 Stefano Gaiarsa1,2
Stefano Gaiarsa1,2 Gherard Batisti Biffignandi2
Gherard Batisti Biffignandi2 Eliana Pia Esposito3
Eliana Pia Esposito3 Michele Castelli4
Michele Castelli4 Keith A. Jolley5
Keith A. Jolley5 Sylvain Brisse6
Sylvain Brisse6 Davide Sassera2*
Davide Sassera2* Raffaele Zarrilli3*
Raffaele Zarrilli3*- 1Microbiology and Virology Unit, Fondazione IRCCS Policlinico San Matteo, Pavia, Italy
- 2Department of Biology and Biotechnology, University of Pavia, Pavia, Italy
- 3Department of Public Health, University of Naples “Federico II”, Naples, Italy
- 4Romeo and Enrica Invernizzi Pediatric Research Center, Department of Biosciences, University of Milan, Milan, Italy
- 5Department of Zoology, University of Oxford, Oxford, United Kingdom
- 6Biodiversity and Epidemiology of Bacterial Pathogens, Institut Pasteur, Paris, France
Acinetobacter species assigned to the Acinetobacter calcoaceticus-baumannii (Acb) complex, are Gram-negative bacteria responsible for a large number of human infections. The population structure of Acb has been studied using two 7-gene MLST schemes, introduced by Bartual and coworkers (Oxford scheme) and by Diancourt and coworkers (Pasteur scheme). The schemes have three genes in common but underlie two coexisting nomenclatures of sequence types and clonal complexes, which complicates communication on A. baumannii genotypes. The aim of this study was to compare the characteristics of the two schemes to make a recommendation about their usage. Using genome sequences of 730 strains of the Acb complex, we evaluated the phylogenetic congruence of MLST schemes, the correspondence between sequence types, their discriminative power and genotyping reliability from genomic sequences. In silico ST re-assignments highlighted the presence of a second copy of the Oxford gdhB locus, present in 553/730 genomes that has led to the creation of artefactual profiles and STs. The reliability of the two MLST schemes was tested statistically comparing MLST-based phylogenies to two reference phylogenies (core-genome genes and genome-wide SNPs) using topology-based and likelihood-based tests. Additionally, each MLST gene fragment was evaluated by correlating the pairwise nucleotide distances between each pair of genomes calculated on the core-genome and on each single gene fragment. The Pasteur scheme appears to be less discriminant among closely related isolates, but less affected by homologous recombination and more appropriate for precise strain classification in clonal groups, which within this scheme are more often correctly monophyletic. Statistical tests evaluate the tree deriving from the Oxford scheme as more similar to the reference genome trees. Our results, together with previous work, indicate that the Oxford scheme has important issues: gdhB paralogy, recombination, primers sequences, position of the genes on the genome. While there is no complete agreement in all analyses, when considered as a whole the above results indicate that the Pasteur scheme is more appropriate for population biology and epidemiological studies of A. baumannii and related species and we propose that it should be the scheme of choice during the transition toward, and in parallel with, core genome MLST.
Introduction
Bacteria belonging to the genus Acinetobacter are glucose non-fermentative Gram-negative coccobacilli that are a frequent cause of health-care associated infections and hospital outbreaks, especially in intensive-care unit patients (Dijkshoorn et al., 2007; Zarrilli et al., 2013). A. baumannii, A. nosocomialis, A. pittii, A. seifertii, and A. dijkshoorniae, five of the most clinically relevant species, are genetically and phenotypically similar to the environmental species A. calcoaceticus and are therefore grouped into a species complex called the A. calcoaceticus-A. baumannii (Acb) complex (Dijkshoorn et al., 2007; Zarrilli et al., 2013; Marí-Almirall et al., 2017). Acinetobacter spp. isolates responsible for epidemics, in particular A. baumannii isolates, are frequently multidrug resistant (MDR) or extensively drug resistant (XDR). The majority of these strains are resistant to carbapenems and a fraction of them are resistant to last resource antimicrobial agent colistin (Zarrilli et al., 2013; Pournaras et al., 2017).
The rise of resistant Acb strains prompted the design and execution of epidemiological investigations of A. baumannii epidemics using a variety of molecular typing methods, among which multilocus sequence typing (MLST) has become the reference approach (Dijkshoorn et al., 1996; Dijkshoorn et al., 2007; Zarrilli et al., 2013). Among the advantages of MLST are its excellent reproducibility, its portability that allows global comparisons, and the ease of interpretation of data in evolutionary terms (Maiden et al., 1998; Maiden et al., 2013; Bialek-Davenet et al., 2014). Besides, a prominent benefit of MLST is a derived nomenclature of sequence types (STs), which have been rapidly and largely adopted by the community, allowing expansion of the global collective knowledge on the distribution, spread and biological features of the major clonal groups.
For A. baumannii, the advantages of an MLST nomenclature have been somewhat reduced by the co-existence of two MLST schemes, which are both widely used. Both schemes encompass A. baumannii and non-baumannii Acinetobacter species. The first scheme was introduced by Bartual and coworkers and is referred as the Oxford scheme, after the platform hosting it (Bartual et al., 2005; Wisplinghoff et al., 2008), whereas a second scheme was later published by Diancourt and coworkers (Pasteur scheme) (Diancourt et al., 2010). Although both schemes appeared to provide largely concordant classifications (Zarrilli et al., 2013, 2015), the co-existence of two nomenclatures (Zarrilli et al., 2015) calls for an assessment of their relative merits in terms of reliability, discrimination (which should be optimized for epidemiological purposes) and phylogenetic concordance of their derived classifications with “true” phylogenic relationships. Although the schemes were initially hosted at two different locations (both using first the mlstdbnet; Jolley et al., 2004) then the BIGSdb software (Jolley and Maiden, 2010), in 2013, the two schemes were united into a single database. This move facilitated curation requests (sometimes using both schemes for the same set of isolates) and harmonized the data analysis functionalities. The hosting of both schemes within a single BIGSdb database, which can incorporate genomic sequences, facilitated the joint MLST analysis of genomic sequences using both schemes.
Molecular epidemiology investigations revealed the occurrence of genetically distinct clonal lineages among populations of A. baumannii (Diancourt et al., 2010; Zarrilli et al., 2013). Three of these lineages, which were initially defined as European clones I to III and subsequently regarded as International Clones (IC) I to III, are distributed worldwide (IC I and IC II are also known as Global Clones, GC). The Pasteur scheme genotypes were numbered according to previous denominations, i.e., IC I, II, and III were named, respectively, as CC1, CC2, and CC3, with their dominant ST named ST1, ST2, and ST3, respectively (Dijkshoorn et al., 1996; van Dessel et al., 2004; Diancourt et al., 2010; Zarrilli et al., 2013). Other successful epidemic clonal lineages have been subsequently identified in the population structure of A. baumannii using the Pasteur MLST scheme, including sequence types ST10, ST15, ST25, ST32, ST78, ST79 (Diancourt et al., 2010; Zarrilli et al., 2013; Da Silva et al., 2014; Pournaras et al., 2014; Ou et al., 2015; Sahl et al., 2015). The Oxford MLST scheme was able to identify international clones I, II, and III and has been shown to possess higher discriminatory power than the Pasteur scheme (Feng et al., 2016; Tomaschek et al., 2016), but to suffer from problems due to recombination and technical artifacts (Hamouda et al., 2010; Hamidian et al., 2017). Recombination plays a crucial role in the evolution of the Acb genomes. Several specific loci are interested by this phenomenon. Among them is the gpi gene, which is part of the capsular operon (thus influencing the bacterium virulence) and one of the seven Oxford MLST scheme genes. Several works suggested to exploit this behavior for classification and adopt the Oxford scheme, as it allows to monitor the capsular type (Kenyon and Hall, 2013; Holt et al., 2016; Schultz et al., 2016; Hamidian et al., 2019).
The aims of the present study were to recapitulate the current status of both schemes, determine the characteristics of the Oxford and the Pasteur MLST schemes in terms of reliability of genotyping, denomination correspondence, phylogenetic congruence with genome-based phylogenies and discriminatory power.
Materials and Methods
Python and Perl Scripts
All the scripts specifically developed and used for this work are available at https://github.com/MIDIfactory.
MLST Data
On 14 September 2018, we retrieved all sequence and profile definitions of both schemes from the PubMLST database1 to evaluate them comparatively using different approaches.
Genome Datasets
Bacterial genomes included in the analysis were manually selected from the PubMLST database. In detail, we selected all complete genomes and all high-quality genomes, i.e., in which all loci of the MLST schemes and the ribosomal MLST scheme (Jolley et al., 2012) could be detected. The resulting dataset contains the genomes of 730 strains, belonging to the Acb complex, i.e., A. baumannii (n = 703), A. nosocomialis (n = 13), A. seifertii (n = 1), A. dijkshoorniae (n = 1), A. pittii (n = 7), A. calcoaceticus (n = 3) (see Supplementary Figures S1A,B for geographical and temporal distribution of the isolates, respectively). A complete list of the genomes is available here: https://pubmlst.org/bigsdb?db=pubmlst_abaumannii_isolates&page=projects.
The allelic variants of all gene fragments of both schemes were extracted from all the genomes, using an in-house Python script based on Blast (Altschul et al., 1990), keeping all results above 95% of identity with known alleles and subsequently selecting only perfect matches, procedure that allowed to assign the corresponding STs. The allelic sequences obtained were then aligned with Muscle (Edgar, 2004). The resulting alignments were concatenated using an in-house Perl script, to obtain two multigene alignments (one per MLST scheme) to be used as input for downstream analyses.
A core genome alignment was obtained to be used as a reference for determining the reliable phylogenetic trees. Gene calling was performed using Prodigal software v2.6.1 (Hyatt et al., 2010) on all 730 genomes in the dataset. A Perl script, which uses the double best Blast hit algorithm, was then used to identify genes orthologs to the previously published core genome by Higgins et al. (2017). Groups of ortholog genes were built and aligned using Muscle (v3.8.31, Edgar, 2004). The resulting core alignments were polished for poorly aligned positions and divergent regions using Gblocks software version 0.91b (Castresana, 2000), and merged in a concatenate of all ortholog genes, via another Perl script.
A Single-nucleotide polymorphism (SNP) alignment was built as a second reference. SNPs were detected using the procedure developed by Gaiarsa and coworkers (Gaiarsa et al., 2015) based on the software Mauve (Darling et al., 2004). Each genome was individually aligned to a reference (the complete genome AB307-0294), and alignments were then concatenated. Core SNPs were defined as single-nucleotide mutations flanked by conserved bases present in all the genomes in analysis.
Phylogenetic Analyses
Phylogenies of all four datasets (Oxford, Pasteur, core genes, core SNPs) were inferred using the same approach. The best model of evolution was determined using ModelTest-ng version 0.1.3 (Darriba et al., 2011). The selected model was GTRGAMMAIX for the three gene datasets, while the analysis for the SNP alignment was performed considering the ascertainment bias and using the Lewis correction (Lewis, 2001), thus with model ASC_GTRGAMMAX. Maximum Likelihood phylogeny was performed using RAxML (Stamatakis, 2014) with 100 bootstrap replicates.
Statistical Analysis
Three statistical tests were performed using the Core-genome and SNP phylogenetic trees as references and comparing them to the phylogenetic trees resulting from MLST gene concatenates. Two topology-based tests (Matching clusters, Robinson-Fould), were performed with TreeCmp (Bogdanowicz et al., 2012). The matching clusters test calculates the number of topology changes that should be performed in order to transform a tree into the reference one. The Robinson-Fould (R-F) test instead counts the different bipartitions between the two trees. In both cases, a value of zero indicates that the two analyzed trees are identical.
The other analysis, likelihood-based Shimodaira-Hasegawa test, was performed with RaxML (Stamatakis, 2014). In this test, a null hypothesis is stated, which assumes that two compared trees are both a correct interpretation of an alignment. The tested hypothesis is that one or more trees are a better representation of the data. P-values smaller than 0.05 indicate that two trees are significantly different.
In addition, the Gini-Simpson index was used to determine the discrimination power of both schemes. The index was calculated using the website service comparingpartitions.info on the entire dataset of 730 genomes and on the genomes of the three main International Clones.
Monophyly of Clonal Complexes
Clonal Complexes were defined using eBURST as described previously (Feil et al., 2004); CCs were defined as groups of Sequence Types that differ from one or more members of the group by just one allele. Monophyly of the CCs was checked on the core genome tree using the R environment and the library spider. Minimum spanning trees of sequence types (STs) were built using Phyloviz Online using the goeBURST algorithm (Ribeiro-Gonçalves et al., 2016). Minimum spanning trees were generated from the 7 alleles of each MLST scheme and species were assigned based on clustering with reference STs. The species field was updated in the PubMLST database for all STs with no ambiguous assignment.
Nucleotide Distance Analysis
To evaluate how well each gene fragment variation correlates with the genome variation, we compared the pairwise nucleotide distances between each pair of genomes in our dataset with the corresponding distances between each gene fragment pairs. The MLST gene fragment sequences, as well as all the concatenated core genome gene sequences, were aligned with Muscle (Edgar, 2004) and then used to calculate pairwise genetic distances via the function “dist.dna” of the R package APE (Paradis et al., 2004).
In order to test the discrimination resolution of each MLST scheme, we plotted the pairwise sequence distance between each MLST locus for each pair of genomes, against the corresponding core genome-wide sequence distance. The correlation was determined adopting the regression linear model in the R environment. Due to an uneven large distribution of genomic distances, we decided to split each dataset in three blocks (as in Bleidorn and Gerth, 2018), based on the genomic distance (first block: 0.0–0.05, second block: 0.05–0.1, third: >0.1). Finally, a heatmap was generated to evaluate R2 and slopes of all regression lines.
Recombination Rate
The recombination rate was calculated for each locus of both MLST schemes, using the RDP4 Software on the alignments of all alleles present the 730 genomes of the dataset (Martin et al., 2015), employing all available algorithms. This same analysis was also performed on a reduced dataset containing only the A. baumannii genome subset, to refine the recombination rate detection within this species.
GdhB Analysis
To investigate the Oxford scheme gene gdhB putative duplication, we extracted all variants of both gene copies from the 730 genomes in our dataset, including sequences not registered in the PubMLST database, through a custom approach based on Blast (identity > 95% with the gdhB-1 or the gdhB-182 allele).
To check primer alignment, the flanking region was extracted for all alleles found, and aligned with the PCR primers used to sequence the gdhB locus. Mismatches between the aligned sequences and the primers were calculated for the entire primer length and for the ten bases at the three-prime end.
Finally, all gdhB and gdhB2 variants were analyzed with a phylogenetic approach. Variants were aligned with the software muscle (Edgar, 2004) and used as input for a Maximum Likelihood phylogeny, executed with fasttree (Price et al., 2010). The genes surrounding the gdhB and gdhB2 sites were extracted using a python script and the software Prodigal (Hyatt et al., 2010). Functions were predicted using COGnitor (Galperin et al., 2015). Codon adaptation index was calculated for all coding sequences in all 730 genomes in the dataset using the CAIcalc script (Puigbo et al., 2008).
Results
Status of the Database Contents of the Pasteur and Oxford MLST Schemes
In order to evaluate the two MLST schemes available for A. baumannii, we start by describing them in detail. Both schemes are built on 7 genes. The concatenation of allele 1 of the 7 Oxford and Pasteur genes yields 2895 and 2976 nucleotides, respectively. Three genes are shared between the two schemes: cpn60, gltA, and recA. The subsequences used for typing, though, differ between the two schemes, as depicted in Figure 1A.
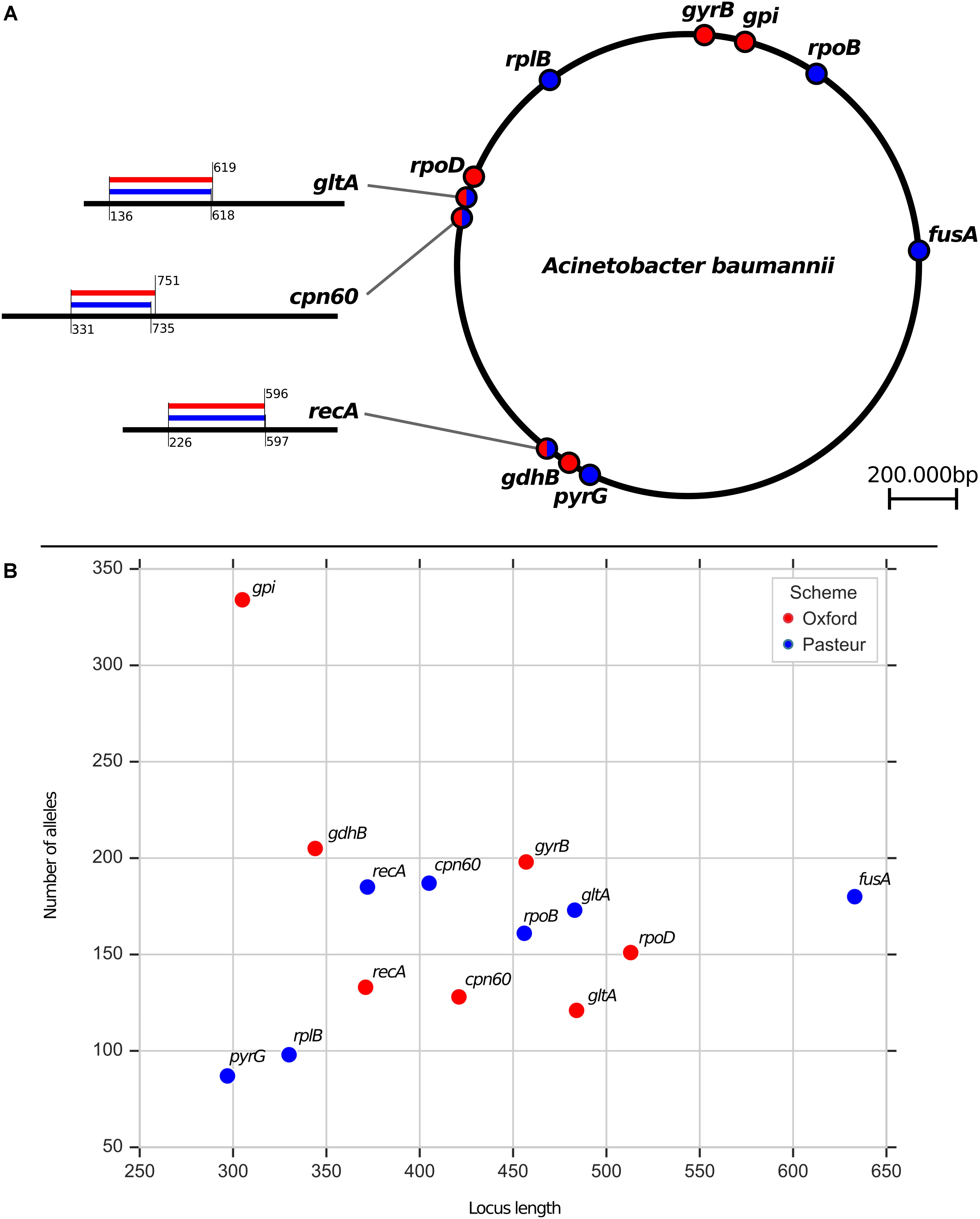
Figure 1. (A) Distribution of the MLST loci on the genome. A panel is used to highlight the relative position of two loci whenever they come from the same gene. (B) Scatterplot comparing loci length and variability in terms of number of alleles registered on the Pubmlst database (https://pubmlst.org/abaumannii).
We downloaded all sequence and profiles definitions on date 14 September 2018. Oxford contained 1866 profiles (STs), while the Pasteur scheme had 1234 STs defined. The number of alleles ranged from 121 to 334 for Oxford, 87–187 for Pasteur. Regarding the three common genes, the allele numbers were 128(Oxford)/187(Pasteur) for cpn60, 121(Oxford)/173(Pasteur) for gltA, 133(Oxford)/185(Pasteur) for recA. Thus, the Pasteur scheme seemed to encompass more diversity in the common genes, although more STs were defined by Oxford overall (Figure 1B).
A gdhB Paralog Complicates in silico Determination of the Oxford ST
730 genomes of the Acb complex were selected and downloaded to be used as dataset. The database included mostly genomes of A. baumannii isolates (703, 96.3%), but also genomes of A. nosocomialis, A. seifertii, A. dijkshoorniae, A. pittii, and A. calcoaceticus isolates. MLST alleles of both schemes were first defined from genomic sequences. While extracting alleles, a consistent proportion of the genomes appeared to have two variants of the gdhB gene. This issue was investigated further, revealing that an alternative gdhB locus (corresponding to alleles 182, 189 and variants of them) was present in 553 (76%) of 730 strains, all belonging to the A. baumannii species. This locus is often annotated in these genomes as gdhB2 and has a sequence similarity with allele 1 of gdhB ranging from 73.98 to 77.94%. Primers used for molecular MLST were aligned to both genomic regions, showing a low affinity for the alternative gdhB2 locus (Table 1). For this reason, we can hypothesize that the gdhB2 locus cannot be amplified with these PCR primers, and indeed all alleles of the paralogous locus were defined only using in-silico methods for ST determination from genomic sequences. In order to evaluate the relationships between gdhB and gdhB2, a phylogeny of all 64 gdhB and the 40 gdhB2 alleles was determined. The resulting tree clearly showed two main clusters, one containing only the putative gdhB2 sequences, i.e., allele 182 and related variants, only found in A. baumannii (Figure 2). The other main cluster, containing the original gdhB variants, on the other hand presents genomes from all the analyzed Acb species, and each species appears grouped in a monophyletic cluster. The genomic surroundings of the two variants (i.e., the three genes upstream and downstream the gdhB and gdhB2 sites) are clearly different. Nucleotide composition analysis was performed on gdhB and gdhB2, showing that both genes have a codon composition that is significantly different from the average of the respective genomes. A subset of isolates that are characterized in the Oxford scheme with STs that include alleles of gdhB2 instead of the original locus where manually analyzed. When replacing the artefactual gdhB2 allele with the correct gdhB locus, the obtained profiles correspond to existing STs, including ST231, a strain of epidemiological importance due to the report of the presence of the carbapenemase gene blaOXA-23. Therefore, the wrong calling of alleles at gdhB2 locus has artifactually inflated the diversity recorded using the Oxford scheme.
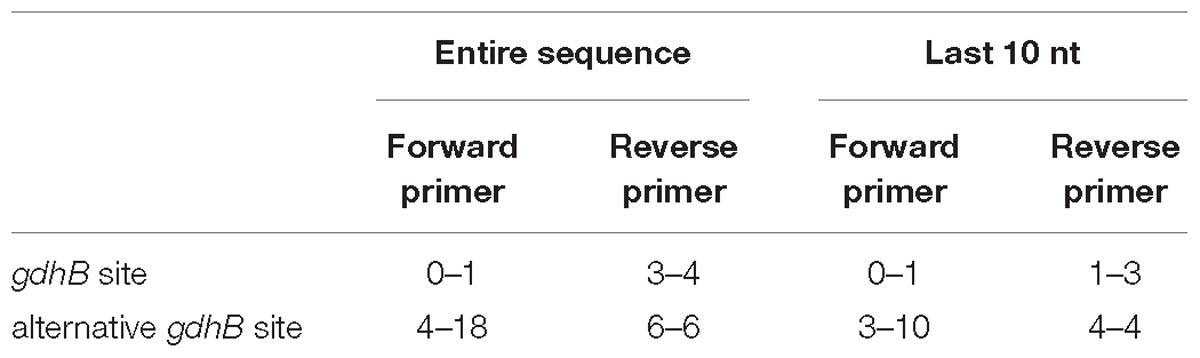
Table 1. Minimum and maximum number of mutations obtained when sequencing primers are aligned to all gdhB loci in the dataset.
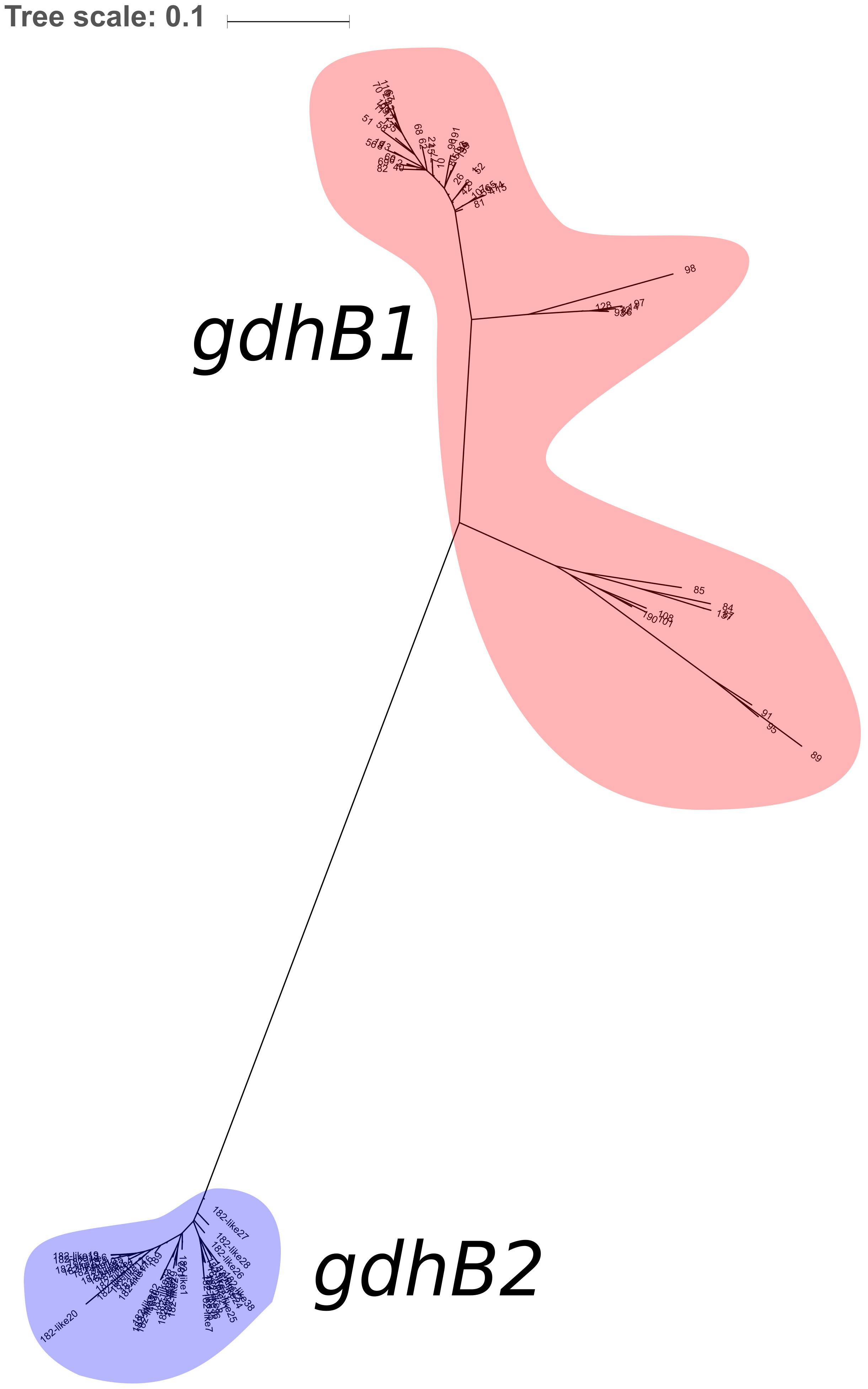
Figure 2. Maximum Likelihood phylogeny of the 104 gdhB variants detected in the dataset. Alleles of the traditional gdhB locus are highlighted in red; alleles of the alternative locus (including 38 non-registered alleles) are highlighted in blue.
Oxford and Pasteur Comparison
Minimum spanning tree analysis demonstrated that both MLST schemes gene sets discriminate the existing species within the Acb complex (Figures 3A,B, MLST-based species identification). ST assignments and eBURST analyses using our 730 genomes dataset generated a convenient and expandable table of correspondence between the two MLST schemes, represented in Table 2 and in Figure 4 and Supplementary Figure S2 as Sankey diagrams.
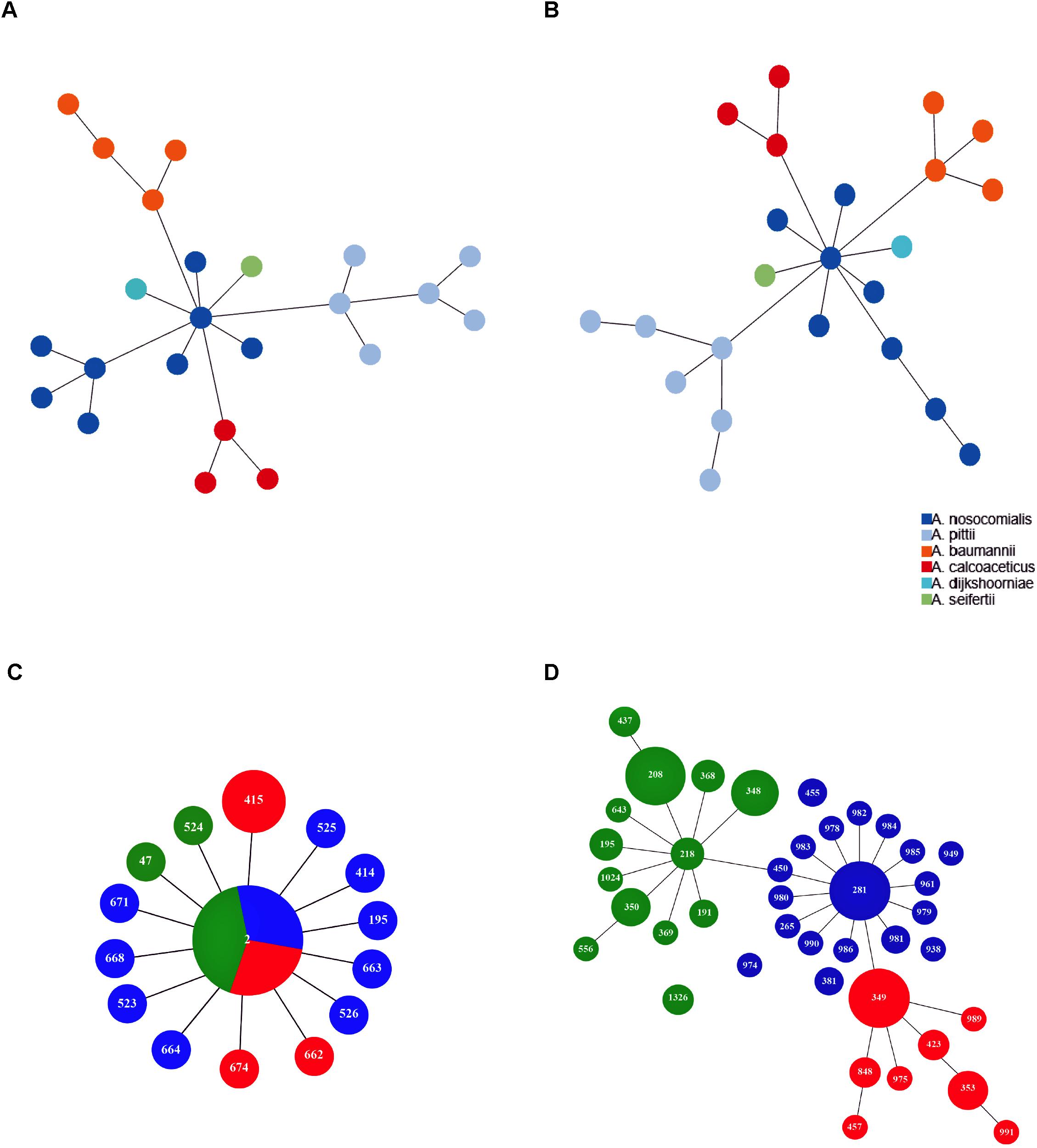
Figure 3. Minimum spanning trees of A. baumannii, A. nosocomialis, A. pittii, A. seifertii, and A. dijkshoorniae (23 isolates) using (A) Pasteur and (B) Oxford MLST scheme. The colors corresponding to Acinetobacter species are shown in the legend. Minimum spanning trees representing the structure of the A. baumannii international clone II (422 isolates) as reconstructed using Pasteur (C) and Oxford (D) MLST schemes. Numbers inside each circle indicate the ST types. Circle size is proportional to the number of isolates belonging to the same ST type. Colors in (C,D) represent sub-branches identified by eBURST using the Oxford MLST scheme.
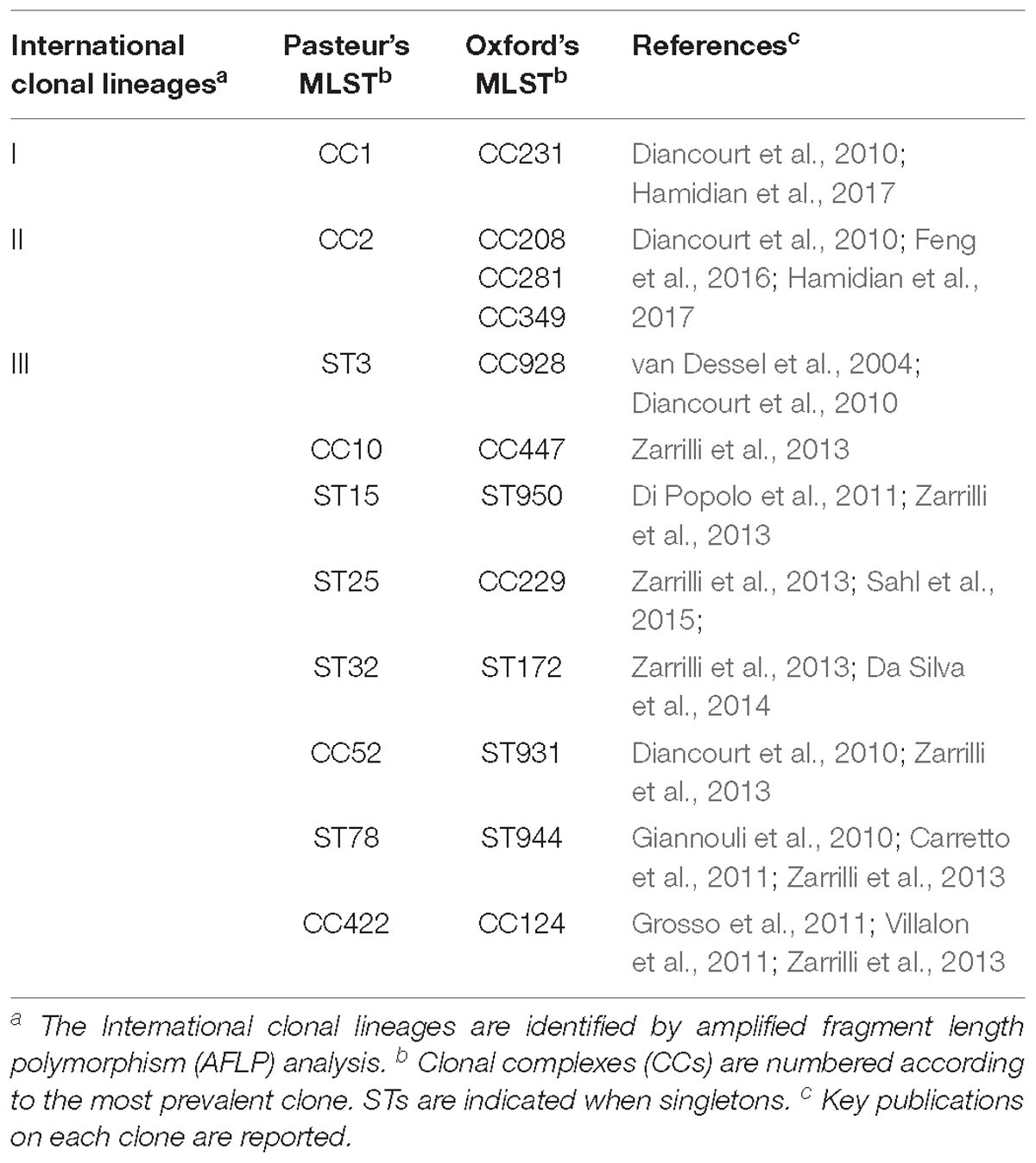
Table 2. Correspondence of A. baumannii clonal lineages as assessed by Oxford and Pasteur MLST schemes.
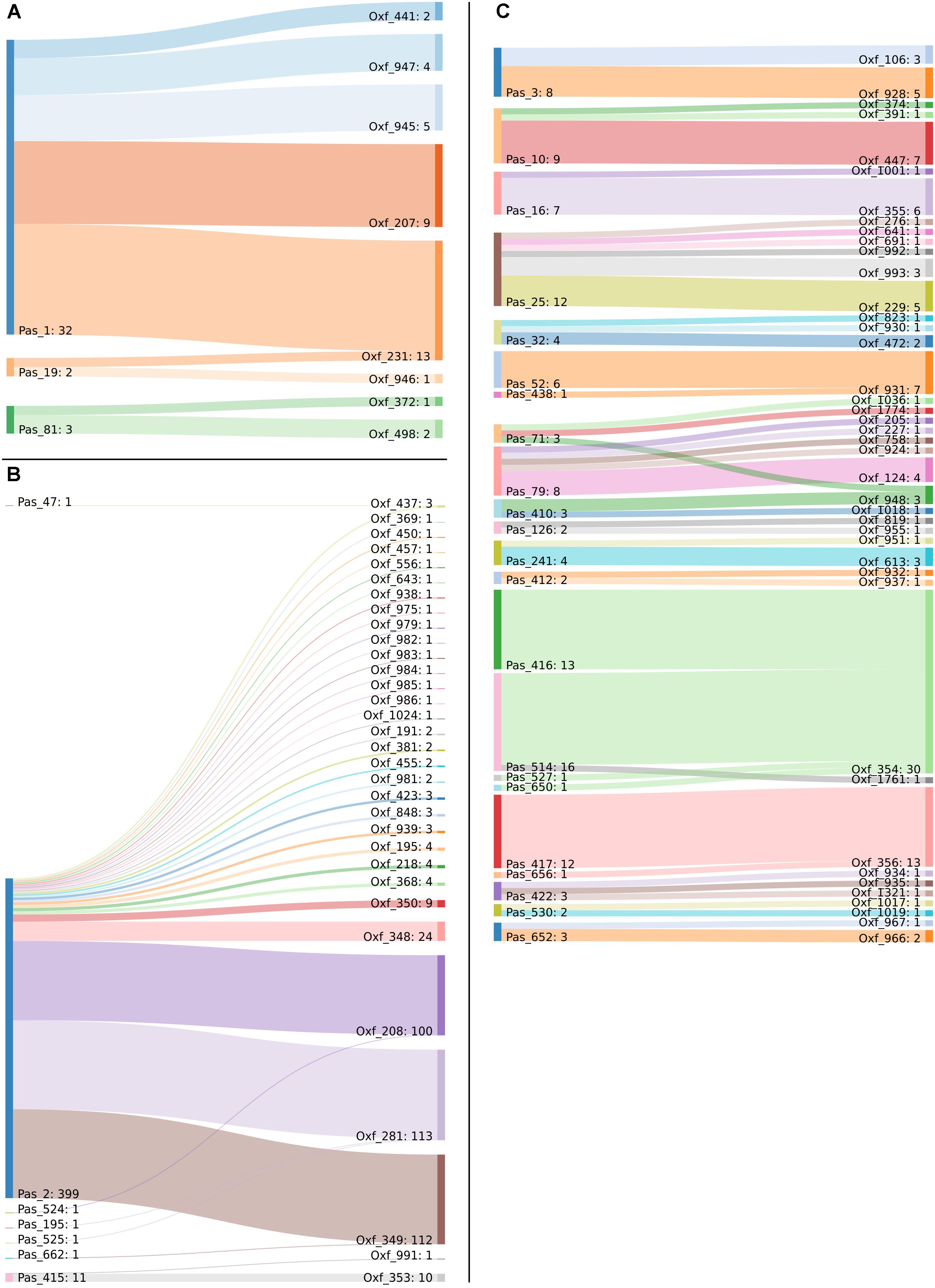
Figure 4. Sankey diagram of the MLST classification of the 730 genomes in use, as performed with the Pasteur and Oxford schemes. Two-way corresponding STs were removed to improve image clarity. Captions show the corresponding STs belonging to (A) International Clone 1, (B) International Clone 2, and (C) all the other genomes.
Then, we extracted CCs from both schemes and checked their monophyly on the core genome phylogeny. The Pasteur scheme had 12 CCs comprising a total of 47 STs, i.e., 35.34% of all STs. Additionally, there were 86 singleton Pasteur STs, while nine CCs are monophyletic. The Oxford scheme had 16 CCs comprising in total 82 STs, i.e., 44.56% of all STs. Additionally, there were 102 singleton Oxford STs and 11 monophyletic CCs. The Pasteur scheme appeared to be less discriminant, but more appropriate for precise strain classification into clonal groups. On the other hand, the Oxford scheme was able to identify additional genotypes and to differentiate isolates belonging to international clone II into three distinct clonal groups (Table 2 and Figures 3C,D).
The Gini-Simpson index of the 730 genomes was almost one order of magnitude higher when classifying the dataset using the Pasteur STs (0.70–0.93 for Oxford). When repeating the calculation on the genomes of the three International Clones, the score difference was lower but still important. The Pasteur scheme obtained values close to 0, being of low discrimination within the three ICs (Table 3).

Table 3. Gini-Simpson index values obtained using the STs of both schemes on the entire dataset of 730 genomes and separately on the three International Clones.
Topologies Comparison and Statistical Analysis
To evaluate the two MLST schemes with respect to phylogenetic inference, we constructed four phylogenies (Figure 5): two using the concatenated alleles of the Pasteur and Oxford scheme as input, and two references using genome-wide data (a core-genome of 1409 high quality genes, and an alignment of 68,340 SNPs). We then statistically compared the trees to evaluate the reliability of MLST concatenates in relationship with the two references. All tests applied suggested a better congruence between the Oxford scheme and the references. Full results are reported in Table 4, which includes the results of cross check tests between the two reference phylogenies. In Figure 5, International Clones I, II, and III (calculated according to the Pasteur CC 1, 2, and 3) are highlighted in all four trees. In the tree obtained using the Oxford genes, the genomes of IC I are split in two separate, non-monophyletic, clades.
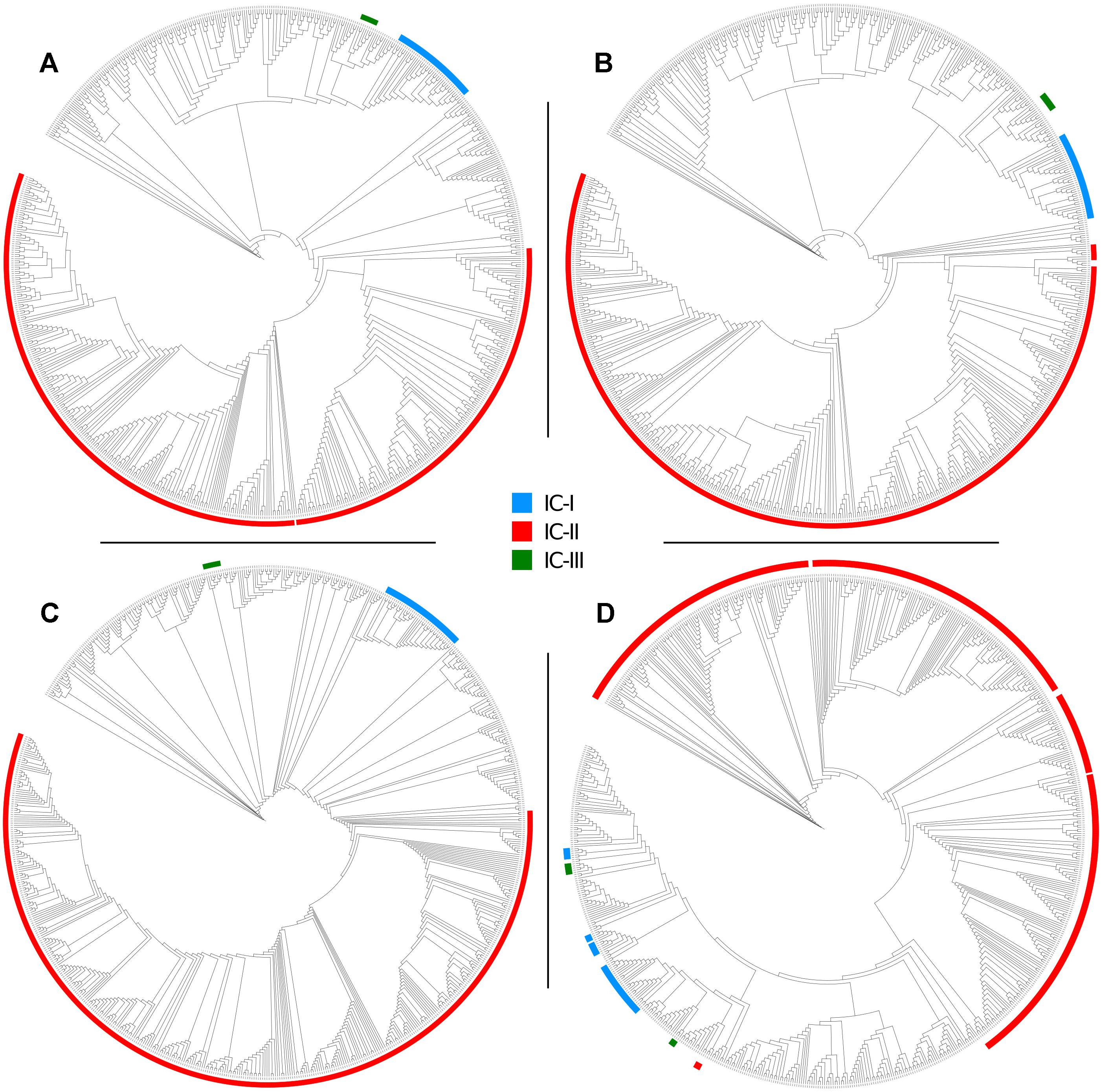
Figure 5. Maximum Likelihood phylogenies of 730 genomes of the Acb complex, inferred from (A) a concatenate of 1409 orthologous core genes, (B) a concatenate of 68,340 SNP positions, (C) a concatenate of the seven alleles used in the Pasteur MLST scheme, and (D) a concatenate of the seven alleles used in the Oxford MLST scheme. Major clonal complexes are highlighted: International clone I in blue, International clone II in red, International clone III in green.
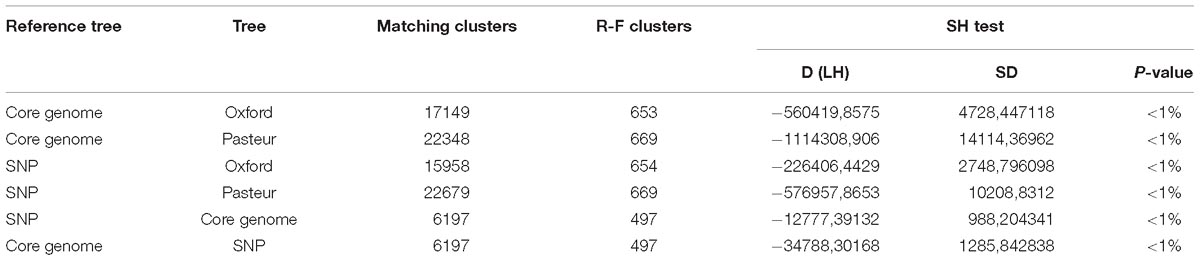
Table 4. Statistical comparison of the phylogenies obtained using the MLST loci of both schemes to the two reference trees obtained with genome-wide SNPS and core genes.
Recombination Analysis
A recombination analysis was run on the alignments of all MLST gene fragments. All gene sequences presented signs of recombination in the non-baumannii genomes. This suggested that the analysis was biased by an uneven evolutionary distance and was repeated only on the 703 A. baumannii genomes. This step allowed detecting a recombination in the gpi locus of the Oxford scheme, while all other loci appeared to be recombination free. These data are in partial agreement with a previous study, which detected recombination in the topologies of the phylogenetic trees generated for the gyrB and gpi genes using the Oxford MLST scheme (Hamouda et al., 2010). High recombinogenicity of the gpi locus was also detected in other studies, which focused on the genomic plasticity of the capsular loci (Kenyon and Hall, 2013; Holt et al., 2016; Schultz et al., 2016; Hamidian et al., 2019). Accordingly, the recombining locus gpi happens to have the highest variability in alleles (Figure 1B). These results suggest that the Pasteur scheme allele diversification is less affected by homologous recombination. Accordingly, the recombining locus gpi happens to have the highest variability in alleles (Figure 1B).
MLST Correlation With Genomic Distance
The pairwise sequence distance between each MLST locus of each pair of genomes was plotted against the corresponding genome-wide distance in order to test its correlation. The test was repeated focusing on three different ranges of genome-wide distances (as in Bleidorn and Gerth, 2018). A total of 56 plots was obtained and reported in Supplementary Figure S3. We expected high quality genes to have a regression line with a positive slope (direct correlation between genomic and locus distance), as close as possible to a value of 1 (this means that the evolution of the MLST sequence follows the same pace as the genome). We thus summarized the slope data in two heatmaps: one with the slope values (Figure 6) and one with the R2-values (Supplementary Figure S4) to assess reliability of the regression. In general, both schemes perform better at lower genomic distances. This trend is highlighted especially in the recA gene, shared between the two schemes. Generally speaking, Pasteur alleles perform well when analyzing high genomic distances, while Oxford alleles have better scores at lower genomic distances. Finally, the gpi gene deserves a particular mention: at low genomic distances, the locus shows a quicker evolution than the genomic reference, while at high genomic distances, it shows an inverted evolution (i.e., at higher genomic distances correspond lower gpi distances). This could be probably due to the recombination detected in the gene sequence: at lower evolutionary distances, recombinations can increase variation, while they can act as equalizers at larger scales, having a cohesive effect (Doroghazi and Buckley, 2011).
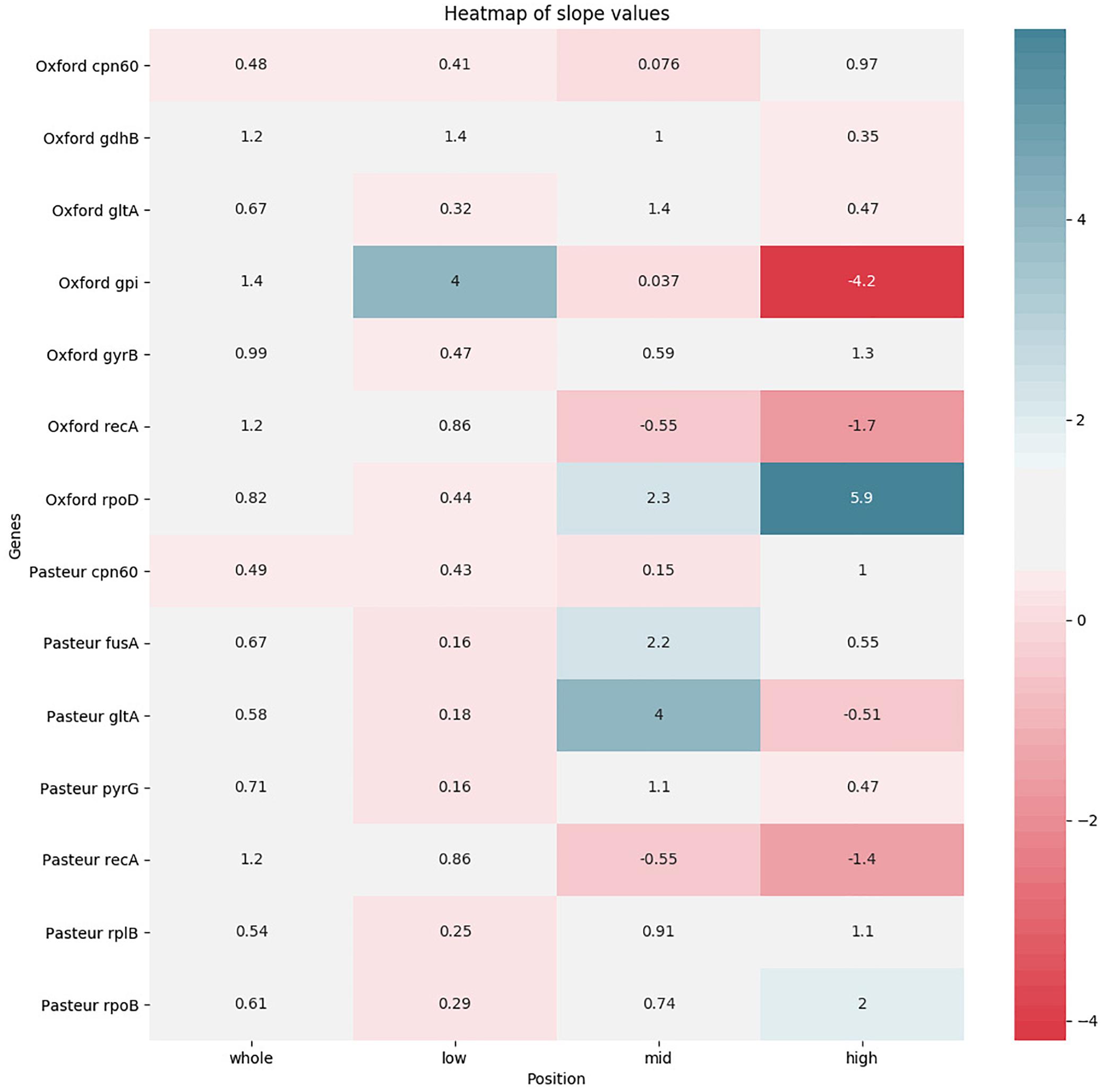
Figure 6. Heatmap showing the levels of agreement of each MLST locus to a reference alignment based on the core genome. Agreement levels are shown in terms of regression line slopes obtained from distance values between genome pairs. For each locus four regression were obtained, based, respectively, on the entirety of genome-wide distances and three subranges. White color indicates that a MLST locus represents the genome-wide distances well, showing a similar evolutionary pace. Red shades (indicating a negative correlation) and blue shades (indicating that the MLST locus shows an evolutionary pace much higher than the genome wide distances) indicate that the locus variation does not well represent genome variation.
Discussion
The aim of this investigation was to compare the two MLST schemes that are widely used to genotype isolates of the Acb species complex. We decided to tackle the problem evaluating the two schemes, their reproducibility, discrimination, strain classification into CCs and compatibility among MLST-based phylogenies and genome-based phylogenies.
Starting from a curated dataset of 730 genomes, two phylogenomic trees were obtained from information collected throughout the whole genomes (core genes and core SNPs). The two resulting trees showed highly similar topologies and can be considered close approximations of the real evolutionary history of the Acb species complex. For this reason, they were used as reliable and unbiased references for the analyses. A phylogeny was obtained from the concatenate alignment of the alleles of each of the two MLST schemes and compared with the two references using three different statistical methods. In all three cases, the tree obtained from the Oxford scheme resulted in a closer approximation of the references.
On the other hand, previous publications described a series of limitations and issues of the Oxford scheme (Hamouda et al., 2010; Hamidian et al., 2017), such as the inclusion of the primers in the registered allele sequences of two of the seven MLST genes. This unusual procedure leads to replacing the true sequence with the primer sequence at these locations, creating mosaic sequences (primerF + internal sequence from isolate + primerR), and removing variation at priming sites when sequenced using primer-based methods. This issue was recognized and corrected previously, so it should not affect future identification, but it remains for all the previously investigated strains that were analyzed by PCR and not in silico (Hamidian et al., 2017).
Here, we detected an entirely novel problem with in silico determination of Oxford profiles, namely the presence of a paralog of the Oxford gene gdhB in a high proportion (553 out of the 730) of Acb genomes, a locus that we found to be often annotated as gdhB2, and that is located in a different genomic region. On multiple occasions, allele sequences resulting from this duplication were incorrectly used to establish new Oxford STs that do not actually exist, as they are based on alleles of the paralog gdhB2. This issue can be explained with an event of gene duplication, or of horizontal gene transfer, the second being more probable considering the low identity between the two paralogs (around 73%). Such event would have occurred early in the evolution of the Acb complex, possibly at the root of the A. baumannii species, followed by a quick sequence divergence of gdhB2 and by the loss of this gene in a number of representatives of the Acb complex (177 out of 730 in our dataset). Nucleotide composition analysis shows that both gdhB and gdhB2 have higher than average codon adaptation index, thus not allowing to understand which of the two events could be more likely.
The incorrect alleles are, to date (14 September 2018) 182 and 189 and have led to the determination of 30 STs: 1567, 1604, 1677, 1678, 1793, 1794, 1796, 1800, 1804, 1805, 1806, 1807, 1808, 1809, 1813, 1815, 1816, 1833, 1834, 1835, 1836, 1837, 1838, 1839, 1840, 1841, 1843, 1851, 1852, 1857. Other 38 unregistered 182-like alleles were found in the dataset used in this project and should not be registered if submitted to the MLST database. We suggest that these gdhB2-based alleles should be removed from the database, and each of the genomes belonging to these STs should be re-analyzed excluding the paralog gdhB2 (a stringent allele calling filter on genetic similarity could be useful for this purpose) to find the correct gdhB allele and subsequently the current ST. The inclusion of the paralog allele in the database is due to the bioinformatics methods used, which did not take into account the possible presence of such paralog. This does not appear to have ever happened in PCR-based classification, as the gdhB primers are sufficiently specific to amplify only the correct locus. While this issue can be solved using bioinformatics, in silico MLST can be performed with different software tools including in-house scripts, so we cannot rule out the possibility of novel gdhB2-based alleles to appear in the future.
Another issue of the Oxford scheme, albeit one that mostly impact phylogenetic analyses, is the presence of possible recombinations, previously reported for two of the loci used, gpi and gyrB (Hamouda et al., 2010). Our analysis does not show a clear signal of recombination in the gyrB gene, but we detected a strong recombination signal in the gpi gene. The fact that recombination in the gyrB locus was detected previously but not within this study could be explained by the use of different methods, as Hamouda and colleagues used a general phylogenetic approach (Hamouda et al., 2010), while we chose an ad hoc recombination detection software.
Clearly, the most reliable classification method for Acinetobacter isolates would be one based on genome-wide information, such as a core genome MLST (cgMLST) (Maiden et al., 2013). Whole genome sequencing, which is required to extract cgMLST data, is now a routine task in many research laboratories, with costs comparable to performing the seven PCR amplifications and Sanger sequencing required for the traditional MLST. Whole genome sequencing has been used to study A. baumannii phylogeny (Snitkin et al., 2011; Sahl et al., 2013; Chan et al., 2015; Wallace et al., 2016), but only two studies so far used cgMLST schemes for A. baumannii (Fitzpatrick et al., 2016; Higgins et al., 2017). The cgMLST scheme by Fitzpatrick and colleagues analyzed genetic similarity based on SNPs in the core genome of a limited number of Acb complex bloodstream isolates, 116 A. baumannii, 28 A. pittii, and 3 A. nosocomialis, and showed higher discriminatory power than PFGE and MLST (Fitzpatrick et al., 2016). Higgins et al. (2017) developed a cgMLST scheme based on 1,339 A. baumannii genomes and validated on 53 A. baumannii genomes. The cgMLST clustering showed a good correlation between PFGE types and also matched the classification of A. baumannii international clones as previously determined by DiversiLab typing or MLST (Higgins et al., 2017). The set of genes by Higgins works well when analyzing strains of A. baumannii sensu stricto but is not fit for the other species of the Acb complex (Higgins et al., 2017) and should thus be restricted to shared genes in order to allow broader use.
As NGS is not yet accessible in all diagnostic laboratories in the world, cgMLST is probably still unfit to be a globally shared typing technique. Additionally, cgMLST classification could in some cases be incompatible with previous works that used MLST classifications, especially if specific STs are found to be polyphyletic and considering that determining STs in silico must be done with caution, as highlighted by our discovery of incorrect gdhB alleles identified based on a paralog sequence. Therefore, 7-gene MLST is likely to continue being used widely in the near future.
Conclusion
In conclusion, the two MLST schemes have complementary characteristics, each with their own advantages: the Pasteur scheme shows lower discrimination, is able to better identify clonal lineages, and in general performs better when comparing evolutionary distant clones. The Oxford scheme in turn shows higher concordance with phylogenies and works better for discrimination among strains at short evolutionary distances. However, a novel and important issue of the Oxford scheme in the genomic era is the presence of an alternative gdhB locus in the majority (533/730) of the A. baumannii genomes. Besides, the presence of primers in sequence templates of two of the genes may have resulted in a few artefactual allele calls.
Previous works recommended the Oxford scheme due to the presence of the gpi gene, which is part of the capsular locus, and thus can provide a link between typing and phenotypic information (Kenyon and Hall, 2013; Holt et al., 2016). Other authors criticize both schemes due to the low level of resolution or polyphyly of Sts (Castillo-Ramírez and Graña-Miraglia, 2019). An opposite view could recommend the use of both schemes to provide a finer characterization. On the other hand, our opinion is to recommend the Pasteur scheme because of the following reasoning: both the link to phenotypic information and the finer characterization will be soon accomplished by the blooming cgMLST method, and will thus not be required for classical MLST schemes in the future. An MLST scheme in the genome era, however, retains great importance as a fundamental nomenclature tool. As such, absence of recombination, absence of wrongly called variants, and better adherence to the main epidemiological clones should be considered the main reasons to choose one of the two available schemes.
To summarize, we recommend the utilization of the Pasteur scheme for 7-gene MLST classification of Acinetobacter isolates of the Acb complex, and that future cgMLST nomenclature of genotypic groups should inherit, as much as possible, the Pasteur MLST denominations that were themselves inherited from pre-MLST international clone nomenclatures.
Author Contributions
SG, SB, DS, and RZ designed the project. EE, KJ, and RZ curated the database and performed eburst analyses. SG and GB performed the bulk of the bioinformatic analyses. MC performed the phylogenetic analyses. SG, KJ, SB, DS, and RZ wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported in part by grants from Wellcome Trust Biomedical Resource (Grant 104992 to KJ), University of Naples “Federico II” (Fondo d’Ateneo per la Ricerca to RZ) and the Italian Ministry of Education, University and Research (MIUR): PRIN2017 (Grant 2017SFBFER to RZ), Dipartimenti di Eccellenza Program (2018–2022) – Department of Biology and Biotechnology “L. Spallanzani,” University of Pavia (to DS).
Conflict of Interest Statement
SB was one of the developers of the Pasteur MLST scheme for Acinetobacter strains of the Acb complex.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank the team of curators of the Pasteur and Oxford Acinetobacter MLST schemes for curating the data and making them publicly available at http://pubmlst.org/abaumannii/. RZ dedicates this study to the memory of Carmelo Bruno Bruni, his mentor of microbial genetics.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00930/full#supplementary-material
FIGURE S1 | (A) Geographic and (B) temporal characterization of the database of 730 genomes used in the present work.
FIGURE S2 | Complete Sankey diagram of the MLST classification of the 730 genomes in use, as performed with the Pasteur and Oxford schemes.
FIGURE S3 | Plots showing the correlation of genetic distances of Acinetobacter genomes between MLST loci and genome-wide distances. Each data-point corresponds to a single pair of Acinetobacter genomes, and shows the divergence of each locus related to core-genome-based distance. For each locus, we show the full range of genome-wide distances and three different subregions representing different genomic distances (first block: 0.0–0.05, second block: 0.05–0.1, third: > 0.1).
FIGURE S4 | Heatmap representing the correlation of each MLST locus in comparison to a reference alignment based on the core genome. Four plots were produced for each locus, based on genome-wide distances (see Supplementary Figure 3). In this figure, we report the R2-values of the interpolation lines.
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bartual, S. G., Seifert, H., Hippler, C., Luzon, M. A., Wisplinghoff, H., and Rodriguez-Valera, F. (2005). Development of a multi-locus sequence typing scheme for characterization of clinical isolates of Acinetobacter baumannii. J. Clin. Microbiol. 43, 4382–4390. doi: 10.1128/jcm.43.9.4382-4390.2005
Bialek-Davenet, S., Criscuolo, A., Ailloud, F., Passet, V., Jones, L., Delannoy-Vieillard, A. S., et al. (2014). Genomic definition of hypervirulent and multidrug-resistant Klebsiella pneumoniae clonal groups. Emerg. Infect. Dis. 20, 1812–1820. doi: 10.3201/eid2011.140206
Bleidorn, C., and Gerth, M. (2018). A critical re-evaluation of multilocus sequence typing (MLST) efforts in Wolbachia. FEMS Microbiol. Ecol. 94:fix163. doi: 10.1093/femsec/fix163
Bogdanowicz, D., Giaro, K., and Wróbel, B. (2012). TreeCmp: comparison of trees in polynomial time. Evol. Bioinform. Online 8, 475–487. doi: 10.4137/EBO.S9657
Carretto, E., Barbarini, D., Dijkshoorn, L., van der Reijden, T. J., Brisse, S., Passet, V., et al. (2011). Widespread carbapenem resistant Acinetobacter baumannii clones in Italian hospitals revealed by a multicenter study. Infect. Genet. Evol. 11, 1319–1326. doi: 10.1016/j.meegid.2011.04.024
Castillo-Ramírez, S., and Graña-Miraglia, L. (2019). Inaccurate multilocus sequence typing of Acinetobacter baumannii. Emerg. Infect. Dis. 25, 186–187. doi: 10.3201/eid2501.180374
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chan, A. P., Sutton, G., DePew, J., Krishnakumar, R., Choi, Y., Huang, X. Z., et al. (2015). A novel method of consensus pan-chromosome assembly and large-scale comparative analysis reveal the highly flexible pan-genome of Acinetobacter baumannii. Genome Biol. 21:143. doi: 10.1186/s13059-015-0701-6
Da Silva, G. J., Van Der Reijden, T., Domingues, S., Mendonça, N., Petersen, K., and Dijkshoorn, L. (2014). Characterization of a novel international clonal complex (CC32) of Acinetobacter baumannii with epidemic potential. Epidemiol. Infect. 142, 1554–1558. doi: 10.1017/S0950268813002288
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2011). Prottest 3: fast selection of best-fit models of protein evolution. Bioinformatics 27, 1164–1165. doi: 10.1093/bioinformatics/btr088
Di Popolo, A., Giannouli, M., Triassi, M., Brisse, S., and Zarrilli, R. (2011). Molecular epidemiology of multidrug-resistant Acinetobacter baumannii strains in four Mediterranean countries with a multilocus sequence typing scheme. Clin. Microbiol. Infect. 17, 197–201. doi: 10.1111/j.1469-0691.2010.03254.x
Diancourt, L., Passet, V., Nemec, A., Dijkshoorn, L., and Brisse, S. (2010). The population structure of Acinetobacter baumannii: expanding multiresistant clones from an ancestral susceptible genetic pool. PLoS One 5:e10034. doi: 10.1371/journal.pone.0010034
Dijkshoorn, L., Aucken, H., Gerner Smidt, P., Janssen, P., Kaufmann, M. E., Garaizar, J., et al. (1996). Comparison of outbreak and nonoutbreak Acinetobacter baumannii strains by genotypic and phenotypic methods. J. Clin. Microbiol. 34, 1519–1525.
Dijkshoorn, L., Nemec, A., and Seifert, H. (2007). An increasing threat in hospitals: multidrug resistant Acinetobacter baumannii. Nat. Rev. Microbiol. 5, 939–951. doi: 10.1038/nrmicro1789
Doroghazi, J. R., and Buckley, D. H. (2011). A model for the effect of homologous recombination on microbial diversification. Genome Biol. Evol. 3, 1349–1356. doi: 10.1093/gbe/evr110
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Feil, E. J., Li, B. C., Aanensen, D. M., Hanage, W. P., and Spratt, B. G. (2004). eBURST: inferring patterns of evolutionary descent among clusters of related bacterial genotypes from multilocus sequence typing data. J. Bacteriol. 186, 1518–1530. doi: 10.1128/JB.186.5.1518-1530.2004
Feng, Y., Ruan, Z., Shu, J., Chen, C. L., and Chiu, C. H. (2016). A glimpse into evolution and dissemination of multidrug-resistant Acinetobacter baumannii isolates in East Asia: a comparative genomics study. Sci. Rep. 6:24342. doi: 10.1038/srep24342
Fitzpatrick, M. A., Ozer, E. A., and Hauser, A. R. (2016). Utility of whole-genome sequencing in characterizing Acinetobacter epidemiology and analyzing hospital outbreaks. J. Clin. Microbiol. 54, 593–612. doi: 10.1128/JCM.01818-15
Gaiarsa, S., Comandatore, F., Gaibani, P., Corbella, M., Dalla Valle, C., Epis, S., et al. (2015). Genomic epidemiology of Klebsiella pneumoniae in Italy and novel insights into the origin and global evolution of its resistance to carbapenem antibiotics. Antimicrob. Agents Chemother. 59, 389–396. doi: 10.1128/AAC.04224-14
Galperin, M. Y., Makarova, K. S., Wolf, Y. I., and Koonin, E. V. (2015). Expanded microbial genome coverage and improved protein family annotation in the COG database. Nucleic Acids Res. 43, D261–D269. doi: 10.1093/nar/gku1223
Giannouli, M., Cuccurullo, S., Crivaro, V., Di Popolo, A., Bernardo, M., Tomasone, F., et al. (2010). Molecular epidemiology of multidrug-resistant Acinetobacter baumannii in a tertiary care hospital in Naples, Italy, shows the emergence of a novel epidemic clone. J. Clin. Microbiol. 48, 1223–1230. doi: 10.1128/JCM.02263-09
Grosso, F., Carvalho, K. R., Quinteira, S., Ramos, A., Carvalho-Assef, A. P., Asensi, M. D., et al. (2011). OXA-23-producing Acinetobacter baumannii: a new hotspot of diversity in Rio de Janeiro? J. Antimicrob. Chemother. 66, 62–65. doi: 10.1093/jac/dkq406
Hamidian, M., Hawkey, J., Wick, R., Holt, K. E., and Hall, R. M. (2019). Evolution of a clade of Acinetobacter baumannii global clone 1, lineage 1 via acquisition of carbapenem- and aminoglycoside-resistance genes and dispersion of ISAba1. Microb. Genom. 5:e000242. doi: 10.1099/mgen.0.000242
Hamidian, M., Nigro, S. J., and Hall, R. M. (2017). Problems with the Oxford multilocus sequence typing scheme for Acinetobacter baumannii: do sequence type 92 (ST92) and ST109 exist? J. Clin. Microbiol. 55, 2287–2289. doi: 10.1128/JCM.00533-17
Hamouda, A., Evans, B. A., Towner, K. J., and Amyes, S. G. (2010). Characterization of epidemiologically unrelated Acinetobacter baumannii isolates from four continents by use of multilocus sequence typing, pulsed-field gel electrophoresis, and sequence-based typing of bla (OXA-51-like) genes. J. Clin. Microbiol. 48, 2476–2483. doi: 10.1128/JCM.02431-09
Higgins, P. G., Prior, K., Harmsen, D., and Seifert, H. (2017). Development and evaluation of a core genome multilocus typing scheme for whole-genome sequence-based typing of Acinetobacter baumannii. PLoS One 12:e0179228. doi: 10.1371/journal.pone.0179228
Holt, K., Kenyon, J. J., Hamidian, M., Schultz, M. B., Pickard, D. J., Dougan, G., et al. (2016). Five decades of genome evolution in the globally distributed, extensively antibiotic-resistant Acinetobacter baumannii global clone 1. Microb. Genom. 2:e000052. doi: 10.1099/mgen.0.000052
Hyatt, D., Chen, G. L., Locascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jolley, K. A., Bliss, C. M., Bennett, J. S., Bratcher, H. B., Brehony, C., Colles, F. M., et al. (2012). Ribosomal multilocus sequence typing: universal characterization of bacteria from domain to strain. Microbiology 158(Pt 4), 1005–1015. doi: 10.1099/mic.0.055459-0
Jolley, K. A., Chan, M. S., and Maiden, M. C. (2004). mlstdbNet - distributed multi-locus sequence typing (MLST) databases. BMC Bioinformatics 5:86. doi: 10.1186/1471-2105-5-86
Jolley, K. A., and Maiden, M. C. J. (2010). BIGSdb: scalable analysis of bacterial genome variation at the population level. BMC Bioinformatics 11:595. doi: 10.1186/1471-2105-11-595
Kenyon, J. J., and Hall, R. M. (2013). Variation in the complex carbohydrate biosynthesis loci of Acinetobacter baumannii genomes. PLoS One 8:e62160. doi: 10.1371/journal.pone.0062160
Lewis, P. O. (2001). A likelihood approach to estimating phylogeny from discrete morphological character data. Syst. Biol. 50, 913–925. doi: 10.1080/106351501753462876
Maiden, M. C., Bygraves, J. A., Feil, E., Morelli, G., Russell, J. E., Urwin, R., et al. (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc. Natl. Acad. Sci. U.S.A. 95, 3140–3145. doi: 10.1073/pnas.95.6.3140
Maiden, M. C., Jansen van Rensburg, M. J., Bray, J. E., Earle, S. G., Ford, S. A., Jolley, K. A., et al. (2013). MLST revisited: the gene-by-gene approach to bacterial genomics. Nat. Rev. Microbiol. 11, 728–736. doi: 10.1038/nrmicro3093
Marí-Almirall, M., Cosgaya, C., Higgins, P. G., Van Assche, A., Telli, M., Huys, G., et al. (2017). MALDI-TOF/MS identification of species from the Acinetobacter baumannii (Ab) group revisited: inclusion of the novel A. seifertii and A. dijkshoorniae species. Clin. Microbiol. Infect. 23:210.e1-210.e9. doi: 10.1016/j.cmi.2016.11.020
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003. doi: 10.1093/ve/vev003
Ou, H. Y., Kuang, S. N., He, X., Molgora, B. M., Ewing, P. J., Deng, Z., et al. (2015). Complete genome sequence of hypervirulent and outbreak-associated Acinetobacter baumannii strain LAC-4: epidemiology, resistance genetic determinants and potential virulence factors. Sci. Rep. 5:8643. doi: 10.1038/srep08643
Paradis, E., Claude, J., and Strimmer, K. (2004). APE: analyses of phylogenetics and evolution in R language. Bioinformatics 20, 289–290. doi: 10.1093/bioinformatics/btg412
Pournaras, S., Dafopoulou, K., Del Franco, M., Zarkotou, O., Dimitroulia, E., Protonotariou, E., et al. (2017). Predominance of international clone 2 OXA-23-1 producing-Acinetobacter baumannii clinical isolates in Greece, 2015: results of a nationwide study. Int. J. Antimicrob. Agents 49, 749–753. doi: 10.1016/j.ijantimicag.2017.01.028
Pournaras, S., Gogou, V., Giannouli, M., Dimitroulia, E., Dafopoulou, K., Tsakris, A., et al. (2014). Single-locus-sequence-based typing of blaOXA-51-like genes for rapid assignment of Acinetobacter baumannii clinical isolates to international clonal lineages. J. Clin. Microbiol. 52, 1653–1657. doi: 10.1128/JCM.03565-13
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Puigbo, P., Bravo, I. G., and Garcia-Vallve, S. (2008). CAIcal: a combined set of tools to assess codon usage adaptation. Biol. Direct. 3:38. doi: 10.1186/1745-6150-3-38
Ribeiro-Gonçalves, B., Francisco, A. P., Vaz, C., Ramirez, M., and Carriço, J. A. (2016). PHYLOViZ online: web-based tool for visualization, phylogenetic inference, analysis and sharing of minimum spanning trees. Nucleic Acids Res. 44, W246–W251. doi: 10.1093/nar/gkw359
Sahl, J. W., Del Franco, M., Pournaras, S., Colman, R. E., Karah, N., Dijkshoorn, L., et al. (2015). Phylogenetic and genomic diversity in isolates from the globally distributed Acinetobacter baumannii ST25 lineage. Sci. Rep. 5:15188. doi: 10.1038/srep15188
Sahl, J. W., Gillece, J. D., Schupp, J. M., Waddell, V. G., Driebe, E. M., Engelthaler, D. M., et al. (2013). Evolution of a pathogen: a comparative genomics analysis identifies a genetic pathway to pathogenesis in Acinetobacter. PLoS One 8:e54287. doi: 10.1371/journal.pone.0054287
Schultz, M. B., Pham Thanh, D., Tran Do Hoan, N., Wick, R. R., Ingle, D. J., Hawkey, J., et al. (2016). Repeated local emergence of carbapenem-resistant Acinetobacter baumannii in a single hospital ward. Microb. Genom. 2:e000050. doi: 10.1099/mgen.0.000050
Snitkin, E. S., Zelazny, A. M., Montero, C. I., Stock, F., Mijares, L., Nisc Comparative Sequence Program, et al. (2011). Genome-wide recombination drives diversification of epidemic strains of Acinetobacter baumannii. Proc. Natl. Acad. Sci. U.S.A. 108, 13758–13763. doi: 10.1073/pnas.1104404108
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Tomaschek, F., Higgins, P. G., Stefanik, D., Wisplinghoff, H., and Seifert, H. (2016). Head-to-head comparison of two multi-locus sequence typing (MLST) schemes for characterization of Acinetobacter baumannii outbreak and sporadic isolates. PLoS One 11:e0153014. doi: 10.1371/journal.pone.0153014
van Dessel, H., Dijkshoorn, L., van der Reijden, T., Bakker, N., Paauw, A., van den Broek, P., et al. (2004). Identification of a new geographically widespread multiresistant Acinetobacter baumannii clone from European hospitals. Res. Microbiol. 155, 105–112. doi: 10.1016/j.resmic.2003.10.003
Villalon, P., Valdezate, S., Medina-Pascual, M. J., Rubio, V., Vindel, A., and Saez-Nieto, J. A. (2011). Clonal diversity of nosocomial epidemic Acinetobacter baumannii strains isolated in Spain. J. Clin. Microbiol. 49, 875–882. doi: 10.1128/JCM.01026-10
Wallace, L., Daugherty, S. C., Nagaraj, S., Johnson, J. K., Harris, A. D., and Rasko, D. A. (2016). Use of comparative genomics to characterize the diversity of Acinetobacter baumannii surveillance isolates in a health care institution. Antimicrob. Agents Chemother. 60, 5933–5941. doi: 10.1128/AAC.00477-16
Wisplinghoff, H., Hippler, C., Bartual, S. G., Haefs, C., Stefanik, D., Higgins, P. G., et al. (2008). Molecular epidemiology of clinical Acinetobacter baumannii and Acinetobacter genomic species 13TU isolates using a multilocus sequencing typing scheme. Clin. Microbiol. Infect. 14, 708–715. doi: 10.1111/j.1469-0691.2008.02010.x
Zarrilli, R., Pournaras, S., Giannouli, M., and Tsakris, A. (2013). Global evolution of multidrug-resistant Acinetobacter baumannii clonal lineages. Int. J. Antimicrob. Agents 41, 11–19. doi: 10.1016/j.ijantimicag.2012.09.008
Keywords: multilocus sequence typing, Acinetobacter baumannii, comparative genomics, phylogeny, sequence types, clonal complexes
Citation: Gaiarsa S, Batisti Biffignandi G, Esposito EP, Castelli M, Jolley KA, Brisse S, Sassera D and Zarrilli R (2019) Comparative Analysis of the Two Acinetobacter baumannii Multilocus Sequence Typing (MLST) Schemes. Front. Microbiol. 10:930. doi: 10.3389/fmicb.2019.00930
Received: 10 January 2019; Accepted: 12 April 2019;
Published: 03 May 2019.
Edited by:
Maria Soledad Ramirez, California State University, Fullerton, United StatesReviewed by:
Ruth M. Hall, University of Sydney, AustraliaSpyros Pournaras, National and Kapodistrian University of Athens Medical School, Greece
Copyright © 2019 Gaiarsa, Batisti Biffignandi, Esposito, Castelli, Jolley, Brisse, Sassera and Zarrilli. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Davide Sassera, ZGF2aWRlLnNhc3NlcmFAdW5pbnB2Lml0 Raffaele Zarrilli, cmFmemFycmlAdW5pbmEuaXQ=