbe or a community of microbes (Tatusov et al., 1997) that may be relevant to their function in the environment. The eggNOG database (evolutionary genealogy of genes: Non-supervised Orthologous Groups) contains orthologous groups of genes that were annotated with functional categories derived from COG and KOG (Eukaryotic Orthologous Groups) categories using a graph-based unsupervised clustering algorithm that extended the COG methodology (Jensen et al., 2008). To assess the level of functional diversity among the 22 Novosphingobium strains, we assigned the protein-coding genes in the 22 genomes to COG categories by searches against eggNOG v4.5.1 (Huerta-Cepas et al., 2016) using the eggNOG-mapper tool (Huerta-Cepas et al., 2017). The abundance of each COG category was normalized against the average abundance for each strain and used to generate a function heatmap of Novosphingobium strains. Core and flexible genes of each strain were extracted and annotated with COG categories as described above for the protein-coding genes.
Predicted proteins in the Novosphingobium genomes were also annotated using KAAS (KEGG Automatic Annotation Server) (Moriya et al., 2007). Metabolic pathways were deduced by manual inspection of KEGG Orthology (KO), which was predicted based on comparisons to the KEGG database (Kanehisa and Goto, 2000; Kanehisa et al., 2014).
Comparative Analysis of the Microcystin-Degrading mlr Gene Cluster
The sphingomonads that belong to family Sphingomonadaceae have been used in bioremediation because of their ability to degrade natural and anthropogenic compounds (Stolz, 2009), an ability that has allowed them to adjust well to contaminated environments. Sphingomonads belong to four genera, Sphingomonas (sensu strictu), Sphingobium, Novosphingobium, and Sphingopyxis (Takeuchi et al., 2001). A large number of species that can degrade MCs have been isolated in genera Sphingomonas, Sphingobium, and Sphingopyxis, whereas THN1 is the only known MC-degrading strain in genus Novosphingobium and the only one found so far to harbor the MC-degrading mlr gene cluster. We suspected that the mlr gene cluster in THN1 was horizontally transferred from other MC-degrading species. To test this idea, we retrieved mlr gene cluster sequences of species in Sphingomonads from GenBank and compared the mlr genes of each pair of microcystin-degrading strains by BLAST alignments. We analyzed the codon usage differences of mlr genes and core gens to further clarify this idea. Within the core genes computed in section Identification of orthologous proteins, we selected five genes that are involved in the same COG category with mlr gene cluster to analyze codon usage differences. Codon usage of core genes and mlr genes was computed and analyzed by graphical codon usage analyser platform (http://gcua.schoedl.de).
Results and Discussion
Genome Sequencing, Assembly, and Annotation
The THN1 genome was assembled into two contigs: a 3,487,514 bp chromosome with 63.6% GC content and a 1,161,559 bp plasmid with 63.2% GC content (Figure S1). We detected 4747 protein-coding sequences in the assembled sequences, as well as 9 rRNAs, 58 tRNAs, and 9 miscRNAs. Functional analysis based on the COG categories assigned a large number of the protein-coding genes to energy production and conservation (9.39%), amino acid transport and metabolism (9.28%), general function prediction (8.69%), replication, recombination and repair (7.35%), inorganic ion transport and metabolism (6.09%), translation, ribosomal structure and biogenesis and replication (5.94%), and lipid transport and metabolism (5.86%). A number of genes were assigned to the unknown function category (8.69%). Further, the mlr gene cluster responsible for MCs degradation was identified in the THN1 genome.
Phylogenetic Analysis of Novosphingobium Strains and Niche Adaptation
We collected 22 Novosphingobium genomes that can degrade substrates. Phylogenetic trees were constructed using 16S rRNAs, core genes, whole-genome composition vectors (CVs), and ANIs (Figure 1). In all four phylogenetic trees, THN1 grouped with N. subterraneum NBRC 16086, indicating that THN1 may belong to the species N. subterraneum. The topologies of the four phylogenetic trees exhibited some differences. In the phylogenetic tree based on the 16S rRNA sequences, Novosphingobium sp. Fuku2-ISO-50 was separated as an individual branch, whereas, in the whole-genome-based CV trees and core gene trees, Novosphingobium sp. Chol11 was separated as an individual branch. To confirm the findings from the phylogenetic analysis, we constructed a heatmap based on ANI values. The pairwise ANI values ranged from 82.95% (between THN1 and Novosphingobium sp. Chol11) to 95.8% (between Novosphingobium sp. SCN63-17 and Novosphingobium sp. 63-713). In the phylogenetic trees based on ANIs, Novosphingobium sp. 63-713 and Novosphingobium sp. SCN63-17 grouped together in a separate clade.
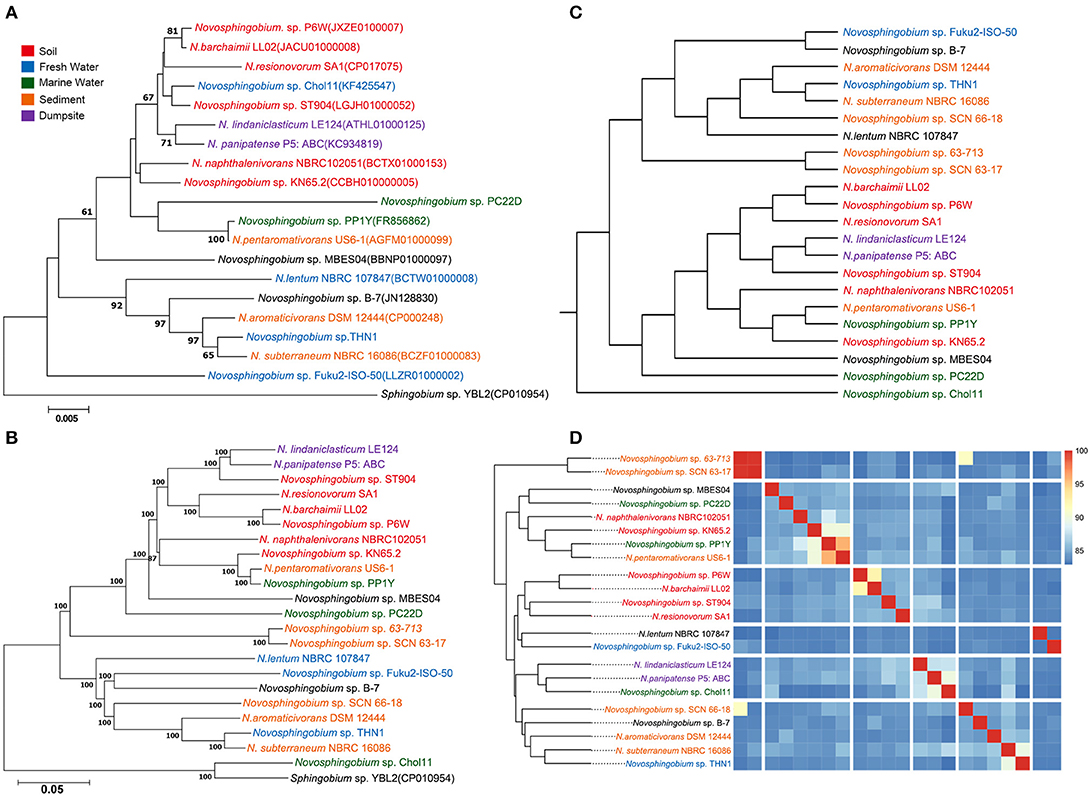
Figure 1. Phylogenetic relationships of 22 Novosphingobium strains. Phylogenetic trees based on (A) 16S rRNA gene sequences and (B) 674 core genes with 1000 bootstraps with Sphingobium sp. YBL2 as an outgroup. The bars represent the number of substitutions per nucleotide position. Percentage bootstrap values (≥50%) are shown next to the nodes. (C) Whole-genome-based phylogeny trees using a composition vector (CV) approach. (D) Average nucleotide identity (ANI)-based phylogenetic dendrograms showing hierarchical clustering of species.
The differences between the whole-genome-based CV and core gene phylogenetic trees revealed that the flexible genes may be important in changing the genome content and shaping the topology of the trees. Several strains in phylogenetic trees clustered together according to their niche specificity. Novosphingobium sp. P6W, N. barchaimii LL02, and N. resinovorum SA1, which were isolated from soil, formed a single clade; N. lindaniclasticum LE124, which was isolated from a contaminated dumpsite, clustered with N. panipatense P5:ABC from a refinery; and sludge-isolated Novosphingobium sp. 63-713 and Novosphingobium sp. SCN63-17 clustered together. This result is consistent with the results of an earlier study that found six Novosphingobium strains were correlated with their isolation sources (Gan et al., 2013), and in a CV tree of Acidithiobacillus caldus, three strains from a copper mine clustered together (Zhang et al., 2016). However, most of the subclades in the phylogenetic trees had more ambiguity in habitat specificity. For example, lake-sourced Novosphingobium sp. Fuku2-ISO-50 clustered with Novosphingobium sp. B-7 from bamboo slips; and THN1, which was isolated from a lake, clustered with N. aromaticivorans DSM12444 from sediment. This result is consistent with previous comparisons of Novosphingobium strains from different habitats (Kumar et al., 2017). Further analysis showed that when more strains were included in phylogenetic trees, the trend to cluster according to habitat was more mixed. This suggests that specific environment variations also may influence genome structure (Lin et al., 2013; Ji et al., 2014; Zhang et al., 2016).
Conserved and Variable Gene Repertoire of Novosphingobium Genomes
Pan-genome information indicates the entire genetic potential of a group (Tettelin et al., 2005), where core genes denote conserved functions and flexible genes (or non-core genes) are either unique to an individual genome (unique genes) or shared by a subset of genomes (distributed genes). We carried out pan-genomic studies to investigate the genomic variations among the 22 Novosphingobium genomes. Comparative analyses based on groups of orthologous proteins revealed 674 core genes that were shared by the 22 Novosphingobium genomes, and over 96% of them were distributed in the chromosome with the remainder in the plasmids (Table 2). The percentages of core genes in each genome varied from 12.89 to 22.49%, which revealed that the core genes made up a small proportion of the total genes in each genome (Figure 2). Therefore, the 22 Novosphingobium strains shared a low percentage of common functional proteins. In a previous study, a comparison of six Novosphingobium strains revealed a higher number of core orthologous groups (929) (Gan et al., 2013), whereas, in a pan-genomic analysis of 26 sphingomonad genomes, 268 core genes were detected (Aylward et al., 2013). A comparative genomic analysis of 27 habitat-specific Novosphingobium strains showed all 27 strains contained 220 core genes, whereas the genomes of Novosphingobium strains from specific habitat shared a higher number of core genes (Kumar et al., 2017). Further analysis indicated that the core genome was asymptotic, and more Novosphingobium genomes will result in minor changes in the core genome of Novosphingobium (Kumar et al., 2017).
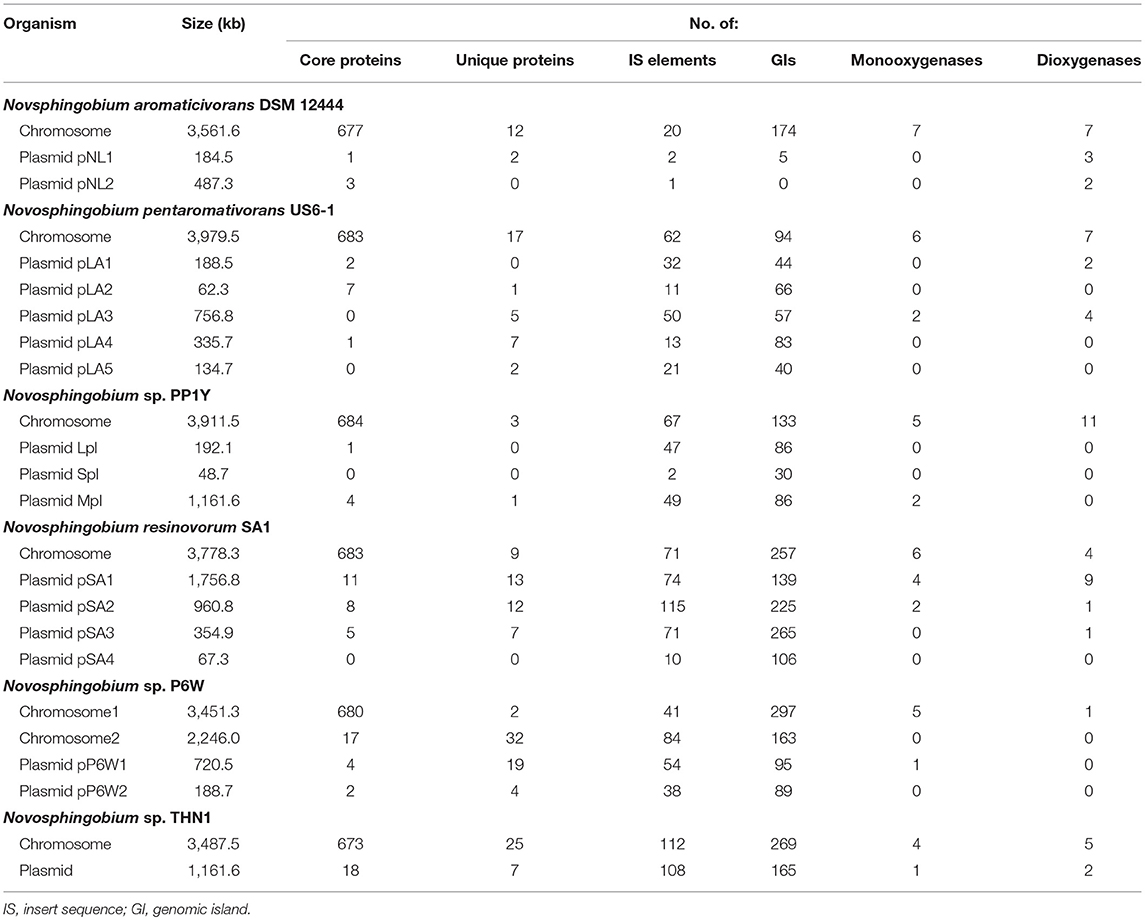
Table 2. Size and number of genetic elements and biodegradative enzymes in the six complete Novosphingobium genomes.
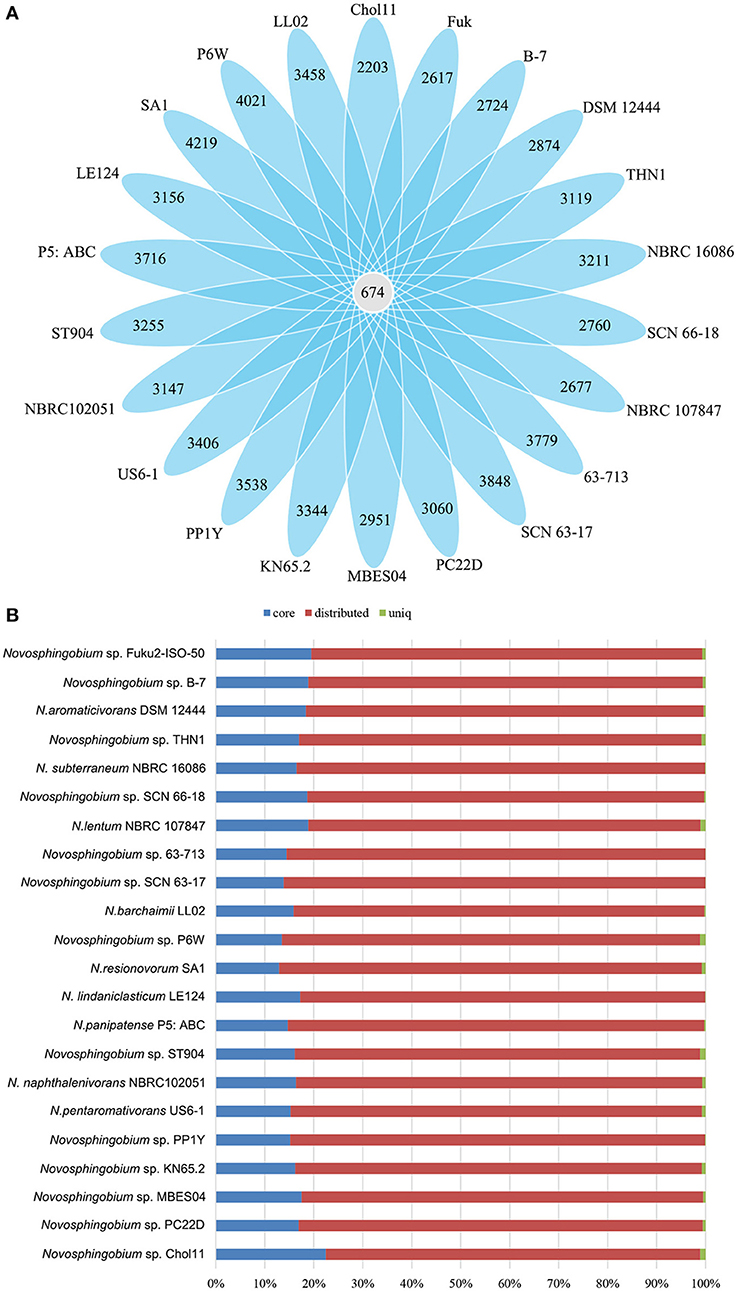
Figure 2. Comparisons of Novosphingobium orthologous protein groups in 22 Novosphingobium genomes. (A) Venn diagram displaying the numbers of core gene families and flexible genes for each of the 22 Novosphingobium strains. (B) Percentage of core, distributed, and unique genes in each of the 22 genomes.
The percentages of unique genes (singletons) in each genome ranged from 0.04 to 1.18% and they were distributed on both the chromosomes (21.95–85.71%) and the plasmids (14.29–78.05%) in six complete genomes (Table 2). The percentages of unique genes on the plasmids were 78.05% for N. resinovorum SA1, 40.35% for Novosphingobium sp. P6W, 46.88% for N. pentaromativorans US6-1, 25% for Novosphingobium sp. PP1Y, 21.88% for THN1, and 14.29% for N. aromaticivorans DSM12444. The percentage of distributed genes in the each genome varied from 76.43 to 86.36%. Similarly, the alignments of 27 Streptococcus agalactiae genomes revealed a great number of distributed genes (72.15%) (Wolf et al., 2018). However, the comparative analyses revealed a relative low proportion of distributed genes in 24 Shewanella strains (42.7%), five drug-resistance Aeromonas hydrophila strains (14.02–32.75%), 23 Pasteurella multocida strains (33.47%) and Microcystis aeruginosa strains (38–48%) (Humbert et al., 2013; Hurtado et al., 2018; Zhang et al., 2018; Zhong et al., 2018). The high proportion of flexible genes indicated there was extensive genomic variation among the Novosphingobium genomes, which may reflect their variable metabolic profiles. The high percentage of flexible genes and low percentage of unique genes derived from the pan-genome analysis led us to look at their functional classification (section Functional annotation of orthologous groups and comparisons among the Novosphingobium strains) for clues into their metabolic diversification.
Mobile Gene Elements and Crisprs in the Novosphingobium Genomes
Analysis of the transposable elements predicted by ISFinder indicated that a large number of insertion sequence (IS) elements from various families were distributed over the genomes of the 22 Novosphingobium strains (Table S1), indicating these strains had high genomic plasticity to adapt to the various environments. High numbers of IS elements (834, 557, and 487) were identified in the genomes of N. lindaniclasticum LE124, N. naphthalenivorans NBRC 102051, and Novosphingobium sp. ST904, respectively. These elements were distributed on both chromosomes and plasmids and the percentages varied in the six complete genomes (Table 2). A large number of prophage and transposons have been identified previously in the sphingomonads (Aylward et al., 2013). Transposable elements could allow gene exchange between Novosphingobium species and other organisms, thereby facilitating their adaption to different habitats.
Besides the ISs, we identified GIs in the genomes of the 22 Novosphingobium strains. A large number of these GIs were widespread in the genomes (Table S2), which also suggested high plasticity. Many of the protein-coding sequences in the GIs were annotated as hypothetical proteins. More GIs were identified on chromosomes than on the plasmids in the six complete genomes (Table 2). Previous studies have shown that GIs were relevant to niche-specific adaptation (Wu et al., 2011; Zhang et al., 2016), so the prevalence of GIs in the 22 Novosphingobium genomes may imply rapid adaptation occurred to give them a survival advantage in diverse environments.
Surprisingly, our analysis of CRISPRs revealed only a few CRISPR loci in the Novosphingobium genomes. One CRISPR array with four spacers was detected in N. resinovorum SA1, one with five spacers was detected in Novosphingobium sp. B-7, and one with 40 spacers was detected in Novosphingobium sp. PC22D, and all three were identified as true CRISPRs. The direct repeat lengths in N. resinovorum SA1, Novosphingobium sp. B-7, and Novosphingobium sp. PC22D were 23, 29, and 29 bp, respectively, and the spacer lengths ranged from 28 bp to 58 bp. The presence of Cas3 in the vicinity of these three CRISPR arrays indicated that they are type I CRISPR systems. CRISPR is a defense mechanism against bacteriophages and the number of spacers within CRISPR arrays is an indicator of the frequency of viral invasions. Therefore, the low number of spacers in the N. resinovorum SA1 and Novosphingobium sp. B-7 indicated a low frequency of viral attacks, and the low number of CRISPR arrays reflected vulnerability of the Novosphingobium viral defense system.
Functional Annotation of Orthologous Groups and Comparisons Among the Novosphingobium Strains
COG annotations were assigned to the protein-coding and flexible genes by searches against the eggNOG database. As expected, the annotations of the protein-coding and flexible genes shared the same COG dendrogram. The abundance of annotated COG categories differed among the 22 strains and the difference between two of the clusters in the functional heatmap was significant (P < 0.05) (Figure 3). Functional analysis of unique proteins revealed that many COG categories were present in specific species and their abundances were diverse (Figure S2).
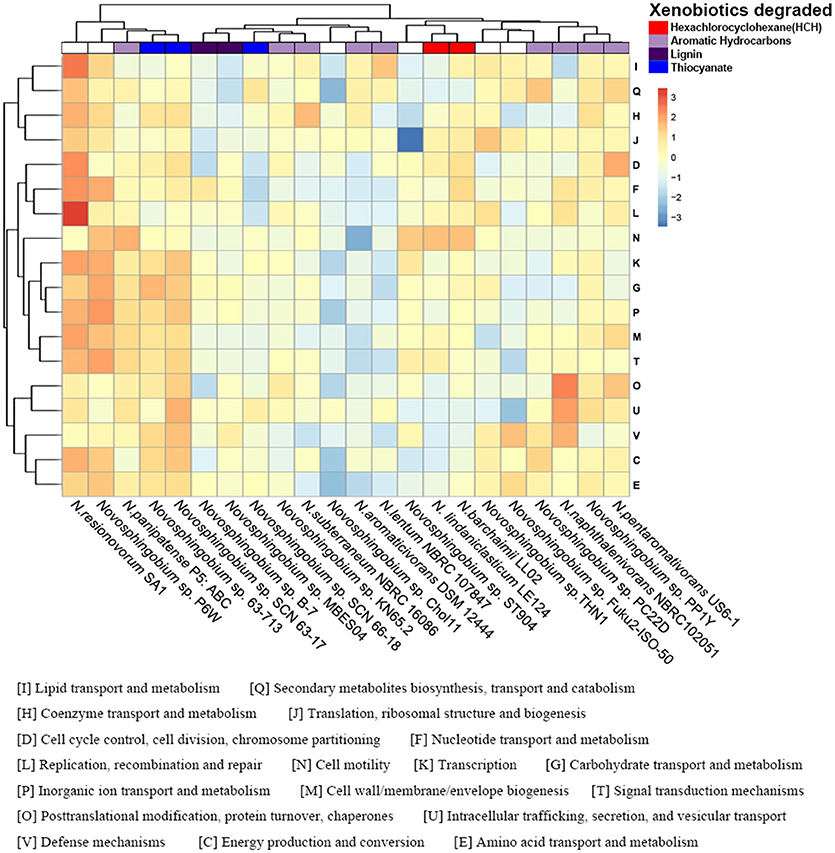
Figure 3. Functional profiling of the Novosphingobium genomes. Heatmap showing the normalized relative abundance of the clusters of orthologous groups (COG) categories enriched in the protein-coding genes in the 22 Novosphingobium genomes. The strains and COG categories were clustered using the Euclidean distance. The color scale represents the relative abundance of gene content for each category, normalized by sample mean.
The phylogenetic trees of several selected Novosphingobium strains were concordant with their habitat specificity, leading us to consider whether the COG functional profiles reflected the habitats or the degradation attributes of the 22 strains. Unlike the phylogenetic clusters, the COG clusters correlated better with the substrate that each strain could degrade than with their environment (Figure 3). This finding suggested a possible link between the functional profiles and substrate-specific traits. For example, N. barchaimii LL02, Novosphingobium sp. P6W, and N. resinovorum SA1 from soil clustered together in the phylogenetic trees but, in the functional heatmap, N. barchaimii LL02, which was isolated from a site contaminated with hexachlorocyclohexane (HCH), clustered with N. lindaniclasticum LE124, another HCH-degrading strain, and not with Novosphingobium sp. P6W and N. resinovorum SA1. Consistently, N. naphthalenivorans NBRC 102051, Novosphingobium sp. PP1Y, and N. pentaromativorans US6-1, all with ability to degrade a wide range of aromatic hydrocarbons, clustered into one subgroup, and Novosphingobium sp. KN65.2 and N. subterraneum NBRC 16086, both of which also degrade aromatic compounds, clustered together. N. aromaticivorans DSM12444, which can degrade aromatic compounds, clustered with N. lentum NBRC107847, which can degrade chloraromatic compounds. Novosphingobium sp. MBES04 and Novosphingobium sp. B-7, both of which can degrade lignin, formed one subclade. Thiocyanate-degrading Novosphingobium sp. SCN63-17 and Novosphingobium sp. 63-713 clustered together. MC-degrading THN1 clustered with Novosphingobium sp. Fuk2-ISO-50, which can degrade phenol and humic matter consisting of acetonitrile and H3PO4. THN1 was isolated from Lake Taihu, China where cyanobacterial bloom breaks out frequently, leading to high concentrations of MCs produced by cyanobacteria in the water (Shen et al., 2003; Duan et al., 2009). To protect itself from damage caused by MC exposure, THN1 has developed the capability of degrading MCs. Similarly, other Novosphingobium strains may have rearranged their metabolic profiles to better adapt to specific habitats and to utilize the compounds to which they were exposed.
Comparison of Central Metabolism and Degradation Pathways Among the Novosphingobium Strains
We assigned metabolic profiles predicted from KEGG pathways to the genes to identify shared metabolic features as well as specific metabolic traits among the 22 Novosphingobium strains. In all 22 strains, the genes were assigned to central carbon, nitrogen, energy, cell mobility, and major degradation metabolism pathways (Figure 4, Table S3). All the strains were found to harbor a core set of genes involved in carbon metabolism. They all had a complete glycolysis pathway, pentose phosphate pathway, and tricarboxylic acid cycle, but an incomplete Calvin-Benson-Bassham cycle, lacking genes that encode phosphoribulokinase (prbK) and ribulose-bisphosphate carboxylase (rbcL/rbcS). For nitrogen metabolism, all 22 strains were capable of ammonia uptake via a core set of genes encoding the Amt family of ammonium transporters and transfer ammonia to glutamate using a glutamine synthetase. All strains had genes that could encode assimilatory nitrite reductase (nirB/nirD), which reduces nitrite to ammonia, as well as a nitronate monooxygenase encoding gene (Ncd2) that converts nitroalkane to nitrite. Furthermore, all 22 strains shared a similar core set of genes that encoded the proteins for uptake and transfer of phosphate and sulfur. The majority of genes involved in central carbon metabolism, nitrogen, phosphate, sulfate metabolism, energy metabolism and cell mobility (above 62.5%) are located on chromosomes. Whereas, a great many of genes involved in degradation pathways (21–50%) are located on plasmids (Table 3). More studies shoud be performed on the intracellular distribution of the genes and their possible exchange/uptake in future.
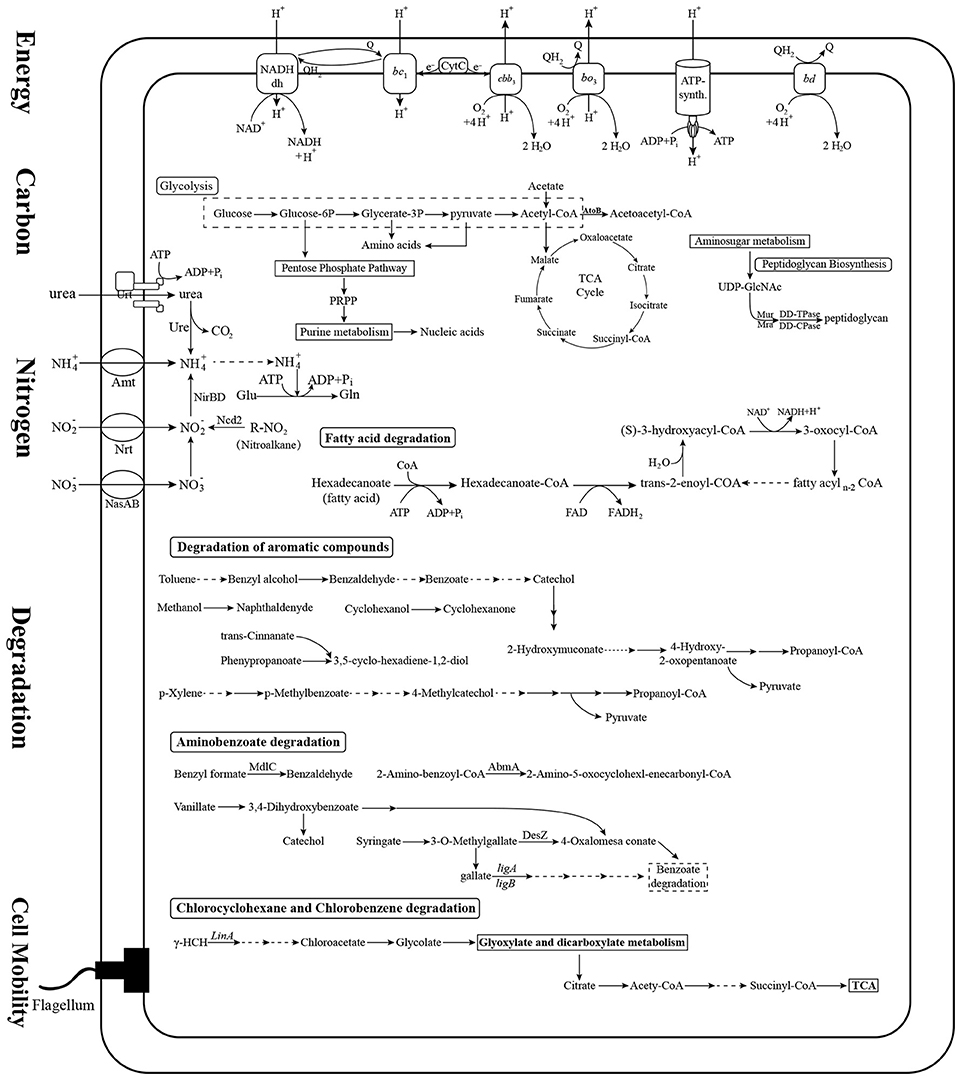
Figure 4. Prediction of the central metabolic potential of 22 Novosphingobium strains. Potential metabolic traits associated with carbon, nitrogen, energy metabolism, and degradation pathways were analyzed. The genes predicted to be involved in these metabolic pathways are listed in Table S3.
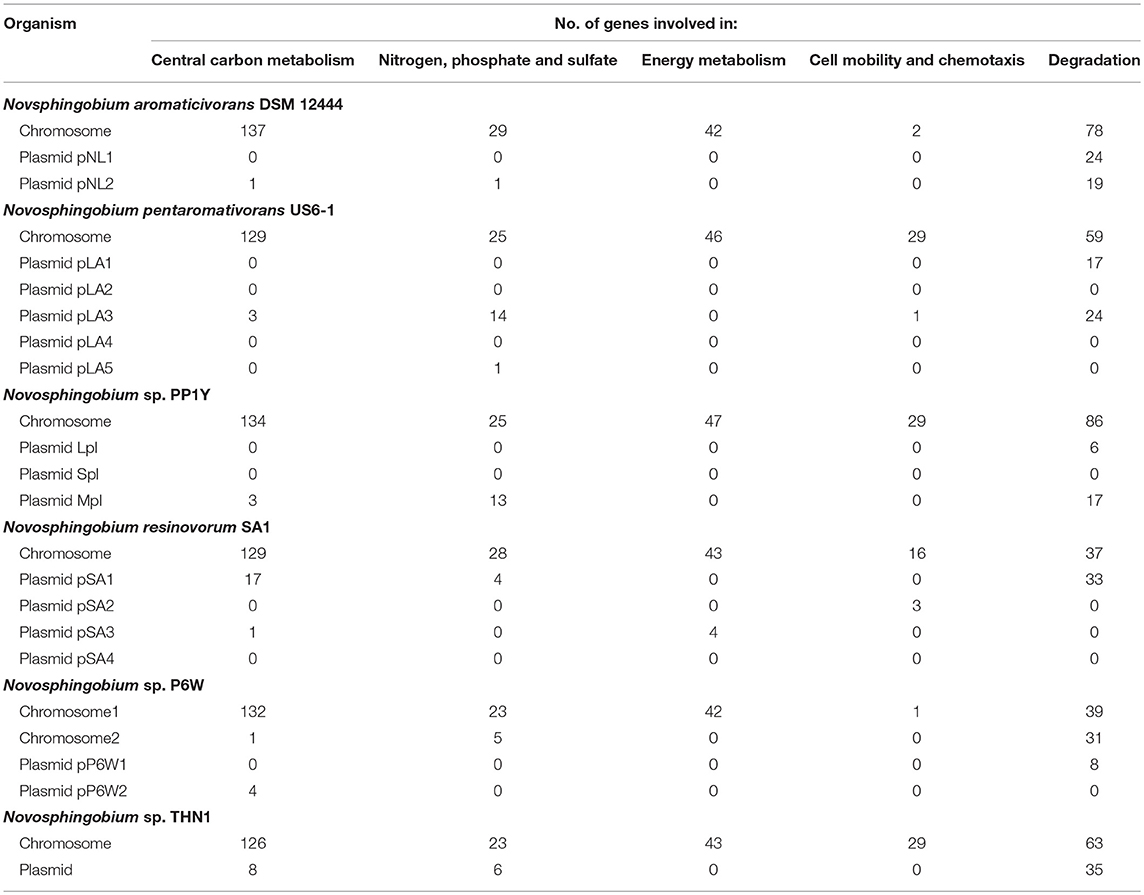
Table 3. Number of genetic elements involved in metabolic pathways in the six complete Novosphingobium genomes.
Variations in the degradation pathways among these strains were a focus of attention in this study. The 22 Novosphingobium strains shared most degradative pathways, including degradation of aromatic compounds, benzoate degradation, and glyoxylate and dicarboxylate metabolism. However, they varied in specific degradative enzymes relevant to the substrate they degrade. The 22 Novosphingobium strains contained a large number of genes involved in the degradation of aromatic compounds. We analyzed the encoded mono- and dioxygenases enzymes that catalyze the ring cleavage step critical to aromatic compound degradation, as described previously (Harayama et al., 1992; Aylward et al., 2013). The distribution of genes encoding mono- and dioxygenases varied among the strains (Table S4). For example, N. resinovorum SA1 and N. panipatense P5:ABC encoded the largest number of monooxygenases and N. pentaromativorans US6-1 and N. resinovorum SA1 encoded the largest number of dioxygenases. The most abundant monooxygenase family identified was nitronate monooxygenase (NMO, K00459) and the most abundant dioxygenase families included taurine dioxygenase (TauD, K03119) and 4-hydroxyphenylpyruvate dioxygenase (HppD, K00457) (Table S4). Most of the identified genes encoding mono- and dioxygenases were located in the chromosome of N. aromaticivorans DSM12444, Novosphingobium sp. P6W, Novosphingobium sp. PP1Y, N. pentaromativorans US6-1, and THN1. However, in N. resinovorum SA1, more of the dioxygenase-encoding genes were distributed in plasmid pSA1 rather than the chromosome (Table 2). NMO preferentially acts on nitroalkanes, so its presence in all the genomes suggests that all of the strains studied could be involved in the utilization of nitroalkane. Our central metabolic profile also revealed that the 22 Novosphingobium strains could metabolize nitroalkane to nitrite in nitrogen metabolic pathways.
The degradation pathway mediated by the lin genes has been associated with HCH degradation (Pearce et al., 2015). The initial steps in the degradation of HCH involve dehydrochlorination mediated by LinA or hydrolytic dechlorinations mediated by LinB, followed by LinC-catalyzed dehydrogenation (Nagata et al., 1993, 1994; Trantirek et al., 2001). The downstream pathway involves reductive dechlorination by the glutathione S-transferase LinD, followed by ring cleavage and conversion by LinE and LinF, respectively (Miyauchi et al., 1998, 1999). However, a growing number of newly sequenced strains have been found to have missing key lin components of the pathway (Dogra et al., 2004; Kaur et al., 2013; Kohli et al., 2013; Kumar Singh et al., 2013; Mukherjee et al., 2013). In our analysis of the HCH degradation pathway, linA was present only in N. barchaimii LL02 but absent in another HCH-degrading strain N. lindaniclasticum LE124. Because N. lindaniclasticum LE124 was confirmed to be able to degrade HCH (Saxena et al., 2013), the initial degradation-related gene remains to be characterized in this strain. linB was present in N. resinovorum SA1, Novosphingobium sp. SCN 66-18, and Novosphingobium sp. Chol11, suggesting that these strains could degrade HCH. linC was identified in N. lindaniclasticum LE124, N. panipatense P5:ABC, N. pentaromativorans US6-1, and Novosphingobium sp. ST904. linD–F were absent in the genomes of the 22 strains. Previous genomic comparisons of Sphingomonadaceae strains also found considerable variations in the presence of lin genes (linA–F) among HCH-degrading bacteria and it was speculated that the absence of some genes may reflect early stages in the acquisition of the HCH-degrading pathway (Pearce et al., 2015).
Overall, the metabolic analysis revealed that the 22 strains shared genes involved in central carbon, nitrogen, energy metabolism, and cell mobility, but varied in specific degradation enzymes and pathways, which may be related to the substrates they were exposed to.
Comparative Analysis of the Mc-Degrading mlr Gene Cluster in THN1 and Other Strains in Family Sphingomonadaceae
THN1 was isolated from a water sample of Lake Taihu with high concentrations of MCs (Jiang et al., 2011). The THN1 genome contained all the functional genes for MC degradation (section Genome sequencing, assembly, and annotation). We compared the mlr cluster in THN1 with that in other MC-degrading strains from other genera to investigate the acquisition of mlr genes and the capacity of THN1 to degrade MCs. The mlr cluster was largely syntenic across all the strains tested with the same organization and with gene sequence identity ≥85% (Figure 5). Among these strains, the whole genome of Sphingopyxis sp. C-1 was sequenced and thus the mlr genes in the genome showed the highest coverage with the mlr genes of THN1. Whereas the mlr genes in Sphingomonas sp. ACM3962, Sphingomonas sp. NV3, Sphingopyxis sp. USTB-05, Sphingopyxis sp. MB-E, and Sphingopyxis sp. IM-2 were obtained by PCR amplifications and were incomplete and thus showed low coverage with the mlr genes of THN1. MC-degradation activity is prevalent in cyanobacteria blooming environments where the concentration of MCs is high. All the strains with a characterized mlr gene cluster in Figure 5, including THN1, were isolated from lakes or reservoirs with cyanobacterial blooms and high concentrations of MCs (Jones et al., 1994; Okano et al., 2009; Wang et al., 2010; Somdee et al., 2013; Lezcano et al., 2016; Maghsoudi et al., 2016). So far, no other Novosphingobium isolates have been found to possess mlr genes and the capability of degrading MCs; therefore, the mlr gene cluster in THN1 may have been obtained from other bacterial genera in the same niche by horizontal gene transfer. The codon usage differences between core genes and mlr genes of THN1 may explain this inference (Figure S3). The acquisition of the MC-degradation ability guaranteed the survival of this Novosphingobium strain. Novosphingobium species can rearrange their genomes and functional profiles to adapt to local environments, which may explain their high survival rates and distribution diversity.
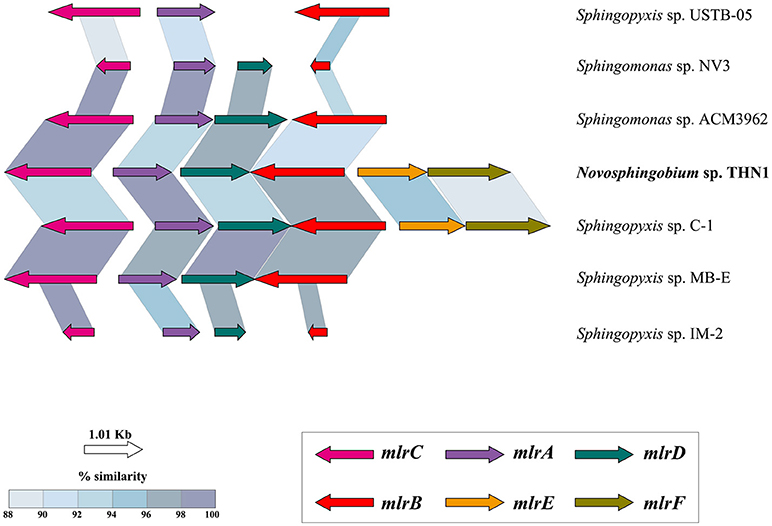
Figure 5. Structure and similarity of the mlr gene cluster in THN1 and other species in family Sphingomonadaceae. TBLASTX was used to identify the mlr sequences and to determine similarities and alignment lengths. mlrA, mlrB, and mlrC encode proteins involved in the degradation of microcystins, and mlrD encodes a transporter protein. The functions of mlrE and mlrF are unknown.
Conclusions
Orthologous protein analysis revealed the presence of a large number of flexible genes and mobile gene elements in 22 Novosphingobium strains, suggesting these strains had high plasticity of genomic contents. Analysis of the COG functional profiles showed that the COG clusters correlated with the substrate that each strain could degrade. Further, the metabolic profiles predicted from the KEGG pathways analysis showed that although all 22 strains shared genes involved in central carbon, nitrogen, phosphate, sulfate, energy metabolism, and cell mobility pathways, specific degradative enzymes and reactions varied. Thus, we propose that environment variation shaped the general genome structure of these Novosphingobium strains as a result of the compounds they were exposed to and redirected the degradation profile to facilitate beneficial substrate utilization. For example, to degrade the high concentration of MCs in Lake Taihu, THN1 may have acquired a mlr gene cluster from other genera, which was integrated into the genome. These genome-guided findings, to some extent, enhance current knowledge of the genomic and metabolic diversity of Novosphingobium species.
Data Availability
The raw PacBio sequence of the Novosphingobium sp. THN1 genome has been deposited in the Sequence Read Archive of the National Center for Biotechnology Information under accession number SRP143911. The complete chromosome and plasmid genome sequences have been deposited in DDBJ/ENA/GenBank under accession numbers CP028347 and CP028348, respectively. Source codes, example softwares and genomic data are available at https://github.com/shenmengyuan/Novo_comparison.
Author Contributions
JW carried out the data analysis and prepared the manuscript draft. TL, RL, and JZ helped design the project and revised the manuscript. CW and JL assisted in formatting figures. PB and MS helped revised the manuscript. QL assisted in data analysis. All authors discussed the manuscript draft and agreed to the final content.
Funding
This research was supported by the State Key Laboratory of Freshwater Ecology and Biotechnology (Grant No. 2016FBZ08), the Chinese Academy of Sciences (Grant No. QYZDY-SSW-SMC004), the Key Research Program of the Chinese Academy of Sciences (Grant No. KFZD-SW-219), and the Science and Technology Basic Resources Investigation Program of China (Grant No. 2017FY100300).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the Laboratory of Biology of Harmful Algae, Institute of Hydrobiology, Chinese Academy of Sciences for providing the Novosphingobium sp. THN1 sample. We thank Margaret Biswas, PhD, from Edanz Group (www.edanzediting.com/ac) for editing a draft of this manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02238/full#supplementary-material
References
Aylward, F. O., McDonald, B. R., Adams, S. M., Valenzuela, A., Schmidt, R. A., Goodwin, L. A., et al. (2013). Comparison of 26 Sphingomonad genomes reveals diverse environmental adaptations and biodegradative capabilities. Appl. Environ. Microbiol. 79, 3724–3733. doi: 10.1128/AEM.00518-13
Balkwill, D. L., Drake, G. R., Reeves, R. H., Fredrickson, J. K., White, D. C., Ringelberg, D. B., et al. (1997). Taxonomic study of aromatic-degrading bacteria from deep-terrestrial-subsurface sediments and description of Sphingomonas aromaticivorans sp nov, Sphingomonas subterranea sp nov, and Sphingomonas stygia sp nov. Int. J. Syst. Bacteriol. 47, 191–201. doi: 10.1099/00207713-47-1-191
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bourne, D. G., Jones, G. J., Blakeley, R. L., Jones, A., Negri, A. P., and Riddles, P. (1996). Enzymatic pathway for the bacterial degradation of the cyanobacterial cyclic peptide toxin microcystin LR. Appl. Environ. Microbiol. 62, 4086–4094.
Bourne, D. G., Riddles, P., Jones, G. J., Smith, W., and Blakeley, R. L. (2001). Characterisation of a gene cluster involved in bacterial degradation of the cyanobacterial toxin microcystin LR. Environ. Toxicol. 16, 523–534. doi: 10.1002/tox.10013
Campos, A., and Vasconcelos, V. (2010). Molecular mechanisms of microcystin toxicity in animal cells. Int. J. Mol. Sci. 11, 268–287. doi: 10.3390/ijms11010268
Chan, J. Z., Halachev, M. R., Loman, N. J., Constantinidou, C., and Pallen, M. J. (2012). Defining bacterial species in the genomic era: insights from the genus Acinetobacter. BMC Microbiol. 12:302. doi: 10.1186/1471-2180-12-302
Chen, W. M., Huang, C. W., Chen, J. C., Chen, Z. H., and Sheu, S. Y. (2017). Novosphingobium ipomoeae sp nov., isolated from a water convolvulus field. Int. J. Syst. Evol. Microbiol. 67, 3590–3596. doi: 10.1099/ijsem.0.002174
Chen, Y. H., Chai, L. Y., Tang, C. J., Yang, Z. H., Zheng, Y., Shi, Y., et al. (2012). Kraft lignin biodegradation by Novosphingobium sp B-7 and analysis of the degradation process. Bioresour. Technol. 123, 682–685. doi: 10.1016/j.biortech.2012.07.028
Chien, J. T., Pakala, S. B., Geraldo, J. A., Lapp, S. A., Humphrey, J. C., Barnwell, J. W., et al. (2016). High-quality genome assembly and annotation for Plasmodium coatneyi, generated using single-molecule real-time PacBio technology. Genome Announc. 4:e00883–16. doi: 10.1128/genomeA.00883-16
Choi, D. H., Kwon, Y. M., Kwon, K. K., and Kim, S. J. (2015). Complete genome sequence of Novosphingobium pentaromativorans US6-1(T). Stand. Genomic Sci. 10:107. doi: 10.1186/s40793-015-0102-1
D'Argenio, V., Petrillo, M., Cantiello, P., Naso, B., Cozzuto, L., Notomista, E., et al. (2011). De Novo sequencing and assembly of the whole genome of Novosphingobium sp strain PP1Y. J. Bacteriol. 193, 4296–4296. doi: 10.1128/JB.05349-11
Dean, B. J. (1985). Recent findings on the genetic toxicology of benzene, toluene, xylenes and phenols. Mutat. Res. 154, 153–181. doi: 10.1016/0165-1110(85)90016-8
Dogra, C., Raina, V., Pal, R., Suar, M., Lal, S., Gartemann, K. H., et al. (2004). Organization of lin genes and IS6100 among different strains of hexachlorocyclohexane-degrading Sphingomonas paucimobilis: evidence for horizontal gene transfer. J. Bacteriol. 186, 2225–2235. doi: 10.1128/JB.186.8.2225-2235.2004
Duan, H., Ma, R., Xu, X., Kong, F., Zhang, S., Kong, W., et al. (2009). Two-decade reconstruction of algal blooms in China's Lake Taihu. Environ. Sci. Technol. 43, 3522–3528. doi: 10.1021/es8031852
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Gan, H. M., Hudson, A. O., Rahman, A. Y. A., Chan, K. G., and Savka, M. A. (2013). Comparative genomic analysis of six bacteria belonging to the genus Novosphingobium: insights into marine adaptation, cell-cell signaling and bioremediation. BMC Genomics 14:431. doi: 10.1186/1471-2164-14-431
Griffiths-Jones, S., Moxon, S., Marshall, M., Khanna, A., Eddy, S. R., and Bateman, A. (2005). Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 33, D121–D124. doi: 10.1093/nar/gki081
Grissa, I., Vergnaud, G., and Pourcel, C. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 35, W52–W57. doi: 10.1093/nar/gkm360
Gupta, P. K. (2008). Single-molecule DNA sequencing technologies for future genomics research. Trends Biotechnol. 26, 602–611. doi: 10.1016/j.tibtech.2008.07.003
Harayama, S., Kok, M., and Neidle, E. L. (1992). Functional and evolutionary relationships among diverse oxygenases. Annu. Rev. Microbiol. 46, 565–601. doi: 10.1146/annurev.mi.46.100192.003025
Hashimoto, T., Onda, K., Morita, T., Luxmy, B. S., Tada, K., Miya, A., et al. (2010). Contribution of the estrogen-degrading bacterium Novosphingobium sp strain JEM-1 to estrogen removal in wastewater treatment. J. Environ. Eng. Asce 136, 890–896. doi: 10.1061/(ASCE)EE.1943-7870.0000218
Hegedus, B., Kos, P. B., Balint, B., Maroti, G., Gan, H. M., Perei, K., et al. (2017). Complete genome sequence of Novosphingobium resinovorum SA1, a versatile xenobiotic-degrading bacterium capable of utilizing sulfanilic acid. J. Biotechnol. 241, 76–80. doi: 10.1016/j.jbiotec.2016.11.013
Hsiao, W., Wan, I., Jones, S. J., and Brinkman, F. S. L. (2003). IslandPath: aiding detection of genomic islands in prokaryotes. Bioinformatics 19, 418–420. doi: 10.1093/bioinformatics/btg004
Hu, J., Adrion, A. C., Nakamura, J., Shea, D., and Aitken, M. D. (2014). Bioavailability of (geno)toxic contaminants in polycyclic aromatic hydrocarbon-contaminated soil before and after biological treatment. Environ. Eng. Sci. 31, 176–182. doi: 10.1089/ees.2013.0409
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Szklarczyk, D., Jensen, L. J., von Mering, C., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). eggNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Humbert, J. F., Barbe, V., Latifi, A., Gugger, M., Calteau, A., Coursin, T., et al. (2013). A tribute to disorder in the genome of the bloom-forming freshwater cyanobacterium microcystis aeruginosa. PLoS ONE 8:e70747. doi: 10.1371/journal.pone.0070747
Hurtado, R., Carhuaricra, D., Soare, S., Viana, M. V. C., Azevedo, V., Maturrano, L., et al. (2018). Pan-genomic approach shows insight of genetic divergence and pathogenic-adaptation of Pasteurella multocida. Gene 670, 193–206. doi: 10.1016/j.gene.2018.05.084
Hutalle-Schmelzer, K. M. L., Zwirnmann, E., Kruger, A., and Grossart, H. P. (2010). Enrichment and cultivation of pelagic bacteria from a humic lake using phenol and humic matter additions. FEMS Microbiol. Ecol. 72, 58–73. doi: 10.1111/j.1574-6941.2009.00831.x
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 11:119. doi: 10.1186/1471-2105-11-119
Hyeon, J. W., Kim, K., Son, A. R., Choi, E., Lee, S. K., and Jeon, C. O. (2017). Novosphingobium humi sp nov., isolated from soil of a military shooting range. Int. J. Syst. Evol. Microbiol. 67, 3083–3088. doi: 10.1099/ijsem.0.002089
Jariyal, M., Jindal, V., Mandal, K., Gupta, V. K., and Singh, B. (2018). Bioremediation of organophosphorus pesticide phorate in soil by microbial consortia. Ecotoxicol. Environ. Saf. 159, 310–316. doi: 10.1016/j.ecoenv.2018.04.063
Jensen, L. J., Julien, P., Kuhn, M., von Mering, C., Muller, J., Doerks, T., et al. (2008). eggNOG: automated construction and annotation of orthologous groups of genes. Nucleic Acids Res. 36, D250–D254. doi: 10.1093/nar/gkm796
Ji, B. Y., Zhang, S. D., Arnoux, P., Rouy, Z., Alberto, F., Philippe, N., et al. (2014). Comparative genomic analysis provides insights into the evolution and niche adaptation of marine Magnetospira sp QH-2 strain. Environ. Microbiol. 16, 525–544. doi: 10.1111/1462-2920.12180
Jiang, Y., Shao, J., Wu, X., Xu, Y., and Li, R. (2011). Active and silent members in the mlr gene cluster of a microcystin-degrading bacterium isolated from Lake Taihu, China. FEMS Microbiol. Lett. 322, 108–114. doi: 10.1111/j.1574-6968.2011.02337.x
Jones, G. J., Bourne, D. G., Blakeley, R. L., and Doelle, H. (1994). Degradation of the cyanobacterial hepatotoxin microcystin by aquatic bacteria. Nat. Toxins 2, 228–235. doi: 10.1002/nt.2620020412
Kampfer, P., Busse, H. J., and Glaeser, S. P. (2018). Novosphingobium lubricantis sp. nov., isolated from a coolant lubricant emulsion. Int. J. Syst. Evol. Microbiol. 68, 1560–1564. doi: 10.1099/ijsem.0.002702
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Kanehisa, M., and Goto, S. (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 28, 27–30. doi: 10.1093/nar/28.1.27
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Kantor, R. S., van Zyl, A. W., van Hille, R. P., Thomas, B. C., Harrison, S. T. L., and Banfield, J. F. (2015). Bioreactor microbial ecosystems for thiocyanate and cyanide degradation unravelled with genome-resolved metagenomics. Environ. Microbiol. 17, 4929–4941. doi: 10.1111/1462-2920.12936
Kaur, J., Verma, H., Tripathi, C., Khurana, J. P., and Lal, R. (2013). Draft genome sequence of a hexachlorocyclohexane-degrading bacterium, Sphingobium baderi strain LL03T. Genome Announc. 1:e00751–e00713. doi: 10.1128/genomeA.00751-13
Kohli, P., Dua, A., Sangwan, N., Oldach, P., Khurana, J. P., and Lal, R. (2013). Draft genome sequence of Sphingobium ummariense strain RL-3, a hexachlorocyclohexane-degrading bacterium. Genome Announc. 1:e00956–13. doi: 10.1128/genomeA.00956-13
Krishnan, R., Menon, R. R., Likhitha Busse, H.-J., Tanaka, N., Krishnamurthi, S., et al. (2017). Novosphingobium pokkalii sp nov, a novel rhizosphere-associated bacterium with plant beneficial properties isolated from saline-tolerant pokkali rice. Res. Microbiol. 168, 113–121. doi: 10.1016/j.resmic.2016.09.001
Kumar Singh, A., Sangwan, N., Sharma, A., Gupta, V., Khurana, J. P., and Lal, R. (2013). Draft genome sequence of Sphingobium quisquiliarum strain P25T, a novel hexachlorocyclohexane (HCH)-degrading bacterium isolated from an HCH dumpsite. Genome Announc. 1:e00717–13. doi: 10.1128/genomeA.00717-13
Kumar, R., Verma, H., Haider, S., Bajaj, A., Sood, U., Ponnusamy, K., et al. (2017). Comparative genomic analysis reveals habitat-specific genes and regulatory hubs within the genus Novosphingobium. Msystems 2:e00020–17. doi: 10.1128/mSystems.00020-17
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Phillippy, A., Delcher, A. L., Smoot, M., Shumway, M., Antonescu, C., et al. (2004). Versatile and open software for comparing large genomes. Genome Biol. 5:R12. doi: 10.1186/gb-2004-5-2-r12
Lagesen, K., Hallin, P., Rodland, E. A., Staerfeldt, H. H., Rognes, T., and Ussery, D. W. (2007). RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108. doi: 10.1093/nar/gkm160
Langille, M. G. I., Hsiao, W. W. L., and Brinkman, F. S. L. (2008). Evaluation of genomic island predictors using a comparative genomics approach. BMC Bioinform. 9:329. doi: 10.1186/1471-2105-9-329
Lezcano, M. A., Moron-Lopez, J., Agha, R., Lopez-Heras, I., Nozal, L., Quesada, A., et al. (2016). Presence or absence of mlr genes and nutrient concentrations co-determine the microcystin biodegradation efficiency of a natural bacterial community. Toxins 8:318. doi: 10.3390/toxins8110318
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, L., Stoeckert, C. J., and Roos, D. S. (2003). OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. doi: 10.1101/gr.1224503
Lin, W., Wang, Y. Z., Gorby, Y., Nealson, K., and Pan, Y. X. (2013). Integrating niche-based process and spatial process in biogeography of magnetotactic bacteria. Sci. Rep. 3:1643. doi: 10.1038/srep01643
Mackintosh, C., Beattie, K. A., Klumpp, S., Cohen, P., and Codd, G. A. (1990). Cyanobacterial microcystin-LR is a potent and specific inhibitor of protein phosphatase-1 and phosphatase-2a from both mammals and higher-plants. FEBS Lett. 264, 187–192. doi: 10.1016/0014-5793(90)80245-E
Maghsoudi, E., Fortin, N., Greer, C., Maynard, C., Page, A., Duy, S. V., et al. (2016). Cyanotoxin degradation activity and mlr gene expression profiles of a Sphingopyxis sp isolated from Lake Champlain, Canada. Environ. Sci. Processes Impacts 18, 1417–1426. doi: 10.1039/C6EM00001K
McCarthy, A. (2010). Third generation DNA sequencing: pacific biosciences' single molecule real time technology. Chem. Biol. 17, 675–676. doi: 10.1016/j.chembiol.2010.07.004
Miyauchi, K., Adachi, Y., Nagata, Y., and Takagi, M. (1999). Cloning and sequencing of a novel meta-cleavage dioxygenase gene whose product is involved in degradation of gamma-hexachlorocyclohexane in Sphingomonas paucimobilis. J. Bacteriol. 181, 6712–6719.
Miyauchi, K., Suh, S. K., Nagata, Y., and Takagi, M. (1998). Cloning and sequencing of a 2,5-dichlorohydroquinone reductive dehalogenase gene whose product is involved in degradation of gamma-hexachlorocyclohexane by Sphingomonas paucimobilis. J. Bacteriol. 180, 1354–1359.
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C., and Kanehisa, M. (2007). KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185. doi: 10.1093/nar/gkm321
Mukherjee, U., Kumar, R., Mahato, N. K., Khurana, J. P., and Lal, R. (2013). Draft genome sequence of sphingobium sp. strain HDIPO4, an avid degrader of hexachlorocyclohexane. Genome Announc. 1:e00749–13. doi: 10.1128/genomeA.00749-13
Nagata, Y., Hatta, T., Imai, R., Kimbara, K., Fukuda, M., Yano, K., et al. (1993). Purification and characterization of gamma-hexachlorocyclohexane (gamma-HCH) dehydrochlorinase (LinA) from Pseudomonas paucimobilis. Biosci. Biotechnol. Biochem. 57, 1582–1583. doi: 10.1271/bbb.57.1582
Nagata, Y., Ohtomo, R., Miyauchi, K., Fukuda, M., Yano, K., and Takagi, M. (1994). Cloning and sequencing of a 2,5-dichloro-2,5-cyclohexadiene-1,4-diol dehydrogenase gene involved in the degradation of gamma-hexachlorocyclohexane in Pseudomonas paucimobilis. J. Bacteriol. 176, 3117–3125. doi: 10.1128/jb.176.11.3117-3125.1994
Nawrocki, E. P., Kolbe, D. L., and Eddy, S. R. (2009). Infernal 1.0: inference of RNA alignments. Bioinformatics 25, 1335–1337. doi: 10.1093/bioinformatics/btp157
Nguyen, T. C., Loganathan, P., Nguyen, T. V., Vigneswaran, S., Kandasamy, J., Slee, D., et al. (2014). Polycyclic aromatic hydrocarbons in road-deposited sediments, water sediments, and soils in Sydney, Australia: comparisons of concentration distribution, sources and potential toxicity. Ecotoxicol. Environ. Saf. 104, 339–348. doi: 10.1016/j.ecoenv.2014.03.010
Nguyen, T. P. O., De Mot, R., and Springael, D. (2015). Draft genome sequence of the carbofuran-mineralizing Novosphingobium sp strain KN65.2. Genome Announc. 3:e00764–15. doi: 10.1128/genomeA.00764-15
Notomista, E., Pennacchio, F., Cafaro, V., Smaldone, G., Izzo, V., Troncone, L., et al. (2011). The marine isolate Novosphingobium sp PP1Y shows specific adaptation to use the aromatic fraction of fuels as the sole carbon and energy source. Microb. Ecol. 61, 582–594. doi: 10.1007/s00248-010-9786-3
Ohta, Y., Nishi, S., Kobayashi, K., Tsubouchi, T., Iida, K., Tanizaki, A., et al. (2015). Draft genome sequence of Novosphingobium sp strain MBES04, isolated from sunken wood from Suruga Bay, Japan. Genome Announc. 3:e01373–14. doi: 10.1128/genomeA.01373-14
Okano, K., Shimizu, K., Kawauchi, Y., Maseda, H., Utsumi, M., Zhang, Z., et al. (2009). Characteristics of a microcystin-degrading bacterium under alkaline environmental conditions. J. Toxicol. 2009:954291. doi: 10.1155/2009/954291
Okano, K., Shimizu, K., Maseda, H., Kawauchi, Y., Utsumi, M., Itayama, T., et al. (2015). Whole-genome sequence of the microcystin-degrading bacterium Sphingopyxis sp. strain C-1. Genome Announc. 3:e00838–15. doi: 10.1128/genomeA.00838-15
Pearce, S. L., Oakeshott, J. G., and Pandey, G. (2015). Insights into ongoing evolution of the hexachlorocyclohexane catabolic pathway from comparative genomics of ten Sphingomonadaceae strains. G3-Genes Genomes Genet. 5, 1081–1094. doi: 10.1534/g3.114.015933
Qi, J., Wang, B., and Hao, B. I. (2004). Whole proteome prokaryote phylogeny without sequence alignment: a K-string composition approach. J. Molecul. Evol. 58, 1–11. doi: 10.1007/s00239-003-2493-7
Retief, J. D. (2000). Phylogenetic analysis using PHYLIP. Methods Mol. Biol. 132, 243–258. doi: 10.1385/1-59259-192-2:243
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Saxena, A., Anand, S., Dua, A., Sangwan, N., Khan, F., and Lal, R. (2013). Novosphingobium lindaniclasticum sp. nov., a hexachlorocyclohexane (HCH)-degrading bacterium isolated from an HCH dumpsite. Intl. J. Syst. Evol. Microbiol. 63, 2160–2167. doi: 10.1099/ijs.0.045443-0
Shen, P. P., Shi, Q., Hua, Z. C., Kong, F. X., Wang, Z. G., Zhuang, S. X., et al. (2003). Analysis of microcystins in cyanobacteria blooms and surface water samples from Meiliang Bay, Taihu Lake, China. Environ. Int. 29, 641–647. doi: 10.1016/S0160-4120(03)00047-3
Sheu, S. Y., Huang, C. W., Chen, J. C., Chen, Z. H., and Chen, W. M. (2018). Novosphingobium arvoryzae sp. nov., isolated from a flooded rice field. Int. J Syst Evol Microbiol. 68, 2151–2157. doi: 10.1099/ijsem.0.002756
Sheu, S. Y., Liu, L. P., Young, C. C., and Chen, W. M. (2017). Novosphingobium fontis sp nov., isolated from a spring. Int. J. Syst. Evol. Microbiol. 67, 2423–2429. doi: 10.1099/ijsem.0.001973
Sievers, F., Wilm, A., Dineen, D., Gibson, T. J., Karplus, K., Li, W. Z., et al. (2011). Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Molecul. Syst. Biol. 7:539. doi: 10.1038/msb.2011.75
Siguier, P., Perochon, J., Lestrade, L., Mahillon, J., and Chandler, M. (2006). ISfinder: the reference centre for bacterial insertion sequences. Nucleic Acids Res. 34, D32–D36. doi: 10.1093/nar/gkj014
Singh, A. K., Chettri, B., Ghosh, A., Chikara, S. K., and Tripathi, T. (2017). Draft genome dequence of Novosphingobium panipatense strain P5:ABC,iIsolated from hydrocarbon-contaminated soil from Noonmati Refinery, Assam, India. Genome Announc. 5:e01265–17. doi: 10.1128/genomeA.01265-17
Snipen, L., and Ussery, D. W. (2010). Standard operating procedure for computing pangenome trees. Stand. Genomic Sci. 2, 135–141. doi: 10.4056/sigs.38923
Somdee, T., Thunders, M., Ruck, J., Lys, I., Allison, M., and Page, R. (2013). Degradation of [Dha(7)]MC-LR by a microcystin degrading bacterium isolated from Lake Rotoiti, New Zealand. ISRN Microbiol. 2013:596429. doi: 10.1155/2013/596429
Stolz, A. (2009). Molecular characteristics of xenobiotic-degrading Sphingomonads. Appl. Microbiol. Biotechnol. 81, 793–811. doi: 10.1007/s00253-008-1752-3
Suzuki, S., and Hiraishi, A. (2007). Novosphingobium naphthalenivorans sp nov., a naphthalene-degrading bacterium isolated from polychlorinated-dioxin-contaminated environments. J. Gen. Appl. Microbiol. 53, 221–228. doi: 10.2323/jgam.53.221
Takeuchi, M., Hamana, K., and Hiraishi, A. (2001). Proposal of the genus Sphingomonas sensu stricto and three new genera, Sphingobium, Novosphingobium and Sphingopyxis, on the basis of phylogenetic and chemotaxonomic analyses. Int. J. Syst. Evol. Microbiol. 51(Pt 4), 1405–1417. doi: 10.1099/00207713-51-4-1405
Tatusov, R. L., Koonin, E. V., and Lipman, D. J. (1997). A genomic perspective on protein families. Science 278, 631–637. doi: 10.1126/science.278.5338.631
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tiirola, M. A., Busse, H. J., Kampfer, P., and Mannisto, M. K. (2005). Novosphingobium lentum sp nov., a psychrotolerant bacterium from a polychlorophenol bioremediation process. Int. J. Syst. Evol. Microbiol. 55, 583–588. doi: 10.1099/ijs.0.63386-0
Tiirola, M. A., Mannisto, M. K., Puhakka, J. A., and Kulomaa, M. S. (2002). Isolation and characterization of Novosphingobium sp strain MT1, a dominant polychlorophenol-degrading strain in a groundwater bioremediation system. Appl. Environ. Microbiol. 68, 173–180. doi: 10.1128/AEM.68.1.173-180.2002
Trantirek, L., Hynkova, K., Nagata, Y., Murzin, A., Ansorgova, A., Sklenar, V., et al. (2001). Reaction mechanism and stereochemistry of gamma-hexachlorocyclohexane dehydrochlorinase LinA. J. Biol. Chem. 276, 7734–7740. doi: 10.1074/jbc.M007452200
Treangen, T. J., Sommer, D. D., Angly, F. E., Koren, S., and Pop, M. (2011). Next generation sequence assembly with AMOS. Curr. Protoc. Bioinform. Chapter 11, Unit 11 18. doi: 10.1002/0471250953.bi1108s33
Waack, S., Keller, O., Asper, R., Brodag, T., Damm, C., Fricke, W. F., et al. (2006). Score-based prediction of genomic islands in prokaryotic genomes using hidden Markov models. BMC Bioinform. 7:142. doi: 10.1186/1471-2105-7-142
Wang, J. F., Wu, P. F., Chen, J., and Yan, H. (2010). Biodegradation of microcystin-RR by a new isolated Sphingopyxis sp USTB-05. Chin. J. Chem. Eng. 18, 108–112. doi: 10.1016/S1004-9541(08)60330-4
Watanabe, K., and Harayama, S. (2001). SWISS-PROT: the curated protein sequence database on Internet. Tanpakushitsu Kakusan Koso 46, 80–86.
Wolf, I. R., Paschoal, A. R., Quiroga, C., Domingues, D. S., de Souza, R. F., Pretto-Giordano, L. G., et al. (2018). Functional annotation and distribution overview of RNA families in 27 Streptococcus agalactiae genomes. BMC Genomics 19:556. doi: 10.1186/s12864-018-4951-z
Wu, X. A., Monchy, S., Taghavi, S., Zhu, W., Ramos, J., and van der Lelie, D. (2011). Comparative genomics and functional analysis of niche-specific adaptation in Pseudomonas putida. FEMS Microbiol. Rev. 35, 299–323. doi: 10.1111/j.1574-6976.2010.00249.x
Yoshikawa, T., Ruhr, L. P., Flory, W., Giamalva, D., Church, D. F., and Pryor, W. A. (1985). Toxicity of polycyclic aromatic hydrocarbons.I. Effect of phenanthrene, pyrene, and their ozonized products on blood chemistry in rats. Toxicol. Appl. Pharmacol. 79, 218–226. doi: 10.1016/0041-008X(85)90343-6
Yucel, O., Wibberg, D., Philipp, B., and Kalinowski, J. (2018). Genome sequence of the bile salt-degrading bacterium Novosphingobium sp. Strain Chol11, a model organism for bacterial steroid catabolism. Genome Announc. 6:e01372–17. doi: 10.1128/genomeA.01372-17
Zhang, D. C., Liu, Y. X., and Huang, H. J. (2017). Novosphingobium profundi sp nov isolated from a deep-sea seamount. Antonie Van Leeuwenhoek International J. Gen. Molecul. Microbiol. 110, 19–25. doi: 10.1007/s10482-016-0769-3
Zhang, G. L., Zhang, Y., Wang, H., Xu, L., and Lv, L. Q. (2018). Comparative genomic analysis of five high drug-resistance Aeromonas hydrophila strains induced by doxycycline in laboratory and nine reference strains in Genbank. Aquacul. Res. 49, 2553–2559. doi: 10.1111/are.13717
Zhang, X., Liu, X., He, Q., Dong, W., Zhang, X., Fan, F., et al. (2016). Gene turnover contributes to the evolutionary adaptation of Acidithiobacillus caldus: insights from comparative genomics. Front. Microbiol. 7:1960. doi: 10.3389/fmicb.2016.01960
Zhao, Q., Yue, S. J., Bilal, M., Hu, H. B., Wang, W., and Zhang, X. H. (2017). Comparative genomic analysis of 26 Sphingomonas and Sphingobium strains: dissemination of bioremediation capabilities, biodegradation potential and horizontal gene transfer. Sci. Total Environ. 609, 1238–1247. doi: 10.1016/j.scitotenv.2017.07.249
Keywords: Novosphingobium, degradation, comparative genomics, genomic variability, metabolic profile
Citation: Wang J, Wang C, Li J, Bai P, Li Q, Shen M, Li R, Li T and Zhao J (2018) Comparative Genomics of Degradative Novosphingobium Strains With Special Reference to Microcystin-Degrading Novosphingobium sp. THN1. Front. Microbiol. 9:2238. doi: 10.3389/fmicb.2018.02238
Received: 22 June 2018; Accepted: 03 September 2018;
Published: 25 September 2018.
Edited by:
Mariusz Cycon, Medical University of Silesia, PolandReviewed by:
Gabor Rakhely, University of Szeged, HungaryHelianthous Verma, University of Delhi, India
Copyright © 2018 Wang, Wang, Li, Bai, Li, Shen, Li, Li and Zhao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Renhui Li, cmVsaUBpaGIuYWMuY24=
Tao Li, bGl0YW9AaWhiLmFjLmNu
†These authors have contributed equally to this work