 Linnea C. Hjelm
Linnea C. Hjelm Johan Nilvebrant
Johan Nilvebrant Per-Åke Nygren1
Per-Åke Nygren1 Anders S. Nilsson
Anders S. Nilsson Johan Seijsing
Johan Seijsing- 1Department of Protein Science, School of Engineering Sciences in Chemistry, Biotechnology and Health, KTH Royal Institute of Technology, Stockholm, Sweden
- 2Department of Molecular Biosciences, The Wenner-Gren Institute, Stockholm University, Stockholm, Sweden
Bacteriophage endolysins and bacterial exolysins are capable of enzymatic degradation of the cell wall peptidoglycan layer and thus show promise as a new class of antimicrobials. Both exolysins and endolysins often consist of different modules, which are responsible for enzymatic functions and cell wall binding, respectively. Individual modules from different endo- or exolysins with different binding and enzymatic activities, can via gene fusion technology be re-combined into novel variants for investigations of arrangements of potential clinical interest. The aim of this study was to investigate if separately produced cell wall binding and enzyme modules could be assembled into a functional lysin via a non-covalent affinity interaction bridge composed of the barnase ribonuclease from Bacillus amyloliquefaciens and its cognate inhibitor barstar, known to form a stable heterodimeric complex. In a proof-of-principle study, using surface plasmon resonance, flow cytometry and turbidity reduction assays, we show that separately produced modules of a lysin cysteine/histidine-dependent amidohydrolase/peptidase (CHAP) from Staphylococcus aureus bacteriophage K endolysin (LysK) fused to barnase and a cell wall binding Src homology 3 domain (SH3b) from the S. simulans exolysin lysostaphin fused to barstar can be non-covalently assembled into a functional lysin showing both cell wall binding and staphylolytic activity. We hypothesize that the described principle for assembly of functional lysins from separate modules through appended hetero-dimerization domains has a potential for investigations of also other combinations of enzymatically active and cell wall binding domains for desired applications.
Introduction
In the exploration of antimicrobials to combat resistant bacterial infections, endolysins have gained attention as alternatives to conventional small molecular antibiotics (Schuch et al., 2014; Czaplewski et al., 2016; Jun et al., 2017; Totté et al., 2017). Endolysins are peptidoglycan hydrolases used by bacteriophages to release replicated progeny by degrading the cell wall of their bacterial host, resulting in cell lysis and death. Endolysins are functionally and structurally similar to both bacterial exolysins and autolysins. The exolysins are produced by certain bacteria with the function of causing lysis of alien bacterial species (Schindler and Schuhardt, 1964), and autolysins regulate cell wall metabolism and population size (Rogers et al., 1980). Development of resistance to endolysins is considered unlikely since these enzymes cleave evolutionary conserved and essential structures in the cell wall with high specificity for particular bacteria (Pastagia et al., 2011; Rodríguez-Rubio et al., 2013). At present, there are four endolysin-based products in ongoing clinical trials against staphylococcal infections (Totté et al., 2017), and one endolysin-based formulation has reached market approval as a medical device for topical application in treatment of less severe skin conditions such as rosacea, acne, and eczema (Herpers et al., 2014).
Endolysins need both to bind the surface of the target bacterium and to be able to cleave the peptidoglycan substrate. For endolysins targeting Gram-positive bacteria, these two functions are commonly mediated by separate domains referred to as cell wall binding domains (CBD) and enzymatically active domains (EAD). Moreover, individual endolysins may contain several EADs (Diaz et al., 1990). The modular properties of endolysins has inspired protein engineering efforts to find new combinations of EADs and CBDs with, from a pharmacological point of view, improved properties like altered strain specificity and/or higher catalytic efficacy (Croux et al., 1993; Schmelcher et al., 2011). Searches for effective new EAD-CBD combinations from collections of individual moieties may, however, involve construction of hundreds of gene fusions that need to be expressed individually before evaluation (Yang et al., 2015, 2017; Verbree et al., 2017). Thus, more effective means for linking sets of EADs and CBDs into various combinations for evaluation of their efficacies to lyse bacteria are called for.
Barnase is a small Bacillus amyloliquefaciens ribonuclease of 110 amino acids showing a high affinity to its own inhibitor, the 89 amino acid barstar (Hartley, 1993; Schreiber and Fersht, 1993). The strong interaction between barnase and barstar has previously been exploited to achieve both stoichiometrically and structurally ordered assemblies of designed multivalent antibody-fragment protein complexes (Deyev et al., 2003). In the present study, we have investigated the possibility to utilize the barnase-barstar system in a modular approach for non-covalent, yet robust, linking of EADs and CBDs into functional cell wall binding and enzymatically active lysins (Figures 1A,B). We report the results from endolysin assembly of the EAD cysteine/histidine-dependent amidohydrolase/peptidase (CHAP) from Staphylococcus aureus bacteriophage K endolysin (LysK) with the CBD Src homology 3 domain (SH3b) from the S. simulans exolysin lysostaphin.
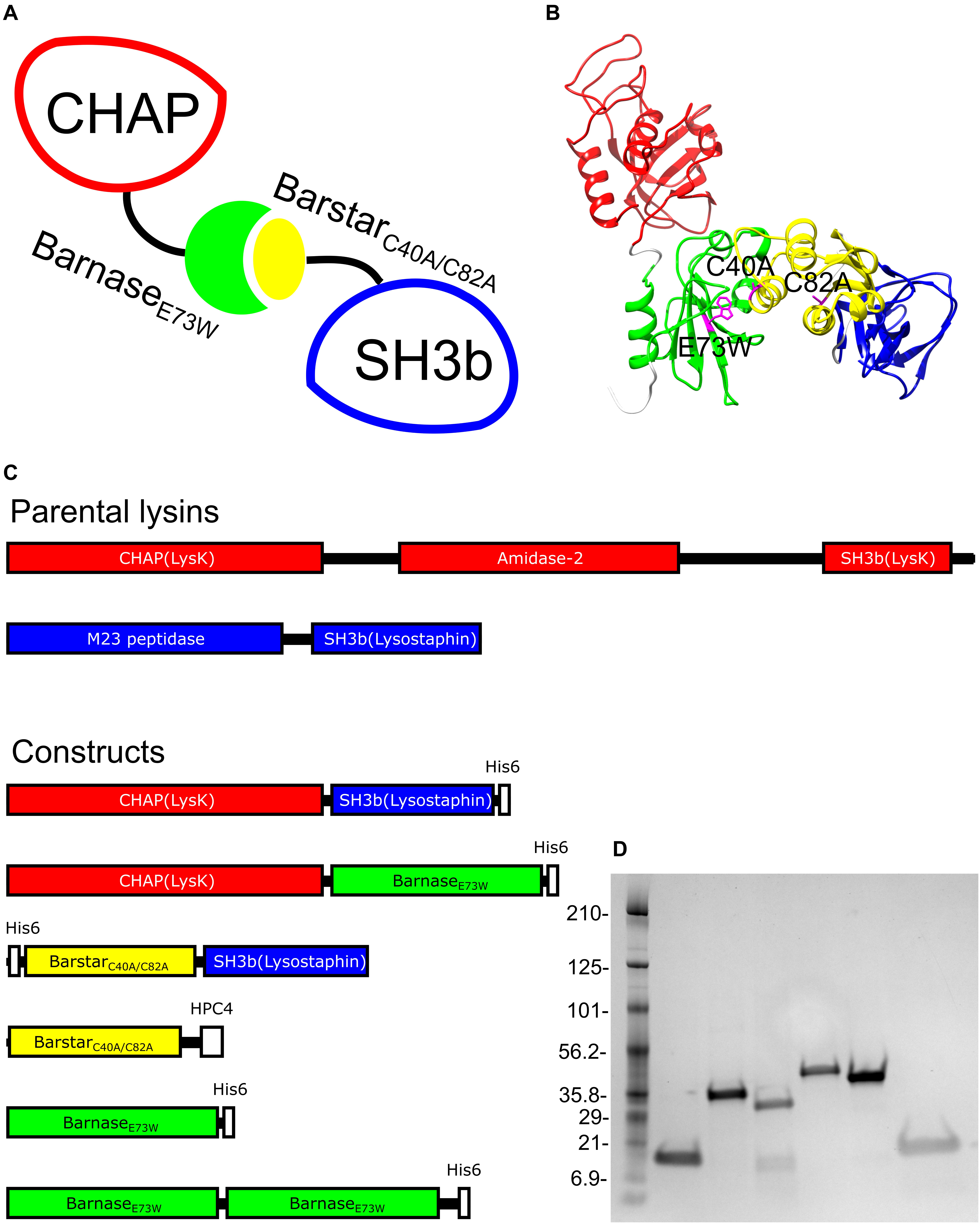
Figure 1. (A) Schematic sketch of protein complex, (B) structure prediction of complex made using I-TASSER (Zhang, 2008) with mutations highlighted in magenta. (C) Representations of parental lysins and protein constructs designed for this study. The parental endolysin LysK and the derived CHAPK domain is in red. The parental Lysostaphin and its derived SH3b domain are in blue. Barnase and barstar are in green and yellow, respectively. (D). SDS–PAGE of protein constructs. From left to right: ladder, BarnaseE73W, BarnaseE73W-BarnaseE73W, BarstarC40A/C82A-SH3b, CHAP-BarnaseE73W, CHAP-SH3b, and BarstarC40A/C82A.
Results and Discussion
Design, Production and Biochemical Characterization of CHAP-BarnaseE73W and BarstarC40A/C82A-SH3b Fusion Proteins
Due to the RNAse activity of wild type barnase, which has been described to be toxic to bacterial cells used for production, a previously described enzymatically inactive variant (E73W) of barnase was used as fusion partner for the construction of a CHAP-BarnaseE73W fusion protein (Hartley, 1993; Ramachandran and Udgaonkar, 1996; Figures 1A–C). For construction of the cognate Barstar-SH3b fusion protein, a double cysteine mutant (C40A/C82A) of barstar was used to avoid potential problems with artefactual disulfide bridges between protein constructs, potentially interfering with the interpretation of the results (Hartley, 1988; Schreiber et al., 1997; Khait and Schreiber, 2012). Constructs for prokaryotic expression of the CHAP-BarnaseE73W and BarstarC40A/C82A-SH3b lysin modules were assembled (Figure 1C). For use as controls, expression constructs for the single domains of BarstarC40A/C82A and BarnaseE73W, a BarnaseE73W-BarnaseE73W dimer to be used as a non-sterically hindered ligand in surface plasmon resonance measurements and a CHAP-SH3b direct fusion protein were also assembled.
In initial constructs, a GGGS linker was used to connect the domains in the different fusion proteins. However, after expression of protein constructs containing CHAP it was realized that this endopeptidase, known to cleave between the pentaglycine and the D-alanine of the staphylococcal peptidoglycan (Becker et al., 2009), was also able to degrade the GGGS linker (data not shown). Thus, the linker in fusion constructs containing CHAP was changed into GSSG, which resulted in proteolytically stable constructs.
All proteins were produced as His6-tagged constructs intracellularly in Escherichia coli and purified from the soluble cytoplasmic fraction using immobilized metal ion affinity chromatography or, in the case of the single domain BarstarC40A/C82A construct which was produced in fusion with a short HPC4 peptide (Rezaie et al., 1992), using an anti-protein C immunoaffinity column (Figure 1C). The purified proteins were analyzed by SDS–PAGE (Figure 1D), circular dichroism (Table 1) and mass spectrometry (Table 1).
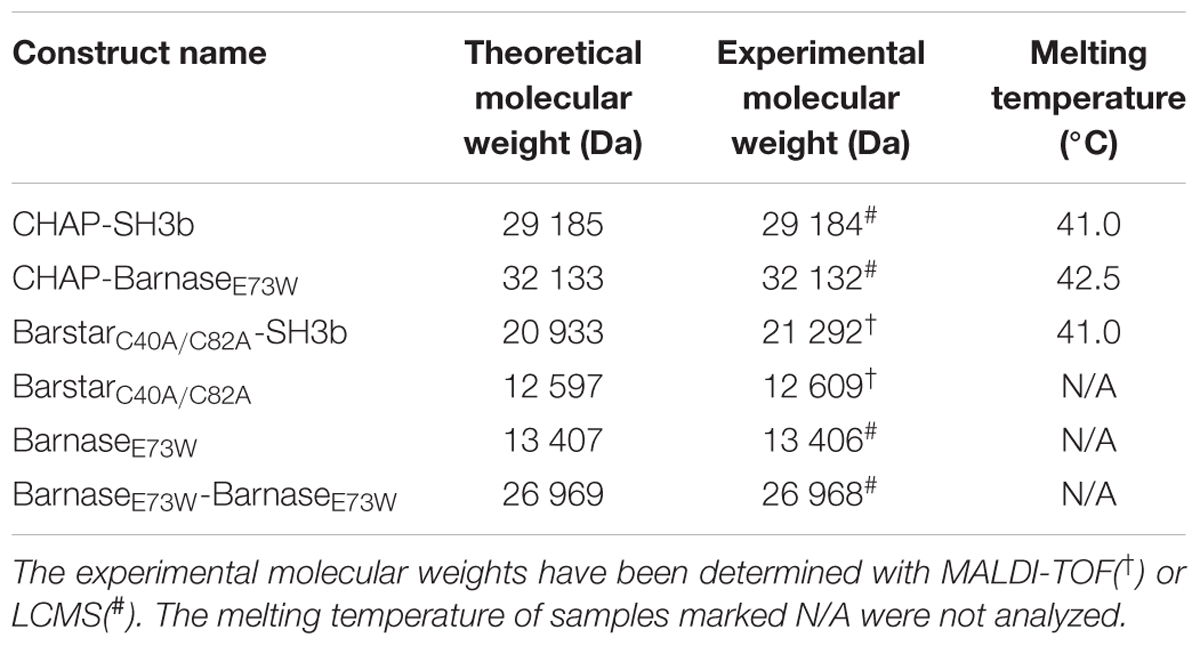
Table 1. Melting temperatures, theoretical and experimental molecular weights of constructs.
CHAP-BarnaseE73W Binds BarstarC40A/C82A-SH3b
The binding affinity between the barnaseE73W and the barstarC40A/C82A domains of the different constructs was investigated using surface plasmon resonance (SPR) technology. Here, BarnaseE73W-BarnaseE73W or CHAP-BarnaseE73W proteins were immobilized on the sensor chip followed by injection of BarstarC40A/C82A or BarstarC40A/C82A-SH3b proteins at different concentrations. The equilibrium dissociation constants (KD) of the interactions were determined from observed equilibrium responses (Figure 2 and Table 2). The affinity between BarstarC40A/C82A and BarnaseE73W-BarnaseE73W (Figure 2A) was determined to 59 nM. The BarstarC40A/C82A-SH3b fusion protein (Figure 2B) displayed a similar affinity (82 nM) as BarstarC40A/C82A suggesting that the interaction was not influenced by fusion to the SH3b moiety. Injections of BarstarC40A/C82A (Figure 2A) or the BarstarC40A/C82A-SH3b fusion protein (Figure 2B) over immobilized CHAP-BarnaseE73W showed that these interactions were of similar strength (108 and 73 nM, respectively). Injection of the negative controls bovine serum albumin (BSA), human serum albumin (HSA) and the monoclonal IgG antibody trastuzumab gave no responses (data not shown). Taken together, these results indicate that fusion of the CHAP or SH3b domains did not have a significant influence on the interaction strength between the barstarC40A/C82A and barnaseE73W domains. Neither did the fusions cause any detectable unspecific binding response.
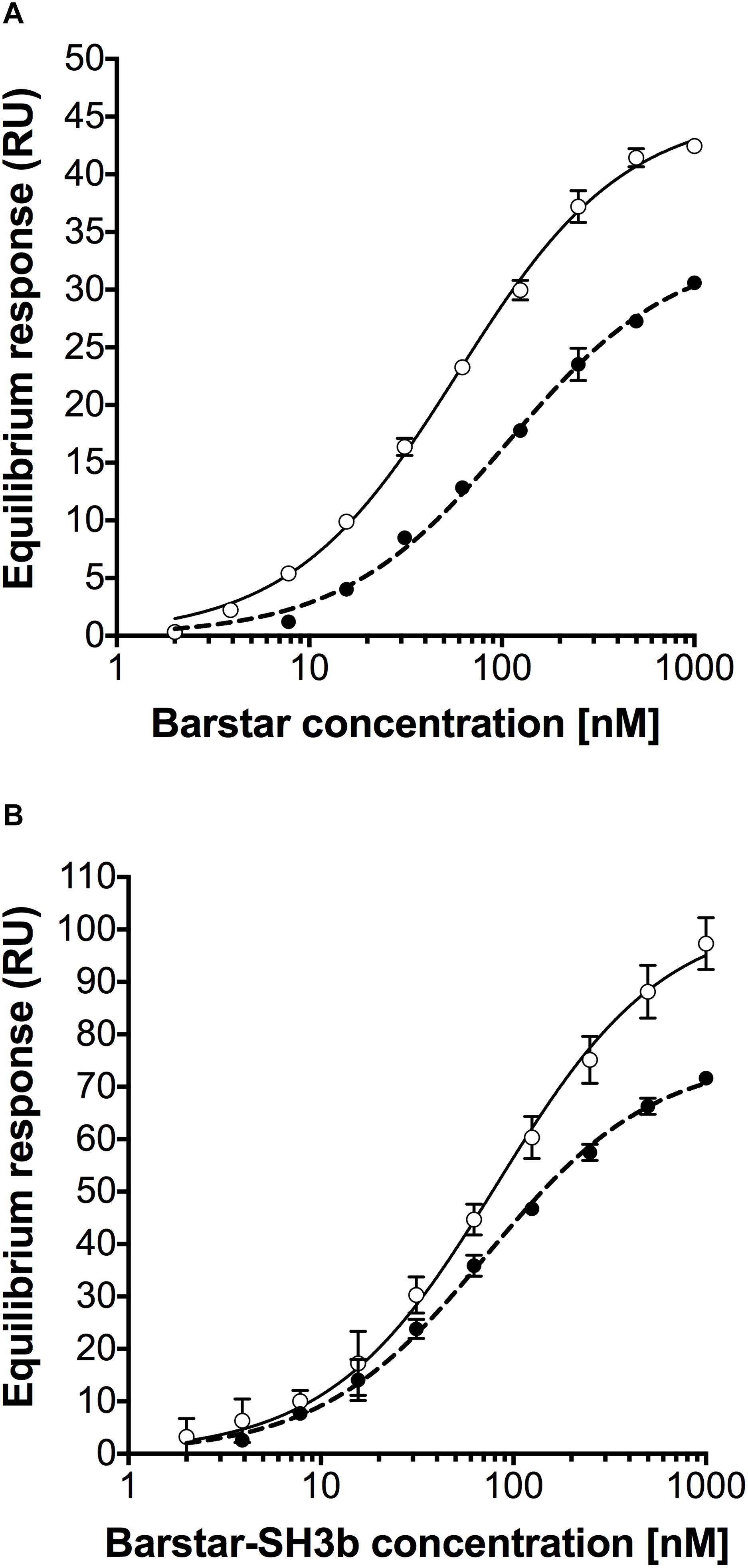
Figure 2. Equilibrium response curves from injection of (A) BarstarC40A/C82A over BarnaseE73W-BarnaseE73W surface (open circles), BarstarC40A/C82A over CHAP-BarnaseE73W surface (black circles), (B) BarstarC40A/C82A-SH3b over BarnaseE73W-BarnaseE73W surface (open circles), and BarstarC40A/C82A-SH3b over CHAP-BarnaseE73W surface (black circles). Error bars represent the standard error of the mean from two replicates.

Table 2. Affinities from surface plasmon resonance analyses (mean from two replicate series of measurements and standard deviation).
BarstarC40A/C82A-SH3b Binds Staphylococcus carnosus
Flow cytometry was used to investigate the cell wall binding properties of the BarstarC40A/C82A-SH3b fusion protein module. Here, different concentrations of biotinylated BarstarC40A/C82A-SH3b protein were incubated with Gram-positive S. carnosus. After washing, any cell-bound protein construct was detected using a streptavidin-phycoerythrin conjugate. The results showed that the BarstarC40A/C82A-SH3b fusion protein bound to S. carnosus in a concentration-dependent manner (Figure 3). This confirms that the recombinantly produced SH3b domain is able to bind to the Staphylococcus peptidoglycan layer (Gründling et al., 2006) and retains its binding after fusion to the barstarC40A/C82A moiety. In control experiments, E. coli cells were used and yielded no signal shift (Supplementary Figure S1) indicating no unspecific binding to outer membrane components of the surface of Gram-negative E. coli cells.
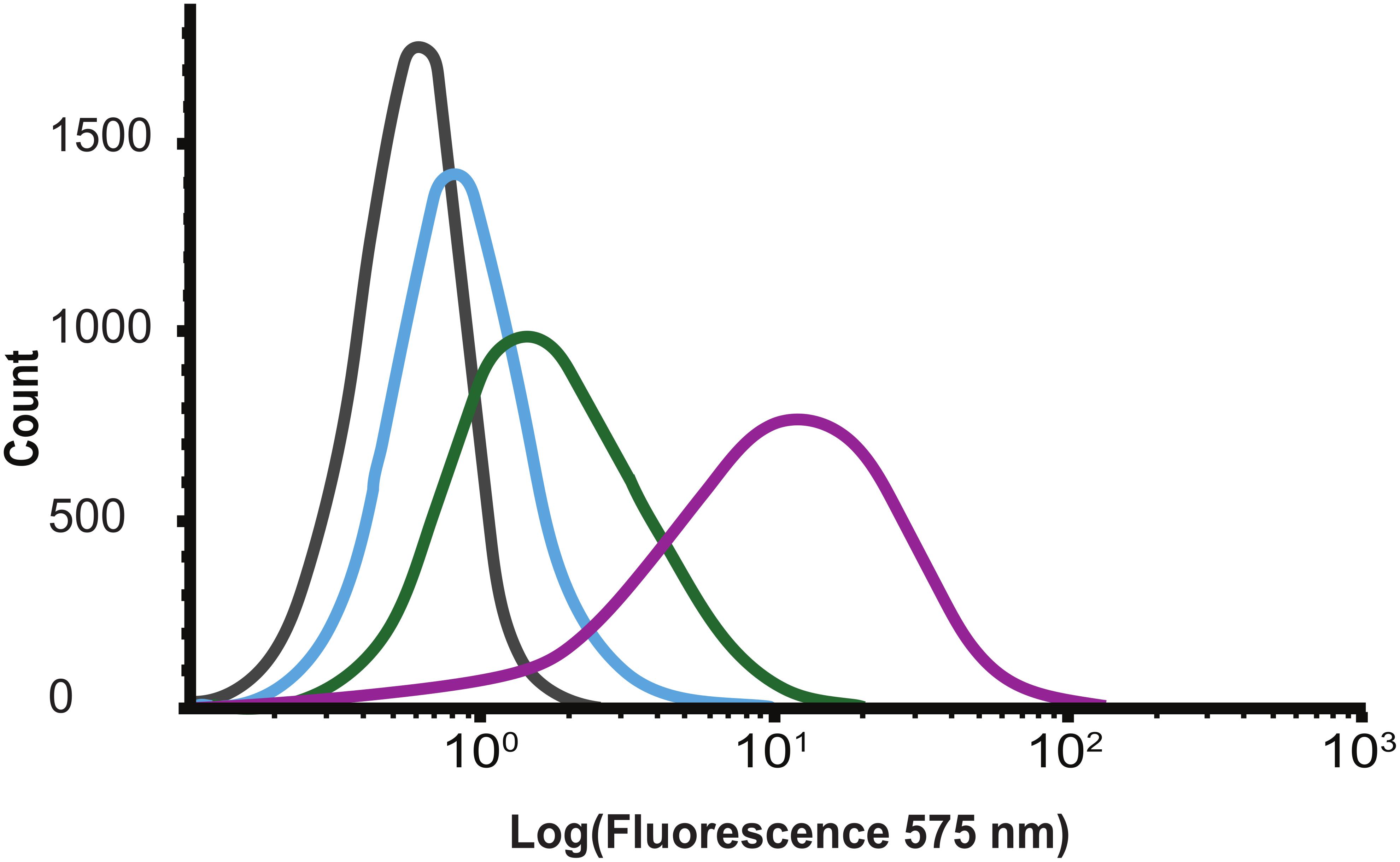
Figure 3. Flow cytometry histograms showing concentration dependent (gray 0, blue 110, green 330, and purple 1000 nM) binding of biotinylated BarstarC40A/C82A-SH3b to Staphylococcus carnosus.
The Assembled CHAP-BarnaseE73W/BarstarC40A/C82A-SH3b Lysin Shows Staphylolytic Activity
To investigate if a complex between the CHAP-BarnaseE73W and BarstarC40A/C82A-SH3b modules could result in a functional lysin, a turbidity reduction assay (TRA) involving S. carnosus cells was performed in which the individual BarstarC40A/C82A-SH3b and CHAP-BarnaseE73W modules were used as controls. As expected, no reduction of the turbidity was observed when cells were incubated with the BarstarC40A/C82A-SH3b module alone whereas a low reduction of the turbidity was observed for the CHAP-BarnaseE73W construct (Figure 4A and Table 3). This is in agreement with previously reported lytic effects of CHAP from LysK also in the absence of a CBD (Horgan et al., 2009). However, when cells were incubated with the heterodimerized CHAP-BarnaseE73W/BarstarC40A/C82A-SH3b complex, a significantly enhanced and concentration-dependent reduction in turbidity was observed (Figure 4A and Table 3). For comparison in the experiment, the CHAP-SH3b gene fusion reference construct was included. As could be expected, the cell lysis capacity observed for this covalently linked construct was considerably higher. Nevertheless, the fact that a cell lysis effect was seen for the non-covalently linked CHAP-BarnaseE73W/BarstarC40A/C82A-SH3b complex holds promise that the barnase/barstar-based system for modular lysin assembly has a potential to be used for qualitative assessment of the compatibility also of other individual EADs and CBDs. Once compatible combinations are identified, direct gene fusion between these could provide leads for further studies, including pre-clinical and clinical tests.
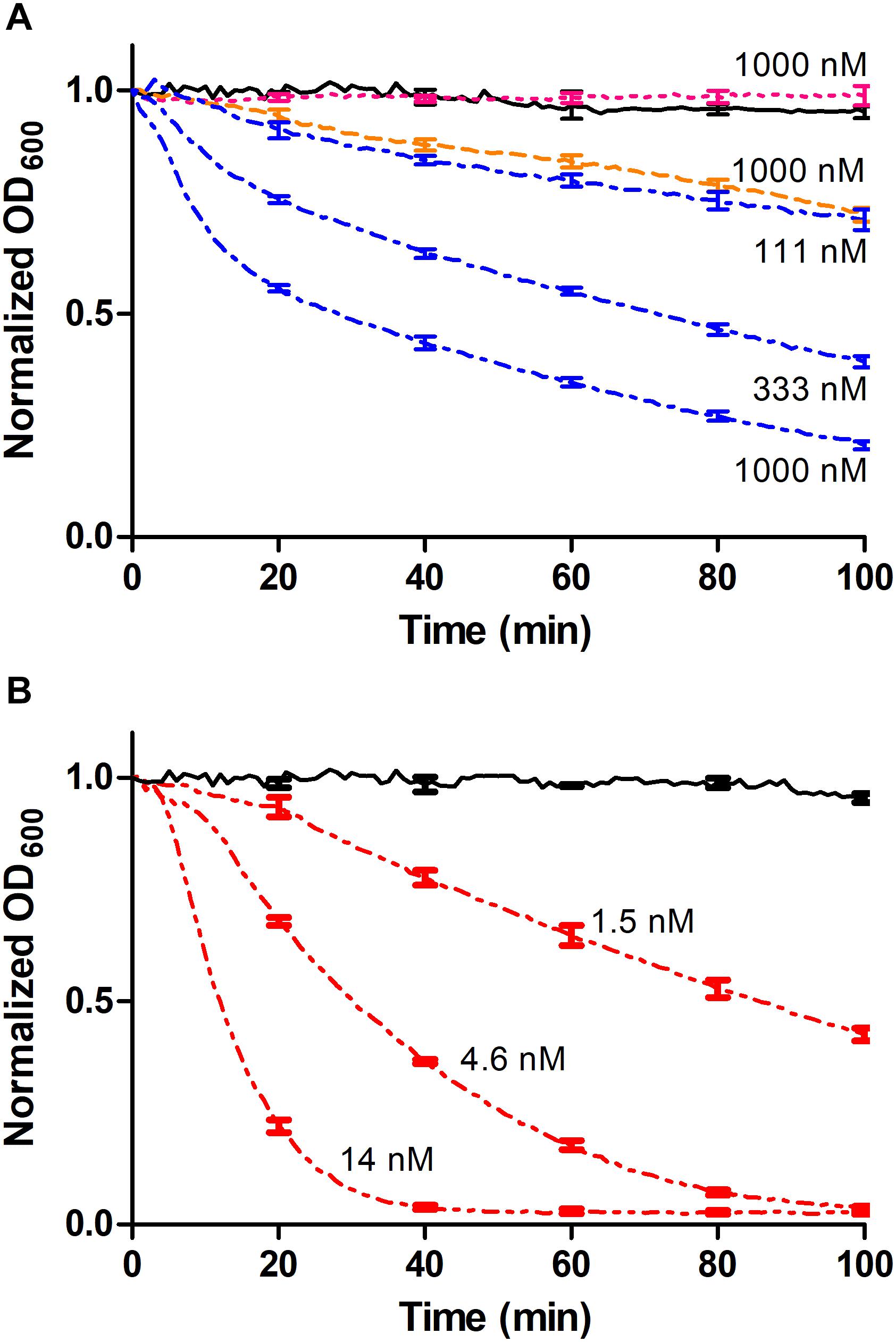
Figure 4. Turbidity reduction assays (TRA). Data points were collected every minute and error bars representing standard deviation is shown each 20 min for intelligibility. (A) The CHAP-BarnaseE73W/BarstarC40A/C82A-SH3b complex was used at concentrations of 1000, 333, and 111 nM, respectively (blue dashed/dotted line). The non-complexed controls BarstarC40A/C82A-SH3b (cyan dotted line) and CHAP-BarnaseE73W (orange dashed line) were used at a concentration of 1 μM and PBS (black continuous line) was used as negative control. (B) The CHAP-SH3b gene fusion had a concentration of 14, 4.6, and 1.5 nM, respectively (red dashed/dotted/dotted line) and PBS (black continuous line) was used as negative control.

Table 3. Specific activities of individual protein constructs and non-covalent protein complexes (mean and standard deviations from triplicate measurements).
The described modular assay concept would potentially benefit from investigating different linker types and lengths between included moieties and a stronger affinity between the appended dimerization domains to increase the fraction of modules being in complex to each other at a given concentration. For instance, in the TRA data presented in Figure 4A, the concentrations of the individual modules used were in the range 111 nM to 1000 nM, corresponding to approximately 1.5–14 times the equilibrium dissociation constant. In this concentration range only 45–76% of the individual modules could under ideal conditions be expected to be present in heterodimeric complexes, limiting the assay sensitivity. By mutating barnaseE73W and/or barstarC40A/C82A back to wild type, a significantly higher affinity between the modules could be expected, although this would be associated with difficulties during recombinant production due to toxicity from the barnase activity (Hartley, 1989). However, it has been shown to be possible to produce also the wild type barnase in E. coli, if co-expressed with the barstar inhibitor and having it removed by denaturation in situ during IMAC purification (Deyev et al., 2003). Alternatively, other cognate fusion partners as leucine zippers (Kostelny et al., 1992), anti-idiotypic affibody molecules (Eklund et al., 2002; Lindborg et al., 2013), or different fusion partner pairs capable of forming a covalent bond between themselves after binding, including intein moieties (Shah and Muir, 2014) or the SpyCatcher/SpyTag domains (Zakeri et al., 2012) could be used. Enzymatic coupling of separately produced modules using the transpeptidase sortase may also be considered (Mao et al., 2004).
Alternative means to screen for novel chimeric EAD-CBD combinations with desired lytic activities have been described, based on combinatorial assembly of lysin modules at the genetic level, rather than on the protein level as in the present study. Here, host cells expressing the different combinations are lysed enzymatically or chemically to release the lysin for assessment of the activity toward the investigated target bacterial cells, after which hits are identified via DNA sequencing (Yang et al., 2015, 2017; Verbree et al., 2017). Compared to the addition of controlled amounts of pre-purified lysins to target cells, these approaches may experience biases from variations in expression levels between different chimeras and a more limited control of the exact assay conditions. Nevertheless, such approaches are very interesting and add to the available means for investigating novel EAD-CBD combinations.
In conclusion, the described experiments show that CHAP and SH3b modules can be physically linked through the non-covalent barnase-barstar interaction and that formed complexes retain both the cell wall binding and the staphylolytic activity of the separate modules. Although only showed for one specific case, the results demonstrate for the first time the interesting principle that a functional lysin can be assembled from separately produced and affinity domain-tagged cell wall binding and catalytic modules, respectively. This holds promise for investigations of other EAD and CBD lysin modules using similar principles.
Materials and Methods
Chemicals and Reagents
All chemicals and reagents were bought from Sigma-Aldrich if not otherwise stated.
Cloning and Mutagenesis
DNA constructs were synthesized and cloned into pET-24a(+) or pET-26b(+) vectors (Merck, Darmstadt, Germany) by BioCat (Heidelberg, Germany) or BioBasic (Markham, ON, Canada). Site directed mutagenesis was performed by Biozilla (Sacramento, CA, United States).
Structure Prediction of Fusion Proteins
Amino acid sequences of the two fusion proteins CHAP-BarnaseE73W and BarstarC40A/C82A-SH3b were uploaded and run with standard settings in the protein structure predictor I-TASSER (Zhang, 2008). Returned predicted structures were aligned to the barnase/barstar complex (2ZA4) using the PyMOL Molecular Graphics System (Version 1.3, Schrödinger, LLC) and secondary structure elements were corrected to what has previously been seen in the barnase/barstar complex, LysK, and Lysostaphin (2ZA4, 4CSH, 5LEO) using UCSF Chimera 1.11.2 (Pettersen et al., 2004; Urakubo et al., 2008; Sanz-Gaitero et al., 2014).
Recombinant Expression and Purification
Plasmids were transformed into BL21 (DE3) competent E. coli cells (Merck, Darmstadt, Germany), and grown to an OD600 of 0.6 after which the culture was cooled on ice and induced with 0.5 mM IPTG (ThermoFisher Scientific, Waltham, MA, United States). Protein expression was performed overnight at 20°C and 200 rpm.
Protein expressing bacteria were pelleted using centrifugation, resuspended in lysis buffer [50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole, 30% glycerol, pH 8.0], homogenized using an EmulsiFlex-C3 (Avestin, Mannheim, Germany) and sonicated by a Vibra-Cell VCX 130 sonicator (Sonics, CT, United States). Cell debris was removed by centrifugation and lysates loaded on His GraviTrap columns (GE Healthcare, Uppsala, Sweden). Columns were washed with lysis buffer and protein eluted with elution buffer [50 mM NaH2PO4, 300 mM NaCl, 250 mM imidazole, 30% glycerol, pH 8.0]. The monomeric construct BarstarC40A/C82A with a HPC4-tag was purified on Anti-Protein C Affinity Matrix (Roche, Penzberg, Upper Bavaria, Germany) according to the manufacturer’s instructions. Size and purity of the produced protein constructs was verified using Mini-Protean TGX Gels (Bio-Rad, Hercules, CA, United States) and SCIEX 4200 MALDI-TOF (SCIEX, Framingham, MA, United States) or LCMS mass spectrometry (UltiMate 3000, ThermoFisher Scientific, Waltham, MA, United States; Impact II, Bruker, Billerica, MA, United States) according to manufacturer’s instructions.
Circular Dichroism Spectroscopy
CHAP-SH3b, CHAP-BarnaseE73W and BarstarC40A/C82A-SH3b were buffer exchanged to PBS and diluted to a final concentration of 0.3–0.5 mg/ml for analysis by circular dichroism. Using the Chirascan system (Applied Photophysics, Surrey, United Kingdom) with a 1 mm High precision cell (110-1P-40 cuvettes, Hellma Analytics, Germany). Ten wavelength scans were recorded between 190 and 280 nm at 20°C. All analyzed constructs gave signal at 210 nm and this wavelength was used to determine the melting point using a temperature gradient of 1°C/min.
Surface Plasmon Resonance
Proteins were buffer exchanged to PBST [150 mM NaCl, 8 mM Na2HPO4, 2 mM NaH2PO4, 0.005% Tween 20, pH 7.4] on PD10 columns (GE Healthcare, Uppsala, Sweden) and filtered prior to SPR analysis using a Biacore T200 system (GE Healthcare, Uppsala, Sweden) at 25°C. Concentrations of the constructs were determined by absorbance measurements.
Approximately 700 RU of BarnaseE73W-BarnaseE73W, and CHAP-BarnaseE73W dissolved in 10 mM NaOAc pH 4.5 were immobilized by amine coupling in individual flow cells on a CM5 Series S sensor chip (GE Healthcare, Uppsala, Sweden). BarstarC40A/C82A and BarstarC40A/C82A-SH3b were injected in duplicates using two-fold dilution series, spanning between 2 and 1000 nM at 30 μl/min with an association time of 500 and 1000 s dissociation time. Surfaces were regenerated between samples with two short (30 s) pulses of 10 mM HCl. Three control samples were injected at 1000 nM; HSA (Sigma-Aldrich, St. Louis, MO, United States), BSA (New England Biolabs, Ipswich, MA, United States) and the monoclonal antibody trastuzumab (Apoteket AB, Stockholm, Sweden).
Sensorgrams were double referenced toward the blank surface and a buffer injection. Report points were collected at the end of each injection for affinity calculations. In contrast to low analyte concentrations, responses at high concentrations did not level off at an equilibrium level in the end of the injections. This effect, probably caused by unspecific binding, gave a concentration-dependent linear contribution to the response curves, which was calculated and subtracted using GraphPad Prism (Version 5, San Diego, CA, United States). Equilibrium dissociation constants (KD) were calculated from plots of log(c) versus adjusted response signals.
Flow Cytometry
The specific binding of biotinylated BarstarC40A/C82A-SH3b to the cell wall of S. carnosus TM300 was evaluated by using a Gallios flow cytometer (Beckman Coulter, Brea, CA, United States). BarstarC40A/C82A-SH3b was labeled with EZ-Link NHS-LC-LC-Biotin (ThermoFisher Scientific, Waltham, MA, United States) according to manufacturer’s instructions. Successful biotinylation was verified by capture on streptavidin-coated magnetic beads (Dynabeads M280, ThermoFisher Scientific, Waltham, MA, United States) followed by analysis by SDS–PAGE (Bio-Rad Laboratories, Hercules, CA, United States) as well as on an SCIEX 4200 MALDI-TOF Mass Spectrometry system (SCIEX, Framingham, MA, United States) (data not shown). Bacteria for analysis were grown in Terrific Soy Broth supplemented with yeast extract (TSB+Y) overnight at 37°C, 150 rpm. The OD600 of the cultures was used to normalize the bacterial amount between experiments. Cells were pre-washed twice in PBS supplemented with 0.1 % (w/v) Pluronic F 108 NF Prill Poloxamer (PBS-P) before incubating with the biotinylated BarstarC40A/C82A-SH3b at the final concentration of 110, 330, or 1000 nM. Following the 45 min long incubation at room temperature, samples were washed twice and resuspended in Streptavidin R-Phycoerythrin conjugate solution (SA-PE; 0.5 μg/ml, #S866, ThermoFisher Scientific; Waltham, MA, United States) and continued to be incubated for 20 min in the dark and on ice. Cells were washed once and resuspended in cold PBS-P prior to flow-cytometric analysis. Fluorescent signal from the R-phycoerythrin was recorded for 200,000 cells per sample at 575 nm. The experiment was performed in duplicate and the data were analyzed using Kaluza (Version 2.1, Beckman Coulter, Brea, CA, United States). Controls included S. carnosus cells only, S. carnosus cells incubated with; biotinylated BarstarC40A/C82A-SH3b, secondary reagent (SA-PE), or BarstarC40A/C82A and secondary reagent (SA-PE). As well as a negative control composed of E. coli (BL21) cells and E. coli (BL21) cells stained with 1000 nM BarstarC40A/C82A-SH3b and secondary reagent (SA-PE).
Turbidity Reduction Assay
Staphylococcus carnosus TM300 substrate cells were grown to an OD600 of 0.4 as previously described by Schmelcher et al. (2012). The cells were pelleted, washed twice in cold buffer [10 mM Na2HPO4, 150 mM NaCl, 25% glycerol, pH 7.5], resuspended and stored at -80°C.
Triplicates of substrate cells and lysin modules were prepared in PBS in a 96-well plate to a total volume of 200 μl. Final OD600 was equal to 1 for the substrate cells and concentrations of the lysin modules ranged from 0.15 to 1000 nM in three-fold dilution steps. PBS was used as negative control. Reduction in OD600 was monitored over time at room temperature using a POLARstar Omega plate reader (BMG Labtech, Cary, NC, United States) and used to calculate the enzymatic activity.
The specific enzymatic activities of the individual constructs and the non-covalent complexes were calculated. First, the steepest slope [ΔOD600/min] of the lysis curves was identified in sigmoid [A], exponential [B] or linear [C] functions to best fit the lysis data. The slope of the PBS controls was subtracted. The resulting values were plotted against the respective construct concentration and function [D] was fitted to the emerging curve. The specific enzymatic activity, namely the slope [ΔOD600/(min∗μM)] of the curve in origo, was calculated as [k ∗a] of function [D].
Data Availability
All datasets generated for this study are included in the manuscript and/or the Supplementary Files.
Author Contributions
LH, JN, P-ÅN, and JS conceived and designed the experiments. LH and JS performed the experiments. LH, JN, P-ÅN, AN, and JS analyzed the data. JN contributed to reagents, materials, and analysis tools. LH, JN, P-ÅN, AN, and JS wrote the manuscript. AN and JS provided the funding.
Funding
This work was supported by the Sixten Gemzeus Foundation (to JS), the Hans Werthén Foundation of the Royal Swedish Academy of Engineering Sciences (to JS), the Sven and Lilly Lawski Foundation (to JS), the Olle Engkvist Byggmästare Foundation [2015/419] (to AN and JS), and the Skolar Award (to JS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Erik Fasterius and Jan-Olov Persson for help with analysis tools and Anne-Sophie Svensson for assistance with LCMS analyses.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00558/full#supplementary-material
References
Becker, S. C., Dong, S., Baker, J. R., Foster-Frey, J., Pritchard, D. G., and Donovan, D. M. (2009). LysK CHAP endopeptidase domain is required for lysis of live staphylococcal cells. FEMS Microbiol. Lett. 294, 52–60. doi: 10.1111/j.1574-6968.2009.01541.x
Croux, C., Ronda, C., López, R., and García, J. L. (1993). Role of the C-terminal domain of the lysozyme of Clostridium acetobutylicum ATCC 824 in a chimeric pneumococcal-clostridial cell wall lytic enzyme. FEBS Lett. 336, 111–114. doi: 10.1016/0014-5793(93)81621-6
Czaplewski, L., Bax, R., Clokie, M., Dawson, M., Fairhead, H., Fischetti, V. A., et al. (2016). Alternatives to antibiotics — a pipeline portfolio review. Lancet Infect. Dis. 16, 239–251. doi: 10.1016/S1473-3099(15)00466-1
Deyev, S. M., Waibel, R., Lebedenko, E. N., Schubiger, A. P., and Plückthun, A. (2003). Design of multivalent complexes using the barnase∗barstar module. Nat. Biotechnol. 21, 1486–1492. doi: 10.1038/nbt916
Diaz, E., Lopez, R., and Garcia, J. L. (1990). Chimeric phage-bacterial enzymes: a clue to the modular evolution of genes. Proc. NatI. Acad. Sci. U.S.A. 87, 8125–8129. doi: 10.1073/pnas.87.20.8125
Eklund, M., Axelsson, L., Uhlén, M., and Nygren, P. Å (2002). Anti-idiotypic protein domains selected from protein A-based affibody libraries. Proteins 48, 454–462. doi: 10.1002/prot.10169
Gründling, A., Schneewind, O., and Gru, A. (2006). Cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus cross-linked peptidoglycan mediates lysostaphin binding to the cell wall envelope of Staphylococcus aureus †. J. Bacteriol. 188, 2463–2472. doi: 10.1128/JB.188.7.2463
Hartley, R. W. (1988). Barnase and barstar. expression of its cloned inhibitor permits expression of a cloned ribonuclease. J. Mol. Biol. 202, 913–915. doi: 10.1016/0022-2836(88)90568-2
Hartley, R. W. (1989). Barnase and barstar: two small proteins to fold and fit together. Trends Biochem. Sci. 14, 450–454.
Hartley, R. W. (1993). Directed mutagenesis and barnase-barstar recognition. Biochemistry 32, 5978–5984. doi: 10.1021/bi00074a008
Herpers, B. L., Badoux, P., Totté, J. E. E., Pietersma, F., Eichenseher, F., and Loessner, M. J. (2014). Specific Lysis of Staphylococcus Aureus by the Bacteriophage Endolysin Staphefekt Sa.100: in Vitro Studies and Human Case Series. in Antibiotic Alternatives for the New Millennium 5th - 7th Nov 2014 London, UK. London: EuroSciCon.
Horgan, M., O’Flynn, G., Garry, J., Cooney, J., Coffey, A., Fitzgerald, G. F., et al. (2009). Phage lysin LysK can be truncated to its CHAP domain and retain lytic activity against live antibiotic-resistant staphylococci. Appl. Environ. Microbiol. 75, 872–874. doi: 10.1128/AEM.01831-08
Jun, S. Y., Jang, I. J., Yoon, S., Jang, K., Yu, K., Cho, J. Y., et al. (2017). Pharmacokinetics and tolerance of the phage endolysin-based candidate drug SAL200 after a single intravenous administration among healthy volunteers. Antimicrob. Agents Chemother. 61:e02629-16. doi: 10.1128/AAC.02629-16
Khait, R., and Schreiber, G. (2012). FRETex: a FRET-based, high-throughput technique to analyze protein-protein interactions. Protein Eng. Des. Sel. 25, 681–687. doi: 10.1093/protein/gzs067
Kostelny, S. A., Cole, M. S., and Tso, J. Y. (1992). Formation of a bispecific antibody by the use of leucine zippers. J. Immunol. 148, 1547–1553.
Lindborg, M., Dubnovitsky, A., Olesen, K., Björkman, T., Abrahmsén, L., Feldwisch, J., et al. (2013). High-affinity binding to staphylococcal protein A by an engineered dimeric affibody molecule. Protein Eng. Des. Sel. 26, 635–644. doi: 10.1093/protein/gzt038
Mao, H., Hart, S. A., Schink, A., and Pollok, B. A. (2004). Sortase-mediated protein ligation: a new method for protein engineering. J. Am. Chem. Soc. 126, 2670–2671. doi: 10.1021/ja039915e
Pastagia, M., Euler, C., Chahales, P., Fuentes-duculan, J., Krueger, J. G., and Fischetti, V. A. (2011). A novel chimeric lysin shows superiority to mupirocin for skin decolonization of methicillin-resistant and -sensitive staphylococcus aureus strains. Antimicrob. Agents Chemother. 55, 738–744. doi: 10.1128/AAC.00890-10
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF chimera - a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Ramachandran, S., and Udgaonkar, J. B. (1996). Stabilization of barstar by chemical modification of the buried Cysteines. Biochemistry 35, 8776–8785. doi: 10.1021/bi9600759
Rezaie, A. R., Fiore, M. M., Neuenschwander, P. F., Esmon, C. T., and Morrissey, J. H. (1992). Expression and purification of a soluble tissue factor fusion protein with an epitope for an unusual calcium-dependent antibody. Protein Expr. Purif. 3, 453–460. doi: 10.1016/1046-5928(92)90062-2
Rodríguez-Rubio, L., Martínez, B., Rodríguez, A., Donovan, D. M., Götz, F., and García, P. (2013). The phage lytic proteins from the Staphylococcus aureus bacteriophage vB_SauS-phiIPLA88 display multiple active catalytic domains and do not trigger staphylococcal resistance. PLoS One 8:e64671. doi: 10.1371/journal.pone.0064671
Rogers, H., Perkins, H., and Ward, J. (eds) (1980). “The bacterial autolysins,” in Microbial Cell Walls and Membranes, (London: Chapman & Hall Ltd).
Sanz-Gaitero, M., Keary, R., Garcia-Doval, C., Coffey, A., and van Raaij, M. J. (2014). Crystal structure of the lytic CHAP(K) domain of the endolysin LysK from Staphylococcus aureus bacteriophage K. Virol. J. 11:133. doi: 10.1186/1743-422X-11-133
Schindler, C. A., and Schuhardt, V. T. (1964). Lysostaphin: a new bacteriolytic agent for the Staphylococcus. Proc. Natl. Acad. Sci. U. S. A. 51, 414–421. doi: 10.1073/pnas.51.3.414
Schmelcher, M., Korobova, O., Schischkova, N., Kiseleva, N., Kopylov, P., Pryamchuk, S., et al. (2012). Staphylococcus haemolyticus prophage ΦSH2 endolysin relies on cysteine, histidine-dependent amidohydrolases/peptidases activity for lysis “from without.”. J. Biotechnol. 162, 289–298. doi: 10.1016/j.jbiotec.2012.09.010
Schmelcher, M., Tchang, V. S., and Loessner, M. J. (2011). Domain shuffling and module engineering of Listeria phage endolysins for enhanced lytic activity and binding affinity. Microb. Biotechnol. 4, 651–662. doi: 10.1111/j.1751-7915.2011.00263.x
Schreiber, G., and Fersht, A. R. (1993). Interaction of barnase with its polypeptide inhibitor barstar studied by protein engineering. Biochemistry 32, 5145–5150. doi: 10.1021/bi00070a025
Schreiber, G., Frisch, C., and Fersht, A. R. (1997). The role of Glu73 of barnase in catalysis and the binding of barstar. J. Mol. Biol. 270, 111–122. doi: 10.1006/jmbi.1997.1080
Schuch, R., Lee, H. M., Schneider, B. C., Sauve, K. L., Law, C., Khan, B. K., et al. (2014). Combination therapy with lysin CF-301 and antibiotic is superior to antibiotic alone for treating methicillin-resistant staphylococcus aureus-induced murine bacteremia. J. Infect. Dis. 209, 1469–1478. doi: 10.1093/infdis/jit637
Shah, N. H., and Muir, T. W. (2014). Inteins: nature’s gift to protein chemists. Chem. Sci. 5, 446–461. doi: 10.1039/C3SC52951G
Totté, J., de Wit, J., Pardo, L., Schuren, F., van Doorn, M., and Pasmans, S. (2017). Targeted anti-staphylococcal therapy with endolysins in atopic dermatitis and the effect on steroid use, disease severity and the microbiome: study protocol for a randomized controlled trial (MAAS trial). Trials 18:404. doi: 10.1186/s13063-017-2118-x
Urakubo, Y., Ikura, T., and Ito, N. (2008). Crystal structural analysis of protein-protein interactions drastically destabilized by a single mutation. Protein Sci. 17, 1055–1065. doi: 10.1110/ps.073322508
Verbree, C. T., Dätwyler, S. M., Meile, S., Eichenseher, F., Donovan, D. M., Loessner, M. J., et al. (2017). Identification of peptidoglycan hydrolase constructs with synergistic staphylolytic activity in cow milk. Appl. Environ. Microbiol. 83:e03445-16. doi: 10.1128/AEM.03445-16
Yang, H., Linden, S. B., Wang, J., Yu, J., Nelson, D. C., and Wei, H. (2015). A chimeolysin with extended-spectrum streptococcal host range found by an induced lysis-based rapid screening method. Sci. Rep. 5:17257. doi: 10.1038/srep17257
Yang, H., Zhang, H., Wang, J., Yu, J., and Wei, H. (2017). A novel chimeric lysin with robust antibacterial activity against planktonic and biofilm methicillin- resistant Staphylococcus aureus. Sci. Rep. 7:40182. doi: 10.1038/srep40182
Zakeri, B., Fierer, J. O., Celik, E., Chittock, E. C., Schwarz-Linek, U., Moy, V. T., et al. (2012). Peptide tag forming a rapid covalent bond to a protein, through engineering a bacterial adhesin. Proc. Natl. Acad. Sci. 109, E690–E697. doi: 10.1073/pnas.1115485109
Keywords: endolysin, exolysin, barnase, barstar, fusion protein, non-covalent interaction, Staphylococcus, antibiotic alternative
Citation: Hjelm LC, Nilvebrant J, Nygren P-Å, Nilsson AS and Seijsing J (2019) Lysis of Staphylococcal Cells by Modular Lysin Domains Linked via a Non-covalent Barnase-Barstar Interaction Bridge. Front. Microbiol. 10:558. doi: 10.3389/fmicb.2019.00558
Received: 18 December 2018; Accepted: 04 March 2019;
Published: 22 March 2019.
Edited by:
Bart Devreese, Ghent University, BelgiumReviewed by:
Konstantin Anatolievich Miroshnikov, Shemyakin–Ovchinnikov Institute of Bioorganic Chemistry (RAS), RussiaHongping Wei, Wuhan Institute of Virology (CAS), China
Copyright © 2019 Hjelm, Nilvebrant, Nygren, Nilsson and Seijsing. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Johan Seijsing, am9oYW4uc2VpanNpbmdAc3Uuc2U=