 Anoop Alex1,2*
Anoop Alex1,2* Agostinho Antunes1,2*
Agostinho Antunes1,2*- 1CIIMAR/CIMAR, Interdisciplinary Centre of Marine and Environmental Research, University of Porto, Porto, Portugal
- 2Department of Biology, Faculty of Sciences, University of Porto, Porto, Portugal
The bacterial members of the genus Shewanella are widely distributed and inhabit both freshwater and marine environments. Some members of Shewanella have gained considerable attention due to its ability to survive in redox-stratified environments. However, a gap of knowledge exists on the key genomic features of the sponge-associated Shewanella sp. involving the successful host-bacteria interaction, as sponge-symbiotic Shewanella are largely underrepresented in the public repositories. With the aim of identifying the genomic signatures of sponge-Shewanella association, we generated a high-quality genome data of a sponge-associated, Shewanella sp. OPT22, isolated from the intertidal marine sponge Ophlitaspongia papilla and performed comprehensive comparative analyses of 68 genome strains of the genus Shewanella including two previously reported genomes of sponge-associated bacteria, Shewanella spongiae KCTC 22492 and Shewanella sp. Alg231_23. The 16S rRNA-based phylogenetic reconstruction showed the well-supported affiliation of OPT22 and KCTC 22492 with previously reported sponge-associated bacteria, affirming the “sponge-specific” nature of these two bacterial strains isolated from different marine sponge species from the Atlantic and Pacific (East Sea) Oceans, respectively. The genome comparison of the 68 strains of Shewanella inhabiting different habitats revealed the unusual/previously unreported abundance of genes encoding for ankyrin-repeat containing proteins (ANKs) in the genomes of the two sponge-associated strains, OPT22 (ANKs; n = 45) and KCTC 22492 (ANKs; n = 52), which might be involved in sponge-Shewanella interactions. Focused analyses detected the syntenic organization of the gene cluster encoding major secretion system (type III/IV/VI) components and the presence of effector homologs in OPT22 and KCTC 22492 that seem to play a role in the virulence of the sponge bacteria. The genomic island (GI) of Shewanella sp. OPT22 was identified to localize a gene cluster encoding T4SS components and ANK (n = 1), whereas S. spongiae KCTC 22492 harbored a total of seven ANKs within multiple GIs. GIs may play a pivotal role in the dissemination of symbioses-related genes (ANKs) through the horizontal gene transfer, contributing to the diversification and adaptation of sponge-associated Shewanella. Overall, the genome analyses of Shewanella isolates from marine sponges revealed genomic repertoires that might be involved in establishing successful symbiotic relationships with the sponge hosts.
Introduction
Members of the genus Shewanella (class Gammaproteobacteria) are facultative anaerobic bacteria widely detected in both marine and freshwater ecosystems. Ability to utilize various electron acceptors in the absence of oxygen is the hallmark feature of the genus Shewanella. It enabled them to adapt and thrive successfully in diverse habitats, ranging from varied salt concentrations, barometric pressures, deep-cold marine environments, and hydrothermal vents. Shewanella genus gained much attention from the scientific community, and has been of great interest due to the ability of several members of the genus Shewanella to convert heavy metals and toxic substances (e.g., uranium, chromium, mercury, arsenic, technetium, and cobalt) to least harmful products, thus making them suitable candidates for bioremediation (Hau and Gralnick, 2007). Moreover, they play a significant role in global biogeochemical cycles of C, N, and S in redox-stratified marine environments (Brettar et al., 2001).
Shewanella species that exist in different ecosystems are reported to have several lifestyles, mainly, the most common free-living state, pathogens (infectious agents in humans, fishes), and epibionts (living on the surface of other organisms), such as S. marinintestina, S. schlegeliana, and S. sairae isolated from the squid intestine and several fishes. The ecological roles of Shewanella living in different habitats are described in detail elsewhere (Hau and Gralnick, 2007; Janda and Abbott, 2014). Symbiotic associations among the members of the genus Shewanella were reported between S. pealeana and accessory nidamental glands (ANG) of the cephalopod, Loligopealei (squid). This symbiotic association may provide protection to eggs from predation or infection through the production of protective compounds (Barbieri et al., 2001). In marine environments, Shewanella are known to colonize other invertebrate hosts like the sea cucumber Apostichopus japonicus (Enomoto et al., 2012; Hong et al., 2017), the marine hydroid Hydractinia echinata (Rischer et al., 2016), the sipuncula Phascolosoma japonicum (“peanut worm”) and hydrocoral species (Ivanova et al., 2004), the red sea coral of the genus Favia (Shnit-Orland et al., 2010), and sponges (Lee et al., 2006; Yang et al., 2006).
Sponges (phylum Porifera) are known to harbor abundant and diverse communities of symbiotic microorganisms. Several approaches including the whole-genome sequencing of symbiotic microbes isolated from sponges, metagenomics/metatranscriptomics, and single-cell genomics have provided insight into the genomic mechanisms of sponge-microbe association. For instance, the abundance and role of transposable insertion elements in the evolution of symbiotic bacterial genomes (Thomas et al., 2010); frequent detection of the genes encoding for adhesion-related proteins and eukaryotic-like proteins (ELPs) and the role of secretion systems (SSs) and effector molecules (Fan et al., 2012; Liu et al., 2012; Bondarev et al., 2013; Alex and Antunes, 2015; Romano et al., 2016) were described as key features.
Despite these advances in the understanding of symbiotic bacteria associated with several sponge species, no comparative genomic evidence is yet available for the most widely studied members of the genus Shewanella, particularly those living in close association with sponge hosts. So far, the genomes of only two strains- Shewanella sp. Alg231_23 (PRJEB13410) isolated from an Atlantic sponge Spongia officinalis and Shewanella spongiae KCTC 22492 isolated from a marine sponge living at 20 m water depth of the East Sea, Korea (Yang et al., 2006) are available.
Here, we performed comprehensive comparative analyses of 68 genome strains of the genus Shewanella including three sponge-associated bacteria, the previously reported S. spongiae KCTC 22492 and Shewanella sp. Alg231_23, and we further generated a high-quality genome data of a sponge-associated bacterium, Shewanella sp. OPT22, to investigate the genomic features responsible for establishing a successful symbiotic sponge-bacteria relationship. Our study showed: (i) the sponge-specific clustering of two sponge-associated Shewanella (OPT22 and KCTC 22492), (ii) an abundance of the genes encoding for ankyrin-repeat containing proteins (ANKs) responsible for evading the host immune response in the genome of OPT22 (ANKs; n = 45) and KCTC 22492 (ANKs; n = 52), (iii) localization of ANKs within the genomic islands (GIs) suggesting the role of horizontal gene transfer to enhance the ecological fitness of the symbiotic bacteria, and (iv) several SS components/effector proteins that act as virulence factors to modulate the bacteria-host interactions. To our knowledge, this is the first detailed genomic analyses of Shewanella spp. isolated from marine sponges unraveling the unique genomic repertoire for its successful colonization and symbiotic lifestyle.
Materials and Methods
Isolation of Shewanella
Shewanella sp. OPT22 was isolated from the intertidal marine sponge Ophlitaspongia papilla (class Demospongiae) collected from the Atlantic coast (41.2308206N 8.7216926W) of Portugal. Sponges were collected in Ziploc®bags and transported to the lab in natural seawater (in a cooling box) within 1 h, and processed immediately. In the lab, ≈1 cm3 of the sponge tissue was carefully excised using a sterile surgical blade and processed under dissection microscope to remove any external debris or other macroscopic materials. Furthermore, sponge tissue was washed using natural sterile seawater (NSW) for 3–4 times on a horizontal platform shaker for 5 min at room temperature in order to remove the loosely associated microbes and other debris. Tissue sample was grinded and homogenized using a sterile pestle and mortar with 10 ml NSW. The homogenate was serially diluted (100 μl of the 10-1 through 10-5 dilutions) and spread plated on DifcoTM Marine Agar 2216 medium containing amphotericin B (2.5 mg/L). Amphotericin B was used to prevent the fugal growth. All plates were incubated in dark at 28°C for 3–4 days. Single colonies were obtained after repeated streaking. Bacterial colonies were inoculated into 5 ml tubes containing DifcoTM Marine Broth 2216 and kept under constant shaking at 28°C. Genomic DNA was extracted from the bacterial cultures in stationary phase using PureLinkTM Genomic DNA kit (Invitrogen) according to the manufacturer’s protocol for bacterial DNA isolation.
Genome Sequencing, Assembly, and Annotation
Paired-end (PE) read library (2 x 100nt) of Shewanella sp. OPT22 genomic DNA was constructed with an insert size of ∼350 bp and sequencing was performed using the Illumina’s HiSeq 2500 Sequencing System. Low quality reads (Phred score < 30) and adapter sequences were removed using cutadapt v1.12 (Martin, 2011). The processed reads of > 1700-fold coverage (based on a 5 Mb genome size) was assembled using Velvet v1.2.10 (Zerbino and Birney, 2008) with the best possible k-mer coverage value (k = 99) obtained from VelvetK. Gene prediction was performed using PROKKA v1.12 (Seemann, 2014). Hidden Markov Model (HMM) profiles of Pfam31.0 and TIGRFAMs15.0, and curated dataset constituting bacterial protein sequences from the UniProt Knowledgebase (UniProtKB Release 2017_3) were compiled locally for the annotation of predicted coding sequences (CDS). Genome completeness was estimated using Alteromonadales marker gene sets defined by CheckM (Parks et al., 2015).
Proteins were categorized in clusters of orthologous groups (COGs) with standalone RPS-BLAST v2.2.31 (Reverse Position-Specific BLAST) against NCBI preformatted CDD (conserved domains database1) with an E-value cutoff of 1e-03. The output was parsed-out and classified into different categories using cdd2cog.pl script v0.1 (Andreas, 2016). Significant differences in proportion of COG categories were determined using z-test. A total of 67 bacterial genomes of the genus Shewanella were retrieved using NCBI Genome Downloading Scripts2. The genome of S. spongiae KCTC 22492 was accessed on October 2018. The list of the genomes of the 67 Shewanella spp. isolated from different ecological niches and assembly version of the genomes used for our comparative analyses is given in Supplementary Table S1. The genomes were re-annotated using PROKKA v1.12 (Seemann, 2014) in order to avoid the inconsistencies with different annotation pipelines.
Phylogenetic Analyses of the Genus Shewanella
The 16S ribosomal RNA (16S rRNA) gene sequences were retrieved from the genomes of Shewanella considered in the present study (the 16S rRNA genes were identified only in the genomes of 57 Shewanella spp.) along with the 16S rRNA sequences of the sponge-associated Shewanella (retrieved by BLAST; n = 11) and were aligned with Clustal Omega (Sievers and Higgins, 2014) implemented in SeaView v4.4.2 (Gouy et al., 2010), with ambiguous regions removed by Gblocks v0.91b (Talavera and Castresana, 2007) using “less stringent” options. The final alignment (∼1,458 bps) was used to construct a maximum likelihood phylogenetic tree in PhyML (Guindon et al., 2010); with 500 bootstrap replicates using nearest-neighbor interchanges (NNIs) tree search criteria. Evolutionary model GTR + I + G adopted under Akaike Information Criterion with correction (AICc) implemented in MrAIC v1.4.4 (Nylander, 2004) was used as a best-fit model of nucleotide substitution. The Pyani (Pritchard et al., 2016) package was used to measure the average nucleotide identity (ANI) and the genome-scale relatedness of bacterial members.
The whole-genome phylogenetic tree was constructed using protein sequences of 114 conserved core single-copy orthologous genes (encompassing 28,356 amino acids). Briefly, the “core” genes of the members of the genus Shewanella was estimated by clustering the CDS using the bidirectional best-hit (BDBH), COGtriangles, and OrthoMCL clustering algorithms implemented in GET_HOMOLOGUES v18042017 (Contreras-Moreira and Vinuesa, 2013) with 75% pairwise alignment coverage and E-value (expectation value for DIAMOND BLASTP alignments) set at 1e-03. Inparalogs were excluded from the clusters (option “-e”). A total of 114 single-copy orthologous genes were aligned using MUSCLE v3.8.31 (Edgar, 2004) and low quality alignment regions were removed using trimAl v1.4 with “automated1” option (Capella-Gutiérrez et al., 2009). Alignment files were concatenated to a super-alignment using FASconCAT v1.1 (Kück and Meusemann, 2010). A phylogenetic tree was inferred using maximum likelihood method implemented in IQ-TREE v1.6.1 (Nguyen et al., 2015) under automated model selection option “TEST” with 1000 bootstrap replicates statistical support.
Detection of Eukaryotic-Like Proteins (ELPs)
Eukaryotic-like proteins (ELPs) containing motifs- tetratrico peptide repeats (TPRs: IPR011990, IPR019734, IPR013105, IPR001440, and IPR011717), ankyrin repeats (ANKs: IPR020683 and IPR002110), and Sel1 repeats (IPR006597)-were detected using InterProScan v5.24.63 (Jones et al., 2014). Presence of TPRs and Sel1 was further validated using TPRpred with E-value cutoffs of 1e-03 and 1e-02. Significant differences in proportion of ANKs were determined using z-test.
Genomic islands were predicted using an integrated approach implemented in IsandViewer 4 (Bertelli et al., 2017). Pre-computed complete reference genome of Shewanella oneidensis MR-1 chromosome available in the IslandViewer webserver was selected to re-order the contigs. Subcellular localization of the genes in the GI was performed using PSORTb v3.0.2 (Yu et al., 2010).
Identification of Secretion System Components
Secretion system components were identified by uploading the whole proteome of Shewanella sp. OPT22 as a query and performed BLAST search using BlastKOALA tool implemented in KEGG v2.1 (Kanehisa et al., 2016, 2017) against the taxonomic group “Bacteria” and “genus_prokaryote” database. T346Hunter, a web based tool was also used to predict the encoded SSs (Martínez-García et al., 2015). The predicted genes were further confirmed by performing NCBI BLASTP service. Easyfig v2.2.2 (Sullivan et al., 2011) was used to generate the gene clusters of the SSs and the GI.
Results and Discussion
Phylogeny of the Genus Shewanella and the Evidence for Sponge-Specific Clustering of Two Symbiotic Strains
Initial similarity search using BLASTn revealed a higher relatedness (sequence identity ranging from 97 to 99%) of the 16S rRNA gene sequences of Shewanella sp. OPT22 sequenced in this study with uncultured bacteria isolated from the sponge Ircinia spp. (Northwestern Mediterranean Sea; JX206530, JX206726, JX206689, and JX206721), Shewanella strains isolated from the sponge Ircinia dendroides (Mediterranean Sea; NR_043617), the Micronesian sponge (GU289647–GU289648), I. strobilina (KC429938), and the Sea of Japan (NR_043591); and Shewanella strains isolated from the coral Montastrea annularis collected in the Florida keys (Table 1). Phylogenetic analysis using the 16S rRNA gene sequences revealed the clustering of Shewanella sp. OPT22 isolated from the Atlantic sponge O. papilla and S. spongiae KCTC 22492 isolated from a marine sponge collected in Korea with other geographically separated sponge-and coral-associated bacteria, which favor the sponge-specific nature of Shewanella sp. OPT22 and S. spongiae KCTC 22492, and likely to represent true sponge-associated bacteria (Figure 1 and Supplementary Figure S1). Clustering of 16S rRNA gene sequences recovered from geographically isolated sponges and/or from different sponge species was reported in many studies (Simister et al., 2012; Alex et al., 2013; Thomas et al., 2016).
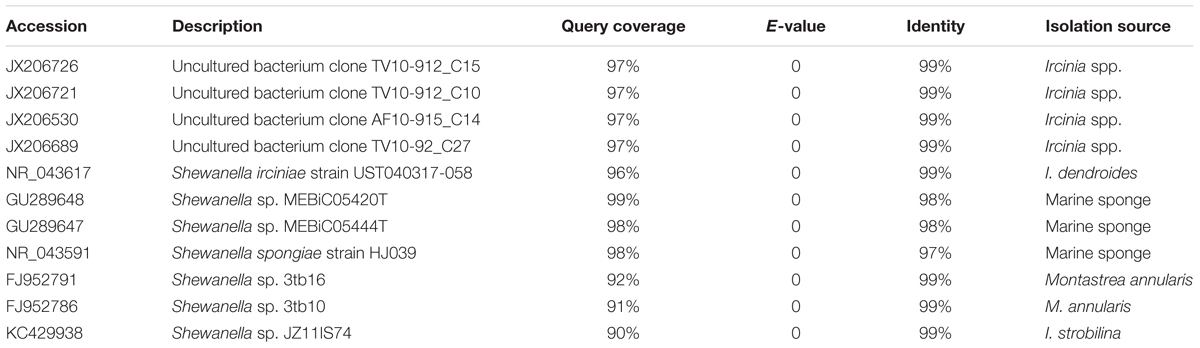
Table 1. Best BLAST hit identities of the queried 16S rRNA gene of Shewanella sp. OPT22.
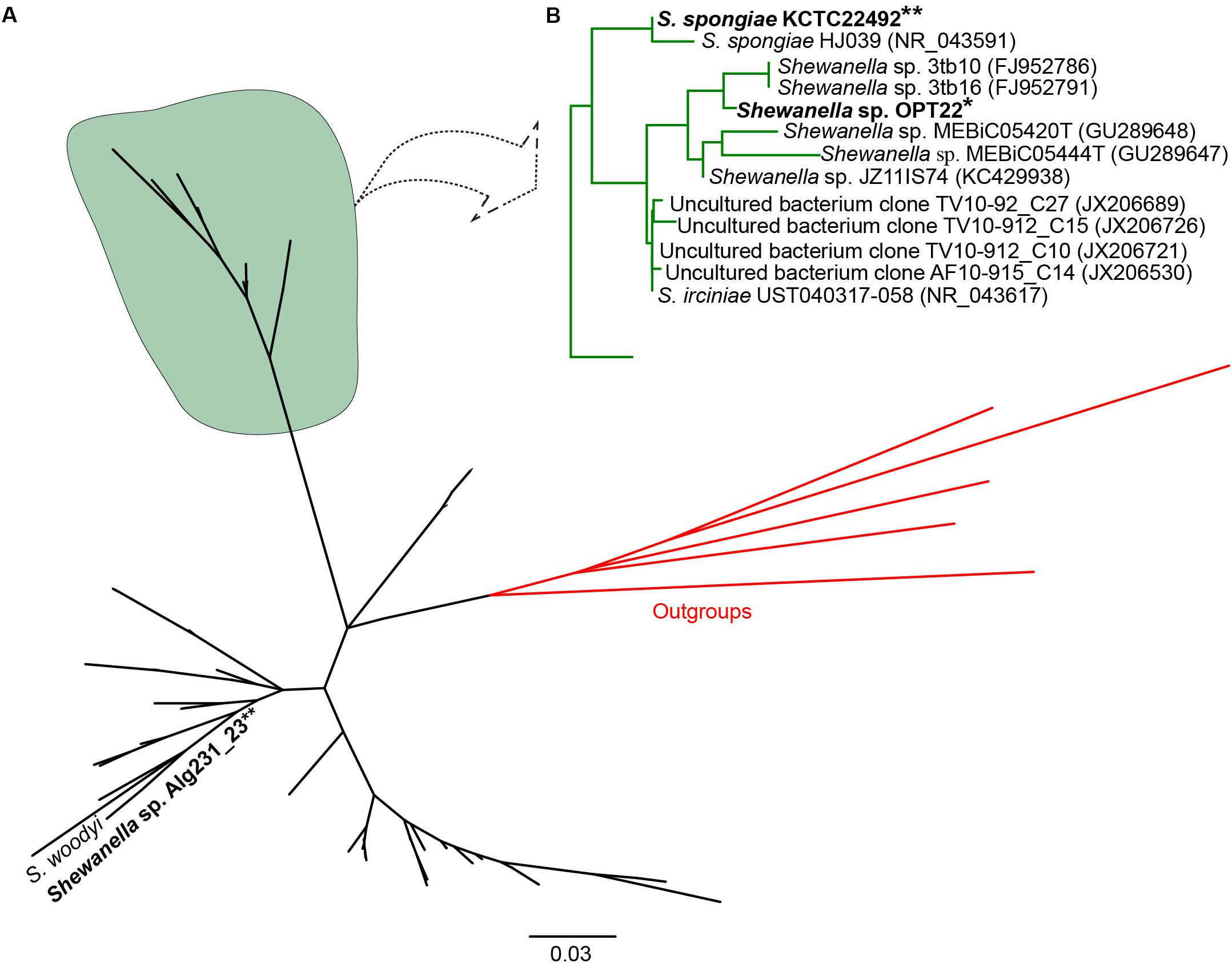
Figure 1. Phylogeny of the genus Shewanella. (A) Phylogenetic tree inferred using the 16S rRNA gene sequences of Shewanella species used for the comparative genomics and the closely related species retrieved from best BLAST hit (shown in Table 1). The sponge-specific cluster is shaded in green. Outgroup species are labeled in red. (B) Detailed view of the sponge-specific cluster. Strains in bold denote the only available genomes of sponge-associated Shewanella that are used in this study. The Shewanella sp. OPT22 sequenced in this study is highlighted by a single asterisk, grouped with other sponge-associated Shewanella strains. Shewanella sp. 3tb10 and Shewanella sp. 3tb16 were isolated from a coral host. Strains with two asterisks denote previously reported sponge-associated Shewanella. The complete phylogenetic tree is provided in Supplementary Figure S1.
However, Shewanella sp. Alg231_23 isolated from the sponge Spongia officinalis sampled from the Atlantic coast of Portugal grouped with S. woodyi ATCC 51908 isolated from a squid/seawater from depths of 200–300 m in the Alboran Sea (Figure 1 and Supplementary Figure S1). The whole-genome phylogeny using a concatenated set of 114 single-copy orthologous genes also grouped Shewanella sp. Alg231_23 and S. woodyi ATCC 51908 together, whereas Shewanella sp. OPT22 and S. spongiae KCTC 22492 were clustered together (Figure 2). Supporting the observed phylogenetic pattern, the ANI analysis also revealed the close evolutionary relatedness of Shewanella sp. Alg231_23 and S. woodyi ATCC 51908, with their ANI values being ∼97%, thus above 95% ANI that corresponds to the 70% DNA-DNA hybridization cutoff value widely used to delineate species (Goris et al., 2007). Shewanella sp. Alg231_23 isolated from a sponge might be an opportunistic or transient bacterium detected during sampling. Furthermore, comparison of Shewanella sp. OPT22 with other Shewanella strains showed an average ANI value ≤ 72% (Supplementary Table S2). Evidence suggests that the sponge-associated Shewanella sp. OPT22 sequenced in this study could be a novel strain closely related to S. irciniae.
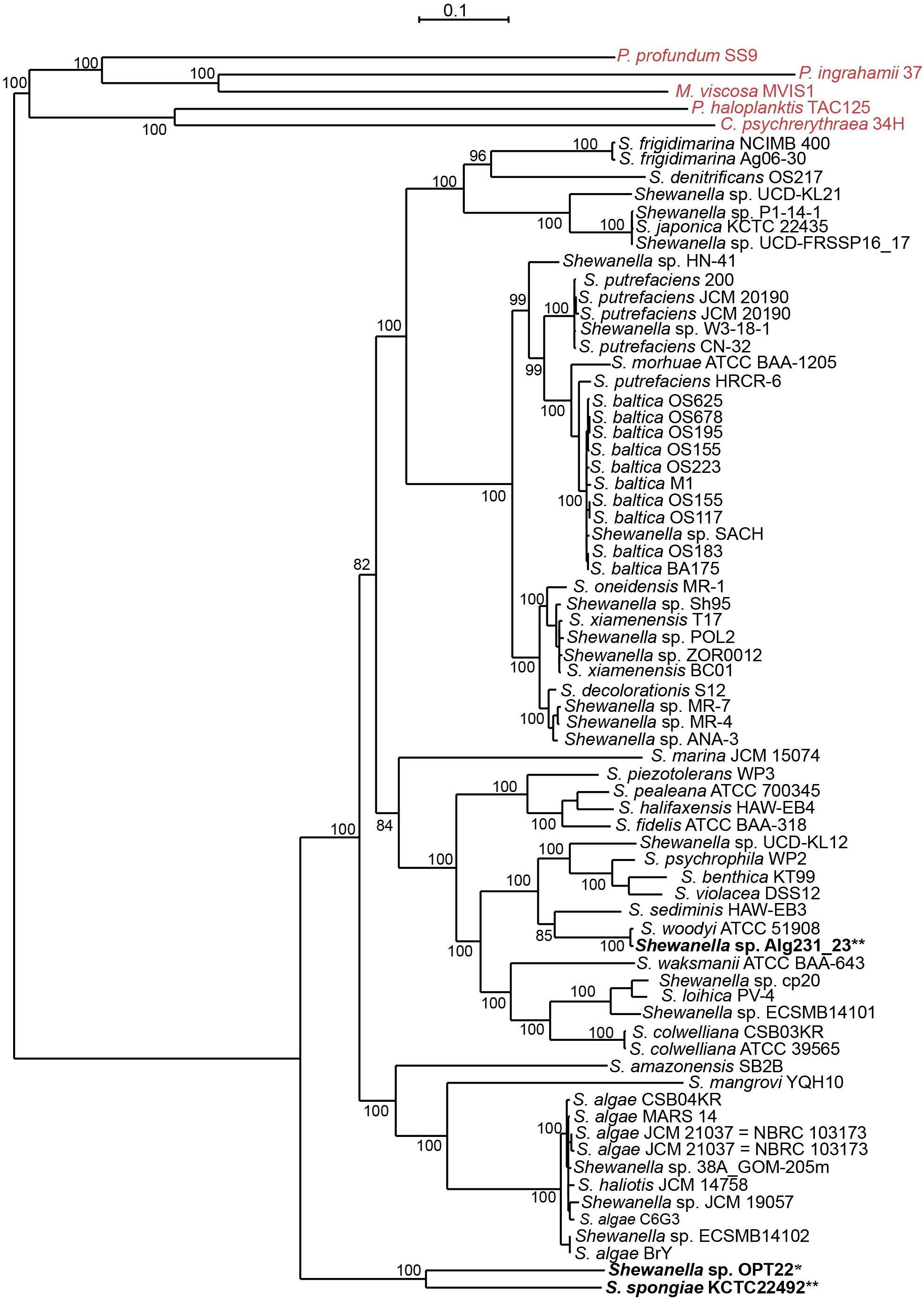
Figure 2. Whole-genome phylogeny of the genus Shewanella. A maximum likelihood tree was inferred from a concatenated protein dataset of 114 single-copy orthologous genes. Strains in bold denote the only available genomes of sponge-associated Shewanella that are used in this study. The genomes of the Shewanella strains sequenced here and from previous studies are shown as single and double asterisks, respectively. Outgroup species are labeled in red. Bootstrap node support values above 80 are shown.
General Genome Characters of Sponge-Associated Shewanella
The genome assembly of the strain Shewanella sp. OPT22 retrieved a total of 69 contigs (N50 of > 0.30 Mb) and a chromosomal genome size of 4.4 Mbp with a low G + C content of 39.6% (Figure 3A). The genome was near complete (93.6%) and had low evidence of possible contamination (0.37%) based on a battery of 519 Alteromonadales marker genes. The genome encoded a total of 3,971 genes, of which 3,840 (96.7%) were protein-coding and 108 were RNA-coding. The functional annotation of genes based on cluster of orthologous (COG) groups is shown in Supplementary Table S3. The detailed genome characteristic features of the three sponge-associated bacteria (Shewanella sp. OPT22, S. spongiae KCTC 22492, and Shewanella sp. Alg231_23) are given in Table 2. Comparison of COG categories among three sponge-associated Shewanella species revealed a significant overrepresentation of functional categories (Z-test, p < 0.01) – COG ‘C’ (Energy production and conversion), ‘E’ (Amino acid transport and metabolism), ‘G’ (Carbohydrate transport and metabolism), ‘H’ (Coenzyme transport and metabolism), ‘K’ (Transcription), ‘M’ (Cell wall/membrane/envelope biogenesis), ‘O’ (Posttranslational modification, protein turnover, chaperones), ‘P’ (Inorganic ion transport and metabolism), ‘Q’ (Secondary metabolites biosynthesis, transport and catabolism), ‘R’ (General function prediction only), ‘S’ (Function unknown), and ‘T’ (Signal transduction mechanisms) in the genome of another sponge-associated bacterium, Shewanella sp. Alg231_23 isolated from a sponge, S. officinalis (Figure 4). This trend is obvious considering a larger genome size of Shewanella sp. Alg231_23 (5.8 Mbp) when compared to Shewanella sp. OPT22 (4.4 Mbp) and S. spongiae KCTC 22492 (4.9 Mbp). Many evidences suggest the existence of trends between functional gene content and genome size (Stover et al., 2000; Jordan et al., 2001; Bentley et al., 2002), and disproportionate enrichment of the genome attributes responsible for regulation and secondary metabolism genes in larger genomes (Konstantinidis and Tiedje, 2004). Furthermore, we detected a uniform distribution of genes belonging to COG functional categories between two closely related sponge-associated bacteria: Shewanella sp. OPT22 and S. spongiae KCTC 22492.
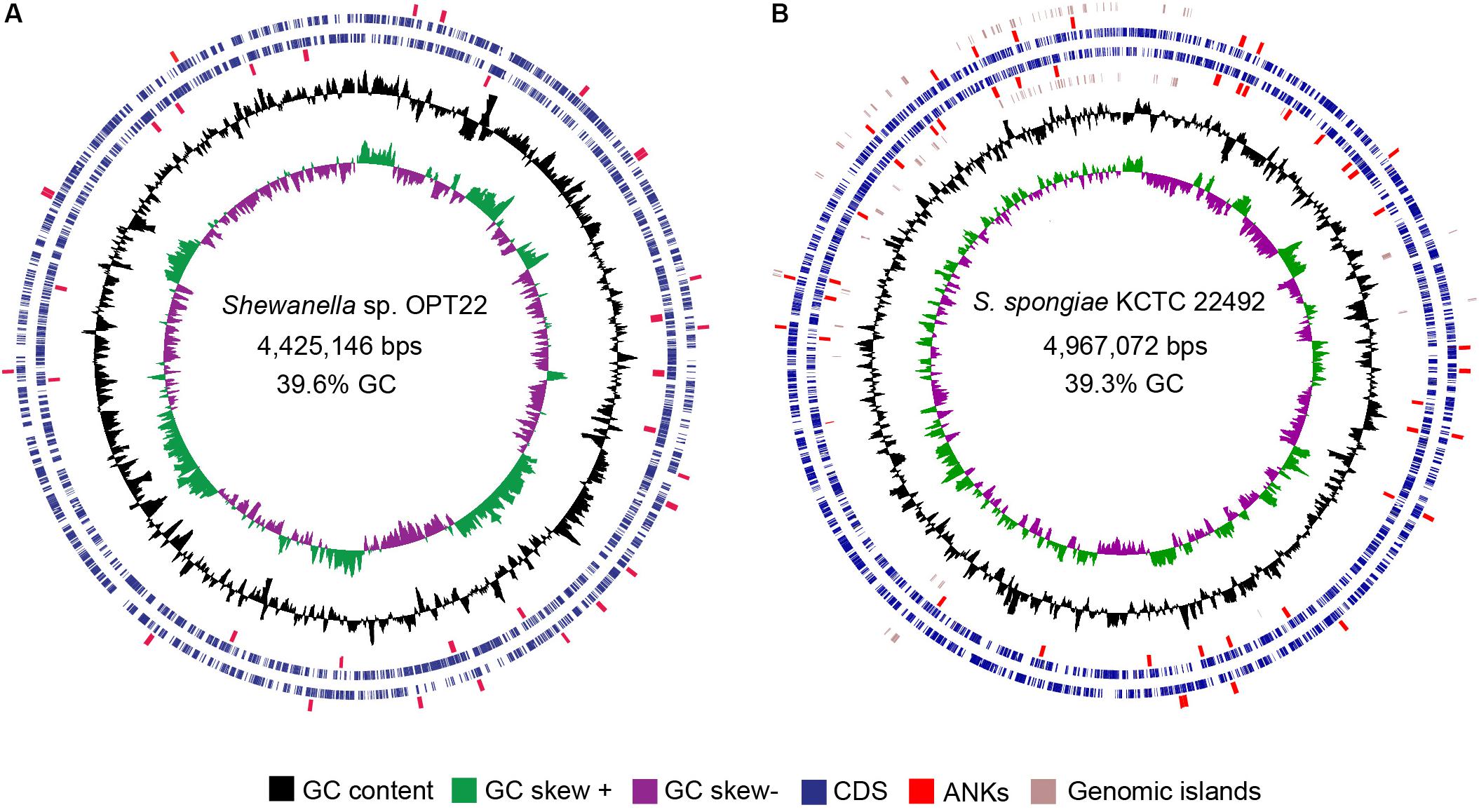
Figure 3. Circular view of the genomes of two sponge-associated Shewanella species, generated with CGview (Stothard and Wishart, 2005). The genome map of (A) Shewanella sp. OPT22 sequenced in this study, isolated from an Atlantic sponge, O. papilla (B) S. spongiae KCTC 22492 isolated from an Eastern Sea (Korea) marine sponge. Circles from interior to exterior represent GC skew and GC content (black circle). Blue circles denote the coding sequences on forward and reverse strands. GIs are represented as brown circles in the genome of S. spongiae KCTC 22492. The ankyrin-repeat domains containing proteins detected in the genomes are highlighted in red.
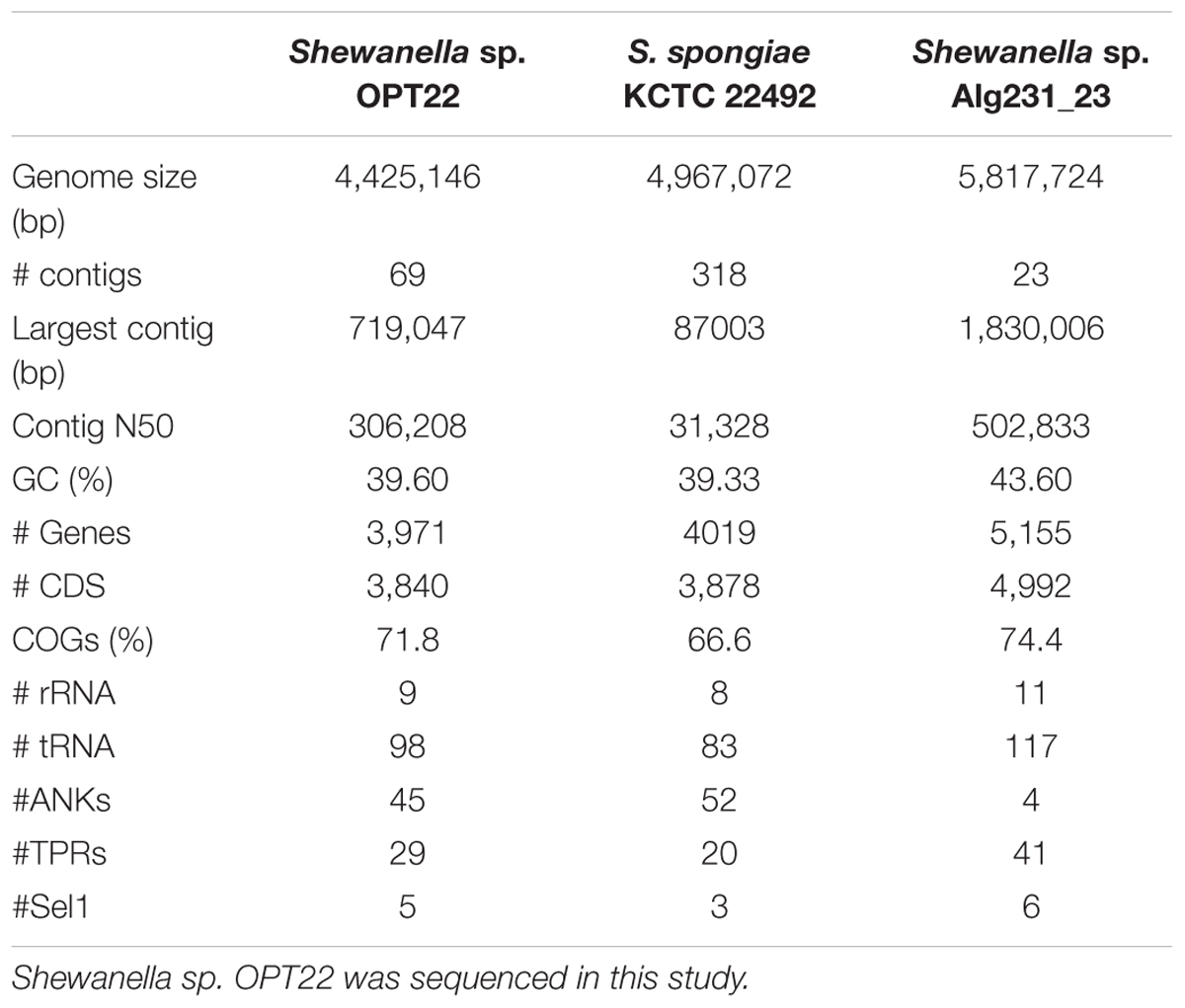
Table 2. Genome characteristics of the three sponge-associated Shewanella species.
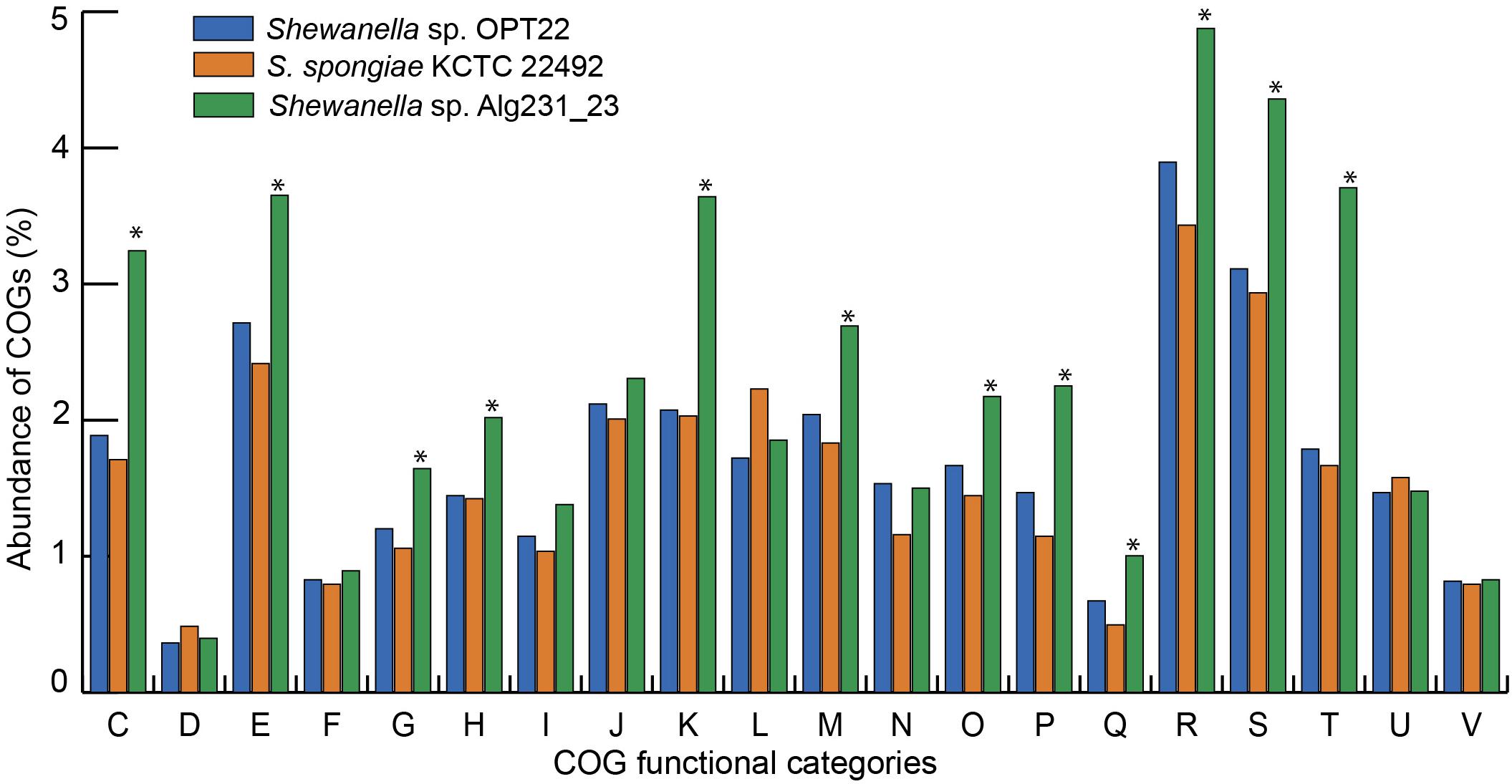
Figure 4. Bar graph representing the relative abundance of COGs in each of the three sponge-associated Shewanella species. COG functional classes from C-V were shown. Shewanella sp. OPT22 sequenced in this study was represented in blue and other two genomes of sponge-associated Shewanella species- KCTC 22492 and Alg231_23 compared in this analysis were represented in light-brown and green, respectively. The asterisks denote significant abundance of COG categories (p < 0.01).
Predicted Genomic Architecture of Sponge-Associated Shewanella for the Successful Association With the Marine Sponge
Eukaryotic-Like Proteins and Abundance of Ankyrin-Like Protein Domains
The genome of Shewanella sp. OPT22 was encoded with certain ELPs containing motifs such as ANKs, TPRs, and Sel1 repeats. ELPs (symbiosis factors) have been commonly detected in pathogenic and symbiotic microbes, playing a vital role in intracellular survival and pathogenicity by interfering with the host protein-protein interactions (Habyarimana et al., 2008; Gomez-Valero et al., 2011). An abundance of proteins containing ankyrin-like domains (ANKs; n = 45; p < 0.01) was detected in the genome of sponge-associated Shewanella sp. OPT22 sequenced in this study when compared to other bacterial members of the genus Shewanella (Figure 5 and Supplementary Tables S4, S5). All the other strains encoded less than 10 ANKs (six strains were devoid of ANKs) in their genomes (Figure 5 and Supplementary Table S5). Overrepresentation of ANKs in Shewanella sp. OPT22 could be a key feature of the sponge-associated lifestyle of this bacterium. Previous study reported a frequent detection of ANKs in symbiotic microbes and suggested that lifestyle is a determinant factor for the abundance of ANKs (Jernigan and Bordenstein, 2014). Affirming the previous statement, the genome of another closely related bacterium- S. spongiae KCTC 22492 also harbored a high number of ANKs (n = 52). ANKs detected in Shewanella sp. OPT22 and S. spongiae KCTC 22492 might facilitate the host immune evasion and the bacterial survival inside the sponge host. Metagenomics of microbial consortium associated with a sponge Cymbastela concentrica (Thomas et al., 2010; Liu et al., 2012), metaproteogenomics of several sponge-associated microbes (Fan et al., 2012), and the genome analyses of symbiont-Poribacteria (Siegl et al., 2011), Deltaproteobacteria (Liu et al., 2011), and Pseudovibrio spp. isolated from different sponges (Bondarev et al., 2013; Alex and Antunes, 2015, 2018; Versluis et al., 2018) revealed the presence of ELPs. Abundance of ANKs in the microbiome of healthy sponges (Fan et al., 2012; Liu et al., 2012) and their loss in sponges experiencing thermal stress and bleaching (Fan et al., 2013) indicate that ANKs play a crucial role in sponge-bacteria symbioses. The role of ANKs in modulating phagocytosis by amoeba and intracellular survival was experimentally validated by heterologous expression of sponge symbiont encoded ANKs, promoting bacteria-eukaryote symbiosis (Nguyen et al., 2014).
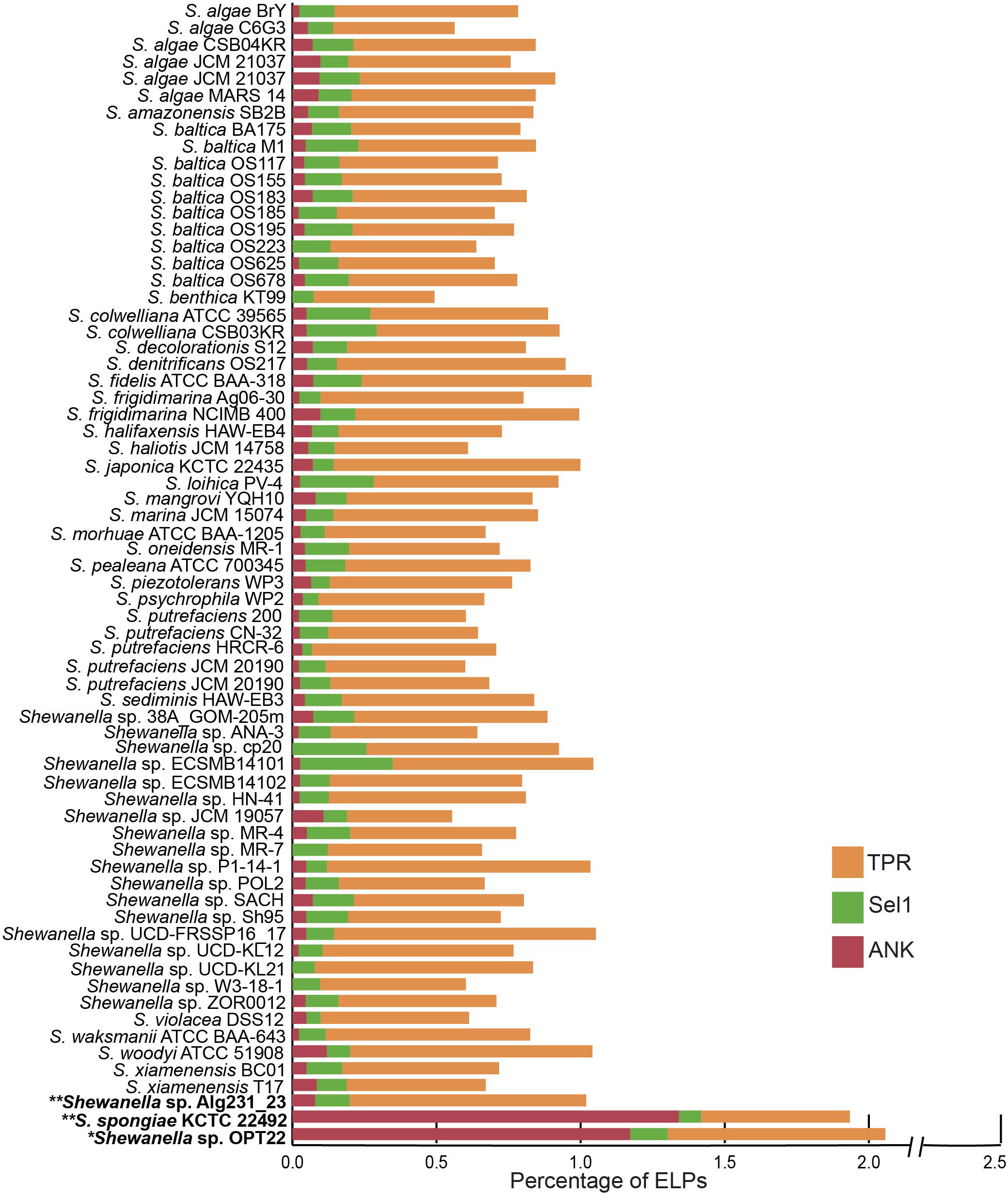
Figure 5. Staked bar plot showing the percentage abundance of eukaryotic-like proteins (ELPs) detected in the genus Shewanella. A total of 68 genomes were compared for the presence of proteins-containing ELPs- ankyrin-repeat (ANKs), tetratrico peptide repeats (TPRs), and Sel1 repeats. The strains isolated from the sponges are shown in bold. Single and double asterisks denote the genomes sequenced in this study and in previous studies, respectively.
Interestingly other ELPs, like TPRs and Sel1, were widespread in all the genomes analyzed here, independent of the lifestyle and habitat (Figure 5 and Supplementary Table S5). The genome of Shewanella sp. OPT22 encoded fewer proteins with TPRs (n = 29) and Sel1 repeats (n = 5). The genome of another sponge-associated bacterium, Shewanella sp. Alg231_23showed less number of ANKs (n = 4), and Sel1 repeats (n = 6); and an overabundance of TPRs (n = 41). A similar trend was detected in S. woodyi ATCC 51908, a closely related strain isolated from the squid (ANKs; n = 6, Sel1; n = 4, TPRs; n = 42). Our analyses (see previous section) showed that Alg231_23 is phylogenetically closer to S. woodyi ATCC 51908 than to OPT22, affirming the statement that rather than lifestyle; phylogenetic history is responsible for the abundance of TPRs (Jernigan and Bordenstein, 2015). However, the aforementioned statement should be interpreted with caution due to the limited number of the genomic data of sponge-associated Shewanella species (n = 3) currently available. Isolation of more Shewanella strains from sponges might give further insight of the role of ELPs in the sponge-associated Shewanella.
Role of Secretion Systems in Shewanella-Sponge Symbioses
Symbiotic microbes utilize multitude of methods to colonize and invade eukaryotic hosts. One such mechanism involves the transport of proteins across the membrane using specialized translocation systems called secretions systems (SSs) to thwart the host immune response and facilitate the bacterial invasion (Green and Mecsas, 2016). KEGG analysis detected the genes coding for type III SS (T3SS) (M00332), type IV SS (T4SS) (M00333), and type VI SS (T6SS) (M00334) in the genome of Shewanella sp. OPT22 (Figures 6, 7 and Supplementary Table S6) suggesting the likely ability of the sequenced bacterial strain to export and inject the effector/virulence molecules into the host cell for the establishment of sponge-bacteria interaction. The details of each SSs are discussed below.
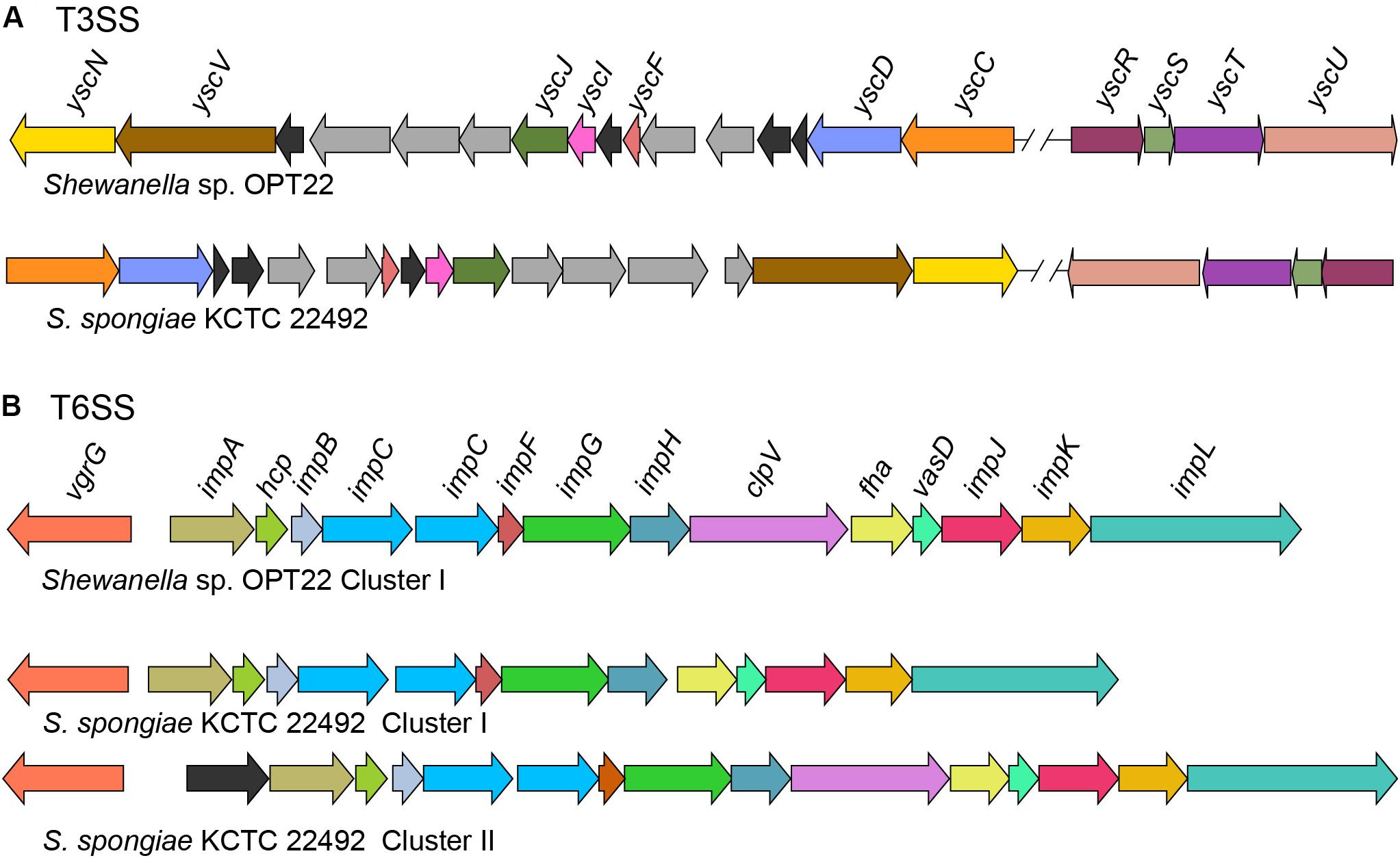
Figure 6. Genetic organization of major SSs in Shewanella sp. OPT22 sequenced in this study and S. spongiae KCTC 22492. (A) Predicted type III secretion system (T3SS) core components in the genome of OPT22 and KCTC 22492. Detected T3SS clusters are shown as cluster I and cluster II. Gene clusters detected in different contigs are separated by double cross-hatched lines. (B) Predicted genes coding for type VI secretion system (T6SS). One gene cluster was detected in the OPT22, whereas two clusters (cluster I and cluster II) in KCTC 22492. Different color schemes are given for each predicted genes in all the SSs. Functionally related group of genes are indicated with the same color in each SS. Black (T3SSs and T6SSs) and dark-gray (T3SS) colors denote predicted hypothetical and unrelated genes. The genes are not shown to scale.
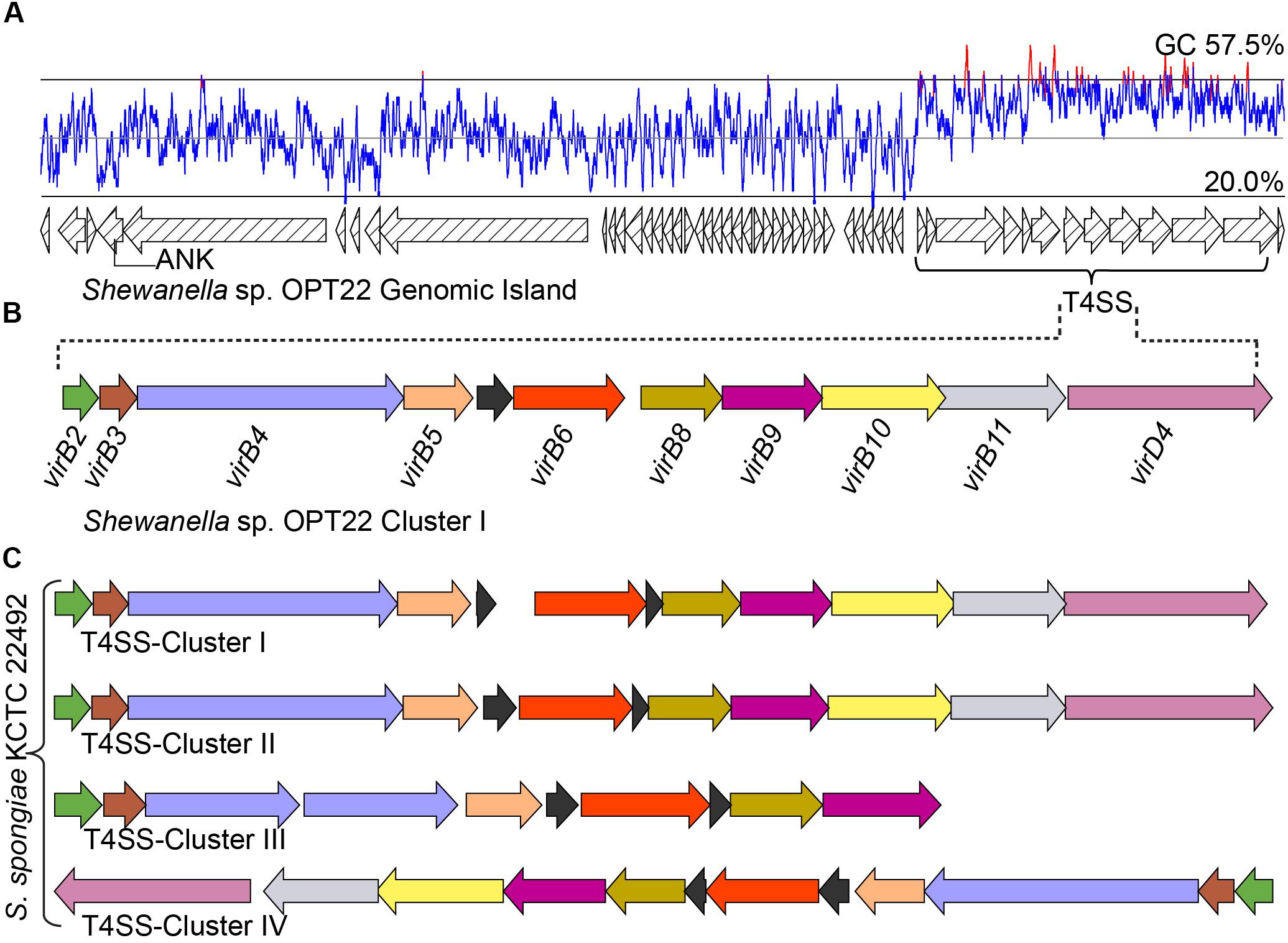
Figure 7. Genomic island (GI) and type IV SS. (A) Predicted GI encoding T4SS gene cluster in Shewanella sp. OPT22. The line graph above the GI represents the GC content. A gene encoding for ankyrin-repeat containing protein is also shown. (B) Genetic organization of the type IV SS in Shewanella sp. OPT22 (cluster I). (C) T4SS gene clusters detected in S. spongiae KCTC 22492 (cluster I, II, III, and IV). Different color schemes are given for each predicted SS genes. Functionally related group of genes are indicated with the same color in each SS in both species. Black color denotes predicted hypothetical genes. The genes are not shown to scale.
Type III Secretion System
Type III secretion systems or “injectisomes” are complex machineries, that provide commensal and pathogenic bacteria with a unique virulence mechanism enabling them to inject the effector from the bacterial cytosol to the cytoplasm/ plasma membrane of the target (host) cells (Puhar and Sansonetti, 2014). In the genome of Shewanella sp. OPT22, we detected the genes encoding for T3SS (non-flagellar T3SS) core components (Figure 6A) and four copies of homologs of T3SS effector (T3E) molecules EspA-like secreted protein (pfam03433) and HopW1-1 (Hrp outer protein). Enteropathogenic Escherichia coli (EPEC) secreted protein A (EspA) is a hydrophilic translocon component responsible for the attachment of the bacterium to the host cell surface by forming a sheath-like filament (Knutton et al., 1998; Sekiya et al., 2001), which aids to penetrate the host mucous barrier (Daniell et al., 2001). The role of other T3Es in the disruption of actin cytoskeleton and inhibition of endocytosis was reported among the plant pathogen Pseudomonas syringae (Kang et al., 2014). Further analyses revealed syntenic organization of T3SS gene cluster of two sponge-associated bacteria, Shewanella sp. OPT22 and S. spongiae KCTC 22492 (Figure 6A). The genome of S. spongiae KCTC 22492 was also encoded with the homologs of genes responsible for T3E molecules-EspA, pathogenicity island two effector protein SseD, and PipB2 (Pathogenicity island-encoded protein). SseD and PipB2 are reported to function as virulence factors in Salmonella, required for establishing infection of the mouse (Klein and Jones, 2001) and maintenance of intracellular pathogenic lifestyle (Henry et al., 2006). Non-flagellar T3SS and genes encoding effector molecules were not detected in another sponge-associated bacterium-Shewanella sp. Alg231_23. Absence of an effective T3SS could possibly widen the range of marine host species on which Shewanella strains can colonize. A similar trend was observed among the plant pathogen Pseudomonas syringae, as an adaptation mechanism to reduce the fitness costs of host-specific virulence in the presence of other ephemeral host plants (Kniskern et al., 2011).
Type VI Secretion System
Type VI secretion systems (T6SS), are widespread in Gram-negative bacteria and have been shown to be involved in pathogenicity, inter- and intra-species competition, bacterial communication (quorum sensing), and biofilm formation (Alteri and Mobley, 2016; Dang and Lovell, 2016; Gallique et al., 2017). In the genome of Shewanella sp. OPT22, we detected a 21 Kbp T6SS gene cluster that encodes genes that are predicted to the 13 core T6SS proteins (Figure 6B and Supplementary Table S6). In the case of S. spongiae KCTC 22492, the T6SS-associated genes are located in two clusters with different sizes-T6SS-I (18 Kbp) and T6SS-II (23 Kbp) (Figure 6B). The prevalence of extra gene cluster coding for T6SS might be advantageous for unknown function, as suggested in Pantoea ananatis strains inhabiting different environments (Shyntum et al., 2014). Both T6SS-I and II clusters of S. spongiae KCTC 22492 encoded the core T6SS proteins, which if expressed could produce the complete machinery for T6SS. Further experimental validation is required to test whether T6SS encoded genes are transcribed in both the clusters. Moreover, the genetic organization of T6SS gene cluster is conserved in Shewanella sp. OPT22 and S. spongiae KCTC 22492. Predicted T6SS gene clusters in the genomes of two sponge-associated Shewanella spp., also encoded the hcpI (COG3157) and vgrG (COG3501) genes with possible effector functions (Bönemann et al., 2010). Homologs of hcpI and vgrG in multiple copies were identified in the genomes of sponge-associated Pseudovibrio spp. (Bondarev et al., 2013; Alex and Antunes, 2015, 2018; Romano et al., 2016). However, we did not identify extra copies of T6SS effector molecules- hcpI and vgrG in the genomes of both sponge-associated Shewanella species. Analysis of Shewanella sp. Alg231_23, isolated from a sponge, revealed syntenic genetic organization of T6SS gene cluster with closely related S. woodyi ATCC 51908 (Supplementary Figure S2). We assume that T6SSs of both Shewanella species are functional, due to the presence of all structural components of the T6SS apparatus that presumably act as a virulence factor by delivering the effector proteins into the sponge host cell.
T4SS and Localization of ANKs Within the GIs of Sponge-Associated Shewanella
In addition to the T3 and T6SSs, we identified a T4SS in the genome of Shewanella sp. OPT22 (Figures 7A,B and Supplementary Table S7). T4SS is homologous to bacterial conjugation system and perform pivotal biological functions, namely DNA exchange mediating horizontal gene transfer (HGT) and translocation of proteins, protein-DNA complexes, effector/virulence molecules into the target cells (Wallden et al., 2010). T4SSs loci of Shewanella sp. OPT22 encoded the homologs of virulence genes: virB2 (K03197), virB3 (K03198), virB4 (K03199), virB5 (K03200), virB6 (K03201), virB8 (K03203), virB9 (K03204), virB10 (K03195), virB11 (K03196), and virD4 (K03205) (Figure 7B). These VirB proteins synthesized from the virB operon, functions to form the pilus and translocation channels spanning the cell envelope of bacteria (Voth et al., 2012). It may also contribute significantly to the development of pathogenesis/symbiosis by promoting the surface adhesion, colonization, and biofilm formation.
Interestingly, T4SS was encoded in a 45 Kbp GI in the chromosomal genome of Shewanella sp. OPT22 (Figures 7A,B and Supplementary Table S7) suggesting that the identified T4SS belong to GI type or GI-associated T4SS and possible role of T4SS in the dissemination of GIs in sponge-associated Shewanella spp. We detected the GIs in all three sponge-associated Shewanella species (Supplementary Figure S3). These self transmissible GIs are capable of excision, replication, conjugal transfer, replication, and integration into recipient bacterial chromosome. In addition, GI-T4SSs also carry many cargo genes, apparently not related/essential for the conjugative transfer, but are interspersed within the GIs (Johnson and Grossman, 2015). The role of T4SSs in the mobilization of GIs, enabling the dissemination of cargo genes namely, antibiotic resistance or virulence genes and catabolic genes, was reported among pathogenic bacteria Haemophilus influenza (Juhas et al., 2007) and Pseudomonas sp. strain B13 (Gaillard et al., 2006). Most of the GI encoded genes of Shewanella sp. OPT22 were predicted as hypothetical. Strikingly, one gene encoding for proteins containing ankyrin-like domain was detected within the GI (Figure 7A). The detected ANK-containing protein lacked signal sequence for general SS pathway; GI-encoded ANK could be a possible candidate for T4SS effector molecule and may be involved in the modulation of host-cell functions (Nakayama et al., 2008). Our subcellular localization analyses of this particular GI, detected a higher number of genes belonging to “unknown” category. It is noteworthy that more genes in GI with “unknown” subcellular localization indicate the absence of orthologous matches in the database, suggesting the presence of the novel/uncharacterized genes or genes acquired recently through the horizontal gene transfer (Cortez et al., 2009). A similar trend was detected in the genomes of the members of the genus Pseudovibrio isolated from different marine invertebrate hosts (Alex and Antunes, 2018).
The genome of S. spongiae KCTC 22492, another sponge-associated bacterium carried multiple T4SS gene clusters, with ten or more genes per cluster (Figure 7C). Genomes of obligate intracellular Rickettsia spp. were reported to carry multiple T4SS gene clusters flanked by integrase or tRNA genes, likely representing the remnants of ancient GIs (Cho et al., 2007). However, we did not observe integrase or tRNA genes flanking the T4SS gene clusters in the genome of S. spongiae KCTC 22492. Though, GI type T4SS was not detected, the GIs of S. spongiae KCTC 22492 harbored seven genes encoding for ankyrin-like domains (Figure 3B and Supplementary Table S8). Enrichment of genes coding for ANKs involved in host-parasite interaction has been reported in the GIs of obligate intracellular parasites-Babela massiliensis (lives in Acanthamoeba castellaniiparasitic) (Pagnier et al., 2015), Orientia tsutsugamushi (lives in trombiculid mites) (Nakayama et al., 2008), and in a bacterium, Burkholderia cenocepacia, for successful pathogenic lifestyle (Patil et al., 2017). The detection of ANks within the GIs of both sponge-associated Shewanella spp. clearly suggest the procurement of certain adaptive traits such as evasion of the bacteria against the host immunity, and thus enhance the chances of survival within the sponge hosts. However, the GIs of another sponge-associated bacterium, Shewanella sp. Alg231_23 did not encode any predicted ANKs, indicating the possible procurement of some of the ANKs through HGT in the sponge-associated bacteria, Shewanella sp. OPT22 and S. spongiae KCTC 22492.
Conclusion
In this study, the genomic features of sponge-associated Shewanella were investigated by comparing them with the genomes of other members of the genus Shewanella isolated from different habitats. The phylogenetic clustering of a sponge-associated Shewanella sp. OPT22 sequenced in this study and previously reported sponge bacterium S. spongiae KCTC 22492 with the 16S rRNA sequences of bacteria isolated from geographically separated different sponges (e.g., Atlantic vs. Pacific - East Sea – regions) reveals the “sponge-specific clustering” of these two strains of Shewanella. Comparative genomic analyses with other members of the genus Shewanella showed an overabundance of the ankyrin-repeat domains containing proteins (ANKs) in the genome of Shewanella sp. OPT22 and S. spongiae KCTC 22492, a key genomic feature which might interfere with phagocytosis and facilitate the bacterium to evade the digestion in a sponge host. Moreover, the major SSs machineries detected in the Shewanella sp. OPT22 and S. spongiae KCTC 22492 might be involved in delivering the lethal effector proteins across the bacterial membrane to the target cell in order to gain control over the host system. Localization of T4SS on a GI of Shewanella sp. OPT22 and cargo genes, particularly the ANKs in both Shewanella sp. OPT22 and S. spongiae KCTC 22492, suggest the possible role of horizontal gene transfer that might contribute to the evolution of sponge-associated Shewanella species and its adaptation to sponge-specific niches. Conclusively, the genome of true symbiotic Shewanella strains isolated from sponges described here has the genomic signatures for symbiotic lifestyle and may further offer insights into the molecular strategies for niche-specific lifestyle.
Data Deposition
This project has been deposited at GenBank under the accession number PGVH00000000.
Author Contributions
AAn analyzed the data, contributed the reagents, and drafted the manuscript. AAl designed the work, executed the experiments, analyzed the data, and drafted the manuscript.
Funding
AAl was supported by a grant (SFRH/BPD/99251/2013) from the Fundação para a Ciência e a Tecnologia (FCT) and PTDC/BIA-BMA/29985/2017 (POCI-01-0145-FEDER-029985) from the European Regional Development Fund (ERDF) through COMPETE 2020 – Operational Program for Competitiveness and Internationalization (POCI) and National Funds through FCT/MCTES. AAn was funded in part by the Strategic Funding UID/Multi/04423/2013 through National Funds provided by FCT and the ERDF in the framework of the program PT2020, by the European Structural and Investment Funds (ESIF) through the Competitiveness and Internationalization Operational Program–COMPETE 2020 and by National Funds through the FCT under the project PTDC/AAG-GLO/6887/2014 (POCI-01-0124-FEDER-016845).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00005/full#supplementary-material
Footnotes
- ^ftp://ftp.ncbi.nih.gov/pub/mmdb/cdd/little_endian/, accessed on June 2017
- ^https://github.com/kblin/ncbi-genome-download, accessed on June 2017
References
Alex, A., and Antunes, A. (2015). Whole genome sequencing of the symbiont Pseudovibrio sp. from the intertidal marine sponge polymastia penicillus revealed a gene repertoire for host-switching permissive lifestyle. Genome Biol. Evol. 7, 3022–3032. doi: 10.1093/gbe/evv199
Alex, A., and Antunes, A. (2018). Genus-wide comparison of Pseudovibrio bacterial genomes reveal diverse adaptations to different marine invertebrate hosts. PLoS One 13:e0194368. doi: 10.1371/journal.pone.0194368
Alex, A., Silva, V., Vasconcelos, V., and Antunes, A. (2013). Evidence of unique and generalist microbes in distantly related sympatric intertidal marine sponges (porifera: demospongiae). PLoS One 8:e80653. doi: 10.1371/journal.pone.0080653
Alteri, C. J., and Mobley, H. L. T. (2016). The versatile type VI secretion system. Microbiol. Spectr. 4 doi: 10.1128/microbiolspec.VMBF-0026-2015
Andreas, L. (2016). Bac-genomics-Scripts: Bovine E. coli Mastitis Comparative Genomics Edition. Available at: https://github.com/aleimba/bac-genomics-scripts.
Barbieri, E., Paster, B. J., Hughes, D., Zurek, L., Moser, D. P., Teske, A., et al. (2001). Phylogenetic characterization of epibiotic bacteria in the accessory nidamental gland and egg capsules of the squid Loligo pealei (Cephalopoda:Loliginidae). Environ. Microbiol. 3, 151–167.
Bentley, S. D., Chater, K. F., Cerdeño-Tárraga, A.-M., Challis, G. L., Thomson, N. R., James, K. D., et al. (2002). Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147. doi: 10.1038/417141a
Bertelli, C., Laird, M. R., Williams, K. P., Lau, B. Y., Hoad, G., Winsor, G. L., et al. (2017). IslandViewer 4: expanded prediction of genomic islands for larger-scale datasets. Nucleic Acids Res. 45, W30–W35. doi: 10.1093/nar/gkx343
Bondarev, V., Richter, M., Romano, S., Piel, J., Schwedt, A., and Schulz-Vogt, H. N. (2013). The genus Pseudovibrio contains metabolically versatile bacteria adapted for symbiosis. Environ. Microbiol. 15, 2095–2113. doi: 10.1111/1462-2920.12123
Bönemann, G., Pietrosiuk, A., and Mogk, A. (2010). Tubules and donuts: a type VI secretion story. Mol. Microbiol. 76, 815–821. doi: 10.1111/j.1365-2958.2010.07171.x
Brettar, I., Moore, E. R. B., and Höfle, M. G. (2001). Phylogeny and abundance of novel denitrifying bacteria isolated from the water column of the central baltic sea. Microb. Ecol. 42, 295–305. doi: 10.1007/s00248-001-0011-2
Capella-Gutiérrez, S., Silla-Martínez, J. M., and Gabaldón, T. (2009). trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Cho, N.-H., Kim, H.-R., Lee, J.-H., Kim, S.-Y., Kim, J., Cha, S., et al. (2007). The Orientia tsutsugamushi genome reveals massive proliferation of conjugative type IV secretion system and host-cell interaction genes. Proc. Natl. Acad. Sci. U.S.A. 104, 7981–7986. doi: 10.1073/pnas.0611553104
Contreras-Moreira, B., and Vinuesa, P. (2013). GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysis. Appl. Environ. Microbiol. 79, 7696–7701. doi: 10.1128/AEM.02411-13
Cortez, D., Forterre, P., and Gribaldo, S. (2009). A hidden reservoir of integrative elements is the major source of recently acquired foreign genes and ORFans in archaeal and bacterial genomes. Genome Biol. 10:R65. doi: 10.1186/gb-2009-10-6-r65
Dang, H., and Lovell, C. R. (2016). Microbial surface colonization and biofilm development in marine environments. Microbiol. Mol. Biol. Rev. 80, 91–138. doi: 10.1128/MMBR.0003715
Daniell, S. J., Takahashi, N., Wilson, R., Friedberg, D., Rosenshine, I., Booy, F. P., et al. (2001). The filamentous type III secretion translocon of enteropathogenic Escherichia coli. Cell. Microbiol. 3, 865–871. doi: 10.1046/j.1462-5822.2001.00168.x
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Enomoto, M., Nakagawa, S., and Sawabe, T. (2012). Microbial communities associated with holothurians: presence of unique bacteria in the coelomic fluid. Microbes Environ. 27, 300–305. doi: 10.1264/jsme2.ME12020
Fan, L., Liu, M., Simister, R., Webster, N. S., and Thomas, T. (2013). Marine microbial symbiosis heats up: the phylogenetic and functional response of a sponge holobiont to thermal stress. ISME J. 7, 991–1002. doi: 10.1038/ismej.2012.165
Fan, L., Reynolds, D., Liu, M., Stark, M., Kjelleberg, S., Webster, N. S., et al. (2012). Functional equivalence and evolutionary convergence in complex communities of microbial sponge symbionts. Proc. Natl. Acad. Sci. U.S.A. 109, E1878–E1887. doi: 10.1073/pnas.1203287109
Gaillard, M., Vallaeys, T., Vorhölter, F. J., Minoia, M., Werlen, C., Sentchilo, V., et al. (2006). The clc element of Pseudomonas sp. strain B13, a genomic island with various catabolic properties. J. Bacteriol. 188, 1999–2013. doi: 10.1128/JB.188.5.1999-2013.2006
Gallique, M., Bouteiller, M., and Merieau, A. (2017). The type vi secretion system: a dynamic system for bacterial communication? Front. Microbiol. 8:1454. doi: 10.3389/fmicb.2017.01454
Gomez-Valero, L., Rusniok, C., Cazalet, C., and Buchrieser, C. (2011). Comparative and functional genomics of legionella identified eukaryotic like proteins as key players in host–pathogen interactions. Front. Microbiol. 2:208. doi: 10.3389/fmicb.2011.00208
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA–DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.644830
Gouy, M., Guindon, S., and Gascuel, O. (2010). Seaview version 4: a multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 27, 221–224. doi: 10.1093/molbev/msp259
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems – an overview. Microbiol. Spectr. 4, doi: 10.1128/microbiolspec.VMBF-0012-2015
Guindon, S., Dufayard, J.-F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Habyarimana, F., Al-Khodor, S., Kalia, A., Graham, J. E., Price, C. T., Garcia, M. T., et al. (2008). Role for the ankyrin eukaryotic-like genes of Legionella pneumophila in parasitism of protozoan hosts and human macrophages. Environ. Microbiol. 10, 1460–1474. doi: 10.1111/j.1462-2920.2007.01560.x
Hau, H. H., and Gralnick, J. A. (2007). Ecology and biotechnology of the genus Shewanella. Annu. Rev. Microbiol. 61, 237–258. doi: 10.1146/annurev.micro.61.080706.093257
Henry, T., Couillault, C., Rockenfeller, P., Boucrot, E., Dumont, A., Schroeder, N., et al. (2006). The Salmonella effector protein PipB2 is a linker for kinesin-1. Proc. Natl. Acad. Sci. U.S.A. 103, 13497–13502. doi: 10.1073/pnas.0605443103
Hong, H.-H., Choi, H., Cheon, S., Lee, H.-G., and Park, C. (2017). Genome sequences of two shewanella spp. Isolated from the gut of the sea cucumber apostichopus japonicus (Selenka, 1867). Genome Announc. 5:e00674–17. doi: 10.1128/genomeA.0067417
Ivanova, E. P., Nedashkovskaya, O. I., Sawabe, T., Zhukova, N. V., Frolova, G. M., Nicolau, D. V., et al. (2004). Shewanella affinis sp. nov., isolated from marine invertebrates. Int. J. Syst. Evol. Microbiol. 54, 1089–1093. doi: 10.1099/ijs.0.029920
Janda, J. M., and Abbott, S. L. (2014). The genus Shewanella: from the briny depths below to human pathogen. Crit. Rev. Microbiol. 40, 293–312. doi: 10.3109/1040841X.2012.726209
Jernigan, K. K., and Bordenstein, S. R. (2014). Ankyrin domains across the tree of life. PeerJ 2:e264. doi: 10.7717/peerj.264
Jernigan, K. K., and Bordenstein, S. R. (2015). Tandem-repeat protein domains across the tree of life. PeerJ 3:e732. doi: 10.7717/peerj.732
Johnson, C. M., and Grossman, A. D. (2015). Integrative and conjugative elements (ICEs): what they do and how they work. Annu. Rev. Genet. 49, 577–601. doi: 10.1146/annurev-genet-112414-055018
Jones, P., Binns, D., Chang, H.-Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Jordan, I. K., Makarova, K. S., Spouge, J. L., Wolf, Y. I., and Koonin, E. V. (2001). Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 11, 555–565. doi: 10.1101/gr.166001
Juhas, M., Crook, D. W., Dimopoulou, I. D., Lunter, G., Harding, R. M., Ferguson, D. J. P., et al. (2007). Novel type IV secretion system involved in propagation of genomic islands. J. Bacteriol. 189, 761–771. doi: 10.1128/JB.01327-06
Kanehisa, M., Furumichi, M., Tanabe, M., Sato, Y., and Morishima, K. (2017). KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 45, D353–D361. doi: 10.1093/nar/gkw1092
Kanehisa, M., Sato, Y., and Morishima, K. (2016). BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J. Mol. Biol. 428, 726–731. doi: 10.1016/j.jmb.2015.11.006
Kang, Y., Jelenska, J., Cecchini, N. M., Li, Y., Lee, M. W., Kovar, D. R., et al. (2014). HopW1 from Pseudomonas syringae disrupts the actin cytoskeleton to promote virulence in Arabidopsis. PLoS Pathog. 10:e1004232. doi: 10.1371/journal.ppat.1004232
Klein, J. R., and Jones, B. D. (2001). Salmonella pathogenicity island 2-encoded proteins SseC and SseD are essential for virulence and are substrates of the type III secretion system. Infect. Immun. 69, 737–743. doi: 10.1128/IAI.69.2.737-743.2001
Kniskern, J. M., Barrett, L. G., and Bergelson, J. (2011). Maladaptation in wild populations of the generalist plant pathogen Pseudomonas syringae. Evol. Int. J. Org. Evol. 65, 818–830. doi: 10.1111/j.1558-5646.2010.01157.x
Knutton, S., Rosenshine, I., Pallen, M. J., Nisan, I., Neves, B. C., Bain, C., et al. (1998). A novel EspA-associated surface organelle of enteropathogenic Escherichia coli involved in protein translocation into epithelial cells. EMBO J. 17, 2166–2176. doi: 10.1093/emboj/17.8.2166
Konstantinidis, K. T., and Tiedje, J. M. (2004). Trends between gene content and genome size in prokaryotic species with larger genomes. Proc. Natl. Acad. Sci. U.S.A. 101, 3160–3165. doi: 10.1073/pnas.0308653100
Kück, P., and Meusemann, K. (2010). FASconCAT: convenient handling of data matrices. Mol. Phylogenet. Evol. 56, 1115–1118. doi: 10.1016/j.ympev.2010.04.024
Lee, O. O., Lau, S. C. K., Tsoi, M. M. Y., Li, X., Plakhotnikova, I., Dobretsov, S., et al. (2006). Shewanella irciniae sp. nov., a novel member of the family Shewanellaceae, isolated from the marine sponge ircinia dendroides in the bay of villefranche, mediterranean sea. Int. J. Syst. Evol. Microbiol. 56, 2871–2877. doi: 10.1099/ijs.0.645620
Liu, M., Fan, L., Zhong, L., Kjelleberg, S., and Thomas, T. (2012). Metaproteogenomic analysis of a community of sponge symbionts. ISME J. 6, 1515–1525. doi: 10.1038/ismej.2012.1
Liu, M. Y., Kjelleberg, S., and Thomas, T. (2011). Functional genomic analysis of an uncultured?-proteobacterium in the sponge Cymbastela concentrica. ISME J. 5, 427–435. doi: 10.1038/ismej.2010.139
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J. 17, 10–12. doi: 10.14806/ej.17.1.200
Martínez-García, P. M., Ramos, C., and Rodríguez-Palenzuela, P. (2015). T346Hunter: a novel web-based tool for the prediction of type III, type IV and type VI secretion systems in bacterial genomes. PLoS One 10:e0119317. doi: 10.1371/journal.pone.0119317
Nakayama, K., Yamashita, A., Kurokawa, K., Morimoto, T., Ogawa, M., Fukuhara, M., et al. (2008). The whole-genome sequencing of the obligate intracellular bacterium Orientia tsutsugamushi revealed massive gene amplification during reductive genome evolution. DNA Res. 15, 185–199. doi: 10.1093/dnares/dsn011
Nguyen, L.-T., Schmidt, H. A., von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nguyen, M. T., Liu, M., and Thomas, T. (2014). Ankyrin-repeat proteins from sponge symbionts modulate amoebal phagocytosis. Mol. Ecol. 23, 1635–1645. doi: 10.1111/mec.12384
Nylander, J. A. (2004). MrAIC.pl. Program Distributed by the Author. Uppsala: Evolutionary Biology Centre.
Pagnier, I., Yutin, N., Croce, O., Makarova, K. S., Wolf, Y. I., Benamar, S., et al. (2015). Babela massiliensis, a representative of a widespread bacterial phylum with unusual adaptations to parasitism in amoebae. Biol. Direct 10:13. doi: 10.1186/s13062-015-0043-z
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Patil, P. P., Mali, S., Midha, S., Gautam, V., Dash, L., Kumar, S., et al. (2017). Genomics reveals a unique clone of burkholderia cenocepacia harboring an actively excising novel genomic island. Front. Microbiol. 8:590. doi: 10.3389/fmicb.2017.00590
Pritchard, L., Glover, R. H., Humphris, S., Elphinstone, J. G., and Toth, I. K. (2016). Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal. Methods 8, 12–24. doi: 10.1039/C5AY02550H
Puhar, A., and Sansonetti, P. J. (2014). Type III secretion system. Curr. Biol. 24, R784–R791. doi: 10.1016/j.cub.2014.07.016
Rischer, M., Klassen, J. L., Wolf, T., Guo, H., Shelest, E., Clardy, J., et al. (2016). Draft genome sequence of shewanella sp. strain P1-14-1, a bacterial inducer of settlement and morphogenesis in larvae of the marine hydroid hydractinia echinata. Genome Announc. 4:e00003–e16. doi: 10.1128/genomeA.0000316
Romano, S., Fernàndez-Guerra, A., Reen, F. J., Glöckner, F. O., Crowley, S. P., O’Sullivan, O., et al. (2016). Comparative genomic analysis reveals a diverse repertoire of genes involved in prokaryote-eukaryote interactions within the pseudovibrio genus. Front. Microbiol. 7:387. doi: 10.3389/fmicb.2016.00387
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sekiya, K., Ohishi, M., Ogino, T., Tamano, K., Sasakawa, C., and Abe, A. (2001). Supermolecular structure of the enteropathogenic Escherichia coli type III secretion system and its direct interaction with the EspA-sheath-like structure. Proc. Natl. Acad. Sci. U.S.A. 98, 11638–11643. doi: 10.1073/pnas.191378598
Shnit-Orland, M., Sivan, A., and Kushmaro, A. (2010). Shewanella corallii sp. nov., a marine bacterium isolated from a red sea coral. Int. J. Syst. Evol. Microbiol. 60, 2293–2297. doi: 10.1099/ijs.0.0157680
Shyntum, D. Y., Venter, S. N., Moleleki, L. N., Toth, I., and Coutinho, T. A. (2014). Comparative genomics of type VI secretion systems in strains of pantoea ananatis from different environments. BMC Genomics 15:163. doi: 10.1186/1471-2164-15-163
Siegl, A., Kamke, J., Hochmuth, T., Piel, J., Richter, M., Liang, C., et al. (2011). Single-cell genomics reveals the lifestyle of poribacteria, a candidate phylum symbiotically associated with marine sponges. ISME J. 5, 61–70. doi: 10.1038/ismej.2010.95
Sievers, F., and Higgins, D. G. (2014). Clustal Omega, accurate alignment of very large numbers of sequences. Methods Mol. Biol. 1079, 105–116. doi: 10.1007/978-1-62703-646-7_6
Simister, R. L., Deines, P., Botté, E. S., Webster, N. S., and Taylor, M. W. (2012). Sponge-specific clusters revisited: a comprehensive phylogeny of sponge-associated microorganisms. Environ. Microbiol. 14, 517–524. doi: 10.1111/j.1462-2920.2011.02664.x
Stothard, P., and Wishart, D. S. (2005). Circular genome visualization and exploration using CGView. Bioinformatics 21, 537–539. doi: 10.1093/bioinformatics/bti054
Stover, C. K., Pham, X. Q., Erwin, A. L., Mizoguchi, S. D., Warrener, P., Hickey, M. J., et al. (2000). Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 406, 959–964. doi: 10.1038/35023079
Sullivan, M. J., Petty, N. K., and Beatson, S. A. (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. doi: 10.1093/bioinformatics/btr039
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Thomas, T., Moitinho-Silva, L., Lurgi, M., Björk, J. R., Easson, C., Astudillo-García, C., et al. (2016). Diversity, structure and convergent evolution of the global sponge microbiome. Nat. Commun. 7:11870. doi: 10.1038/ncomms11870
Thomas, T., Rusch, D., DeMaere, M. Z., Yung, P. Y., Lewis, M., Halpern, A., et al. (2010). Functional genomic signatures of sponge bacteria reveal unique and shared features of symbiosis. ISME J. 4, 1557–1567. doi: 10.1038/ismej.2010.74
Versluis, D., Nijsse, B., Naim, M. A., Koehorst, J. J., Wiese, J., Imhoff, J. F., et al. (2018). Comparative genomics highlights symbiotic capacities and high metabolic flexibility of the marine genus pseudovibrio. Genome Biol. Evol. 10, 125–142. doi: 10.1093/gbe/evx271
Voth, D. E., Broederdorf, L. J., and Graham, J. G. (2012). Bacterial type IV secretion systems: versatile virulence machines. Fut. Microbiol. 7, 241–257. doi: 10.2217/fmb.11.150
Wallden, K., Rivera-Calzada, A., and Waksman, G. (2010). Type IV secretion systems: versatility and diversity in function. Cell. Microbiol. 12, 1203–1212. doi: 10.1111/j.1462-5822.2010.01499.x
Yang, S.-H., Kwon, K. K., Lee, H.-S., and Kim, S.-J. (2006). Shewanella spongiae sp. nov., isolated from a marine sponge. Int. J. Syst. Evol. Microbiol. 56, 2879–2882. doi: 10.1099/ijs.0.645400
Yu, N. Y., Wagner, J. R., Laird, M. R., Melli, G., Rey, S., Lo, R., et al. (2010). PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics 26, 1608–1615. doi: 10.1093/bioinformatics/btq249
Keywords: sponge-microbe symbiosis, Shewanella, adaptation, eukaryotic-like proteins, ankyrin repeat
Citation: Alex A and Antunes A (2019) Whole-Genome Comparisons Among the Genus Shewanella Reveal the Enrichment of Genes Encoding Ankyrin-Repeats Containing Proteins in Sponge-Associated Bacteria. Front. Microbiol. 10:5. doi: 10.3389/fmicb.2019.00005
Received: 20 March 2018; Accepted: 07 January 2019;
Published: 06 February 2019.
Edited by:
Hongyue Dang, Xiamen University, ChinaReviewed by:
Xavier Bellanger, Université de Lorraine, FranceJohannes Gescher, Karlsruhe Institute of Technology (KIT), Germany
Copyright © 2019 Alex and Antunes. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Anoop Alex, YW5vb3B0aHljYXVkQGdtYWlsLmNvbQ== Agostinho Antunes, YWFudHVuZXNAY2lpbWFyLnVwLnB0