 Yoshiki Fujii
Yoshiki Fujii Yen Hai Doan
Yen Hai Doan Yoshiyuki Suzuki
Yoshiyuki Suzuki Toyoko Nakagomi3
Toyoko Nakagomi3 Osamu Nakagomi
Osamu Nakagomi Kazuhiko Katayama
Kazuhiko Katayama- 1Department of Virology II, National Institute of Infectious Diseases, Tokyo, Japan
- 2Graduate School of Natural Sciences, Nagoya City University, Nagoya, Japan
- 3Department of Hygiene and Molecular Epidemiology, Nagasaki University, Nagasaki, Japan
- 4Laboratory of Viral Infection I, Kitasato University, Tokyo, Japan
A comprehensive molecular epidemiological study using next-generation sequencing technology was conducted on 333 rotavirus A (RVA)-positive specimens collected from six sentinel hospitals across Japan over three consecutive seasons (2012–2014). The majority of the RVA isolates were grouped into five genotype constellations: Wa-like G1P[8], DS-1-like G1P[8], G2P[4], G3P[8] and G9P[8]. Phylogenetic analysis showed that the distribution of strains varied by geographical locations and epidemic seasons. The VP7 genes of different G types were estimated to evolve at 7.26 × 10-4–1.04 × 10-3 nucleotide substitutions per site per year. The Bayesian time-scaled tree of VP7 showed that the time to the most recent common ancestor of epidemic strains within a region was 1–3 years, whereas that of the epidemic strains across the country was 2–6 years. This study provided, for the first time, the timeframe during which an epidemic strain spread locally and within the country and baseline information needed to predict how rapidly RVAs spread.
Introduction
Rotavirus A (RVA) is a major cause of gastroenteritis in infants and young children worldwide. In 2013, RVA caused 215,000 deaths in children under 5 years of age globally (Tate et al., 2016). RVA imposes a huge burden even on developed countries, including Japan (Sanchez-Fauquier et al., 2006; Hull et al., 2011; Banyai et al., 2012; McDermid et al., 2012; Lanata et al., 2013; Nakagomi et al., 2013; Collins et al., 2015).
The RVA genome contains 11 gene segments of double-stranded RNA that encode six structural (VPs) and six non-structural proteins (NSPs) (Estes and Greenberg, 2013). To classify the RVAs, a specific genotype is assigned to each of the 11 genome segments, according to predefined nucleotide sequence identity cutoff values (Matthijnssens et al., 2008a,b, 2011). The classification system denotes the VP7-VP4-VP6-VP1-VP2-VP3-NSP1-NSP2-NSP3-NSP4-NSP5/6 genes of an RVA strain as a descriptor Gx-P[x]-Ix-Rx-Cx-Mx-Ax-Nx-Tx-Ex-Hx (x indicates genotype number), respectively. Human RVAs have been mostly classified into three genogroups, based on the genotype constellations (Nakagomi et al., 1989). The Wa, DS-1 and AU-1 genogroups are described as G1-P[8]-I1-R1-C1-M1-A1-N1-T1-E1-H1, G2-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2, and G3-P[9]-I3-R3-C3-M3-A3-N3-T3-E3-H3, respectively (Matthijnssens et al., 2008a,b, 2011). The G1, G3, G4, G9, and G12 viruses are usually grouped into Wa-like constellation, and G2 and G8 viruses are usually grouped into DS-1-like constellation (Heiman et al., 2008).
Recently, two live attenuated RVA vaccines were introduced in Japan (Rotarix®, GlaxoSmithKline Biologicals, Belgium in November 2011 and RotaTeq®, Merck & Co., Inc., United States in July 2012). Although RVA vaccines were not part of a routine immunization schedule in Japan, coverage was estimated to be at least 39% in 2012–2013 (Hashizume et al., 2015). The current RVA vaccines are effective (Lambert et al., 2009; Leshem et al., 2014b; Karafillakis et al., 2015), but the selective pressure of a vaccine may induce an epidemic strain shift. Therefore, we began a comprehensive molecular epidemiological study to monitor the RVA epidemics in Japan since 2012. In the first year of surveillance (2012), we reported the predominance of the unusual G1P[8] strain, which possesses a DS-1-like genotype constellation (DS-1-like G1P[8]: G1-P[8]-I2-R2-C2-M2-A2-N2-T2-E2-H2), in Japan (Fujii et al., 2014). Surprisingly, this strain with an unusual genotype constellation spread nationwide to become predominant (46.7%), and thus, this DS-1-like G1P[8] strain should be monitored continuously. A previous report (Fujii et al., 2014) focused on an emerging DS-1-like G1P[8] strain. In the present study, we performed comprehensive molecular epidemiological research, based on next-generation sequencing (NGS) of RVA strains in 2013–2014 and added published results from 2012, to generate a phylogenetic tree. Moreover, a Bayesian time scale analyses was conducted to better understand the epidemics in Japan and the evolutionary history of RVA.
Materials and Methods
Sample Collection
Surveillance was conducted at six sentinel hospitals in Hokkaido prefecture (NTT East Japan Sapporo Hospital and Otaru Kyokai Hospital), Akita Prefecture (Yuri Kumiai General Hospital), Aichi Prefecture (Konan Kosei Hospital), Kyoto Prefecture (Nantan General Hospital) and Yamaguchi Prefecture (Yamaguchi University Hospital) during three seasons (2012–2014) (Supplementary Figure S1). Three hospitals in Akita, Aichi and Kyoto participated throughout the entire period. The hospital in Yamaguchi Prefecture participated in 2013 season and into 2014. Two hospitals in Hokkaido Prefecture participated only in 2013. A total of 789 stool specimens were collected from children aged < 5 years who were hospitalized for acute gastroenteritis. Gastroenteritis was defined as three or more passages of watery diarrhea or looser-than-normal stool within 24 h. Specimens were collected after written informed consent was obtained from the patients or their guardians for the donation of samples. The typical RVA season in Japan is January to July. No RVA-positive stool was found from August to December in this study. In 2012, 237 specimens were collected from February to July as previously described (Fujii et al., 2014). In the 2013 season, 427 specimens were collected from January to July. In the last season (2014), surveillance began in January. Although the epidemic peak in Japan is March and April, sample collection was stopped by March 2014 because of the end of the fiscal year. Thus, only 125 specimens were collected from January to March in the 2014 season. Approximately 10% stool suspensions in PBS (10 mM, pH 7.2) were examined for RVA antigen with an ELISA kit, Rotaclone® (Meridian Diagnostics, Cincinnati, OH, United States). ELISA-negative samples were excluded from analysis.
cDNA Library and Nucleotide Sequencing
Viral RNA was extracted from stool suspensions with the Direct-zol RNA MiniPrep kit (Zymo Research, Irvine, CA, United States) according to manufacturer’s instructions as previously described (Fujii et al., 2014). NGS was performed as described (Dennis et al., 2014; Doan et al., 2016). Briefly, a 200-bp fragment library was constructed for each sample with the NEBNext Ultra RNA Library Prep Kit for Illumina v1.2 (New England Biolabs, Ipswich, MA, United States), according to manufacturer’s instructions. A 151-cycle paired-end-read sequencing run was conducted on a MiSeq desktop sequencer (Illumina, San Diego, CA, United States) using the MiSeq Reagent Kit v2 (300 cycles). Sequence data were analyzed using CLC Genomics Workbench Software v7.0.3 (CLC Bio, Aarhus, Denmark) and deposited in the DDBJ/GenBank databases (LC172271-LC174338). The NGS analysis yielded several sequences that lacked one or both ends of segments. This result usually depends on amount of virus in the stool specimen.
Genotyping and Phylogenetic Analysis
Genotypes of the 11 genome segments were determined by the RotaC 2.0 automated genotyping tool for RVA (Maes et al., 2009). Near-full-length genome sequences were aligned with reference sequences using CLUSTAL W that was included in the MEGA software package, version 7.0.18 and the MAFFT multiple sequence alignment software program, version 7.0 (Katoh et al., 2009). Sequence alignments and phylogenetic analysis were performed using MEGA 7 software (Tamura et al., 2013). Genetic distances were calculated with the Tamura three-parameter at a nucleotide level, and phylogenetic trees were constructed by the Maximum Likelihood Method with 1000 bootstrap replicates. Reference sequences were retrieved from GenBank and the lineage designations defined based on previous studies for G1 (Arista et al., 2006; Le et al., 2010), G2 (Doan et al., 2015), G3 (Wang et al., 2014), and G9 trees (Phan et al., 2007).
Bayesian Evolutionary Analysis by BEAST
Evolutionary rates and time of most recent common ancestors (tMRCA) were determined for the VP7 gene of RVAs by the Bayesian Markov chain Monte Carlo (MCMC) method implemented in BEAST v1.8.1 (Drummond et al., 2012). The model used for BEAST analysis was T93+G. A strict clock and coalescent exponential growth model (Drummond et al., 2002) were used. MCMC runs were carried out for 200 million generations to achieve convergence with sampling every 1000 steps. Convergence was assessed from effective sample size after a 10% burn-in using Tracer software v1.6.1 Only parameters with an effective sample size > 200 were accepted. Maximum clade credibility trees were annotated with Treeannotator and viewed with FigTree v1.4.22.
Ethics Statement
This study protocols were approved by the medical research ethics committee of the National Institute of Infectious Diseases for the use of human subjects (No. 335).
Results
Genotype Distribution Based on Full-Genome Sequencing
A total of 789 stool specimens were collected, and 333 RVA-positive samples were identified by ELISA screening: the detection rates were 45.6% (108/237) in 2012, 49.4% (211/427) in 2013, and 11.2% (14/125) in 2014. The genotype data of 108 samples in 2012 were previously reported (Fujii et al., 2014). In the present study, 225 RVA strains collected in 2013 and 2014 were analyzed by NGS, and the genotype constellations of all samples were determined. Most of the samples were grouped into five genotype constellations: G1-P[8]-I1-R1-C1-M1-A1-N1-T1-E1-H1 (Wa-like G1P[8]), G1-P[8]-I2-R2-C2-M2-A2-N2-T2-E2-H2 (DS-1-like G1P[8]), G2-P[4]-I2-R2-C2-M2-A2-N2-T2-E2-H2 (DS-1-like G2P[4]), G3-P[8]-I1-R1-C1-M1-A1-N1-T1-E1-H1 (Wa-like G3P[8]), and G9-P[8]-I1-R1-C1-M1-A1-N1-T1-E1-H1 (Wa-like G9P[8]). No atypical genotype constellation was detected except DS-1-like G1P[8] strains. Three mixed infection samples were detected: two seemed to be mixed infections of Wa-like G1P[8] and DS-1-like G1P[8], and one seemed to be a mixed infection of the Wa-like G1P[8] and G9P[8] strains.
During 2012–2014, DS-1-like G1P[8] (51.7%) was the most prevalent strain, followed by Wa-like G9P[8] (25.8%) (Table 1). Wa-like G1P[8], DS-1 like G2P[4], and Wa-like G3P[8] strains were detected in 12.6, 5.4, and 3.6% of cases, respectively. The reassortant DS-1-like G1P[8] strain is a newly emerged dominant genotype constellation, initially detected in 2012. This strain continued as to be dominant in 2013 and was detected in all six regions. In Aichi Prefecture, DS-1-like G1P[8] viruses were detected in 96.6% (86/89) of specimens. No DS-1-like G1P[8] was detected in 2014. However, only 14 RVA-positive specimens were detected in 2014. In particular, in Kyoto, no RVA-positive sample was detected in 2014 (0/15). On the whole, the genotype distributions varied drastically by region. For example, in 2013, DS-1-like G1P[8] was most prevalent in Sapporo, Akita and Aichi. Wa-like G1P[8] predominated in Kyoto and Yamaguchi, and G9P[8] predominated in Otaru (Table 1).
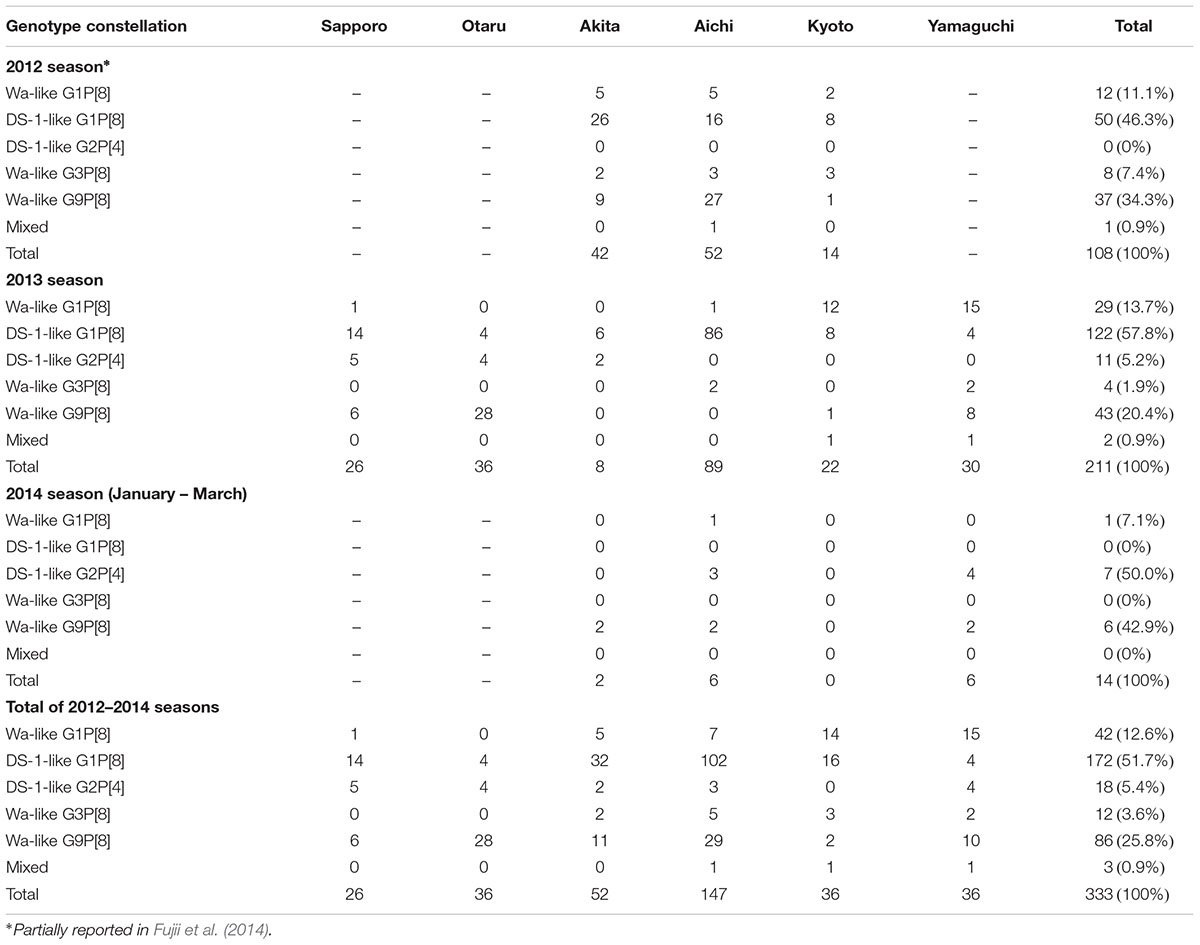
Table 1. Genotype distribution of RVAs in Japan (2012–2014).
Phylogenetic Analysis
Of 225 analyzed samples detected in 2013 and 2014, we completed nearly full-genome sequencing of 188 RNA samples (Supplementary Table S1). The remaining samples were excluded from phylogenetic analyses because the sequence data were incomplete (less than 80% length of at least one segment). With the 103 samples from 2012 (Fujii et al., 2014), 291 samples were used for phylogenetic analysis.
To construct a VP7 tree (Figure 1A), all samples were classified into four genotypes: G1, G2, G3, and G9. Moreover, the G1 strains were divided into lineages 1 and 2, and G9 strains were divided into lineages 3 and 6. All of the DS-1-like G1P[8] strains were concentrated in one cluster of G1 lineage 1 (Figure 1A). When a detailed tree of each genotype was generated (Figures 1B–E), viruses of each genotype were further divided into multiple subclusters. Then, the subclusters were divided by region and season of collection. For example, Wa-like G1P[8] was divided into four subclusters (Figure 1B). One included nine strains from Kyoto in 2013 (NT-2013) and belongs to lineage 2. The other three subclusters belong to lineage 1; one consists of 149 DS-1-like G1P[8] strains, another is mainly composed of strains from Akita in 2012 (YR-2012), and the third is mainly composed of strains from Yamaguchi in 2013 (YM-2013). For the G2 tree (Figure 1C), SP011 and four strains from Otaru in 2013 were separated from other G2 strains, and four strains from Yamaguchi in 2014 were also different from other strains. For G3 tree (Figure 1D), NT009 and three strains from Aichi in 2012 were separated from other G3 strains. For lineage 3 of the G9 tree (Figure 1E), strains from Aichi in 2012 (KN-2012), Akita in 2012 (YR-2012) and Otaru in 2013 (OT-2013), including NT062, had similar sequences in each subcluster (nucleotide identities were > 99.8%).
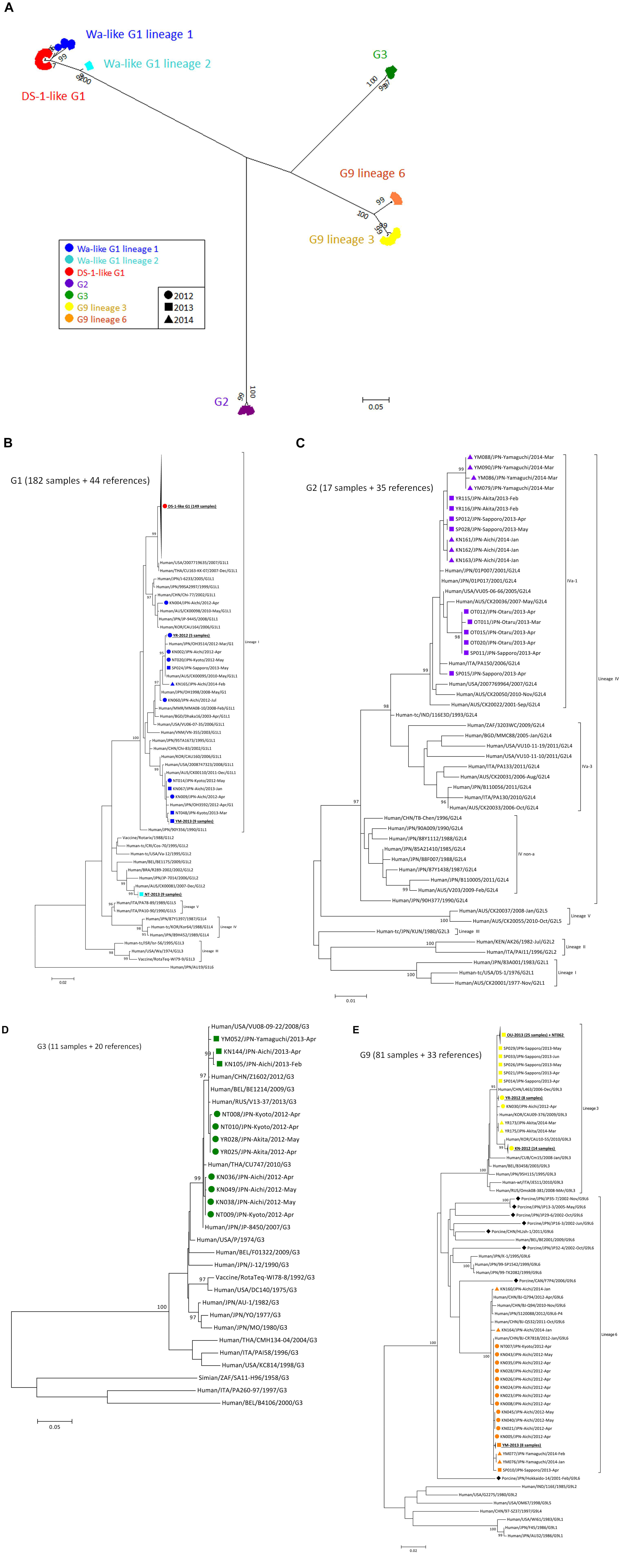
Figure 1. Phylogenetic tree of VP7 gene of RVA detected in Japan (2012–2014). A phylogenetic tree for VP7 was constructed from the nucleotide sequences of 291 RVA strains obtained in Japan in 2012–2014 and shown in radiation form (A). All of Japanese strains were classified into seven major groups, and their markers were colored according to the groups: Wa-like G1P[8] lineage 1 (blue), Wa-like G1P[8] lineage 2 (light blue), DS-1-like G1P[8] (red), G2P[4] (purple), G3P[8] (green), G9 lineage 3 (yellow) and G9 lineage 6 (orange). The shapes of markers relate to the season of detection: 2012 (circle), 2013 (square) and 2014 (triangle). The tree of each VP7 genotype is shown with representative strains as references: G1 (B), G2 (C), G3 (D), and G9 (E). Similar strains are compressed to a triangle. Porcine-derived strains are marked by a diamond shape. The phylogenetic trees were constructed by using the Maximum Likelihood Method with 1000 bootstrap replicates. Percent bootstrap support is indicated by the value at each node when the value was 95% or larger.
The phylogenetic trees of other gene segments are shown in Figure 2. Color dispersions show that intra-genogroup reassortments seemed to occur frequently, whereas inter-genogroup reassortment was not found except for the presence of DS-1-like G1P[8]. Reassortments among the Wa-genogroup strains (Wa-like G1P[8], Wa-like G3P[8], and Wa-like G9P[8]) were frequently found. Meanwhile, no reassortment was found in all segments of the G1 lineage 2 strains (light blue), which indicates a local epidemic in Kyoto in 2013. Some further reassortments were found between DS-1-like G1P[8] strains (red) and other strains. A total of six further reassortants including genes derived from DS-1-like G1P[8] strains were identified (Figure 3), and of these, three were already reported (Fujii et al., 2014). Of the rest, one was DS-1-like G1P[8] (KN152), and the two were G2P[4] strains (SP028 and YR115). KN152 reassorted with VP6 and NSP4 of G2P[4] strain, SP028 reassorted with VP1 and NSP4 of DS-1-like G1P[8] strain, and YR115 reassorted with NSP4 of DS-1-like G1P[8] strain. Reassortments seem to occur with greater frequency in NSP4 gene.
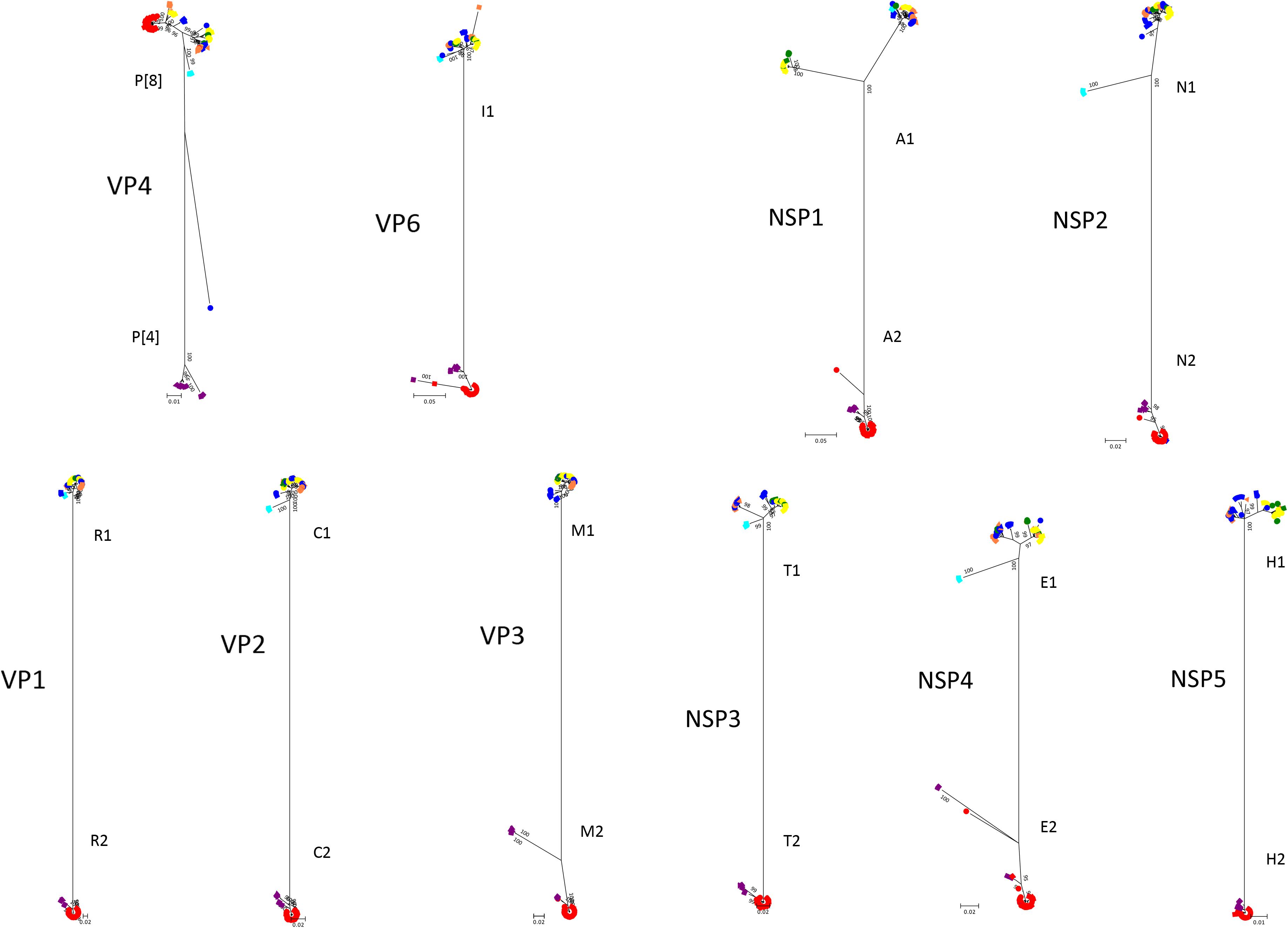
Figure 2. Phylogenetic trees of RVA genes other than VP7. Phylogenetic trees of 10 segments (VP4, VP6, VP1, VP2, VP3, NSP1, NSP2, NSP3, NSP4, and NSP5) were constructed from the nucleotide sequences of 291 RVA strains detected in this study, and shown in radiation form. The colors of markers indicate the groups of VP7 sequences, and the shapes of markers indicate the season in which they were detected (as in Figure 1). The phylogenetic trees were constructed by using the Maximum Likelihood Method with 1000 bootstrap replicates. Percent bootstrap support is indicated by the value at each node when the value was 95% or larger.
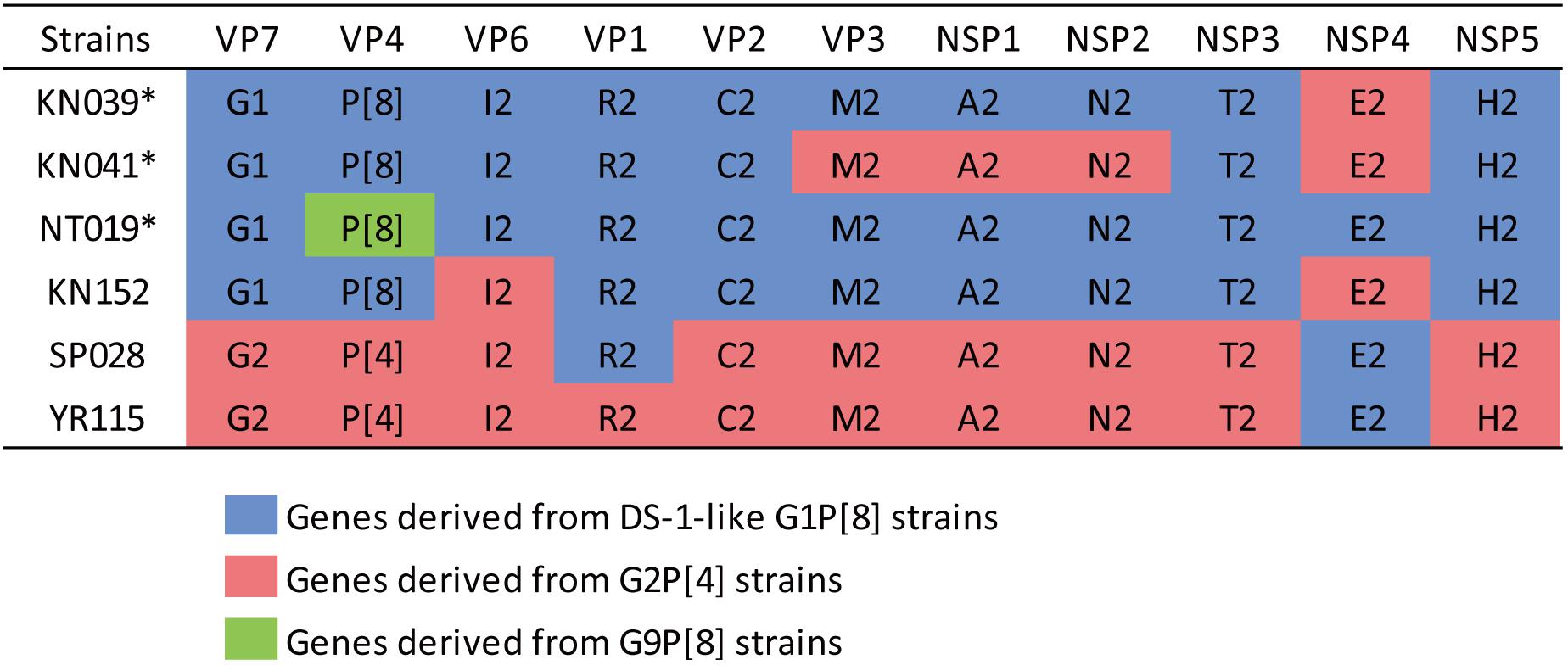
Figure 3. Further reassortant strains between DS-1-like G1P[8] and other strains. Six further reassortant strains were identified in 2012–2014, and their genotype constellations are shown. Asterisks indicate strains listed in our previous report (Fujii et al., 2014). Blue, red, or green color indicate the genes derived from DS-1-like G1P[8], G2P[4], or G9P[8] strains, respectively.
Bayesian Analysis
A Bayesian time-scaled tree of VP7 was generated for each genotype with our 291 samples and the other 121 representative strains retrieved from GenBank (44 G1 references, 35 G2 references, 20 G3 references, and 26 G9 references) (Figure 4). The estimated evolution rates were 7.26 × 10-4 nucleotide substitutions per site per year (the 95% highest posterior density (HPD) was 5.99–8.63 × 10-4) for G1 strains, 7.41 × 10-4 (5.66–9.196 × 10-4) for G2 strains, 7.34 × 10-4 (4.69 × 10-4–1.01 × 10-3) for G3 strains, and 1.03 × 10-3 (7.46 × 10-4–1.38 × 10-3) for G9 strains.
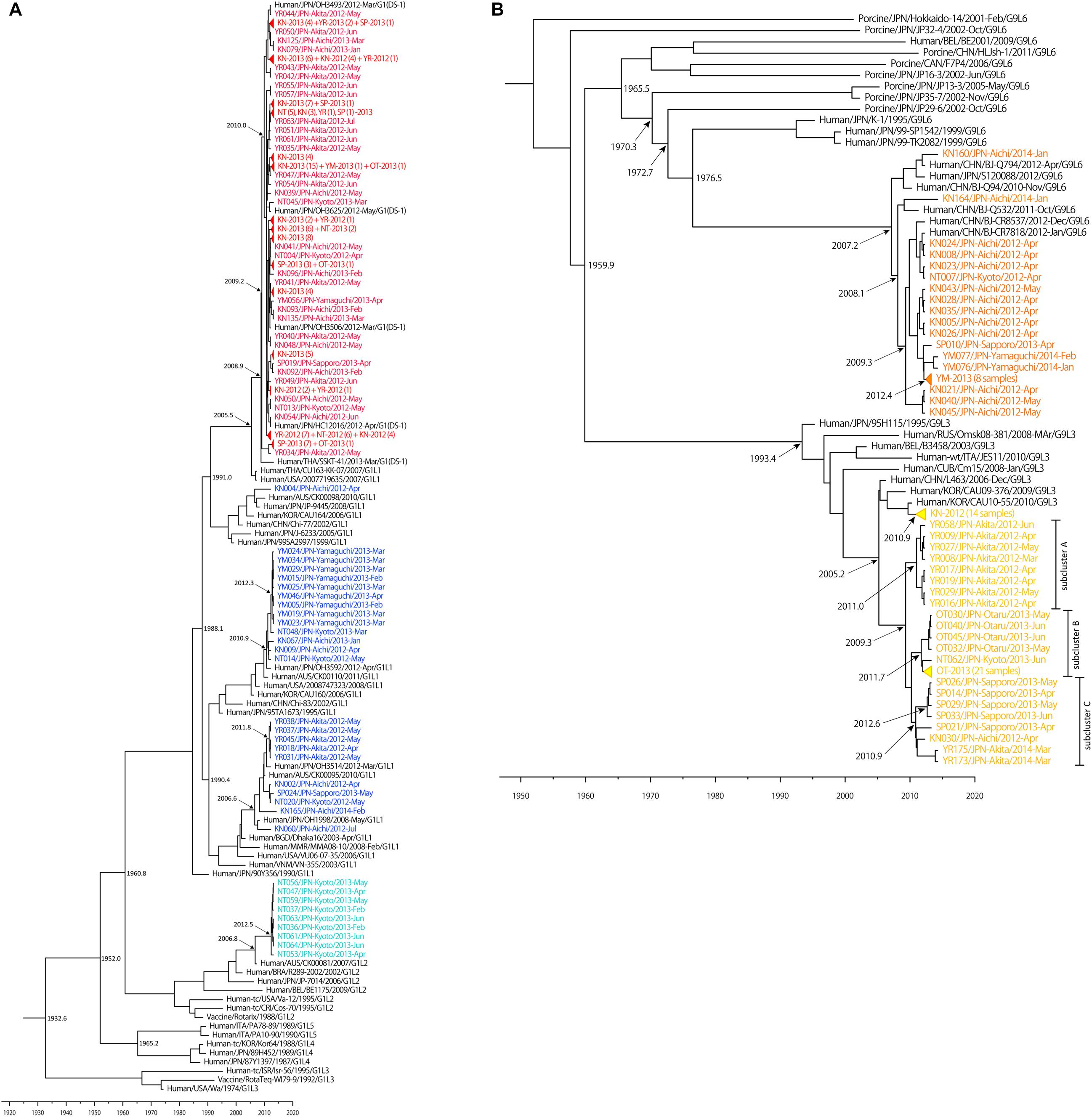
Figure 4. Maximum clade credibility (MCC) tree of VP7 constructed using the Bayesian MCMC method. Bayesian time-scaled trees of VP7 of G1 (A) and G9 (B) strains were generated, based on same data set as Figure 1. The estimated ages are indicated for particular nodes. Wa-like G1P[8] lineage 1 strains, Wa-like G1P[8] lineage 2 strains, DS-1-like G1P[8] strains, G9 lineage 3 strains and G9 lineage 6 strains detected in our study were colored: blue, light blue, red, yellow, and orange, respectively. Some similar strains were compressed to triangles.
The Bayesian time-scaled trees of G1 and G9 genotypes are presented in Figure 4. For the G2 and G3 trees, the sample numbers were too low to investigate. For the G1 tree (Figure 4A), all of the 149 DS-1-like G1P[8] strains were concentrated in one subcluster, and the DS-1-like G1P[8] strains detected in Okayama (Kuzuya et al., 2014), Osaka (Yamamoto et al., 2014), and Thailand (Komoto et al., 2015) were in same subcluster. The common ancestry of the DS-1-like G1P[8] strains was estimated to trace back to 2009.2 (2007.8–2010.5). This is 3 years (2–5 years considering 95% HPD) before this strain became a national epidemic in 2012. Meanwhile, the Wa-like G1 type was divided into three subclusters as described above (Figure 1B). For example, the time of most recent common ancestors (tMRCA) of the strains collected in Yamaguchi in 2013 was estimated 2012.3 (2011.5–2012.9). Thus, it was less than 1–2 years before the ancestor virus became epidemic in Yamaguchi. Two other subclusters (Akita in 2012 and Kyoto in 2013) showed similar patterns.
While the G9 genotype is divided into six lineages (Phan et al., 2007), only lineages 3 and 6 were circulating in Japan. Lineages 3 and 6 were estimated to have separated around 1960 (Figure 4B). Most of lineage 3 strains detected in Aichi in 2012 were clustered away from other strains detected in other places, and the tMRCA was estimated as 2010.9 (2010.0–2011.6). For descriptive purpose, the other lineage 3 strains were further divided into three subclusters (i.e., A, B, and C) (Figure 4B). For subcluster A, the tMRCA of eight strains from Akita in 2012 was 2011.0 (2009.6–2011.9). For subcluster B, the tMRCA of 26 strains from Otaru in 2013 except for NT062 was 2011.7 (2010.7–2012.5). Subcluster C consists of strains from Sapporo (2013), Akita (2014), and Aichi (2012), and the tMRCA was 2010.9 (2009.5–2012.0). In each case, tMRCAs were 1–3 years before the local epidemics. Their common ancestor was traced back to 2009.3 (2006.9–2010.9). This is 2–6 years before the national epidemic as well for DS-1-like G1P[8]. G9 lineage 6 strains have a common ancestry with human and porcine RVA until around 1970. After that, human G9 lineage 6 seemed to evolve independently. The now-circulating G9 lineage 6 strains in Japan were traced back to 2007.2 (2004.7–2009.0). Because these G9 lineage 6 strains were already detected nationwide before we started this study in 2012, it seemed to be less than 5 years (95% HPD: 3–8 years) before the national epidemic as well.
Discussion
In this study, NGS analysis revealed information about RVA epidemics in Japan from 2012 to 2014. Although there are limitations of collection period and non-homogeneous populations, 333 RVA-positive samples were collected from six sentinel hospitals across the country. All RVA strains were grouped into five patterns of genotype constellation except for three mixed infection cases (Figure 1). Using phylogenetic analysis, viruses were found to be further divided into multiple subclusters, and the strain distributions were different, depending on when and where they were collected (Figures 1B–E). This shows the obvious seasonal and geographical differences of RVA strain distribution at the sequence level. The evolutionary history and epidemics of RVA in Japan in 2014 are illustrated in Figure 5. Although more complex genotype distributions are found in developing and developed countries (Reidy et al., 2005; Chitambar et al., 2014; Dennis et al., 2014; Heylen et al., 2014; Nyaga et al., 2015), only five patterns of genotype constellation were detected in Japan. Without unusual strains, Japanese RVA epidemics can be explained with a simple model.
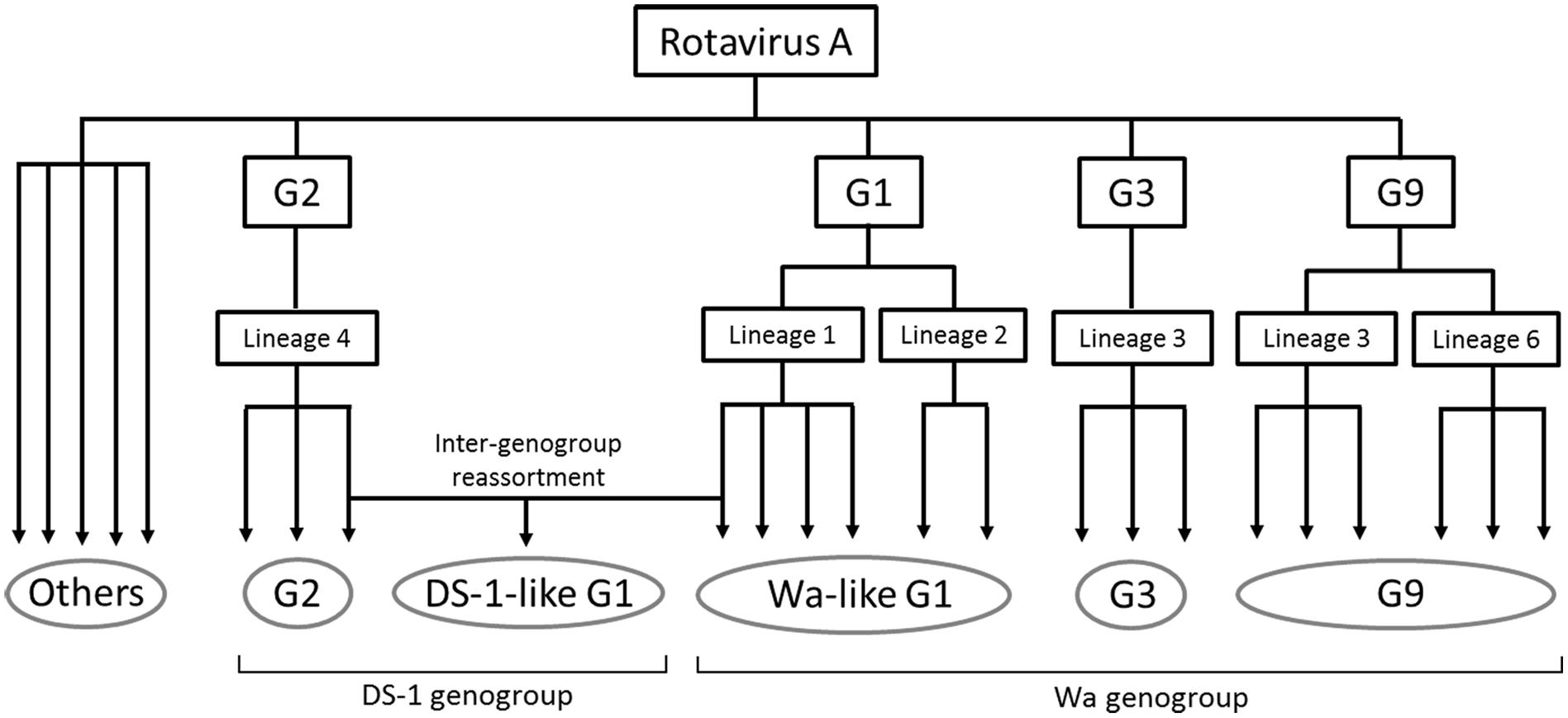
Figure 5. Simplified illustration indicating the RVA evolutionary history and epidemics in Japan in 2014. The five grouped RVAs based on genotype constellation are commonly circulating in Japan and are evolving by further dividing into multiple subclusters and sometimes by intra-genotype reassortment. DS-1-like G1P[8] strain exceptionally seemed to have generated by inter-genogroup reassortment between Wa-like G1P[8] strain and G2P[4] strain. “Others” means minor strains, such as G4, G8, G12, and so on.
In 2012, DS-1-like G1P[8] was initially detected in Japan and seemed to be generated by reassortment of typical Wa-like G1P[8] and DS-1-like G2P[4] as previously described (Fujii et al., 2014). DS-1-like G1P[8] viruses were reported in Thailand (Komoto et al., 2015) and the Philippines (Yamamoto et al., 2017) in 2012, but it is difficult to determine where this strain came from. In Japan, DS-1-like G1P[8] was the predominant strain in 2012–2013, and six further reassortants between DS-1-like G1P[8] and others were detected (Figure 3). Of these, five occurred in the NSP4 segment. Thus, reassortment seems to occur more frequently in the NSP4 segment than other segments, and its evolutionary rate is higher than other segments (Tatsumi et al., 2014; Zeller et al., 2015). Therefore, the exceptional tendency for substitutions of NSP4 might affect the frequency of reassortments. DS-1-like G1P[8] was not detected in 2014, and G2P[4] accounted for half of RVA strains instead (Table 1). Several countries reported increased rates of G2P[4] after vaccine introduction (Leshem et al., 2014a; Wu et al., 2014; Montenegro et al., 2015). However, further investigation is required as the number of specimens in 2014 was small due partly to a shorter collection period and partly to an increasing use of RVA vaccine (Hashizume et al., 2015).
We found substantial differences in the distributions of genotypes, depending on where they were collected (Table 1). For example, in 2013, the predominant strains were DS-1-like G1P[8] in Sapporo and G9 lineage 3 in Otaru, even though these cities are only 30 km apart. Thus, RVAs might spread more slowly than noroviruses, another cause of gastroenteritis (Green, 2013). One reason may be that most RVA-associated gastroenteritis patients are infants. Infants are usually much less active than adults, and so, a viral strain has less chance to spread. Actually, the age distribution of norovirus infection cases is generally wider than that of RVAs, and noroviruses spread rapidly and show little difference in geographical distribution (Lindesmith et al., 2008; Siebenga et al., 2009; Green, 2013). Once an RVA strain is prevalent in a region, the population of infants there will acquire herd immunity in a few years, and the epidemic of that strain will subside. When the population without immunity to that strain (i.e., newborns) increases sufficiently, the strain could become prevalent again.
Next, a Bayesian time-scaled phylogenetic tree was constructed to understand the evolution of RVAs (Figure 4). The evolutionary rate was estimated to be 7.26 × 10-4 to 1.04 × 10-3 nucleotide substitutions per site per year for the G1, G2, G3, and G9 strains, which is similar to previous reports (Nagaoka et al., 2012; Afrad et al., 2014; Doan et al., 2015; Zeller et al., 2015). Analyses of tMRCA of prevalent RVA strains provided us a prediction of the spread speed of RVA strains. It takes 1–3 years from generation of parent viruses to be epidemic in a region and 1–3 years more to spread nationwide. Therefore, it takes 2–6 years from the generation of an ancestor virus for it to be an epidemic strain across Japan. Because DS-1-like G1P[8] strain emerged only in 2012, this method is appropriate for evaluating the evolutionary behavior of RVA. The tMRCA of DS-1-like G1P[8] strains was 2009.2 (2007.8–2010.5). Thus, it fits the pattern described above. Because RVAs were not comprehensively examined in Japan until 2012, the DS-1-like G1P[8] viruses might have been missed by the detection network. Although the DS-1-like G1P[8] virus was not detected again in 2014 in our study, this strain was circulating in Japan at least until 2015, according to another report (Sugimoto et al., 2017).
Here, we described the epidemics of RVA in Japan and elucidated the evolutionary behavior of RVAs. These findings will be important to predict epidemics of RVA. To monitor RVA epidemics reliably, a continuous nationwide surveillance is needed. As RVA gastroenteritis cases are decreasing owing to vaccine effect, the burden of analyses will become easier. Meanwhile, efficient sample collection will become more important in the future.
Author Contributions
YF performed all experiments and was responsible for writing the manuscript. YD and YS gave assistance to perform phylogenetic analysis and Bayesian evolutionary analysis. TN and ON organized the sample collection network with hospitals. KK gave assistance for the research and helped to draft the manuscript.
Funding
This work was supported by a commissioned project for the Research on Emerging and Re-emerging Infectious Diseases from the Japanese Ministry of Health, Labour and Welfare (H23-Shinko-Ippan-005) and for the Research Program on Emerging and Re-emerging Infectious Diseases from AMED under grant numbers 16fk0108304j0403, 18fk0108034j0202, and 18fk0108078j0001.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We sincerely thank the staffs at the six sentinel hospitals for their work to collect stool specimens and to provide the epidemiological information.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00038/full#supplementary-material
FIGURE S1 | A map of Japan showing the sample collection sites and periods. The sites of hospitals joining in this study are indicated as black circles. The names of hospitals, prefectures, and collection periods (in parentheses) are shown.
TABLE S1 | Sample list used for phylogenetic analysis.
Footnotes
References
Afrad, M. H., Matthijnssens, J., Afroz, S. F., Rudra, P., Nahar, L., Rahman, R., et al. (2014). Differences in lineage replacement dynamics of G1 and G2 rotavirus strains versus G9 strain over a period of 22 years in Bangladesh. Infect. Genet. Evol. 28, 214–222. doi: 10.1016/j.meegid.2014.10.002
Arista, S., Giammanco, G. M., De Grazia, S., Ramirez, S., Lo Biundo, C., Colomba, C., et al. (2006). Heterogeneity and temporal dynamics of evolution of G1 human rotaviruses in a settled population. J. Virol. 80, 10724–10733. doi: 10.1128/jvi.00340-06
Banyai, K., Laszlo, B., Duque, J., Steele, A. D., Nelson, E. A., Gentsch, J. R., et al. (2012). Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: insights for understanding the impact of rotavirus vaccination programs. Vaccine 30(Suppl. 1), A122–A130. doi: 10.1016/j.vaccine.2011.09.111
Chitambar, S. D., Ranshing, S. S., Pradhan, G. N., Kalrao, V. R., Dhongde, R. K., and Bavdekar, A. R. (2014). Changing trends in circulating rotavirus strains in Pune, western India in 2009-2012: emergence of a rare G9P[4] rotavirus strain. Vaccine 32(Suppl. 1), A29–A32. doi: 10.1016/j.vaccine.2014.03.027
Collins, P. J., Mulherin, E., O’Shea, H., Cashman, O., Lennon, G., Pidgeon, E., et al. (2015). Changing patterns of rotavirus strains circulating in Ireland: re-emergence of G2P[4] and identification of novel genotypes in Ireland. J. Med. Virol. 87, 764–773. doi: 10.1002/jmv.24095
Dennis, F. E., Fujii, Y., Haga, K., Damanka, S., Lartey, B., Agbemabiese, C. A., et al. (2014). Identification of novel Ghanaian G8P[6] human-bovine reassortant rotavirus strain by next generation sequencing. PLoS One 9:e100699. doi: 10.1371/journal.pone.0100699
Doan, Y. H., Haga, K., Fujimoto, A., Fujii, Y., Takai-Todaka, R., Oka, T., et al. (2016). Genetic analysis of human rotavirus C: the appearance of Indian-Bangladeshi strain in Far East Asian countries. Infect. Genet. Evol. 41, 160–173. doi: 10.1016/j.meegid.2016.03.027
Doan, Y. H., Nakagomi, T., Agbemabiese, C. A., and Nakagomi, O. (2015). Changes in the distribution of lineage constellations of G2P[4] Rotavirus A strains detected in Japan over 32 years (1980-2011). Infect. Genet. Evol. 34, 423–433. doi: 10.1016/j.meegid.2015.05.026
Drummond, A. J., Nicholls, G. K., Rodrigo, A. G., and Solomon, W. (2002). Estimating mutation parameters, population history and genealogy simultaneously from temporally spaced sequence data. Genetics 161, 1307–1320.
Drummond, A. J., Suchard, M. A., Xie, D., and Rambaut, A. (2012). Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973. doi: 10.1093/molbev/mss075
Estes, M. K., and Greenberg, H. B. (2013). “Rotaviruses,” in Fields virology, eds P. M. H. David and M. Knipe (Philadelphia, PA: Lippincott Williams & Wilkins), 1347–1401.
Fujii, Y., Nakagomi, T., Nishimura, N., Noguchi, A., Miura, S., Ito, H., et al. (2014). Spread and predominance in Japan of novel G1P[8] double-reassortant rotavirus strains possessing a DS-1-like genotype constellation typical of G2P[4] strains. Infect. Genet. Evol. 28, 426–433. doi: 10.1016/j.meegid.2014.08.001
Green, K. Y. (2013). “Caliciviridae: the noroviruses,” in Fields Virology, eds P. M. H. David and M. Knipe (Philadelphia, PA: Lippincott Williams & Wilkins), 582–608.
Hashizume, M., Nakagomi, T., and Nakagomi, O. (2015). An early detection of decline in rotavirus cases during the 2013/2014 season in Japan as revealed by time-series analysis of national surveillance data. Trop. Med. Health 43, 177–181. doi: 10.2149/tmh.2015-23
Heiman, E. M., McDonald, S. M., Barro, M., Taraporewala, Z. F., Bar-Magen, T., and Patton, J. T. (2008). Group A human rotavirus genomics: evidence that gene constellations are influenced by viral protein interactions. J. Virol. 82, 11106–11116. doi: 10.1128/JVI.01402-08
Heylen, E., Batoko Likele, B., Zeller, M., Stevens, S., De Coster, S., Conceicao-Neto, N., et al. (2014). Rotavirus surveillance in Kisangani, the Democratic Republic of the Congo, reveals a high number of unusual genotypes and gene segments of animal origin in non-vaccinated symptomatic children. PLoS One 9:e100953. doi: 10.1371/journal.pone.0100953
Hull, J. J., Teel, E. N., Kerin, T. K., Freeman, M. M., Esona, M. D., Gentsch, J. R., et al. (2011). United States rotavirus strain surveillance from 2005 to 2008: genotype prevalence before and after vaccine introduction. Pediatr. Infect. Dis. J. 30, S42–S47. doi: 10.1097/INF.0b013e3181fefd78
Karafillakis, E., Hassounah, S., and Atchison, C. (2015). Effectiveness and impact of rotavirus vaccines in Europe, 2006-2014. Vaccine 33, 2097–2107. doi: 10.1016/j.vaccine.2015.03.016
Katoh, K., Asimenos, G., and Toh, H. (2009). Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 537, 39–64. doi: 10.1007/978-1-59745-251-9_3
Komoto, S., Tacharoenmuang, R., Guntapong, R., Ide, T., Haga, K., Katayama, K., et al. (2015). Emergence and characterization of unusual DS-1-like G1P[8] rotavirus strains in children with diarrhea in Thailand. PLoS One 10:e0141739. doi: 10.1371/journal.pone.0141739
Kuzuya, M., Fujii, R., Hamano, M., Kida, K., Mizoguchi, Y., Kanadani, T., et al. (2014). Prevalence and molecular characterization of G1P[8] human rotaviruses possessing DS-1-like VP6, NSP4, and NSP5/6 in Japan. J. Med. Virol. 86, 1056–1064. doi: 10.1002/jmv.23746
Lambert, S. B., Faux, C. E., Hall, L., Birrell, F. A., Peterson, K. V., Selvey, C. E., et al. (2009). Early evidence for direct and indirect effects of the infant rotavirus vaccine program in Queensland. Med. J. Aust. 191, 157–160.
Lanata, C. F., Fischer-Walker, C. L., Olascoaga, A. C., Torres, C. X., Aryee, M. J., and Black, R. E. (2013). Global causes of diarrheal disease mortality in children < 5 years of age: a systematic review. PLoS One 8:e72788. doi: 10.1371/journal.pone.0072788
Le, V. P., Chung, Y. C., Kim, K., Chung, S. I., Lim, I., and Kim, W. (2010). Genetic variation of prevalent G1P[8] human rotaviruses in South Korea. J. Med. Virol. 82, 886–896. doi: 10.1002/jmv.21653
Leshem, E., Lopman, B., Glass, R., Gentsch, J., Banyai, K., Parashar, U., et al. (2014a). Distribution of rotavirus strains and strain-specific effectiveness of the rotavirus vaccine after its introduction: a systematic review and meta-analysis. Lancet Infect. Dis. 14, 847–856. doi: 10.1016/s1473-3099(14)70832-1
Leshem, E., Moritz, R. E., Curns, A. T., Zhou, F., Tate, J. E., Lopman, B. A., et al. (2014b). Rotavirus vaccines and health care utilization for diarrhea in the United States (2007-2011). Pediatrics 134, 15–23. doi: 10.1542/peds.2013-3849
Lindesmith, L. C., Donaldson, E. F., Lobue, A. D., Cannon, J. L., Zheng, D. P., Vinje, J., et al. (2008). Mechanisms of GII.4 norovirus persistence in human populations. PLoS Med. 5:e31. doi: 10.1371/journal.pmed.0050031
Maes, P., Matthijnssens, J., Rahman, M., and Van Ranst, M. (2009). RotaC: a web-based tool for the complete genome classification of group A rotaviruses. BMC Microbiol. 9:238. doi: 10.1186/1471-2180-9-238
Matthijnssens, J., Ciarlet, M., Heiman, E., Arijs, I., Delbeke, T., McDonald, S. M., et al. (2008a). Full genome-based classification of rotaviruses reveals a common origin between human Wa-Like and porcine rotavirus strains and human DS-1-like and bovine rotavirus strains. J. Virol. 82, 3204–3219. doi: 10.1128/JVI.02257-07
Matthijnssens, J., Ciarlet, M., Rahman, M., Attoui, H., Banyai, K., Estes, M. K., et al. (2008b). Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 153, 1621–1629. doi: 10.1007/s00705-008-0155-1
Matthijnssens, J., Ciarlet, M., McDonald, S. M., Attoui, H., Banyai, K., Brister, J. R., et al. (2011). Uniformity of rotavirus strain nomenclature proposed by the Rotavirus Classification Working Group (RCWG). Arch. Virol. 156, 1397–1413. doi: 10.1007/s00705-011-1006-z
McDermid, A., Le Saux, N., Grudeski, E., Bettinger, J. A., Manguiat, K., Halperin, S. A., et al. (2012). Molecular characterization of rotavirus isolates from select Canadian pediatric hospitals. BMC Infect. Dis. 12:306. doi: 10.1186/1471-2334-12-306
Montenegro, F. M., Falbo, A. R., Germano, E. M., Correia, N. B., Souza Eda, S., Nakagomi, O., et al. (2015). Reduction in rotavirus disease and sustained predominance of G2P[4] rotavirus strain following introduction of rotavirus vaccine in Recife, Brazil. J. Trop. Pediatr. 61, 206–209. doi: 10.1093/tropej/fmv006
Nagaoka, Y., Tatsumi, M., Tsugawa, T., Yoto, Y., and Tsutsumi, H. (2012). Phylogenetic and computational structural analysis of VP7 gene of group a human rotavirus G1P[8] strains obtained in Sapporo, Japan from 1987 to 2000. J. Med. Virol. 84, 832–838. doi: 10.1002/jmv.23247
Nakagomi, O., Nakagomi, T., Akatani, K., and Ikegami, N. (1989). Identification of rotavirus genogroups by RNA-RNA hybridization. Mol. Cell. Probes 3, 251–261. doi: 10.1016/0890-8508(89)90006-6
Nakagomi, T., Kato, K., Tsutsumi, H., and Nakagomi, O. (2013). The burden of rotavirus gastroenteritis among Japanese children during its peak months: an internet survey. Jpn. J. Infect. Dis. 66, 269–275. doi: 10.7883/yoken.66.269
Nyaga, M. M., Jere, K. C., Esona, M. D., Seheri, M. L., Stucker, K. M., Halpin, R. A., et al. (2015). Whole genome detection of rotavirus mixed infections in human, porcine and bovine samples co-infected with various rotavirus strains collected from sub-Saharan Africa. Infect. Genet. Evol. 31, 321–334. doi: 10.1016/j.meegid.2015.02.011
Phan, T. G., Okitsu, S., Maneekarn, N., and Ushijima, H. (2007). Genetic heterogeneity, evolution and recombination in emerging G9 rotaviruses. Infect. Genet. Evol. 7, 656–663. doi: 10.1016/j.meegid.2007.05.001
Reidy, N., O’Halloran, F., Fanning, S., Cryan, B., and O’Shea, H. (2005). Emergence of G3 and G9 rotavirus and increased incidence of mixed infections in the southern region of Ireland 2001-2004. J. Med. Virol. 77, 571–578. doi: 10.1002/jmv.20494
Sanchez-Fauquier, A., Montero, V., Moreno, S., Sole, M., Colomina, J., Iturriza-Gomara, M., et al. (2006). Human rotavirus G9 and G3 as major cause of diarrhea in hospitalized children, Spain. Emerg. Infect. Dis. 12, 1536–1541. doi: 10.3201/eid1210.060384
Siebenga, J. J., Vennema, H., Zheng, D. P., Vinje, J., Lee, B. E., Pang, X. L., et al. (2009). Norovirus illness is a global problem: emergence and spread of norovirus GII.4 variants, 2001-2007. J. Infect. Dis. 200, 802–812. doi: 10.1086/605127
Sugimoto, D., Nakano, M., Inada, M., Fujitani, M., Chiba, S., and Sakai, T. (2017). Distribution of rotavirus genotypes from the 2008/2009 to the 2015/2016 season in Nara prefecture, Japan. Jpn. J. Infect. Dis. 70, 593–594. doi: 10.7883/yoken.JJID.2016.419
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Tate, J. E., Burton, A. H., Boschi-Pinto, C., and Parashar, U. D. (2016). Global, regional, and national estimates of rotavirus mortality in children < 5 Years of Age, 2000-2013. Clin. Infect. Dis. 62(Suppl. 2), S96–S105. doi: 10.1093/cid/civ1013
Tatsumi, M., Nagaoka, Y., Tsugawa, T., Yoto, Y., Hori, T., and Tsutsumi, H. (2014). Characterization of the NSP4 gene of group A human rotavirus G1P[8] strains circulating in Sapporo, Japan from 1987 to 2000. J. Med. Virol. 86, 354–359. doi: 10.1002/jmv.23723
Wang, Y. H., Pang, B. B., Ghosh, S., Zhou, X., Shintani, T., Urushibara, N., et al. (2014). Molecular epidemiology and genetic evolution of the whole genome of G3P[8] human rotavirus in Wuhan, China, from 2000 through 2013. PLoS One 9:e88850. doi: 10.1371/journal.pone.0088850
Wu, F. T., Banyai, K., Jiang, B., Wu, C. Y., Chen, H. C., Feher, E., et al. (2014). Molecular epidemiology of human G2P[4] rotaviruses in Taiwan, 2004-2011. Infect. Genet. Evol. 28, 530–536. doi: 10.1016/j.meegid.2014.09.033
Yamamoto, D., Tandoc, A. III, Mercado, E., Quicho, F., Lupisan, S., Obata-Saito, M., et al. (2017). First detection of DS-1-like G1P[8] human rotavirus strains from children with diarrhoea in the Philippines. New Microbes New Infect. 18, 54–57. doi: 10.1016/j.nmni.2017.04.001
Yamamoto, S. P., Kaida, A., Kubo, H., and Iritani, N. (2014). Gastroenteritis outbreaks caused by a DS-1-like G1P[8] rotavirus strain, Japan, 2012-2013. Emerg. Infect. Dis. 20, 1030–1033. doi: 10.3201/eid2006.131326
Keywords: rotavirus, Japan, full-genome, next-generation sequencing, reassortment, Bayesian analysis
Citation: Fujii Y, Doan YH, Suzuki Y, Nakagomi T, Nakagomi O and Katayama K (2019) Study of Complete Genome Sequences of Rotavirus A Epidemics and Evolution in Japan in 2012–2014. Front. Microbiol. 10:38. doi: 10.3389/fmicb.2019.00038
Received: 12 October 2018; Accepted: 10 January 2019;
Published: 31 January 2019.
Edited by:
Souvik Ghosh, Ross University School of Veterinary Medicine, Saint Kitts and NevisReviewed by:
Helen O’Shea, Cork Institute of Technology, IrelandNobumichi Kobayashi, Sapporo Medical University, Japan
Copyright © 2019 Fujii, Doan, Suzuki, Nakagomi, Nakagomi and Katayama. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhiko Katayama, a2F0YXlhbWFAbGlzY2kua2l0YXNhdG8tdS5hYy5qcA==; a2F0YXlhbWFAbmloLmdvLmpw