 Maria João Catalão
Maria João Catalão Sérgio R. Filipe
Sérgio R. Filipe Madalena Pimentel
Madalena Pimentel- 1Research Institute for Medicines (iMed.ULisboa), Faculty of Pharmacy, Universidade de Lisboa, Lisbon, Portugal
- 2UCIBIO-REQUIMTE, Departamento de Ciências da Vida, Faculdade de Ciências e Tecnologia, Caparica, Portugal
- 3Laboratory of Bacterial Cell Surfaces and Pathogenesis, Instituto de Tecnologia Química e Biológica António Xavier, Universidade Nova de Lisboa, Oeiras, Portugal
Tuberculosis (TB), which is caused by Mycobacterium tuberculosis (Mtb), is one of the leading cause of death by an infectious diseases. The biosynthesis of the mycobacterial cell wall (CW) is an area of increasing research significance, as numerous antibiotics used to treat TB target biosynthesis pathways of essential CW components. The main feature of the mycobacterial cell envelope is an intricate structure, the mycolyl-arabinogalactan-peptidoglycan (mAGP) complex responsible for its innate resistance to many commonly used antibiotics and involved in virulence. A hallmark of mAGP is its unusual peptidoglycan (PG) layer, which has subtleties that play a key role in virulence by enabling pathogenic species to survive inside the host and resist antibiotic pressure. This dynamic and essential structure is not a target of currently used therapeutics as Mtb is considered naturally resistant to most β-lactam antibiotics due to a highly active β-lactamase (BlaC) that efficiently hydrolyses many β-lactam drugs to render them ineffective. The emergence of multidrug- and extensive drug-resistant strains to the available antibiotics has become a serious health threat, places an immense burden on health care systems, and poses particular therapeutic challenges. Therefore, it is crucial to explore additional Mtb vulnerabilities that can be used to combat TB. Remodeling PG enzymes that catalyze biosynthesis and recycling of the PG are essential to the viability of Mtb and are therefore attractive targets for novel antibiotics research. This article reviews PG as an alternative antibiotic target for TB treatment, how Mtb has developed resistance to currently available antibiotics directed to PG biosynthesis, and the potential of targeting this essential structure to tackle TB by attacking alternative enzymatic activities involved in Mtb PG modifications and metabolism.
Introduction
According to the latest report available from the World Health Organization (WHO), it is estimated that in 2017, there were about 10.3 million new cases of TB worldwide and about 1.8 million people died from this infection. The emergence of multidrug-resistant (MDR) and extensive drug-resistant (XDR) strains to the available antibiotics is a worldwide public health problem of increasing importance, with a treatment success rate of only about 50%, which decreases to 23% in the case of XDR-TB (World Health Organization, 2010, 2011, 2014, 2017; Horsburgh et al., 2015). The lack of effective treatment regimens against MDR-TB and XDR-TB isolates has highlighted the potential of repurposing existing antibiotic options in alternative and innovative ways (Mainardi et al., 2011) as all drugs, except for bedaquiline and delamanid, which are currently used to treat TB, were approved several years ago, demonstrating the complexity of TB drug development (Wong et al., 2013; Keener, 2014; Diacon et al., 2016).
A hallmark of Mtb, the causative agent of TB, as a successful pathogen is its intricate CW (Brennan and Nikaido, 1995; Jankute et al., 2015) that has been associated with the genetic differences among human lineages of Mtb (Portevin et al., 2011). The core of the mycobacteria cell envelope is composed of three main structures: (1) the characteristic long-chain mycolic acids (MA); (2) a highly branched arabinogalactan (AG) polysaccharide; and (3) a very cross-linked and modified meshwork of PG. The entire complex, referred to as mycolyl-arabinogalactan-peptidoglycan (mAGP) (Brennan and Nikaido, 1995; Alderwick et al., 2015; Jankute et al., 2015), is essential for Mtb viability, virulence, and persistence and can modulate the innate immune response (Brennan and Nikaido, 1995; Stanley and Cox, 2013; Jankute et al., 2015). In addition, it acts as an impregnable external barrier responsible for the intrinsic resistance of Mtb to several drugs (Nikaido, 1994; Gygli et al., 2017; Nasiri et al., 2017). The essential nature of CW synthesis and assembly has rendered the mycobacterial CW as the most extensively exploited target of anti-TB drugs (Wong et al., 2013; Bhat et al., 2017). Ethambutol, isoniazid, and ethionamide successfully target the synthesis of the various components of mAGP (Jackson et al., 2013), and resistance to these drugs, which is mediated by the accumulation of chromosomal mutations in genes involved in CW biosynthesis pathways, can arise under selective pressure of antibiotic use (Eldholm and Balloux, 2016; Gygli et al., 2017; Nasiri et al., 2017). Mtb has been considered innately resistant to most β-lactam antibiotics that target PG biosynthesis due to (1) a highly active β-lactamase (BlaC) that efficiently inactivates many β-lactams (Wang et al., 2006; Hugonnet and Blanchard, 2007) and (2) the fact that a large proportion of the CW PG is cross-linked by non-classical L,D-transpeptidases, which are intrinsically impervious to these antibiotics (Lavollay et al., 2008; Cordillot et al., 2013). Widespread antibiotic resistance in Mtb, in combination with the lack of progress in developing new effective treatments, is threatening the ability of tackling the outcomes caused by highly resistant Mtb strains. This highlights the need of considering alternative therapeutic schemes to combat the global increase in resistance to the current anti-TB regimens. This review summarizes the current knowledge about the mechanisms employed by mycobacteria to circumvent the activity of currently available antibiotics that target PG biosynthesis with an emphasis on recent advancements regarding the efficacy of carbapenems, a more recent class of extended-spectrum β-lactams against highly drug-resistant Mtb clinical strains, and the potential application of mycobacteriophage-encoded lysis proteins to kill mycobacteria by weakening the CW.
Impact of the Atypical Mycobacterial PG Structure on Resistance to Antibiotics that Target PG Biosynthesis
A distinctive feature of the mycobacterial CW is its unusual PG layer (Alderwick et al., 2015; Jankute et al., 2015), which is essential for survival of Mtb and that is linked with the exceptional immunogenic activity associated with the CW. The PG macromolecule contains a number of unique subtleties that enable Mtb to survive inside the host and resist different antibiotics (Gygli et al., 2017; Nasiri et al., 2017). The PG layer of Mtb is surrounded by other layers dominated by lipids, carbohydrates, and phosphatidyl-myo-inositol-based lipoglycans that provide a permeability barrier against hydrophilic drugs (Nikaido, 1994; Brennan and Nikaido, 1995; Hoffmann et al., 2008). PG acts as a pro-inflammatory inducer that is hypothetically masked within the mAGP complex (Brennan and Nikaido, 1995; Jankute et al., 2015), which constitutes the major structural component of the cell envelope. Access of antibiotics that target PG biosynthesis is critical for their efficacy, and it is now assumed that several pathogenic bacteria have developed different strategies to hide PG (Atilano et al., 2011, 2014), thus circumventing their antibacterial activity. Mycobacterial PG forms the basal layer of the mAGP complex, where glycan chains composed of alternating N-acetylglucosamine (GlcNAc) and modified N-acetylmuramic acid (MurNAc) residues, linked in a β (1 → 4) configuration (Alderwick et al., 2015), are interconnected through oligopeptides. The muramic acid residues in Mtb are found containing a combination of N-acetyl and N-glycolyl derivatizations. In the latter case, the N-acetyl group present in MurNAc residues has been oxidized to an N-glycolyl group through the action of the enzyme N-acetyl muramic acid hydroxylase (NamH) to form MurNGly (Raymond et al., 2005). Although the precise function of the N-glycolyl modification, a structural modification that is unique to mycobacteria (and closely related genera) is yet to be elucidated, it has been hypothesized that it contributes to: (1) the stability of the mycobacterial CW, by strengthening the mesh-like structure of the PG layer providing sites for additional hydrogen bonding between different parts of the PG macromolecule (Brennan and Nikaido, 1995); (2) the increase of β-lactam resistance (Raymond et al., 2005); (3) the protection of bacteria from degradation via lysozyme (Raymond et al., 2005); and (4) the overall innate immune response triggered by the CW of mycobacteria, as the glycolylated form of the muramyl dipeptide is an important contributor to the unusual immunogenicity of mycobacteria. This component of the mycobacterial PG is a strong inducer of NOD2-mediated host responses (Coulombe et al., 2009; Schenk et al., 2016), although playing a limited role in the pathogenesis of Mtb infection (Hansen et al., 2014). Beside the contribution of glycolylated muramic acid residues to the overall host-mycobacteria interaction, Mtb PG-derived muropeptides released by the action of a group of enzymes called “resuscitation-promoting factors,” encoded by the rpf genes have also been associated with β-lactam and vancomycin tolerance and increased outer membrane (OM) impermeability (Kana et al., 2010; Wivagg and Hung, 2012). The pentapeptide chains of the mycobacterial PG can also be modified by amidation, glycylation, or methylation (Mahapatra et al., 2005), which contributes to its resistance to endopeptidase activity of PG hydrolases (Lavollay et al., 2008). However, the functional significance of these modifications for Mtb drug resistance is unknown.
The mature PG architecture is also marked by a high degree of direct peptide cross-links, a characteristic that is not frequently found in other bacteria. Overall, 80% of the peptides are cross-linked in two types of linkages in order to maintain the complexity of the mycobacterial cell envelope during growth and under non-replicating conditions (Lavollay et al., 2008). Mycobacterial PG cross-linking is catalyzed by D,D-transpeptidases (penicillin-binding proteins) and typically by the combined action of non-classical L,D-transpeptidases (Ldts) and D,D-carboxypeptidases. The action of these enzymes results in PG peptides, which connect neighboring glycan chains, that are linked through 4 → 3 (D-Ala-mDAP) and 3 → 3 (mDAP-mDAP) linkages, respectively (Figure 1; Lavollay et al., 2008). The latter set of proteins contributes to the intrinsic resistance to β-lactams and provides protection from PG endopeptidases (Lavollay et al., 2008; Cordillot et al., 2013). Another unique feature of mycobacterial PG is that it provides the attachment site for AG (which is catalyzed by the Lcp1 phosphotransferase) (McNeil et al., 1990; Baumgart et al., 2016; Grzegorzewicz et al., 2016; Harrison et al., 2016), a highly branched molecule assembled from arabinofuranose and galactofuranose monosaccharides, which overlays the PG and that can also preclude PG synthesis from being targeted by β-lactams (Schubert et al., 2017).
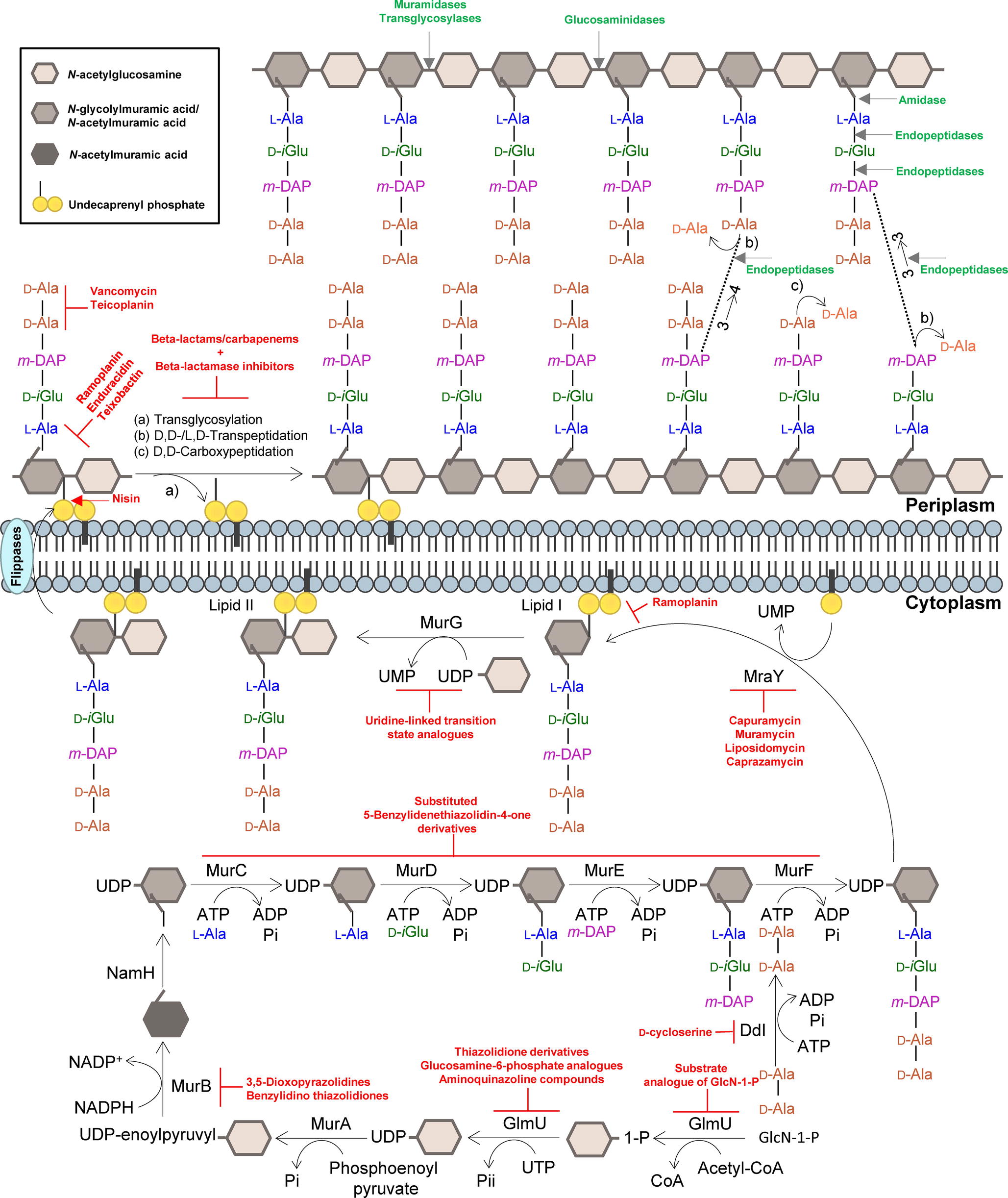
Figure 1. Summary of the mycobacterial peptidoglycan biosynthesis pathway. The peptidoglycan precursors are produced in the cytoplasm, and peptidoglycan monomeric units are assembled in the inner leaflet of the cytoplasmic membrane. Polymerization and cross-linking of tetrapeptide side chains take place at the periplasm. Inhibitors of the peptidoglycan biosynthetic enzymes are colored in red, and peptidoglycan bonds that are targeted by mycobacteriophage endolysins are colored in green. Adapted from Abrahams and Besra, 2018, with permission.
Mycobacterial Intrinsic Resistance to Antibiotics that Target PG Biosynthesis: A New Trick for an Old Dogma
PG biosynthesis (Figure 1) represents the site of action of the most widely used class of antibacterial agents for infection treatment (Vollmer et al., 2008; Bugg et al., 2011; Cho et al., 2014; Pavelka et al., 2014). However, except for D-cycloserine, an oral antimycobacterial agent that is specifically recommended by the WHO as a second-line anti-TB agent used as a last option for the treatment of TB (Hwang et al., 2013), antibiotics that target PG synthesis such as the β-lactams are only rarely used in the treatment of TB (Wong et al., 2013; Wivagg et al., 2014). This lack of efficacy against Mtb has primarily been attributed to a chromosomally encoded broad spectrum class A β-lactamase enzyme BlaC (Flores et al., 2005; Wang et al., 2006; Hugonnet and Blanchard, 2007), which hydrolyses the core β-lactam ring and deactivates the antibiotic, to different drug efflux pumps, to low affinity penicillin-binding proteins (PBPs) and to the expression of PG-biosynthetic enzymes insensitive to β-lactams (non-classical transpeptidases) (Wivagg et al., 2014; Gygli et al., 2017; Nasiri et al., 2017). In addition, the PG is camouflaged by the MA-rich mycobacterial OM that limits penetration of antibiotics (Figure 2; Brennan and Nikaido, 1995; Jankute et al., 2015).
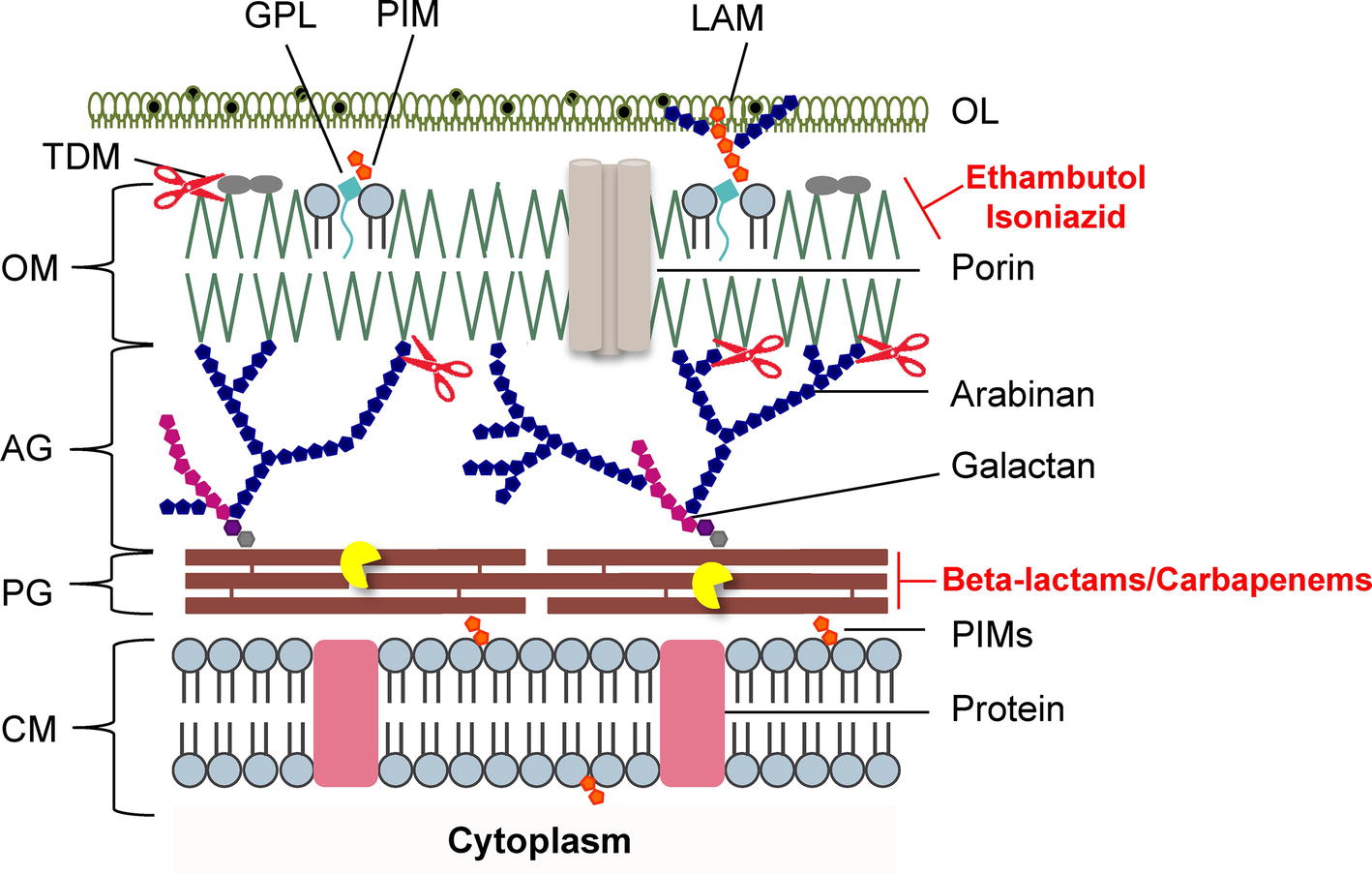
Figure 2. Schematic representation of the mycobacterial cell envelope layers. Inhibitors of mycolic acids and peptidoglycan biosynthesis are indicated in red. The mycobacteriophage lysis protein targets are indicated as follows: the pacman cartoon represents digestion of the PG by the endolysins; scissors illustrate LysB detachment of the OM. AG, arabinogalactan; CM, cytoplasmic membrane; GLP, glycolipids; LAM, lipoarabinomannan; OL, outer layer; OM, outer membrane; PG, peptidoglycan; PIMs, phosphatidylinositol mannosides; PLs, phospholipids; TDM, trehalose dimycolate. Adapted from Catalão and Pimentel, 2018 with permission from the authors.
Resistance to D-Cycloserine in Mtb
D-cycloserine is a structural analog of D-alanine and interferes with the formation of PG biosynthesis, by acting as a competitive inhibitor of alanine racemase (Alr) and D-alanine-D-alanine ligase (Ddl) enzymes, which are involved in PG synthesis (Prosser and de Carvalho, 2013a,b,c). Ddl is the main target of D-cycloserine and is preferentially inhibited over Alr in Mtb (Prosser and de Carvalho, 2013a). Resistance to this antibiotic has been associated with loss-of-function mutations in metabolism-related genes of ubiquinone and menaquinone and ald (Rv2780), which encodes an L-alanine dehydrogenase (Hong et al., 2014; Desjardins et al., 2016). A recent study has identified novel mutations connected with D-cycloserine resistance in MDR and XDR Mtb strains, which demonstrate that resistance to this antibiotic is highly complex and involves diverse genes associated with different cellular processes such as lipid metabolism, methyltransferase, stress response, and transport systems (Chen et al., 2017). In another study, a genomic screening of more than 1,500 drug-resistant strains of Mtb revealed the presence of three main alr mutations (alrMtb M319 T, alrMtb Y364D, alrMtb R373L) that confer D-cycloserine resistance (Nakatani et al., 2017). Despite the importance of D-cycloserine as a second-line drug used to treat MDR- and XDR-TB, the mechanisms underlying D-cycloserine resistance in Mtb clinical strains are still undetermined.
The emergence of MDR and XDR Mtb strains has become a serious health threat and has initiated the search for new therapeutic strategies. Some of those strategies include revisiting the potential use of β-lactams as an alternative therapeutic approach to tackle drug-resistant TB when no acceptable alternative exists (Hugonnet et al., 2009; Keener, 2014; Diacon et al., 2016).
Resistance to β-Lactams in Mtb
Recent developments have led to the suggestion of using carbapenems, a modern class of extended-spectrum β-lactams, as the last line of defense against recalcitrant drug-resistant TB (Hugonnet et al., 2009; Payen et al., 2012; Gonzalo and Drobniewski, 2013; Davies Forsman et al., 2015; Jaganath et al., 2016; Payen et al., 2018). Among β-lactams, carbapenems are unique as they are not only relatively resistant to the hydrolytic activity of BlaC, but also act as potent inhibitors of this enzyme (Tremblay et al., 2010). The efficacy of carbapenems in killing Mtb is further increased by the ability of these compounds to inhibit the different enzymes that contribute to the assembly of mycobacterial PG (Gupta et al., 2010; Dubée et al., 2012; Erdemli et al., 2012; Cordillot et al., 2013; Bianchet et al., 2017; Kumar et al., 2017a). While most β-lactams inhibit D,D-transpeptidases (PBPs), which are the enzymes that catalyze the formation of 4 → 3 transpeptide linkages in the PG network (Zapun et al., 2008), they are unable to inhibit the L,D-transpeptidases (Ldts) that catalyze the formation of 3 → 3 transpeptide linkages. As the PG of mycobacteria contains a high proportion (up to 80%) of 3 → 3 cross-links (Lavollay et al., 2008; Cordillot et al., 2013), β-lactams will not fully prevent the assembly of the mycobacterial PG. Carbapenems inhibit not only D,D-transpeptidases but also L,D-transpeptidases (as well as D,D-carboxypeptidases) (Baranowski et al., 2018; García-Heredia et al., 2018).
Ldt and PBP enzymes are structurally unrelated and contain cysteine and serine residues in their active sites, respectively. Mtb genome encodes five L,D-transpeptidases, designated by LdtMt1 to LdtMt5 (Cordillot et al., 2013). It was shown that the presence of L,D-transpeptidases can markedly alter β-lactam susceptibility (Lavollay et al., 2008; Gupta et al., 2010; Dubée et al., 2012; Kumar et al., 2012; Cordillot et al., 2013; Kieser et al., 2015; Wivagg et al., 2016). In addition, recent studies indicate that Mtb strains that lack both ldtMt1 and ldtMt2 display enhanced susceptibility not only to amoxicillin, a β-lactam antibiotic, but also to vancomycin, a glycopeptide antibiotic (Schoonmaker et al., 2014). Furthermore, a synergistic effect of carbapenem with rifampicin was observed against rifampicin-resistant clinical isolates of Mtb (Kaushik et al., 2015, 2017).
Most of the anti-TB drugs associated with CW biosynthesis inhibition lack the ability to reduce treatment duration of TB drug regimens. This is related to the fact that some bacteria can withstand the presence of the antibiotics by becoming dormant, i.e., being unable to replicate, as dormant bacteria do not actively synthesize the CW and are presumably not affected by the presence of inhibitors of the CW synthesis. Recent research has shown that a combinatorial treatment that is based on the use of the β-lactamase inhibitor clavulanate and meropenem is effective against both actively replicating and non-replicating XDR Mtb isolates (Solapure et al., 2013). However, its high cost and intravenous administration present challenges to its widespread use. According to the WHO anti-tuberculosis classification, the carbapenems are included in Group D3, which indicates that safety and efficacy information to support its use against TB is restricted and should not be considered as an alternative regimen designated to treat TB (WHO, 2011, 2014). The existing in vivo and clinical studies suggest that there are advantages in carbapenem use as they are usually well-tolerated, although the variance in the extent of the treatment, dosing, and the absence of pharmacokinetic data limit interpretation of the effectiveness of these antibiotics against TB. Information regarding carbapenem resistance is scarce; mutations in CW biosynthesis genes and in crfA have been associated with resistance to different carbapenem antibiotics such as imipenem, meropenem, and biapenem (Lun et al., 2014; Kumar et al., 2017b). Nevertheless, these studies have been an enormous contribution to the recent and increased effort for repurposing β-lactams as an ultimate therapeutic option to treat life-threatening TB-infected patients and to unveil to what extent the wider Mtb human clinical isolates population may be susceptible to these antibiotics (Tiberi et al., 2016).
Mycobacterial PG Assembly Enzymes as Targets for Antibiotics
The PG layer provides shape and rigidity to an individual cell of Mtb (Brennan, 2003). Since it is mainly restricted to bacterial cells, the enzymes that are involved in the biosynthesis of PG offer an attractive target for the development of new antibiotics against TB. In addition, the enzymes that catalyze the PG biosynthesis pathway in mycobacteria are essential, and therefore, their inhibition is expected to result in selective destruction of the bacteria (Moraes et al., 2015; Bhat et al., 2017; Abrahams and Besra, 2018). The biosynthesis process of mycobacterial PG is similar to other bacteria (Figure 1). The first step is catalyzed by the acetyltransferase and uridyltransferase activities of GlmU (Rv1018c), to yield UDP-GlcNAc (Zhang et al., 2009). The functional resemblance of the GlmU uridyltransferase with human enzymes (Peneff et al., 2001) turns this enzyme into an unsuitable target (Rani and Khan, 2016). However, the lack of GlcN-1-P from mammals makes the acetyltransferase domain a promising target, and different substrate analogs of GlcN-1-P have been designed and shown to exhibit an inhibitory effect against GlmU by blocking synthesis of UDP-GlcNAc (Figure 1; Li et al., 2011; Tran et al., 2013; Rani et al., 2015). The sequential MurA-F ligase pathway involves the formation of the UDP-N-acetylmuramic acid (UDP-MurNAc)-pentapeptide. MurA (Rv1315), a UDP-N-acetylglucosamine 1-carboxyvinyltransferase, and MurB (Rv0482), a UDP-N-acetylenolpyruvoylglucosamine reductase, are implicated in the formation of UDP-MurNAc. NamH (Rv3808), a UDP-N-acetylmuramic acid hydroxylase, hydroxylates UDP-MurNAc to UDP-N-glycolylmuramic acid (UDP-MurNGlyc) in the cytoplasm to generate both types of UDP-muramyl substrates, although Mtb PG is enriched in the latter (Mahapatra et al., 2005; Raymond et al., 2005). Specific inhibitors of Mtb MurA and MurB have not been described to date. The broad-spectrum antibiotic, fosfomycin, which targets Gram-negative MurA, has no activity against Mtb since the critical cysteine (Cys117) residue, which is required for inhibition by the drug, is replaced in Mtb by an aspartic acid residue, contributing to the intrinsic resistance against this antibiotic (Kim et al., 1996). A limited number of inhibitors have been reported against MurB, specifically the 3,5-dioxopyrazolidine and benzylidene thiazolidinedione derivatives which can competitively inhibit the formation of UDP-MurNAc (Figure 1; Kumar et al., 2011; Rana et al., 2014). Inhibitors of NamH have not been reported, probably due to the fact that namH is not essential in Mtb (Hansen et al., 2014). Therefore, NamH may not be a key target for anti-TB therapy. However, Mtb strains that lack namH are hypersusceptible to β-lactam antibiotics, and therefore, inhibitors of NamH could potentiate the effect of carbapenems (Raymond et al., 2005; Hansen et al., 2014). From this point, the pentapeptide chain is attached to UDP-MurNAc/Glyc by the ATP-dependent Mur ligases (Figure 1), beginning with UDP-N-acetylmuramoyl-L-alanine addition by MurC (Rv2151c). This is followed by D-isoglutamate addition by MurD (Rv2155c), m-DAP addition by MurE (Rv2158c), and finally D-alanyl-D-alanine addition by MurF (Rv2157c). This generates the muramyl-pentapeptide product UDP-MurNAc/Glyc-L-Ala-D-isoGlu-m-DAP-D-Ala-D-Ala, also known as Park’s nucleotide (Figure 1; Pavelka et al., 2014). Several inhibitors of the Mur ligases have been identified (Hrast et al., 2014). One example is the substituted 5-benzylidenethiazolidin-4-one derivatives that inhibit the formation of the pentapetide chains (Tomasic et al., 2010). However, their utilization is limited against Mtb Mur ligases given that only MurC and MurE have been biochemically characterized (Mahapatra et al., 2000; Li et al., 2011). Ddl is the target of D-cycloserine, a structural analog of D-Ala, inhibiting the binding of either the two D-Ala substrates to Ddl (Bruning et al., 2011; Prosser and de Carvalho, 2013c). The assembled Park’s nucleotide is then transferred to undecaprenyl phosphate present at the membrane by MraY (Rv2156c) generating Lipid I. Nucleoside-peptide antibiotics that inhibit MraY have been described, including muramycin, liposidomycin, caprazamycin, and capuramycin (Dini, 2005; Wiegmann et al., 2016; Tran et al., 2017). Remarkably, capuramycin has been shown to kill non-replicating Mtb, an uncommon characteristic of the majority of CW biosynthesis inhibitors (Koga et al., 2004; Reddy et al., 2008; Nikonenko et al., 2009; Siricilla et al., 2015). The final intracellular step of PG synthesis is catalyzed by MurG, a glycosyltransferase that is responsible for producing lipid II, the final monomeric block of PG. An Escherichia coli designed inhibitor of MurG was tested against Mtb with limited success and has become the first inhibitor identified against the Mtb glycosyltransferase (Trunkfield et al., 2010).
Translocation of lipid II across the plasma membrane is carried out by a flippase. This was initially thought to be an FtsW-like protein, Rv2154c (Mohammadi et al., 2011). However, recent research has shown that FtsW/RodA enzymes elongate PG chains through a transglycosylase activity (Meeske et al., 2016; Emami et al., 2017), and therefore, the best candidate for the PG precursor flippase is currently MurJ (Rv3910). Following the transport of PG precursor across the mycobacterial membrane, the bifunctional PonA1/PBP1 (Rv0050) and PonA2/PBP2 (Rv3682) enzymes, PBPs that possess both the transglycosylase and transpeptidase domains attach the GlcNAc moiety to the muramyl moiety of the nascent PG chain (Figure 1). Lipid II inhibitors, such as the depsipeptide antibiotics ramoplanin and enduracidin (Fang et al., 2006) and teixobactin (Ling et al., 2015), that prevent the transglycosylation of the translocated lipid II by binding to it have been described recently. The transpeptidase activity of PonA1 and PonA2 catalyzes the classical 4 → 3 cross-linkages between m-DAP and D-Ala of the adjacent pentapeptide chains present in neighboring glycan chains, with the cleavage of the terminal D-Ala. Other D,D-transpeptidation and D,D-carboxypeptidation reactions are catalyzed by the monofunctional PBPs, both resulting in the cleavage of the terminal D-Ala of the peptide stem (Zapun et al., 2008). Among the muropeptides present in the Mtb PG, up to 80% of the cross-links are 3 → 3 links between m-DAP residues of two adjacent tetrapeptide stems, with the release of the fourth position D-Ala (Lavollay et al., 2008), performed by non-classical L,D-transpeptidases, LdtMt1 (Rv0116c), LdtMt2 (Rv2518c), LdtMt3 (Rv1433), LdtMt4 (Rv0192), and LdtMt5 (Rv0483) (Lavollay et al., 2008; Cordillot et al., 2013). As mentioned before, the L,D-transpeptidase and D,D-carboxypeptidase activities are unaffected by most β-lactam antibiotics, except the carbapenems (Gupta et al., 2010; Dubée et al., 2012; Kumar et al., 2012; Cordillot et al., 2013; Rullas et al., 2015; Bianchet et al., 2017; Kumar et al., 2017a). Moenomycin, a glycolipid that inhibits the transglycosylase activity of PBPs (van Heijenoort et al., 1987) is yet to have recognized efficacy against Mtb. The existence of other antibiotics that act on the availability of PG precursors: (1) the glycopeptides, vancomycin and teicoplanin, that bind to the D-Ala-D-Ala terminus of the pentapeptide stem and prevent PG polymerization (Reynolds, 1989); (2) the lantibiotic family of antibiotics, such as nisin, that interact with the pyrophosphate moiety of lipid II, with the consequent delocalization of this molecule that can form a pore in the cytoplasmic membrane and inhibit PG biosynthesis (Wiedemann et al., 2001), opens new avenues to find suitable synergistic antibiotic combination schemes for effective treatments.
Prospective use of Mycobacteriophage Endolysins to Degrade the Mycobacterial PG
The mycobacterial PG is modified by several enzymes, which confer resistance to some widely used antibiotics (Mahapatra et al., 2005; Raymond et al., 2005). Mycobacteriophages, the viruses of mycobacteria, synthesize enzymes to eliminate each layer of the cell envelope (recently reviewed in Catalão and Pimentel, 2018), so that phage particles can escape from the bacterial cell at the end of a replicating cycle. Mycobacteriophage-encoded PG hydrolases (endolysins) are predicted to target and degrade nearly every bond in mycobacterial PG (Figure 1; Payne and Hatfull, 2012; Catalão et al., 2013; Pimentel, 2014). Given the essentiality of the mycobacterial cell envelope (Brennan and Nikaido, 1995; Jankute et al., 2015; Chiaradia et al., 2017), it is reasonable to consider that the enzymatic degradation of mycobacterial CW by the mycobacteriophage lytic enzymes (Gil et al., 2008, 2010; Payne et al., 2009; Catalão et al., 2011; Gigante et al., 2017) may be a promising therapeutic approach to kill extracellular pathogenic mycobacteria (Grover et al., 2014) or after their internalization by macrophages (Lai et al., 2015). However, access of mycobacteriophage endolysins to their substrate, the PG, is hindered by the MA-rich mycobacterial OM, which restrains their use as anti-TB therapeutic agents (Figure 2). Therefore, transport of phage enzymes and/or antibiotics that target the PG metabolism through this OM remains the major constraint in the application of these compounds in therapy (Catalão and Pimentel, 2018). As the enzymes involved in MA and AG biosynthesis and integrity play an important role in the development of drug resistance in Mtb, inhibition of the synthesis of these CW layers would damage the CW as a barrier, increase its permeability, and increase the susceptibility of bacteria to various anti-mycobacterial drugs (Figure 2). Mycobacteriophage-encoded LysB proteins are specific lysis proteins that act enzymatically, not only hydrolyzing lipids on the outer leaflet of the OM, but also, importantly, detaching it from the CW by cleaving the ester linkage to the AG polymer due to a mycolyl-arabinogalactan esterase activity (Figure 2; Gil et al., 2008, 2010; Payne et al., 2009; Gigante et al., 2017). Interestingly, it has been recently reported that ethambutol, one of the first-line drugs for TB treatment, leads to the loss of the MA layer by blocking polymerization of arabinose in AG, which impairs de novo synthesis of the outer envelope layers (Schubert et al., 2017). As cell division seems to be unaffected by ethambutol, the authors proposed that the inhibition of MA synthesis generates a defective CW composed predominately of exposed PG. Inactivation of the Ag85 complex (fbpA, fbpB, and fbpC) proteins that possess mycolyltransferase activity and are involved in biogenesis of trehalose dimycolate (TDM), a glycolipid that has been proposed to be present in the outer leaflet of the mycobacterial OM, increased sensitivity both to first-line TB drugs and to erythromycin, imipenem, rifampicin, and vancomycin (Lingaraju et al., 2016). Since production of TDM by Ag85 is essential for the intrinsic antibiotic resistance of mycobacteria (Morris et al., 2005), Ag85-specific inhibitors or TDM hydrolysis by LysB (Figure 2; Gil et al., 2010) can have a positive impact on the fight to control mycobacterial drug resistance.
Understanding the mechanisms used by mycobacteriophages to deteriorate each layer of the extremely complex mycobacterial cell envelope is highly relevant for the design of new strategies against mycobacteria. Given the abundance of isolated mycobacteriophages, which constitute an enormous reservoir of CW degrading enzymes capable of hydrolyzing each specific linkage of the mycobacterial cell envelope (Hatfull, 2006; Payne and Hatfull, 2012), it is worth to consider the possibility of using these enzymes in synergistic combinations with CW targeting antibiotics, which have a limited access to their target, the PG in normally growing bacteria (Figure 2).
Concluding Remarks
Inhibition of the assembly of the bacterial CW by anti-mycobacterial agents that successfully target the synthesis of its various components has proven useful in tackling TB (Wong et al., 2013; Bhat et al., 2017). However, modification of CW targets mediated by specific enzymes or the accumulation of chromosomal mutations and degradation/modification of drugs by production of antibiotic inactivating enzymes has rendered Mtb resistant to most classes of antimicrobials (Eldholm and Balloux, 2016; Gygli et al., 2017; Nasiri et al., 2017). Infections due to Mtb are an increasing problem worldwide, and the emergence of XDR-TB suggests that Mtb may become refractory to any chemotherapeutic agent in the future (Horsburgh et al., 2015; World Health Organization, 2017). The limited number of new anti-mycobacterial agents approved for therapy and the wide variety of Mtb intrinsic and acquired drug resistance mechanisms to the available drugs have contributed to an increased effort to repurpose the use of antibiotics that are not commonly used in anti-TB therapy and to find suitable synergistic antibiotic combinations for effective treatment of life-risk TB (Mainardi et al., 2011; Wong et al., 2013; Keener, 2014; Diacon et al., 2016). Recent studies have uncovered the possibility of targeting the mycobacterial PG biosynthesis and degradation as an alternative option for anti-TB therapy (Tomasic et al., 2010; Trunkfield et al., 2010; Li et al., 2011; Rana et al., 2014; Ling et al., 2015; Rani et al., 2015; Rullas et al., 2015; Tran et al., 2017). In addition, several observations suggest that inhibition of PG synthesis by transpeptidase inhibitors such as the carbapenems or glycopeptide antibiotics could synergize with other CW inhibitors and increase their efficacy (Figures 1 and 2; Hugonnet et al., 2009; Kumar et al., 2012; Kieser et al., 2015; Schubert et al., 2017). The recent developments toward the potential application of mycobacteriophage-dedicated enzymes targeting the complex mycobacterial CW arrangement have also renewed the interest of repurposing mycobacterial PG metabolism as an anti-TB therapy target (Gil et al., 2008, 2010; Payne and Hatfull, 2012; Catalão and Pimentel, 2018). More research is needed in the near future that could lead to the design and development of therapeutics that increase the efficacy of currently available antibiotics and enzymes that target PG metabolism, which is not currently considered as an alternative to treat TB.
Author Contributions
MC and MP conceived and designed the study and wrote the manuscript. MC, SF, and MP participated in manuscript revising and editing.
Funding
This work was funded by a Research Grant 2018 of the European Society of Clinical Microbiology and Infectious Diseases (ESCMID) and Fundação para a Ciência e Tecnologia (FCT), Lisbon, Portugal, through research grant PTDC/BIA-MIC/31233/2017 awarded to MC.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
References
Abrahams, K. A., and Besra, G. S. (2018). Mycobacterial cell wall biosynthesis: a multifaceted antibiotic target. Parasitology 145, 116–133. doi: 10.1017/S0031182016002377
Alderwick, L. J., Harrison, J., Lloyd, G. S., and Birch, H. L. (2015). The mycobacterial cell wall-peptidoglycan and arabinogalactan. Cold Spring Harb. Perspect. Med. 5:a021113. doi: 10.1101/cshperspect.a021113
Atilano, M. L., Yates, J., Glittenberg, M., Filipe, S. R., and Ligoxygakis, P. (2011). Wall teichoic acids of Staphylococcus aureus limit recognition by the Drosophila peptidoglycan recognition protein-SA to promote pathogenicity. PLoS Pathog. 7:e1002421. doi: 10.1371/journal.ppat.1002421
Atilano, M. L., Pereira, P. M., Vaz, F., Catalão, M. J., Reed, P., Grilo, I. R., et al. (2014). Bacterial autolysins trim cell surface peptidoglycan to prevent detection by the Drosophila innate immune system. elife 3:e02277. doi: 10.7554/eLife.02277
Baranowski, C., Welsh, M. A., Sham, L. T., Eskandarian, H. A., Lim, H. C., Kieser, K. J., et al. (2018). Maturing Mycobacterium smegmatis peptidoglycan requires non-canonical crosslinks to maintain shape. elife 7:e37516. doi: 10.7554/eLife.37516
Baumgart, M., Schubert, K., Bramkamp, M., and Frunzke, J. (2016). Impact of LytR-CpsA-Psr proteins on cell wall biosynthesis in Corynebacterium glutamicum. J. Bacteriol. 198, 3045–3059. doi: 10.1128/JB.00406-16
Bhat, Z. S., Rather, M. A., Maqbool, M., Lah, H. U., Yousuf, S. K., and Ahmad, Z. (2017). Cell wall: a versatile fountain of drug targets in Mycobacterium tuberculosis. Biomed. Pharmacother. 95, 1520–1534. doi: 10.1016/j.biopha.2017.09.036
Bianchet, M. A., Pan, Y. H., Basta, L. A. B., Saavedra, H., Lloyd, E. P., Kumar, P., et al. (2017). Structural insight into the inactivation of Mycobacterium tuberculosis non-classical transpeptidase LdtMt2 by biapenem and tebipenem. BMC Biochem. 18:8. doi: 10.1186/s12858-017-0082-4
Brennan, P. J. (2003). Structure, function, and biogenesis of the cell wall of Mycobacterium tuberculosis. Tuberculosis 83, 91–97. doi: 10.1016/S1472-9792(02)00089-6
Brennan, P. J., and Nikaido, H. (1995). The envelope of mycobacteria. Annu. Rev. Biochem. 64, 29–63. doi: 10.1146/annurev.bi.64.070195.000333
Bruning, J. B., Murillo, A. C., Chacon, O., Barletta, R. G., and Sacchettini, J. C. (2011). Structure of the Mycobacterium tuberculosis D-alanine:D-alanine ligase, a target of the antituberculosis drug D-cycloserine. Antimicrob. Agents Chemother. 55, 291–301. doi: 10.1128/AAC.00558-10
Bugg, T. D., Braddick, D., Dowson, C. G., and Roper, D. I. (2011). Bacterial cell wall assembly: still an attractive antibacterial target. Trends Biotechnol. 29, 167–173. doi: 10.1016/j.tibtech.2010.12.006
Catalão, M. J., Milho, C., Gil, F., Moniz-Pereira, J., and Pimentel, M. (2011). A second endolysin gene is fully embedded in-frame with the lysA gene of mycobacteriophage Ms6. PLoS One 6:e20515. doi: 10.1371/journal.pone.0020515
Catalão, M. J., Gil, F., Moniz-Pereira, J., São-José, C., and Pimentel, M. (2013). Diversity in bacterial lysis systems: bacteriophages show the way. FEMS Microbiol. Rev. 37, 554–571. doi: 10.1111/1574-6976.12006
Catalão, M. J., and Pimentel, M. (2018). Mycobacteriophage lysis enzymes: targeting the mycobacterial cell envelope. Viruses 10:E428. doi: 10.3390/v10080428
Chen, J., Zhang, S., Cui, P., Shi, W., Zhang, W., and Zhang, Y. (2017). Identification of novel mutations associated with cycloserine resistance in Mycobacterium tuberculosis. J. Antimicrob. Chemother. 72, 3272–3276. doi: 10.1093/jac/dkx316
Chiaradia, L., Lefebvre, C., Parra, J., Marcoux, J., Burlet-Schiltz, O., Etienne, G., et al. (2017). Dissecting the mycobacterial cell envelope and defining the composition of the native mycomembrane. Sci. Rep. 7:12807. doi: 10.1038/s41598-017-12718-4
Cho, H., Uehara, T., and Bernhardt, T. G. (2014). Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery. Cell 4, 1300–1311. doi: 10.1016/j.cell.2014.11.017
Cordillot, M., Dubée, V., Triboulet, S., Dubost, L., Marie, A., Hugonnet, J. E., et al. (2013). In vitro cross-linking of Mycobacterium tuberculosis peptidoglycan by L,D-transpeptidases and inactivation of these enzymes by carbapenems. Antimicrob. Agents Chemother. 57, 5940–5945. doi: 10.1128/AAC.01663-13
Coulombe, F., Divangahi, M., Veyrier, F., de Léséleuc, L., Gleason, J. L., Yang, Y., et al. (2009). Increased NOD2-mediated recognition of N-glycolyl muramyl dipeptide. J. Exp. Med. 206, 1709–1716. doi: 10.1084/jem.20081779
Davies Forsman, L., Giske, C. G., Bruchfeld, J., Schön, T., Juréen, P., and Ängeby, K. (2015). Meropenem-clavulanic acid has high in vitro activity against multidrug-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 59, 3630–3632. doi: 10.1128/AAC.00171-15
Desjardins, C. A., Cohen, K. A., Munsamy, V., Abeel, T., Maharaj, K., Walker, B. J., et al. (2016). Genomic and functional analyses of Mycobacterium tuberculosis strains implicate ald in D-cycloserine resistance. Nat. Genet. 48, 544–551. doi: 10.1038/ng.3548
Diacon, A. H., van der Merwe, L., Barnard, M., von Groote-Bidlingmaier, F., Lange, C., García-Basteiro, A. L., et al. (2016). β-Lactams against tuberculosis-new trick for an old dog? N. Engl. J. Med. 375, 393–394. doi: 10.1056/NEJMc1513236
Dini, C. (2005). MraY inhibitors as novel antibacterial agents. Curr. Top. Med. Chem. 5, 1221–1236. doi: 10.2174/156802605774463042
Dubée, V., Triboulet, S., Mainardi, J. L., Ethève-Quelquejeu, M., Gutmann, L., Marie, A., et al. (2012). Inactivation of Mycobacterium tuberculosis L,D-transpeptidase LdtMt₁ by carbapenems and cephalosporins. Antimicrob. Agents Chemother. 56, 4189–4195. doi: 10.1128/AAC.00665-12
Eldholm, V., and Balloux, F. (2016). Antimicrobial resistance in Mycobacterium tuberculosis: the odd one out. Trends Microbiol. 24, 637–648. doi: 10.1016/j.tim.2016.03.007
Emami, K., Guyet, A., Kawai, Y., Devi, J., Wu, L. J., Allenby, N., et al. (2017). RodA as the missing glycosyltransferase in Bacillus subtilis and antibiotic discovery for the peptidoglycan polymerase pathway. Nat. Microbiol. 2:16253. doi: 10.1038/nmicrobiol.2016.253
Erdemli, S. B., Gupta, R., Bishai, W. R., Lamichhane, G., Amzel, L. M., and Bianchet, M. A. (2012). Targeting the cell wall of Mycobacterium tuberculosis: structure and mechanism of L,D-transpeptidase 2. Structure 20, 2103–2115. doi: 10.1016/j.str.2012.09.016
Fang, X., Tiyanont, K., Zhang, Y., Wanner, J., Boger, D., and Walker, S. (2006). The mechanism of action of ramoplanin and enduracidin. Mol. BioSyst. 2, 69–76. doi: 10.1039/b515328j
Flores, A. R., Parsons, L. M., and Pavelka, M. S. Jr. (2005). Characterization of novel Mycobacterium tuberculosis and Mycobacterium smegmatis mutants hypersusceptible to beta-lactam antibiotics. J. Bacteriol. 187, 1892–1900. doi: 10.1128/JB.187.6.1892-1900.2005
Gigante, A. M., Hampton, C. H., Dillard, R. S., Gil, F., Catalão, M. J., Moniz-Pereira, J., et al. (2017). The Ms6 Mycolyl-Arabinogalactan Esterase LysB is Essential for an Efficient Mycobacteriophage-Induced Lysis. Viruses 9:E343. doi: 10.3390/v9110343
García-Heredia, A., Pohane, A. A., Melzer, E. S., Carr, C. R., Fiolek, T. J., Rundell, S. R., et al. (2018). Peptidoglycan precursor synthesis along the sidewall of pole-growing mycobacteria. elife 7:e37243. doi: 10.7554/eLife.37243
Gil, F., Catalão, M. J., Moniz-Pereira, J., Leandro, P., McNeil, M., and Pimentel, M. (2008). The lytic cassette of mycobacteriophage Ms6 encodes an enzyme with lipolytic activity. Microbiology 154, 1364–1371. doi: 10.1099/mic.0.2007/014621-0
Gil, F., Grzegorzewicz, A. E., Catalão, M. J., Vital, J., McNeil, M. R., and Pimentel, M. (2010). Mycobacteriophage Ms6 LysB specifically targets the outer membrane of Mycobacterium smegmatis. Microbiology 156, 1497–1504. doi: 10.1099/mic.0.032821-0
Gonzalo, X., and Drobniewski, F. (2013). Is there a place for β-lactams in the treatment of multidrug-resistant/extensively drug-resistant tuberculosis? Synergy between meropenem and amoxicillin/clavulanate. J. Antimicrob. Chemother. 68, 366–369. doi: 10.1093/jac/dks395
Grover, N., Paskaleva, E. E., Mehta, K. K., Dordick, J. S., and Kane, R. S. (2014). Growth inhibition of Mycobacterium smegmatis by mycobacteriophage-derived enzymes. Enzym. Microb. Technol. 63, 1–6. doi: 10.1016/j.enzmictec.2014.04.018
Grzegorzewicz, A. E., de Sousa-d’Auria, C., McNeil, M. R., Huc-Claustre, E., Jones , V., Petit, C., et al. (2016). Assembling of the Mycobacterium tuberculosis cell wall core. J. Biol. Chem. 291, 18867–18879. doi: 10.1074/jbc.M116.739227
Gupta, R., Lavollay, M., Mainardi, J. L., Arthur, M., Bishai, W. R., and Lamichhanem, G. (2010). The Mycobacterium tuberculosis protein LdtMt2 is a nonclassical transpeptidase required for virulence and resistance to amoxicillin. Nat. Med. 16, 466–469. doi: 10.1038/nm.2120
Gygli, S. M., Borrell, S., Trauner, A., and Gagneux, S. (2017). Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectives. FEMS Microbiol. Rev. 41, 354–373. doi: 10.1093/femsre/fux011
Hansen, J. M., Golchin, S. A., Veyrier, F. J., Domenech, P., Boneca, I. G., Azad, A. K., et al. (2014). N-glycolylated peptidoglycan contributes to the immunogenicity but not pathogenicity of Mycobacterium tuberculosis. J. Infect. Dis. 209, 1045–1054. doi: 10.1093/infdis/jit622
Harrison, J., Lloyd, G., Joe, M., Lowary, T. L., Reynolds, E., Walters-Morgan, H., et al. (2016). Lcp1 is a phosphotransferase responsible for ligating arabinogalactan to peptidoglycan in Mycobacterium tuberculosis. MBio 7:e00972–16. doi: 10.1128/mBio.00972-16
Hatfull, G. F., Pedulla, M. L., Jacobs-Sera, D., Cichon, P. M., Foley, A., Ford, M. E., et al. (2006). Exploring the mycobacteriophage metaproteome: phage genomics as an educational platform. PLoS Genet. 2:e92. doi: 10.1371/journal.pgen.0020092
Hoffmann, C., Leis, A., Niederweis, M., Plitzko, J. M., and Engelhardt, H. (2008). Disclosure of the mycobacterial outer membrane: cryo-electron tomography and vitreous sections reveal the lipid bilayer structure. Proc. Natl. Acad. Sci. USA 105, 3963–3967. doi: 10.1073/pnas.0709530105
Hong, W., Chen, L., and Xie, J. (2014). Molecular basis underlying Mycobacterium tuberculosis D-cycloserine resistance. Is there a role for ubiquinone and menaquinone metabolic pathways? Expert Opin. Ther. Targets 18, 691–701. doi: 10.1517/14728222.2014.902937
Horsburgh, C. R. Jr, Barry, C. III., and Lange, C. (2015). Treatment of tuberculosis. New Engl. J. Med. 373, 2149–2160. doi: 10.1056/NEJMra1413919
Hrast, M., Sosic, I., Sink, R., and Gobec, S. (2014). Inhibitors of the peptidoglycan biosynthesis enzymes MurA-F. Bioorg. Chem. 55, 2–15. doi: 10.1016/j.bioorg.2014.03.008
Hugonnet, J. E., and Blanchard, J. S. (2007). Irreversible inhibition of the Mycobacterium tuberculosis beta-lactamase by clavulanate. Biochemistry 46, 11998–12004. doi: 10.1021/bi701506h
Hugonnet, J. E., Tremblay, L. W., Boshoff, H. I., Barry, C. E. 3rd., and Blanchard, J. S. (2009). Meropenem-clavulanate is effective against extensively drug-resistant Mycobacterium tuberculosis. Science 323, 1215–1218. doi: 10.1126/science.1167498
Hwang, T. J., Wares, D. F., Jafarov, A., Jakubowiak, W., Nunn, P., and Keshavjee, S. (2013). Safety of cycloserine and terizidone for the treatment of drug-resistant tuberculosis: a meta-analysis. Int. J. Tuberc. Lung Dis. 17, 1257–1266. doi: 10.5588/ijtld.12.0863
Jackson, M., McNeil, M. R., and Brennan, P. J. (2013). Progress in targeting cell envelope biogenesis in Mycobacterium tuberculosis. Future Microbiol. 8, 855–875. doi: 10.2217/fmb.13.52
Jaganath, D., Lamichhane, G., and Shah, M. (2016). Carbapenems against Mycobacterium tuberculosis: a review of the evidence. Int. J. Tuberc. Lung Dis. 20, 1436–1447. doi: 10.5588/ijtld.16.0498
Jankute, M., Cox, J. A., Harrison, J., and Besra, G. S. (2015). Assembly of the mycobacterial cell wall. Annu. Rev. Microbiol. 69, 405–423. doi: 10.1146/annurev-micro-091014-104121
Kana, B. D., Mizrahi, V., and Gordhan, B. G. (2010). Depletion of resuscitation-promoting factors has limited impact on the drug susceptibility of Mycobacterium tuberculosis. J. Antimicrob. Chemother. 65, 1583–1585. doi: 10.1093/jac/dkq199
Kaushik, A., Makkar, N., Pandey, P., Parrish, N., Singh, U., and Lamichhane, G. (2015). Carbapenems and rifampin exhibit synergy against Mycobacterium tuberculosis and Mycobacterium abscessus. Antimicrob. Agents Chemother. 59, 6561–6567. doi: 10.1128/AAC.01158-15
Kaushik, A., Ammerman, N. C., Tasneen, R., Story-Roller, E., Dooley, K. E., Dorman, S. E., et al. (2017). In vitro and in vivo activity of biapenem against drug-susceptible and rifampicin-resistant Mycobacterium tuberculosis. J. Antimicrob. Chemother. 72, 2320–2325. doi: 10.1093/jac/dkx152
Keener, A. B. (2014). Oldie but goodie: repurposing penicillin for tuberculosis. Nat. Med. 20, 976–978. doi: 10.1038/nm0914-976
Kieser, K. J., Baranowski, C., Chao, M. C., Long, J. E., Sassetti, C. M., Waldor, M. K., et al. (2015). Peptidoglycan synthesis in Mycobacterium tuberculosis is organized into networks with varying drug susceptibility. Proc. Natl. Acad. Sci. USA 112, 13087–13092. doi: 10.1073/pnas.1514135112
Kim, D. H., Lees, W. J., Kempsell, K. E., Lane, W. S., Duncan, K., and Walsh, C. T. (1996). Characterization of a Cys115 to Asp substitution in the Escherichia coli cell wall biosynthetic enzyme UDP-GlcNAc enolpyruvyl transferase (MurA) that confers resistance to inactivation by the antibiotic fosfomycin. Biochemistry 35, 4923–4928. doi: 10.1021/bi952937w
Koga, T., Fukuoka, T., Doi, N., Harasaki, T., Inoue, H., Hotoda, H., et al. (2004). Activity of capuramycin analogues against Mycobacterium tuberculosis, Mycobacterium avium and Mycobacterium intracellulare in vitro and in vivo. J. Antimicrob. Chemother. 54, 755–760. doi: 10.1093/jac/dkh417
Kumar, V., Saravanan, P., Arvind, A., and Mohan, C. G. (2011). Identification of hotspot regions of MurB oxidoreductase enzyme using homology modeling, molecular dynamics and molecular docking techniques. J. Mol. Model 17, 939–953. doi: 10.1007/s00894-010-0788-3
Kumar, P., Arora, K., Lloyd, J. R., Lee, I. Y., Nair, V., Fischer, E., et al. (2012). Meropenem inhibits D,D-carboxypeptidase activity in Mycobacterium tuberculosis. Mol. Microbiol. 86, 367–381. doi: 10.1111/j.1365-2958.2012.08199.x
Kumar, P., Kaushik, A., Lloyd, E. P., Li, S. G., Mattoo, R., Ammerman, N. C., et al. (2017a). Non-classical transpeptidases yield insight into new antibacterials. Nat. Chem. Biol. 13, 54–61. doi: 10.1038/nchembio.2237
Kumar, P., Kaushik, A., Bell, D. T., Chauhan, V., Xia, F., Stevens, R. L., et al. (2017b). Mutation in an unannotated protein confers carbapenem resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 61:e02234–16. doi: 10.1128/AAC.02234-16
Lai, M. J., Liu, C. C., Jiang, S. J., Soo, P. C., Tu, M. H., and Lee, J. J. (2015). Antimycobacterial activities of endolysins derived from a mycobacteriophage, BTCU-1. Molecules 20, 19277–19290. doi: 10.3390/molecules201019277
Lavollay, M., Arthur, M., Fourgeaud, M., Dubost, L., Marie, A., Veziris, N., et al. (2008). The peptidoglycan of stationary-phase Mycobacterium tuberculosis predominantly contains cross-links generated by L,D-transpeptidation. J. Bacteriol. 190, 4360–4366. doi: 10.1128/JB.00239-08
Li, Y., Zhou, Y., Ma, Y., and Li, X. (2011). Design and synthesis of novel cell wall inhibitors of Mycobacterium tuberculosis GlmM and GlmU. Carbohydr. Res. 346, 1714–1720. doi: 10.1016/j.carres.2011.05.024
Ling, L. L., Schneider, T., Peoples, A. J., Spoering, A. L., Engels, I., Conlon, B. P., et al. (2015). A new antibiotic kills pathogens without detectable resistance. Nature 517, 455–459. doi: 10.1038/nature14098
Lingaraju, S., Rigouts, L., Gupta, A., Lee, J., Umubyeyi, A. N., Davidow, A. L., et al. (2016). Geographic differences in the contribution of ubiA mutations to high-level ethambutol resistance in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 60, 4101–4105. doi: 10.1128/AAC.03002-15
Lun, S., Miranda, D., Kubler, A., Guo, H., Maiga, M. C., Winglee, K., et al. (2014). Synthetic lethality reveals mechanisms of Mycobacterium tuberculosis resistance to β-lactams. MBio 5:e01767–14. doi: 10.1128/mBio.01767-14
Mahapatra, S., Crick, D. C., and Brennan, P. J. (2000). Comparison of the UDP-N-acetylmuramate-L-alanine ligase enzymes from Mycobacterium tuberculosis and Mycobacterium leprae. J. Bacteriol. 182, 6827–6830. doi: 10.1128/JB.182.23.6827-6830.2000
Mahapatra, S., Scherman, H., Brennan, P. J., and Crick, D. C. (2005). N-glycolylation of the nucleotide precursors of peptidoglycan biosynthesis of Mycobacterium spp. is altered by drug treatment. J. Bacteriol. 187, 2341–2347. doi: 10.1128/JB.187.7.2341-2347.2005
Mainardi, J., Hugonnet, J., Gutmann, L., and Arthur, M. (2011). Fighting resistant tuberculosis with old compounds: the carbapenem paradigm. Clin. Microbiol. Infect. 17, 1755–1756. doi: 10.1111/j.1469-0691.2011.03699.x
McNeil, M., Daffe, M., and Brennan, P. J. (1990). Evidence for the nature of the link between the arabinogalactan and peptidoglycan of mycobacterial cell walls. J. Biol. Chem. 265, 18200–18206.
Meeske, A. J., Riley, E. P., Robins, W. P., Uehara, T., Mekalanos, J. J., Kahne, D., et al. (2016). SEDS proteins are a widespread family of bacterial cell wall polymerases. Nature 537, 634–638. doi: 10.1038/nature19331
Mohammadi, T., van Dam, V., Sijbrandi, R., Vernet, T., Zapun, A., Bouhss, A., et al. (2011). Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 30, 1425–1432. doi: 10.1038/emboj.2011.61
Moraes, G. L., Gomes, G. C., Monteiro de Sousa, P. R., Alves, C. N., Govender, T., Kruger, H. G., et al. (2015). Structural and functional features of enzymes of Mycobacterium tuberculosis peptidoglycan biosynthesis as targets for drug development. Tuberculosis 95, 95–111. doi: 10.1016/j.tube.2015.01.006
Morris, R. P., Nguyen, L., Gatfield, J., Visconti, K., Nguyen, K., Schnappinger, D., et al. (2005). Ancestral antibiotic resistance in Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. USA 102, 12200–12205. doi: 10.1073/pnas.0505446102
Nasiri, M. J., Haeili, M., Ghazi, M., Goudarzi, H., Pormohammad, A., Imani Fooladi, A. A., et al. (2017). New insights in to the intrinsic and acquired drug resistance mechanisms in mycobacteria. Front. Microbiol. 8:681. doi: 10.3389/fmicb.2017.00681
Nakatani, Y., Opel-Reading, H. K., Merker, M., Machado, D., Andres, S., Kumar, S. S., et al. (2017). Role of alanine racemase mutations in Mycobacterium tuberculosis D-cycloserine resistance. Antimicrob. Agents Chemother. 61, e01575–e01517. doi: 10.1128/AAC.01575-17
Nikaido, H. (1994). Prevention of drug access to bacterial targets: permeability barriers and active efflux. Science 264, 382–388. doi: 10.1126/science.8153625
Nikonenko, B. V., Reddy, V. M., Protopopova, M., Bogatcheva, E., Einck, L., and Nacy, C. A. (2009). Activity of SQ641, a capuramycin analog, in a murine model of tuberculosis. Antimicrob. Agents Chemother. 53, 3138–3139. doi: 10.1128/AAC.00366-09
Pavelka, M. S. Jr., Mahapatra, S., and Crick, D. C. (2014). Genetics of peptidoglycan biosynthesis. Microbiol Spectr. 2:MGM2-0034-2013. doi: 10.1128/microbiolspec.MGM2-0034-2013
Payen, M. C., De Wit, S., Martin, C., Sergysels, R., Muylle, I., Van Laethem, Y., et al. (2012). Clinical use of the meropenem-clavulanate combination for extensively drug-resistant tuberculosis. Int. J. Tuberc. Lung Dis. 16, 558–560. doi: 10.5588/ijtld.11.0414
Payen, M. C., Muylle, I., Vandenberg, O., Mathys, V., Delforge, M., Van den Wijngaert, S., et al. (2018). Meropenem-clavulanate for drug-resistant tuberculosis: a follow-up of relapse-free cases. Int. J. Tuberc. Lung Dis. 22, 34–39. doi: 10.5588/ijtld.17.0352
Payne, K., Sun, Q., Sacchettini, J., and Hatfull, G. F. (2009). Mycobacteriophage Lysin B is a novel mycolylarabinogalactan esterase. Mol. Microbiol. 73, 367–381. doi: 10.1111/j.1365-2958.2009.06775.x
Payne, K. M., and Hatfull, G. F. (2012). Mycobacteriophage endolysins: diverse and modular enzymes with multiple catalytic activities. PLoS One 7:e34052. doi: 10.1371/journal.pone.0034052
Peneff, C., Ferrari, P., Charrier, V., Taburet, Y., Monnier, C., Zamboni, V., et al. (2001). Crystal structures of two human pyrophosphorylase isoforms in complexes with UDPGlc(Gal)NAc: role of the alternatively spliced insert in the enzyme oligomeric assembly and active site architecture. EMBO J. 20, 6191–6202. doi: 10.1093/emboj/20.22.6191
Pimentel, M. (2014). Genetics of phage lysis. Microbiol. Spectr. 2, 1–13. doi: 10.1128/microbiolspec.MGM2-0017-2013
Portevin, D., Gagneux, S., Comas, I., and Young, D. (2011). Human macrophage responses to clinical isolates from the Mycobacterium tuberculosis complex discriminate between ancient and modern lineages. PLoS Pathog. 7:e1001307. doi: 10.1371/journal.ppat.1001307
Prosser, G. A., and de Carvalho, L. P. (2013a). Metabolomics reveal D-alanine-D-alanine ligase as the target of D-cycloserine in Mycobacterium tuberculosis. ACS Med. Chem. Lett. 4, 1233–1237. doi: 10.1021/ml400349n
Prosser, G. A., and de Carvalho, L. P. (2013b). Reinterpreting the mechanism of inhibition of Mycobacterium tuberculosis D-alanine-D-alanine ligase by D-cycloserine. Biochemistry 52, 7145–7149. doi: 10.1021/bi400839f
Prosser, G. A., and de Carvalho, L. P. (2013c). Kinetic mechanism and inhibition of Mycobacterium tuberculosis D-alanine-D-alanine ligase by the antibiotic D-cycloserine. FEBS J. 280, 1150–1166. doi: 10.1111/febs.12108
Rana, A. M., Trivedi, P., Desai, K. R., and Jauhari, S. (2014). Novel S-triazine accommodated 5-benzylidino-4-thiazolidinones: synthesis and in vitro biological evaluations. Med. Chem. Res. 23, 4320–4336. doi: 10.1007/s00044-014-0995-z
Rani, C., Mehra, R., Sharma, R., Chib, R., Wazir, P., Nargotra, A., et al. (2015). High-throughput screen identifies small molecule inhibitors targeting acetyltransferase activity of Mycobacterium tuberculosis GlmU. Tuberculosis 95, 664–677. doi: 10.1016/j.tube.2015.06.003
Rani, C., and Khan, I. A. (2016). UDP-GlcNAc pathway: potential target for inhibitor discovery against Mycobacterium tuberculosis. Eur. J. Pharm. Sci. 83, 62–70. doi: 10.1016/j.ejps.2015.12.013
Raymond, J. B., Mahapatra, S., Crick, D. C., and Pavelka, M. S. Jr. (2005). Identification of the namH gene, encoding the hydroxylase responsible for the N-glycolylation of the mycobacterial peptidoglycan. J. Biol. Chem. 280, 326–333. doi: 10.1074/jbc.M411006200
Reddy, V. M., Einck, L., and Nacy, C. A. (2008). In vitro antimycobacterial activities of capuramycin analogues. Antimicrob. Agents Chemother. 52, 719–721. doi: 10.1128/AAC.01469-07
Reynolds, P. E. (1989). Structure, biochemistry and mechanism of action of glycopeptide antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 8, 943–950. doi: 10.1007/BF01967563
Rullas, J., Dhar, N., McKinney, J. D., Garcia-Perez, A., Lelievre, J., Diacon, A. H., et al. (2015). Combinations of beta-lactam antibiotics–currently in clinical trials are efficacious in a DHP-I-deficient mouse model of tuberculosis infection. Antimicrob. Agents Chemother. 59, 4997–4999. doi: 10.1128/AAC.01063-15
Schenk, M., Mahapatra, S., Le, P., Kim, H. J., Choi, A. W., Brennan, P. J., et al. (2016). Human NOD2 recognizes structurally unique muramyl dipeptides from Mycobacterium leprae. Infect. Immun. 84, 2429–2438. doi: 10.1128/IAI.00334-16
Schoonmaker, M. K., Bishai, W. R., and Lamichhane, G. (2014). Nonclassical transpeptidases of Mycobacterium tuberculosis alter cell size, morphology, the cytosolic matrix, protein localization, virulence, and resistance to β-lactams. J. Bacteriol. 196, 1394–1402. doi: 10.1128/JB.01396-13
Schubert, K., Sieger, B., Meyer, F., Giacomelli, G., Böhm, K., Rieblinger, A., et al. (2017). The antituberculosis drug ethambutol selectively blocks apical growth in CMN group bacteria. MBio 8:e02213–16. doi: 10.1128/mBio.02213-16
Siricilla, S., Mitachi, K., Wan, B., Franzblau, S. G., and Kurosu, M. (2015). Discovery of a capuramycin analog that kills non-replicating Mycobacterium tuberculosis and its synergistic effects with translocase I inhibitors. J. Antibiot. 68, 271–278. doi: 10.1038/ja.2014.133
Solapure, S., Dinesh, N., Shandil, R., Ramachandran, V., Sharma, S., Bhattacharjee, D., et al. (2013). In vitro and in vivo efficacy of β-lactams against replicating and slowly growing/non-replicating Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 57, 2506–2510. doi: 10.1128/AAC.00023-13
Stanley, S. A., and Cox, J. S. (2013). Host-pathogen interactions during Mycobacterium tuberculosis infections. Curr. Top. Microbiol. Immunol. 374, 211–241. doi: 10.1007/82_2013_332
Tiberi, S., Payen, M. C., Sotgiu, G., D’Ambrosio, L., Alarcon Guizado, V., Alffenaar, J. W., et al. (2016). Effectiveness and safety of meropenem/clavulanate-containing regimens in the treatment of MDR- and XDR-TB. Eur. Respir. J. 47, 1235–1243. doi: 10.1183/13993003.02146-2015
Tomasic, T., Zidar, N., Kovac, A., Turk, S., Simcic, M., Blanot, D., et al. (2010). 5-Benzylidenethiazolidin-4-ones as multi-target inhibitors of bacterial Mur ligases. Chem. Med. Chem. 5, 286–295. doi: 10.1002/cmdc.200900449
Tran, A. T., Wen, D., West, N. P., Baker, E. N., Britton, W. J., and Payne, R. J. (2013). Inhibition studies on Mycobacterium tuberculosis N-acetylglucosamine-1-phosphate uridyltransferase (GlmU). Org. Biomol. Chem. 11, 8113–8126. doi: 10.1039/c3ob41896k
Tran, A. T., Watson, E. E., Pujari, V., Conroy, T., Dowman, L. J., Giltrap, A. M., et al. (2017). Sansanmycin natural product analogues as potent and selective anti-mycobacterials that inhibit lipid I biosynthesis. Nat. Commun. 8:14414. doi: 10.1038/ncomms14414
Tremblay, L. W., Fan, F., and Blanchard, J. S. (2010). Biochemical and structural characterization of Mycobacterium tuberculosis β-lactamase with the carbapenems ertapenem and doripenem. Biochemistry 49, 3766–3773. doi: 10.1021/bi100232q
Trunkfield, A. E., Gurcha, S. S., Besra, G. S., and Bugg, T. D. (2010). Inhibition of Escherichia coli glycosyltransferase MurG and Mycobacterium tuberculosis Gal transferase by uridine-linked transition state mimics. Bioorg. Med. Chem. 18, 2651–2663. doi: 10.1016/j.bmc.2010.02.026
van Heijenoort, Y., Leduc, M., Singer, H., and van Heijenoort, J. (1987). Effects of moenomycin on Escherichia coli. J. Gen. Microbiol. 133, 667–674. doi: 10.1099/00221287-133-3-667
Vollmer, W., Blanot, D., and de Pedro, M. A. (2008). Peptidoglycan structure and architecture. FEMS Microbiol. Rev. 32, 149–167. doi: 10.1111/j.1574-6976.2007.00094.x
Wang, F., Cassidy, C., and Sacchettini, J. C. (2006). Crystal structure and activity studies of the Mycobacterium tuberculosis beta-lactamase reveal its critical role in resistance to beta-lactam antibiotics. Antimicrob. Agents Chemother. 50, 2762–2771. doi: 10.1128/AAC.00320-06
Wiedemann, I., Breukink, E., van Kraaij, C., Kuipers, O. P., Bierbaum, G., de Kruijff, B., et al. (2001). Specific binding of nisin to the peptidoglycan precursor lipid II combines pore formation and inhibition of cell wall biosynthesis for potent antibiotic activity. J. Biol. Chem. 276, 1772–1779. doi: 10.1074/jbc.M006770200
Wiegmann, D., Koppermann, S., Wirth, M., Niro, G., Leyerer, K., and Ducho, C. (2016). Muraymycin nucleoside peptide antibiotics: uridine derived natural products as lead structures for the development of novel antibacterial agents. Beilstein J. Org. Chem. 12, 769–795. doi: 10.3762/bjoc.12.77
Wivagg, C. N., and Hung, D. T. (2012). Resuscitation-promoting factors are required for β-lactam tolerance and the permeability barrier in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 56, 1591–1594. doi: 10.1128/AAC.06027-11
Wivagg, C. N., Bhattacharyya, R. P., and Hung, D. T. (2014). Mechanisms of β-lactam killing and resistance in the context of Mycobacterium tuberculosis. J. Antibiot. 67, 645–654. doi: 10.1038/ja.2014.94
Wivagg, C. N., Wellington, S., Gomez, J. E., and Hung, D. T. (2016). Loss of a class A penicillin-binding protein alters β-lactam susceptibilities in Mycobacterium tuberculosis. ACS Infect. Dis. 2, 104–110. doi: 10.1021/acsinfecdis.5b00119
World Health Organization. (2011). Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis-2011 Update. Geneva.
World Health Organization. (2014). Companion Handbook to the WHO Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis. Geneva.
Wong, E. B., Cohen, K. A., and Bishai, W. R. (2013). Rising to the challenge: new therapies for tuberculosis. Trends Microbiol. 21, 493–501. doi: 10.1016/j.tim.2013.05.002
Zapun, A., Contreras-Martel, C., and Vernet, T. (2008). Penicillin-binding proteins and beta-lactam resistance. FEMS Microbiol. Rev. 32, 361–385. doi: 10.1111/j.1574-6976.2007.00095.x
Keywords: mycobacteria, cell wall, tuberculosis, antibiotic resistance, peptidoglycan, β-lactams, mycobacteriophage lysis enzymes
Citation: Catalão MJ, Filipe SR and Pimentel M (2019) Revisiting Anti-tuberculosis Therapeutic Strategies That Target the Peptidoglycan Structure and Synthesis. Front. Microbiol. 10:190. doi: 10.3389/fmicb.2019.00190
Edited by:
Christoph Mayer, University of Tübingen, GermanyReviewed by:
Frédéric J. Veyrier, Institut National de la Recherche Scientifique (INRS), CanadaPatrick Joseph Moynihan, University of Birmingham, United Kingdom
Nicolas Gisch, Forschungszentrum Borstel (LG), Germany
Copyright © 2019 Catalão, Filipe and Pimentel. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria João Catalão, bWpjYXRhbGFvQGZmLnVsaXNib2EucHQ=