 Lei Ren1,2
Lei Ren1,2 Yanchun Yan
Yanchun Yan- 1Agricultural College, Guangdong Ocean University, Zhanjiang, China
- 2Graduate School of Chinese Academy of Agricultural Sciences, Beijing, China
- 3Shenzhen Research Institute of Guangdong Ocean University, Shenzhen, China
- 4Faculty of Chemistry and Environmental Science, Guangdong Ocean University, Zhanjiang, China
The genus Arthrobacter is ubiquitously distributed in different natural environments. Many xenobiotic-degrading Arthrobacter strains have been isolated and described; however, few have been systematically characterized with regard to multiple interrelated metabolic pathways and the genes that encode them. In this study, the biodegradability of seven aromatic compounds by Arthrobacter sp. YC-RL1 was investigated. Strain YC-RL1 could efficiently degrade p-xylene (PX), naphthalene, phenanthrene, biphenyl, p-nitrophenol (PNP), and bisphenol A (BPA) under both separated and mixed conditions. Based on the detected metabolic intermediates, metabolic pathways of naphthalene, biphenyl, PNP, and BPA were proposed, which indicated that strain YC-RL1 harbors systematic metabolic pathways toward aromatic compounds. Further, genomic analysis uncovered part of genes involved in the proposed pathways. Both intradiol and extradiol ring-cleavage dioxygenase genes were identified in the genome of strain YC-RL1. Meanwhile, gene clusters predicted to encode the degradation of biphenyl (bph), para-substituted phenols (npd) and protocatechuate (pca) were identified, and bphA1A2BCD was proposed to be a novel biphenyl-degrading gene cluster. The complete metabolic pathway of biphenyl was deduced via intermediates and functional gene analysis (bph and pca gene clusters). One of the these genes encoding ring-cleavage dioxygenase in bph gene cluster, a predicted 2,3-dihydroxybiphenyl 1,2-dioxygenase (BphC) gene, was cloned and its activity was confirmed by heterologous expression. This work systematically illuminated the metabolic versatility of aromatic compounds in strain YC-RL1 via the combination of metabolites identification, genomics analysis and laboratory experiments. These results suggested that strain YC-RL1 might be a promising candidate for the bioremediation of aromatic compounds pollution sites.
Introduction
Widespread environmental pollution and tightening environmental regulations have increased demand for environmental remediation and driven research on pollutant degradation. Biodegradation is recognized as a promising approach for eliminating pollutants from some environmental settings (Boxall et al., 2004; Singh, 2008). Whereas eukaryota mostly transform xenobiotics for detoxification or via fortuitous transformation by broad-spectrum enzymes, prokaryota typically metabolize them for assimilation as essential nutrients and energy (Copley, 2009; Fenner et al., 2013). Numerous bacteria have been isolated and characterized with respect to the molecular mechanisms underlying their metabolic potential for the degradation of environmental pollutants (Copley, 2009; Fenner et al., 2013). Despite early optimism, biodegradation has not been a panacea for environmental pollution. Many issues require further investigation before its full potential can be revealed. For example challenges remain regarding the degradation of pollutants present at low concentrations, present in extreme conditions (i.e., saline-alkali soil, deserts, etc.), present in mixtures with other compounds, and those that might be toxic or metabolized to toxic intermediates (Yagi et al., 2009; Laffin et al., 2010; Narancic et al., 2012).
As one of the most common structural units of organic compounds in nature, the benzene ring and its derivatives are extensively used in industrial production, including daily supplies, agro-products, energy products, etc. Although some structural properties of these compounds make them recalcitrant to degradation, many aromatic-degrading microbes have been isolated (Fuchs et al., 2011; Xu et al., 2015), with metabolic pathways and mechanisms reported for some (Chauhan et al., 2000; Zhang et al., 2012; Shah et al., 2014; Gran-Scheuch et al., 2017), but not all (Goncalves et al., 2006; Zhou et al., 2015). These works and hundreds of others have advanced our knowledge of aromatic compounds degradation. Some of these isolates could degrade several kinds of xenobiotics and have attracted much attention from researchers (Supplementary Table S1). This topic is particularly relevant to environmental remediation since pollutants rarely occur in isolation at contaminated sites, but are rather more likely to part of mixture of compounds. There is, therefore a pressing need to study the related mechanisms of metabolic versatility which is more relevant to the in situ situation.
The genus Arthrobacter was firstly isolated from soil in 1889 and established within the family Micrococcaceae in 1947 (Conn and Dimmick, 1947; Koch et al., 1995). Arthrobacter have been widely isolated from diverse environments including air, soil, and fresh water. They were recognized for nutritional versatility with respect to the decomposition of organic matter and environmental pollutants (Katznelson and Sirois, 1961; Gunner, 1963). Specific reports of the metabolic versatility of Arthrobacter strains have detailed their ability of degrade xenobiotics, such as pesticides, haloalkanes, polycyclic aromatic compounds, and benzene derivatives (Chauhan et al., 2000; Rong et al., 2009; Husserl et al., 2010; Wang et al., 2012; Yao et al., 2015).
In our previous study, a versatile aromatic-degrading bacterium, Arthrobacter sp. YC-RL1, was isolated from petroleum contaminated soil, and its genome was sequenced and published, but without further analysis (Ren et al., 2016a,b). In this study, the metabolic versatility of strain YC-RL1 was investigated by: (i) analyzing its ability to degrade seven aromatic compounds, (ii) detection of the intermediates produced during metabolism, and (iii) investigating the genetic basis likely to encode the observed activities. Our results revealed that: (I) strain YC-RL1 could utilize PX, PNP, naphthalene, phenanthrene, biphenyl and BPA for growth, separately and simultaneously, (II) the metabolic pathways of selected aromatic compounds were deduced via metabolic intermediates analysis which suggested that strain YC-RL1 possesses systematic metabolic pathways toward aromatic compounds, (III) several genes and gene clusters (including a novel gene cluster encoding the transformation of biphenyl to benzoate) contributed to the utilization of these aromatic compounds were identified via comparative genomic analysis, and the activity of a 2,3-dihydroxybiphenyl 1,2-dioxygenase was confirmed via heterologous expression.
Materials and Methods
Bacterial Strains, Growth Conditions, and Chemicals
Arthrobacter sp. YC-RL1 was grown in Luria-Bertani (LB) medium and mineral salt medium (MSM) with PNP or other aromatic compounds at 30°C. Strain YC-RL1 is available from China General Microbiological Culture Collection Center (CGMCC) with accession number CGMCC 10611. Escherichia coli DH5α and E. coli BL21 were used for gene cloning and gene expression, respectively (the detail information was presented in Supplementary Table S2).
The detailed information about aromatic compounds and other reagents used in this study was presented in Supplementary Table S3. All the substrates were dissolved in methanol as stock solution (2 × 104 mg/L) separately, sterilized by membrane filtration (0.22 μm), and then supplemented into MSM to obtain the target concentration. LB and MSM media were prepared as previously described (Ren et al., 2016a).
Biodegradability of Different Aromatic Compounds
An inoculum of strain YC-RL1 was prepared as previously described and then used to determine the biodegradability of different aromatic compounds (Ren et al., 2016a). Two series of measurements were performed: (i) 200 μL of inoculum were added into 20 mL MSM medium supplemented with one of the following substrates: PX, PNP, naphthalene, phenanthrene, biphenyl, BPA or di 2-ethyl hexyl phthalate (DEHP) (50 mg/L, respectively), and cultures without inoculation were set as control; (ii) the substrates above were simultaneously added into 20 mL MSM medium to 50 mg/L each (inoculation of 200 μL inoculum was set as treatment and culture without inoculation was set as control). Meanwhile, inoculation of 200 μL inoculum with 25 μL menthol in 20 mL MSM medium was performed as control too.
Cells were cultivated at 30°C and 180 rpm. After 5 days incubation, samples were extracted twice by an equal volume of ethyl acetate. Extracts were dried by nitrogen-blow and finally dissolved in methanol (equal to the initial volume). The residual concentration of each substrate was then quantified by high-performance liquid chromatography–mass spectrometry (HPLC–MS) analysis. The percent recovery of each substrate was also measured (all above 98.0%). All the experiments were conducted in triplicate.
The calculation of degradation rates described as below:
where Cck represents the final concentration of each substrate in the control treatment, Cf represents the final concentration of each substrate with inoculation of strain YC-RL1.
Identification of Metabolic Pathways
Naphthalene, BP, BPA, and PNP were selected as representative substrates for further analysis. Strain YC-RL1 was precultured as previously described (Ren et al., 2016a) and inoculated into fresh MSM supplemented with one of the selected substrates (50 mg/L) as sole carbon source. All cultures were cultivated under 30°C and 180 rpm. Subcultures were withdrawn every 12 h and extracted as described above.
The quantification of selected aromatic compounds and identification of metabolic intermediates were accomplished by HPLC–MS analysis. A triple quadrupole mass spectrometer (Agilent 6420, United States) equipped with an electrospray ionization (ESI) was employed in this study. Samples were directly injected into detector (10 μL) and the mobile phase was methanol (100%, 0.2 mL/min). The scan mode with a capillary voltage of 3.5 kV for negative ionization was applied and the carrier gas (nitrogen, 99.999%) was heated to 325 (gas flow: 8 L/min). Negative ions were acquired from 100 to 600 Da and analyzed by Mass hunter (version A.02.00, Agilent, United States).
In order to distinguish some easily confused intermediates (benzoate vs. salicylaldehyde and protocatechuate vs. gentisate), standard chemicals were applied to HPLC analysis. An Agilent 1260 system equipped with an Eclipse Plus C18 column (4.6 × 75 mm × 3.5 μm), a column oven, a quaternary pump, a mixer and a diode array detector was used. The mobile phase was composed of methanol and 50 mM acetic acid solution (pH = 3.0) with a constant ratio (9:1) and the flow rate was 2 mL/min. The injection volume was 20 μL.
Based on detected metabolic intermediates and known reports, the metabolic pathways of selected aromatic compounds were deduced. The multiple interrelated metabolic pathways of different aromatic compounds were built thereafter.
Genes and Gene Clusters Involved in Aromatics Metabolism
The whole-genome sequencing of strain YC-RL1 was accomplished using PacBio RS II platform to provide further insights into molecular mechanisms of metabolic versatility. The genome of strain YC-RL1 was annotated and deposited into GenBank. The circular representation of genome and plasmids was generated by circos (version 0.69) (Krzywinski et al., 2009). The genome circular comparison between strain YC-RL1 and 5 Arthrobacter strains (including three aromatic compounds-degrading strain) were performed using BLAST Ring Image Generator (BRIG, version 0.95) (Alikhan et al., 2011). A summary of genome features of strain YC-RL1 and five other Arthrobacter strains were shown in Supplementary Table S4. Further, nucleotide-based genome alignment between YC-RL1 and the Arthrobacter sp. RE117 was performed to study genome rearrangements using the progressive MAUVE algorithm in the MAUVE genome alignment software (version 2.3.1) (Darling et al., 2010).
For the decomposition of aromatic compounds under oxic conditions, ring-cleavage is known as the key step which is mostly mediated by dioxygenases (Vaillancourt et al., 2008). Based on obtained genome sequence and annotation results, all dioxygenase coding sequences were retrieved and manually screened to identify dioxygenases involved in the catabolism of aromatic compounds. Basic Local Alignment Search Tools (BLAST) of NCBI1 was used to locate the target sequences (dioxygenase genes). Their genomic neighborhood were further analyzed to identify possible gene clusters. The identified genes and gene clusters were also further analyzed by RAST web service2 to show their similarity with known genomes (Overbeek et al., 2013; Brettin et al., 2015).
The sequence alignment was performed with Clustal X2 program (version 2.1) (Larkin et al., 2007). Phylogenetic relationship of dioxygenases was demonstrated by MEGA 7.0 using neighbor-joining method (the bootstrap value was set as 1000) (Kumar et al., 2016). The genome sequence of strain YC-RL1 has been deposited into GenBank under accession number NZ_CP013297 and also available from RAST database with accession number 1654525.
Characterization of 2,3-Dihydroxybiphenyl 1,2-Dioxygenase (BphC)
Bacterial strains, plasmids and primers for the amplification and expression of bphC were listed in Supplementary Table S2. The total DNA of strain YC-RL1 was extracted by Bacterial Genomic DNA Extraction kit (Takara, Japan). The targeted gene was obtained by PCR. The Ex Taq® DNA Polymerase (Takara, Japan) was used for the PCR following the protocol. The obtained fragments were inserted into pMD19-T vector and then transformed into E. coli DH5α competent cell. Positive transformants were selected for plasmid extraction. The recombinant plasmid was sequenced by Sangon Biotech Co., Ltd. For the gene expression, the target sequence was inserted into pET32a(+) vector and then transformed into E. coli BL21. Positive transformants were confirmed by sequencing and then cultured in LB media at 37°C. Targeted protein was induced by isopropyl-D-thiogalactopyranoside (IPTG). The cells were harvested by centrifuging at 10,000 rpm for 3 min, washed, and resuspended in the PBS buffer (pH 7.8). The cell suspension was disrupted by sonication and centrifuged at 8,000 rpm for 5 min to obtain the supernatant. The targeted protein was purified from the supernatant by immobilized metal ion affinity chromatography (ÄKTA, GE Healthcare, United States). The purification was confirmed by SDS-PAGE. Finally, the purified protein was used for functional confirmation. The function of BphC was confirmed by measuring the transformation of 2,3-dihydroxy biphenyl, briefly: (i) monitoring the products 2-hydroxy-6-oxo-6-phenylhexa-2,4-dienoate (HOPDA) at 434 nm with a scanning spectrophotometer, (ii) observing the change color of the reaction mixture, (iii) analyzing the generated products by HPLC-MS.
Results
Biodegradability of Different Aromatic Compounds
The degradation rates of selected aromatic compounds, alone and as part of a mixture, is shown in Figure 1. PX was completely degraded when alone and as part of a mixture. The percent removal of PNP, naphthalene, BPA and biphenyl were all above 90.0% under both situations. The percent removal of phenanthrene was 87.9 ± 3.6% and 82.3 ± 4.2% when alone and as part of a mixture, respectively. However, the degradation rates of DEHP was very low and no cell growth was observed which indicated that strain YC-RL1 could not utilize DEHP as carbon source for growth. However, very little DEHP was degraded (12.7 ± 4.1% and 3.6 ± 2.5% under separated and mixed conditions, respectively) due to fortuitous hydrolysis by broad-spectrum hydrolases. These results demonstrated that strain YC-RL1 could utilize different kinds of aromatic compound for growth. As to the inability of DEHP utilization, the lack of necessary hydrolases might be the reason. The decomposition of DEHP consists two main steps: transformation of DEHP into benzoate and utilization of benzoate (Net et al., 2015; Ren et al., 2018). Two ester bond hydrolases, DEHP hydrolase and mono-(2-ethylhexyl) phthalate (MEHP) hydrolase, contributed to the first step (Latorre et al., 2012; Nahurira et al., 2017). These results also indicated that strain YC-RL1 possess systematic metabolic pathways for aromatic compounds and related molecular mechanism.
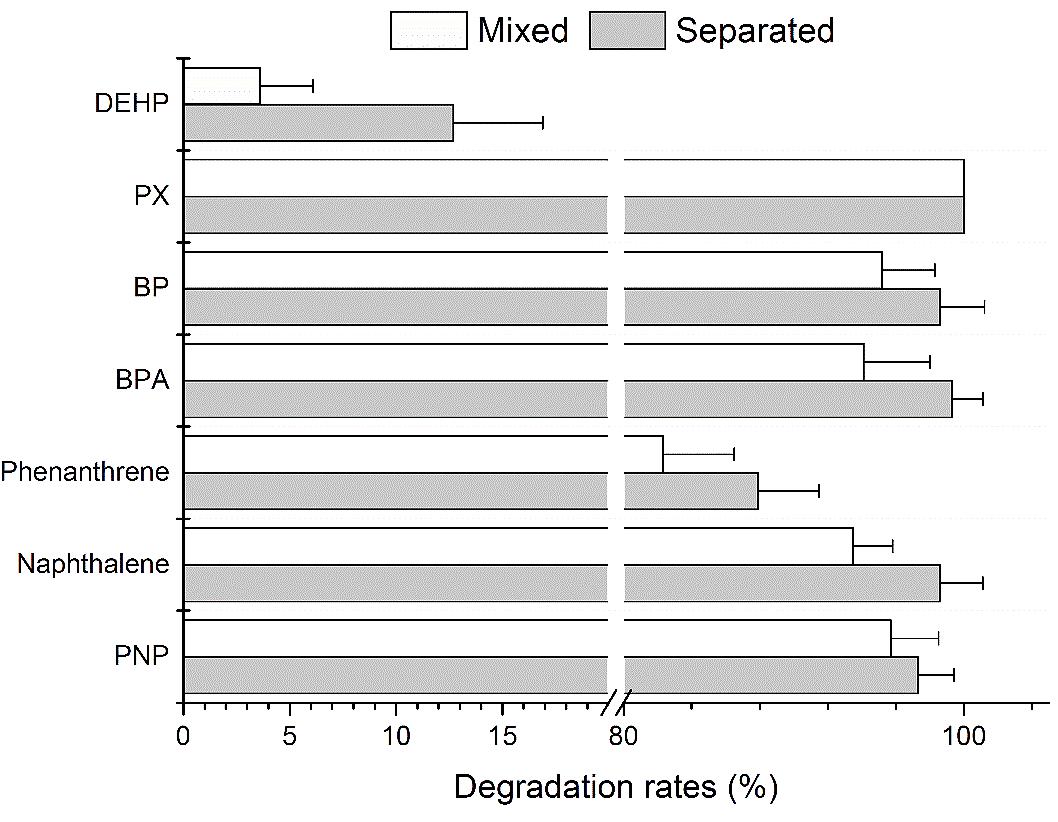
FIGURE 1. The degradation rates of different aromatic compounds under separated and mixed situations.
Identification of Metabolic Pathways
On the basis of HPLC and HPLC-MS analysis (Supplementary Figure S1) and known reports, the potential metabolic intermediates of PNP, naphthalene, biphenyl and BPA were listed in Table 1. Detailed information of detected substances, including m/z, proposed intermediates, molecular formula and molecular weight, were presented too. Some intermediates were simultaneously detected in the degradation of two or more kinds of aromatic compounds (Table 1, marked with gray background), including protocatechuate, hydroxyquinol, and maleylacetate. Given the huge variety of aromatic compounds, it is not surprising that there is a diversity of metabolic pathways for the degradation of these compounds. As reviewed by Fuchs et al. (2011), microbes transform different aromatic compounds via a limited number of intermediates, for example, benzoate, hydroxyquinol, and protocatechuate, which are then susceptible to ring-cleavage reactions. The metabolic pathways of selected aromatic compounds were deduced according to the obtained intermediates information (Figure 2).
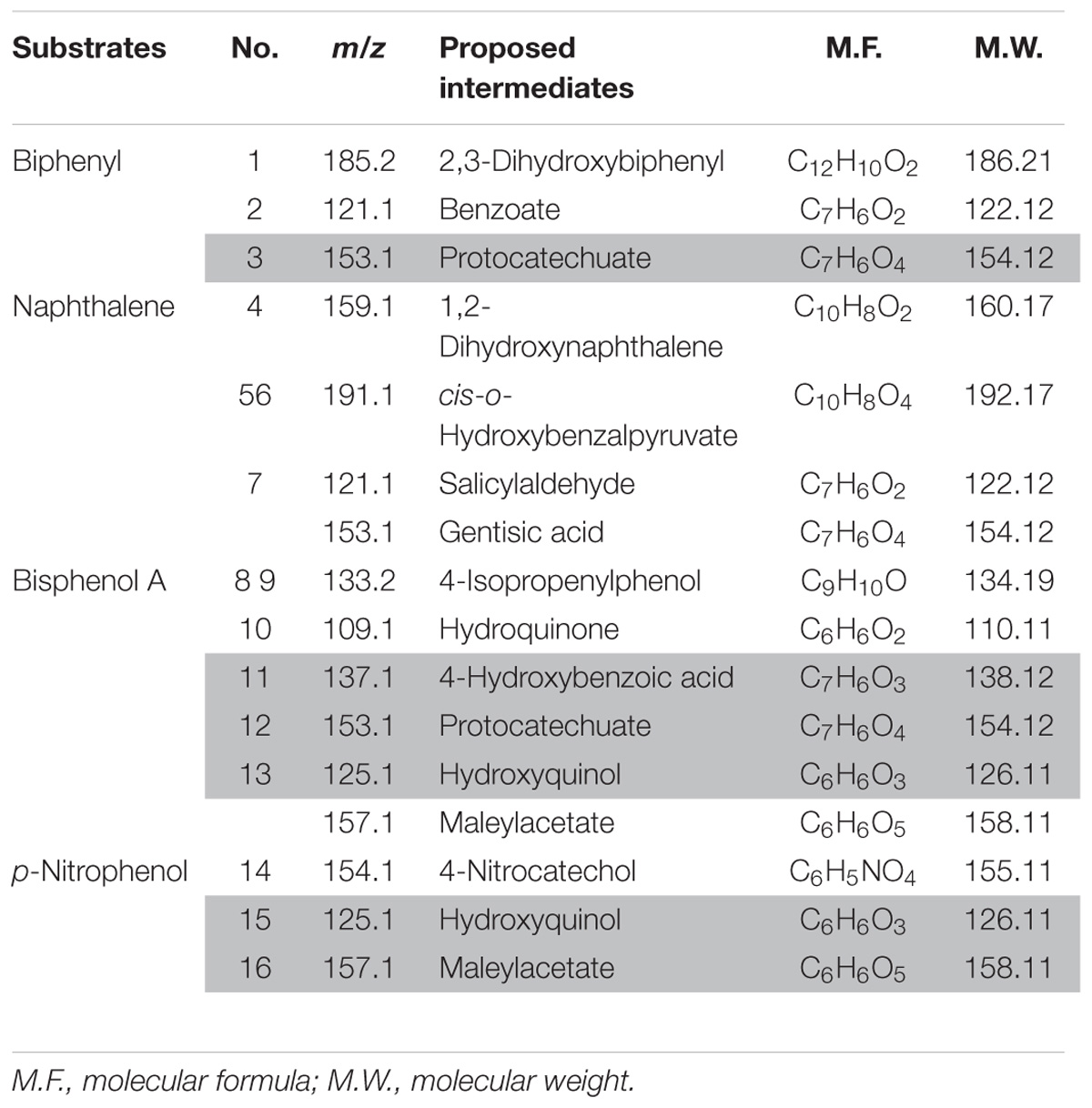
TABLE 1. The metabolic intermediates of aromatic compounds.
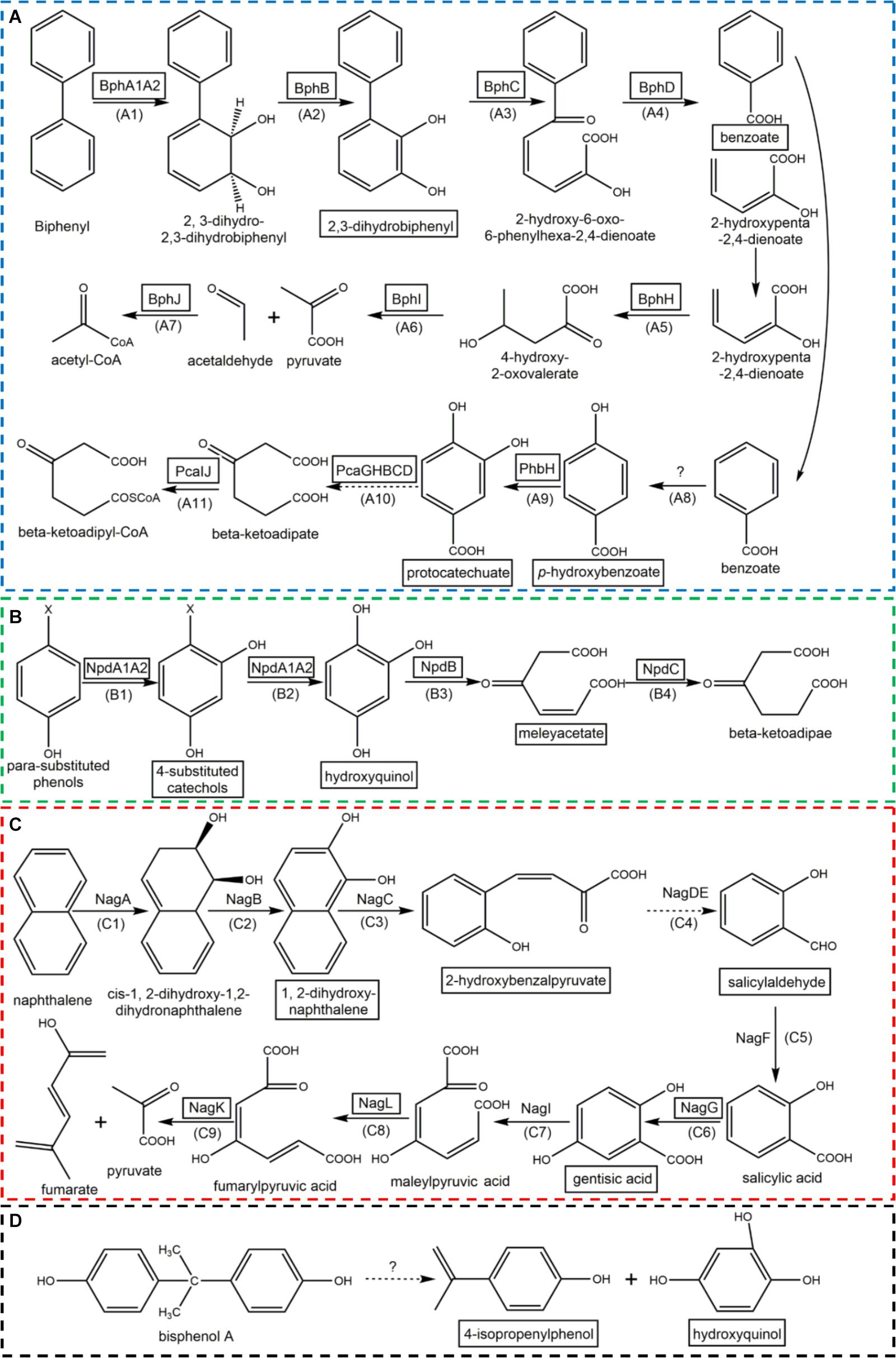
FIGURE 2. The metabolic pathways of biphenyl (A), para-substituted phenols (B), naphthalene (C), and bisphenol A (D). (Enzymes in block diagram indicate that they were identified in strain YC-RL1; metabolic intermediates in block diagram indicate that they were detected by HPLC-MS; the dotted arrows mean there are more than one enzyme contributed to this process; the detailed information of each step (under each arrow) were described in Table 2; ‘?’ means the enzymes involved in were still unknown).
For the metabolism of biphenyl (Figure 2A), biphenyl was transformed into benzoate and 2-hydroxypenta-2,4-dienoate via 2,3-dihydroxybiphenyl (2,3-DHBP, through ring hydroxylation) and HOPDA (through ring-cleavage). Benzoate was then transformed into protocatechuate and further utilized through ring-cleavage (benzoate pathway). The metabolism of PNP (Figure 2B) belongs to well-known 4-nitrocatechol (4-NC) pathway since PNP was transformed into hydroxyquinol via 4-NC. Then, hydroxyquinol entered the hydroxyquinol pathway. The degradation of naphthalene (Figure 2C) was similar with the process of biphenyl. Firstly, naphthalene was transformed into salicylaldehyde via 1,2-dihydroxynaphthalene. Salicylic acid was then generated with the dehydrogenation of salicylaldehyde. Salicylic acid was hydroxylated into gentisic acid. The ring-cleavage of gentisic acid formed maleylpyruvic acid which was then transformed into fumarate and pyruvate via fumaryl-pyruvic acid. For the metabolism of BPA (Figure 2D), hydroxyquinol and 4-isopropenylphenol were identified which indicated that two aromatic rings were separated before ring-cleavage. Hydroxyquinol was utilized via hydroxyquinol pathway. 4-Isopropenylphenol was transformed into protocatechuate via 4-hydroxybenzoate and then entered benzoate pathway.
Genes and Gene Clusters Contributed to the Metabolic Versatility
The genome of strain YC-RL1 is consisted of a circular chromosome (3,846,272 base pairs with an average G + C content of 64.24%) and two plasmids [plasmid 1 (112,763 bp, G + C = 57.81%) and plasmid 2 (59,604 bp, G + C = 58.96%)] (Supplementary Figure S2). A visual inspection of the circular alignment of genomes highlighted sequence similarity between query genomes and reference genome (Figure 3A) and the results indicated that strain YC-RL1 showed high genome similarity with Arthrobacter sp. RE117. The chromosomal alignments between strain YC-RL1 and RE117 indicated that the genome structures between two strains were almost identical (Figure 3B). A total of 170 homologous locally collinear blocks (LCBs) were recognized and these blocks sequentially located on the genome.
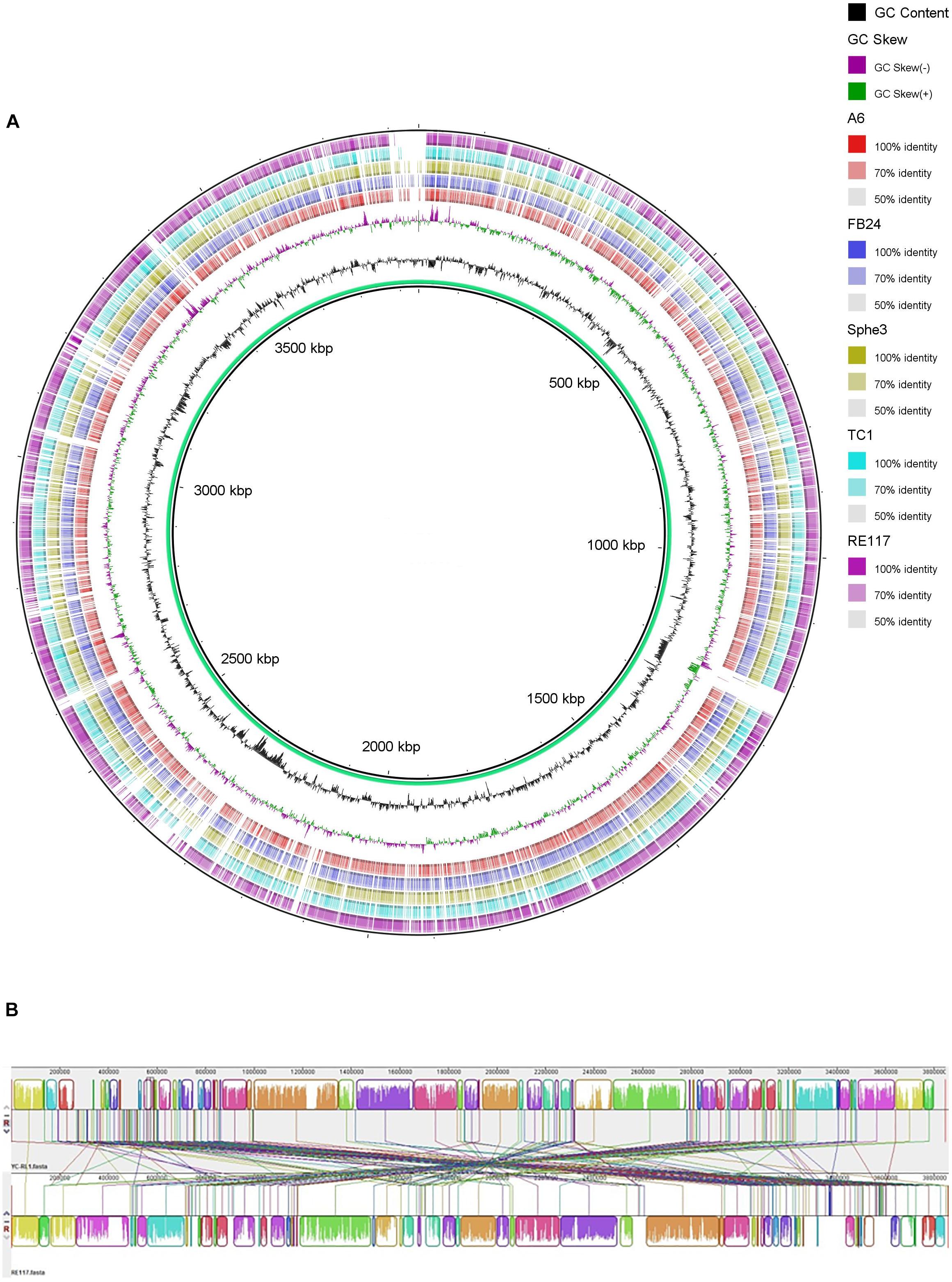
FIGURE 3. Whole-genome comparisons of strain YC-RL1 with five Arthrobacter strains (A) and nucleotide-based genome alignment between strain YC-RL1 and RE117 (B). [(A) The color intensity in each ring represents the BLAST match identity. (B) Homologous blocks were presented with identically colored regions and linked across the sequences].
Genome annotation and comparative genomics analysis of YC-RL1 revealed a variety of genes involved in the metabolism of aromatic compounds (Supplementary Dataset S1). A total of eight dioxygenase genes were found in the genome and plasmids, three of which had homology to ring-cleavage dioxygenase genes (Supplementary Table S5).
Typically, there are two types of dioxygenase involved in the ring-cleavage of aromatic compounds, intradiol and extradiol dioxygenases (Wang et al., 2017). The diversity of dioxygenases significantly affects the metabolic versatility because the substituted groups on the benzene ring decide the position of ring-cleavage and potential toxicity of the substrate to the enzyme itself. To have an in-depth understanding of the metabolic versatility, three ring-cleavage dioxygenases were selected for further analysis. Reported ring-cleavage dioxygenases were downloaded from GenBank. As shown in Figure 4, both intradiol and extradiol ring-cleavage dioxygenases were detected in YC-RL1. As the substituted groups on the benzene ring decide the position of ring-cleavage, the diversity of ring-cleavage dioxygenases partially explained the metabolic versatility of strain YC-RL1.
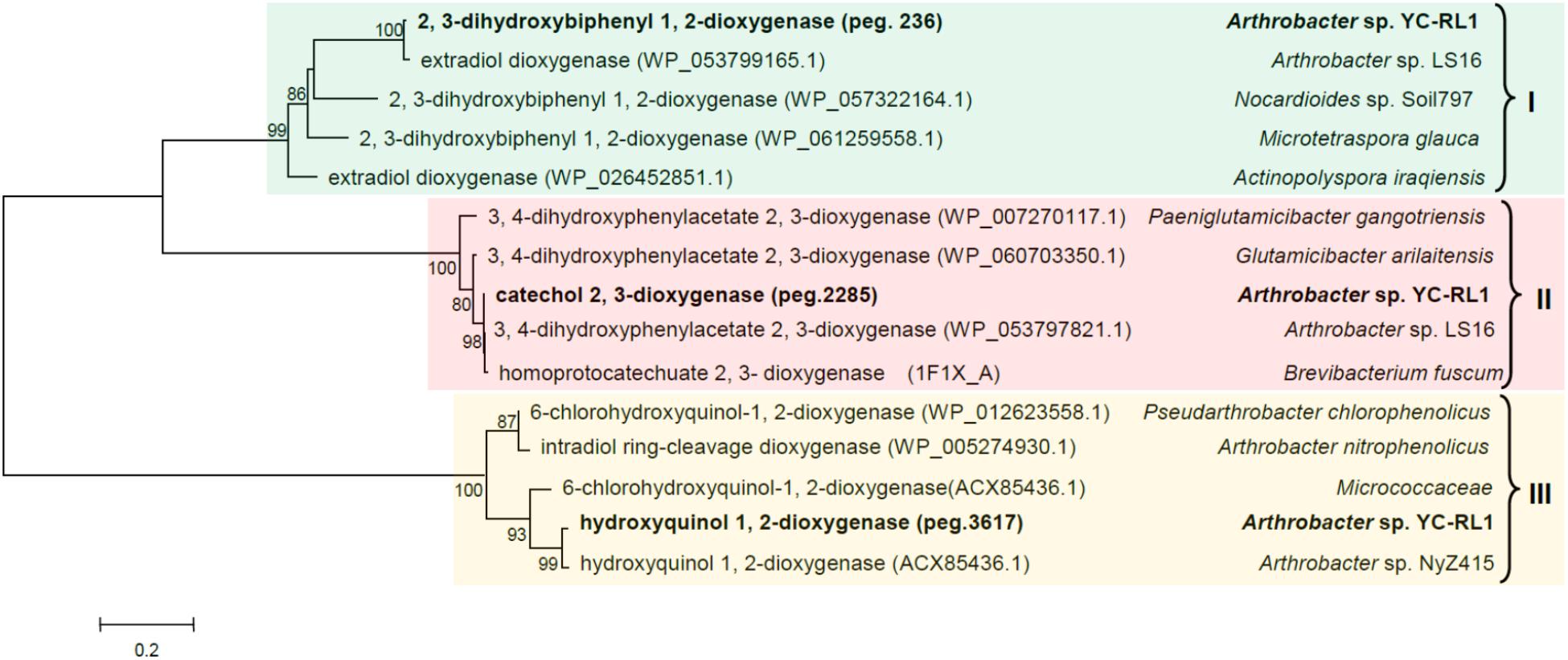
FIGURE 4. Phylogenetic tree of three ring-cleavage dioxygenases in YC-RL1 with known ring-cleavage dioxygenases. (Three different colors suggested three different categories of dioxygenase; type I and type II are extradiol dioxygenase, type III is intradiol dioxygenase; the accession numbers of each sequence were included in the parentheses. The phylogenetic tree was constructed by MEGA 7.0 software using neighbor-joining method and the bootstrap value was set as 1000. Gaps and hypervariable regions were remained for the final alignment and the number of positions used for the final alignment is 381. The scale bar represents the number of expected changes per site).
Genes flanking ring-cleavage dioxygenase genes were searched and conserved clusters were identified by RAST server (Supplementary Dataset S2). All the identified genes and gene clusters were manually checked. Three conserved gene clusters involved in aromatic-degradation were identified by comparative genomics analysis, including npd (encoding para-substituted phenols metabolic pathway), bph (a novel gene cluster encoding biphenyl degradation), and pca (encoding protocatechuate branch of β-ketoadipate pathway) (Table 2) (Perry and Zylstra, 2007; Liu et al., 2010; Pham and Sylvestre, 2013). The sequence similarities of npd and pca genes with known reports were all above 90%. As the pca gene cluster has been extensively investigated, we would not describe and discuss it here (Harwood and Parales, 1996).
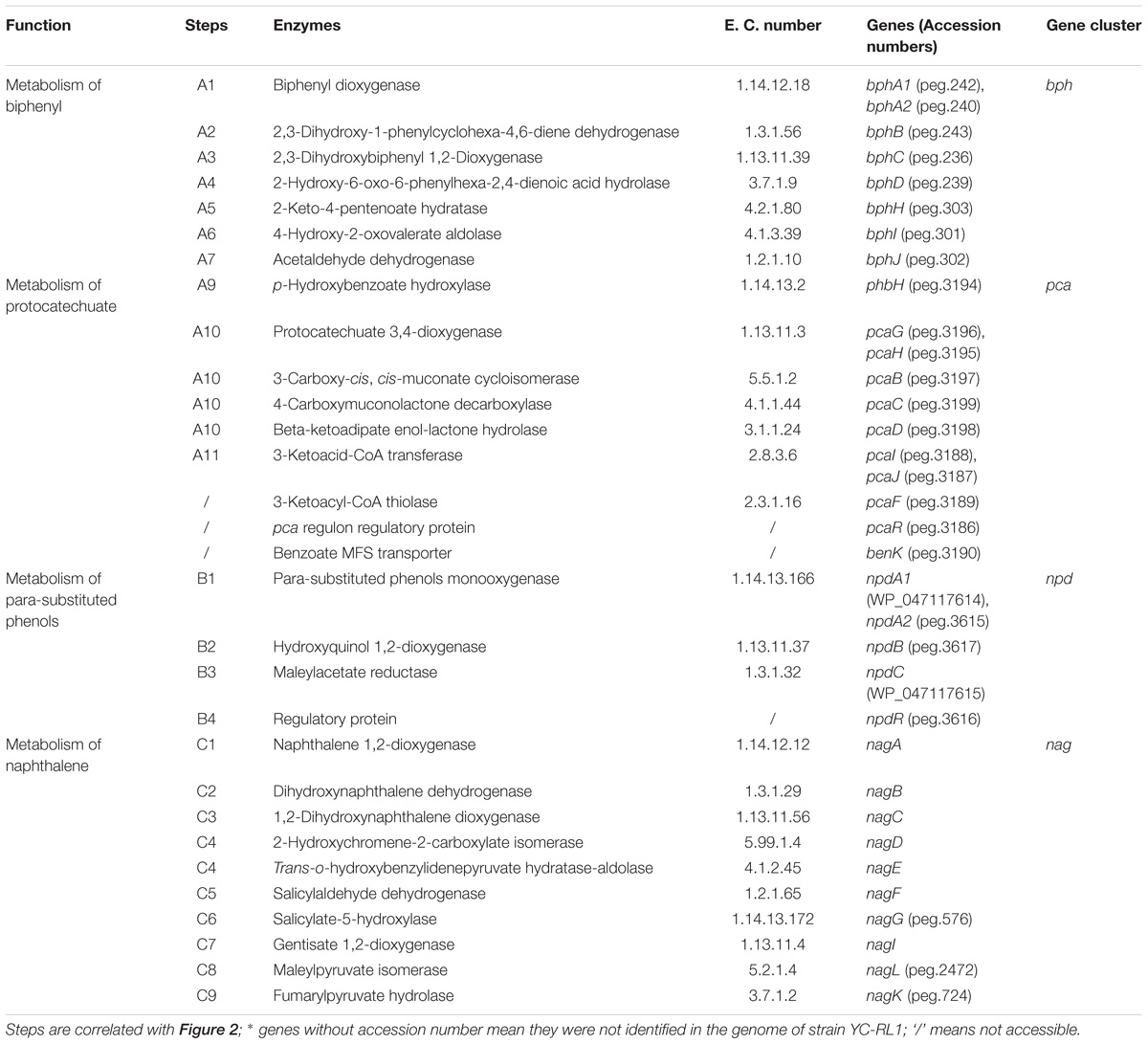
TABLE 2. Enzymes involved in the metabolism of biphenyl, protocatechuate, para-substituted phenols, and naphthalene.
The complete metabolic pathway of biphenyl includes three main steps (Figure 2): (I) transformation of biphenyl into 2,3-dihydroxybiphenyl, in which O2 was introduced at the 2,3-position of biphenyl to produce dihydrodiol and then dehydrogenated to 2,3-dihydroxybiphenyl; (II) ring-cleavage of 2,3-dihydroxybiphenyl and then the hydrolysis of the product (HOPDA) to produce benzoate and 2-hydroxypenta-2,4-dienoate; (III) utilization of benzoate and 2-hydroxypenta-2,4-dienoate. Based on homology to well characterized biphenyl degradation genes, three gene clusters were identified in the genome of strain YC-RL1 that are likely to encode the complete degradation of biphenyl (Figure 5A: bphA1A2BCD, bphHIJ, and pcaGHBCD). The homology analysis of Bph enzymes were performed with known records (Figure 5B) and the results indicated that gene cluster bphA1A2BCD was a novel biphenyl-degrading gene cluster. This novel gene cluster encodes enzymes initialed the degradation of biphenyl, including a biphenyl dioxygenase genes (bphA1A2), a dihydrodiol dehydrogenase gene (bphB), a 2,3-dihydroxybiphenyl dioxygenase gene (bphC), a HOPDA hydroxylase gene (bphD), and a potential transcriptional regulator gene (bphR) (Figure 5A). Gene cluster bphHIJ was identified next to bphA1A2BCD and likely encodes for the transformation of 2-hydroxypenta-2,4-dienoate into acetyl-CoA. For the utilization of benzoate, one widely reported pca gene cluster was identified and located on the chromosome. Benzoate was utilized via protocatechuate branch of β-ketoadipate pathway, which was encoded by pcaGHBCD. Genes (pcaIJF), responsible for the utilization of β-ketoadipate, were also identified in pca gene cluster. Meanwhile, p-hydroxybenzoate hydroxylase gene (phbH) and benzoate MFS transporter gene (benK) were also identified in pca gene cluster. This is the first report of the complete biphenyl metabolic pathway and related genes in genus Arthrobacter.
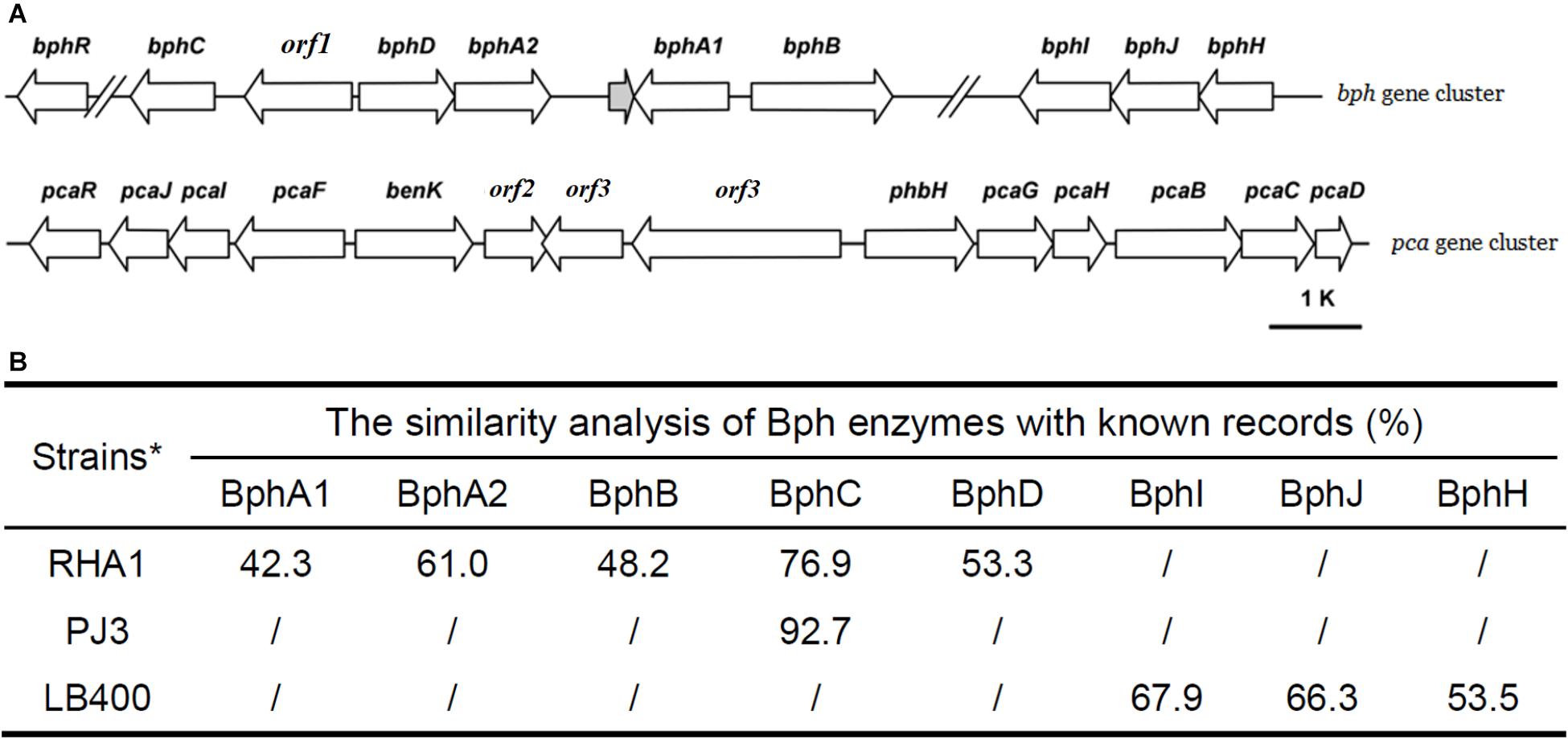
FIGURE 5. The genetic organization of bph and pca gene cluster (A) and the similarity analysis of Bph enzymes with known records (B). [‘//’ means a long distance between two genes; the distance between bphC and bphR is 7192 bp; the distance between bphB and bphI is 62,400 bp; RHA1: Rhodococcus sp. RHA1 (Masai et al., 1997), LB400: Pseudomonas sp. LB400 (Hofer et al., 1994)].
There are three key steps in the degradation of para-substituted phenols (Figure 2B): (i) addition of hydroxyl groups to the substrates, (ii) cleavage of the aromatic ring, and (iii) reduction of maleylacetate. In strain YC-RL1, three enzymes and one transcription regulator involved in the degradation of PNP were identified. Known gene clusters (in genus Arthrobacter) involved in the degradation of para-substituted phenols were presented in Figure 6A. The degradation of para-substituted phenols was initialed by a two-component monooxygenase (NpdA1 and NpdA2, reductase and oxygenase components, respectively). This monooxygenase system is a typical class D flavoprotein monooxygenases (two-component flavin-diffusible monooxygenase, TC-FDM). The reductase component (NpdA1) of monooxygenase is able to reduce FAD/FMN at the expense of NAD(P)H. NADH is the preferred electron donor and FAD is the preferred electron acceptor. Then, the oxygenase component (NpdA2) of monooxygenase receives electrons from the FADH2 and hydroxylates the substrates with O2. The generated hydroxyquinol is further applied for ring-cleavage which is mediated by a hydroxyquinol 1,2-dioxygenase (NpdB). The product (maleyacetate) is further hydrolyzed to β-ketoadipate and then enters the TCA cycle.
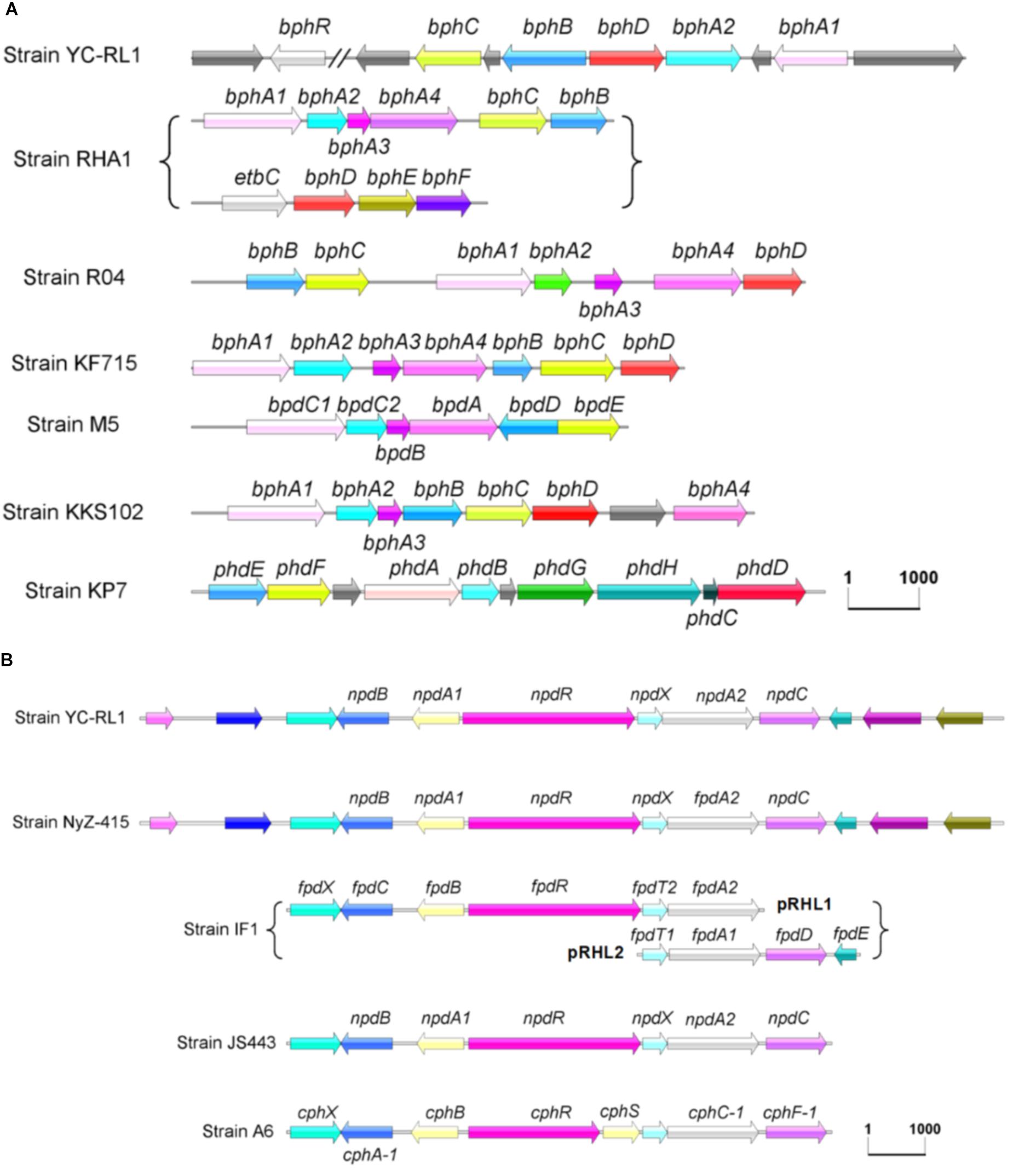
FIGURE 6. Gene clusters involved in the degradation of biphenyl (A) and para-substituted phenols (B). (Strain RHA1: Rhodococcus sp. RHA1, Strain R04: Rhodococcus sp. R04, Strain KF715: Pseudomonas putida KF715, Strain M5: Rhodococcus sp. M5, Strain KKS102: Achromobacter georgiopolitanum KKS102, Strain KP7: Nocardioides sp. KP7, Strain NyZ-415: Arthrobacter sp. NyZ415, Strain IF1: Arthrobacter sp. IF1, Strain JS443: Arthrobacter sp. JS443, Strain A6: Arthrobacter chlorophenolicus A6).
For the degradation of naphthalene, genes contributed to the peripheral pathway were not found and three genes (nagG, nagL, and nagK) involved in the utilization of salicylic acid were identified. Same situation occurred with BPA degradation and one cytochrome P450 protein (peg.429) potentially involved in the degradation of BPA was identified, which could transform BPA into 1, 2-bis(4-hydroxyphenyl)-2-propanol and 2,2-bis(4-hydroxyphenyl)-1-propanol. The similarity of the identified cytochrome P450 protein with known BPA-degrading cytochrome P450 protein (BAG15884) is 47% (Sasaki et al., 2008). Other enzymes related to BPA metabolism are still unknown and further investigation is needed.
Functional Confirmation of 2,3-Dihydroxybiphenyl 1,2-Dioxygenase (BphC)
The bphC gene with an open reading frame of 930 bp was cloned and inserted into a pET32a(+) vector. The expression and purification of BphC was confirmed by SDS-PAGE (Supplementary Figure S3). The calculated molecular weight (M.W.) of BphC is 34.14 kDa and the tagged-sequence has a M.W. of approximately 20 kDa. The recombinant BphC protein with tagged-sequence was successfully purified and appeared as a 54 kDa protein on the SDS gel. The purified BphC was applied to the transformation of 2,3-dihydroxybiphenyl. The activity was measured by monitoring the products (HOPDA) with a scanning spectrophotometer. The specific absorption of HOPDA was observed at 434 nm and the maximum absorption value was at 30 min. Results shown that, the absorption values at 434 nm increased with the time (Figure 7A). That means the concentration of HOPDA increased with the reaction. BphD was able to degrade 86.3 ± 4.2% of 50 mg/L 2,3-dihydroxybiphenyl in 30 min at 25°C accompanied with color change (colorless to yellow, Figure 7B). The generated yellow products were further analyzed by HPLC-MS and showed a m/z of 217 (M.W. = 218) which suggested the yellow product was HOPDA (Figure 7C).
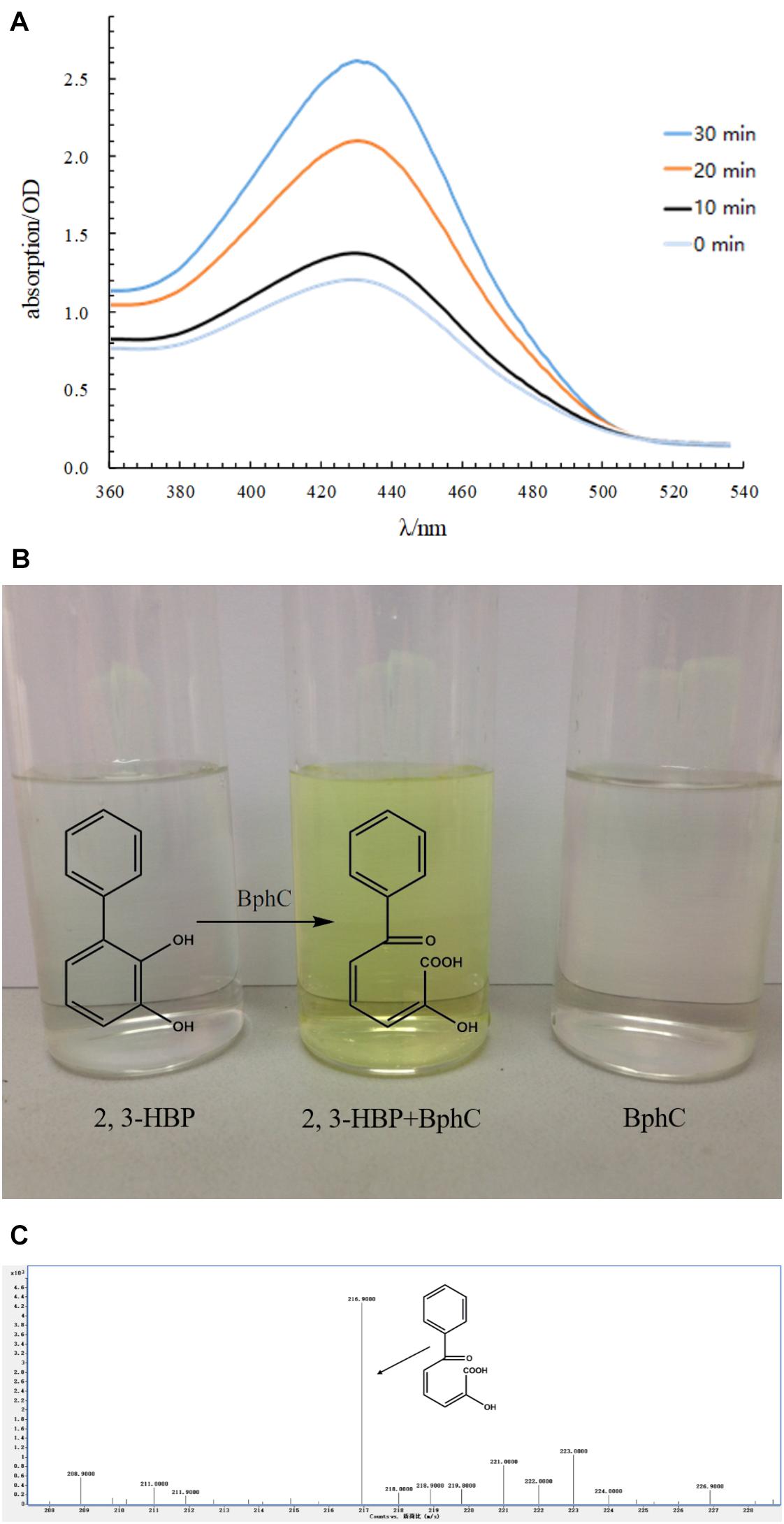
FIGURE 7. Functional characterization of BphC. (A: Monitoring the production of HOPDA using a scanning spectrophotometer; B: Reaction of BphC and 2,3-dihydroxy biphenyl (2,3-HBP); C: HPLC-MS analysis of the yellow products).
Discussion
The metabolic versatility of microbes is an important characteristic of their role in the cycling of matter and the detoxification of hazardous compounds (Larimer et al., 2003; Llamas et al., 2006). Most reports about genus Arthrobacter have focused on their role in the metabolism of xenobiotics, especially benzenoids and their derivatives, as well as heterocyclic compounds and hydrocarbons. For example, nicotine degrading strain M2012083 was isolated from tobacco waste and identified as Arthrobacter aurescens. The comparative genome analysis of strain M2012083 revealed the molecular basis of nicotine degradation and survival capacities of Arthrobacter species (Yao et al., 2015). Many Arthrobacter species were found to be involved in the catabolism of substituted phenols, such as 4-nitrophenol, 4-chlorophenol, and 4-fluorophenol (Nordin et al., 2005; Perry and Zylstra, 2007; Ferreira et al., 2009). For strain YC-RL1, the efficient utilization of several kinds of aromatic compounds was confirmed which suggested its metabolic versatility and application potential.
As to the failure of DEHP utilization, the lack of necessary esterase might be the reason. To verify this hypothesis, two works were further conducted: (i) all the annotations of hydrolase and esterase were manually checked to identify potential enzymes involved in the hydrolysis of ester bond in DEHP; (ii) the sequences of all reported esterase/hydrolase involved in the hydrolysis of phthalic acid esters (PAEs) were downloaded and searched against the genome of strain YC-RL1 to identify related enzymes. All annotated esterases/hydrolases were shown in Supplementary Dataset S3. A total of 33 hydrolases and 8 esterases were identified and only 7 hydrolases were related to metabolism of aromatic compounds. All these esterases and hydrolases were manually checked by BLAST and the results indicated that none of these was related to the metabolism of DEHP or PAEs. As reviewed by Ren et al. (2018), 14 esterases/hydrolases, belonging to four families of esterase and one unknown family, were reported to be involved in the metabolism of PAEs until now. All these 14 esterases/hydrolases were searched against the genome of strain YC-RL1 and no positive result was obtained. Meanwhile, the reports of Arthrobacter isolates with the ability of DEHP (or PAEs) utilization were limited too (Chatterjee and Dutta, 2008; Wen et al., 2014). These indicated that the lack of esterase (which contributed to the hydrolysis of ester bonds in PAEs) in genus Arthrobacter might be a general situation.
As described by Fuchs et al. (2011), biodegradation of aromatic compounds is typically divided into two main steps, peripheral (upper) pathways and central (lower) pathways: microbes metabolize a diversity of aromatic compounds via peripheral pathways into a few key common intermediates such as catechol, protocatechuate and benzoate, etc. Central pathways then cleave the aromatic ring and convert these intermediate products into central metabolites such as acetyl-CoA, succinyl-CoA and pyruvate, which can be used for microbial growth. This metabolic convergence is a typical and useful strategy for microbial growth that limits the genetic burden necessary to encode the metabolism of a vast array of natural and man-made products. In strain YC-RL1, different kinds aromatics were transformed into several kinds of metabolic intermediates (salicylic acid, protocatechuate, hydroxyquinol, etc.) via different peripheral pathways. As to the central pathway, the utilization of these intermediates has been widely and systematically investigated such as benzoate pathway, hydroxyquinol pathway, and beta-ketoadipate pathway.
Based on the whole-genome analysis, three dioxygenases involved in the ring-cleavage were identified, including both intradiol and extradiol dioxygenases. Gene clusters contributed to the catabolism of para-substituted phenols, biphenyl, and protocatechuate were identified. Of these, gene cluster bphA1A2BCD (contributed to the peripheral pathway of biphenyl catabolism) was a novel biphenyl-degrading gene cluster.
For the degradation of para-substituted phenols, Arthrobacter was known as one of the predominant degraders. The degradation pathways have been proposed in some isolated strains, including Arthrobacter sp. IF1 (4-fluorophenol), Arthrobacter sp. JS443 (4-nitrophenol), Arthrobacter chlorophenolicus A6 (4-chlorophenol and 4-nitrophenol), and Arthrobacter sp. NyZ415 (4-nitrophenol) (Nordin et al., 2005; Perry and Zylstra, 2007; Ferreira et al., 2009; Liu et al., 2010). Although the location of gene clusters and the gene names were different, this gene cluster (npd gene cluster, Figure 6A) is highly conserved and only reported in Arthrobacter species. However, some differences exist: (i) this gene cluster was located on genome except for YC-RL1 (plasmid2), (ii) genes in this gene cluster were located in a same region except for strain IF1, in which genes were separated in two regions, and (iii) the transcriptional regulator NpdR in strain YC-RL1 contains a conserved bacterial transcriptional activator domain (BTAD), which belongs to the DNRI/REDD/AFSR family of regulators (obtained from SMART database, data not shown), but the transcriptional regulator in strain A6 was divided into two [CphR (AAN08756) and CphS (AAN08755)]. The identification of npd gene cluster explained the degrading ability of para-substituted phenols in strain YC-RL1. Further, we compared the G + C content (mol%) of genome (64.26%), plasmid2 (58.96%) and npd gene cluster (59.42%). This indicated that plasmid2, including npd gene cluster, was possibly obtained by horizontal transfer. These results may also provide important information to evaluate the origin and evolution of npd gene cluster.
Although several Arthrobacter strains which could degrade biphenyl have been isolated, the complete gene clusters contributed to the metabolism of biphenyl have not been identified in genus Arthrobacter. Péloquin and Greer (1993) obtained the gene cluster (DNA fragment, not the complete sequence information) contributed to the degradation of biphenyl and sequenced the partial sequence of bphC. Yang M. et al. (2007) identified and characterized BphC in Arthrobacter PJ3. The identification of complete gene clusters involved in the degradation of biphenyl, especially the identification of the novel bph gene cluster, would advance the investigation of biphenyl metabolism. And different from npd gene cluster, bph gene cluster showed high diversity (both sequence similarity and structure of gene cluster, Figure 6B), even the catabolic pathways were same.
For the metabolism of biphenyl, a multi-component biphenyl dioxygenase, Rieske-type oxygenase (ROs), initials the process by adding hydroxy group to generate dihydrodiol compound (Colbert et al., 2000). The ROs contains a large group of aromatic ring-hydroxylating dioxygenases, which could stereoselectively introduce two hydroxy groups in one enzymatic step (Fernández de las Heras et al., 2012; Chen et al., 2014; Gally et al., 2015). The ROs is always comprised of an oxygenase (BphA1) and a reductase (BphA2). The oxygenase contains a Rieske domain with a [2Fe–2S] cluster and a catalytic domain with a mononuclear Fe(II) binding site. The Rieske [2Fe–2S] cluster accepts electrons from the reductase and transfers them to the mononuclear iron for catalysis. The product, 2,3-dihydroxy-1-phenylcyclohexa-4,6-diene (dihydrodiol compound), is then dehydrogenated to 2,3-dihydroxybiphenyl by a dehydrogenase. The gene coding for the dehydrogenase varies from different species and shows high similarity with 3-ketosteroid-Δ-dehydrogenase, which is involved in the transformation of steroid compounds (Itagaki et al., 1990; Choi et al., 1995; Fernández de las Heras et al., 2012). The dioxygenases which mediated the ring-cleavage of 2,3-dihydroxybiphenyl at the 1, 2-position shows high similarity. Genes coding for 2,3-dihydroxybiphenyl dioxygenases are always selected as the probe for the detection of bph gene cluster and become the most extensively investigated (Hauschild et al., 1996; Sugimoto et al., 1997; Sakai et al., 2002; Yang X. et al., 2007). Then, the product (HOPDA) is hydrolyzed by a C–C bond hydrolase (BphD). The location of the hydrolase gene (bphD) varies from different species. In some species, bphD is clustered with bphABC like Rhodococcus sp. R04, Pseudomonas putida KF715, Achromobacter georgiopolitanum KKS102, Rhodococcus sp. M5, and Nocardioides sp. KP7 (Kikuchi et al., 1994; Labbe et al., 1997; Nishi et al., 2000; Saito et al., 2000; Yang X. et al., 2007). In some other species, bphD is separated from bphABC and even locates on different plasmids such as Rhodococcus sp. RHA1, one of the most extensively investigated biphenyl degrading strain (Masai et al., 1997). The products of C-C bond hydrolysis, benzoate and 2-hydroxypenta-2,4-dienoate, are further utilized via protocatechuate branch of beta-ketoadipate pathway (benzoate) and BphHIJ mediated pathway (2-hydroxypenta-2,4-dienoate).
In strain YC-RL1, the biphenyl dioxygenase system was consisted of an oxygenase component (BphA1) and a reductase component (BphA2). The conserved domain analysis showed that BphA2 comprised a ferredoxin reductase like oxidoreductase domain and a 2Fe–2S iron–sulfur cluster binding domain. The 2Fe–2S iron–sulfur cluster binding domain transferred electrons from NADH to FAD to form FADH2. The electrons in FADH2 were further transferred by the ferredoxin reductase like oxidoreductase domain. The oxygenase component (BphA1), containing a Rieske (2Fe–2S) domain, accepted electrons from the reductase component and transferred them to the mononuclear iron for catalysis, which transformed biphenyl into dihydrodiol compound. The dihydrodiol dehydrogenase (BphB) showed high similarity to 3-ketosteroid-Δ1-dehydrogenase, which catalyzed the introduction of a double bond into the position of carbon 1 and 2 of ring A of 3-ketosteroid. The dihydrodiol compound was dehydrogenated into 2,3-dihydroxybiphenyl by BphB. Then, 2,3-dihydroxybiphenyl was cleaved at the meta- position by a highly conserved dioxygenase (BphC), which belongs to the extradiol dioxygenase. The C–C bond hydrolase (BphD) worked on the meta-cleavage product (HOPDA). The hydrolysis product of HOPDA, benzoate, was further utilized via protocatechuate branch of beta-ketoadipate pathway. The protocatechuate and hydroxyquinol branches of beta-ketoadipate pathways showed significant differences and they are responsible for benzoic and phenolic intermediates. Another hydrolysis product of HOPDA, 2-hydroxypenta-2,4-dienoate, was transformed into 4-hydroxy-2-oxovalerate by a 2-keto-4-pentenoate hydratase (BphH). Then, 2-keto-4-pentenoate was degraded into pyruvate and acetaldehyde via 2-keto-4-pentenoate aldolase (BphI). Acetaldehyde was further transformed into acetyl-CoA by acetaldehyde dehydrogenase (BphJ). Based on the above results, the complete metabolic pathways and enzymes involved in the metabolism of biphenyl were identified via metabolic and genomic analysis.
This work systematically demonstrated the metabolic versatility of aromatic compounds in Arthrobacter sp. YC-RL1 via metabolic and genomic analysis. Future characterization of the identified enzymes presented in this work, to understand their function in the metabolism of aromatic compounds, as well as their contribution to the metabolic versatility of strain YC-RL1, will help to the development of efficient strategies for the in situ bioremediation of aromatic compounds contaminated sites.
Author Contributions
LR and YY designed the research. LR, YJ, ZL, and RZ contributed to experimental work. LR, ZZ, and HH finished the data analysis. LR and YY wrote and revised the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (31700097, 41301252, 31170119, 31800109, and 31540067), The Future Industrial Development Fund of Shenzhen (JCYJ20170413111950426), Natural Science Foundation of Guangdong (2016A030310330 and 2018A03 0313131), Guangdong Scientific and Technological Research Program (2013B020309010), and Program for Scientific Research start-up Funds of Guangdong Ocean University (R18013).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We appreciate Anthony G. Hay (Department of Microbiology, Cornell University) for his kind help on the revision of the manuscript. We also appreciate the reviewers for their kind comments and insightful suggestions to improve the earlier version of manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02438/full#supplementary-material
Footnotes
References
Alikhan, N. F., Petty, N. K., Zakour, B. N. L., and Beatson, S. A. (2011). BLAST Ring Image Generator (BRIG): simple prokaryote genome comparisons. BMC Genomics 12:402. doi: 10.1186/1471-2164-12-402
Boxall, A. B., Sinclair, C. J., Fenner, K., Kolpin, D., and Maund, S. J. (2004). When synthetic chemicals degrade in the environment. Environ. Sci. Technol. 38, 368A–375A. doi: 10.1021/es040624v
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Chatterjee, S., and Dutta, T. K. (2008). Metabolic cooperation of Gordonia sp. strain MTCC 4818 and Arthrobacter sp. strain WY in the utilization of butyl benzyl phthalate: effect of a novel co-culture in the degradation of a mixture of phthalates. Microbiology 154, 3338–3346. doi: 10.1099/mic.0.2008/021881-0
Chauhan, A., Chakraborti, A. K., and Jain, R. K. (2000). Plasmid-Encoded degradation of p-nitrophenol and 4-nitrocatechol by Arthrobacter protophormiae. Biochem. Biophys. Res. Commun. 270, 733–740. doi: 10.1006/bbrc.2000.2500
Chen, Q., Wang, C. H., Deng, S. K., Wu, Y. D., Li, Y., Yao, L., et al. (2014). Novel three-component Rieske non-heme iron oxygenase system catalyzing the N-dealkylation of chloroacetanilide herbicides in sphingomonads DC-6 and DC-2. Appl. Environ. Microbiol. 80, 5078–5085. doi: 10.1128/AEM.00659-14
Choi, K. P., Molnar, I., Yamashita, M., and Murooka, Y. (1995). Purification and characterization of the 3-ketosteroid-Δ1-dehydrogenase of Arthrobacter simplex produced in Streptomyces lividans. J. Biochem. 117, 1043–1049. doi: 10.1093/oxfordjournals.jbchem.a124804
Colbert, C. L., Couture, M., Eltis, L. D., and Bolin, J. T. (2000). A cluster exposed: structure of the Rieske ferredoxin from biphenyl dioxygenase and the redox properties of Rieske Fe-S proteins. Structure 8, 1267–1278. doi: 10.1016/S0969-2126(00)00536-0
Conn, H. J., and Dimmick, I. (1947). Soil bacteria similar in morphology to Mycobacterium and Corynebacterium. J. Bacteriol. 54, 291–303.
Copley, S. D. (2009). Evolution of efficient pathways for degradation of anthropogenic chemicals. Nat. Chem. Biol. 5, 559–566. doi: 10.1038/nchembio.197
Darling, E. A., Mau, B., and Perna, T. N. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
Fenner, K., Canonica, S., Wackett, L. P., and Elsner, M. (2013). Evaluating pesticide degradation in the environment: blind spots and emerging opportunities. Science 341, 752–758. doi: 10.1126/science.1236281
Fernández de las Heras, L., van der Geize, R., Drzyzga, O., Perera, J., and María Navarro Llorens, J. (2012). Molecular characterization of three 3-ketosteroid-Δ1-dehydrogenase isoenzymes of Rhodococcus ruber strain Chol-4. J. Steroid. Biochem. Mol. Biol. 132, 271–281. doi: 10.1016/j.jsbmb.2012.06.005
Ferreira, M. I. M., Iida, T., Hasan, S. A., Nakamura, K., Fraaije, M. W., Janssen, D. B., et al. (2009). Analysis of two gene clusters involved in the degradation of 4-fluorophenol by Arthrobacter sp. strain IF1. Appl. Environ. Microbiol. 75, 7767–7773. doi: 10.1128/AEM.00171-09
Fuchs, G., Boll, M., and Heider, J. (2011). Microbial degradation of aromatic compounds - from one strategy to four. Nat. Rev. Microbiol. 9, 803–816. doi: 10.1038/nrmicro2652
Gally, C., Nestl, B. M., and Hauer, B. (2015). Engineering Rieske non-heme iron oxygenases for the asymmetric dihydroxylation of alkenes. Angew. Chem. Int. Ed. Engl. 54, 12952–12956. doi: 10.1002/anie.201506527
Goncalves, E. R., Hara, H., Miyazawa, D., Davies, J. E., Eltis, L. D., and Mohn, W. W. (2006). Transcriptomic assessment of isozymes in the biphenyl pathway of Rhodococcus sp. strain RHA1. Appl. Environ. Microbiol. 72, 6183–6193. doi: 10.1128/AEM.00947-06
Gran-Scheuch, A., Fuentes, E., Bravo, D. M., Jimenez, J. C., and Perez-Donoso, J. M. (2017). Isolation and characterization of phenanthrene degrading bacteria from diesel fuel-contaminated Antarctic soils. Front. Microbiol. 8:1634. doi: 10.3389/fmicb.2017.01634
Gunner, H. B. (1963). Nitrification by Arthrobacter globiformis. Nature 197, 1127–1128. doi: 10.1038/1971127a0
Harwood, C. S., and Parales, R. E. (1996). The beta-ketoadipate pathway and the biology of self-identity. Annu. Rev. Microbiol. 50, 553–590. doi: 10.1146/annurev.micro.50.1.553
Hauschild, J. E., Masai, E., Sugiyama, K., Hatta, T., Kimbara, K., Fukuda, M., et al. (1996). Identification of an alternative 2, 3-dihydroxybiphenyl 1, 2-dioxygenase in Rhodococcus sp. strain RHA1 and cloning of the gene. Appl. Environ. Microbiol. 62, 2940–2946.
Hofer, B., Backhaus, S., and Timmis, K. N. (1994). The biphenyl/polychlorinated biphenyl-degradation locus (bph) of Pseudomonas sp. LB400 encodes four additional metabolic enzymes. Gene 144, 9–16. doi: 10.1016/0378-1119(94)90196-1
Husserl, J., Spain, J. C., and Hughes, J. B. (2010). Growth of Arthrobacter sp. strain JBH1 on nitroglycerin as the sole source of carbon and nitrogen. Appl. Environ. Microbiol. 76, 1689–1691. doi: 10.1128/AEM.02400-09
Itagaki, E., Wakabayashi, T., and Hatta, T. (1990). Purification and characterization of 3-ketosteroid-delta1-dehydrogenase from Nocardia corallina. Biochim. Biophys. Acta 1038, 60–67. doi: 10.1016/0167-4838(90)90010-D
Katznelson, H., and Sirois, J. C. (1961). Auxin production by species of Arthrobacter. Nature 191, 1323–1324. doi: 10.1038/1911323a0
Kikuchi, Y., Yasukochi, Y., Nagata, Y., Fukuda, M., and Takagi, M. (1994). Nucleotide sequence and functional analysis of the meta-cleavage pathway involved in biphenyl and polychlorinated biphenyl degradation in Pseudomonas sp. strain KKS102. J. Bacteriol. 176, 4269–4276. doi: 10.1128/jb.176.14.4269-4276.1994
Koch, C., Schumann, P., and Stackebrandt, E. (1995). Reclassification of Micrococcus agilis (Ali-Cohen 1889) to the genus Arthrobacter as Arthrobacter agilis comb-nov. and Emendation of the genus Arthrobacter. Int. J. Syst. Bacteriol. 45, 837–839. doi: 10.1099/00207713-45-4-837
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Labbe, D., Garnon, J., and Lau, P. (1997). Characterization of the genes encoding a receptor-like histidine kinase and a cognate response regulator from a biphenyl/polychlorobiphenyl-degrading bacterium, Rhodococcus sp. strain M5. J. Bacteriol. 179, 2772–2776. doi: 10.1128/jb.179.8.2772-2776.1997
Laffin, B., Chavez, M., and Pine, M. (2010). The pyrethroid metabolites 3-phenoxybenzoic acid and 3-phenoxybenzyl alcohol do not exhibit estrogenic activity in the MCF-7 human breast carcinoma cell line or Sprague–Dawley rats. Toxicology 267, 39–44. doi: 10.1016/j.tox.2009.10.003
Larimer, F. W., Chain, P., Hauser, L., Lamerdin, J., Malfatti, S., Do, L., et al. (2003). Complete genome sequence of the metabolically versatile photosynthetic bacterium Rhodopseudomonas palustris. Nat. Biotechnol. 22, 55–61. doi: 10.1038/nbt923
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., McGettigan, P. A., McWilliam, H., et al. (2007). Clustal W and clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Latorre, I., Hwang, S., Sevillano, M., and Montalvo-Rodríguez, R. (2012). PVC biodeterioration and DEHP leaching by DEHP-degrading bacteria. Int. Biodeterior. Biodegradation 69, 73–81. doi: 10.1016/j.ibiod.2011.12.011
Liu, P., Zhang, J., and Zhou, N. (2010). Characterization and mutagenesis of a two-component monooxygenase involved in para-nitrophenol degradation by an Arthrobacter strain. Int. Biodeterior. Biodegradation 64, 293–299. doi: 10.1016/j.ibiod.2010.03.001
Llamas, I., Moral, A. D., Martínez-Checa, F., Arco, Y., Arias, S., and Quesada, E. (2006). Halomonas maura is a physiologically versatile bacterium of both ecological and biotechnological interest. Antonie Van Leeuwenhoek 89, 395–403. doi: 10.1007/s10482-005-9043-9
Masai, E., Sugiyama, K., Iwashita, N., Shimizu, S., Hauschild, J. E., Hatta, T., et al. (1997). The bphDEF meta-cleavage pathway genes involved in biphenyl/polychlorinated biphenyl degradation are located on a linear plasmid and separated from the initial bphACB genes in Rhodococcus sp. strain RHA1. Gene 187, 141–149. doi: 10.1016/S0378-1119(96)00748-2
Nahurira, R., Ren, L., Song, J., Jia, Y., Wang, J., Fan, S., et al. (2017). Degradation of di(2-ethylhexyl) phthalate by a novel Gordonia alkanivorans strain YC-RL2. Curr. Microbiol. 74, 309–319. doi: 10.1007/s00284-016-1159-9
Narancic, T., Djokic, L., Kenny, S. T., O Connor, K. E., Radulovic, V., Nikodinovic-Runic, J., et al. (2012). Metabolic versatility of Gram-positive microbial isolates from contaminated river sediments. J. Hazard. Mater. 215-216, 243–251. doi: 10.1016/j.jhazmat.2012.02.059
Net, S., Sempéré, R., Delmont, A., Paluselli, A., and Ouddane, B. (2015). Occurrence, fate, behavior and ecotoxicological state of phthalates in different environmental matrices. Environ. Sci. Technol. 49, 4019–4035. doi: 10.1021/es505233b
Nishi, A., Tominaga, K., and Furukawa, K. (2000). A 90-kilobase conjugative chromosomal element coding for biphenyl and salicylate catabolism in Pseudomonas putida KF715. J. Bacteriol. 182, 1949–1955. doi: 10.1128/JB.182.7.1949-1955.2000
Nordin, K., Unell, M., and Jansson, J. K. (2005). Novel 4-chlorophenol degradation gene cluster and degradation route via hydroxyquinol in Arthrobacter chlorophenolicus A6. Appl. Environ. Microbiol. 71, 6538–6544. doi: 10.1128/AEM.71.11.6538-6544.2005
Overbeek, R., Olson, R., Pusch, G. D., Olsen, G. J., Davis, J. J., Disz, T., et al. (2013). The seed and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. doi: 10.1093/nar/gkt1226
Péloquin, L., and Greer, C. W. (1993). Cloning and expression of the polychlorinated biphenyl-degradation gene cluster from Arthrobacter M5 and comparison to analogous genes from gram-negative bacteria. Gene 125, 35–40. doi: 10.1016/0378-1119(93)90742-L
Perry, L. L., and Zylstra, G. J. (2007). Cloning of a gene cluster involved in the catabolism of p-nitrophenol by Arthrobacter sp. strain JS443 and characterization of the p-nitrophenol monooxygenase. J. Bacteriol. 189, 7563–7572. doi: 10.1128/JB.01849-06
Pham, T. T. M., and Sylvestre, M. (2013). Has the bacterial biphenyl catabolic pathway evolved primarily to degrade biphenyl? The diphenylmethane case. J. Bacteriol. 195, 3563–3574. doi: 10.1128/JB.00161-13
Ren, L., Jia, Y., Ruth, N., Shi, Y., Wang, J., Qiao, C., et al. (2016a). Biotransformation of bisphenols mediated by a novel Arthrobacter sp. strain YC-RL1. Appl. Microbiol. Biotechnol. 100, 1967–1976. doi: 10.1007/s00253-015-7076-1
Ren, L., Jia, Y., Ruth, N., Zhao, B., and Yan, Y. (2016b). Complete genome sequence of an aromatic compound degrader Arthrobacter sp. YC-RL1. J. Biotechnol. 219, 34–35. doi: 10.1016/j.jbiotec.2015.12.008
Ren, L., Lin, Z., Liu, H., and Hu, H. (2018). Bacteria-mediated phthalic acid esters degradation and related molecular mechanisms. Appl. Microbiol. Biotechnol. 102, 1085–1096. doi: 10.1007/s00253-017-8687-5
Rong, L., Guo, X., Chen, K., Zhu, J., Li, S., and Jiang, J. (2009). Isolation of an isocarbophos-degrading strain of Arthrobacter sp. scl-2 and identification of the degradation pathway. J. Microbiol. Biotechnol. 19, 1439–1446. doi: 10.4014/jmb.0811.0626
Saito, A., Iwabuchi, T., and Harayama, S. (2000). A novel phenanthrene dioxygenase from Nocardioides sp. strain KP7: expression in Escherichia coli. J. Bacteriol. 182, 2134–2141. doi: 10.1128/JB.182.8.2134-2141.2000
Sakai, M., Masai, E., Asami, H., Sugiyama, K., Kimbara, K., and Fukuda, M. (2002). Diversity of 2, 3-dihydroxybiphenyl dioxygenase genes in a strong PCB degrader, Rhodococcus sp. strain RHA1. J. Biosci. Bioeng. 93, 421–427. doi: 10.1016/S1389-1723(02)80078-0
Sasaki, M., Tsuchido, T., and Matsumura, Y. (2008). Molecular cloning and characterization of cytochrome P450 and ferredoxin genes involved in bisphenol A degradation in Sphingomonas bisphenolicum strain AO1. J. Appl. Microbiol. 105, 1158–1169. doi: 10.1111/j.1365-2672.2008.03843.x
Shah, A. A., Kato, S., Shintani, N., Kamini, N. R., and Nakajima-Kambe, T. (2014). Microbial degradation of aliphatic and aliphatic-aromatic co-polyesters. Appl. Microbiol. Biotechnol. 98, 3437–3447. doi: 10.1007/s00253-014-5558-1
Singh, B. K. (2008). Organophosphorus-degrading bacteria: ecology and industrial applications. Nat. Rev. Microbiol. 7, 156–164. doi: 10.1038/nrmicro2050
Sugimoto, K., Yamada, T., Hauschild, J. E., Senda, T., Masai, E., Fukuda, M., et al. (1997). Purification and crystallization of a 2, 3-dihydroxybiphenyl dioxygenase Etbc from Rhodococcus sp. strain RHA1. Protein Peptide Lett. 4, 415–418.
Vaillancourt, F. H., Bolin, J. T., and Eltis, L. D. (2008). The ins and outs of ring-cleaving dioxygenases. Crit. Rev. Biochem. Mol. 41, 241–267. doi: 10.1080/10409230600817422
Wang, Y., Li, J., and Liu, A. (2017). Oxygen activation by mononuclear nonheme iron dioxygenases involved in the degradation of aromatics. J. Biol. Inorg. Chem. 22, 395–405. doi: 10.1007/s00775-017-1436-5
Wang, Y., Miao, B., Hou, D., Wu, X., and Peng, B. (2012). Biodegradation of di-n-butyl phthalate and expression of the 3, 4-phthalate dioxygenase gene in Arthrobacter sp. ZH2 strain. Process Biochem. 47, 936–940. doi: 10.1016/j.procbio.2012.02.027
Wen, Z., Gao, D., and Wu, W. (2014). Biodegradation and kinetic analysis of phthalates by an Arthrobacter strain isolated from constructed wetland soil. Appl. Microbiol. Biotechnol. 98, 4683–4690. doi: 10.1007/s00253-014-5568-z
Xu, M., He, Z., Zhang, Q., Liu, J., Guo, J., Sun, G., et al. (2015). Responses of aromatic-degrading microbial communities to elevated nitrate in sediments. Environ. Sci. Technol. 49, 12422–12431. doi: 10.1021/acs.est.5b03442
Yagi, J. M., Sims, D., Brettin, T., Bruce, D., and Madsen, E. L. (2009). The genome of Polaromonas naphthalenivorans strain CJ2, isolated from coal tar-contaminated sediment, reveals physiological and metabolic versatility and evolution through extensive horizontal gene transfer. Environ. Microbiol. 11, 2253–2270. doi: 10.1111/j.1462-2920.2009.01947.x
Yang, M., Ma, P., Li, W., Liu, J., Li, L., Zhu, X., et al. (2007). 2, 3-Dihydroxybiphenyl dioxygenase gene was first discovered in Arthrobacter sp. strain PJ3. Chin. Sci. Bull. 52, 1205–1211. doi: 10.1007/s11434-007-0191-3
Yang, X., Liu, X., Song, L., Xie, F., Zhang, G., and Qian, S. (2007). Characterization and functional analysis of a novel gene cluster involved in biphenyl degradation in Rhodococcus sp. strain R04. J. Appl. Microbiol. 103, 2214–2224. doi: 10.1111/j.1365-2672.2007.03461.x
Yao, Y., Tang, H., Su, F., and Xu, P. (2015). Comparative genome analysis reveals the molecular basis of nicotine degradation and survival capacities of Arthrobacter. Sci. Rep. 5:8642. doi: 10.1038/srep08642
Zhang, S., Sun, W., Xu, L., Zheng, X., Chu, X., Tian, J., et al. (2012). Identification of the para-nitrophenol catabolic pathway, and characterization of three enzymes involved in the hydroquinone pathway, in Pseudomonas sp. 1-7. BMC Microbiol. 12:27. doi: 10.1186/1471-2180-12-27
Keywords: aromatic compounds, biodegradation, metabolic versatility, dioxygenase, biphenyl
Citation: Ren L, Jia Y, Zhang R, Lin Z, Zhen Z, Hu H and Yan Y (2018) Insight Into Metabolic Versatility of an Aromatic Compounds-Degrading Arthrobacter sp. YC-RL1. Front. Microbiol. 9:2438. doi: 10.3389/fmicb.2018.02438
Received: 28 June 2018; Accepted: 24 September 2018;
Published: 11 October 2018.
Edited by:
Mariusz Cycoń, Medical University of Silesia, PolandReviewed by:
Lukasz Dziewit, University of Warsaw, PolandRafael Bosch, Universidad de les Illes Balears, Spain
Copyright © 2018 Ren, Jia, Zhang, Lin, Zhen, Hu and Yan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yanchun Yan, eWFueWFuY2h1bjIwMTJAcXEuY29t; eWFueWFuY2h1bkBjYWFzLmNu