 Mitsuo Ogura
Mitsuo Ogura Tsutomu Sato2
Tsutomu Sato2 Kimihiro Abe
Kimihiro Abe- 1Institute of Oceanic Research and Development, Tokai University, Shizuoka, Japan
- 2Department of Frontier Bioscience, Hosei University, Koganei, Japan
- 3Research Center for Micro-Nano Technology, Hosei University, Koganei, Japan
Glucose is the most favorable carbon source for many bacteria, which have several glucose-responsive gene networks. Recently, we found that in Bacillus subtilis glucose induces the expression of the extracellular sigma factor genes sigX and sigM through the acetylation of CshA (RNA helicase), which associates with RNA polymerase (RNAP). We performed a transposon mutagenesis screen for mutants with no glucose induction (GI) of sigX-lacZ. While screening for such mutants, we recently found that the GI of sigX/M involves YlxR, a nucleoid-associated protein (NAP) that regulates nearly 400 genes, including metabolic genes. It has been shown that acetylated CshA positively regulates expression of ylxR-containing operon. Here, we report additional mutations in yqfO or tsaD required for the GI of sigX. YqfO contains a universally conserved domain with unknown function. YqfO and YlxR were found to regulate expression of the tsaEBD-containing operon. Mutational analysis using lacZ fusions revealed the adenine-rich cis-element for YlxR. TsaD is a component of the TsaEBD enzyme required for the synthesis of threonylcarbamoyl adenosine (t6A). The t6A modification of tRNA is universal across the three domains of life. Western blot analysis showed that the tsaD mutation in the presence of glucose reduced levels of soluble PdhA, PdhB, and PdhD, which are subunits of the pyruvate dehydrogenase complex (PDHc). This resulted in severely defective PDHc function and thus reduced concentrations of cellular acetyl-CoA, a reaction product of PDHc and plausible source for CshA acetylation. Thus, we discuss a suggested glucose-responsive system (GRS) involving self-reinforcing CshA acetylation. This self-reinforcing pathway may contribute to the maintenance of the acetyl-CoA pool for protein acetylation.
Introduction
Glucose is the most favorable carbon source for many bacteria, and these bacteria have several glucose-responsive networks (Deutscher, 2008). In Gram-positive bacteria, including Bacillus subtilis, the transcription factor CcpA is the master regulator for the carbon catabolite regulation (Deutscher, 2008; Fujita, 2009). The incorporation of glucose in the bacterial culture medium results in an increase of the metabolite fructose 1,6-bisphosphate, which triggers the phosphorylation of Ser46 of HPr, a phosphocarrier protein in the sugar phosphotransferase system (P-Ser-HPr). P-Ser-HPr associates with and activates CcpA, leading to global positive and negative effects on the transcriptional network. Moreover, there are several additional glucose-responsive transcription factors, such as CcpC, CcpN, CggR, and GlcT (Fujita, 2009). In Escherichia coli catabolite gene-activator protein CAP has been conventionally considered a transcription factor responding to glucose. However, recent genomic analyses led to an idea, that CAP is also a nucleoid-associated protein (NAP, Dillon and Dorman, 2010; Sandhya et al., 2015).
Accumulated studies identified proteins called as NAP which are not structurally related to histones but have similar functions to histones in bacteria (Drlica and Rouviere-Yaniv, 1987; Browning et al., 2010; Dillon and Dorman, 2010). NAPs have many roles in transcription, recombination including phage-infection, and chromosome condensation, rearrangement, maintenance, and segregation (Dillon and Dorman, 2010). NAPs generally have non-specific DNA-binding activity or recognize local DNA structure (Browning et al., 2010). However, NAPs, such as Fis and IHF bind to specific DNA sequences (Azam and Ishihama, 1999). The modes of transcriptional regulation of NAPs are diverse, for example, H-NS inhibits RNA polymerase (RNAP) progression on DNA, while Fis regulates transcription through various modes of interaction with RNAP (Dillon and Dorman, 2010). YlxR is a NAP of B. subtilis, which either positively or negatively regulates approximately 400 genes (Ogura and Kanesaki, 2018). Furthermore, YlxR is shown to be involved in the glucose induction (GI) of various genes, including sigX and sigM, which encode extracellular function (ECF) sigma factors SigX and SigM, respectively (Helmann, 2016; Ogura and Asai, 2016; Ogura and Kanesaki, 2018).
Several hundreds of lysine-acetylated proteins have been identified in many bacteria (Ouidir et al., 2016; Carabetta and Cristea, 2017). Glucose addition to the medium often induced protein acetylation in bacteria, such as E. coli and B. subtilis (Lima et al., 2011; Kosono et al., 2015; Schilling et al., 2015). Proteomic analysis of B. subtilis revealed that CshA, one of the DEAD-box helicases, is acetylated (Lehnik-Habrink et al., 2013; Kosono et al., 2015). We recently found that addition of glucose stimulated lysine acetylation of CshA (Ogura and Asai, 2016). CshA is also known to associate with RNAP (Delumeau et al., 2011). The association of acetylated CshA with RNAP would enhance its affinity to SigX and SigM (Figure 1; Helmann, 2016; Ogura and Asai, 2016). This leads to GI of sigX and sigM (Shiwa et al., 2015; Ogura and Asai, 2016). In most cases, ECF sigma factors are subject to membrane-embedded anti-sigma factors, which trap a cognate ECF sigma factor, leading to inactivation of the ECF sigma factor (Helmann, 2016). However, CshA-dependent GI of SigX/M is not under control of anti-sigma factors (Ogura and Asai, 2016). The GI of sigX caused by acetylation of CshA was susceptible to disruption by the mutation of genes encoding pyruvate dehydrogenase (PDH), namely pdhABCD (Gao et al., 2002; Ogura and Asai, 2016). PDH consists of the multi-enzyme subunit (PDHc) and is an enormously large protein complex. The disruption of the pdh genes would result in the reduction of the intracellular acetyl-CoA pool, which is affected by the activity of PDH, that is, the conversion of pyruvate to acetyl-CoA. PDH has three enzymatic activities and three components: PDH [E1 (PdhA and PdhB)], dihydrolipoamide acetyltransferase [E2 (PdhC)], and lipoamide dehydrogenase [E3 (PdhD)] (Hodgson et al., 1983). Additionally, two genes are involved in acetyl-CoA metabolism through synthesis of acetyl-phosphate, pta encoding phosphotranacetylase and ackA encoding acetyl kinase. Moreover, with glucose as the carbon source cellular concentrations of acetyl-phosphate decrease in the pta strain, while it accumulates in the ackA strain (Klein et al., 2007) and both mutations have severe effects on the B. subtilis acetylated proteome (Kosono et al., 2015).
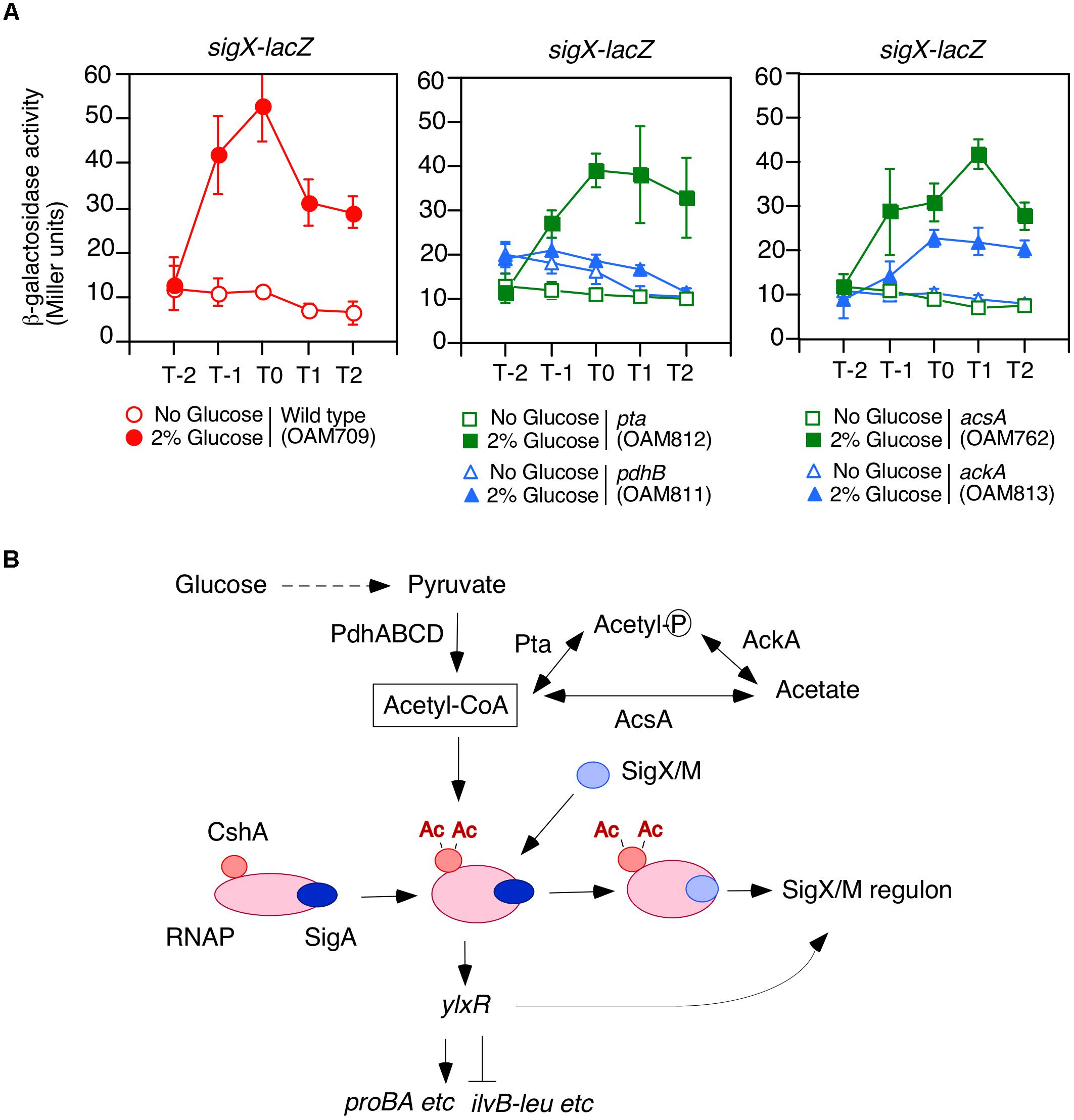
Figure 1. Requirement of acetyl-CoA produced by PDHc for GI of sigX-lacZ. (A) Expression of sigX-lacZ in the mutants. Means of the β-Gal activities from three independent experiments and the standard deviations are shown. The x axis represents the growth time in hours relative to the end of vegetative growth (T0). All strains are the derivatives of wild-type OAM709 and the relevant genotype is indicated below the panel. (B) Model of GI of sigX/sigM and acetyl-CoA metabolism. Glucose addition stimulates the acetylation of CshA (Ogura and Asai, 2016). CshA has been shown to associate with RNAP. RNAP with acetylated CshA may stimulate the replacement of σX/M for σA in the RNAP holoenzyme. σA -associated RNAP holoenzyme with acetylated CshA stimulate the transcription of ylxR (Ogura and Kanesaki, 2018). YlxR regulates many genes and changes the cellular metabolic state, which may lead to induction of the SigX/M regulons. Ac, acetyl moiety; PdhABCD, pyruvate dehydrogenase (PDH); Pta, phosphotransacetylase; AcsA, acetyl-CoA syntheatse; AckA, acetate kinase. “P” in the circle indicates phosphate residue.
The transposon mutagenesis screen for mutants with no GI of sigX-lacZ revealed yqfO and tsaD in addition to the previously analyzed cshA and ylxR genes (Ogura and Asai, 2016; Ogura and Kanesaki, 2018). Here we analyzed yqfO and tsaD in detail. TsaD is a component of a tRNA modification enzyme that is required for the synthesis of threonylcarbamoyl adenosine (t6A) (Thiaville et al., 2014, 2015). The CshA acetylation induced ylxR-containing operon expression (Ogura and Kanesaki, 2018) and YlxR and YqfO regulated the tsaD-containing operon. In the tsaD mutant, the soluble PDHc subunits were markedly reduced in the presence of glucose. This would contribute to the observed low intracellular acetyl-CoA pool and result in reduced CshA acetylation. The finding that the disruption of tsaD decreases the soluble PDHc subunits suggests a relationship between the lack of t6A and protein quality control.
Materials and Methods
Strains, Media, and β-Galactosidase Analysis
All B. subtilis strains used in this study are listed in Table 1 and Supplementary Table S1. One-step competence medium (MC; Kunst et al., 1994), Schaeffer’s sporulation medium (Schaeffer et al., 1965), and Luria-Bertani (LB) medium (Difco, Lennox) were used. Antibiotic concentrations were described previously (Ogura and Tanaka, 1996; Ogura et al., 1997). Synthetic oligonucleotides were commercially prepared by Tsukuba Oligo Service (Ibaraki, Japan) and are listed in Supplementary Table S2. Methods of β-galactosidase analysis using ONPG(2-Nitrophenyl-β-D-galactopyranoside) were previously described (Ogura and Tanaka, 1996). β-galactosidase analysis using CRPG (Chlorophenol red β-D-galactopyranoside, Roche, Germany) was performed using method similar to those used for ONPG. Since CRPG is the red pigment, 0.7 ml of Z-buffer with 0.2 ml of CRPG solution (4 mg/ml in Z-buffer) and 0.5 ml of 1 M Na2CO3 in water was mixed and used for measurement of OD570 as a blank control. To calculate Miller units, OD550 values were measured using cell suspension samples processed in parallel.
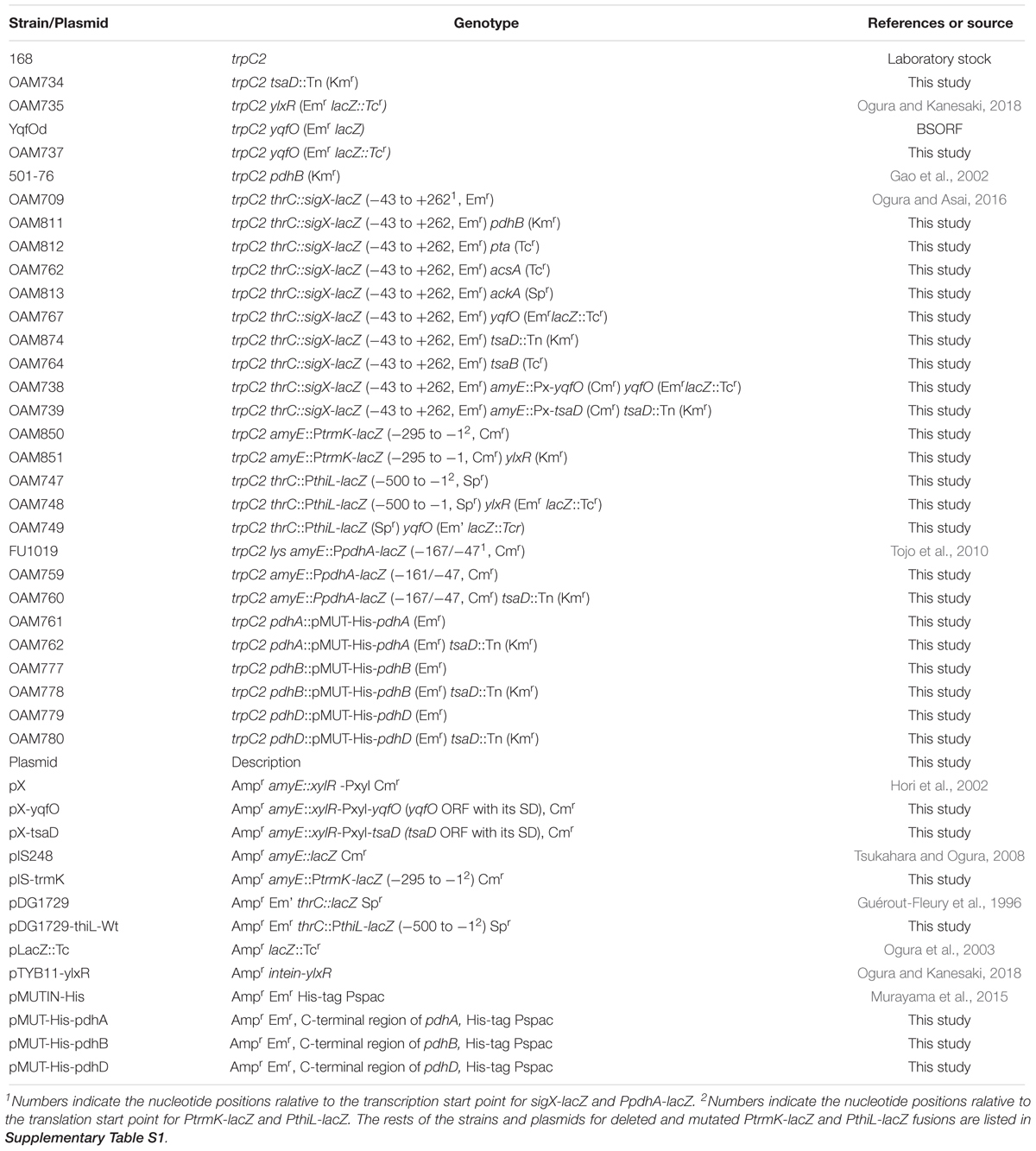
Table 1. Strains and plasmids used in the study.
Growth Condition
Strains were grown on a LB agar plate containing appropriate antibiotics at 37°C overnight. The cells were scraped and suspended in the sporulation medium. The suspension was inoculated into 50 ml sporulation medium (with or without 2% glucose) without antibiotics in a 200 ml flask. Klett value was adjusted around 10 units. The flask was gently shaken (110 reciprocation/min) at 37°C. Cell growth was monitored with Klett calorimeter (Klett Mfg. Co., Inc., New York, NY).
Strain Construction
The pta::Tcr, acsA::Tcr, tsaB::Tcr, and ackA::Spr units were constructed using PCR. Briefly the upstream and downstream regions of the concerned genes and Tcr from pBEST304 (Itaya, 1992) and Spr from pDG1729 (Guérout-Fleury et al., 1996) were amplified using the indicated primers (Supplementary Table S2) and then combined by PCR. These units were directly used for transformation of B. subtilis 168. From the resultant Tcr and Spr strains total DNAs were taken. Those were used in PCR to confirm the expected chromosomal structure as template.
Plasmid Construction
The plasmids used in this study are listed in Table 1 and Supplementary Table S1. pX-yqfO and pX-tsaD were constructed by cloning of the PCR products amplified with the oligonucleotide pairs, pX-yqfO-Spe/pX-yqfO-Bgl(SpeI/BglI digestion) and pX-gcp-Spe/pX-gcp-Bam (SpeI/BamHI digestion), respectively, into pX treated with SpeI/BamHI (Hori et al., 2002). To construct pDG1729-thiL, the PCR product amplified with the oligonucleotide pair pDG1729-gcp-E/pDG1729-gcp-B was digested with EcoRI/BamHI and cloned into pDG1729 treated with the same enzymes (Guérout-Fleury et al., 1996). To construct pIS-trmK, the PCR product amplified with the oligonucleotide pair yqfO-Eco/pIS-trmK-B was digested with EcoRI/BamHI and cloned into pIS284 treated with the same enzymes (Tsukahara and Ogura, 2008). To construct pMUT-His-pdhA, pMUT-His-pdhB, and pMUT-His-pdhD, the PCR products amplified with the oligonucleotide pairs, pdhA-F-E/pdhA-R-Xh, pdhB-F-E/pdhB-R-Xh, and pdhD-F-E/pdhD-R-Xh were digested with EcoRI/XhoI and cloned into pMUTIN-His treated with the same enzymes (Murayama et al., 2015).
Western Blot Analysis
Cells were grown in 50 ml of sporulation medium with or without 2% glucose. At T1, cells were harvested and washed with 1 ml of TBS buffer (10 mM Tris–HCl pH 7.5, 150 mM NaCl) containing 1 mM PMSF. Cells were disrupted with French Pressure Cell and centrifuged (15000 × g) to obtain a cleared lysate. Western blot analysis was performed by a method similar to that described previously (Hata et al., 2001). Monoclonal mouse anti-His tag antibody was purchased from Medical and Biological Laboratories (Nagoya, Japan). Polyclonal rabbit anti-SigA antibody was previously described (Ogura, 2016). These antibodies were diluted (1/1000) in Can Get Signal solution 1 (ToYoBo, Tokyo, Japan), and Can Get Signal solution 2 (ToYoBo) was used for secondary antibody.
Fractionation of Membrane and Aggregated Proteins
During preparation of cleared cell lysate, the cell pellets were obtained after centrifugation. The pellets were solubilized in 0.3 ml of 1% Triton X-100 in TBS buffer at 4°C for 30 min and then centrifuged (10000 × g) for 5 min. The obtained pellets were again solubilized in 0.3 ml of 0.5% Triton X-100 in TBS buffer at 4°C for 30 min and then centrifuged (10000 × g) for 5 min. Both solubilized fractions were mixed (membrane protein fractions). The obtained pellets were fractions of aggregated proteins and solubilized in 0.6 ml of 7 M urea, 100 mM DTT and 4% CHAPS at 4°C for 10 min. These fractions were examined by SDS-PAGE and western bolt analysis. The methods were similar to those described in a previous paper (Runde et al., 2014). In the study, mass spectrometric analysis showed only 1–4% contaminated membrane proteins in the aggregates fraction.
Examination of the Concentration of Intracellular Acetyl-CoA
Cells were grown in 50 ml of sporulation medium with or without 2% glucose. At T0, 30 ml of cells was harvested and washed with TEN buffer (20 mM Tris–HCl pH 7.5, 1 mM EDTA, 150 mM NaCl, and 1 mM PMSF). Cells were disrupted with French Pressure Cell and centrifuged (15000 × g). The resulting cleared cell lysate was treated with a Deproteinizing Sample Preparation kit (Biovision, CA, United States). Measurement of the acetyl CoA concentration was performed with a PicoProbe Acetyl CoA Fluorometric Assay kit (Biovision).
Results
Requirement of Acetyl-CoA Produced by PDHc for Glucose Induction of sigX-lacZ
We observed that a mutation that disrupts pdhC abolishes the GI of sigX-lacZ (Ogura and Asai, 2016). A similar result was obtained for pdhB (Figure 1A). B. subtilis cells may utilize an additional acetyl-CoA producer for CshA acetylation, that is, acetyl-CoA synthetase encoded by acsA (Grundy et al., 1993). Thus, we tested whether a mutation of acsA affected the GI of sigX-lacZ. The strain bearing both sigX-lacZ and acsA showed GI of sigX-lacZ, indicating no involvement of acsA in the GI of sigX-lacZ in our condition, i.e., without acetate in the medium (right, Figure 1A). Furthermore, addition of acetate to the culture, which would enhance PDH-independent acetyl-CoA production, resulted in no induction of sigX-lacZ (left, Supplementary Figure S1A). This is a surprising observation, which has to be further explored.
Acetyl-phosphate could be the acetyl moiety source for CshA acetylation (Figure 1B). To investigate the influence of acetyl-phosphate on GI of sigX-lacZ, we examined the effects of both ackA and pta mutations, since both genes are involved in acetyl-phosphate production. GI of sigX-lacZ was observed in the both mutated cells, although it was moderately attenuated in the ackA strain for unknown reasons (Figure 1A). These suggest that acetyl-phosphate would not be involved in GI of sigX. Taken together, the results confirmed that acetyl-CoA produced by PDH is involved in acetylated CshA-dependent GI of sigX.
CshA is known to be associated with RNase J1 as well as RNase Y (Cascante-Estepa et al., 2016), and RNase Y is not involved in the GI of sigX (Ogura and Asai, 2016), and thus it is possible that CshA causes the GI of sigX through RNase J1. However, examination of sigX-lacZ expression in the strain bearing sigX-lacZ with the rnjA mutation revealed, however, that this is not the case (right, Supplementary Figure S1A).
The YqfO and TsaD Genes Involved in Glucose Induction of sigX-lacZ
Previously we performed transposon mutagenesis using sigX-lacZ (Ogura and Asai, 2016) and identified Tn-insertion into trmK and tsaD. Tn insertion into trmK encoding tRNA methyltranferase (Roovers et al., 2008) resulted in loss of GI of sigX-lacZ (left, Supplementary Figure S1B). To examine the effect of trmK disruption alone on GI, we used a trmK disruption mutant in which the expression of downstream yqfO encoding a conserved protein regulating gene transcription (Tascou et al., 2003) was ensured through an IPTG-inducible promoter. In the presence of IPTG, the strain showed reduced sigX-lacZ expression compared to that in the wild type, and thus GI was partially impaired (right, Supplementary Figure S1B). The observation suggested that trmK contributes to GI of sigX to some extent and the possible role of trmK will be analyzed elsewhere. When compared to the extent of impairment of GI between trmK::Tn and trmK with Pspac-yqfO mutants, a complete loss of GI was observed in the Tn-inserted trmK mutant. This suggested that Tn-insertion into trmK has a polar effect on downstream yqfO. We observed that without IPTG, similar expression pattern was observed, which suggested that leaky expression of the IPTG-dependent promoter would ensure the yqfO expression (data not shown). To solve the question, a disruption mutant of yqfO was constructed, and we examined the β-Gal activity of the sigX-lacZ strain with yqfO. The result showed that yqfO disruption caused elimination of GI (Figure 2A). Glucose addition to the wild-type strain showed enhancement of cell mass in the stationary phase, which was also observed in the yqfO mutant (Figure 2A). These indicate that it is disruption of yqfO that causes the elimination of GI.
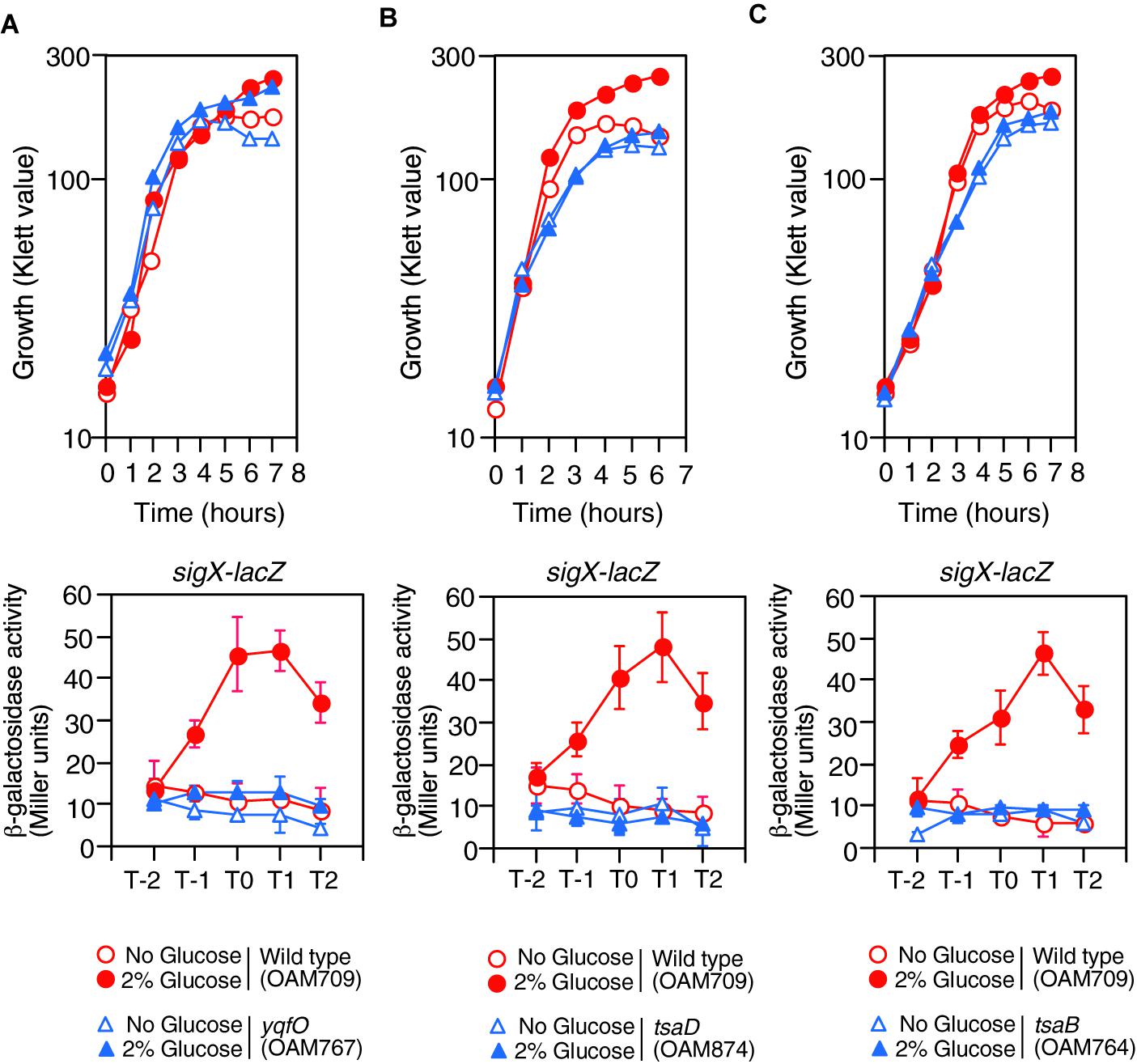
Figure 2. Growth and expression of sigX-lacZ in the mutants. (A) yqfO. (B) tsaD. (C) tsaB. The relevant genotypes are indicated below the panel. Growth curves of the wild-type and each mutant strain monitored by Klett colorimeter are shown (upper panel). Means of the β-Gal activities from three independent experiments and the standard deviations are shown (lower panel). The x axis is the same as in the legend to Figure 1.
The tsaD gene is located within a five-membered operon, of which the first gene is thiL, encoding a thiamine monophosphate kinase (Schyns et al., 2005). The product, thiamine pyrophosphate, is a cofactor for PDH. The following ORFs are tsaE, tsaB, rimI (ribosomal alanine N-acetyl transferase), and tsaD. We noted that rimI was not involved in CshA acetylation, because GI of sigX-lacZ was still observed in the rimI mutant (left, Supplementary Figure S1C). Without IPTG, similar expression pattern of sigX-lacZ was observed, suggesting leaky expression of the IPTG-dependent promoter would ensure the tsaD expression (data not shown). The tsaD gene encodes a component of enzyme complex, and the other components are encoded by tsaB and tsaE (Thiaville et al., 2015). With respect to tsaD, there is no possibility of a polar effect, as the tsaD ORF is located at the last position in the operon (Supplementary Figure S1C). TsaEBD is required for the synthesis of threonylcarbamoyl adenosine (t6A), which is used to modify tRNAs in bacteria. Disruption of tsaD resulted in decreased sigX-lacZ expression and loss of GI (Figure 2B). Irrespective of glucose addition, the tsaD mutant showed approximately one hour-delay of growth and we observed that glucose did not enhance cell mass at stationary phase (Figure 2B). We successfully disrupted tsaB by double crossover recombination. In the tsaB mutant, the GI of sigX-lacZ was also abolished and similar growth profiles were observed to those in the tsaD cells, as expected (Figure 2C); these are consistent with the TsaEBD requirement for GI.
Next, yqfO and tsaD strains with corresponding xylose-inducible genes at the amyE locus were constructed, and their β-Gal activities were examined. Without xylose, the yqfO strain showed some GI, probably due to leaky expression of the xylose-inducible promoter, whereas the tsaD strain showed no GI (Figure 3). In the presence of xylose, both strains showed significant GI of sigX-lacZ (Figure 3). These results indicate that yqfO and tsaD are involved in the GI of sigX. To understand whether these mutations influence the expression and GI of sigX independently or in the same regulatory cascade, we constructed a strain with disruption of both tsaD and yqfO. Then we examined the expression of sigX-lacZ in this double mutant. The expression and GI of sigX-lacZ in the double mutant were similar to the expression in either of the single mutants (right, Supplementary Figure S1C). Moreover, we tested the effect of the other double mutant, ylxR and tsaD, and obtained similar results (right, Supplementary Figure S1C). These results suggested that these genes might be in the same regulatory cascade. Subsequently we examined the possible relationship among these genes as described below.
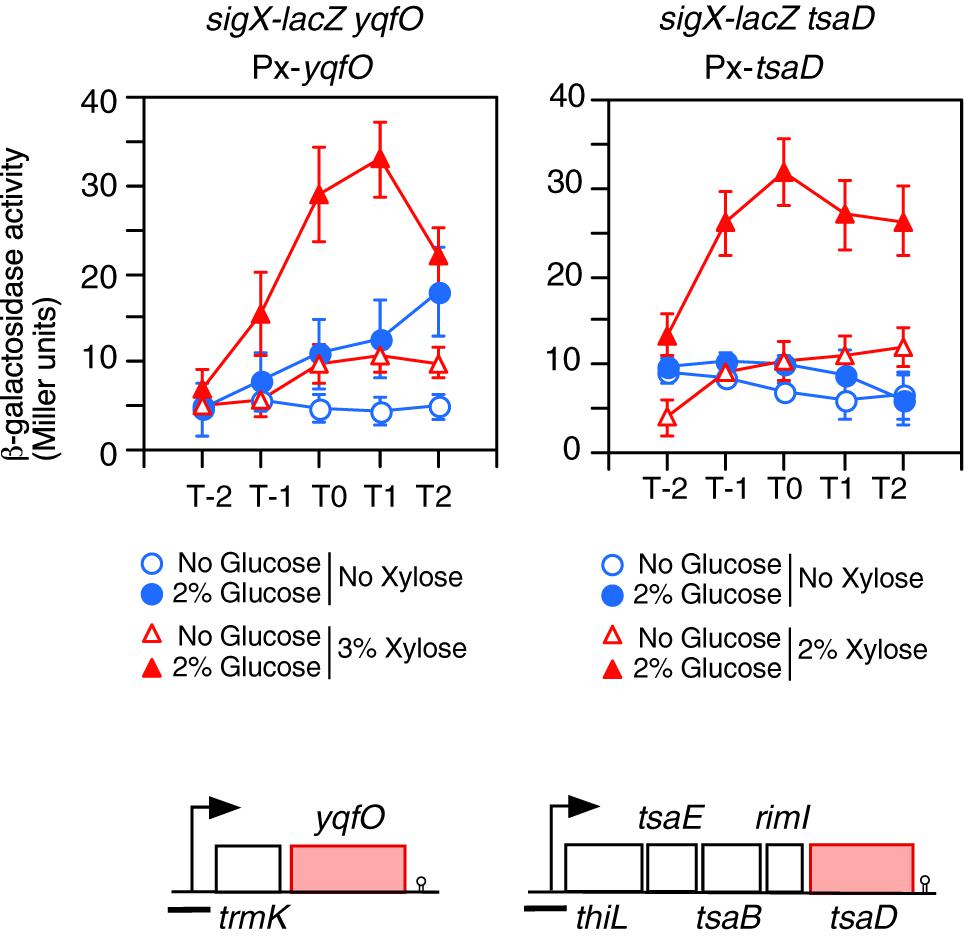
Figure 3. Complementation of the GI of sigX-lacZ in each gene disruption mutant by artificial induction of the gene. Means of the β-Gal activities from three independent experiments and the standard deviations are shown. The x axis is the same as in the legend to Figure 1. The relevant genotypes are indicated above the panel. Strains are as follows: left, OAM738; right, OAM739. The chromosomal structure of the operon containing the corresponding gene is shown under the panel. Boxes and bent arrows show open-reading frames and promoters, respectively. The gene names are shown along the boxes. The black bar shows the cloned promoter region for analyzing promoter expression.
Positive Regulation of PtrmK by YlxR
YlxR is the NAP regulating many gene expression in early stationary phase (Ogura and Kanesaki, 2018), thus we examined the effect of the ylxR disruption on the expression of PtrmK. As a result, we observed decreased PtrmK expression in the ylxR disruption mutant (Figure 4A). Glucose addition resulted in the induction of PtrmK in the wild type but not in the ylxR mutant. Based on these results, it was concluded that PtrmK expression is positively regulated by ylxR.
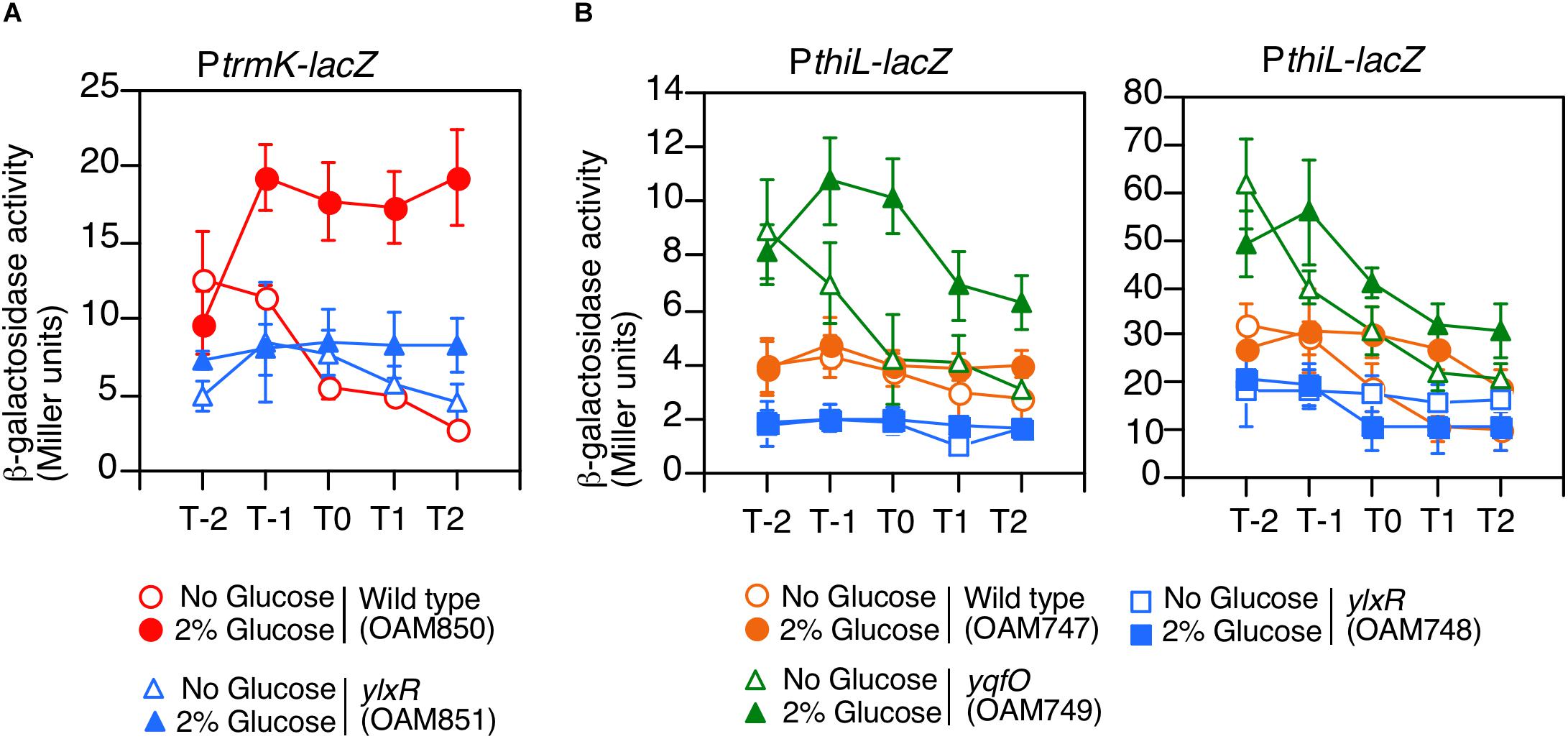
Figure 4. Expression of PtrmK-lacZ and PthiL-lacZ. Means of the β-Gal activities from three independent experiments and the standard deviations are shown. The x axis is the same as in the legend to Figure 1. (A) PtrmK-lacZ. (B) PthiL-lacZ. β-galactosidase activities measured using CRPG (right) instead of ONPG (left) as a substrate are shown.
Regulation of PthiL by YlxR and YqfO
We examined the effects of disruption of yqfO and ylxR on the expression of PthiL, which drives tsaD. This was based on the assumption that there might be some regulatory relationship between the yqfO, ylxR, and tsaD. As a result, we observed that the expression of PthiL decreased moderately in the ylxR disruptant (left, Figure 4B). Thus, it was concluded that PthiL expression is positively regulated by ylxR. Moreover, PthiL expression was observed to increase and to be induced by the addition of glucose in the yqfO disruptant (left, Figure 4B). Therefore, yqfO plays negative regulatory role in PthiL expression and it was suggested that YqfO might repress GI of PthiL. The expression of PthiL was relatively low (around 4 Miller units), and thus, we used a highly sensitive substrate for β-galactosidase, CRPG. Then we observed similar results to those obtained by commonly used substrate, ONPG (right, Figure 4B). It should be noted that the decrease of PthiL expression was observed to a lesser extent in the ylxR strain when CRPG was used.
Cis-Element for YlxR Revealed by Mutational Analysis of PtrmK
We performed mutational analysis of the YlxR-dependent promoter, PtrmK, using its lacZ fusions (Figure 5A). The time course data of the Wt fusion are shown in Figure 4A. We observed significant expression of del3 (-147/-1 relative to the translation start site) and del5 (-220/-95) but not del4 (-94/-1) both in the presence and absence of glucose, suggesting that the promoter is within the -147/-95 region carrying the putative promoter-like sequence TTGGAT-N17-TATGAT (-134/-106). These observations are consistent with those of previous genome-wide transcription analysis, where transcription initiation was observed immediately upstream of the trmK ORF, i.e., approximately 100 base pairs (Nicolas et al., 2012, subtiwiki). Decreased β-Gal activities in the ylxR disruptants in the presence of glucose were detected in the Wt (-295/-1), del1 (-220/-1) and del2 fusions (-174/-1) but not del3 (-147/-1), suggesting that the cis-element(s) for YlxR is within the -174/-147 region containing the candidate tandemly repeated sequences, ATCAAAA (-170/-164) and TTCAAAA (-154/-148). Indeed, introduction of nucleotide changes into the sequences resulted in dramatic loss of β-Gal activities in addition to loss of GI, indicating that these sequences are positive cis-elements for YlxR (M2 and M3). Notably, disruption of the downstream element had more profound effects compared to disruption of the upstream element. Next, we observed that in the wild-type background deletion of the -295/-220 region resulted in decreased and increased β-Gal activities between the Wt and del1 fusion pair and del5 and del6 pair, respectively. This may be because of the presence or absence of the downstream -95/-1 region in the fusions. Because the -295/-220 region contains a possible cis-element for YlxR, GTCAAAA (-272/-266), we introduced a nucleotide change into this sequence and examined its β-Gal activities (M1). As a result, the fusion showed greatly enhanced activities compared to the wild-type, demonstrating that this sequence would be a negative cis-element for YlxR. In the del5, del6, del7, M1, and M2 fusions but not in the M3 fusion, the disruption of ylxR resulted in the decreased fusion expression, compared to the expression of the corresponding fusion in the wild-type background, especially in the presence of glucose. This is consistent with the properties of these fusions carrying the positive cis-element for YlxR (-154/-148). We note that the expression of the del5, del6, and M1 fusions in the ylxR background was still induced by glucose at a lesser rate compared to that in the wild-type background. The cause of these observations remains unknown. Taken together, we detected positive two cis-elements for YlxR on the PtrmK promoter.

Figure 5. Expression of various PtrmK-lacZ and PthiL-lacZ fusions. (A) PtrmK-lacZ. (B) PthiL-lacZ. Strains were grown in sporulation medium with or without 2% glucose and sampled hourly from T-2 to T2. The averages and standard deviations of the observed peak values of β-Gal activities from the three independent cultures are shown. Numbers in parentheses show standard deviations. Structures of various lacZ fusions are depicted. Numbers indicate the nucleotide positions relative to the translation start nucleotide. The boxes on the line show the genes. The relevant genotype and the addition of glucose are shown (G- or G+). The construction of the mutated fusions is described in Supplementary Methods. Time-course data in the strains with wild-type fusion are shown in Figure 4.
Cis-Element for YlxR Revealed by Mutational Analysis of PthiL
To clarify the possible cis-element for YlxR and YqfO on the PthiL promoter, we performed mutational analysis of this promoter (Figure 5B). The time course data of the Wt fusion are shown in Figure 4B. The –10 and –35 elements of PthiL have not been determined, but a putative terminator (stem-loop structure with U-tract) has been identified upstream of the thiL ORF (Rudner et al., 1993). To examine the location of transcription initiation activity in the upstream region of thiL, PthiL-lacZ fusions with various regions deleted were constructed, and their β-Gal activities were examined. The Wt (-500/-1 relative to the translation start site), del1 (-297/-1), and del2 (-210/-1) fusions showed β-Gal activity in the wild-type background, while the del3 (-138/-1) and del4 (-500/-278) fusions showed no β-Gal activity (Figure 5B). These results strongly suggested that the -35 and -10 elements for the thiL promoter are in the -210/-138 region. This region contains TCGAGT-N16-ATTAAT (-203/-176), the candidate -35 and -10 elements. It was observed that in the del8 (-500/-39) fusion a comparable expression was observed similar to that observed in the Wt fusion, while in the del5 (-500/-170), del6 (-500/-137), and del7 (-500/-68) fusions no expression was observed in the wild-type background. This suggested that there might be a positive cis-element for YlxR within the -68/-39 region, where there is no candidate promoter sequence. Importantly, this region contains similar cis-element for YlxR identified in PtrmK, AGCAAAA (-67/-61). The introduction of nucleotide changes into this sequence resulted in the complete loss of β-Gal activity in the wild-type background (M1). These results suggested that the AGCAAAA (-67/-61) sequence serves as a positive cis-element for YlxR.
Next, we examined the expression of these mutant fusions in the yqfO-disruption background. We observed that yqfO disruption resulted in enhanced expression of the Wt fusion. Similarly, the disruption of yqfO enhanced the β-Gal activity in both del2 and del5 fusions, suggesting that cis-element for yqfO is in the core promoter region (-210/-170). However, the further construction of scanning mutants within the region required to identify the cis-element is beyond the scope of this study. The del6 and del7 fusions with the promoter in addition to the terminator showed no β-Gal activity even in the yqfO background, although del5 expression was observed in the yqfO strain, suggesting that yqfO disruption could increase the expression due to the lack of the terminator. It should be noted that in the yqfO background, del8 fusion lacking cis-element like sequence AGCACAA (-43/-37) showed no enhancement of β-Gal activity. This suggested that this sequence may be required for enhancement by disruption of yqfO, the exact role of this sequence, however, remains unclear.
In the Wt, del1, del2, and del8 fusions with the cis-element for YlxR, ylxR disruption decreased the expression in both the presence and absence of glucose to various extents. This is consistent with the positive role of YlxR in PthiL expression. We note that ylxR disruption did not cause complete loss of fusion expression in the Wt fusion, suggesting a secondary and weak positive effect on PthiL expression by the ylxR disruption. In fact, M1 showed low levels of expression in the ylxR disruptant. This may be due to the negative effect of ylxR disruption on yqfO expression (Figure 4A), leading to enhancement of PthiL expression.
YlxR-Binding to PthiL and PtrmK
YlxR was first identified as a non-specific DNA-binding protein, but it may contain a preferential binding site in the nucleotide sequence as well as other NAPs (Sandhya et al., 2015; Ogura and Kanesaki, 2018). As expected, YlxR bound to the DNA probes with PtrmK and PthiL in the EMSA (Supplementary Figure S2). To examine preferential binding of YlxR to the identified the three putative cis-elements, we performed EMSA using the wild-type and mutated PtrmK probes for YlxR. The probe contains three cis-elements for YlxR. When M1 and M3 were independently introduced into the probe, similar DNA-binding affinity of YlxR was observed, although the retardation of the YlxR-DNA complex was slightly lowered, indicating lower molecular mass of the complex (compare lane 4 to lane 12, left, Supplementary Figure S2A), or affinity of the probe to YlxR was lowered (compare lane 2 to lane 6, left, Supplementary Figure S2A). Next, we used the probe with both M1/M3 mutations (right, Supplementary Figure S2A). In lane 2 of Supplementary Figure S2A, a sharp band corresponding to the probable YlxR/wild-type DNA complex was observed, indicating that 0.1 μM was critical concentration of YlxR, where DNA-binding was partially observed. On the other hand, no band was observed for the M1/M3 probe at 0.1 μM YlxR (lane 7). Moreover, when lane 5 and lane 10 were compared, all probes were retarded due to DNA-binding of YlxR. However, slightly lowered mobility was observed for the mutant probe. These show the differential binding of YlxR to the wild-type and mutated probes. Finally, we tested whether YlxR binds preferentially the cis-element in PthiL. When lane 3 and lane 7 were compared, YlxR showed weak DNA-binding activities to the probe with a mutated cis-element compared to the wild-type probe (Supplementary Figure S2B). These results suggest that YlxR preferentially binds to its cis-elements, although the difference in affinity between preferential binding sites and non-specific ones was small.
Transcription of PpdhABCD
TsaD is a component of TsaEBD required for the synthesis of t6A-modified tRNA; thus TsaD might be involved in translational control of several proteins. As an acetyl-CoA producer, PDHc is required for CshA acetylation (Figure 1B). Subunits of PDHc are known to be most actively synthesized proteins in the growing B. subtilis cells (Eymann et al., 2004), suggesting that PDHc synthesis would be under transcriptional, translational and post-translational control. Therefore, it is possible that subunits of PDHc are under translational or post-translational control involving TsaD. First, we examined the transcription of the pdhABCD operon. A previous study showed that this operon transcription increases in the presence of glucose (Blencke et al., 2003). We confirmed this observation using PpdhA-lacZ (Figure 6A). Furthermore, we observed that this increase is not dependent of TsaD, YlxR, CcpA, CcpC, CcpN, or CggR (Figure 6A, data not shown). These results suggest an unknown mechanism for the GI of PpdhA. The pdhABCD operon has an internal promoter (Nicolas et al., 2012). To test possible effects of the tsaD mutation on the internal promoter, pdhD-lacZ was constructed (Supplementary Figure S3). The expression and GI of pdhD-lacZ were similar in both wild-type and tsaD cells. Thus, it was confirmed that the tsaD disruption also plays no role in the GI of pdhD-lacZ.
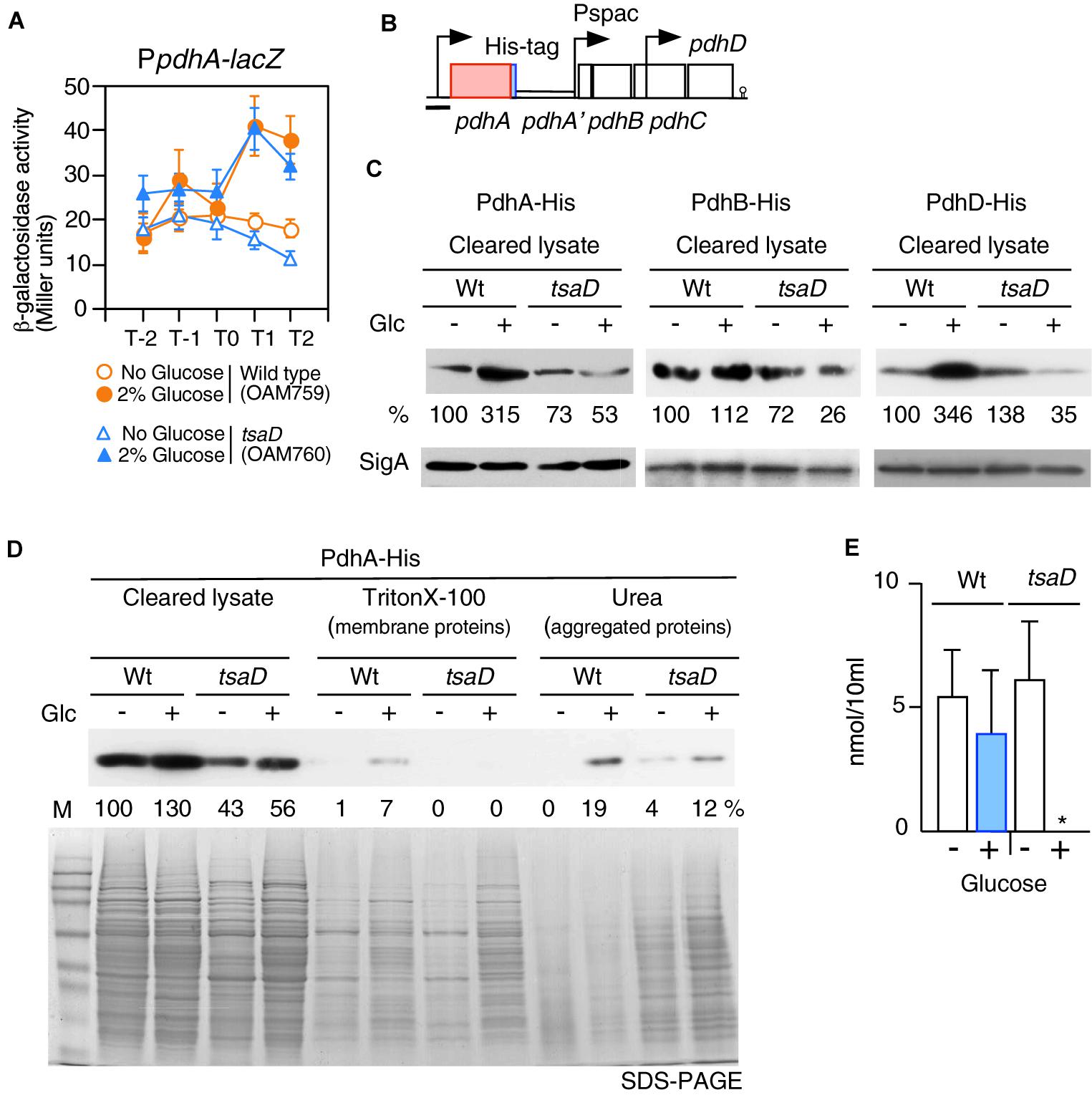
Figure 6. Expression of PpdhA-lacZ and measurement of soluble PDH subunits in the tsaD mutant. (A) Expression of PpdhA-lacZ. Means of the β-Gal activities from three independent experiments and the standard deviations are shown. The x axis is the same as in the legend to Figure 1. (B). The chromosomal structure of the strain with pdhA-His (OAM761) is shown. The legend for graphics is the same as that of Figure 3. The black bar shows the cloned promoter region for analyzing promoter expression. The chromosomal structures of the strains bearing PdhB-His and PdhD-His are similar to that of OAM761 (not shown). (C) Western analysis of PdhA-His, PdhB-His and PdhD-His. For the culture of each strain bearing His-tagged Pdh subunit [PdhA-His, wild type (OAM761), tsaD (OAM762); PdhB-His, wild type (OAM777), tsaD (OAM778)], 1 mM IPTG was added in sporulation medium with or without 2% glucose (“Glc” means glucose addition). For the cultures of strains with PdhD-His, wild type (OAM779) and tsaD (OAM780), no IPTG was added. Equal protein amounts of soluble fractions were analyzed in western blot using anti-His-tag monoclonal antibody for detection of each His-tagged protein. As a control, SigA detected by anti-SigA antibody is shown. (D) Western blot analysis of cleared cell lysates, “membrane protein” fractions (Triton X-100 extracts), and “aggregated protein” fractions (insoluble fractions by Triton × -100, solubilized by urea-containing buffer) from the wild type and tsaD cells for PdhA-His. OAM761 and OAM762 cells were cultivated and treated as described in the Materials and Methods. Cell mass equivalents were analyzed using both western blot and SDS-PAGE stained by Coomassie. “M” indicates molecular weights marker (Amersham full range rainbow marker). (E) Examination of cellular acetyl-CoA pool. Samples from two independent cultures (wild type, 168; tsaD, OAM734) were analyzed three times, and standard deviations are shown. ∗indicates that the value is less than the detection limit (0.5 nmol/10 ml).
Decrease of PDHc Subunits in the tsaD Disruptant With Glucose
To test the influence of tsaD disruption on PdhA at the protein level, western analysis of PdhA was performed. For the detection of PdhA, a His-tag was genetically introduced into the C-terminal end of pdhA (Figure 6B). To sustain downstream pdhBCD transcription, IPTG was added to the culture of the pdhA-His strain. The strain grew normally, suggesting no harmful influence of the His-tag addition to PdhA (data not shown). After cells were lysed, soluble fractions were obtained by centrifugation. Western analysis of soluble fractions with equal protein amounts of the wild-type strain using anti-His-tag antibody confirmed that glucose addition increased PdhA-His at the protein level (Figure 6C). A similar western analysis of tsaD cells revealed that glucose addition significantly decreased the amount of soluble PdhA-His, compared to that of the wild-type strain with glucose, even though transcription was similarly enhanced (Figure 6). This suggested some post-transcriptional defects at the protein level.
During preparation of the cleared cell lysate, the insoluble fractions contained membrane- and aggregated-proteins. Thus, we separated these insoluble fractions to “membrane protein” and “aggregated protein” fractions. Notably, each fraction was derived from the equal cell amounts, which is different from the method used for the results shown in Figure 6C. These fractions were analyzed by western blot to detect PdhA-His (Figure 6D). With this method, however, we also obtained similar results in soluble fractions to those shown in Figure 6C. Despite the presence of PdhA-His in the “membrane protein” fraction, we wanted to verify its localization, since PdhA-YFP has previously been identified in the cytoplasm (Monahan et al., 2014). We similarly observed localization of PdhA-GFP in the cytoplasm (Supplementary Figure S4), which refutes evidence for membrane localization of PdhA. Thus, this detection may be due to the contaminated cytosolic PdhA because of cellular abundance. In the “aggregated protein” fraction from wild type cells with glucose, PdhA-His were detected. Perhaps an abundance of pdh mRNA in the cell creates a burden for the translational machinery, leading to generation of aggregated PdhA-His. Overexpression of protein has often caused protein aggregation (Bednarska et al., 2013).
From tsaD cells with and without glucose, significant and minor levels of PdhA-His, respectively, were detected in the urea fractions (Figure 6D). These results showed that in the tsaD cells, misfolded or aggregated PdhA was generated. We noted using SDS-PAGE that the “aggregated protein” fraction was enriched in the tsaD cells, suggesting that the tsaD mutation has a global effect on protein quality control (Figure 6D). In the “membrane proteins” fraction from the tsaD cells with glucose, slightly more amount of proteins was observed compared to that without glucose. The detected proteins initially aggregated or misfolded and thus were fractioned into the insoluble pellets, which were solubilized by a weak detergent like Triton X-100. This experiment was further performed five times, with three trials yielding similar results to that shown in Figure 6D. In two cases, PdhA-His was scarcely detected in the urea fractions of wild type or tsaD cells (data not shown). Although it is not well understood how the fate of misfolded proteins is determined (i.e., refolding, amyloid formation, or degradation), aggregates are considered pathway intermediates (Supplementary Figure S5; Bednarska et al., 2013; Balchin et al., 2016). Thus, in the former cases, relatively small amounts of aggregated PdhA-His in the tsaD cells could be detected because of protein aggregation. In contrast, in the latter cases most of insoluble PdhA-His could be degraded through the aggregation state. Taken together, these results strongly suggest that protein quality control of PdhA requires TsaD especially in the glucose-added condition.
There remained the possibility that TsaD is needed for the protein quality control of the other PDHc components. To examine this, similar western analyses were performed; however, the antigenic activity of PdhC-His was too low, and thus no information was obtained (data not shown). Western analysis of soluble PdhB-His and PdhD-His showed significant decreases in the tsaD cells with glucose like PdhA-His (Figure 6C). These findings also strongly suggested that protein quality control of PdhB and PdhD requires TsaD in the glucose-added condition. In western analysis of insoluble fractions His-tagged Pdh proteins were scarcely detected (data not shown). These results suggested that probable protein aggregates from PdhB-His and PdhD-His might be susceptible to protein degradation.
Acetyl-CoA Pool in tsaD Cells
When glucose was added to culture of the tsaD disruption mutant, levels of soluble PDHc subunits were severely decreased. Thus, the activity of PDHc may be inhibited by the decrease of protein amount as well as disturbances in the stoichiometry of PDHc in the mutant. To examine this, intracellular acetyl-CoA concentrations were determined (Figure 6E). Glucose addition increased PDHc in wild-type cells, but the size of the acetyl-CoA pool remained similar. There was also a similar level of acetyl-CoA observed in tsaD cells without glucose. In contrast, in tsaD cells with glucose, a severe decrease in the levels of acetyl-CoA was observed. This should decrease the acetylation of CshA in the presence of glucose, leading to the elimination of the GI of sigX/M.
Discussion
Based on the data obtained in this study, we suggest a new glucose-responsive system (GRS) that includes protein lysine acetylation, transcriptional regulation, and protein quality control. We present a model for the regulation of the acetylation of CshA, which stimulates the formation of sigma X/M-bound RNAP (Figures 1B, 7).
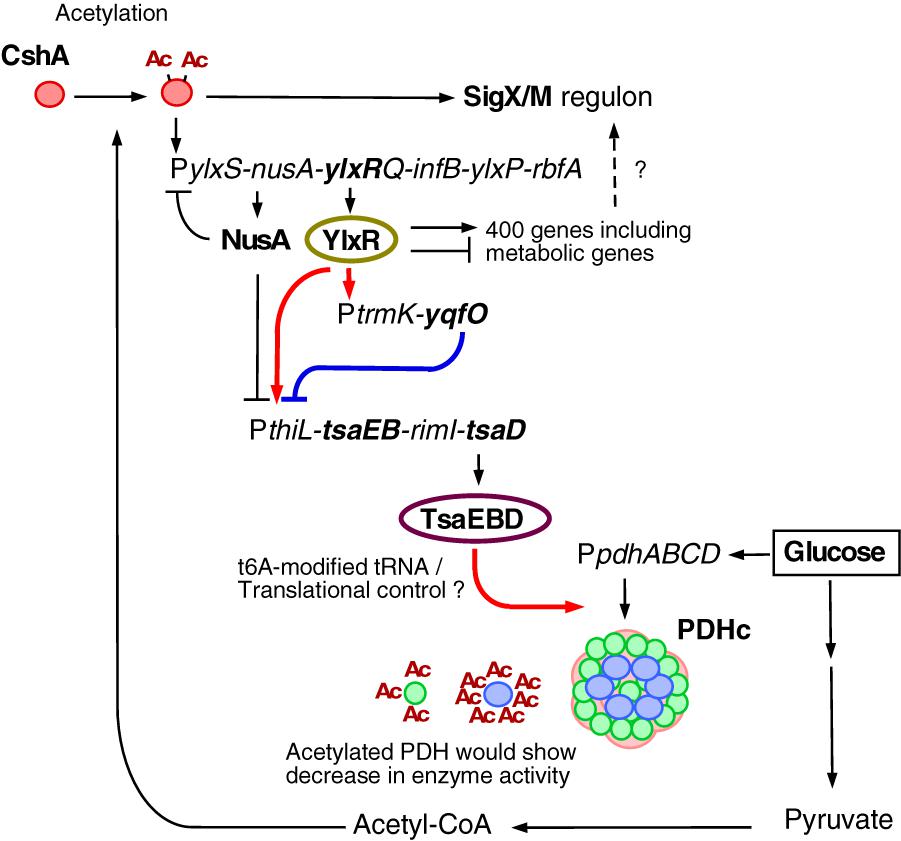
Figure 7. Suggested model of glucose-responsive system (GRS). The thick lines show the regulatory pathways identified and analyzed in this paper. The other pathways have been analyzed in Ogura and Asai, 2016 (acetylation of CshA and sigX/M regulation), Ogura and Kanesaki, 2018 (CshA-dependent regulation of PylxS driving expression of YlxR, which regulates metabolic genes), and Mondal et al., 2016 (NusA inhibits PylxS and PthiL). With respect to PDHc, this cartoon does not correctly reflect the actual structure (Zhou et al., 2001). Green circle, PdhE1 (pdhA and pdhB); blue circle, PdhE3 (pdhD); pink moiety, PdhE2 core (pdhC). PDHc is also known to be acetylated (under glucose-rich conditions, at 3 moieties of PdhA and 7 moieties of PdhD, Kosono et al., 2015), and acetylation reduces enzyme activity. Arrows indicate transcription, translation, acetylation, enzyme reaction, protein association or transcriptional activation. T-bars indicate transcriptional repression or attenuation. Ac, acetyl moiety; PDHc, pyruvate dehydrogenase complex; RNAP, RNA polymerase.
YqfO is a conserved protein among Firmicutes and bears a nif3 conserved domain with a suggested function related to transcriptional regulation (Tascou et al., 2003). The structure of the Bacillus cereus YqfO has been resolved and suggests that YqfO may have a ligand-binding domain (Godsey et al., 2007). YqfO was demonstrated to be involved in PthiL expression, and thus probably yqfO disruption may have impaired the regulation of tsaEDB, leading to loss of GI of sigX through the dysregulation of PDHc expression. However, YqfO may be indirectly involved in regulation of GI of sigX.
YlxR is also involved in the expression of thiL. According to this model, the acetylation of CshA is a self-reinforcing system. The disruptions of ylxR and yqfO result in the elimination of the GI of sigX/M. This is due to the disruption of robust and fine-tuned expression of the tsaEBD-containing operon, the products of which play a role in the quality control of PDHc subunits. However, in the earlier genome-wide analysis using ylxR cells in the presence of glucose the thiL operon was not detected as a target for YlxR (Ogura and Kanesaki, 2018). This seems to be an inconsistent result according to the lacZ analysis. These are inherent biases in this type of genome-wide analysis, which may be resulting from specific mRNA degradation during the sample preparation. Thus, the results in this study are plausible.
According to the expression landscape data (Nicolas et al., 2012, subtiwiki), under some conditions transcription initiation of the thiL operon was observed immediately upstream of the thiL gene; however, the more upstream region was not analyzed because this region contains rRNA operons. On the other hand, the PthiL promoter is known to be one of the target promoters for the NusA-dependent termination/attenuation system. Genome-wide analysis of the effects of NusA-depletion revealed enhanced expression of the thiL operon, indicating that NusA serves as a negative regulator for PthiL (Mondal et al., 2016). The detected promoter in our analysis associated with its downstream terminator is likely involved in NusA-dependent regulation. Taken together, it is likely that the proposed thiL promoter is functional.
One of the outputs of this newly suggested GRS is GI of sigX/M, which shows wider roles of SigX/M beyond the nature of ECF sigma factor in surface stress response. In other words, expression of the SigX/M regulons responds to nutritional changes. This system would regulate the cellular acetyl-CoA pool, which is one of the sources for protein lysine acetylation, through PDHc under glucose-rich growth conditions. PDHc would play a critical role in CshA acetylation as a supplier of acetyl-CoA. PdhA and PdhD were acetylated under glucose-rich conditions in B. subtilis (Kosono et al., 2015). Another study reported that all components of PDHc were acetylated (Carabetta et al., 2016). The transcription of pdhABCD is stimulated by glucose addition, leading to larger amounts of PDH, while we observed similar levels of intracellular acetyl-CoA. In many bacteria including Salmonella, the acetylation of many enzymes for glycolysis has been reported to reduce their enzymatic activities (Wang et al., 2010; Nakayasu et al., 2017), and in B. subtilis, PdhC and PdhD activities were controlled at the protein modification or activity level but not at the enzyme concentration level (Chubukov et al., 2013). Considering these data, B. subtilis PDHc activity may be down regulated by its acetylation.
We found that TsaD required for the synthesis of t6A in tRNA plays some role in the protein quality control of PDHc subunits in the presence of glucose. Under such a condition, transcription of mRNA encoding Pdh subunits is enhanced, which could burden the translational machinery without t6A-modified tRNA. t6A is located at position 37 of the anticodon loop in tRNAs that decode ANN codons. t6A is universally conserved across the three domains of life (Thiaville et al., 2014). In the most bacterial genomes sequenced to date, homologs of TsaE, TsaB, and TsaD have been found. In many bacteria, these genes are essential (Thiaville et al., 2014, 2015). However, in Deinococcus radiodurans R1, tsaB and tsaD are non-essential, and in Synechocystis sp. PCC6308 tsaD is non-essential. In both bacteria, the essentiality of the rest of the genes in TsaEBD has not been experimentally determined. All three genes were reported to be essential in B. subtilis, as initial attempts for constructing these gene disruptions were not successful (Kobayashi et al., 2003). Later two groups have reported that tsaE is non-essential (Hunt et al., 2006; Koo et al., 2017). Tn insertion into tsaD was confirmed by PCR analysis and tsaB was able to be disrupted in this study, demonstrating that these genes were not essential. However, it should be noted that some unknown suppressor mutation for lethality within the genome could help the generation of the tsaD or tsaB disruptant, though there were no observations such as low transformation rate during gene disruption or transfer process to suggest suppressor mutation.
In the depletion or disruption mutants of the genes involved in the synthesis of t6A, various phenotypes have been observed, such as those about transcriptional control, cell division, and cyanophycin accumulation (Zuther et al., 1998; Lei et al., 2012; Bitoun et al., 2014). The mechanism by which these phenotypes are caused by TsaEBD is currently unknown. In the B. subtilis tsaD cells with glucose, the intracellular acetyl-CoA pool was not detected. This observation does not indicate that the tsaD cells lack acetyl-CoA, because the tsaD cells was able to grow with about one-h delay compared to growth of wild-type cells. Perhaps smaller amount of Pdh components impaired acetyl-CoA pool in the tsaD cells with glucose, but active acetyl-CoA flux may function to some extent. In the E. coli tsaD-depleted mutant, a reduction in assembled PDHc was observed and this was attributed to the accumulation of glycated components of PDHc (Katz et al., 2010). Protein glycation is thought to eventually produce toxic compounds and the authors pointed out that TsaD is a possible glycopeptidase for glycated PDHc. However, the archaeal TsaD homolog Kae1 showed no protease activity (Hecker et al., 2007), and there are no other reports for the protease activity of TsaD. Thus, the speculation that removal of glycated PDHc by TsaD in B. subtilis may contribute to quality control of PDHc is not likely. Moreover, the depletion or deficiency of t6A synthesis should affect translation, although the direct protein target of t6A-modified tRNA is unknown in bacteria (Thiaville et al., 2015). Recently, the idea has emerged that translational speed is evolutionarily optimized for folding of each protein (Balchin et al., 2016). In yeast and nematoda, the lack of anticodon loop modifications in tRNA results in ribosome pausing and slower translation rate, leading to the misfolding of proteins (Patil et al., 2012; Nedialkova and Leidel, 2015). Since ribosome surface is associated with the chaperons for nascent polypeptide folding, leading to cotranslational protein-folding, operon-coded proteins may be concomitantly susceptible to protein misfolding (Balchin et al., 2016). Thus, it is reasonable that PDHc subunits showed similar responses in the tsaD cells with glucose as to the protein quality control. Our finding that a deficiency of t6A synthesis in tsaD cells resulted in decreased soluble PDHc subunits in B. subtilis is consistent with these former observations. This discussion premised the direct translational control of PDHc by the TsaEBD complex. However, there are the other possibilities, including impaired global protein quality control indirectly resulting in the decreased PDHc.
The suggestion of this GRS revealed that the cellular glucose response was not completely clarified in previous studies. Additional detailed studies of this system will provide insight into the physiology of bacteria in adapting to glucose-rich conditions.
Author Contributions
MO designed the study, contributed to acquisition, analysis and interpretation of the data, and wrote the manuscript. TS and KA contributed to the acquisition of the data.
Funding
This work was supported by JSPS KAKENHI Grant Nos. 15K07367 and 18K05415, and the Research Program of the Institute of Oceanic Research and Development, Tokai University.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Y. Fujita (Fukuyama University), S. Kosono (Tokyo University), S. Matsuoka (Saitama University), and T. Oshima (Toyama Prefect. University) for kindly supplying the bacterial strains and plasmids used in Toyama this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00923/full#supplementary-material
References
Azam, T. A., and Ishihama, A. (1999). Twelve species of the nucleoid-associated protein from Escherichia coli. Sequence recognition specificity and DNA binding affinity. J. Biol. Chem. 274, 33105–33113. doi: 10.1074/jbc.274.46.33105
Balchin, D., Hayer-Hartl, M., and Hartl, F. U. (2016). In vivo aspects of protein folding and quality control. Science 353:aac4354. doi: 10.1126/science.aac4354
Bednarska, N. G., Schymkowitz, J., Rousseau, F., and Van Eldere, J. (2013). Protein aggregation in bacteria: the thin boundary between functionality and toxicity. Microbiology 159, 1795–1806. doi: 10.1099/mic.0.069575-0
Bitoun, J. P., Liao, S., Xie, G. G., Beatty, W. L., and Wen, Z. T. (2014). Deficiency of BrpB causes major defects in cell division, stress responses and biofilm formation by Streptococcus mutans. Microbiology 160, 67–78. doi: 10.1099/mic.0.072884-0
Blencke, H. M., Homuth, G., Ludwig, H., Mäder, U., Hecker, M., and Stülke, J. (2003). Transcriptional profiling of gene expression in response to glucose in Bacillus subtilis: regulation of the central metabolic pathways. Metab. Eng. 5, 133–149.
Browning, D. F., Grainger, D. C., and Busby, S. J. (2010). Effects of nucleoid-associated proteins on bacterial chromosome structure and gene expression. Curr. Opin. Microbiol. 13, 773–780. doi: 10.1016/j.mib.2010.09.013
Carabetta, V. J., and Cristea, I. M. (2017). The regulation, function, and detection of protein acetylation in bacteria. J. Bacteriol. 199, 107–117.
Carabetta, V. J., Greco, T. M., Tanner, A. W., Cristea, I. M., and Dubnau, D. (2016). Temporal regulation of the Bacillus subtilis acetylome and evidence for a role of MreB acetylation in cell wall growth. mSystems 1, e00005-16.
Cascante-Estepa, N., Gunka, K., and Stülke, J. (2016). Localization of components of the RNA-degrading machine in Bacillus subtilis. Front. Microbiol. 7:1492. doi: 10.3389/fmicb.2016.01492
Chubukov, V., Uhr, M., Le Chat, L., Kleijn, R. J., Jules, M., Link, H., et al. (2013). Transcriptional regulation is insufficient to explain substrate-induced flux changes in Bacillus subtilis. Mol. Syst. Biol. 9:709. doi: 10.1038/msb.2013.66
Delumeau, O., Lecointe, F., Muntel, J., Guillot, A., Guédon, E., Monnet, V., et al. (2011). The dynamic protein partnership of RNA polymerase in Bacillus subtilis. Proteomics 11, 2992–3001. doi: 10.1002/pmic.201000790
Deutscher, J. (2008). The mechanisms of carbon catabolite repression in bacteria. Curr. Opin. Microbiol. 11, 87–93. doi: 10.1016/j.mib.2008.02.007
Dillon, S. C., and Dorman, C. J. (2010). Bacterial nucleoid-associated proteins, nucleoid structure and gene expression. Nat. Rev. Microbiol. 8, 185–195. doi: 10.1038/nrmicro2261
Drlica, K., and Rouviere-Yaniv, J. (1987). Histonelike proteins of bacteria. Microbiol. Rev. 51, 301–319.
Eymann, C., Dreisbach, A., Albrecht, D., Bernhardt, J., Becher, D., Gentner, S., et al. (2004). A comprehensive proteome map of growing Bacillus subtilis cells. Proteomics 4, 2849–2876.
Fujita, Y. (2009). Carbon catabolite control of the metabolic network in Bacillus subtilis. Biosci. Biotechnol. Biochem. 73, 245–259.
Gao, H., Jiang, X., Pogliano, K., and Aronson, A. I. (2002). The E1beta and E2 subunits of the Bacillus subtilis pyruvate dehydrogenase complex are involved in regulation of sporulation. J. Bacteriol. 184, 2780–2788.
Godsey, M. H., Minasov, G., Shuvalova, L., Brunzelle, J. S., Vorontsov, I. I., Collart, F. R., et al. (2007). The 2.2 A resolution crystal structure of Bacillus cereus Nif3-family protein YqfO reveals a conserved dimetal-binding motif and a regulatory domain. Protein Sci. 16, 1285–1293.
Grundy, F. J., Waters, D. A., Takova, T. Y., and Henkin, T. M. (1993). Identification of genes involved in utilization of acetate and acetoin in Bacillus subtilis. Mol. Microbiol. 10, 259–271.
Guérout-Fleury, A. M., Frandsen, N., and Stragier, P. (1996). Plasmids for ectopic integration in Bacillus subtilis. Gene 180, 57–61.
Hata, M., Ogura, M., and Tanaka, T. (2001). Involvement of stringent factor RelA in expression of the alkaline protease gene aprE in Bacillus subtilis. J. Bacteriol. 183, 4648–4651.
Hecker, A., Leulliot, N., Gadelle, D., Graille, M., Justome, A., Dorlet, P., et al. (2007). An archaeal orthologue of the universal protein Kae1 is an iron metalloprotein which exhibits atypical DNA-binding properties and apurinic-endonuclease activity in vitro. Nucleic Acids Res. 35, 6042–6051.
Helmann, J. D. (2016). Bacillus subtilis extracytoplasmic function (ECF) sigma factors and defense of the cell envelope. Curr. Opin. Microbiol. 30, 122–132. doi: 10.1016/j.mib.2016.02.002
Hodgson, J. A., Lowe, P. N., and Perham, R. N. (1983). Wild-type and mutant forms of the pyruvate dehydrogenase multienzyme complex from Bacillus subtilis. Biochem. J. 211, 463–472.
Hori, K., Kaneko, M., Tanji, Y., Xing, X. H., and Unno, H. (2002). Construction of self-disruptive Bacillus megaterium in response to substrate exhaustion for polyhydroxybutyrate production. Appl. Microbiol. Biotechnol. 59, 211–216.
Hunt, A., Rawlins, J. P., Thomaides, H. B., and Errington, J. (2006). Functional analysis of 11 putative essential genes in Bacillus subtilis. Microbiology 152, 2895–2907.
Itaya, M. (1992). Construction of a novel tetracycline resistance gene cassette useful as a marker on the Bacillus subtilis chromosome. Biosci. Biotechnol. Biochem. 56, 685–686.
Katz, C., Cohen-Or, I., Gophna, U., and Ron, E. Z. (2010). The ubiquitous conserved glycopeptidase Gcp prevents accumulation of toxic glycated proteins. mBio 1, e00195-10. doi: 10.1128/mBio.00195-10
Klein, A. H., Shulla, A., Reimann, S. A., Keating, D. H., and Wolfe, A. J. (2007). The intracellular concentration of acetyl phosphate in Escherichia coli is sufficient for direct phosphorylation of two-component response regulators. J. Bacteriol. 189, 5574–5581.
Kobayashi, K., Ehrlich, S. D., Albertini, A., Amati, G., Andersen, K. K., Arnaud, M., et al. (2003). Essential Bacillus subtilis genes. Proc. Natl. Acad. Sci. U.S.A. 100, 4678–4683.
Koo, B. M., Kritikos, G., Farelli, J. D., Todor, H., Tong, K., Kimsey, H., et al. (2017). Construction and analysis of two genome-scale deletion libraries for Bacillus subtilis. Cell Syst. 4, 291–305. doi: 10.1016/j.cels.2016.12.013
Kosono, S., Tamura, M., Suzuki, S., Kawamura, Y., Yoshida, A., Nishiyama, M., et al. (2015). Changes in the acetylome and succinylome of Bacillus subtilis in response to carbon source. PLoS One 10:e0131169. doi: 10.1371/journal.pone.0131169
Kunst, F., Msadek, T., and Rapoport, G. (1994). “Signal transduction network controlling degradative enzyme synthesis and competence in Bacillus subtilis,” in Regulation of Bacterial Differentiation, eds P. J. Piggot, cpsfnmC.P. Jr.cpefnm Moran, and P. Youngman (Washington, DC: ASM Press), 1–20.
Lehnik-Habrink, M., Rempeters, L., Kovács,ÁT., Wrede, C., Baierlein, C., Krebber, H., et al. (2013). DEAD-Box RNA helicases in Bacillus subtilis have multiple functions and act independently from each other. J. Bacteriol. 195, 534–544. doi: 10.1128/JB.01475-12
Lei, T., Yang, J., Zheng, L., Markowski, T., Witthuhn, B. A., and Ji, Y. (2012). The essentiality of staphylococcal Gcp is independent of its repression of branched-chain amino acids biosynthesis. PLoS One 7:e46836. doi: 10.1371/journal.pone.0046836
Lima, B. P., Antelmann, H., Gronau, K., Chi, B. K., Becher, D., Brinsmade, S. R., et al. (2011). Involvement of protein acetylation in glucose-induced transcription of a stress-responsive promoter. Mol. Microbiol. 81, 1190–1204. doi: 10.1111/j.1365-2958.2011.07742.x
Monahan, L. G., Hajduk, I. V., Blaber, S. P., Charles, I. G., and Harry, E. J. (2014). Coordinating bacterial cell division with nutrient availability: a role for glycolysis. mBio 5, e00935-14. doi: 10.1128/mBio.00935-14
Mondal, S., Yakhnin, A. V., Sebastian, A., Albert, I., and Babitzke, P. (2016). NusA-dependent transcription termination prevents misregulation of global gene expression. Nat. Microbiol. 1:15007. doi: 10.1038/nmicrobiol.2015.7
Murayama, S., Ishikawa, S., Chumsakul, O., Ogasawara, N., and Oshima, T. (2015). The role of α-CTD in the genome-wide transcriptional regulation of the Bacillus subtilis cells. PLoS One 10:e0131588. doi: 10.1371/journal.pone.0131588
Nakayasu, E. S., Burnet, M. C., Walukiewicz, H. E., Wilkins, C. S., Shukla, A. K., Brooks, S., et al. (2017). Ancient regulatory role of lysine acetylation in central metabolism. mBio 8, e01894-17. doi: 10.1128/mBio.01894-17
Nedialkova, D. D., and Leidel, S. A. (2015). Optimization of codon translation rates via tRNA modifications maintains proteome integrity. Cell 161, 1606–1618. doi: 10.1016/j.cell.2015.05.022
Nicolas, P., Mäder, U., Dervyn, E., Rochat, T., Leduc, A., Pigeonneau, N., et al. (2012). Condition-dependent transcriptome reveals high-level regulatory architecture in Bacillus subtilis. Science 335, 1103–1106. doi: 10.1126/science.1206848
Ogura, M. (2016). Post-transcriptionally generated cell heterogeneity regulates biofilm formation in Bacillus subtilis. Genes Cells 21, 335–349. doi: 10.1111/gtc.12343
Ogura, M., and Asai, K. (2016). Glucose induces ECF sigma factor genes, sigX and sigM, independent of cognate anti-sigma factors through acetylation of CshA in Bacillus subtilis. Front. Microbiol. 7:1918. doi: 10.3389/fmicb.2016.01918
Ogura, M., and Kanesaki, Y. (2018). Newly identified nucleoid-associated-like protein YlxR regulates metabolic gene expression in Bacillus subtilis. mSphere 3, e00501-18. doi: 10.1128/mSphere.00501-18
Ogura, M., Ohshiro, Y., Hirao, S., and Tanaka, T. (1997). A new Bacillus subtilis gene, med, encodes a positive regulator of comK. J. Bacteriol. 179, 6244–6253.
Ogura, M., Shimane, K., Asai, K., Ogasawara, N., and Tanaka, T. (2003). Binding of response regulator DegU to the aprE promoter is inhibited by RapG, which is counteracted by extracellular PhrG in Bacillus subtilis. Mol. Microbiol. 49, 1685–1697.
Ogura, M., and Tanaka, T. (1996). Transcription of Bacillus subtilis degR is D-dependent and suppressed by multicopy proB through D. J. Bacteriol. 178, 216–222.
Ouidir, T., Kentache, T., and Hardouin, J. (2016). Protein lysine acetylation in bacteria: current state of the art. Proteomics 16, 301–309. doi: 10.1002/pmic.201500258
Patil, A., Chan, C. T., Dyavaiah, M., Rooney, J. P., Dedon, P. C., and Begley, T. J. (2012). Translational infidelity-induced protein stress results from a deficiency in Trm9-catalyzed tRNA modifications. RNA Biol. 9, 990–1001. doi: 10.4161/rna.20531
Roovers, M., Kaminska, K. H., Tkaczuk, K. L., Gigot, D., Droogmans, L., and Bujnicki, J. M. (2008). The YqfN protein of Bacillus subtilis is the tRNA: m1A22 methyltransferase (TrmK). Nucleic Acids Res. 36, 3252–3262. doi: 10.1093/nar/gkn169
Rudner, R., Chevrestt, A., Buchholz, S. R., Studamire, B., White, A. M., and Jarvis, E. D. (1993). Two tRNA gene clusters associated with rRNA operons rrnD and rrnE in Bacillus subtilis. J. Bacteriol. 175, 503–509.
Runde, S., Molière, N., Heinz, A., Maisonneuve, E., Janczikowski, A., Elsholz, A. K., et al. (2014). The role of thiol oxidative stress response in heat-induced protein aggregate formation during thermotolerance in Bacillus subtilis. Mol. Microbiol. 91, 1036–1052. doi: 10.1111/mmi.12521
Sandhya, S., Visweswariah, S., and Busby, J. W. (2015). Evolution of bacterial transcription factors: how proteins take on new tasks, but do not always stop doing the old ones. Trends Microbiol. 23, 463–467. doi: 10.1016/j.tim.2015.04.009
Schaeffer, P., Millet, J., and Aubert, J. (1965). Catabolite repression of bacterial sporulation. Proc. Natl. Acad. Sci. U.S.A. 54, 704–711.
Schilling, B., Christensen, D., Davis, R., Sahu, A. K., Hu, L. I., Walker-Peddakotla, A., et al. (2015). Protein acetylation dynamics in response to carbon overflow in Escherichia coli. Mol. Microbiol. 98, 847–863. doi: 10.1111/mmi.13161
Schyns, G., Potot, S., Geng, Y., Barbosa, T. M., Henriques, A., and Perkins, J. B. (2005). Isolation and characterization of new thiamine-deregulated mutants of Bacillus subtilis. J. Bacteriol. 187, 8127–8136.
Shiwa, Y., Yoshikawa, H., Tanaka, T., and Ogura, M. (2015). Bacillus subtilis degSU operon is regulated by the ClpXP-Spx regulated proteolysis system. J. Biochem. 157, 321–330. doi: 10.1093/jb/mvu076
Tascou, S., Kang, T. W., Trappe, R., Engel, W., and Burfeind, P. (2003). Identification and characterization of NIF3L1 BP1, a novel cytoplasmic interaction partner of the NIF3L1 protein. Biochem. Biophys. Res. Commun. 309, 440–448.
Thiaville, P. C., El Yacoubi, B., Köhrer, C., Thiaville, J. J., Deutsch, C., Iwata-Reuyl, D., et al. (2015). Essentiality of threonylcarbamoyladenosine (t(6)A), a universal tRNA modification, in bacteria. Mol. Microbiol. 98, 1199–1221. doi: 10.1111/mmi.13209
Thiaville, P. C., Iwata-Reuyl, D., and de Crécy-Lagard, V. (2014). Diversity of the biosynthesis pathway for threonylcarbamoyladenosine (t(6)A), a universal modification of tRNA. RNA Biol. 11, 1529–1539. doi: 10.4161/15476286.2014.992277
Tojo, S., Kumamoto, K., Hirooka, K., and Fujita, Y. (2010). Heavy involvement of stringent transcription control depending on the adenine or guanine species of the transcription initiation site in glucose and pyruvate metabolism in Bacillus subtilis. J. Bacteriol. 192, 1573–1585. doi: 10.1128/JB.01394-09
Tsukahara, K., and Ogura, M. (2008). Promoter selectivity of the Bacillus subtilis response regulator DegU, a positive regulator of the fla/che operon and sacB. BMC Microbiol. 8:8. doi: 10.1186/1471-2180-8-8
Wang, Q., Zhang, Y., Yang, C., Xiong, H., Lin, Y., Yao, J., et al. (2010). Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science 327, 1004–1007. doi: 10.1126/science.1179687
Zhou, Z. H., McCarthy, D. B., O’Connor, C. M., Reed, L. J., and Stoops, J. K. (2001). The remarkable structural and functional organization of the eukaryotic pyruvate dehydrogenase complexes. Proc. Natl. Acad. Sci. U.S.A. 98, 14802–14807.
Keywords: protein lysine acetylation, transposon mutagenesis, translational control, RNA polymerase, universal tRNA modification
Citation: Ogura M, Sato T and Abe K (2019) Bacillus subtilis YlxR, Which Is Involved in Glucose-Responsive Metabolic Changes, Regulates Expression of tsaD for Protein Quality Control of Pyruvate Dehydrogenase. Front. Microbiol. 10:923. doi: 10.3389/fmicb.2019.00923
Received: 30 October 2018; Accepted: 11 April 2019;
Published: 01 May 2019.
Edited by:
Ivan Mijakovic, Chalmers University of Technology, SwedenReviewed by:
Alan J. Wolfe, Loyola University Chicago, United StatesValerie De Crecy-Lagard, University of Florida, United States
Copyright © 2019 Ogura, Sato and Abe. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Mitsuo Ogura, b2d1cmFtQHNjYy51LXRva2FpLmFjLmpw
†Present address: Kimihiro Abe, Graduate School of Life and Environmental Sciences, University of Tsukuba, Tsukuba, Japan