 Vartul Sangal1*
Vartul Sangal1* Michael Goodfellow2
Michael Goodfellow2 Jochen Blom3
Jochen Blom3 Geok Yuan Annie Tan4
Geok Yuan Annie Tan4 Hans-Peter Klenk2
Hans-Peter Klenk2 Iain C. Sutcliffe1
Iain C. Sutcliffe1- 1Faculty of Health and Life Sciences, Northumbria University, Newcastle upon Tyne, United Kingdom
- 2School of Natural and Environmental Sciences, Newcastle University, Newcastle upon Tyne, United Kingdom
- 3Bioinformatics and Systems Biology, Justus-Liebig-Universität, Gießen, Germany
- 4Institute of Biological Sciences, Faculty of Science, University of Malaya, Kuala Lumpur, Malaysia
Strains belonging to the genus Amycolatopsis are well known for the production of a number of important antimicrobials and other bioactive molecules. In this study, we have sequenced the genomes of five Amycolatopsis strains including Amycolatopsis circi DSM 45561T, Amycolatopsis palatopharyngis DSM 44832T and Amycolatopsis thermalba NRRL B-24845T. The genome sequences were analyzed with 52 other publically available Amycolatopsis genomes, representing 34 species, and 12 representatives from related genera including Saccharomonospora, Saccharopolyspora, Saccharothrix, Pseudonocardia and Thermobispora. Based on the core genome phylogeny, Amycolatopsis strains were subdivided into four major clades and several singletons. The genus Amycolatopsis is homogeneous with only three strains noted to group with other genera. Amycolatopsis halophila YIM93223T is quite distinct from other Amycolatopsis strains, both phylogenetically and taxonomically, and belongs to a distinct genus. In addition, Amycolatopsis palatopharyngis DSM 44832T and Amycolatopsis marina CGMCC4 3568T grouped in a clade with Saccharomonospora strains and showed similar taxogenomic differences to this genus as well as other Amycolatopsis strains. The study found a number of strains, particularly those identified as Amycolatopsis orientalis, whose incorrect identification could be resolved by taxogenomic analyses. Similarly, some unclassified strains could be assigned with species designations. The genome sequences of some strains that were independently sequenced by different laboratories were almost identical (99–100% average nucleotide and amino acid identities) consistent with them being the same strain, and confirming the reproducibility and robustness of genomic data. These analyses further demonstrate that whole genome sequencing can reliably resolve intra- and, inter-generic structures and should be incorporated into prokaryotic systematics.
Introduction
The genus Amycolatopsis is well known for the commercial production of multiple antibiotics, including the important broad spectrum antibiotics rifamycin and, vancomycin (Xu et al., 2014; Chen et al., 2016). These strains also have the potential to produce a number of other secondary metabolites and bioactive molecules (Adamek et al., 2018) and, can be exploited for a range of biotechnological applications (Davila Costa and Amoroso, 2014). The genus currently includes 72 validly named species (List of prokaryotic names with standing in nomenclature1) that may vary in their phenotypic and morphological characteristics (Tan and Goodfellow, 2015). Amycolatopsis strains commonly reside in arid, or hyper-arid soil and have chemo-organotrophic to facultatively autotrophic lifestyles (Tan and Goodfellow, 2015). However, some species have also been isolated from activated sludge, equine placentas and from clinical and plant material (Tan and Goodfellow, 2015). Amycolatopsis strains can be mesophilic or thermophilic, with a DNA GC content of 66–75 mol%. They can form branching substrate hyphae that fragment into square, or rod-shaped elements and carry aerial hyphae (Saintpierre-Bonaccio et al., 2005; Tan and Goodfellow, 2015).
A recent multilocus sequence analysis (MLSA) based on seven housekeeping genes (atpD, clpB, gapA, gyrB, nuoD, pyrH and rpoB) revealed the presence of four major groups of species within the genus, with some singletons (Adamek et al., 2018), whilst Sanchez-Hidalgo et al. (2018) described 11 subgroups in a 16 S rRNA analysis, and four major groupings based on an MLSA with four housekeeping genes (atpD, dnaK, recA and rpoB). The type strains of Amycolatopsis halophila and Amycolatopsis marina were found to be quite distinct from other Amycolatopsis strains. Although average nucleotide identities based on MUMmer (ANIm) supported the classification of these strains within the genus, the percentage of conserved proteins indicated that A. halophila might belong to a different genus (Adamek et al., 2018). A five gene MLSA (with atpI, gyrA, ftsZ, secA and dnaK) also indicated that A. halophila is more similar to members of the genus Prauserella than to Amycolatopsis species (Bose et al., 2016). Therefore, we have applied a more comprehensive phylogenetic and taxogenomic approach to the genus (Sangal et al., 2016) and analyzed 57 Amycolatopsis genome sequences belonging to 34 species, including five sequenced by us in this study. Twelve genome sequences representing other genera, including Saccharomonospora, Saccharopolyspora, Saccharothrix, Pseudonocardia, and Thermobispora, were included for the comparative analyses (Supplementary Table 1). This study highlights the subgeneric structure within the genus and supports the separation of A. halophila as a member of a different genus. In addition, it is clear that a number of strains have been misidentified and misclassified using traditional approaches. Some strains sequenced independently by different laboratories grouped together, suggesting that genome based approaches are more reliable and reproducible than techniques such as DNA–DNA hybridisation, which suffers from a lack of reproducibility and compatibility of results between different laboratories (Achtman and Wagner, 2008; Moore et al., 2010).
Materials and Methods
Bacterial Strains and Genome Sequencing
Five strains, Amycolatopsis circi DSM 45561T, Amycolatopsis palatopharyngis DSM 44832T, Amycolatopsis ruanii 49.3a, Amycolatopsis thermalba 50.9 b and A. thermalba NRRL B-24845T were grown in 5 ml Brain-Heart Infusion broth (Oxoid) at 28°C for 48 h. Genomic DNA from each strain was extracted from 1.5 ml culture using the UltraClean® Microbial DNA Isolation Kit (MoBio).
The genome sequencing was performed on an Illumina MiSeq instrument and the paired-end reads were assembled using SPAdes 3.9.0 (Bankevich et al., 2012). The draft genome sequences have been submitted to the DDBJ/EMBL/GenBank databases and are publicly available (Supplementary Table 1).
The genome sequences of 52 Amycolatopsis strains and 12 representative strains of related genera were obtained from GenBank (Supplementary Table 1). Two independent genome assemblies were available for seven Amycolatopsis strains in GenBank and both of them were included to test the reliability and reproducibility of the genomic data.
Computational Analyses
BLAST-based pairwise average nucleotide identity (ANIb) and pairwise fragment similarity scores (fragment size of 500-bp) were calculated from the nucleotide sequences using Jspecies (Richter and Rosselló-Móra, 2009) and GEGENEES (Agren et al., 2012), respectively. The genome sequences were annotated using Prokka (Seemann, 2014) and were compared using EDGAR (Blom et al., 2016) for calculation of the core- and pan- genomes and the number of genes shared within each phylogenetic cluster. Pairwise amino acid identity (AAI) was also calculated using EDGAR (Blom et al., 2016) and pairwise digital DNA–DNA hybridization values were calculated using GGDC 2.1 (Auch et al., 2010a,b). A maximum-likelihood (ML) tree was constructed from the concatenated protein sequences of the core genes after removing sites with missing data using the best-fit amino acid substitution model (LG + F + I + G4) with 100,000 SH-aLRT and 100,000 ultrafast bootstrap replicates using IQ-Tree (Nguyen et al., 2015). A Neighbor-Joining (NJ) tree was generated from the pairwise GGDC distance matrix using MEGA (Kumar et al., 2016). The tree was re-rooted on Thermobispora bispora DSM 43833T. Pairwise percentage of conserved proteins (POCP) were calculated using the scripts, data_file_4.sh (Moose, 2017) and runPOCP.sh (Pantiukh and Grouzdev, 2017) that are based on the previously described approach (Qin et al., 2014).
Results
Phylogenetic and Taxogenomic Groups Within the Genus
A total of 602 genes were conserved (core genome) across the 69 genomes, including the strains representing related genera (Supplementary Table 1). A ML tree generated from concatenated core proteins resolved Amycolatopsis strains, representing 34 species (including 29 type strains), into four major groups and several singletons (Figure 1). These groupings are consistent with both the MLSA based phylogenies albeit with minor exceptions (Adamek et al., 2018; Sanchez-Hidalgo et al., 2018). We have applied the same group designations as used by Adamek et al. (2018). Group A is the largest group with 19 isolates assigned to nine species, including two without formal species designations. Group B encompasses 16 isolates (nine species) while groups C and D are relatively smaller with eight isolates (6 species and 1 undefined) and seven isolates (four species and one undefined), respectively (Figure 1). Amycolatopsis taiwanensis DSM 45107T, Amycolatopsis sacchari DSM 44468T, Amycolatopsis nigrescens CSC17Ta 90T and Amycolatopsis xylanica CPCC202699T are present as singletons within the Amycolatopsis clade (Figure 1). These strains were also recovered as singletons in the MLSA analysis of Sanchez-Hidalgo et al. (2018), except for A. xylanica which was located at the periphery of the strains in our cluster A. Of the other singletons, A. sacchari DSM 44468T is consistently associated with A. dongchuanensis in 16S rRNA gene trees [e.g. (Wang et al., 2018); group I in Sanchez-Hidalgo et al. (2018) and Tang et al. (2016)], whilst the A. nigrescens is probably closely related to Amycolatopsis minnesotensis (Tang et al., 2016; Sanchez-Hidalgo et al., 2018;Wang et al., 2018). Similarly, A. taiwanensis is consistently associated with Amycolatopsis helveola and Amycolatopsis pigmentata (Tang et al., 2016; Sanchez-Hidalgo et al., 2018; Wang et al., 2018). These associations suggest that these singletons may expand into species groups as more whole genomes become available.
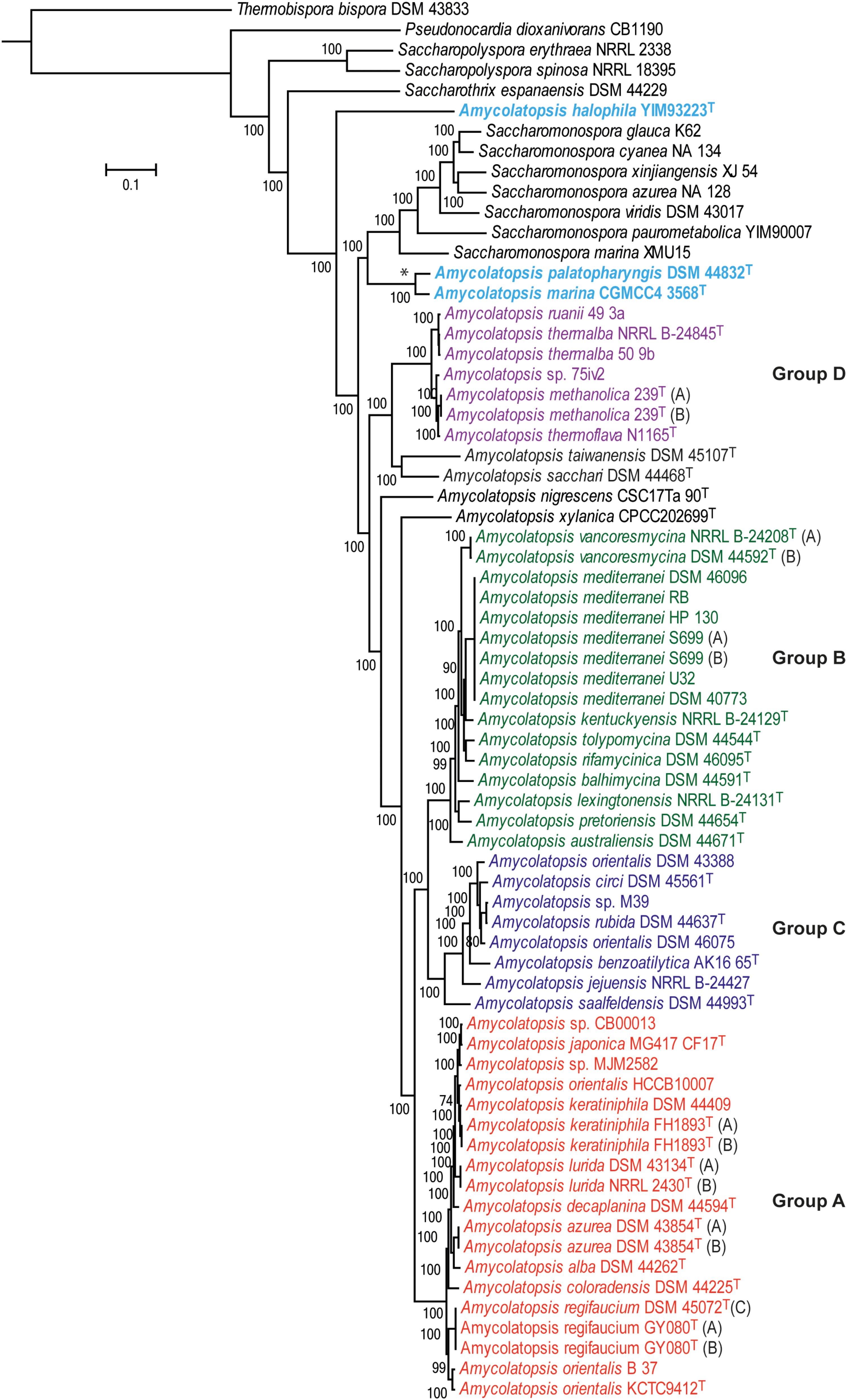
FIGURE 1. A maximum-likelihood tree derived from concatenated core protein sequences. The scale bar represents amino acid substitutions per site. Amycolatopsis groups are labeled and shown in different colors. The clade with the two Amycolatopsis strains that are more close to Saccharomonospora strains is labeled with a star (∗) sign.
Notably, A. palatopharyngis DSM 44832T and A. marina CGMCC4 3568T [16S and MLSA group G (Sanchez-Hidalgo et al., 2018)] formed a group that is more closely related to Saccharomonospora strains than to other members of Amycolatopsis while A. halophila YIM93223T [16S group J (Sanchez-Hidalgo et al., 2018)] is quite distant to all other strains and forms a single member clade (Figure 1). Overall, group structure is also consistent with the NJ tree obtained from the BLAST-based genome-to-genome distances (Supplementary Figure 1).
The results of fragmented genome BLAST searches are consistent with the phylogenetic groupings with a mean fragmented BLAST similarity (FBS) within the Amycolatopsis groups varying from 35.9 ± 18.8 for Group C to 65.1 ± 17.0 for Group D (Figure 2A). The mean FBS score between the Amycolatopsis groups varied from 8.6 ± 1.8 to 11.4 ± 2.1 and between Amycolatopsis (excluding the three anomalous strains A. palatopharyngis DSM 44832T, A. marina CGMCC4 3568T, and A. halophila YIM93223T) and strains of the other genera from 3.9 ± 1.6. Although the group of A. palatopharyngis, and A. marina is closely related to Saccharomonospora strains, the mean FBS score (5.3 ± 1.2) between these taxa was comparable to the score with the other Amycolatopsis strains (5.6 ± 1.1). Similarly, A. halophila was also equidistant from the Amycolatopsis strains (4.1 ± 1.1) and from the other genera (including A. marina and A. palatopharyngis; 3.5 ± 1.0).
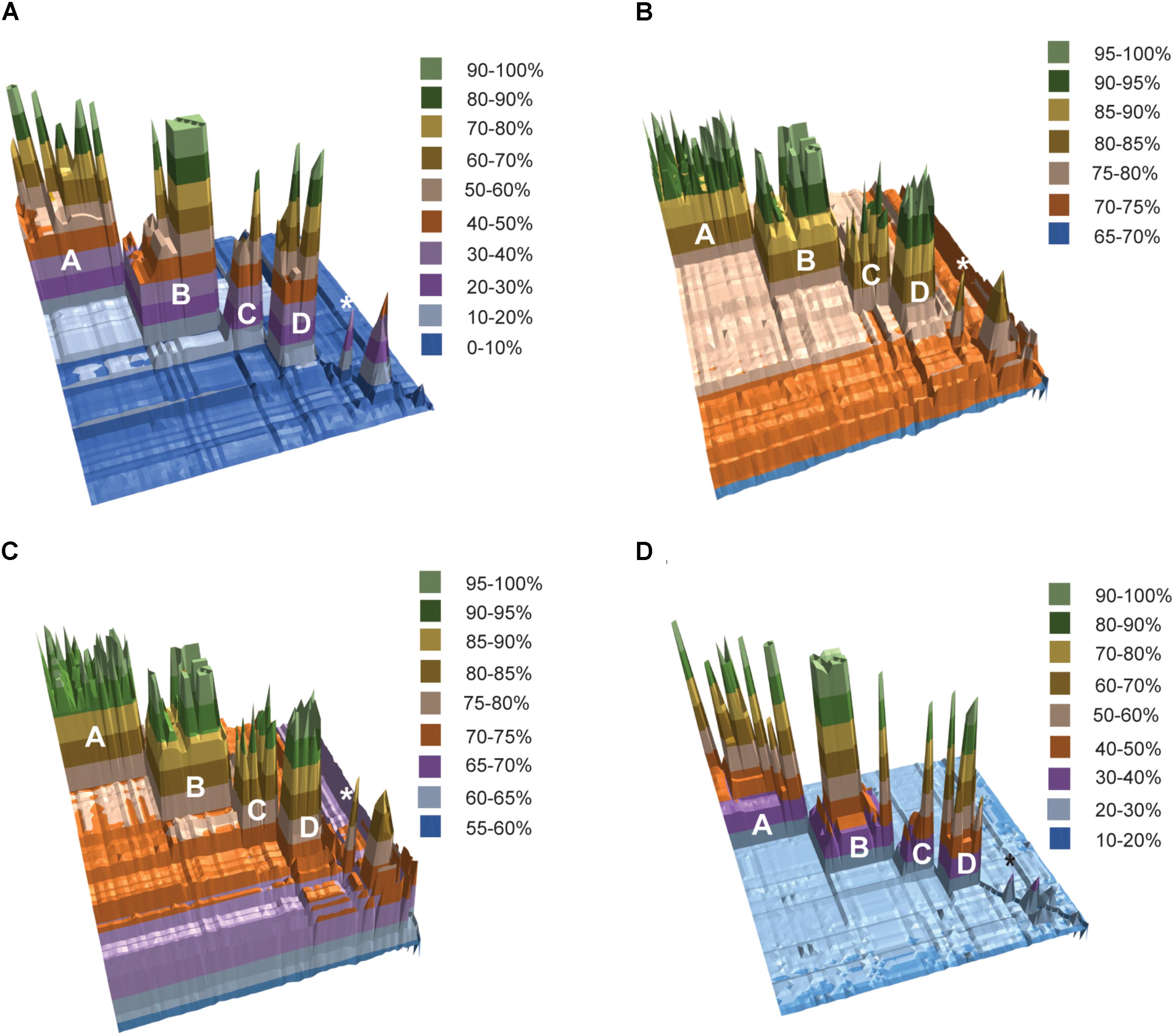
FIGURE 2. 3D plots of pairwise matrices obtained from (A) fragmented BLAST searches (FBS values), (B) average nucleotide identities (ANIb values), (C) average amino-acid identities (AAI values) and (D) digital DNA-DNA hybridisation (dDDH values). The Amycolatopsis groups are labeled. The group of A. palatopharyngis DSM 44832T and A. marina CGMCC4 3568T is labeled with the star (∗) sign.
Average nucleotide identity and AAI values also support the phylogenetic groupings (Figures 2B,C). Average ANIb values within and between the Amycolatopsis groups are >80% (85.94 ± 5.24 - 93.65 ± 3.41) and >75% (75.68 ± 0.69 - 76.97 ± 0.91), respectively. The strains of the other genera and the three anomalous strains shared lower ANIb values when compared with the Amycolatopsis strains (72.52 ± 1.52 and 72.77 ± 1.77, respectively). Similarly, average AAI values are >80% (86.69 ± 6.11 - 94.48 ± 3.06) within the Amycolatopsis groups and approximately 70%, or higher (70.99 ± 1.02 - 73.93 ± 1.98) between these groups. Average AAI values between Amycolatopsis strains and the three anomalous strains and the other genera are 67.54 ± 4.27 and 66.45 ± 3.70, respectively.
Pairwise dDDH values were also calculated for the dataset which varied between 36.2 ± 14.4 and 59.5 ± 19.7 within each Amycolatopsis group (Figure 2D). The average dDDH values ranged from 21.4 ± 0.5 to 22.2 ± 0.5 between the Amycolatopsis groups which is comparable to the values between the Amycolatopsis strains and the strains from the other genera (20.1 ± 0.7). This confirms that dDDH is useful in identifying strains belonging to the same species but has limited resolution at the intergeneric level.
A POCP value of 50% is often used as the genus boundary where two strains with more than 50% conserved proteins are considered to belong to the same genus (Qin et al., 2014). Consistent with this, pairwise POCP values among the Amycolatopsis strains (excluding anomalous strains) varied between 50.71 and 99.96% (67.38 ± 9.75; Supplementary Table 2). However, POCP could not resolve the status of the anomalous strains as A. marina and A. palatopharyngis type strains showed POCP values of 57.09 ± 2.95 and 58.53 ± 3.13 against the strains in the Amycolatopsis clade and the Saccharomonospora strains, respectively. POCP values between A. halophila YIM93223T, and other Amycolatopsis strain was <50% except for some of the strains in group D, A. marina and A. palatopharyngis (Supplementary Table 2). All of these results suggest that 54 of the 57 Amycolatopsis strains belong to the same genus while A. halophila YIM93223T should be assigned to a different genus. A. palatopharyngis DSM 44832T and A. marina CGMCC4 3568T grouped in the clade with Saccharomonospora strains; however, they showed comparable distances to both Saccharomonospora and Amycolatopsis strains. A larger analysis including more species from the genus Saccharomonospora and other representatives of the family Pseudonocardiaceae is required to clarify the status of these two species. However, it is notable that the recent phylogenomic study of Nouioui et al. (2018) recovered A. marina within the genus Amycolatopsis, although this study did not include A. palatopharyngis.
Genomic Features of Amycolatopsis Strains Are Consistent With the Phylogenetic Groups
The genome sizes of the Amycolatopsis strains varied between 7 and 11 Mb with a GC content of 68 – 72 mol% (Figure 3 and Supplementary Table 1). A. halophila YIM93223T is clearly an outlier with a genome size of 5.6 Mb and a 67.8 mol% GC content. Amycolatopsis salitolerans consistently clusters with A. halophila in 16S rRNA gene trees (Guan et al., 2012; Tang et al., 2016; Sanchez-Hidalgo et al., 2018; Wang et al., 2018) and thus likely belongs to the same genus. A. salitolerans is reported to have a GC content of 66.4% (Guan et al., 2012); whilst this value needs confirmation from whole genome sequence data, it is similar to that for A. halophila YIM93223T and is outwith the range for strains of Amycolatopsis sensu stricto (Figure 3). Cumulatively, these data are consistent with the recent proposal that A. halophila and A. salitolerans should be reclassified into the genus Haloechinothrix (Tang et al., 2010) as Haloechinothrix halophila comb. nov. and Haloechinothrix salitolerans comb. nov., respectively (Nouioui et al., 2018). A. palatopharyngis DSM 44832T and A. marina CGMCC4 3568T are also separable due to genomes sizes and GC content being at the lower end of the respective range for the Amycolatopsis strains (Figure 3 and Supplementary Table 1). Although the range of the genome size and GC content is quite broad for Amycolatopsis sensu stricto strains, some patterns are visible at the group level. For instance, the strains in Group A have a genome size of 8.27 – 9.81 Mb, and a GC content of 68.5 – 69 mol%. The genomes of strains in Group B varied between 9.04 and 10.86 Mb with a GC content ranging from 70.8 to 72 mol%.
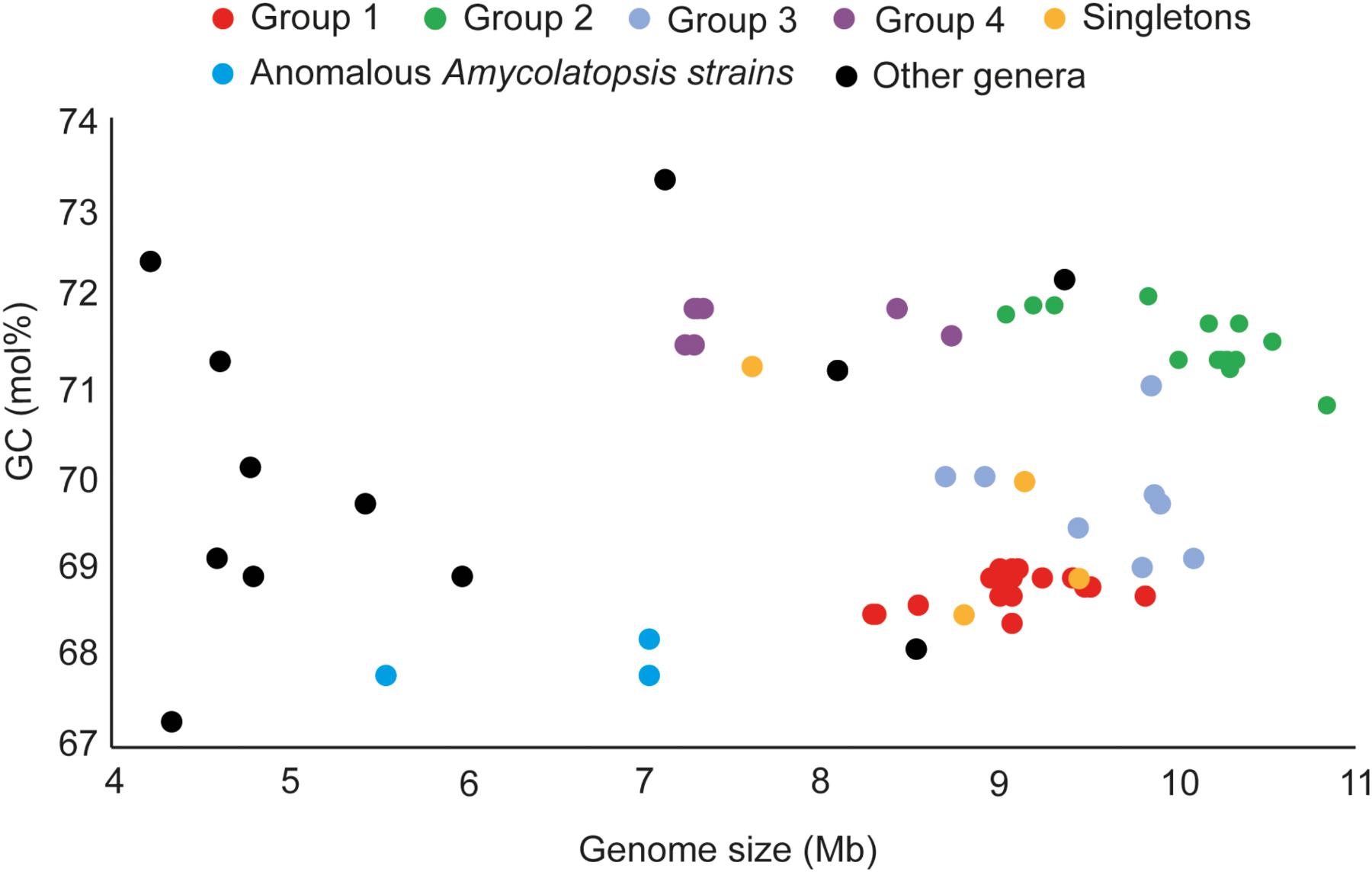
FIGURE 3. Distribution of genome sizes and GC contents among the different phylogenetic groups.
In order to establish whether the size of the core genome can help identify intergeneric boundaries, core genes were calculated for the entire dataset followed by a sequential removal of the other genera and the outlier strains A. halophila, A. palatopharyngis and A. marina. The core genome of the entire dataset contained 602 genes (6.1–16.9%). The number of annotated genes among the Amycolatopsis strains ranged between 5,098 and 9,890 after excluding those of other genera (Supplementary Table 1) with 1,382 of them (14.0 – 27.1%) shared by all of the strains, which compares well with the 1,212 core genes identified by Adamek et al. (2018). The number of core genes increased to 1,634 (17.5 – 24.7% of 6,606 – 9,890 genes) after removing the A. halophila strain from the dataset and only slightly to 1,739 genes (17.6 – 25%) after exclusion of the genomes of the A. palatopharyngis and A. marina strains. Therefore, the proportion of genes in the core genome may not be reliable for separating members of different genera due to overlap in values within a genus and when strain(s) from other genera are included (Figure 4). As expected, the number of core genes is much higher within each Amycolatopsis group (43.1 – 69.5%).
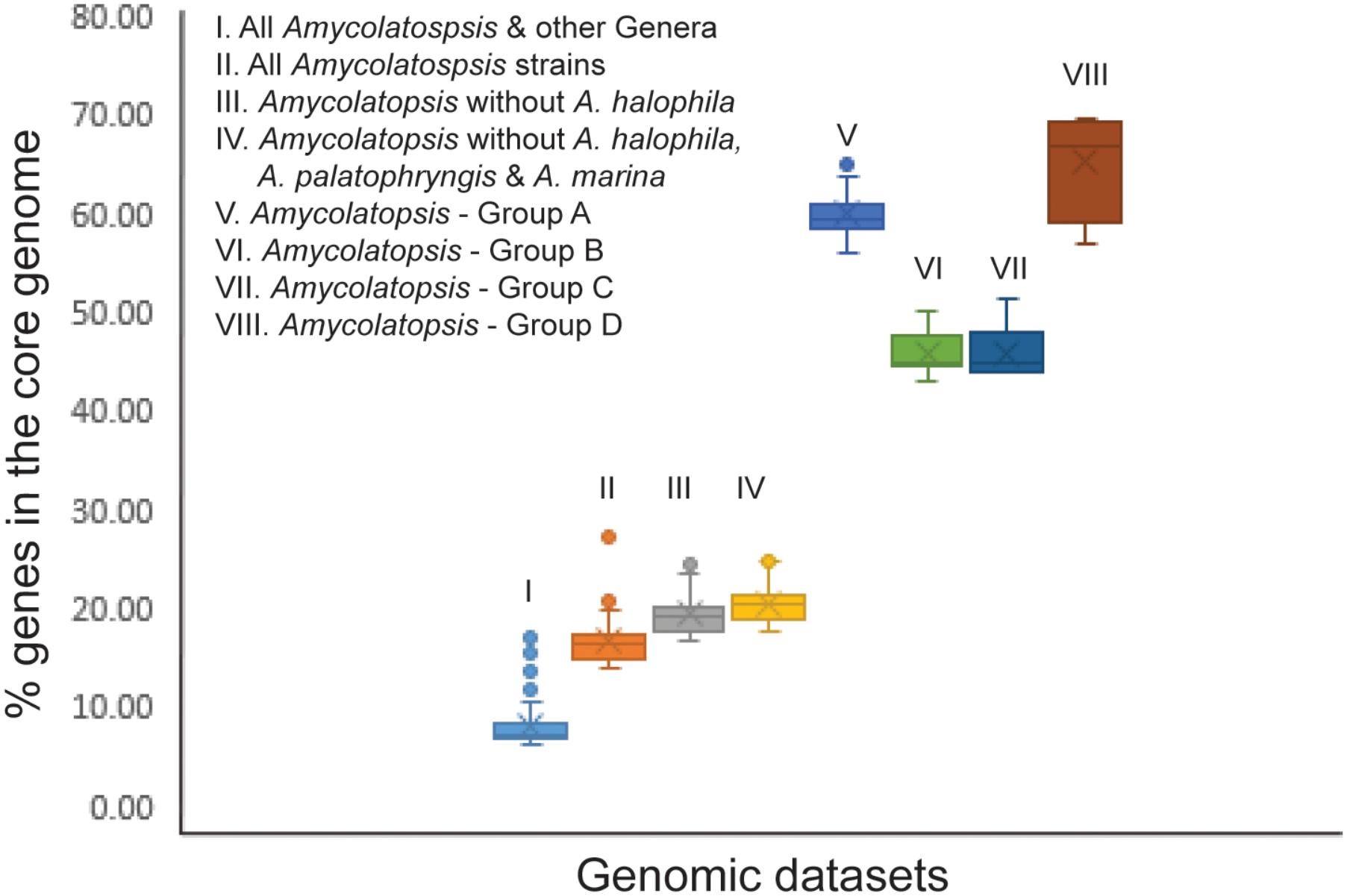
FIGURE 4. A box plot showing the fraction of genes in the core genome for the different genomic datasets.
Species-Group A
Group A is the largest of the Amycolatopsis groups including nine type strains (Figure 1 and Supplementary Table 1), many of which are known to produce different antibiotics and other bioactive molecules (Tan and Goodfellow, 2015). Average FBS, ANIb, and AAI values within this group are 55.9 ± 12.4, 90.76 ± 2.88, and 92.48 ± 2.56, respectively. dDDH values are consistent with the recognition of nine species, namely Amycolatopsis alba, Amycolatopsis azurea, Amycolatopsis coloradensis, Amycolatopsis decaplanina, Amycolatopsis japonica, Amycolatopsis keratiniphila, Amycolatopsis lurida, Amycolatopsis orientalis, and Amycolatopsis regifaucium (Figure 1). This analysis identified a potential case of misidentification of the industrial vancomycin producer A. orientalis strain HCCB10007 (Xu et al., 2014), which, as noted previously (Adamek et al., 2018; Sanchez-Hidalgo et al., 2018), is notably distant from A. orientalis KCTC9412T (dDDH 38.4%; ANIb 50.8%) and shows a dDDH of 72% and ANIb of 96.4% with A. keratiniphila FH1893T. Therefore, this strain should be reclassified as A. keratiniphila. Two strains, Amycolatopsis sp. CB00013 and Amycolatopsis sp. MJM252 could be assigned to A. japonica based on the dDDH cut-off value of 70%, as noted previously (Sanchez-Hidalgo et al., 2018). These assignments are also consistent with the previously suggested FBS score of >66.8% and ANIb and AAI values of ∼95% or higher (Konstantinidis and Tiedje, 2005b; Sangal et al., 2016). While A. keratiniphila, and A. japonica strains are clearly separated into two species by a dDDH value of ∼60%, other matrices suggest that they may belong to the same species.
Multiple assemblies were available in GenBank for four of the type strains in Group A (Figure 1 and Supplementary Table 1) and all of them were included to check the reproducibility of the sequence data between different laboratories and the robustness of the approach. Pairwise FBS, ANIb, AAI, and dDDH values between these assembles were >99.7%, confirming the authenticity of these strains albeit with a minor exception. The dDDH value between the genome sequences of A. keratiniphila FH1893T (Assembly accession numbers CA_900105855.1, and GCA_001953855.1) was 97.7%. These assemblies were submitted by two different laboratories and this variation may reflect the quality of the sequences. Other taxogenomic values are consistent with them being associated with the same strain (Supplementary Tables 3A–D).
Species-Group B
Group B is also quite diverse encompassing 16 of the 57 Amycolatopsis strains (Figure 1 and Supplementary Table 1). The mean FBS (55.74 ± 21.36), ANIb (90.45 ± 4.72) and, AAI scores (91.23 ± 4.40) for this group are comparable to the values observed for Group A. All of the pairwise matrices support the presence of nine species including Amycolatopsis australiensis, Amycolatopsis balhimycina, Amycolatopsis kentuckyensis, Amycolatopsis lexingtonensis, Amycolatopsis mediterranei, Amycolatopsis pretoriensis, Amycolatopsis rifamycinica, Amycolatopsis tolypomycina, and Amycolatopsis vancoresmycina in the group, which is consistent with their taxonomic assignment (Figure 1 and Supplementary Table 1). While most of the members of this clade are known to produce antibiotics and/or bioactive molecules (Davila Costa and Amoroso, 2014; Tan and Goodfellow, 2015), the A. kentuckyensis, A. lexingtonensis, and A. pretoriensis strains were isolated from lesions on equine placenta and may be pathogenic (Labeda et al., 2003). A. vancoresmycina DSM 44592T has been sequenced independently by two laboratories and the high genomic similarities are consistent with them being the same strain (Supplementary Tables 3A–D). This group also includes seven A. mediterranei isolates that are grouped together with >97.7% dDDH, and 99.8–100% ANIb and AAI similarities, indicating it to be a highly homogeneous species with very limited genomic diversity.
Species-Group C
Group C shows a slightly higher diversity than was found in Groups A and B (FBS, 35.85 ± 18.83; ANIb, 85.94 ± 5.24; AAI, 86.69 ± 6.11; Supplementary Tables 3A–D). Based on the taxogenomic matrices, eight strains within this group can be assigned to seven species (Figure 1 and Supplementary Table 1). The assignment of five of these species, A. circi, Amycolatopsis rubida, Amycolatopsis benzoatilytica, Amycolatopsis jejuensis and Amycolatopsis saalfeldensis is consistent with the literature (Tan and Goodfellow, 2015). However, two strains, DSM 43388 and DSM 46075, identified as A. orientalis, are clearly misclassified. The type strain of A. orientalis, KCTC9412T, clustered in Group A. Both these strains are quite distinct from each other as well as from other strains in Group C and belong to novel species based on the taxogenomic values. Amycolatopsis sp. strain M39 shares a dDDH value of 93.2, FBS value of 87.25 ± 0.17, ANIb 98.96 ± 0.10, and AAI of 99.1 with A. rubida DSM 44637T suggesting that strain M39 strain should be assigned to this species, as noted previously (Sanchez-Hidalgo et al., 2018). Most of the strains in this group were isolated from diverse soil samples but with some exceptions, e.g., A. benzoatilytica strain AK16 65T, was isolated from a patient with submandibular mycetoma (Tan and Goodfellow, 2015).
Species-Group D
The species group D is the smallest group with an average FBS score of 65.12 ± 16.95, an average ANIb value of 93.65 ± 3.41 and an average AAI score of 94.48 ± 3.06 (Supplementary Tables 3A–D). These strains were assigned to four species based on the pairwise dDDH values including Amycolatopsis methanolica, A. thermalba, Amycolatopsis thermoflava and a potentially novel species with Amycolatopsis sp. strain 75iv2 (Supplementary Table 3D). A. ruanii strain 49.3a probably should be reclassified as A. thermalba due to its high dDDH 93.3%, FBS 92.78 ± 0.12, ANIb 99.19 ± 0.04, and AAI value of 99.23 with the type strain of latter. Both genome sequences of the A. methanolica type strain show ≥99.9% ANIb and AAI similarities but their dDDH is slightly lower (98.3%). Similar to A. keratiniphila, this may be due to minor variations in the quality of genome sequences generated in different laboratories. Although, Amycolatopsis sp. strain 75iv2 and A. thermoflava strain N1165T can be assigned to two different species based on the dDDH values, they share ANIb (95.08%) and AAI (96.42%) with each other that marginally overlap using the recommended cut-off for defining species (Konstantinidis and Tiedje, 2005a, 2007; Sangal et al., 2016).
Discussion
Whole genome based approaches are now routinely used to resolve the structure of complex prokaryotic taxa (Kyrpides et al., 2014; Sangal et al., 2014, 2016; Schleifer et al., 2015; Sutcliffe, 2015; Mahato et al., 2017; Carro et al., 2018; Chun et al., 2018). In addition to the genome-based phylogenies, calculation of dDDH, ANI and AAI from the genomic sequences have become the gold standard for defining species with cut-off values of 70%, 95% and 95–96%, respectively (Konstantinidis and Tiedje, 2005a, 2007; Auch et al., 2010b). However, the data on separating prokaryotic genera are limited (Qin et al., 2014; Sangal et al., 2016). We have previously suggested that an ANI value of approximately 74.8% can be used to define genera and that FBS values of ∼66.8% and 6.9% can help identifying interspecific and intergeneric boundaries, respectively (Sangal et al., 2016). Using a combination of phylogenomic and taxogenomic approaches, we defined seven species groups in the genus Rhodococcus that were as distant from each other as from representatives of other genera (Sangal et al., 2016). In contrast, strains representing the genus Micromonospora were found to be monophyletic, consistent with their assignment to a single genus (Carro et al., 2018). In this study, we have extended this approach to the industrially and biomedically important genus, Amycolatopsis, and found that the majority of strains assigned to this taxon clustered on a single branch that was separated from the related genus Saccharomonospora (Figure 1). However, the type strains of A. palatopharyngis and A. marina clustered more closely to Saccharomonospora and the type strain of A. halophila formed a single member clade (Figure 1). The taxogenomic matrices are in agreement with the phylogenomic groupings (Figure 2) with minor exceptions. For example, the FBS and ANI values between A. taiwanensis strain DSM 45107T and the other Amycolatopsis strains are slightly below the suggested cut-off values (Supplementary Tables 3A,B); however, this strains clustered close to group D within the Amycolatopsis clade and clearly belongs to this genus. Therefore, the suggested cut-off values should be considered a guide and used in combination with genome based phylogenies (Sangal et al., 2016).
The percentage of conserved proteins (POCP) has been used to define strains at the genus level with ≥50% proteins between a pair of strains with at least 50% alignable region and 40% sequence identity considered to indicate membership of the same genus (Qin et al., 2014). However, the status of the anomalous strains A. marina and A. palatopharyngis remains unresolved due to POCP values of > 50% both with the other Amycolatopsis strains as well as with the Saccharomonospora strains (Supplementary Table S2). As noted above, further analysis of a larger dataset is required not only to clarify the taxonomic status of A. marina and A. palatopharyngis but also that of the effectively named species Amycolatopsis flava which groups with them in 16S rRNA trees (Wei et al., 2015). In agreement with a previous study where POCP was applied to a slightly smaller set of Amycolatopsis genomes (Adamek et al., 2018), less than 50% proteins were conserved between A. halophila YIM93223T and the majority of the Amycolatopsis strains but with some exceptions (Supplementary Table S2). These results are consistent with the classification of A. halophila into a different genus (Nouioui et al., 2018). We also applied a stringent approach to identify conserved proteins by calculating the core genome for the entire dataset with a sequential exclusion of genomes from members of other genera and anomalous Amycolatopsis strains (Figure 4). However, no clear correlation to core conserved proteins and intergeneric boundary could be identified from this analysis.
The majority of the Amycolatopsis strains clustered into four robust groups based on the phylogenomic and taxogenomic analyses (Figures 1, 2A–D). The pan-genomic analyses identified some genes that are conserved within each group but absent in other Amycolatopsis strains (Supplementary Tables S4A–D). 147 genes were specific to Group A, 114 genes to Group B, 54 genes to Group C and 244 genes to group D. A large proportion of these genes (42–59%) encode hypothetical proteins; however, some genes are annotated as transcriptional regulators, Sigma factors and some genes potentially belong to different biosynthetic gene clusters. Indeed, the biosynthetic potential to produce secondary metabolites varied between the Amycolatopsis groups with a strong correlation to the number of biosynthetic gene clusters (Adamek et al., 2018).
Conclusions
Amycolatopsis is a homogeneous genus where most strains conform to the phylogenomic and taxogenomic indices defined for intra-generic boundaries. In contrast, A. palatopharyngis DSM 44832T and A. marina CGMCC4 3568T formed a clade closer to Saccharomonospora strains with comparable taxogenomic distances between them and the other Amycolatopsis strains. We also show that genomic data are robust and reproducible between different laboratories and can help resolve cases of misclassification and misidentification. Some strains identified as A. orientalis should either be assigned to other species or to presumptive novel species. Genomic analyses also assigned some undefined strains to known species. These results provide further evidence that matrices derived from the whole genome sequencing data can provide a robust framework for prokaryotic systematics.
Data Accession
The genome sequence data from this study has been submitted to GenBank/DDBJ/EMBL databases and are publically available with the accession numbers given in Supplementary Table S1.
Author Contributions
VS, MG, IS, and H-PK have designed the study. VS and GT carried out the experimental work. VS and JB analyzed the data. VS drafted the manuscript. All the authors provided intellectual inputs and approved the final version.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank the NU-OMICS facility at Northumbria University for assistance in the genome sequencing and Jimmy Gibson for IT assistance. We also thank Genomic Encyclopedia of Bacteria and Archaea (GEBA) for funding the project CSP 448 for FY2011 that covered sequencing of seven type strains. We acknowledge the BMBF grant FKZ 031A533 within the de.NBI network for supporting the EDGAR platform.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02281/full#supplementary-material
Footnotes
References
Achtman, M., and Wagner, M. (2008). Microbial diversity and the genetic nature of microbial species. Nat. Rev. Microbiol. 6, 431–440. doi: 10.1038/nrmicro1872
Adamek, M., Alanjary, M., Sales-Ortells, H., Goodfellow, M., Bull, A. T., Winkler, A., et al. (2018). Comparative genomics reveals phylogenetic distribution patterns of secondary metabolites in Amycolatopsis species. BMC Genomics 19:426. doi: 10.1186/s12864-018-4809-4
Agren, J., Sundstrom, A., Hafstrom, T., and Segerman, B. (2012). Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS One 7:e39107. doi: 10.1371/journal.pone.0039107
Auch, A. F., Klenk, H. P., and Göker, M. (2010a). Standard operating procedure for calculating genome-to-genome distances based on high-scoring segment pairs. Stand. Genomic Sci. 2, 142–148. doi: 10.4056/sigs.541628
Auch, A. F., Von Jan, M., Klenk, H. P., and Göker, M. (2010b). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Blom, J., Kreis, J., Spanig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Bose, D., Sarkar, I., Labar, R., Oshone, R., Ghazal, S., Morris, K., et al. (2016). Comparative genomics of Prauserella sp. Am3, an actinobacterium isolated from root nodules of Alnus nepalensis in India. Symbiosis 70,49–58. doi: 10.1007/s13199-016-0401-3
Carro, L., Nouioui, I., Sangal, V., Meier-Kolthoff, J. P., Trujillo, M. E., Montero-Calasanz, M. D. C., et al. (2018). Genome-based classification of micromonosporae with a focus on their biotechnological and ecological potential. Sci. Rep. 8:525. doi: 10.1038/s41598-017-17392-0
Chen, S., Wu, Q., Shen, Q., and Wang, H. (2016). Progress in understanding the genetic information and biosynthetic pathways behind Amycolatopsis antibiotics, with implications for the continued discovery of novel drugs. Chembiochem 17, 119–128. doi: 10.1002/cbic.201500542
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., Da Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Davila Costa, J. S., and Amoroso, M. J. (2014). Current biotechnological applications of the genus Amycolatopsis. World J. Microbiol. Biotechnol. 30, 1919–1926. doi: 10.1007/s11274-014-1622-3
Guan, T. W., Xia, Z. F., Tang, S. K., Wu, N., Chen, Z. J., Huang, Y., et al. (2012). Amycolatopsis salitolerans sp. nov., a filamentous actinomycete isolated from a hypersaline habitat. Int. J. Syst. Evol. Microbiol. 62, 23–27. doi: 10.1099/ijs.0.030031-0
Konstantinidis, K. T., and Tiedje, J. M. (2005a). Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. U.S.A. 102, 2567–2572. doi: 10.1073/pnas.0409727102
Konstantinidis, K. T., and Tiedje, J. M. (2005b). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Konstantinidis, K. T., and Tiedje, J. M. (2007). Prokaryotic taxonomy and phylogeny in the genomic era: advancements and challenges ahead. Curr. Opin. Microbiol. 10, 504–509. doi: 10.1016/j.mib.2007.08.006
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kyrpides, N. C., Hugenholtz, P., Eisen, J. A., Woyke, T., Goker, M., Parker, C. T., et al. (2014). Genomic encyclopedia of bacteria and archaea: sequencing a myriad of type strains. PLoS Biol. 12:e1001920. doi: 10.1371/journal.pbio.1001920
Labeda, D. P., Donahue, J. M., Williams, N. M., Sells, S. F., and Henton, M. M. (2003). Amycolatopsis kentuckyensis sp. nov., Amycolatopsis lexingtonensis sp. nov. and Amycolatopsis pretoriensis sp. nov., isolated from equine placentas. Int. J. Syst. Evol. Microbiol. 53, 1601–1605. doi: 10.1099/ijs.0.02691-0
Mahato, N. K., Gupta, V., Singh, P., Kumari, R., Verma, H., Tripathi, C., et al. (2017). Microbial taxonomy in the era of OMICS: application of DNA sequences, computational tools and techniques. Antonie Van Leeuwenhoek 110, 1357–1371. doi: 10.1007/s10482-017-0928-1
Moore, E. R., Mihaylova, S. A., Vandamme, P., Krichevsky, M. I., and Dijkshoorn, L. (2010). Microbial systematics and taxonomy: relevance for a microbial commons. Res. Microbiol. 161, 430–438. doi: 10.1016/j.resmic.2010.05.007
Moose, A. (2017). POCP Calculation for Two Genomes. FigShare. Code. doi: 10.6084/m9.figshare.4577953.v1
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nouioui, I., Carro, L., García-López, M., Meier-Kolthoff, J. P., Woyke, T., Kyrpides, N. C., et al. (2018). Genome-based taxonomic classification of the phylum actinobacteria. Front. Microbiol. 9:2007. doi: 10.3389/fmicb.2018.02007
Pantiukh, K., and Grouzdev, D. (2017). POCP-Matrix Calculation for a Number of Genomes. FigShare. Code. doi: 10.6084/m9.figshare.5602957.v1
Qin, Q. L., Xie, B. B., Zhang, X. Y., Chen, X. L., Zhou, B. C., Zhou, J., et al. (2014). A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215. doi: 10.1128/JB.01688-14
Richter, M., and Rosselló-Móra, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106,19126–19131. doi: 10.1073/pnas.0906412106
Saintpierre-Bonaccio, D., Amir, H., Pineau, R., Tan, G. Y., and Goodfellow, M. (2005). Amycolatopsis plumensis sp. nov., a novel bioactive actinomycete isolated from a New-Caledonian brown hypermagnesian ultramafic soil. Int. J. Syst. Evol. Microbiol. 55, 2057–2061. doi: 10.1099/ijs.0.63630-0
Sanchez-Hidalgo, M., Gonzalez, I., Diaz-Munoz, C., Martinez, G., and Genilloud, O. (2018). Comparative genomics and biosynthetic potential analysis of two lichen-isolated Amycolatopsis strains. Front. Microbiol. 9:369. doi: 10.3389/fmicb.2018.00369
Sangal, V., Goodfellow, M., Jones, A. L., Schwalbe, E. C., Blom, J., Hoskisson, P. A., et al. (2016). Next-generation systematics: an innovative approach to resolve the structure of complex prokaryotic taxa. Sci. Rep. 6:38392. doi: 10.1038/srep38392
Sangal, V., Nieminen, L., Tucker, N. P., and Hoskisson, P. A. (2014). “Revolutionising systematics through next-generation sequencing,” in Methods in Microbiology: Bacterial Taxonomy, eds M. Goodfellow, I. C. Sutcliffe, and J. Chun (Oxford: Elsevier), 75–101.
Schleifer, K. H., Amann, R., and Rossello-Mora, R. (2015). Taxonomy in the age of genomics. Introduction. Syst. Appl. Microbiol. 38, 207–208. doi: 10.1016/j.syapm.2015.05.002
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sutcliffe, I. C. (2015). Challenging the anthropocentric emphasis on phenotypic testing in prokaryotic species descriptions: rip it up and start again. Front. Genet. 6:218. doi: 10.3389/fgene.2015.00218
Tan, G.Y.A., and Goodfellow, M. (2015). “Amycolatopsis,” in Bergey’s Manual of Systematic Bacteriology, ed. W. B. Whitman (Hoboken, NJ: John Wiley & Sons), 1–40.
Tang, B., Xie, F., Zhao, W., Wang, J., Dai, S., Zheng, H., et al. (2016). A systematic study of the whole genome sequence of Amycolatopsis methanolica strain 239(T) provides an insight into its physiological and taxonomic properties which correlate with its position in the genus. Synth. Syst. Biotechnol. 1, 169–186. doi: 10.1016/j.synbio.2016.05.001
Tang, S. K., Wang, Y., Zhang, H., Lee, J. C., Lou, K., Kim, C. J., et al. (2010). Haloechinothrix alba gen. nov., sp. nov., a halophilic, filamentous actinomycete of the suborder Pseudonocardineae. Int. J. Syst. Evol. Microbiol. 60, 2154–2158. doi: 10.1099/ijs.0.018531-0
Wang, J., Leiva, S., Huang, J., and Huang, Y. (2018). Amycolatopsis Antarctica sp. nov., isolated from the surface of an Antarctic brown macroalga. Int. J. Syst. Evol. Microbiol. 68, 2348–2356. doi: 10.1099/ijsem.0.002844
Wei, X. M., Jiang, Y. Y., Chen, X., Jiang, Y., and Lai, H. X. (2015). Amycolatopsis flava sp nov., a halophilic actinomycete isolated from Dead sea. Antonie Van Leeuwenhoek 108, 879–885. doi: 10.1007/s10482-015-0542-z
Keywords: prokaryotic systematics, core genome, average nucleotide identity (ANI), average amino-acid identity (AAI), digital DNA-DNA hybridization, Amycolatopsis
Citation: Sangal V, Goodfellow M, Blom J, Tan GYA, Klenk H-P and Sutcliffe IC (2018) Revisiting the Taxonomic Status of the Biomedically and Industrially Important Genus Amycolatopsis, Using a Phylogenomic Approach. Front. Microbiol. 9:2281. doi: 10.3389/fmicb.2018.02281
Received: 22 June 2018; Accepted: 06 September 2018;
Published: 27 September 2018.
Edited by:
Don A. Cowan, University of Pretoria, South AfricaReviewed by:
Habibu Aliyu, Karlsruher Institut für Technologie (KIT), GermanyVirginia Helena Albarracín, Center for Electron Microscopy (CIME), Argentina
Copyright © 2018 Sangal, Goodfellow, Blom, Tan, Klenk and Sutcliffe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vartul Sangal, dmFydHVsLnNhbmdhbEBub3J0aHVtYnJpYS5hYy51aw==