 Eugenio Mancera1*
Eugenio Mancera1* Corey Frazer2
Corey Frazer2 Allison M. Porman2Susana Ruiz-Castro1Alexander D. Johnson3*
Allison M. Porman2Susana Ruiz-Castro1Alexander D. Johnson3* Richard J. Bennett2*
Richard J. Bennett2*- 1Departamento de Ingeniería Genética, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, Unidad Irapuato, Irapuato, Mexico
- 2Department of Molecular Microbiology and Immunology, Brown University, Providence, RI, United States
- 3Department of Microbiology and Immunology, University of California, San Francisco, San Francisco, CA, United States
Species from the genus Candida are among the most important human fungal pathogens. Several of them are frequent commensals of the human microbiota but are also able to cause a variety of opportunistic infections, especially when the human host becomes immunocompromised. By far, most of the research to understand the molecular underpinnings of the pathogenesis of these species has focused on Candida albicans, the most virulent member of the genus. However, epidemiological data indicates that related Candida species are also clinically important. Here, we describe the generation of a set of strains and plasmids to genetically modify C. dubliniensis and C. tropicalis, the two pathogenic species most closely related to C. albicans. C. dubliniensis is an ideal model to understand C. albicans pathogenesis since it is the closest species to C. albicans but considerably less virulent. On the other hand, C. tropicalis is ranked among the four most common causes of infections by Candida species. Given that C. dubliniensis and C. tropicalis are obligate diploids with no known conventional sexual cycle, we generated strains that are auxotrophic for at least two amino acids which allows the tandem deletion of both alleles of a gene by complementing the two auxotrophies. The strains were generated in two different genetic backgrounds for each species — one for which the genomic sequence is available and a second clinically important one. In addition, we have adapted plasmids developed to delete genes and epitope/fluorophore tag proteins in C. albicans so that they can be employed in C. tropicalis. The tools generated here allow for efficient genetic modification of C. dubliniensis and C. tropicalis, and thus facilitate the study of the molecular basis of pathogenesis in these medically relevant fungi.
Introduction
Fungi from the genus Candida are a heterogeneous group of ascomycete yeasts. Although the human infections caused by Candida species (candidiasis) have been a subject of study since ancient Greece, it was not until 1923 that the name Candida albicans was proposed for the first member of the genus (Lynch, 1994). Nowadays, the genus comprises more than 160 species that were grouped in part because no clearly defined sexual cycle was identified (Turner and Butler, 2014). Therefore, the genus is polyphyletic and quite diverse. In addition, due to recent improvements and standardization in fungal taxonomy, many Candida species are being renamed (Gabaldon et al., 2016). More than 30 species of Candida have been identified as the causative agent of candidiasis. However, around 95% of the infections are caused by only four species: C. albicans, C. glabrata, C. parapsilosis, and C. tropicalis (Turner and Butler, 2014; Gabaldon et al., 2016). These species, except for C. glabrata, belong to a clade whose members translate the CTG codon as serine instead of leucine (Figure 1) (Butler et al., 2009). Although this clade has been traditionally known as the “CTG clade,” recent findings have shown that there are related clades of ascomycetes that also translate the CTG codon in non-standard ways (Krassowski et al., 2018). In addition to the three CTG clade species mentioned above, the CTG clade comprises other species that are rarer etiological disease agents such as Candida dubliniensis, C. guilliermondii, C. lusitaniae, and C. auris, with the latter associated with multidrug resistant infections (Moran et al., 2011; Gabaldon et al., 2016; Sears and Schwartz, 2017). Overall, the CTG clade encompasses most of the pathogenic species of Candida.
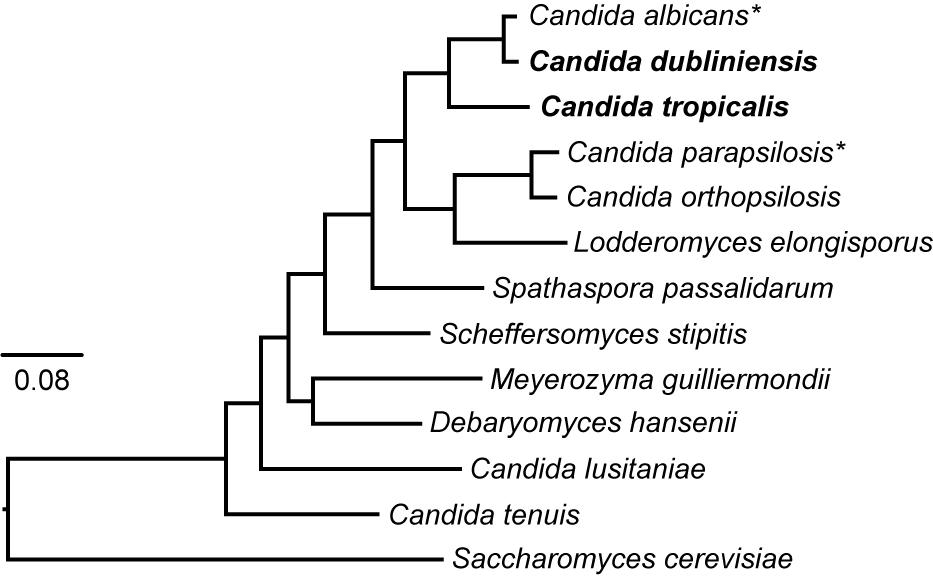
Figure 1. Phylogeny of the CTG clade. The phylogenetic relationships of the species belonging to the clade that has been traditionally known as the “CTG clade” and whose genome has been sequenced is shown. There are related ascomycete clades that also translate the CTG codon in alternative ways and it has therefore been proposed to name the clade CUG-Ser1 (Krassowski et al., 2018). The tree was generated by the Candida Gene Order Browser (CGOB) from 100,000 informative amino acid positions shared by all species as described (Maguire et al., 2013). Candida dubliniensis and C. tropicalis are shown in bold and asterisks denote species for which there are large collections of gene-deletion mutants. The tree was rooted using the budding yeast (Saccharomyces cerevisiae) which is a sister species of the clade. C. glabrata is not shown in the tree, but it is more closely related to S. cerevisiae than to CTG species.
Pathogenic species of Candida are frequent commensals of the human microbiota. Some estimates suggest that as much as 70% of the adult human population is a carrier of some kind of yeast in the gastrointestinal tract, most often Candida species (Schulze and Sonnenborn, 2009). These species are able to asymptomatically colonize many human tissues (Lynch, 1994) with the gastrointestinal and genitourinary tracts of healthy individuals being especially common niches. However, under specific circumstances such as imbalances of the immune system or the local microbiota, Candida species are able to cause a variety of diseases, from superficial mucosal infections to life-threatening systemic conditions. In cases of hematogenously disseminated candidiasis, the mortality rates are as high as 40% (Nobile and Johnson, 2015). The incidence of diseases caused by Candida species has increased since the 1980s, especially of nosocomial infections. Although such an increase could partially be explained by better detection methods, it is also attributed to advances in medical practices. Therapies that alter the immune system, that allow survival of immunocompromised patients, or that involve the implantation of medical devices have broadened the impact of candidiasis in the human population (Gabaldon et al., 2016).
The frequency of infections caused by each Candida species varies depending on the geographical region, although C. albicans is consistently the most frequent cause of candidiasis being associated with more than 70% of cases in some regions (Turner and Butler, 2014). For this reason, most of the research to understand the molecular mechanisms responsible for the pathogenesis of these species has focused on C. albicans. Over the years, several strategies have been developed to genetically modify C. albicans. The most common strategy relies on homologous recombination and involves parental strains with two or more amino acid auxotrophies. The two alleles of a gene can thus be deleted in tandem by performing transformations with the two corresponding nutritional markers (Noble and Johnson, 2005). This strategy overcomes the intrinsic difficulties of genetically modifying this species given that it is diploid with no known conventional sexual cycle. Using such a strategy, several collections of gene deletion mutants have been generated in C. albicans (Homann et al., 2009; Noble et al., 2010). Approaches that employ counterselectable or recyclable markers such as the SAT1 flipper system have also been instrumental for disrupting genes in C. albicans, especially in clinical isolates for which no auxotrophic strains have been generated (Wilson et al., 2000; Reuß et al., 2004). These methods are more time consuming since they involve an extra experimental step to remove the marker before the second allele can be targeted for deletion.
More recently, a number of approaches to genetically modify C. albicans using CRISPR-mediated systems have been developed (Vyas et al., 2015; Min et al., 2016; Grahl et al., 2017; Huang and Mitchell, 2017; Ng and Dean, 2017; Nguyen et al., 2017). While promising, CRISPR-mediated systems can also benefit from using auxotrophic strains that have previously been generated. For example, a recent CRISPR-Cas9 system developed for C. albicans involves insertion of a cassette at the LEU2 locus, which results in an auxotrophic strain when utilizing a heterozygous LEU2/Δleu2 strain. After the target locus has been modified by CRISPR, restoration of the ability to grow in medium lacking leucine is used to select for cells that have lost the CRISPR-Cas9 cassette due to recombination at the LEU2 locus (Nguyen et al., 2017). Transposon-mediated genetic modifications have also been used to disrupt genes in C. albicans to study gene essentiality in bulk (Uhl et al., 2003; Segal et al., 2018). Overall, the use of auxotrophic strains with their corresponding nutritional markers has been the strategy of choice to generate large collections of mutants, and is still the method most routinely used to generate gene-knockout strains in C. albicans.
Recent international surveys have asserted the medical importance of other species aside from C. albicans, especially in some geographic regions. For example, the incidence of C. glabrata and C. parapsilosis is particularly high in Europe and the Americas, respectively. In addition, these surveys have suggested that the incidence of these other species relative to C. albicans is increasing over time (Turner and Butler, 2014). Although this observation can be influenced by improvements in the methods to distinguish different Candida species, it could also be associated with the widespread use of antifungal drugs and the increased drug resistance of these species compared to C. albicans (Turner and Butler, 2014; Gabaldon et al., 2016). Given the interest in C. glabrata and C. parapsilosis, collections of gene deletion mutants have been generated using similar genetic approaches to those used for C. albicans (Holland et al., 2014; Schwarzmuller et al., 2014).
Candida tropicalis is one of the four most important causes of candidiasis worldwide. Its incidence is especially high in Africa and the Middle East and several strains are naturally resistant to fluconazole, the most commonly used antifungal drug (Turner and Butler, 2014; Gabaldon et al., 2016). A number of groups have now reported the generation of gene deletion mutants in this diploid species. Common strategies include the use of dual auxotrophic strains with corresponding genetic markers as in C. albicans, or a combination of single amino acid auxotrophic strains and drug resistance markers to delete the two alleles of a gene (Mancera et al., 2015; Anderson et al., 2016; Zhang et al., 2016; Zheng et al., 2017). In addition, strategies using counterselectable or recyclable markers have also been used to disrupt genes in C. tropicalis, especially when auxotrophic strains are not available for the isolate of interest (Mancera et al., 2015; Anderson et al., 2016; Wang et al., 2018). However, no large collections of gene-deletion strains are available. A variety of genetic backgrounds have been used in C. tropicalis and the genome sequence for only one of them is available to date.
In addition to the three CTG species mentioned above, C. dubliniensis has drawn considerable attention in recent years. Even though the incidence of this species as a cause of candidiasis is low, phylogenetically it is the closest known species to C. albicans (Sullivan et al., 2005) to the extent that they are able to mate with one another (Pujol et al., 2004). It has been estimated that these species diverged from a common ancestor around 20 million years ago and past epidemiological surveys may have often mistaken C. dubliniensis as C. albicans (Moran et al., 2012). Due to its evolutionary proximity but attenuated virulence, C. dubliniensis is being used as a model to identify the species-specific genetic determinants that underlie C. albicans pathogenicity. C. dubliniensis genes have been targeted using reusable drug-resistant markers developed for C. albicans as well as the URA3 counterselectable marker (Staib et al., 2001). However, the use of URA3 as a genetic marker is known to affect virulence and chromosomal stability in C. albicans and so is now avoided where possible (Staab and Sundstrom, 2003; Noble and Johnson, 2005).
In this work, we describe a set of strains and plasmids that allow for the generation of gene deletion mutants in C. dubliniensis and C. tropicalis, the two species most closely related to C. albicans (Figure 1). The strains are auxotrophic for different combinations of histidine, leucine and arginine. These three amino acid biosynthetic genes have been shown to be dispensable for C. albicans in the tail vein mouse model of disseminated infection and in the gastrointestinal mouse model of commensalism (Noble and Johnson, 2005; Pande et al., 2013). Gene deletion cassettes carrying the missing genes can be easily generated by fusion PCR. This is done from a set of plasmids that contain the C. albicans HIS1, LEU2, and ARG4 cloned genes and which complement the auxotrophies of the strains. Since these markers are not the C. dubliniensis or C. tropicalis genes, the probability of insertion into the endogenous loci is diminished, thereby increasing the efficiency of recombination at the intended gene target. The auxotrophic strains have been generated in two different genetic backgrounds for each species. For both species, one of the backgrounds is the one whose genome has been sequenced to date. The other background has been used for molecular studies in these species including those by our groups. In addition, we describe modification of the SAT1 flipper system developed for C. albicans so that it can be more efficiently used in C. tropicalis. Similarly, we have adapted C. albicans vectors to fuse genes with an epitope tag or a fluorescent protein for use in C. tropicalis. Overall, the strategies described here allow for the rapid construction of targeted gene deletions in C. dubliniensis and C. tropicalis, as well as other common genetic manipulations. This will facilitate the generation of collections of strains to better understand the molecular basis of pathogenesis in these medically important fungi.
Materials and Methods
Generation of Auxotrophic Strains
Auxotrophic strains were generated using the SAT1-flipping strategy (Sasse and Morschhauser, 2012). For the deletion of amino acid biosynthetic genes in C. dubliniensis, the SAT1 reusable cassette was PCR amplified from the plasmid pSFS2A with primers containing ∼65 bp of sequence identical to the 5′ and 3′ regions immediately up and downstream of the HIS1, LEU2, and ARG4 open reading frames (ORFs; primer pairs EMO001/EMO002, EMO003/EMO004 and EMO005/EMO006, respectively; Supplementary Table S1). pSFS2A is a plasmid derived from pSFS2 (Reuß et al., 2004) that contains the SAT1 reusable cassette in the backbone of vector pBC SK+ instead of pBluescript II KS and that was kindly provided by Joachim Morschhauser (U. Würzburg). The cassettes were transformed into the CD36 and Wü284 wild type C. dubliniensis strains by electroporation as previously described (Kohler et al., 1997; Staib et al., 2001). For selection of transformants, cells were grown on YEPD medium plates containing 400 μg/ml of nourseothricin (NAT) for 48 h at 30°C. Correct integration of the SAT1 cassette was confirmed by colony PCR of the 5′ and 3′ junctions. For the excision of the SAT1 cassette cells were grown overnight at 30°C in YEP medium containing 2% maltose and then plated on YEPD plates containing 50 μg/ml NAT. After 24 h of growth at 30°C, small colonies were selected and further verified for the loss of NAT resistance by growth on YEPD + 400 μg/ml NAT plates for 48 h at 30°C, as has been described previously (Sasse and Morschhauser, 2012; Porman et al., 2013). Six successive rounds of gene deletion/flipping were needed to obtain the triple auxotrophic mutant. In the final strain, the loss of the three genes was confirmed by PCR with primers that anneal inside the ORF of each gene and by the inability of the strains to grow in synthetic defined (SD) media lacking histidine, leucine or arginine.
For the generation of the equivalent auxotrophic strains in C. tropicalis, we first attempted to use the SAT1-flipping strategy amplifying the deletion cassette with long oligonucleotides that have the 5′ and 3′ homology regions in the same way as for C. dubliniensis. Although we did obtain transformants using this strategy, the integration of the deletion cassette was not in the correct locus. To increase the specificity of the recombination process in this species we used longer homology arms flanking the SAT1 cassette. For this purpose, ∼900 bp regions flanking the HIS1, LEU2, and ARG4 ORFs were PCR amplified from genomic DNA and cloned adjacent to the SAT1 cassette in the pSFS2A or pEM008 plasmids (see below for the description of pEM008 and Supplementary Table S1 for primers used). Preparation of the deletion cassettes for transformation was done either by digesting with the outermost restriction enzymes (see Supplementary Table S1), or by PCR using oligos that anneal to the cloned homology arms as a template. Transformation was performed in the MYA-3404 and AM2005/0093 C. tropicalis strain backgrounds, and selection of transformants and verification of correct integration was done as for C. dubliniensis. Flipping out of the SAT1 marker when using the pSFS2A cassette was achieved by growing C. tropicalis cells for 2 days at 30°C in either liquid or solid YEP medium containing 2% maltose (Porman et al., 2013). Alternatively, flipping out of the markers coming from the pEM008 plasmid involved growing cells overnight at 30°C in either YNB medium with 2% casamino acids (Chauvel et al., 2012) or in synthetic medium containing 2% succinate (Gerami-Nejad et al., 2004). Cells were subsequently screened in YEPD supplemented with 400 μg/ml NAT plates for the loss of NAT resistance before proceeding to delete the second allele. As for C. dubliniensis, the absence of the gene was also verified by colony PCR with primers that amplify part of the ORF and by the inability of the strains to grow in SD media lacking the corresponding amino acid. The details of all generated strains are provided in Table 1.
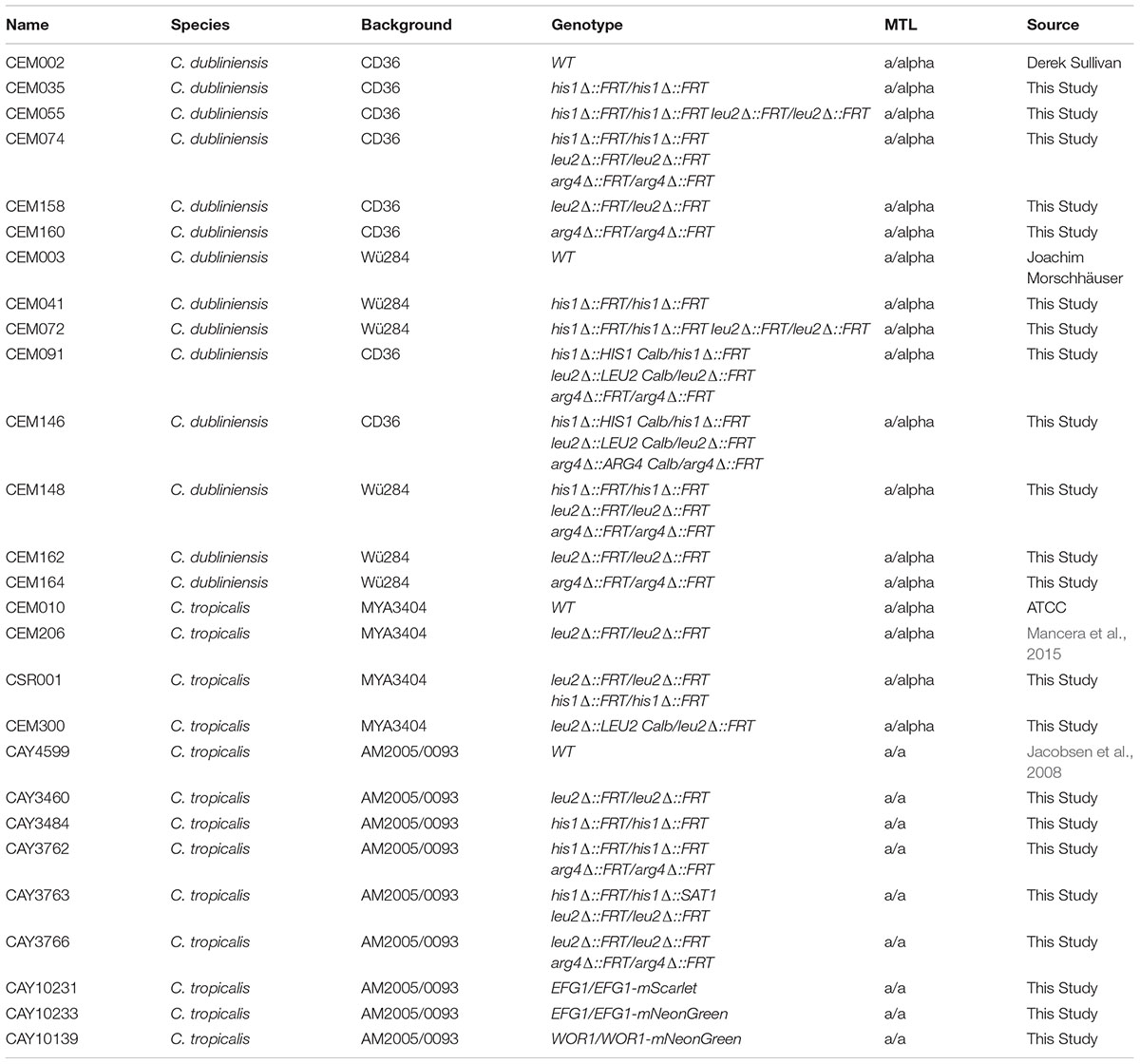
Table 1. C. dubliniensis and C. tropicalis strains used in this study.
Generation of Plasmids Carrying Nutritional and Drug-Resistance Markers
To clone the C. albicans HIS1, LEU2, and ARG4 genes for use as nutritional markers these genes were PCR amplified with their corresponding promoter and terminator using genomic DNA from the wild type SC5314 strain (see Supplementary Table S1 for primers). Each gene was cloned in the pCR-BluntII-TOPO vector using the Zero Blunt TOPO PCR cloning kit (Invitrogen) following the protocol described by the manufacturer (see Table 2 for plasmid names). The SAT1 and CaHygB genes were PCR amplified from plasmids pSFS2A (Reuß et al., 2004) and pYM70 (Basso et al., 2010), respectively (see Supplementary Table S1 for primers). These genes were cloned in the pCR2.1-TOPO vector using the TOPO TA Cloning Kit (Invitrogen) using the manufacturer’s protocol. All cloned genes were verified by sequencing and the name and details of each resulting plasmid (including the GeneBank accession numbers) are shown in Table 2.

Table 2. Plasmids to genetically modify C. dubliniensis and C. tropicalis.
Gene Deletion Strategy and Reintegration of Nutritional Markers
The overall methodology used to delete genes using the auxotrophic strains and markers described here is the same as that previously described for C. albicans (Figure 2) (Noble and Johnson, 2005; Hernday et al., 2010). Gene deletion cassettes were generated by fusion PCR of three fragments: the sequence coding for the nutritional marker, and ∼350 bp sequences flanking the 5′ and 3′ ends of the ORF to be deleted. The PCR amplification of the HIS1, LEU2, and ARG4 nutritional markers was performed using plasmids pEM001, pEM002, and pEM003 as a template, respectively, with primers EMO126 and EMO622. These primers have a region that anneals to the TOPO vectors where the markers are cloned, and a second region that anneals to the primers used for the amplification of the flanks. A specific 20 bp sequence that functions as a “barcode” can be introduced to facilitate the identification of each gene deletion mutant. These barcode sequences can be included as part of the synthesis of primer EMO622 between the sequences GCAGGGATGCGGCCGCTGAC and AGCTCGGATCCACTAGTAACG as has been described (Noble and Johnson, 2005). PCR amplification of the flanks of the gene to be deleted was performed using C. dubliniensis or C. tropicalis genomic DNA as a template, together with primers designed to amplify the ∼350 bp region upstream and downstream of the ORF. The following two sequences were included at the 5′ end of the reverse primer for the 5′ flank amplification and the forward primer of the 3′ flank amplification, respectively: CACGGCGCGCCTAGCAGCGG and GTCAGCGGCCGCATCCCTGC. These sequences allow annealing of the flanks and the auxotrophic markers amplified with primers EMO126 and EMO622 for the fusion PCR reaction. Fusion PCR was performed as described (Hernday et al., 2010) by mixing the three PCR fragments in one final PCR reaction together with the forward primer used to amplify from the 5′ flank and the reverse primer used to amplify from the 3′ flank.
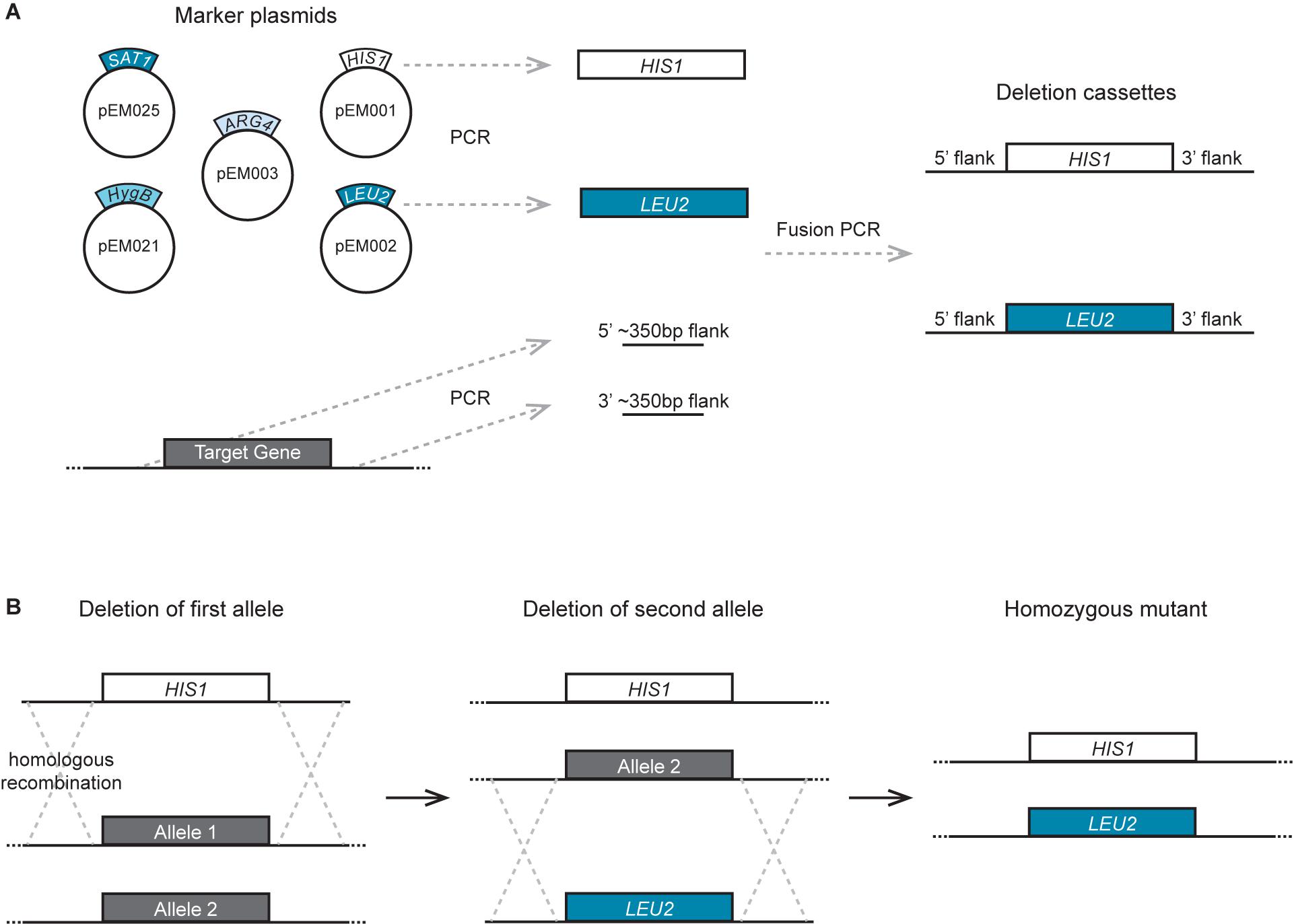
Figure 2. Strategy to generate gene deletion mutants in C. dubliniensis and C. tropicalis. (A) Depiction of the procedure to generate the deletion cassette. The markers are amplified by PCR using as a template one of the plasmids carrying the nutritional markers or the drug-resistant markers. ∼350 bp flanking regions of the target gene are also amplified by PCR using genomic DNA as a template. The three fragments are combined by fusion PCR to generate the deletion cassettes. (B) Scheme of the procedure to delete the two alleles of the gene of interest by transformation and homologous recombination using the deletion cassettes. Each allele is deleted in tandem using a different nutritional marker so that the markers do not need to be eliminated before targeting the second allele.
Generation of the deletion cassettes with the SAT1 and CaHygB drug-resistance markers was performed as for the auxotrophic markers, but using the plasmids pEM025 and pEM021, respectively, as PCR templates. Similarly, the markers for C. tropicalis transformations with the markers from C. maltosa and C. dubliniensis that were originally used for C. albicans were PCR amplified from previously described plasmids (Noble and Johnson, 2005). These plasmids are pSN40 (C. maltosa LEU2), pSN52 (C. dubliniensis HIS1), and pSN69 (C. dubliniensis ARG4).
The products of fusion PCR reactions were purified and concentrated using a QIAGEN MinElute PCR Purification Kit, and between 0.5 and 2.5 μg of DNA were transformed by electroporation as described (Kohler et al., 1997; Staib et al., 2001). Selection of transformants was performed by growing cells on SD medium lacking the corresponding amino acid, or on YEPD plates supplemented with 400 μg/ml NAT or 500 μg/ml hygromycin B. Verification of correct integration into the genome was performed by colony PCR of the 5′ and 3′ junctions. In addition, after deleting the second allele, the absence of the target gene was verified by colony PCR with primers that amplify a region within the target ORF. Re-integration of the C. albicans HIS1, LEU2, and ARG4 nutritional markers in C. dubliniensis and C. tropicalis was performed at the corresponding loci. The fusion PCR protocol described above was used to conjugate the C. albicans nutritional markers with ∼350 bp flanks of the C. dubliniensis and C. tropicalis nutritional genes. The corresponding primers are provided in Supplementary Table S1. Selection of transformants in SD media lacking the corresponding amino acid also provided proof of complementation of the auxotrophies by these markers in C. dubliniensis and C. tropicalis.
Construction of an Improved SAT1 Flipper Cassette for C. tropicalis
In our experiments, the efficiency of recycling of the SAT1 cassette derived from pSFS2A in C. tropicalis cells was much lower than that in C. albicans. To improve the efficiency of SAT1 excision in C. tropicalis, we tested replacing the C. albicans MAL2 promoter with its C. tropicalis homolog, or by using the PCK1 promoter of C. albicans or C. tropicalis (Figure 3). The C. albicans MAL2 promoter was previously cloned using BamHI and SalI sites in the plasmid pSFS2A (Reuß et al., 2004). However, there is a second SalI site in the SAT1 ORF of plasmid pSFS2A that precludes re-using this site. To circumvent this issue, we used PCR fusion to combine a fragment containing the desired promoter and a 359 bp segment of the 5′ end of the SAT1 gene from pSFS2A. The resulting PCR product was then cloned into pSFS2A using the BamHI site and an EcoRV site that is present within SAT1. The primer pairs used to PCR amplify the promoter and the SAT1 fragment are the following: for the C. tropicalis MAL2 promoter, EMO178 – EMO179 and EMO176 – EMO177; for the C. tropicalis PCK1 promoter, EMO197 – EMO210 and EMO177 – EMO211; and for the C. albicans PCK1 promoter, EMO242 – EMO243 and EMO177 – EMO244. For the fusion PCR, the following primer pairs were used: C. tropicalis MAL2 promoter, EMO177 – EMO178; C. tropicalis PCK1 promoter, EMO177 – EMO197; C. albicans PCK1 promoter, EMO177 – EMO242. All primer sequences are shown in Supplementary Table S1. As shown in Table 2, the resulting vectors were named pEM004 (contains C. tropicalis MAL2 promoter), pEM008 (contains C. tropicalis PCK1 promoter), and pEM010 (contains C. albicans PCK1 promoter). Flipping out of the SAT1 cassette when using the C. tropicalis MAL2 promoter was achieved by growing cells overnight at 30°C in YEP medium containing 2% maltose. When using the C. albicans or C. tropicalis PCK1 promoters, cassette flipping was induced using either YNB medium with 2% casamino acids (Chauvel et al., 2012) or synthetic medium containing 2% succinate (Gerami-Nejad et al., 2004). Selection of cells that had lost the SAT1 marker was done by growing isolated colonies in YEPD plates for 24 h and replica plating on YEPD plates supplemented with 400 μg/ml NAT.

Figure 3. SAT1-flipper tools for C. tropicalis. (A) Depiction of the structure of the SAT1 flipper cassette that can be used to delete genes in C. tropicalis. Here, the FLP gene is expressed under the control of the PCK1 promoter. This cassette is cloned into plasmids pEM008 and pEM010 (Table 2). (B) Illustration of the structure of the cassette to C-terminally tag genes with a 13x MYC epitope tag that allows for rapid recycling of the SAT1 marker. This cassette is cloned into plasmids pEM018 and pEM019 (Table 2). For cassettes in (A,B), versions with either the C. tropicalis or the C. albicans PCK1 promoter driving FLP were generated. (C) Depiction of the cassettes to C-terminally tag genes with either the mNeonGreen (pSFS2-mNeonGreen) or mScarlet (pSFS2-mScarlet) fluorophores. In all cassettes, recombination by the FLP recombinase results in the excision of the DNA region located between the two FRT sites from the genome. After recombination, only a single FRT site remains. The cassettes were generated in the pSFS2A plasmid that was itself constructed in a pBC SK+ plasmid backbone. The plasmid backbone, which is the same for all plasmids, is not shown. The sequences of the plasmids have been submitted to GeneBank and the accession numbers are provided in Table 2. pro., promoter; ter., terminator. (D) Micrographs showing the cellular localization of Efg1 C-terminal tagged with the fluorophores mScarlet or mNeonGreen. (E) Images showing the localization of the Wor1-mNeonGreen fusion in white and hybrid/opaque cells. The histogram of the mNeonGreen channel in white and hybrid/opaque cells is equivalent. In (D,E), the left panels show cells by differential interference contrast microscopy (DIC), the central panels show the fluorophore signal, and the right panels the merged images. Scale bars, 5 μm.
To generate a vector that contains the cassette to epitope tag genes with an immunoprecipitable peptide and whose SAT1 marker can be efficiently excised in C. tropicalis, we cloned the sequence coding for a 13x MYC tag into plasmids pEM008 and pEM010. As described above, the SAT1 flippable cassette in these two plasmids uses the PCK1 promoter to express the FLP gene (Figure 3). The 13x MYC tag sequence was amplified from the plasmid pADH34 (Hernday et al., 2010) using primers EMO249 and EMO250. This PCR product, that also contains the FRT site at the 3′ end of the MYC tag, was cloned into the BamH1 site of pEM008 and pEM010 that is immediately upstream of the PCK1 promoter. Thus, induction of the FLP recombinase will excise the SAT1 cassette leaving only the epitope tag at the C-terminus of the gene and the FRT site downstream of it. As shown in Table 2, the resulting plasmids were named pEM018 (C. tropicalis PCK1 promoter) and pEM019 (C. albicans PCK1 promoter).
Tagging of genes using these vectors was performed using a similar procedure to the one described for tagging C. albicans genes with the pADH34 vector (Hernday et al., 2010). For efficient transformations, homology regions for directing homologous recombination reactions were extended as was the case for most genetic modifications in C. tropicalis. We therefore generated ∼350 bp homology regions by fusion PCR of the DNA regions flanking the target gene together with the genetic marker cassette. The cassette was PCR amplified from vectors pEM018 and pEM019 using primers EMO623 and EMO624 (Supplementary Table S1). The primers used to extend the homology flanks were designed to amplify the ∼350 bp immediately up and downstream of the stop codon of the target gene, not including the stop codon. In addition, the reverse complement sequence of primer EMO623 (CCGTTAATTAACCCGGGGATCCG) was added to the 5′ end of the reverse primer to amplify the 5′ flank, and the reverse complement sequence of primer EMO624 (GATCCACTAGTTCTAGAGCGGCCGCC) was added to the 5′ end of the forward primer to amplify the 3′ flank. These two sequences allow the flanks to anneal to the tagging cassette for the fusion PCR. Fusion PCR was performed as before to combine the three PCR fragments, using the forward primer employed to amplify the 5′ flank and the reverse primer used to amplify the 3′ flank. The product of the fusion PCR was purified, concentrated and transformed by electroporation, as described above. Selection of transformants was performed on YEPD plates with 400 μg/ml NAT for 48 h at 30°C, and correct integration verified by colony PCR of junction regions. Flipping out of the SAT1 cassette was performed as described above by growing cells in YNB medium with 2% casamino acids. Verification of gene tagging was performed by PCR amplification and Sanger sequencing of the ORF-tag junction.
C-Terminal Tagging With Next-Generation Fluorophores
The ORFs of mNeonGreen or mScarlet-I fluorophores (Shaner et al., 2013; Bindels et al., 2017) were codon optimized for expression in C. albicans including a GGSG linker on the 5′ end, a stop codon, and flanking XhoI sites1. These sequences were cloned into pSFS2A which had been digested with XhoI and treated with calf intestinal phosphatase. Correct orientation of the insert was confirmed by restriction digest analysis and Sanger sequencing (Figure 3). Details of the resulting plasmids can be seen in Table 2 including GeneBank accession numbers. To tag EFG1 and WOR1 with the mNeonGreen or mScarlet-I fluorophores, the tagging cassette was amplified from plasmid pSFS2-mNeonGreen or pSFS2-mScarlet using primers with ∼80 bp homology sequences to the region immediately up and down of the corresponding ORF stop codon, excluding the stop codon itself (for primers see Supplementary Table S1). The cassette was then transformed by electroporation as described above and transformants were selected on YEPD + 400 μg/ml NAT plates for 48 h at 30°C. Correct integration of the fluorophore tag was verified by colony PCR of integration junctions. WOR1 was tagged in cells in the white state, which were afterwards switched to opaque as previously described (Mancera et al., 2015). For visualization under the microscope, cells of all tagged strains were grown overnight in liquid Spider medium at room temperature. Cells were imaged using a Zeiss Observer.Z1 equipped with a CoolSnap HQ2 camera, Colibi 470 nm LED light source and Zeiss filter set 38 (GFP) and 45 (Texas Red).
Results
A Set of Strains and Plasmids to Genetically Modify C. dubliniensis
Candida dubliniensis is a member of the CTG clade that has only been isolated in the diploid form and for which no conventional sexual cycle has been identified. To be able to delete genes in this species, we generated parental strains that are auxotrophic for histidine, leucine and arginine (Table 1). These strains were generated by deleting the open reading frames (ORFs) of both alleles of the HIS1, LEU2, and ARG4 genes employing the SAT1-flipping strategy previously used for gene deletions in C. albicans (Reuß et al., 2004). In these strains, two of the auxotrophies can be used to select for replacement of each allele of a given target gene with the corresponding amino acid biosynthetic gene. The third auxotrophy is convenient as it can be used to select for reintegration of the gene of interest to verify the phenotype of the mutant, as well as other genetic modifications. For the generation of deletion cassettes, the HIS1, LEU2, and ARG4 genes of C. albicans were independently cloned with their endogenous promoters and terminators in the same plasmid backbone so that they can be amplified with a unique set of primers (Table 2). The amino acid sequence identity of the HIS1, LEU2, and ARG4 genes between C. albicans and C. dubliniensis is 98, 97, and 99%, respectively. Despite the similarity of these genes at the amino acid level, the differences at the nucleotide level (90–94% identity) are expected to lower the probability that the amino acid biosynthetic markers will integrate back into the corresponding endogenous loci in C. dubliniensis.
As shown in Figure 2, the strategy presented here employs fusion PCR to join DNA fragments together to generate the deletion cassettes for transformation. PCR amplicons of ∼350 bp flanking the 5′ and 3′ ends of the target gene are fused to the sequence of the desired amino acid biosynthetic gene. The PCR-amplified deletion cassettes are then transformed into auxotrophic parental strains and selection performed in medium lacking the corresponding amino acid. The correct integration of the amino acid marker into the locus of interest is then verified by colony PCR. As a proof of concept, we integrated the C. albicans HIS1, LEU2, and ARG4 genes into C. dubliniensis strains deleted for the corresponding endogenous genes. Importantly, all three C. albicans genes complemented for the auxotrophies in C. dubliniensis. C. dubliniensis strains carrying the three integrated C. albicans genes are also useful as a control to compare other gene deletion mutants using this method. Overall, the gene deletion strategy presented here is very similar to that successfully used to generate large collections of mutants in C. albicans and C. parapsilosis. We have successfully generated several gene deletion mutants in C. dubliniensis that will be described elsewhere (E. M. et al., manuscript in preparation). The two alleles of target genes were successfully deleted by two consecutive transformations, suggesting that the process to knockout these genes or the nutritional markers did not cause extensive aneuploidies, which is a possible side effect of genetically modifying these species.
The C. dubliniensis auxotrophic strains were generated in two different genetic backgrounds, CD36 and Wü284. CD36 is the type strain and is the only C. dubliniensis strain whose genome has been sequenced to date (Jackson et al., 2009). Wü284 on the other hand is the strain that has been most used to study C. dubliniensis at a molecular level. The first genetic modification systems in C. dubliniensis using the URA3 marker were developed in this genetic background, and even the group that reported the sequence of CD36 used Wü284 for some phenotypic assays (Staib et al., 2001; Jackson et al., 2009). We generated the triple auxotrophic strains in both backgrounds as a starting point for gene deletion construction. As shown in Table 1, we also generated single auxotrophic strains for each amino acid and the double histidine/leucine auxotroph. All of these strains are available and will greatly facilitate genetic studies of C. dubliniensis.
A Set of Strains and Plasmids to Genetically Modify C. tropicalis
As is the case for C. dubliniensis, no conventional sexual cycle or haploid forms are known for C. tropicalis. Thus, the same difficulties exist for genetic manipulation of the species as for other closely related members of the CTG clade. To facilitate genetic modification of C. tropicalis, we generated a set of strains that are auxotrophic for two of the three following amino acids: histidine, leucine and arginine (Table 1). As for C. dubliniensis, these parental strains were generated by deleting the ORFs of both alleles of HIS1, LEU2, and ARG4 using a SAT1-flipping strategy. However, in contrast to strain construction in C. dubliniensis, deletion of the amino acid biosynthetic genes in C. tropicalis utilized longer homology arms for efficient targeting of the cassette into the genome. As described in detail in the Methods, ∼900 bp flanking sequences were cloned into the plasmid containing the SAT1 marker to generate the deletion cassettes for the three genes. The observed differences in the integration efficiency between the two species may reflect dissimilarities in their DNA double-strand break repair mechanisms. For example, non-homologous end-joining (NHEJ) rather than homologous recombination could be the preferred repair pathway in C. tropicalis when the flanking homology regions are short.
The same strategy outlined for C. dubliniensis can be used to delete genes in auxotrophic C. tropicalis strains. The cloned HIS1, LEU2, and ARG4 genes are used as templates for the generation of deletion cassettes which are transformed into the corresponding parental strains. We used either the amino acid biosynthetic genes from C. albicans as described above for C. dubliniensis, or C. maltosa LEU2 and C. dubliniensis HIS1 and ARG4 that have been used for strain construction in C. albicans. The protein sequence identity between C. tropicalis, C. albicans, C. maltosa, and C. dubliniensis amino acid biosynthetic genes ranges between 86 and 99%, and all of the genes tested complemented the corresponding auxotrophy in C. tropicalis. The correct integration of the auxotrophic markers was verified by PCR of the DNA junctions between the deleted gene and the targeting cassette. In addition to the three nutritional markers described above, we also cloned the SAT1 and CaHygB genes into a similar plasmid backbone (Table 2) and used these for providing resistance to nourseothricin and hygromycin B, respectively. These markers can be PCR amplified with the same set of primers used for the nutritional markers to generate gene deletion cassettes. We have successfully used both nutritional markers and the drug resistance markers in different combinations to effectively delete multiple genes in C. tropicalis (Mancera et al., 2015; Anderson et al., 2016; E. M. et al., manuscript in preparation). As with C. dubliniensis, the two alleles of different target genes were successfully deleted in tandem, which suggests that these strains do not contain aneuploid chromosomes.
The C. tropicalis auxotrophic strains were generated in MYA-3404 and AM2005/0093 genetic backgrounds. MYA-3404 is the strain whose genome has been sequenced (Butler et al., 2009), while AM2005/0093 is a strain that was isolated from urine in Colombia and has been phylogenetically placed close to MYA-3404 by multi-locus sequence typing (Jacobsen et al., 2008). However, AM2005/0093 was naturally homozygous for the mating type-like (MTL) locus and thus was one of several strains picked to study the molecular mechanisms that control phenotypic switching, as switching is MTL-regulated in C. albicans (Mancera et al., 2015; Anderson et al., 2016). We had previously reported some of the auxotrophic strains generated in the AM2005/0093 background. Overall, the strains generated in the two genetic backgrounds (Table 1) offer an ideal starting point for the generation of gene deletion mutants in C. tropicalis.
A SAT1-Flipping Strategy to Efficiently Delete or Epitope Tag Genes in C. tropicalis
The SAT1 flippable cassette was originally developed for facilitating genetic manipulation of the C. albicans genome (Reuß et al., 2004). It is a powerful genetic engineering tool for diploid species since it allows the deletion of both alleles of a target gene even in the absence of available nutritional markers. Several variations of the system have been constructed, but in general the system employs the FLP site-specific recombinase to act on two FLP recognition target sites (FRT) that are present in direct repeats flanking the cassette containing the selectable marker. The FLP recombinase catalyzes excision of the region between the FRT sites and removes the selection marker, thus allowing for repeated use of the same marker for other genetic modifications. In a recent version of the system, the FLP gene is conditionally expressed using the promoter of the C. albicans MAL2 gene which is induced when cells are grown in media containing maltose as the carbon source (Sasse and Morschhauser, 2012). Overall, the system combines a drug-resistance marker that allows efficient selection of transformants with an effective system for recycling the marker.
The same SAT1-flipping strategy has also been used successfully in C. dubliniensis and C. tropicalis to delete genes and was used here for the generation of auxotrophic strains. Nevertheless, the FLP-mediated excision of the deletion cassette was much less efficient in C. tropicalis than in C. albicans. Even after several days of growth in maltose-containing media very few cells lost the SAT1 marker, hindering the overall utility of this reusable system. The difficulties in SAT1 recycling likely reflect poor induction of FLP using the CaMAL2 promoter in C. tropicalis cells. We therefore first replaced the CaMAL2 promoter with the endogenous version from C. tropicalis, but FLP-induced recombination was still rare. As a second strategy, we tested driving FLP expression from the promoter of the C. tropicalis PCK1 gene or the C. albicans PCK1 gene (Figure 3). The PCK1 promoter has previously been used to regulate gene expression in C. albicans as it is induced by gluconeogenic carbon sources (Gerami-Nejad et al., 2004; Chauvel et al., 2012). We therefore tested SAT1 recycling by growing cells in succinate or casamino acids as the carbon source. Using either the C. albicans or the C. tropicalis PCK1 promoter we found that excision of SAT1 in C. tropicalis was as efficient as that using the MAL2 promoter-driven FLP flipper system in C. albicans. We have therefore engineered an efficient system for marker recycling in C. tropicalis.
The recyclable SAT1 cassette has also been used as part of a construct to epitope tag genes in C. albicans (Hernday et al., 2010). For this purpose, SAT1 is placed adjacent to the epitope tag so that nourseothricin can be used to select transformants, and the FLP gene is then induced to flip-out the SAT1 cassette leaving only the epitope tag fused to the gene and an FRT site downstream of the tag. Removal of the SAT1 cassette diminishes the potential for disruption of the untranslated region (UTR) to impact gene function. To adapt this system for C. tropicalis, we cloned a 13x MYC tag into the plasmids described above that use the C. tropicalis or C. albicans PCK1 promoter to regulate FLP expression (Figure 3). When tested, this strategy allowed efficient excision of the SAT1 marker after epitope tagging several genes. The results of the immunoprecipitation of these genes will be described elsewhere (E. M. et al., manuscript in preparation). It is important to note that genome-wide sequencing data from the immunoprecipitation experiments does not show any evidence of large-scale copy number variations in these strains, again consistent with them having euploid genomes.
A Set of Plasmids to Tag Candida Genes With Next-Generation Fluorophores
It has not been trivial to use conventional fluorophores such as GFP to visualize the cellular localization of gene products in C. tropicalis. For example, although we are able to generate the appropriate genetically modified strains, a number of protein-GFP fusions or GFP reporters were not visible under the microscope, with the exception of highly expressed histone-GFP and histone-mCherry fusions (Porman et al., 2011). The yeast-enhanced GFP and YFP have also been expressed successfully from a strong constitutive promoter when not fused to a target gene (Defosse et al., 2018). To enable cell biological approaches in C. tropicalis, we engineered plasmids that contain the next-generation fluorophores mNeonGreen and mScarlet that had been codon-optimized for C. albicans2 (Figure 3). These plasmids can be used to generate gene-fluorophore fusions by amplifying the fluorophore cassette using oligonucleotides with sequence identity to the C-terminus of the gene of interest. The fluorophores were cloned in the SAT1 flippable system so that transformants can be selected using nourseothricin, and the drug-resistance marker flipped out by induction of the FLP gene. As a proof of concept, we tagged the transcription factors Efg1 and Wor1 with mNeonGreen, and also tagged Efg1 with mScarlet. As observed in Figure 3, it was possible to visualize the cellular localization of Efg1 when fused to both fluorophores. Efg1 is a transcription factor that regulates morphological transitions in C. tropicalis including filamentation and white-opaque switching (Mancera et al., 2015). The Wor1-mNeonGreen fusion was also detected, and this protein was only visible in cells that were in the hybrid/opaque state and not visible in cells in the white state. This is in agreement with the role of this transcription factor in determining the cell’s phenotypic state (Porman et al., 2013; Anderson et al., 2016). The discrete localization of both transcription factors in the nucleus is consistent with their function as transcriptional regulators. We have therefore successfully engineered tools that can be used to tag proteins with next-generation fluorophores in order to improve the visualization of gene products in C. tropicalis cells.
Discussion
Fungi from the genus Candida are among the most important fungal human pathogens. They are a common source of a variety of diseases and can cause opportunistic infections with high mortality rates. Most research has focused on understanding C. albicans pathogenicity since it is the most prevalent Candida pathogen. However, other members of the genus are of high medical relevance, especially in some regions of the world. In addition, modern medical practices, such as the prophylactic use of antifungal drugs, appear to be favoring the increase in prevalence of alternative Candida species.
Here, we report the generation of a broad set of tools for genetic manipulation of C. dubliniensis and C. tropicalis, the two most closely related species to C. albicans. The strategies described here overcome the intrinsic difficulties of genetically engineering in diploid species that do not undergo a conventional sexual cycle. The parental strains are auxotrophic for HIS1, LEU2, and/or ARG4 nutritional markers that have been used in the past to construct large collections of gene deletion mutants in C. albicans and C. parapsilosis. We also generated vectors containing the HIS1, LEU2, and ARG4 nutritional markers from C. albicans, as well as the SAT1 and CaHygB drug resistance markers in a similar plasmid backbone. This allows the PCR amplification of all the markers with the same set of primers for generation of the deletion cassettes. Two different selectable markers can then be used to delete both alleles of the target gene without the need to recycle the marker between the two rounds of transformation. This improvement could be crucial when attempting to generate large collections of knockout strains. In addition, we have engineered plasmids to create epitope and fluorophore gene fusions in C. tropicalis that allow for the immunoprecipitation of proteins as well as the study of their cellular localization. These tools will be useful in the study of this species at both a cellular and molecular level.
In the process of generating strains, we observed two interesting differences between C. dubliniensis and C. tropicalis in terms of the ease with which they can be genetically modified. The first concerns the specificity of the homologous recombination process as short homology regions (∼60 bp) are enough to specifically target the locus of interest in C. dubliniensis as has been reported for C. albicans (Hernday et al., 2010). In contrast, genetic modification of C. tropicalis often needed longer homology regions (∼350 bp) for efficient gene deletion, although successful integration of tagging constructs has been achieved with homology regions of ∼80 bp (Porman et al., 2011). These differences could be due to how DNA double-strand breaks are repaired in these species, with non-homologous end joining (NHEJ) being used in C. tropicalis when the homology regions are short. The strategies presented here overcome this difficulty since the deletion cassettes are generated by fusion PCR of the marker allowing the use of long homology flanks.
A second major difference observed between C. dubliniensis and C. tropicalis concerned the efficiency of SAT1 marker recycling when using the FLP recombinase under the control of the MAL2 promoter. In C. dubliniensis, FLP-mediated site-specific recombination was efficient, as previously described in C. albicans (Reuß et al., 2004; Sasse and Morschhauser, 2012). In contrast, marker excision was infrequent in C. tropicalis following growth on maltose-containing medium when using the CaMAL2 promoter. We tested replacing the CaMAL2 promoter with the endogenous C. tropicalis promoter yet still observed low SAT1 excision rates, which could indicate low expression of this promoter in C. tropicalis. However, we found that replacing the MAL2 promoter with the PCK1 promoter (from either C. tropicalis or C. albicans) led to efficient excision of the SAT1 marker in C. tropicalis when cells were grown on a gluconeogenic carbon source. This established that low expression of the FLP gene from the MAL2 promoter was the limitation, and may indicate differences in the pathways used by these two Candida species to metabolize maltose.
Overall, the genetic tools presented here for C. dubliniensis and C. tropicalis will facilitate genetic studies of Candida biology and pathogenicity. C. dubliniensis is an ideal model to understand C. albicans pathogenicity given its phylogenetic proximity yet attenuated virulence. C. tropicalis is a sister species of the previous two and is consistently ranked amongst the four most prevalent disease-causing Candida species. Together with C. albicans and C. parapsilosis, these species are also useful for evolutionary studies (Figure 1) as they span a considerable evolutionary range but are close enough phylogenetically to capture subtle changes between their genomes (Butler et al., 2009). The genetic modification tools presented here will further accelerate the establishment of these species as evolutionary models.
Data Availability
The datasets generated for this study can be found in GenBank, MK425745, MK425746, MK425747, MK425748, MK431394, MK431395, MK431396, MK431397, MK431398, MK431399, MK431400, and MK427053.
Author Contributions
AJ, AP, EM, and RB contributed to the conception and design of the study. AP, CF, EM, and SR-C constructed the strains and plasmids. EM wrote the first draft of the manuscript. EM and RB wrote sections of the manuscript. All authors contributed to manuscript revision, and read and approved the submitted version.
Funding
This work was supported by a Human Frontier Science Program grant to EM, a UC-MEXUS grant to EM, National Institutes of Health grants AI083311 and AI049187 to AJ, AI081704 to RB, PRODEP grant 511-6/17-7433 to EM, CONACyT grant CB-2016-01 282511 to EM, a PATH award from the Burroughs Wellcome Fund to RB and a Welcome Trust Seed Award in Science to EM.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Derek Sullivan, Brian Wong and especially Joachim Morschhäuser for providing strains, plasmids and advice, and Sergio Campos and Adriana Espinoza for critical comments on the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00357/full#supplementary-material
Footnotes
- ^https://currentprotocols.onlinelibrary.wiley.com/doi/abs/10.1002/cpmc.76
- ^https://currentprotocols.onlinelibrary.wiley.com/doi/abs/10.1002/cpmc.76
References
Anderson, M. Z., Porman, A. M., Wang, N., Mancera, E., Huang, D., Cuomo, C. A., et al. (2016). A multistate toggle switch defines fungal cell fates and is regulated by synergistic genetic cues. PLoS Genet. 12:e1006353. doi: 10.1371/journal.pgen.1006353
Basso, L. R. Jr., Bartiss, A., Mao, Y., Gast, C. E., Coelho, P. S., Snyder, M., et al. (2010). Transformation of Candida albicans with a synthetic hygromycin B resistance gene. Yeast 27, 1039–1048. doi: 10.1002/yea.1813
Bindels, D. S., Haarbosch, L., van Weeren, L., Postma, M., Wiese, K. E., Mastop, M., et al. (2017). mScarlet: a bright monomeric red fluorescent protein for cellular imaging. Nat. Methods 14, 53–56. doi: 10.1038/nmeth.4074
Butler, G., Rasmussen, M. D., Lin, M. F., Santos, M. A., Sakthikumar, S., Munro, C. A., et al. (2009). Evolution of pathogenicity and sexual reproduction in eight Candida genomes. Nature 459, 657–662. doi: 10.1038/nature08064
Chauvel, M., Nesseir, A., Cabral, V., Znaidi, S., Goyard, S., Bachellier-Bassi, S., et al. (2012). A versatile overexpression strategy in the pathogenic yeast Candida albicans: identification of regulators of morphogenesis and fitness. PLoS One 7:e45912. doi: 10.1371/journal.pone.0045912
Defosse, T. A., Courdavault, V., Coste, A. T., Clastre, M., de Bernonville, T. D., Godon, C., et al. (2018). A standardized toolkit for genetic engineering of CTG clade yeasts. J. Microbiol. Methods 144, 152–156. doi: 10.1016/j.mimet.2017.11.015
Gabaldon, T., Naranjo-Ortiz, M. A., and Marcet-Houben, M. (2016). Evolutionary genomics of yeast pathogens in the Saccharomycotina. FEMS Yeast Res. 16:fow064. doi: 10.1093/femsyr/fow064
Gerami-Nejad, M., Hausauer, D., McClellan, M., Berman, J., and Gale, C. (2004). Cassettes for the PCR-mediated construction of regulatable alleles in Candida albicans. Yeast 21, 429–436. doi: 10.1002/yea.1080
Grahl, N., Demers, E. G., Crocker, A. W., and Hogan, D. A. (2017). Use of RNA-protein complexes for genome editing in non-albicans Candida species. mSphere 2:e218-17. doi: 10.1128/mSphere.00218-17
Hernday, A. D., Noble, S. M., Mitrovich, Q. M., and Johnson, A. D. (2010). Genetics and molecular biology in Candida albicans. Methods Enzymol. 470, 737–758. doi: 10.1016/S0076-6879(10)70031-8
Holland, L. M., Schroder, M. S., Turner, S. A., Taff, H., Andes, D., Grózer, Z., et al. (2014). Comparative phenotypic analysis of the major fungal pathogens Candida parapsilosis and Candida albicans. PLoS Pathog. 10:e1004365. doi: 10.1371/journal.ppat.1004365
Homann, O. R., Dea, J., Noble, S. M., and Johnson, A. D. (2009). A phenotypic profile of the Candida albicans regulatory network. PLoS Genet 5:e1000783. doi: 10.1371/journal.pgen.1000783
Huang, M. Y., and Mitchell, A. P. (2017). Marker recycling in Candida albicans through CRISPR-Cas9-induced marker excision. mSphere 2:e0050-17. doi: 10.1128/mSphere.00050-17
Jackson, A. P., Gamble, J. A., Yeomans, T., Moran, G. P., Saunders, D., Harris, D., et al. (2009). Comparative genomics of the fungal pathogens Candida dubliniensis and Candida albicans. Genome Res. 19, 2231–2244. doi: 10.1101/gr.097501.109
Jacobsen, M. D., Davidson, A. D., Li, S. Y., Shaw, D. J., Gow, N. A., Odds, F. C., et al. (2008). Molecular phylogenetic analysis of Candida tropicalis isolates by multi-locus sequence typing. Fungal Genet. Biol. 45, 1040–1042. doi: 10.1016/j.fgb.2008.03.011
Kohler, G. A., White, T. C., and Agabian, N. (1997). Overexpression of a cloned IMP dehydrogenase gene of Candida albicans confers resistance to the specific inhibitor mycophenolic acid. J. Bacteriol. 179, 2331–2338. doi: 10.1128/jb.179.7.2331-2338.1997
Krassowski, T., Coughlan, A. Y., Shen, X. X., Zhou, X., Kominek, J., Opulente, D. A., et al. (2018). Evolutionary instability of CUG-Leu in the genetic code of budding yeasts. Nat. Commun. 9:1887. doi: 10.1038/s41467-018-04374-7
Lynch, D. P. (1994). Oral candidiasis. History, classification, and clinical presentation. Oral. Surg. Oral. Med. Oral. Pathol. 78, 189–193. doi: 10.1016/0030-4220(94)90146-5
Maguire, S. L., Oheigeartaigh, S. S., Byrne, K. P., Schroder, M. S., O’Gaora, P., Wolfe, K. H., et al. (2013). Comparative genome analysis and gene finding in Candida species using CGOB. Mol. Biol. Evol. 30, 1281–1291. doi: 10.1093/molbev/mst042
Mancera, E., Porman, A. M., Cuomo, C. A., Bennett, R. J., and Johnson, A. D. (2015). Finding a missing gene: EFG1 regulates morphogenesis in Candida tropicalis. G3 5, 849–856. doi: 10.1534/g3.115.017566
Min, K., Ichikawa, Y., Woolford, C. A., and Mitchell, A. P. (2016). Candida albicans gene deletion with a transient CRISPR-Cas9 system. mSphere 1:e130-16. doi: 10.1128/mSphere.00130-16
Moran, G. P., Coleman, D. C., and Sullivan, D. J. (2011). Comparative genomics and the evolution of pathogenicity in human pathogenic fungi. Eukaryot. Cell 10, 34–42. doi: 10.1128/EC.00242-10
Moran, G. P., Coleman, D. C., and Sullivan, D. J. (2012). Candida albicans versus Candida dubliniensis: why is C. albicans more pathogenic? Int. J. Microbiol. 2012:205921. doi: 10.1155/2012/205921
Ng, H., and Dean, N. (2017). Dramatic improvement of CRISPR/Cas9 editing in Candida albicans by increased single guide RNA expression. mSphere 2:e385-16. doi: 10.1128/mSphere.00385-16
Nguyen, N., Quail, M. M. F., and Hernday, A. D. (2017). An efficient, rapid, and recyclable system for CRISPR-mediated genome editing in Candida albicans. mSphere 2:e149-17. doi: 10.1128/mSphereDirect.00149-17
Nobile, C. J., and Johnson, A. D. (2015). Candida albicans biofilms and human disease. Annu. Rev. Microbiol. 69, 71–92. doi: 10.1146/annurev-micro-091014-104330
Noble, S. M., French, S., Kohn, L. A., Chen, V., and Johnson, A. D. (2010). Systematic screens of a Candida albicans homozygous deletion library decouple morphogenetic switching and pathogenicity. Nat. Genet. 42, 590–598. doi: 10.1038/ng.605
Noble, S. M., and Johnson, A. D. (2005). Strains and strategies for large-scale gene deletion studies of the diploid human fungal pathogen Candida albicans. Eukaryot. Cell 4, 298–309. doi: 10.1128/EC.4.2.298-309.2005
Pande, K., Chen, C., and Noble, S. M. (2013). Passage through the mammalian gut triggers a phenotypic switch that promotes Candida albicans commensalism. Nat. Genet. 45, 1088–1091. doi: 10.1038/ng.2710
Porman, A. M., Alby, K., Hirakawa, M. P., and Bennett, R. J. (2011). Discovery of a phenotypic switch regulating sexual mating in the opportunistic fungal pathogen Candida tropicalis. Proc. Natl. Acad. Sci. U.S.A. 108, 21158–21163. doi: 10.1073/pnas.1112076109
Porman, A. M., Hirakawa, M. P., Jones, S. K., Wang, N., and Bennett, R. J. (2013). MTL-independent phenotypic switching in Candida tropicalis and a dual role for Wor1 in regulating switching and filamentation. PLoS Genet. 9:e1003369. doi: 10.1371/journal.pgen.1003369
Pujol, C., Daniels, K. J., Lockhart, S. R., Srikantha, T., Radke, J. B., Geiger, J., et al. (2004). The closely related species Candida albicans and Candida dubliniensis can mate. Eukaryot. Cell 3, 1015–1027. doi: 10.1128/EC.3.4.1015-1027.2004
Reuß, O., Vik, A., Kolter, R., and Morschhauser, J. (2004). The SAT1 flipper, an optimized tool for gene disruption in Candida albicans. Gene 341, 119–127. doi: 10.1016/j.gene.2004.06.021
Sasse, C., and Morschhauser, J. (2012). Gene deletion in Candida albicans wild-type strains using the SAT1-flipping strategy. Methods Mol. Biol. 845, 3–17. doi: 10.1007/978-1-61779-539-8_1
Schulze, J., and Sonnenborn, U. (2009). Yeasts in the gut: from commensals to infectious agents. Dtsch. Arztebl. Int. 106, 837–842. doi: 10.3238/arztebl.2009.0837
Schwarzmuller, T., Ma, B., Hiller, E., Istel, F., Tscherner, M., Brunke, S., et al. (2014). Systematic phenotyping of a large-scale Candida glabrata deletion collection reveals novel antifungal tolerance genes. PLoS Pathog. 10:e1004211. doi: 10.1371/journal.ppat.1004211
Sears, D., and Schwartz, B. S. (2017). Candida auris: an emerging multidrug-resistant pathogen. Int. J. Infect. Dis. 63, 95–98. doi: 10.1016/j.ijid.2017.08.017
Segal, E. S., Gritsenko, V., Levitan, A., Yadav, B., Dror, N., Steenwyk, J. L., et al. (2018). Gene essentiality analyzed by in vivo transposon mutagenesis and machine learning in a stable haploid isolate of Candida albicans. mBio 9:e2048-18. doi: 10.1128/mBio.02048-18
Shaner, N. C., Lambert, G. G., Chammas, A., Ni, Y., Cranfill, P. J., Sell, B. R., et al. (2013). A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nat. Methods 10, 407–409. doi: 10.1038/nmeth.2413
Staab, J. F., and Sundstrom, P. (2003). URA3 as a selectable marker for disruption and virulence assessment of Candida albicans genes. Trends Microbiol. 11, 69–73. doi: 10.1016/S0966-842X(02)00029-X
Staib, P., Moran, G. P., Sullivan, D. J., Coleman, D. C., and Morschhauser, J. (2001). Isogenic strain construction and gene targeting in Candida dubliniensis. J. Bacteriol. 183, 2859–2865. doi: 10.1128/JB.183.9.2859-2865.2001
Sullivan, D. J., Moran, G. P., and Coleman, D. C. (2005). Candida dubliniensis: ten years on. FEMS Microbiol. Lett. 253, 9–17. doi: 10.1016/j.femsle.2005.09.015
Turner, S. A., and Butler, G. (2014). The Candida pathogenic species complex. Cold Spring Harb. Perspect. Med. 4:a019778. doi: 10.1101/cshperspect.a019778
Uhl, M. A., Biery, M., Craig, N., and Johnson, A. D. (2003). Haploinsufficiency-based large-scale forward genetic analysis of filamentous growth in the diploid human fungal pathogen C. albicans. EMBO J. 22, 2668–2678. doi: 10.1093/emboj/cdg256
Vyas, V. K., Barrasa, M. I., and Fink, G. R. (2015). A Candida albicans CRISPR system permits genetic engineering of essential genes and gene families. Sci. Adv. 1:e1500248.
Wang, J., Peng, J., Fan, H., Xiu, X., Xue, L., Wang, L., et al. (2018). Development of mazF-based markerless genome editing system and metabolic pathway engineering in Candida tropicalis for producing long-chain dicarboxylic acids. J. Ind. Microbiol. Biotechnol. 45, 971–981. doi: 10.1007/s10295-018-2074-9
Wilson, R. B., Davis, D., Enloe, B. M., and Mitchell, A. P. (2000). A recyclable Candida albicans URA3 cassette for PCR product-directed gene disruptions. Yeast 16, 65–70. doi: 10.1002/(SICI)1097-0061(20000115)16:1<65::AID-YEA508>3.0.CO;2-M
Zhang, Y., Tao, L., Zhang, Q., Guan, G., Nobile, C. J., Zheng, Q., et al. (2016). The gray phenotype and tristable phenotypic transitions in the human fungal pathogen Candida tropicalis. Fungal Genet. Biol. 93, 10–16. doi: 10.1016/j.fgb.2016.05.006
Keywords: Candida dubliniensis, Candida tropicalis, genetic modification, mNeonGreen, mScarlet
Citation: Mancera E, Frazer C, Porman AM, Ruiz-Castro S, Johnson AD and Bennett RJ (2019) Genetic Modification of Closely Related Candida Species. Front. Microbiol. 10:357. doi: 10.3389/fmicb.2019.00357
Received: 23 December 2018; Accepted: 11 February 2019;
Published: 19 March 2019.
Edited by:
Maurizio Sanguinetti, Catholic University of the Sacred Heart, ItalyReviewed by:
Geraldine Butler, University College Dublin, IrelandMacit Ilkit, Çukurova University, Turkey
Copyright © 2019 Mancera, Frazer, Porman, Ruiz-Castro, Johnson and Bennett. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Eugenio Mancera, ZXVnZW5pby5tYW5jZXJhQGNpbnZlc3Rhdi5teA== Alexander D. Johnson, YWpvaG5zb25AY2dsLnVjc2YuZWR1 Richard J. Bennett, cmljaGFyZF9iZW5uZXR0QGJyb3duLmVkdQ==