 Lauren M. Bradford1
Lauren M. Bradford1 Gisle Vestergaard2,3
Gisle Vestergaard2,3 András Táncsics4
András Táncsics4 Baoli Zhu1
Baoli Zhu1 Michael Schloter4
Michael Schloter4 Tillmann Lueders1*
Tillmann Lueders1*- 1Institute of Groundwater Ecology, Helmholtz Zentrum München, Neuherberg, Germany
- 2Section of Microbiology, University of Copenhagen, Copenhagen, Denmark
- 3Research Unit Comparative Microbiome Analysis, Helmholtz Zentrum München, Neuherberg, Germany
- 4Regional University Center of Excellence in Environmental Industry, Szent István University, Gödöllö, Hungary
While most studies using RNA-stable isotope probing (SIP) to date have focused on ribosomal RNA, the detection of 13C-labeled mRNA has rarely been demonstrated. This approach could alleviate some of the major caveats of current non-target environmental “omics.” Here, we demonstrate the feasibility of total RNA-SIP in an experiment where hydrocarbon-degrading microbes from a BTEX-contaminated aquifer were studied in microcosms with 13C-labeled toluene under microoxic conditions. From the total sequencing reads (∼30 mio. reads per density-resolved RNA fraction), an average of 1.2% of reads per sample were identified as non-rRNA, including mRNA. Members of the Rhodocyclaceae (including those related to Quatrionicoccus spp.) were most abundant and enriched in 13C-rRNA, while well-known aerobic degraders such as Pseudomonas spp. remained unlabeled. Transcripts related to cell motility, secondary metabolite formation and xenobiotics degradation were highly labeled with 13C. mRNA of phenol hydroxylase genes were highly labeled and abundant, while other transcripts of toluene-activation were not detected. Clear labeling of catechol 2,3-dioxygenase transcripts supported previous findings that some of these extradiol dioxygenases were adapted to low oxygen concentrations. We introduce a novel combination of total RNA-SIP with calculation of transcript-specific enrichment factors (EFs) in 13C-RNA, enabling a targeted approach to process-relevant gene expression in complex microbiomes.
Introduction
Unraveling the activity of microorganisms in complex environments is a difficult but essential step toward understanding their metabolism and community interactions in processes like pollutant degradation. Stable isotope probing (SIP) of nucleic acids is widely applied to discover which organisms metabolize a particular substrate, and can provide insights into catabolic pathways used (Coyotzi et al., 2016; Lueders et al., 2016). As microbes metabolize an isotopically labeled substrate, the isotope is incorporated into their biomolecules, including nucleic acids. These can then be extracted and separated into labeled and unlabeled fractions by isopycnic centrifugation (Meselson and Stahl, 1958). Compared to DNA-SIP where DNA labeling only occurs during cell replication, RNA-SIP allows for a more dynamic perspective of specifically active populations (Manefield et al., 2002; Lueders et al., 2016).
The majority of RNA-SIP studies to this point have targeted small subunit ribosomal RNA (SSU rRNA) to taxonomically identify microbes involved in substrate metabolism. However, isopycnic gradients have the capacity to resolve both labeled rRNA and mRNA (Rickwood, 1992). Still, only a small number of studies have actually targeted functional transcripts from SIP gradients, and in most cases researchers looked at the transcripts using reverse-transcription PCR and fingerprinting (Huang et al., 2009; Pratscher et al., 2011; El Zahar Haichar et al., 2012). These methods can only provide insights into the labeling of very specific transcript pools and may suffer from primer bias or even a total lack of suitable PCR targets. Only two studies to date have combined mRNA-SIP with next generation sequencing. Dumont et al. (2013) studied the cycling of 13CH4 in oxic lake sediment cultures. Their workflow involved both rRNA depletion prior to isopycnic centrifugation and linear amplification of the mRNA recovered from gradient fractions to produce sufficient material for 454 sequencing. With the increased sequencing depth of Illumina platforms, it is now possible to directly investigate total RNA from heavy and light gradient fractions instead, recovering both taxonomic and functional information. This was first accomplished by Fortunato and Huber (2016), who used total RNA-SIP to study chemolithoautotrophic communities from a deep-sea hydrothermal vent in hydrogen-enriched incubations. The authors also discussed methodological caveats of sequencing gradient fractions with very little RNA. Total RNA sequencing (RNA-seq) can avoid many of the pitfalls of PCR and allows the discovery of transcripts in an undirected manner. However, since the total amount of RNA which can be loaded into a SIP gradient is typically limited to <1 μg (Whiteley et al., 2007), the low yield of RNA after fractionation represents a challenge for downstream analyses. Post-gradient linear amplification of RNA (Dumont et al., 2013) may provide means of increasing template amounts for sequencing, and has also been hypothesized to increase the proportion of mRNA in total RNA (Cao et al., 2010). Thus, linear amplification could be a viable strategy for improving total mRNA reads from gradient RNA.
Nucleic acid-based SIP has been widely applied to identify the organisms responsible for degrading environmental pollutants (Lueders et al., 2016). Though both aerobic and anaerobic hydrocarbon degraders have been intensively studied, degraders that thrive under microoxic conditions have received comparably little attention. Still, such degraders could be very relevant in habitats with fluctuating oxygen availability, or at redox gradients. They could differ from strictly aerobic or anaerobic degraders by more efficient terminal oxidases (Morris and Schmidt, 2013) and by a flexible use of oxygenase-based catabolic pathways and aerobic or anaerobic respiration (Kukor and Olsen, 1996; Wilson and Bouwer, 1997). Here, we aimed to contribute to this discussion by demonstrating that transcriptome-SIP can be applied to microaerobic degraders. Sediments from a long-term hydrocarbon-contaminated aquifer in Siklós, Hungary have been recently subjected to DNA-SIP with 13C-toluene in a microcosm study to investigate the microbes involved in biodegradation under microoxic conditions (Táncsics et al., 2018). Amplicon sequencing of 16S rRNA genes identified various lineages within the Rhodocyclaceae as the main toluene degraders, and subfamily 1.2.C extradiol dioxygenase genes affiliated to Zoolgoea spp. were found in 13C-labeled DNA. For the present study, we extracted highly 13C-labeled total RNA from the same microcosms after 7 days of microoxic incubation, to enable cross-comparison between amplicon-based DNA-SIP and our transcriptome-SIP method. Our results provide a proof-of-principle that transcriptome-SIP can actually be applied to pollutant-degrading complex microbiomes and that it can provide additional insights, beyond taxonomic identification, into the activity of uncultured degrader populations. We also test whether post-centrifugation linear amplification of gradient RNA offers a viable means of improving mRNA ratios in sequencing results.
Materials and Methods
Study Site and SIP Incubation
Details about the study site in Siklós, Hungary, sediment sampling, and the setup and incubation of microoxic SIP microcosms with 13C-labeled toluene can be found elsewhere (Táncsics et al., 2012, 2018). Briefly, sediment samples were taken from a monitoring well 6 m below ground surface in the center of a BTEX plume. Each microcosm consisted of a 100 ml serum bottle with 5 g (wet weight) sediment material, 50 ml artificial groundwater medium (Winderl et al., 2010), with a N2/CO2 (80/20, v/v) headspace. This amount of sediment was chosen to prevent redox stratification during incubation. Five microliter of non-labeled (12C) or fully labeled (13C7) toluene were added by injection with a glass syringe after headspace exchange. Abiotic control microcosms were set up in the same manner, but autoclaved 3x before addition of non-labeled toluene. These microcosms controlled for possible abiotic toluene loss during incubation (Táncsics et al., 2018). Microcosms were incubated at 16°C with 145 rpm rotary shaking. Dissolved oxygen levels were measured with planar oxygen sensor spots and a Fibox 3 Oxygen Meter (PreSens, Regensburg, Germany) and maintained between 0 and 0.5 mg/L by daily replenishment with air injected through a 0.2 μm filter.
RNA Extraction and Centrifugation
RNA was extracted from duplicate labeled and unlabeled microcosm sediments after 7 days of incubation using the RNA PowerSoil® Total RNA Isolation Kit (Qiagen, Hilden, Germany). Isopycnic density gradient centrifugation was carried out according to Lueders (2015). Gradients were prepared with 5 ml of 2 g/ml cesium trifluroacetate (CsTFA; GE Healthcare, Munich, Germany), 185 μl formamide, and 1 ml of gradient buffer (0.1 M Tris-HCl, 0.1 M KCl, 1 mM EDTA) containing 1 μg of RNA. Gradients were loaded into 5.1 ml Quick Seal centrifuge tubes (Beckman Coulter, Indianapolis, United States), sealed, and spun on a VT1 65.2 rotor in an Optima XE90 ultracentrifuge (Beckman Coulter, Indianapolis, United States) at 125,000 g for ∼60 h. Seven fractions from each tube were collected according to Lueders (2015). Fraction density was measured by weighing known volumes. RNA was recovered by precipitation with 1 vol. of isopropanol, washed with cold 70% EtOH, and resuspended in 25 μl EB (Qiagen, Hilden, Germany). RNA from duplicate microcosms was ultra-centrifuged separately and selected fractions were pooled following RNA quantification.
RNA Quantification
RNA recovered from fractions was quantified with RT-qPCR using the AccessQuick RT-PCR kit (Promega, Madison, WI, United States) and Ba519f and Ba907r universal 16S primers on an Mx3000p machine (Agilent, Santa Clara, CA, United States) as described by Glaubitz et al. (2009). rRNA standards were generated using the Riboprobe in vitro transcription kit (Promega, Madison, WI, United States) from plasmids containing a cloned 16S rRNA gene from Methylobacterium sp., and quantified with the Quant-iT RiboGreen RNA Assay Kit (Thermo Fisher Scientific, Waltham, MA, United States) on an Mx3000p (Agilent, Santa Clara, CA, United States). Each 40 μl RT-qPCR reaction contained: 20 μl 2x AccessQuick Master Mix, 0.4 μl 20 mg/ml bovine serum albumin, 0.2 μl 1/500 diluted SYBRgreen (Thermo Fisher Scientific, Waltham, MA, United States), 0.6 μl 1/500 diluted ROX, 10 μM each primer, and 2 μl of standard or unknown RNA. The temperature program was: 45°C reverse transcription for 30 min, initial denaturation of 95°C for 3 min, then 35 amplification cycles (30 s at 95°C, 30 s at 52°C, 30 s at 68°C), with a final extension step at 68°C for 5 min. After the run, a dissociation curve from 55 to 95°C was recorded to discriminate products from unspecific amplification products.
Based on quantitative rRNA profiles across gradients, selected RNA fractions from duplicate microcosms and gradients were pooled to obtain sufficient RNA amounts for linear amplification and sequencing (Figure 1). These pools were designated unlabeled light (12C-light, density 1.778–1.779 g ml-1), unlabeled heavy (12C-heavy, 1.795–1.798 g ml-1), labeled light (13C-light, 1.776–1.794 g ml-1) and labeled heavy (13C-heavy, 1.824–1.828 g ml-1). RNA amounts present in the heavy fractions of the 12C gradients were too low for transcriptome sequencing. Instead, the “heaviest” fractions from which sufficient RNA could be obtained from the 12C gradients were chosen, in order to control for possible shifts caused by physical characteristics of RNA such as GC content (Figure 1). Pooled RNA was concentrated via precipitation with isopropanol, washed with 70% EtOH, and resuspended in 15 μl EB (Qiagen, Hilden, Germany).
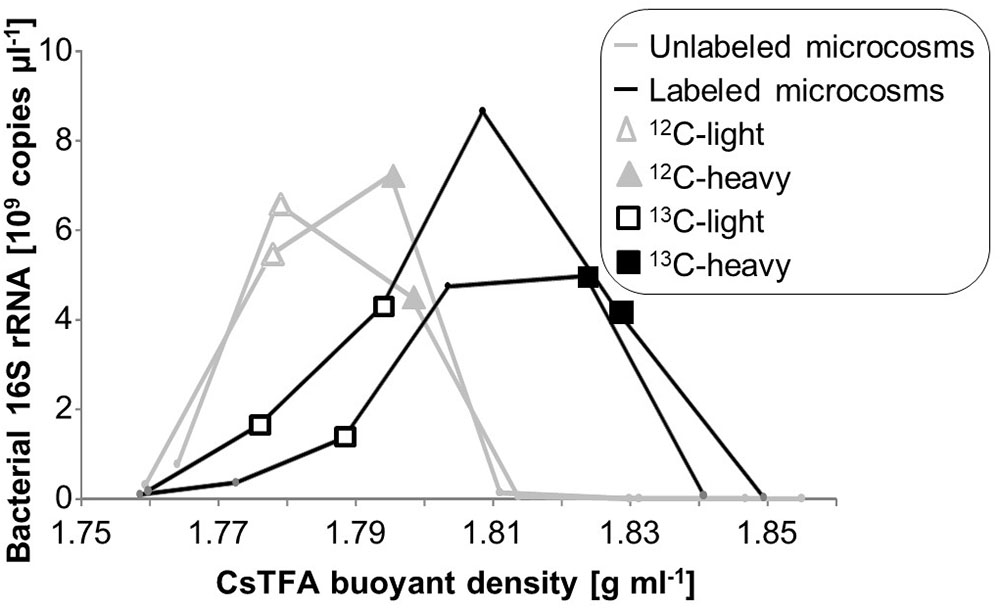
FIGURE 1. Quantitative distribution of rRNA in SIP gradients, as measured by RT-qPCR of bacterial SSU rRNA across buoyant density fractions. RNA fractions pooled from duplicate gradients for direct sequencing are given with respective symbols.
Post-gradient RNA Treatment and Sequencing
Linear amplification was performed on a subset of each of the four RNA pools using the MessageAmp II Bacteria Kit (Thermo Fisher Scientific, Waltham, MA, United States) according to manufacturer instructions. RNA was quantified and purity-checked using a NanoDrop ND-1000 spectral photometer (VWR, Ismaning, Germany) and a 2100 Bioanalyzer (Agilent, Santa Clara, CA, United States) with the RNA 6000 Nano LabChip. Library preparation with input templates ≥20 ng of total RNA was performed with the TruSeq® Stranded mRNA HT technology High Sample kit according to manufacturer protocols. The generated libraries were quality checked with the 2100 Bioanalyzer (Agilent) using a DNA 1000 LabChip, and quantified with the Qubit®dsDNA HS Assay Kit on a Qubit machine (Thermo Fisher Scientific, Waltham, MA, United States). A 1% PhiX v3 control library spike-in (Illumina, San Diego, CA, United States) was added to the library before cluster generation. Cluster generation and sequencing were performed on the NextSeq500 sequencing system (Illumina, San Diego, CA, United States) using a high-output paired-end 150 cycle (2x 150 PE) by IMGM Laboratories GmbH (Munich, Germany). All sequencing raw data generated in this study as well as sequences representing important 16S OTUs and functional transcripts have been submitted to the NCBI sequence read archive (SRA) via the Gene Expression Omnibus under the accession number GSE119644.
Data Handling
An in-house bioinformatics pipeline was developed for transcriptome-SIP data handling. Removal of sequencing adapters and merging of paired-end sequences was performed with Adapterremoval (Lindgreen, 2012) with default settings except: trim ambiguous bases (N) at 5′/3′ termini, trim bases at 5′/3′ termini with quality scores ≤15 and minimum read length of 50. Deconseq (Schmieder and Edwards, 2011) was used to remove PhiX sequences. RNA was sorted into 16S, 23S, and non-rRNA using SortmeRNA v2 (Kopylova et al., 2012). The Biopieces framework (Hansen et al., 2008) was used for command line processes. Non-rRNA tags were compared against the NCBI-nr (downloaded February 2016), COG (Galperin et al., 2014), and KEGG v58.1 (Kanehisa et al., 2015) databases using DIAMOND (Buchfink et al., 2015). Non-rRNA analysis was performed using MEGAN 5 (Huson et al., 2007), which uses a lowest common ancestor (LCA) algorithm to assign reads to phylogenetic or functional groups, which places transcripts at the most specific level possible (Huson et al., 2007).
16S OTU assignment was performed using QIIME v1.9.1 (Caporaso et al., 2010). Preprocessing of reads identified as 16S by SortmeRNA was done with the split_libraries_fastq.py (quality = 2, p = 0.01), truncate_reverse_primer.py, identify_chimeric_seqs.py (method: usearch61), and filter_fasta.py scripts. Minimum Phred score was 2 (Bokulich et al., 2013). OTU calling was performed with a subsampled open-reference based method using RDP classifier (Wang et al., 2007) using the Silva database release 123 (Quast et al., 2013). For this, the scripts pick_open_reference_otus.py (97% similarity) and filter_otus_from_otu_table.py were used. 16S analysis and ordination plotting were performed in R (R Core Team, 2013) using the phyloseq package (McMurdie and Holmes, 2013). Significance tests were performed in Microsoft Excel 2010 using the Student’s t-test (paired, two-tailed).
Calculation of Transcript Enrichment
Enrichment factors (EFs) were calculated as previously done in rRNA-SIP (Kramer et al., 2016; Zhang and Lueders, 2017) using the following equation:
where “heavy 13C” and “light 13C” were the relative abundance of a specific rRNA taxon or mRNA transcript – assigned to the same KEGG/COG category – in heavy and light fractions of the 13C-toluene gradients, respectively, and “heavy 12C” and “light 12C” the same for respective 12C-control gradients. Positive values indicate isotopic labeling of a transcript; negative enrichment indicates more frequent detection in the light fraction of 13C-RNA.
Linear Amplification Testing
A direct test of the effect of linear amplification on the proportion of mRNA in the total RNA was conducted in a SIP-independent manner using total RNA from P. aeruginosa and RT-qPCR of housekeeping genes. Further details can be found in the Supplementary Material.
Results
Total RNA Centrifugation
Total RNA extracted after 7 days of microcosm incubation and active degradation of toluene was subjected to isopycnic centrifugation. A clear shift in rRNA distribution over density gradients was observed between microcosms provided with 13C-labeled vs. unlabeled toluene (Figure 1). As previously also observed for DNA (Táncsics et al., 2018), the small amounts of RNA remaining in light fractions of 13C gradients suggested a community highly enriched in active toluene-degraders. Likewise, very little rRNA was present from unlabeled microcosms at a density >1.81 g ml-1. This complicated the selection of appropriate control fractions for sequencing, as our study, unlike most previous RNA-SIP studies, could not rely on post-gradient PCR amplification. Thus, we selected total RNA from the “heavier” and “lighter” slopes of unlabeled RNA (<1.78; >1.79 g ml-1, respectively) for sequencing of control fractions (Figure 1), in order to identify possible intrinsic density shifts of transcript pools caused by physical properties of RNA such as GC content.
Composition of Total RNA-Seq Libraries
A total of 279 million paired-end reads were obtained by RNA-seq across the eight libraries (4 unamplified, 4 which underwent linear amplification) with an average of ∼30 million per library. After QC, a total of about 1 million reads could be identified as mRNA using the KEGG or COG databases (Table 1). Total SSU and LSU (large subunit) rRNA reads accounted for 35 ± 3 and 64 ± 3% of sequencing reads, respectively. The ratio of non-rRNA reads was slightly but significantly increased in linearly amplified sequencing libraries (p = 0.023). GC-content of SSU rRNA reads appeared unchanged across gradient densities, and was also not significantly affected by linear amplification (Figure 2). However, a slight increase in average GC-content of non-rRNA reads (including mRNA) was observed with buoyant density (∼58% GC at ∼1.78 g ml-1; 59.5% GC at ∼1.83 g ml-1, respectively). Moreover, linear amplification significantly reduced the GC-content of non-rRNA reads by an average of 2.1 ± 0.9% compared to unamplified libraries (p = 0.025).
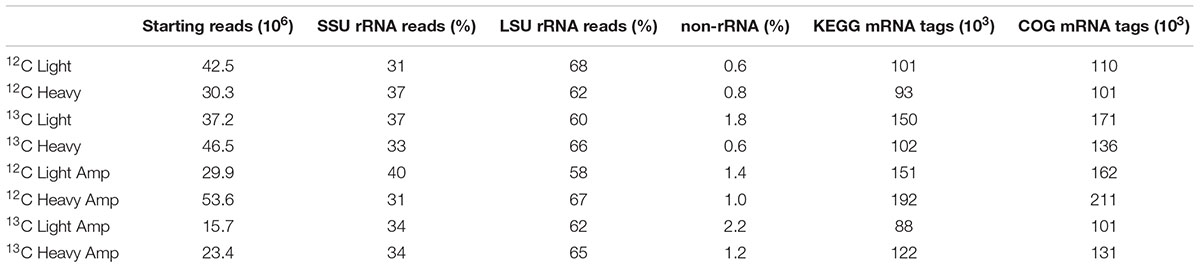
TABLE 1. Number of RNA sequencing reads and proportion of reads identified as rRNA and non-rRNA in RNA-seq libraries of density-resolved total RNA from SIP gradients.
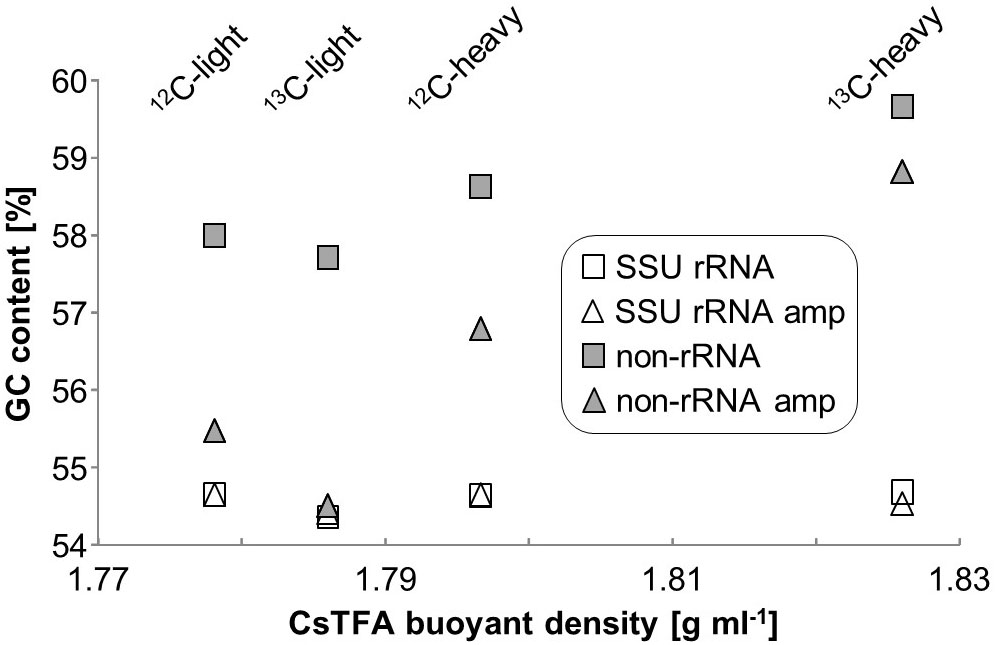
FIGURE 2. GC content of unamplified and amplified RNA-seq libraries of RNA-SIP buoyant density fractions.
Reads Enriched in 13C-Labeled rRNA
Nearly all SSU rRNA sequences in the libraries were bacterial, with a maximum of 0.09% of reads in libraries matching to eukaryotic rRNA and no reads matching archaeal rRNA. The community was overwhelmingly comprised of Proteobacteria (Supplementary Table S1), with an average of 98% of SSU reads, with only small contributions of Bacteroidetes (avg. 1.8%) and other phyla (avg. 0.6%). Most of the transcripts within the Proteobacteria were affiliated to the Betaproteobacteria; of these the majority belonged to members of the Rhodocyclaceae (Figure 3). Five of the six most abundant genera in total rRNA (unidentified Rhodocyclaceae, Dechloromonas, Pseudomonas, Quatrionicoccus, Zoogloea, and Azonexus) belonged to the Rhodocyclaceae. All genera within the Rhodocyclaceae were strongly enriched in 13C-labeled RNA, with the exception of reads affiliated to Azoarcus spp. (Figure 3); no labeling appeared in members outside of this family. About 21% of the total rRNA reads were within the Rhodocyclaceae, whose more precise affiliation remained unclear. rRNA of Pseudomonas spp. comprised an average 10% of the communities and appeared unlabeled.
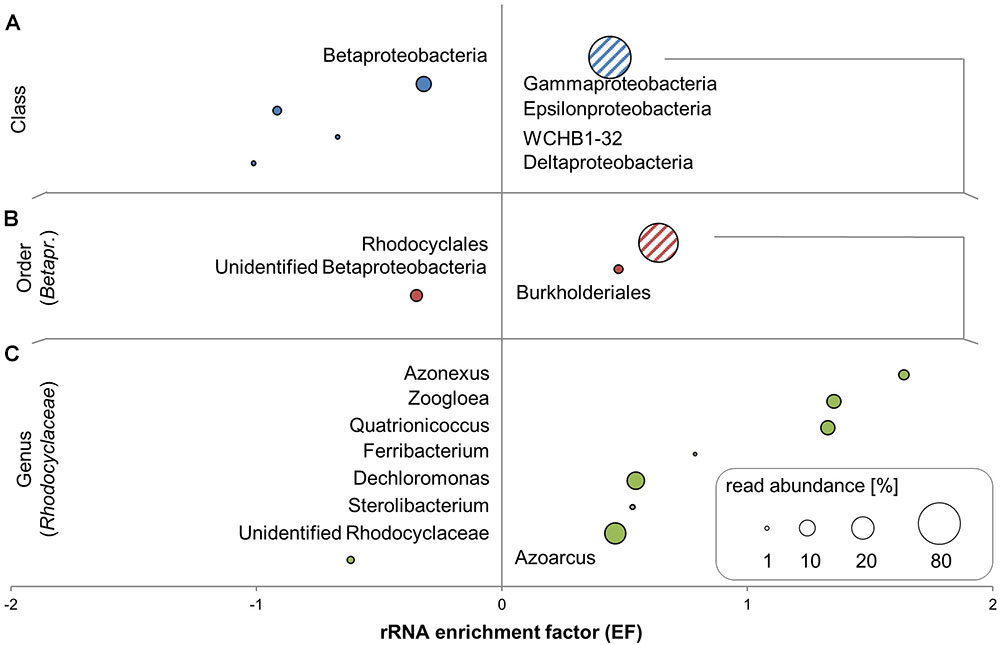
FIGURE 3. 13C-enriched bacterial taxa identified by SIP in unamplified RNA of toluene-degrading microcosms. Rankings of SSU rRNA enrichment factors (EFs) are resolved at the class-level (A), for orders within Betaproteobacteria (B), and for genera within Rhodocyclaceae (C). EFs are shown in combination with relative read abundances averaged across all RNA-seq libraries. Groups shown are those with >1% average read abundance at each level.
Considerable differences in taxon-level abundances were found between unamplified and amplified libraries (Supplementary Table S1). For example, Dechloromonas spp. averaged at 11.4% of the unamplified community but at 18.3% after amplification, while Zoogloea rRNA was detected at 13.4% in the unamplified community and at 6% in the amplified.
Functional Transcripts Enriched in 13C-Labeled RNA
The number of non-rRNA reads identified as mRNA was between 88 and 192 × 103 reads per library (Table 1). The transcript ratios discussed in this section are given as a percentage of those reads, not of total non-rRNA, which also included tRNA and other RNA types. The most abundant category of functional transcripts could not be further identified using KEGG and COG databases (avg. 30% of reads using KEGG and 26% of reads using COG). Among those reads which could be further identified, transcripts linked to genes involved in carbohydrate, amino acid, and energy metabolism were most abundant and were all enriched in 13C-RNA, as calculated via EFs for the different gene categories, pathways and transcripts (Figure 4).
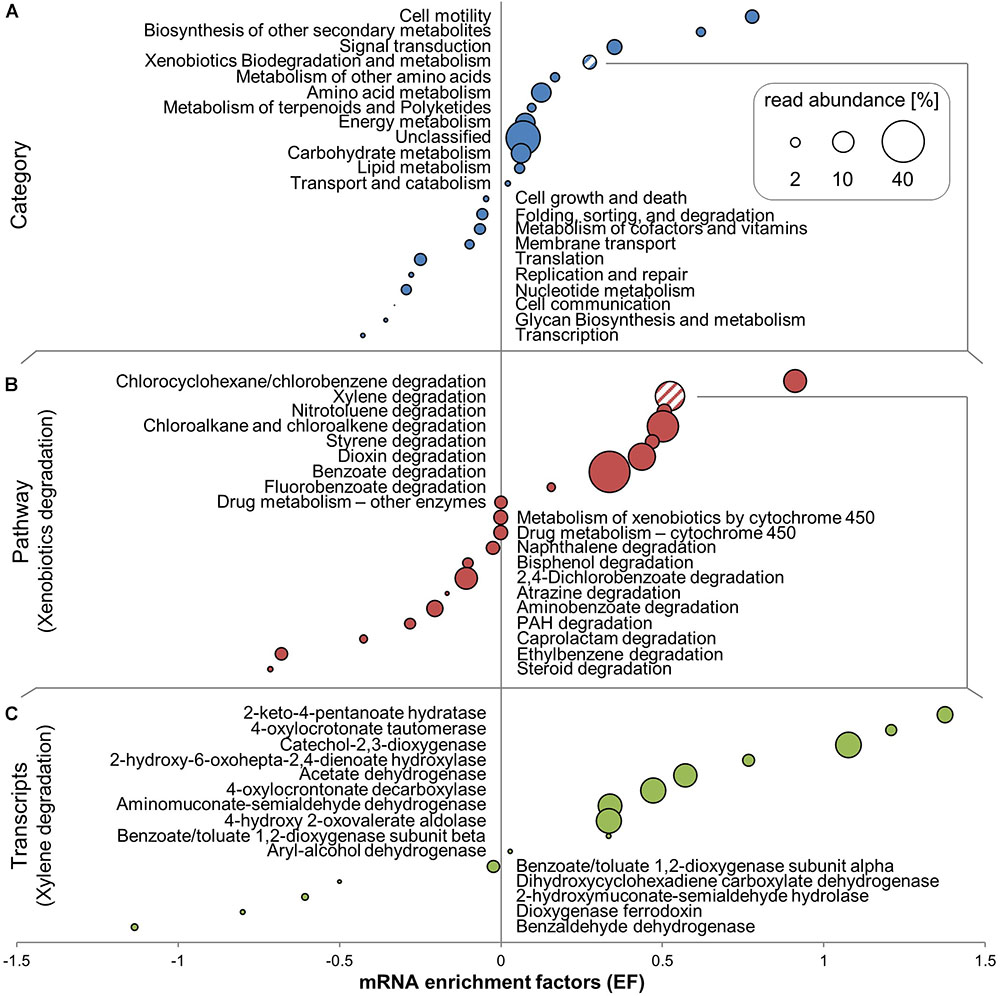
FIGURE 4. 13C-enriched mRNA transcripts identified by SIP in unamplified RNA of toluene-degrading microcosms. Rankings of mRNA enrichment factors (EFs) are resolved at the level of KEGG categories (A), pathways within the xenobiotics biodegradation and metabolism category (B), and individual transcripts within the xylene degradation pathway (C). EFs are shown in combination with relative read abundances averaged across all RNA-seq libraries. Individual transcripts shown are those with ≥ 20 total reads.
Cell motility was the transcript category most highly enriched in 13C-RNA (Figure 4A). Many transcripts linked to motility-associated genes were highly abundant in mRNA (e.g., Flagellin at ∼2.65% of all transcripts in KEGG annotation), and almost all were enriched in 13C-labeled RNA. Supplementary Table S2 shows the phylogenetic affiliation of transcripts of the fliC gene based on MEGAN 5’s LCA algorithm. Between 35 and 40% of these transcripts could only be assigned at the phylum level, at which nearly all (96–100%) belonged to the Proteobacteria. Some could be specifically assigned at the species level, and 10–20% of all transcripts linked to fliC were assigned to Azovibrio restrictus.
The second most clearly labeled transcript category was linked to genes coding for biosynthesis of secondary metabolites (Figure 4A). The main 13C-enriched transcripts in this group were identified as genes coding for a catalase-peroxidase (K03782), which averaged at over 1000 copies per library. Labeling observed for the most abundant COG categories was generally consistent with KEGG results (Supplementary Figure S1), but biodegradation pathways are not resolved as a separate category in this database. The COG category “Secondary metabolite biosynthesis” includes similar pathways as the respective KEGG category, but also includes transcripts linked to genes involved in BTEX degradation, such as phenol hydroxylase and phenol-2-monooxygenase, which were highly enriched in 13C-RNA. Signal transduction was the third most labeled transcript category (Figure 4A). Approximately 20% of transcripts assigned there were for the fliC gene, which besides its role in motility is also part of the two-component signaling system in KEGG.
Xenobiotic Degradation Transcripts
“Xenobiotics degradation” was the fourth most clearly labeled transcript category in KEGG (Figure 4A), though about half of the pathways in that category were themselves not enriched in 13C-RNA (Figure 4B). It should be noted that many of the pathways expressed and enriched in 13C shared enzymes – for example, phenol hydroxylase (K16249), which can be involved in chlorocyclohexane and chlorobenzene degradation, benzoate degradation, toluene degradation, and in the degradation of aromatic compounds in general. Transcripts of the benzoate degradation pathway were the most abundant, though not the most highly labeled (Figure 4B).
Transcripts of catechol-2,3-dioxygenase (C23O), an enzyme previously associated with central pollutant metabolism at the investigated site (Táncsics et al., 2010, 2012, 2013), were identified as very abundant and enriched in 13C in KEGG (Figure 4C). Taxonomic association of these C23O transcripts was done using MEGAN 5 (Supplementary Table S3). Of the transcripts that could be assigned at the family level, the highest percent belonged to the Rhodocyclaceae (avg. 7.8%), followed closely by Pseudomonadaceae (avg. 6.5%). However, most of these transcripts could not be assigned more specifically than at the phylum level. Transcripts of the catechol-1,2-dioxygenase gene (K03381), on the other hand, were barely present, appearing only in very low read numbers (∼1) per library.
Amongst initial hydrocarbon degradation mechanisms, transcripts of the phenol hydroxylase gene (dmpL; K16243) were highly abundant and labeled, with transcript numbers of up to 160 per library. In contrast, transcripts of genes coding for toluene monooxygenase subunits (K15760-K15764, which may code for toluene-2,3, or 4-monooxygenases) were present in low read numbers (∼15), with varying amounts of labeling. Transcripts linked to genes coding for benzylsuccinate synthase (bssA; K07540) were present in all treatments but not very abundant (∼20 reads) and not labeled.
Transcripts of Respiratory Pathways
Transcripts of genes required for prokaryotic oxidative phosphorylation were present in the microoxic microcosms, but the majority were unlabeled. The full operon of nuoA-N, coding for Type I NADH dehydrogenase, was expressed, while no transcripts of the ndh operon for Type II NADH dehydrogenase were detected. Complete pathways of both denitrification and dissimilatory nitrate reduction to ammonium (DNRA) were transcribed, but the only transcript enriched in 13C-RNA found in these pathways codes for NapA (K02567). We were also interested in whether the putative NO-dismutase genes previously detected at the site (Zhu et al., 2017) would be expressed during microaerobic toluene degradation. For this, all mRNA transcripts assigned to “Candidatus Methylomirabilis oxyfera” (between 0 and 62 reads per library) were compared to the NCBI-nr database via nucleotide BLAST. All transcripts annotated as canonical nitric oxide reductases belonging to “Ca. Methylomirabilis oxyfera” actually matched more closely to the putative nitric oxide dismutase (nod). Up to 21% of all transcripts annotated as nitric oxide reductase subunit B (norB; K04561) were putative nod transcripts. These nod transcripts appeared in read numbers of ten or fewer, so EF calculation was not robust.
Effect of Linear Amplification
Linear amplification increased RNA amounts by at least 300-fold. NMDS plots of functional transcript pools (Figure 5A and Supplementary Figure S2B) and of SSU rRNA profiles (Supplementary Figure S2A) in the 8 treatments were used to delineate the impact of linear amplification on total RNA-seq libraries, and on overall labeling patterns. Within the unamplified or amplified data sets, the separation between profiles was consistent with expectations for a typical SIP experiment. For example, heavy and light profiles for the control (12C) microcosm were much more similar, while those of the labeled (13C) microcosm were clearly separated, mainly on NMDS axis 1 (Figure 5A). This separation was observed for both unamplified and amplified libraries. However, linear amplification itself seemed to introduce a further discriminant vector to overall transcript pools, showing that this treatment changed the community profile. A similar skewing pattern was also observed for SSU rRNA (Supplementary Figure S2A).
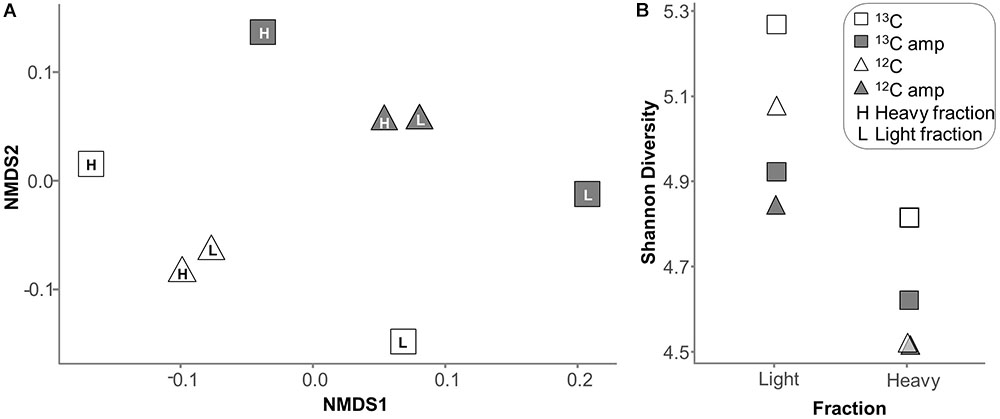
FIGURE 5. (A) NMDS ordination of profiles of functional transcript reads as identified in unamplified and amplified RNA-seq libraries of RNA-SIP fractions using KEGG. (B) Shannon diversity of SSU rRNA reads in the same libraries.
Not unexpectedly, overall transcript diversity was always lower in heavy than in light fractions (Figure 5B). Diversity of transcript libraries after linear amplification was consistently though not significantly (p = 0.073) decreased, suggesting that the amplification process could leave behind some specific transcripts or over-amplify others. Rare transcripts with copy numbers less than ten appeared with equal frequency in amplified and unamplified libraries (Supplementary Figure S3). Additionally, predicted Chao1 richness perfectly matched the richness observed (not shown), suggesting an adequate sequencing depth for sufficient community coverage.
In addition, we conducted a SIP- and sequencing-independent lab experiment with pure culture RNA of P. aeruginosa, to directly test whether linear amplification increased the proportion of mRNA in the total RNA. RT-qPCR results of housekeeping gene transcripts did not indicate that linear amplification systematically increased mRNA ratios (Supplementary Figure S4). In fact, two of the three housekeeping genes tested had lower transcript/total RNA ratios after linear amplification than before, while one remained unchanged.
Discussion
Methodological Considerations
This study demonstrates the value of total-RNA SIP (also termed transcriptome-SIP) in providing targeted insights into microbial activities in processes such as pollutant degradation, on both taxonomic and functional levels. However, the level of detail clearly depended on the number of mRNA reads obtained from gradient fractions. The amount of non-rRNA reads was between 0.6 and 2.2% in our libraries, but this small proportion was not unexpected for total RNA-seq and is consistent with the percent non-RNA or putative mRNA found in other total-RNA-seq studies (Supplementary Table S4). Most transcriptomic studies use mRNA enrichment (eukaryotes) or rRNA depletion (prokaryotes) to increase the depth of mRNA sequencing. However, our interest in SSU-based microbial community composition and desire to avoid potential skewing effects motivated a depletion-free, total-RNA approach. This made deep sequencing necessary for adequate non-rRNA coverage, preventing the sequencing of true biological replicates for RNA fractions. However, the inclusion of both amplified and unamplified libraries, in which overall labeling patterns were generally consistent (Figure 5 and Supplementary Figure S2 and Supplementary Tables S1–S3), provided a certain level of confidence to this end. A further cautioning statement needs to be added with reference to the comparably late time point in microcosm incubation (7 days) chosen for transcriptome-SIP analysis. This was motivated by our need for abundant and highly 13C-labeled RNA in this primary proof-of-concept study, while we are aware that certain patterns of transcript labeling during initial phases of toluene catabolism may have been missed, due to the possibly more rapid turnover of distinct mRNA species.
The interpretation of SIP data strongly relies on comparing sequencing results between 12C and 13C-treatments, and across similar ranges of buoyant densities (Whiteley et al., 2007). This was not fully possible in the present study, mainly because we could not rely on post-gradient PCR amplification of 12C-RNA recovered from heavy fractions and vice versa. Direct sequencing of total RNA and parallel linear amplification necessitated a certain minimum amount (∼120 ng) of total RNA recovered after fractionation. To increase yields for low-RNA fractions, several fractions of similar density across duplicate centrifugation gradients were pooled (Figure 1). In essence, RNA from all meaningful fractions of the density-resolved total RNA obtained were sequenced from both 12C and 13C gradients, which was the best that could be accomplished considering methodological limitations. While the lack of sequencing replicates precludes statistical calculations or differential expression analysis that have been previously used in amplicon-based SIP studies (Pepe-Ranney et al., 2016), the EFs based on differences in labeled and control microcosms offer a semi-quantitative way to interpret labeling (Kramer et al., 2016; Zhang and Lueders, 2017). Nevertheless, the total amounts of RNA recovered from gradient fractions and the net costs of transcriptome sequencing will remain a major limitation of transcriptome-SIP work in the future.
Effect of Linear Amplification
Linear amplification is one potential strategy to alleviate the technical limitations involved in total RNA-seq following SIP gradients. We compared this to direct sequencing in the present study. Linear amplification increased RNA amounts, but also decreased GC content in non-rRNA (Figure 2), decreased library diversity (Figure 5B), and caused profound changes to community profiles for SSU rRNA and functional transcripts (Figure 5A and Supplementary Figure S2). Linearly amplified libraries contained a much higher percentage of small (<50 bp) read fragments which were removed early in the bioinformatic pipeline. Previous studies have also found changes in transcript ratios when using various linear amplification protocols (Nygaard et al., 2003; Degrelle et al., 2008), which may be due to inefficiency or bias in the initial reverse transcription step (Levesque-Sergerie et al., 2007). The equal numbers of rare transcripts in amplified and unamplified libraries (Supplementary Figure S3) indicate that the amplification process did not simply miss less common transcripts, but must have selected based on another characteristic, perhaps GC-content (Figure 2).
While the proportion of non-rRNA was somewhat increased by linear amplification (Table 1), this increase was too small to provide tangible benefits in the face of the negative effects caused by the treatment. Our independent test based on RT-qPCR targeting P. aeruginosa housekeeping genes did not indicate an increase in mRNA ratios as a result of linear amplification (Supplementary Figure S4). Thus we cannot confirm the idea that linear amplification could increase the proportion of mRNA to total RNA reads in sequencing libraries in a meaningful manner (Cao et al., 2010). Because of the marked skewing effects observed in overall library composition (Figure 5A and Supplementary Figure S2), we recommend that linear amplification should be used only if total RNA yield is too low for direct analysis, and samples studied using linear amplification should only be compared to samples treated likewise.
Taxonomic Insights via rRNA of Labeled Degraders
Since contaminated aquifers are well-known to harbor not only bacteria, but also archaea and microeukaryotes (Griebler and Lueders, 2009), the lack of respective rRNA in our libraries came as a surprise. Possibly, the limited availability of oxygen in our microoxic microcosms as well as the substantial toluene loads (∼1 mM; Táncsics et al. (2018)) contributed to selection of specific bacterial degraders during SIP incubation. Betaproteobacteria have been reported previously to be prevalent in oxygen-limited, aromatic hydrocarbon-contaminated groundwater environments (Fahy et al., 2006). Rhodocyclaceae in particular are known to be important at this site, both from previous samplings (Táncsics et al., 2012, 2013) and from DNA-SIP of the same microcosms as used in this study (Táncsics et al., 2018). Members of the Rhodocyclaceae can degrade aromatics both aerobically and anaerobically (Song et al., 2001; Chakraborty et al., 2005). Dechloromonas aromatica, for example, can degrade several aromatic compounds using oxygen, nitrate, perchlorate, or chlorate as electron acceptors (Chakraborty et al., 2005). Dechloromonas rRNA was very abundant and clearly enriched in 13C-rRNA, despite being represented by less than 1% of the DNA amplicons sequenced in DNA-SIP of the same microcosms (Táncsics et al., 2018). Since labeling of DNA requires cellular replication, while RNA labeling can occur independent of cellular growth (Manefield et al., 2002), this could indicate that Dechloromonas was metabolically active despite slow growth during microoxic SIP incubation. Alternately, this differential representation could be a result of primer bias during the production of PCR amplicons for sequencing. Primer bias was even more clearly apparent for the genus Azonexus, which was abundant (∼6%; Supplementary Table S1) and highly enriched in 13C-rRNA, while not detected at all in DNA amplicons (Táncsics et al., 2018).
Quatrionicoccus spp. (a genus within the Rhodocyclaceae – originally called Quadricoccus but reclassified as Quatrionicoccus; Tindall and Euzéby, 2006) – were first isolated from activated sludge biomass in an enhanced biological phosphorus removal reactor with cycling aerobic/anaerobic conditions (Maszenan et al., 2002). Members of this genus are considered to be strict aerobes. This genus comprised an average 10% of the rRNA reads and was highly labeled, indicating effective toluene metabolism under microoxic conditions. This was consistent with labeling results from DNA-SIP of the same microcosms (Táncsics et al., 2018). The genus is also highly abundant in Siklós groundwater itself (Farkas et al., 2016). Before this work, members of the genus have not been described as hydrocarbon degraders. This contrasts other members of the Rhodocyclacaeae such as Zoogloea spp. which were clearly identified as labeled in both rRNA and mRNA in the present study, and which have been frequently reported as aerobic degraders of aromatic hydrocarbons (Jechalke et al., 2013; Farkas et al., 2015).
In contrast, other microbes of the BTEX contaminated sediment were abundant in RNA but without any indication of labeling, including members of the Pseudomonadaceae and Comamonadaceae, which are well-known to degrade hydrocarbons (Fahy et al., 2006; Wald et al., 2015), under either aerobic (Martinez-Lavanchy et al., 2010) or nitrate-reducing conditions (Sun and Cupples, 2012). Thus in our experiment, they must have been active and producing transcripts, potentially thriving on electron donors introduced with the original sediment inoculum, but not receiving labeled carbon from the added toluene.
Functional Insights via Labeled mRNA
The combination of stable isotope probing (SIP) with metatranscriptomic sequencing provided information on a whole range of functional transcripts produced by active degraders during incubation. Here, we discuss transcript labeling patterns which appeared most relevant to us in understanding degrader activity under microoxic conditions. Transcripts linked to genes coding for proteins involved in cell motility were the most highly labeled group in both KEGG and COG analyses (Figure 4A and Supplementary Figure S1). The importance of chemotaxis of degraders toward aromatics, including BTEX, is well-known (Krell et al., 2013). In highly contaminated systems, motile cells have advantages not only in moving toward contaminant sources, but also in escaping toxically high local concentrations (Parales et al., 2000). Still, the apparent importance of motility amongst degraders in our agitated SIP microcosms came as a surprise, and could potentially be connected to a lower availability of toluene or oxygen during the late phase of degradation (Táncsics et al., 2018), triggering a motility response (Lacal, 2017).
Transcripts of genes coding for flagellin, the principle component of bacterial flagella, were the most abundant of the transcripts linked to motility and were highly 13C-enriched. Between 10 and 20% of all transcripts of the fliC gene were assigned to Azovibrio restrictus (Supplementary Table S2), a N2-fixing microaerophile within the Rhodocyclaceae (Reinhold-Hurek and Hurek, 2000), while only ∼0.1% of 16S rRNA reads were affiliated to Azovibrio sp. (Supplementary Table S1). We find it highly unlikely that an organism so rare within the community could have contributed so much of one of the most abundant and highly labeled transcripts in our microcosms. Either lateral gene transfer, or the lack of more appropriate reference genome data for other members of the Rhodocyclaceae, such as Azonexus spp., could potentially explain this finding. No other single species contributed more than 1.6% of transcripts of fliC within each sample, even known motile toluene degraders such Dechloromonas (Chakraborty et al., 2005) or Zoogloea spp. (Farkas et al., 2015), which were much more abundant in rRNA reads than Azovibrio.
Xenobiotic Degradation Transcripts
The relatively high copy numbers and labeling of transcripts for the phenol hydroxylase gene and the low numbers or complete absence of other possible oxygen-dependent toluene-activating gene transcripts indicated that phenol hydroxylase was the primary driver of toluene activation in these microcosms. Phenol hydroxylase has a broad substrate specificity for monoaromatics and its gene has been previously found in microoxic BTEX-contaminated groundwater (Da Silva and Corseuil, 2012) and used as a biomarker for aerobic aromatic degradation (Nebe et al., 2009). However, it is also possible that transcripts of other genes in upper pathways of toluene catabolism were transcribed at earlier time points of microcosm incubation, but may have been subject to more rapid turnover and decay.
Catechol-2,3-dioxygenase (C23O) is an extradiol dioxygenase which has long been used as a conserved functional marker gene for aerobic aromatic degraders (Táncsics et al., 2010, 2012, 2013). Kukor and Olsen (1996) found that C23O in the subfamily 1.2.C of extradiol dioxygenases efficiently used low amounts of dissolved oxygen, and likely evolved in environments with low oxygen and substrate concentrations. Considering its frequent presence in contaminated environments (Hendrickx et al., 2006; Táncsics et al., 2012) and the previously seen high diversity at this site (Táncsics et al., 2010, 2012, 2013), it is not surprising that C23O was highly expressed and the related transcripts highly 13C-labeled in SIP microcosms. The majority of these transcripts could only be identified to the phylum level (Supplementary Table S3). This may be because gene sequences are so similar across different lineages that affiliation by MEGAN’s lowest-common-ancestor algorithm stopped at a higher level. A more likely explanation is the lack of more specifically assigned reference sequences in the KEGG database, since most C23Os could not be linked to any cultured bacterium.
Within the transcripts that could be assigned more specifically, Rhodocyclaceae were most prominent. This included matches to Azovibrio restrictus, previously found in hydrocarbon-contaminated soil (Yang et al., 2014) and to Zoogloea oleivorans, a facultative aerobic hydrocarbon-degrader (Farkas et al., 2015). Transcripts of the C23O gene affiliated to the Pseudomonadaceae were nearly as frequent as that of the Rhodocyclaceae (avg. 6.5%), although Pseudomonadaceae were clearly not isotopically enriched (Supplementary Table S3). Members of this family are well-known as aerobic degraders of BTEX compounds (Martinez-Lavanchy et al., 2010) and have long been known to harbor C23O (Kojima et al., 1961). The results of our labeling experiment suggest that they were not competitive in toluene degradation in the microcosms, or assimilated C from alternative substrates for biomass buildup.
Microoxic sedimentary systems may well contain anoxic micro-niches where anaerobic metabolism may take place. Furthermore, the daily replenishment of oxygen during microcosm incubation (Táncsics et al., 2018) resulted in periods of anoxia. For these reasons, we also searched for transcripts of marker genes of anaerobic aromatic degradation, i.e., benzylsuccinate synthase (Bss) (Von Netzer et al., 2016). While bssA was indeed transcribed in all microcosms, transcripts were not found labeled, indicating that anaerobic degraders were not competitive in the microcosms.
Transcripts of Respiratory Pathways
Genes involved in aerobic respiratory pathways were transcribed in low to moderate copy numbers. However, transcripts of genes coding for a Type I NADH dehydrogenase were present alone, although the majority of Betaproteobacteria, which dominated the system, are known to harbor genes for Type II as well (Marreiros et al., 2016). This may indicate an adaptation to microoxic conditions, as some obligate microaerophiles harbor only Type I (Smith et al., 2000), and expression of Type II in E. coli occurs only under fully oxic conditions (Spiro et al., 1989).
Despite not having added nitrate to microcosms, transcripts of genes coding for all steps of canonical denitrification and DNRA were detected. All related transcripts were unlabeled except for napA, which codes for a nitrate reductase adapted to low-nitrate conditions (Potter et al., 1999) and has been suggested to be involved in aerobic denitrification (Ji et al., 2015), indictating that such physiologies could have been relevant during microcosm incubation. Moreover, a previous study found abundant putative nitric oxide dismutase (nod) genes at this site (Zhu et al., 2017). NO-dismutation is hypothesized to provide a novel mechanism in reductive nitrogen cycling (Ettwig et al., 2012). The enzyme is proposed to dismutate NO into N2 and O2, thus producing intracellular O2 for the oxygenase-dependent activation of hydrocarbons (Ettwig et al., 2010; Zedelius et al., 2011). A recent metatranscriptomics study has indeed found both anaerobic and aerobic catabolic pathways to be expressed in a denitrifying, benzene-degrading enrichment culture dominated by members of the Peptococcaceae, although transcripts related to NO-dismutation were not detected (Atashgahi et al., 2018). Since the discovery of respective nod genes is still recent, it is likely that such sequence reads can still be misannotated as canonical NO-reductases (Nor) in databases. In the present study, transcripts from “Ca. Methylomirabilis oxyfera” that were annotated by KEGG as nors more closely matched putative nod sequences (Zhu et al., 2017) when compared to the NCBI-nr database.
In summary, this study builds on the advantage of metatranscriptome sequencing to provide functional details about microbial activities in a given system, which may be hard to delineate with more traditional, targeted techniques like RT-PCR. At the same time, the combination with isotopic labeling adds a new process-targeted handle to the metatranscriptomics pipeline, with a potential to provide functional context even for constituents of the large fraction of unclassified transcripts, which were detected also in our study. Here, we confirm the central role of Rhodocyclaceae and the respective C23O pathways in toluene degradation in microoxic microcosms (Táncsics et al., 2018). In addition, we reveal the important role of phenol hydroxylase as well as gene categories outside of strict catabolism, such as cell motility, and also of different respiratory pathways in active degrader populations. In the future, this novel transcriptome-SIP approach can be extended to elucidate microbial activities and interactions in processes and systems beyond contaminated aquifers.
Author Contributions
LB, AT, and TL designed the experiments. LB and GV analyzed the data, with specific contributions from BZ and MS. LB and TL wrote the initial manuscript draft with additions and revisions contributed from all authors.
Funding
This research was funded by the European Research Council (ERC) under the European Union’s Seventh Framework Programme (FP7/2007-2013), grant agreement 616644 (POLLOX) to TL. We also acknowledge support by the Helmholtz Society.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2018.02696/full#supplementary-material
References
Atashgahi, S., Hornung, B., Waals, M. J., Rocha, U. N., Hugenholtz, F., Nijsse, B., et al. (2018). A benzene-degrading nitrate-reducing microbial consortium displays aerobic and anaerobic benzene degradation pathways. Sci. Rep. 8:4490. doi: 10.1038/s41598-018-22617-x
Bokulich, N. A., Subramanian, S., Faith, J. J., Gevers, D., Gordon, J. I., Knight, R., et al. (2013). Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10, 57–59. doi: 10.1038/nmeth.2276
Buchfink, B., Xie, C., and Huson, D. H. (2015). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. doi: 10.1038/nmeth.3176
Cao, F.-L., Liu, H.-H., Wang, Y.-H., Liu, Y., Zhang, X.-Y., Zhao, J.-Q., et al. (2010). An optimized RNA amplification method for prokaryotic expression profiling analysis. Appl. Microbiol. Biotechnol. 87, 343–352. doi: 10.1007/s00253-010-2459-9
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Chakraborty, R., O’Connor, S. M., Chan, E., and Coates, J. D. (2005). Anaerobic degradation of benzene, toluene, ethylbenzene, and xylene compounds by Dechloromonas strain RCB. Appl. Environ. Microbiol. 71, 8649–8655.
Coyotzi, S., Pratscher, J., Murrell, J. C., and Neufeld, J. D. (2016). Targeted metagenomics of active microbial populations with stable-isotope probing. Curr. Opin. Biotechnol. 41, 1–8. doi: 10.1016/j.copbio.2016.02.017
Da Silva, M. L. B., and Corseuil, H. X. (2012). Groundwater microbial analysis to assess enhanced BTEX biodegradation by nitrate injection at a gasohol-contaminated site. Int. Biodeterior. Biodegradation 67, 21–27. doi: 10.1016/j.ibiod.2011.11.005
Degrelle, S. A., Hennequet-Antier, C., Chiapello, H., Piot-Kaminski, K., Piumi, F., Robin, S., et al. (2008). Amplification biases: possible differences among deviating gene expressions. BMC Genomics 9:46. doi: 10.1186/1471-2164-9-46
Dumont, M. G., Pommerenke, B., and Casper, P. (2013). Using stable isotope probing to obtain a targeted metatranscriptome of aerobic methanotrophs in lake sediment. Environ. Microbiol. Rep. 5, 757–764. doi: 10.1111/1758-2229.12078
El Zahar Haichar, F., Roncato, M.-A., and Achouak, W. (2012). Stable isotope probing of bacterial community structure and gene expression in the rhizosphere of Arabidopsis thaliana. FEMS Microbiol. Ecol. 81, 291–302. doi: 10.1111/j.1574-6941.2012.01345.x
Ettwig, K. F., Butler, M. K., Le Paslier, D., Pelletier, E., Mangenot, S., Kuypers, M. M., et al. (2010). Nitrite-driven anaerobic methane oxidation by oxygenic bacteria. Nature 464, 543–548. doi: 10.1038/nature08883
Ettwig, K. F., Speth, D. R., Reimann, J., Wu, M. L., Jetten, M. S., and Keltjens, J. T. (2012). Bacterial oxygen production in the dark. Front. Microbiol. 3:273. doi: 10.3389/fmicb.2012.00273
Fahy, A., McGenity, T. J., Timmis, K. N., and Ball, A. S. (2006). Heterogeneous aerobic benzene-degrading communities in oxygen-depleted groundwaters. FEMS Microbiol. Ecol. 58, 260–270. doi: 10.1111/j.1574-6941.2006.00162.x
Farkas, M., Szoboszlay, S., Benedek, T., Révész, F., Veres, P. G., Kriszt, B., et al. (2016). Enrichment of dissimilatory Fe (III)-reducing bacteria from groundwater of the Siklós BTEX-contaminated site (Hungary). Folia Microbiol. 62, 63–71. doi: 10.1007/s12223-016-0473-8
Farkas, M., Táncsics, A., Kriszt, B., Benedek, T., Tóth, E. M., Kéki, Z., et al. (2015). Zoogloea oleivorans sp. nov., a floc-forming, petroleum hydrocarbon-degrading bacterium isolated from biofilm. Int. J. Syst. Evol. Microbiol. 65, 274–279. doi: 10.1099/ijs.0.068486-0
Fortunato, C. S., and Huber, J. A. (2016). Coupled RNA-SIP and metatranscriptomics of active chemolithoautotrophic communities at a deep-sea hydrothermal vent. ISME J. 10, 1925–1938. doi: 10.1038/ismej.2015.258
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2014). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Glaubitz, S., Lueders, T., Abraham, W.-R., Jost, G., Jürgens, K., and Labrenz, M. (2009). 13C-isotope analyses reveal that chemolithoautotrophic Gamma-and Epsilonproteobacteria feed a microbial food web in a pelagic redoxcline of the central Baltic Sea. Environ. Microbiol. 11, 326–337. doi: 10.1111/j.1462-2920.2008.01770.x
Griebler, C., and Lueders, T. (2009). Microbial biodiversity in groundwater ecosystems. Freshw. Biol. 54, 649–677. doi: 10.1111/j.1365-2427.2008.02013.x
Hansen, M. A., Oey, H., Fernandez-Valverde, S., Jung, C.-H., and Mattick, J. S. (2008). “Biopieces: a bioinformatics toolset and framework,” in Proceedings of the 19th International Conference on Genome Informatics, Gold Coast.
Hendrickx, B., Dejonghe, W., Faber, F., Boënne, W., Bastiaens, L., Verstraete, W., et al. (2006). PCR-DGGE method to assess the diversity of BTEX mono-oxygenase genes at contaminated sites. FEMS Microbiol. Ecol. 55, 262–273. doi: 10.1111/j.1574-6941.2005.00018.x
Huang, W. E., Ferguson, A., Singer, A. C., Lawson, K., Thompson, I. P., Kalin, R. M., et al. (2009). Resolving genetic functions within microbial populations: in situ analyses using rRNA and mRNA stable isotope probing coupled with single-cell Raman-fluorescence in situ hybridization. Appl. Environ. Microbiol. 75, 234–241. doi: 10.1128/AEM.01861-08
Huson, D. H., Auch, A. F., Qi, J., and Schuster, S. C. (2007). MEGAN analysis of metagenomic data. Genome Res. 17, 377–386. doi: 10.1101/gr.5969107
Jechalke, S., Franchini, A. G., Bastida, F., Bombach, P., Rosell, M., Seifert, J., et al. (2013). Analysis of structure, function, and activity of a benzene-degrading microbial community. FEMS Microbiol. Ecol. 85, 14–26. doi: 10.1111/1574-6941.12090
Ji, B., Yang, K., Zhu, L., Jiang, Y., Wang, H., Zhou, J., et al. (2015). Aerobic denitrification: a review of important advances of the last 30 years. Biotechnol. Bioprocess Eng. 20, 643–651. doi: 10.1007/s12257-015-0009-0
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2015). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kojima, Y., Itada, N., and Hayaishi, O. (1961). Metapyrocatechase: a new catechol-cleaving enzyme. J. Biol. Chem. 236, 2223–2228.
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Kramer, S., Dibbern, D., Moll, J., Huenninghaus, M., Koller, R., Krueger, D., et al. (2016). Resource partitioning between bacteria, fungi, and protists in the detritusphere of an agricultural soil. Front. Microbiol. 7:1524. doi: 10.3389/fmicb.2016.01524
Krell, T., Lacal, J., Reyes-Darias, J. A., Jimenez-Sanchez, C., Sungthong, R., and Ortega-Calvo, J. J. (2013). Bioavailability of pollutants and chemotaxis. Curr. Opin. Biotechnol. 24, 451–456. doi: 10.1016/j.copbio.2012.08.011
Kukor, J. J., and Olsen, R. H. (1996). Catechol 2, 3-dioxygenases functional in oxygen-limited (hypoxic) environments. Appl. Environ. Microbiol. 62, 1728–1740.
Lacal, J. (2017). “The potential of hydrocarbon chemotaxis to increase bioavailability and biodegradation efficiency,” in Cellular Ecophysiology of Microbe, ed. T. Krell (New York, NY: Springer International Publishing), 1–14. doi: 10.1007/978-3-319-20796-4_3-1
Levesque-Sergerie, J.-P., Duquette, M., Thibault, C., Delbecchi, L., and Bissonnette, N. (2007). Detection limits of several commercial reverse transcriptase enzymes: impact on the low-and high-abundance transcript levels assessed by quantitative RT-PCR. BMC Mol. Biol. 8:93. doi: 10.1186/1471-2199-8-93
Lindgreen, S. (2012). AdapterRemoval: easy cleaning of next-generation sequencing reads. BMC Res. Notes 5:337. doi: 10.1186/1756-0500-5-337
Lueders, T. (2015). “DNA-and RNA-based stable isotope probing of hydrocarbon degraders,” in Hydrocarbon and Lipid Microbiology Protocols, eds T. J. McGenity, K. N. Timmis, and B. Nogales (New York, NY: Humana Press), 181–197. doi: 10.1007/8623_2015_74
Lueders, T., Dumont, M. G., Bradford, L., and Manefield, M. (2016). RNA-stable isotope probing: from carbon flow within key microbiota to targeted transcriptomes. Curr. Opin. Biotechnol. 41, 83–89. doi: 10.1016/j.copbio.2016.05.001
Manefield, M., Whiteley, A. S., Griffiths, R. I., and Bailey, M. J. (2002). RNA stable isotope probing, a novel means of linking microbial community function to phylogeny. Appl. Environ. Microbiol. 68, 5367–5373. doi: 10.1128/AEM.68.11.5367-5373.2002
Marreiros, B. C., Sena, F. V., Sousa, F. M., Batista, A. P., and Pereira, M. M. (2016). Type II NADH: quinone oxidoreductase family: phylogenetic distribution, structural diversity and evolutionary divergences. Environ. Microbiol. 18, 4697–4709. doi: 10.1111/1462-2920.13352
Martinez-Lavanchy, P. M., Müller, C., Nijenhuis, I., Kappelmeyer, U., Buffing, M., McPherson, K., et al. (2010). High stability and fast recovery of expression of the TOL plasmid-carried toluene catabolism genes of Pseudomonas putida mt-2 under conditions of oxygen limitation and oscillation. Appl. Environ. Microbiol. 76, 6715–6723. doi: 10.1128/AEM.01039-10
Maszenan, A., Seviour, R., Patel, B., and Schumann, P. (2002). Quadricoccus australiensis gen. nov., sp. nov., a beta-proteobacterium from activated sludge biomass. Int. J. Syst. Evol. Microbiol. 52, 223–228. doi: 10.1099/00207713-52-1-223
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Meselson, M., and Stahl, F. W. (1958). The replication of DNA in Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 44, 671–682.
Morris, R. L., and Schmidt, T. M. (2013). Shallow breathing: bacterial life at low O2. Nat. Rev. Microbiol. 11, 205–212. doi: 10.1038/nrmicro2970
Nebe, J., Baldwin, B. R., Kassab, R. L., Nies, L., and Nakatsu, C. H. (2009). Quantification of aromatic oxygenase genes to evaluate enhanced bioremediation by oxygen releasing materials at a gasoline-contaminated site. Environ. Sci. Technol. 43, 2029–2034. doi: 10.1021/es900146f
Nygaard, V., Løland, A., Holden, M., Langaas, M., Rue, H., Liu, F., et al. (2003). Effects of mRNA amplification on gene expression ratios in cDNA experiments estimated by analysis of variance. BMC Genomics 4:11. doi: 10.1186/1471-2164-4-11
Parales, R. E., Ditty, J. L., and Harwood, C. S. (2000). Toluene-degrading bacteria are chemotactic towards the environmental pollutants benzene, toluene, and trichloroethylene. Appl. Environ. Microbiol. 66, 4098–4104. doi: 10.1128/AEM.66.9.4098-4104.2000
Pepe-Ranney, C., Campbell, A. N., Koechli, C. N., Berthrong, S., and Buckley, D. H. (2016). Unearthing the ecology of soil microorganisms using a high resolution DNA-SIP approach to explore cellulose and xylose metabolism in soil. Front. Microbiol. 7:703. doi: 10.3389/fmicb.2016.00703
Potter, L. C., Millington, P., Griffiths, L., Thomas, G. H., and Cole, J. A. (1999). Competition between Escherichia coli strains expressing either a periplasmic or a membrane-bound nitrate reductase: does Nap confer a selective advantage during nitrate-limited growth? Biochem. J. 344, 77–84.
Pratscher, J., Dumont, M. G., and Conrad, R. (2011). Assimilation of acetate by the putative atmospheric methane oxidizers belonging to the USCalpha clade. Environ. Microbiol. 13, 2692–2701. doi: 10.1111/j.1462-2920.2011.02537.x
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Reinhold-Hurek, B., and Hurek, T. (2000). Reassessment of the taxonomic structure of the diazotrophic genus Azoarcus sensu lato and description of three new genera and new species, Azovibrio restrictus gen. nov., sp. nov., Azospira oryzae gen. nov., sp. nov. and Azonexus fungiphilus gen. nov., sp. nov. Int. J. Syst. Evol. Microbiol. 50, 649–659. doi: 10.1099/00207713-50-2-649
Rickwood, D. (ed.). (1992). Preparative Centrifugation: A Practical Approach. Oxford: Oxford University Press, 143–186.
Schmieder, R., and Edwards, R. (2011). Fast identification and removal of sequence contamination from genomic and metagenomic datasets. PLoS One 6:e17288. doi: 10.1371/journal.pone.0017288
Smith, M. A., Finel, M., Korolik, V., and Mendz, G. L. (2000). Characteristics of the aerobic respiratory chains of the microaerophiles Campylobacter jejuni and Helicobacter pylori. Arch. Microbiol. 174, 1–10. doi: 10.1007/s002030000174
Song, B., Palleroni, N. J., Kerkhof, L. J., and Häggblom, M. M. (2001). Characterization of halobenzoate-degrading, denitrifying Azoarcus and Thauera isolates and description of Thauera chlorobenzoica sp. nov. Int. J. Syst. Evol. Microbiol. 51, 589–602. doi: 10.1099/00207713-51-2-589
Spiro, S., Roberts, R., and Guest, J. (1989). FNR-dependent repression of the ndh gene of Escherichia coli and metal ion requirement for FNR-regulated gene expression. Mol. Microbiol. 3, 601–608.
Sun, W., and Cupples, A. M. (2012). Diversity of five anaerobic toluene-degrading microbial communities investigated using stable isotope probing. Appl. Environ. Microbiol. 78, 972–980. doi: 10.1128/AEM.06770-11
Táncsics, A., Farkas, M., Szoboszlay, S., Szabó, I., Kukolya, J., Vajna, B., et al. (2013). One-year monitoring of meta-cleavage dioxygenase gene expression and microbial community dynamics reveals the relevance of subfamily I.2.C extradiol dioxygenases in hypoxic, BTEX-contaminated groundwater. Syst. Appl. Microbiol. 36, 339–350. doi: 10.1016/j.syapm.2013.03.008
Táncsics, A., Szabó, I., Baka, E., Szoboszlay, S., Kukolya, J., Kriszt, B., et al. (2010). Investigation of catechol 2, 3-dioxygenase and 16S rRNA gene diversity in hypoxic, petroleum hydrocarbon contaminated groundwater. Syst. Appl. Microbiol. 33, 398–406. doi: 10.1016/j.syapm.2010.08.005
Táncsics, A., Szalay, A. R., Farkas, M., Benedek, T., Szoboszlay, S., Szabó, I., et al. (2018). Stable isotope probing of hypoxic toluene degradation at the Siklós aquifer reveals prominent role of Rhodocyclaceae. FEMS Microbiol. Ecol. 94:fiy088. doi: 10.1093/femsec/fiy088
Táncsics, A., Szoboszlay, S., Szabó, I., Farkas, M., Kovács, B., Kukolya, J., et al. (2012). Quantification of subfamily I.2.C catechol 2, 3-dioxygenase mRNA transcripts in groundwater samples of an oxygen-limited BTEX-contaminated site. Environ. Sci. Technol. 46, 232–240. doi: 10.1021/es201842h
Tindall, B., and Euzéby, J. (2006). Proposal of Parvimonas gen. nov. and Quatrionicoccus gen. nov. as replacements for the illegitimate, prokaryotic, generic names Micromonas Murdoch and Shah 2000 and Quadricoccus Maszenan et al., 2002, respectively. Int. J. Syst. Evol. Microbiol. 56, 2711–2713. doi: 10.1099/ijs.0.64338-0
Von Netzer, F., Kuntze, K., Vogt, C., Richnow, H. H., Boll, M., and Lueders, T. (2016). Functional gene markers for fumarate-adding and dearomatizing key enzymes in anaerobic aromatic hydrocarbon degradation in terrestrial environments. J. Mol. Microbiol. Biotechnol. 26, 180–194. doi: 10.1159/000441946
Wald, J., Hroudova, M., Jansa, J., Vrchotova, B., Macek, T., and Uhlik, O. (2015). Pseudomonads rule degradation of polyaromatic hydrocarbons in aerated sediment. Front. Microbiol. 6:1268. doi: 10.3389/fmicb.2015.01268
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/AEM.00062-07
Whiteley, A. S., Thomson, B., Lueders, T., and Manefield, M. (2007). RNA stable-isotope probing. Nat. Protoc. 2, 838–844. doi: 10.1038/nprot.2007.115
Wilson, L., and Bouwer, E. (1997). Biodegradation of aromatic compounds under mixed oxygen/denitrifying conditions: a review. J. Ind. Microbiol. Biotechnol. 18, 116–130.
Winderl, C., Penning, H., von Netzer, F., Meckenstock, R. U., and Lueders, T. (2010). DNA-SIP identifies sulfate-reducing Clostridia as important toluene degraders in tar-oil-contaminated aquifer sediment. ISME J. 4, 1314–1325. doi: 10.1038/ismej.2010.54
Yang, S., Wen, X., Zhao, L., Shi, Y., and Jin, H. (2014). Crude oil treatment leads to shift of bacterial communities in soils from the deep active layer and upper permafrost along the China-Russia Crude Oil Pipeline route. PLoS One 9:e96552. doi: 10.1371/journal.pone.0096552
Zedelius, J., Rabus, R., Grundmann, O., Werner, I., Brodkorb, D., Schreiber, F., et al. (2011). Alkane degradation under anoxic conditions by a nitrate-reducing bacterium with possible involvement of the electron acceptor in substrate activation. Environ. Microbiol. Rep. 3, 125–135. doi: 10.1111/j.1758-2229.2010.00198.x
Zhang, L., and Lueders, T. (2017). Micropredator niche differentiation between bulk soil and rhizosphere of an agricultural soil depends on bacterial prey. FEMS Microbiol. Ecol. 93:fix103. doi: 10.1093/femsec/fix103
Keywords: RNA-seq, RNA-SIP, metatranscriptomics, hydrocarbon degradation, groundwater, dioxygenases
Citation: Bradford LM, Vestergaard G, Táncsics A, Zhu B, Schloter M and Lueders T (2018) Transcriptome-Stable Isotope Probing Provides Targeted Functional and Taxonomic Insights Into Microaerobic Pollutant-Degrading Aquifer Microbiota. Front. Microbiol. 9:2696. doi: 10.3389/fmicb.2018.02696
Received: 21 August 2018; Accepted: 23 October 2018;
Published: 13 November 2018.
Edited by:
Zofia Piotrowska-Seget, University of Silesia of Katowice, PolandReviewed by:
Siavash Atashgahi, Wageningen University & Research, NetherlandsJan Roelof Van Der Meer, Université de Lausanne, Switzerland
Anna Gałązka, Institute of Soil Science and Plant Cultivation, Poland
Copyright © 2018 Bradford, Vestergaard, Táncsics, Zhu, Schloter and Lueders. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tillmann Lueders, dGlsbG1hbm4ubHVlZGVyc0BoZWxtaG9sdHptdWVuY2hlbi5kZQ==