Abstract
More than two-thirds of the powerful greenhouse gas nitrous oxide (N2O) emissions from soils can be attributed to microbial denitrification and nitrification processes. Bacterial denitrification reactions are catalyzed by the periplasmic (Nap) or membrane-bound (Nar) nitrate reductases, nitrite reductases (NirK/cd1Nir), nitric oxide reductases (cNor, qNor/ CuANor), and nitrous oxide reductase (Nos) encoded by nap/nar, nir, nor and nos genes, respectively. Rhizobium etli CFN42, the microsymbiont of common bean, is unable to respire nitrate under anoxic conditions and to perform a complete denitrification pathway. This bacterium lacks the nap, nar and nos genes but contains genes encoding NirK and cNor. In this work, we demonstrated that R. etli is able to grow with nitrate as the sole nitrogen source under aerobic and microoxic conditions. Genetic and functional characterization of a gene located in the R. etli chromosome and annotated as narB demonstrated that growth under aerobic or microoxic conditions with nitrate as nitrogen source as well as nitrate reductase activity requires NarB. In addition to be involved in nitrate assimilation, NarB is also required for NO and N2O production by NirK and cNor, respectively, in cells grown microoxically with nitrate as the only N source. Furthermore, β-glucuronidase activity from nirK::uidA and norC::uidA fusions, as well as NorC expression and Nir and Nor activities revealed that expression of nor genes under microoxic conditions also depends on nitrate reduction by NarB. Our results suggest that nitrite produced by NarB from assimilatory nitrate reduction is detoxified by NirK and cNor denitrifying enzymes that convert nitrite into NO which in turn is reduced to N2O, respectively.
Introduction
Nitrous oxide (N2O) is a powerful greenhouse gas (GHG) and a major cause of ozone layer depletion (Ravishankara et al., 2009) with an atmospheric lifetime of 114 years and an estimated 300-fold greater potential for global warming compared with that of carbon dioxide (CO2), based on its radiative capacity (Intergovernmental Panel on Climate Change [IPCC], 2014). Human activities such as agriculture, fossil fuel combustion, wastewater management and industrial processes have provoked escalating emissions of N2O which contribute to climate change. More than 60% of N2O emissions globally are emitted from agricultural soils (Smith et al., 2012). This contribution has been amplified through the so-called “green revolution,” which has increased the presence of nitrogen (N) in soil through the application of synthetic nitrogen-based fertilizers mainly fabricated through the Haber-Bosch process. Many processes and microorganisms are sources of N2O, being nitrifiers and denitrifiers the two most important groups of soil microorganisms involved (Pilegaard, 2013; Butterbach-Bahl et al., 2014). Denitrification consists in the respiratory reduction of the nitrate present in many terrestrial and aquatic ecosystems. This process is initiated by a periplasmic (Nap) or membrane-bound (Nar) nitrate reductase depending on the species. The nitrite produced from dissimilatory nitrate reduction is then transformed into nitric oxide (NO), a potent cytotoxic free radical and ozone-depleting gas, through the action of the respiratory Cu-containing (NirK) or the cd1-type nitrite reductase (NirS). Then, NO is reduced to N2O by cytochrome cb-type (cNor/CuANor) or quinol-dependent (qNor) nitric oxide reductases. Finally, a nitrous oxide reductase (Nos) catalyzes the last step of denitrification by producing N2 from N2O (for recent reviews see de Vries et al., 2007; Zumft and Kroneck, 2007; Richardson, 2011; van Spanning, 2011; Simon and Klotz, 2013; Al-Attar and de Vries, 2015; Torres et al., 2016).
Strategies to mitigate N2O emissions from agricultural soils have to be developed in order to decrease current levels of N2O production in particular in the context of the continuing population growth (Thomson et al., 2012). One proposed strategy is to promote a sustainable agriculture reducing the dependence on chemical fertilizers and increasing biological nitrogen fixation (BNF) through the application of nitrogen-fixing bacteria to legume crops. However, legumes also contribute to N2O emissions by providing N-rich residues into the soils or through the denitrification process that is performed by some rhizobia under free-living or symbiotic conditions (Inaba et al., 2009, 2012; Hirayama et al., 2011; Torres et al., 2016). In fact, many rhizobia species contain denitrification genes. Among them, Bradyrhizobium diazoefficiens is considered a model in the study of rhizobial denitrification given its capacity to grow with nitrate as respiratory substrate under anoxic conditions through denitrification, a process that has been extensively investigated in this bacterium (for reviews see Bedmar et al., 2005, 2013; Delgado et al., 2007; Sánchez et al., 2011; Torres et al., 2016). B. diazoefficiens denitrification reactions are catalyzed by Nap, NirK, cNor, and Nos enzymes encoded by napEDABC, nirK, norCBQD, and nosRZDYFLX genes, respectively. In addition to denitrify, B. diazoefficiens is also capable to grow under free-living conditions with nitrate as the sole N source. In this bacterium, the assimilatory nitrate reductase (NasC) constitutes an integrated biochemical system involved in nitrate assimilation and NO detoxification that has been demonstrated to be another source of NO and probably of N2O (Cabrera et al., 2016).
Rhizobium etli CFN42, the endosymbiont of common bean (Phaseolus vulgaris) contains a chromosome and six large plasmids named from pCFN42a to pCFN42f (González et al., 2006). In this bacterium, genes encoding the NirK and cNor denitrification enzymes have been identified on plasmid pCFN42f (Gómez-Hernández et al., 2011). However, genes encoding either a respiratory nitrate reductase (Nar or Nap) or the nitrous oxide reductase enzyme (Nos) are not present in R. etli CFN42 genome. Consequently, this rhizobium species is unable to respire nitrate and to perform complete denitrification pathway. Genetic and functional characterization of the reductases encoded by R. etli nirK and norC suggest a detoxifying role for these enzymes. In fact, phenotypic characterization of R. etli nirK and norC mutants demonstrated that NirK is required for nitrite reduction to NO and that cNor is required to detoxify NO (Bueno et al., 2005; Gómez-Hernández et al., 2011). Under symbiotic conditions, recent analyses of the levels of nitrosylleghemoglobin complexes (LbNO) of the nodules from common bean plants exposed to nitrate clearly demonstrated the capacity of the nodules to produce NO from nitrate present in the nutrient solution (Gómez-Hernández et al., 2011; Calvo-Begueria et al., 2018). However, the capacity of R. etli to produce NO or N2O from nitrate under free-living conditions has not been investigated so far. As mentioned before, R. etli lacks genes encoding the respiratory nitrate reductases (Nap or Nar). Sequence analysis revealed that an open reading frame in the R. etli chromosome (RHE_CHO1780) encodes a putative assimilatory nitrate reductase (NarB). RHE_CHO1780 resides within a cluster of other uncharacterized ORFs (RHE_CHO1781 and RHE_CHO1782) predicted to encode components (NirD and NirB) of an assimilatory nitrite reductase. This genomic context suggests a potential involvement of NarB in nitrate reduction to nitrite that would be further reduced to amonia by NirBD. However, the functional role of R. etli NarB has not been studied to date. Through the phenotypic characterization of a R. etli narB mutant, in this work we demonstrate the dual role of NarB in nitrate assimilation and in denitrification.
Table 1
| Strain or plasmid | Relevant characteristics | References |
|---|---|---|
| Bacteria | ||
| Rhizobium etli | ||
| CFN42 | Nalr (wild-type) | Quinto et al., 1982 |
| CE3 | Smr derivative of CFN42, NalrSmr (wild-type) | Noel et al., 1984 |
| CFNX702 | CE3 derivative, nirK::loxP, NalrSmr | Gómez-Hernández et al., 2011 |
| CFNX701 | CE3 derivative, norC::loxSp, NalrSmr Spr | Gómez-Hernández et al., 2011 |
| DR4000 | CE3 derivative, ΔnarB::ΩSpSm, NalrSmrSpr | This work |
| Escherichia coli | ||
| DH5α | supE44ΔlacU169 (φ80lacZΔM15) hsdR17recA1endA1gyrA96 thi-1relA1 | Sambrook et al., 1989 |
| S17.1 | thi, pro, recA,hsdR, hsdM, RP4Tc::Mu, Km::Tn7, TprSmrSpr | Simon et al., 1983 |
| Plasmids | ||
| pBluescript KS | Cloning vector, Apr | Invitrogen |
| pK18mobsacB | Suicide cloning vector, Kmr | Schäfer et al., 1994 |
| pBBR1MCS-2 | Broad host range cloning vector, Kmr | Kovach et al., 1995 |
| pHP45Ω | Vector carrying an ΩSpSm casette | Prentki and Krisch, 1984 |
| pRK415 | Broad host range plasmid, Tcr | Keen et al., 1988 |
| pNIC-01 | pBBMCS53 derivative norC::uidA, Gmr | Gómez-Hernández et al., 2011 |
| pNIC-03 | pBBMCS53 derivative nirK::uidA, Gmr | Gómez-Hernández et al., 2011 |
| pDR4000 | pK18mobsacB carrying narB with 2287 bp delection and narB::Ω insertion, SmrSprKmr | This work |
| pDR4002 | pBBR1MCS-2 derivative carrying R. etli narB, Kmr | This work |
| pLG4002 | pRK415 derivative carrying R. etli narB, Tcr | This work |
Bacterial strains and plasmids.
Materials and Methods
Bacterial Strains, Plasmids, and Growth Conditions
The bacterial strains and plasmids used in this work are listed in Table 1. Rhizobium etli strains were grown at 30°C in TY rich medium (Tryptone Yeast, Beringer, 1974) or in Y minimal medium (MMY) with succinate (10 mM) and ammonium chloride (10 mM) as carbon and nitrogen sources, respectively (Bravo and Mora, 1988). For growth under microoxic or anoxic conditions, flasks containing cell cultures were sealed with rubber septa, and flushed at the starting point of the incubation with 2% (v/v) O2 and 98% N2 (v/v) or 100% (v/v) N2, respectively. For growth with different nitrogen sources, cells were incubated in MMY with 10 mM ClNH4, KNO3 or NaNO2 as sole N source. Antibiotics were added to R. etli CE3, and narB, nirK, and norC cultures (see Table 1) at the following concentrations (μg ml−1): nalidixic acid (Nal) 20, kanamycin (Km) 30, spectinomycin (Sp) 100, streptomycin (Sm) 100. Escherichia coli DH5α used as receptor in cloning experiments and S17.1 used as donor in conjugation experiments were grown at 37°C in LB medium (Sambrook and Russell, 2001) and the antibiotics were added at the following concentrations (μg ml−1): spectinomycin 25, streptomycin 25, kanamycin 20, and ampicillin 200.
For determination of growth rates and enzymatic activities, cells were firstly grown aerobically in TY medium for 24 h, harvested by centrifugation at 8000 g for 10 min and washed twice with MMY containing ClNH4, KNO3 or NaNO2 as sole N source, depending on the treatment. Then, cells were incubated in the minimal medium for another 24 h under the desired oxygen conditions. Initial optical density at 600 nm of the cultures was around 0.05 for growth rates measurements or around 0.25 for enzymatic activity analyses.
For characterization of narB mutant growth, an additional step under starvation conditions was included before growing cells in the minimal medium. The starvation step consisted of a 24 h incubation of the cells in the minimal medium containing salts (CaCl2 and FeCl3), and lacking any nitrogen or carbon sources.
Construction and Complementation of R. etli narB Mutant
The oligonucleotide primers (Sigma) used in this work are listed in Supplementary Table S1. Genomic and plasmid DNA isolation were carried out using the REALPURE Genomic DNA purification Kit (Real) and Qiagen Plasmid Kit (Qiagen), respectively. PCR was performed using the High Fidelity DNA polymerase Phusion®enzyme (Thermo Fisher Scientific) and DNA digestions were carried out using Fast digest enzymes (Thermo Fisher Scientific).
To generate the narB mutant, the two regions flanking the narB gene (fragments F1 and F2), were amplified by PCR using narB_up_For/narB_up_Rev (in positions 1864715 to 1864732 and 1865293 to 1865310, respectively) and narB_down_For/narB_down_Rev (in positions 1867579 to 1867598 and 1868201 to 1868220, respectively) primer pairs. Then, fragments F1 and F2 were cloned into the pBlueScriptKS (pBSKS) vector (Invitrogen) as XbaI/BamHI and BamHI/EcoRI fragments, respectively, generating plasmid pBKS_F1F2. To construct a suicide plasmid useful for double recombination, the XbaI/EcoRI fragment from pBKS_F1F2 was cloned into the pK18mobsacB suicide vector (Schäfer et al., 1994) yielding plasmid pK18F1F2. This plasmid was further modified by inserting the ΩSp/Sm cassette (Prentki and Krisch, 1984) into the BamHI site of pK18F1F2 plasmid (between F1 and F2 fragments) obtaining plasmid pDR4000 that was analyzed by sequencing. Replacement of the R. etli narB wild type allele with the truncated mutant allele in plasmid pDR4000 was carried out by double recombination. With this purpose, plasmid pDR4000 was transferred via conjugation into R. etli CE3 using E. coli S17-1 as donor. Double recombination events were favored by growth on agar plates containing sucrose using the sacB marker present in plasmid pK18mobsacB. Double recombinants were selected as resistant to Sm and Sp and susceptible to Km. To verify that the gene replacement had occurred, the derivatives were analyzed by PCR using primers narB_EXT-For, narB_EXT-Rev, narB_IN-For, and narB_IN-Rev (Supplementary Table S1) and the narB mutant strain was named DR4000.
A plasmid carrying the R. etli narB gene constitutively expressed from the lacZ promoter was obtained by cloning the narB coding region in the broad-host range cloning vector pBBR1MCS-2 (Kovach et al., 1995). To that end, the narB gene was amplified by PCR using narB_compl_For/narB_compl_Rev primers (Supplementary Table S1) and cloned as an XbaI/HindIII fragment into pBBR1MCS-2. The plasmid obtained (pDR4002) was sequenced and transferred to R. etli CE3 (WT) and DR4000 strains by conjugation using E. coli S17-1 as donor. The strain derivatives containing pDR4002 (WT/pDR4002 and DR4000/pDR4002, respectively) were checked by plasmid isolation and PCR. Concurrently, a WT strain containing pBBR1MCS-2 empty vector was obtained (WT/pBBR1MCS-2), as a control.
An additional plasmid carrying the narB gene constitutively expressed from the lacZ promoter was constructed by cloning the XbaI/HindIII fragment containing narB from pDR4002 into the pRK415 vector (Keen et al., 1988). The plasmid obtained (pLG4002) was introduced by conjugation into R. etli DR4000 strain harboring plasmids pNIC-03 and pNIC-01 that contain a nirK::uidA or norC::uidA transcriptional fusions, respectively. In addition, the pRK415 empty vector was introduced into WT- pNIC-03, WT- pNIC-01, DR4002- pNIC-03, and DR4002- pNIC-01 derivatives.
Extracelullar NO2− Determination
To measure the concentration of NO2− in the medium during growth with NO3− under aerobic or microoxic conditions, aliquots were taken from cultures at different time points. Culture samples were centrifuged at 8000 g for 10 min and nitrite concentration was estimated in the supernatant after diazotization by adding the sulphanilamide-naphthylethylenediamine dihydrochloride reagent (Nicholas and Nason, 1957).
Cell Extract Preparation and Determination of Nitrate and Nitrite Reductase Activities
To analyze nitrate reductase (NR) activity, cells at an initial OD600 of about 0.25 were incubated aerobically with KNO3 as the sole nitrogen source for 24 h. After centrifugation at 8000 g for 10 min, cells were harvested and disrupted by using a French pressure cell (SLM Aminco, Jessup, MD, United States). Then, fractionated cells were centrifuged at 10000 g for 10 min and the supernatant containing the soluble cell extract was used for NR activity. To analyze nitrite reductase (Nir) activity, cells were grown microoxically with KNO3 as the sole nitrogen source for 24 h. Then, cells were harvested by centrifugation, washed twice with 50 mM Tris–HCl pH 7.5 and resuspended in 1 ml of the same buffer.
Methyl-viologen dependent nitrate reductase (MV-NR) and nitrite reductase (MV-Nir) activities were determined by using 105 μl of the soluble cell extract (∼0.5 mg protein) or cell suspension (∼0.1 mg protein), respectively. The reaction mixture also contained 0.2 mM Methyl Viologen and 10 mM KNO3 for NR activity or 0.01 mM NaNO2 for Nir activity. The reaction was started by the addition of 15 μl of freshly prepared 144 mM sodium dithionite solution in 300 mM NaHCO3. After incubation for 20 min at 30°C, the reaction was stopped by vigorous shaking until the samples had lost their blue color. Nitrite produced by NR or consumed by Nir enzymes was estimated after diazotization as described previously for extracellular NO2− determination.
NO Production and Consumption Activities
In order to investigate the capacity of the different mutants to produce or consume NO, cell cultures at an initial OD600 of about 0.25 were incubated microoxically with KNO3 as the sole nitrogen source for 24 h, harvested by centrifugation, washed twice with 25 mM Na2HPO4/NaH2PO4 buffer (pH 7.4), and resuspended in 1.5 ml of the same buffer. NO production and consumption activities were determined by using an ISONOP NO electrode APOLLO 4000®(World Precision Instruments). The reaction chamber (2 ml) was temperature-controlled, magnetically stirred and contained: 1410 μl of 25 mM Na2HPO4/NaH2PO4 buffer (pH 7.4) and 250 μl of cell suspension (0.4–0.7 mg protein) for NO production or 760 μl of 25 mM Na2HPO4/NaH2PO4 buffer (pH 7.4) and 900 μl of cell suspension (1.5–2.5 mg protein) for NO consumption. To generate an anoxic atmosphere, 100 μl of an enzymatic mix containing Aspergillus niger glucose oxidase (40 units⋅ml−1), bovine liver catalase (250 units⋅ml−1) (Sigma-Aldrich), 90 μl of 1 M sodium succinate and 100 μl of 320 mM glucose were added to the chamber. Once a steady base line was obtained, 50 μl of 50 mM NaNO2 (NO production) or 50 μl of 2 mM NO (NO consumption) was added to the chamber to start the reaction.
N2O Production
To measure N2O accumulation, R. etli CE3 and the mutant strains were cultured as indicated above for NO experiments. After 24 h growth, 1 ml was taken from the headspace of cultures, using a Hamilton®Gastight syringe, and manually injected into an HP 4890D gas chromatography instrument equipped with an electron capture detector (ECD) as described by Torres et al. (2014).
Haem-Staining Analysis
To study the expression of the NorC component of cNor, we performed haem c-staining analyses of proteins from membranes of R. etli CE3 and the mutant strains cultured as indicated above for NO and N2O experiments. After 24 h growth, cells were harvested by centrifugation, washed twice with 50 mM Na2HPO4/NaH2PO4 (pH 6.8) buffer containing 1 mM MgCl2, 0.9% NaCl and 0.1 mM CaCl2, and resuspended in 2.5 ml of the same buffer containing 0.1 mM 4-(2-aminoethyl) benzene-sulfonyl fluoride hydrochloride (ABSF), RNase (20 μg ml−1), and DNase I (20 μg ml−1). Cells were disrupted using a French pressure cell (SLM Aminco, Jessup, MD, United States) and the membrane fraction was prepared as described previously (Torres et al., 2013). Then, membrane proteins were separated by SDS-PAGE, transferred to a nitrocellulose membrane and stained for haem-dependent peroxidase activity as described previously (Vargas et al., 1993) using the chemiluminescence detection kit “SuperSignal” (Thermo Fisher Scientific, Pierce, IL, United States). Protein concentration was estimated using the Bio-Rad assay (Bio-Rad Laboratories).
Measurement of β-Glucuronidase Activity
To analyze the expression of nirK and nor genes, R. etli CE3 and the mutant cells containing a nirK::uidA or a norC::uidA transcriptional fusions were incubated microoxically (2% initial O2 concentration) for 14 h in MMY medium containing ClNH4 or KNO3 as sole N source, with exception of the narB mutant that was grown for 21 h. Quantitative GUS activity was determined on 1.0-ml culture samples using 4-nitrophenyl β-D-glucuronide as substrate as described previously (Girard et al., 2000). Data were normalized to total cell protein concentration by the Lowry method over a second set of 1.0-ml samples.
Results
R. etli narB Encodes the Assimilatory Nitrate Reductase
Figure 1 shows that R. etli (WT) is able to grow with NO3− as the sole nitrogen source under oxic or microoxic (2% initial O2 concentration) conditions reaching values of optical densities (OD) at 600 nm of around 0.6 or 0.4, respectively. However, this bacterium was unable to use NO3− for respiration being incapable to grow under anoxic conditions with nitrate as the sole N source (Figure 1). These results suggest that R. etli uses nitrate through the assimilatory pathway under oxic or microoxic conditions where oxygen was used for respiration. However, it is unable to respire nitrate when oxygen is absent. Similar growth rates were reached when the wild-type (WT) cells were grown in minimal medium amended with 10 mM of ClNH4 as the sole N source (Figure 1).
FIGURE 1
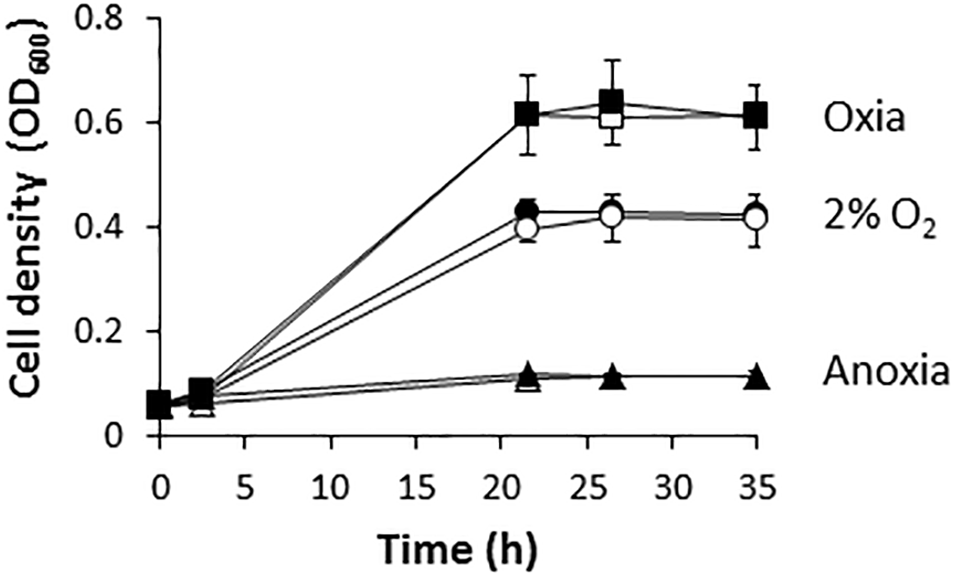
Nitrate-dependent growth of R. etli. Cells were cultured under oxic (squares), microoxic (circles) or anoxic (triangles) conditions, with either NH4+ (white) or NO3− (black), as sole N source. Results are the mean values and error bars from at least two different cultures assayed in triplicate.
In order to determine the implication of RHE_CHO1780 encoding for a putative assimilatory nitrate reductase (NarB) in nitrate assimilation, a R. etli mutant strain lacking the narB gene was constructed. As shown in Figure 2A, aerobic growth of R. etli narB mutant was highly decreased compared to that reached by the WT strain (0.1 and 0.4 OD600, respectively, after 35 h culture). However, when the narB mutant was complemented with plasmid pDR4002 that constitutively expresses narB (narB + NarB+), similar growth rates as those from WT cells were observed (Figure 2A). No significant differences of growth rates were found when the WT strain was complemented with plasmid pDR4002 (WT + NarB+) or with pBBR1MCS-2 (WT + vector) (Figure 2A). Moreover, NO2− was not detected in the culture medium of narB mutant grown oxicallly (Figure 2B). However, WT cells accumulated around 100 μM NO2− in the medium after 35 h growth (Figure 2B). Interestingly, both narB and WT strains containing pDR4002 (narB + NarB+ or WT + NarB+) accumulated about 180 μM NO2− in the medium. These results suggest that R. etli NarB is the enzyme responsible for nitrate reduction to nitrite, the first step of nitrate assimilation. To corroborate this observation, we also measured MV-NR activity of R. etli cells grown under oxic conditions with NO3− as sole N source. As shown in Figure 2C, MV-NR activity was around 10-fold lower in the narB mutant compared to that observed in the WT strain. The constitutive expression of narB in the narB mutant (narB + NarB+), restored NR activity to levels significantly higher (about 15-fold) to those observed in WT cells. A similar increase of NR activity (about 13-fold) was observed in WT cells containing pDR4002 compared to NR levels of WT strain with or without the empty vector pBBR1MCS-2 (Figure 2C). The higher effect of the presence of pDR4002 on nitrite accumulation (Figure 2B) and MV-NR activity (Figure 2C) is due to the over-expression of narB gene by the constitutive lacZ promoter present in pDR4002. Taken together, these results confirm the participation of NarB in nitrate reduction to nitrite when cells are cultured under aerobic conditions with nitrate as the only N source.
FIGURE 2
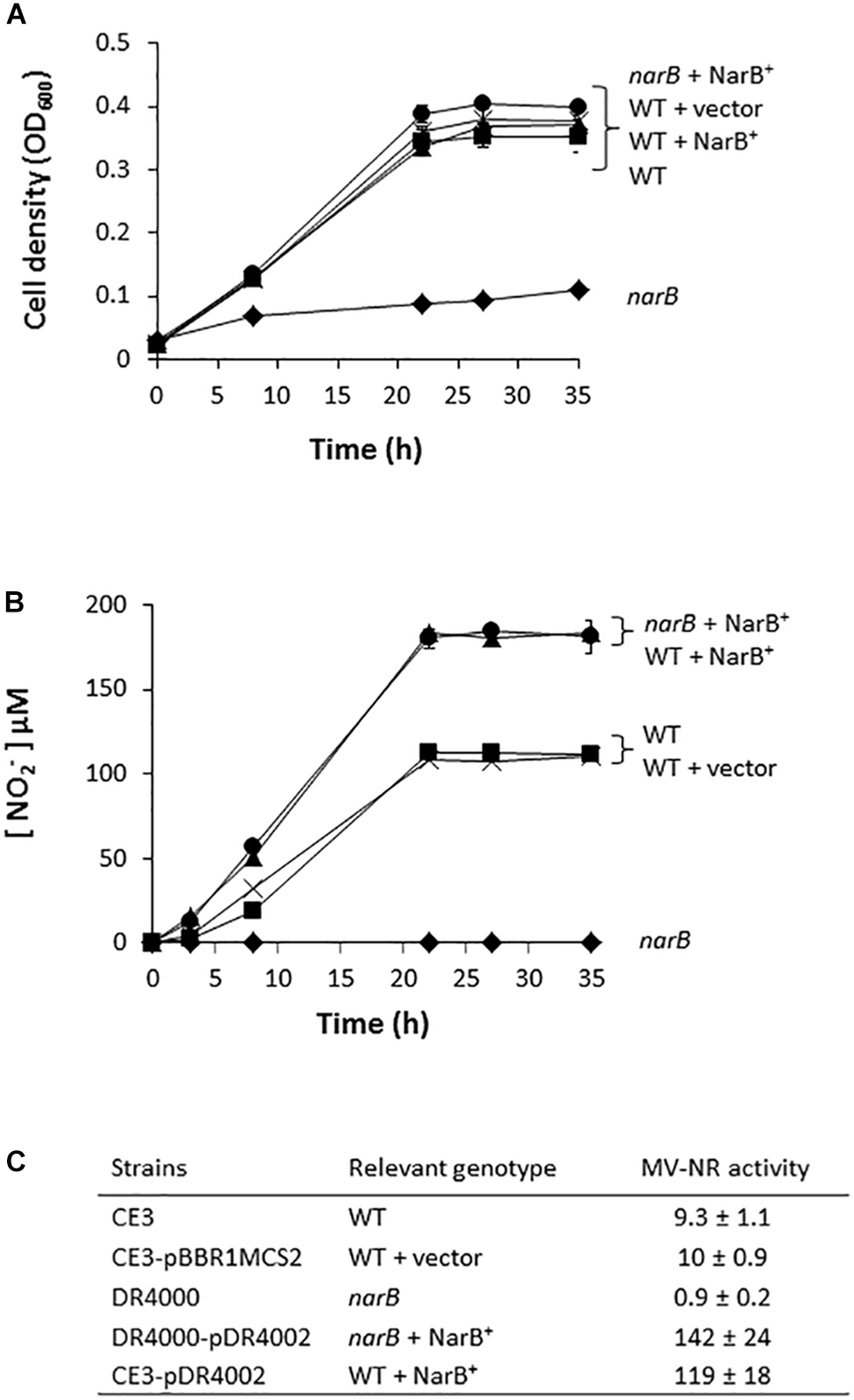
Nitrate-dependent aerobic growth and NR activity of a R. etli narB mutant. (A) Growth curves. (B) Extracellular nitrite concentration in the growth medium. (C) MV-NR activity expressed as nmol NO2− produced mg protein−1 h−1. R. etli WT ( ), WT + vector (X), narB (
), WT + vector (X), narB ( ), narB + NarB+ (
), narB + NarB+ ( ), and WT + NarB+ (
), and WT + NarB+ ( ) strains were incubated aerobically in minimal medium with NO3− as sole N-source. Data are expressed as the mean value ± SD from two different cultures assayed in triplicate.
) strains were incubated aerobically in minimal medium with NO3− as sole N-source. Data are expressed as the mean value ± SD from two different cultures assayed in triplicate.
R. etli narB, nirK, and norC Are Required for Denitrification
To investigate the implication of R. etli narB, nirK and norC genes in the denitrification process, we performed growth rate experiments of the R. etli narB, nirK and norC mutant cells cultured under microoxic conditions and in the presence of nitrate as sole nitrogen source. While narB was obtained in this work, the nirK or norC mutants were previously constructed by Gómez-Hernández et al. (2011). As observed when cells were incubated under oxic conditions (Figure 1A), the narB mutant showed a clear defect in its ability to grow when compare to the WT cells (Figure 3A). Complementation of narB mutant with plasmid pDR4002 expressing constitutively narB (narB+ NarB+) restored the WT ability to grow under microoxic conditions (Figure 3A). The norC mutant was completely unable to grow microoxically with nitrate as unique nitrogen source. However, no differences in nitrate-dependent growth rates were detected between the nirK mutant and WT strain (Figure 3A). The capacity of the nirK mutant to grow with nitrate might be due to its ability to assimilate nitrate and nitrite through the activity of the NarB and NirBD assimilatory nitrate and nitrite reductase enzymes. As shown in Figure 3B, WT cells incubated microoxically with NO3− accumulated low levels of NO2− in the medium after 8 h incubation (5 μM NO2−) that was consumed after 45 h growth. Growth of the R. etli nirK mutant under the same growth conditions resulted in higher levels of NO2− concentration in the medium compared to those observed in WT cultures (20 μM versus 0 μM NO2− after 45 h incubation) (Figure 3B). Furthermore, levels of nitrite detected in the culture medium of narB or norC mutants cultivated under the same conditions were undetectable (Figure 3B). Interestingly, narB mutant containing plasmid pDR4002 (narB + NarB+) accumulated about 11-times more NO2− in the medium compared to WT cells (56 μM versus 5 μM NO2− after 8 h incubation) (Figure 3B).
FIGURE 3
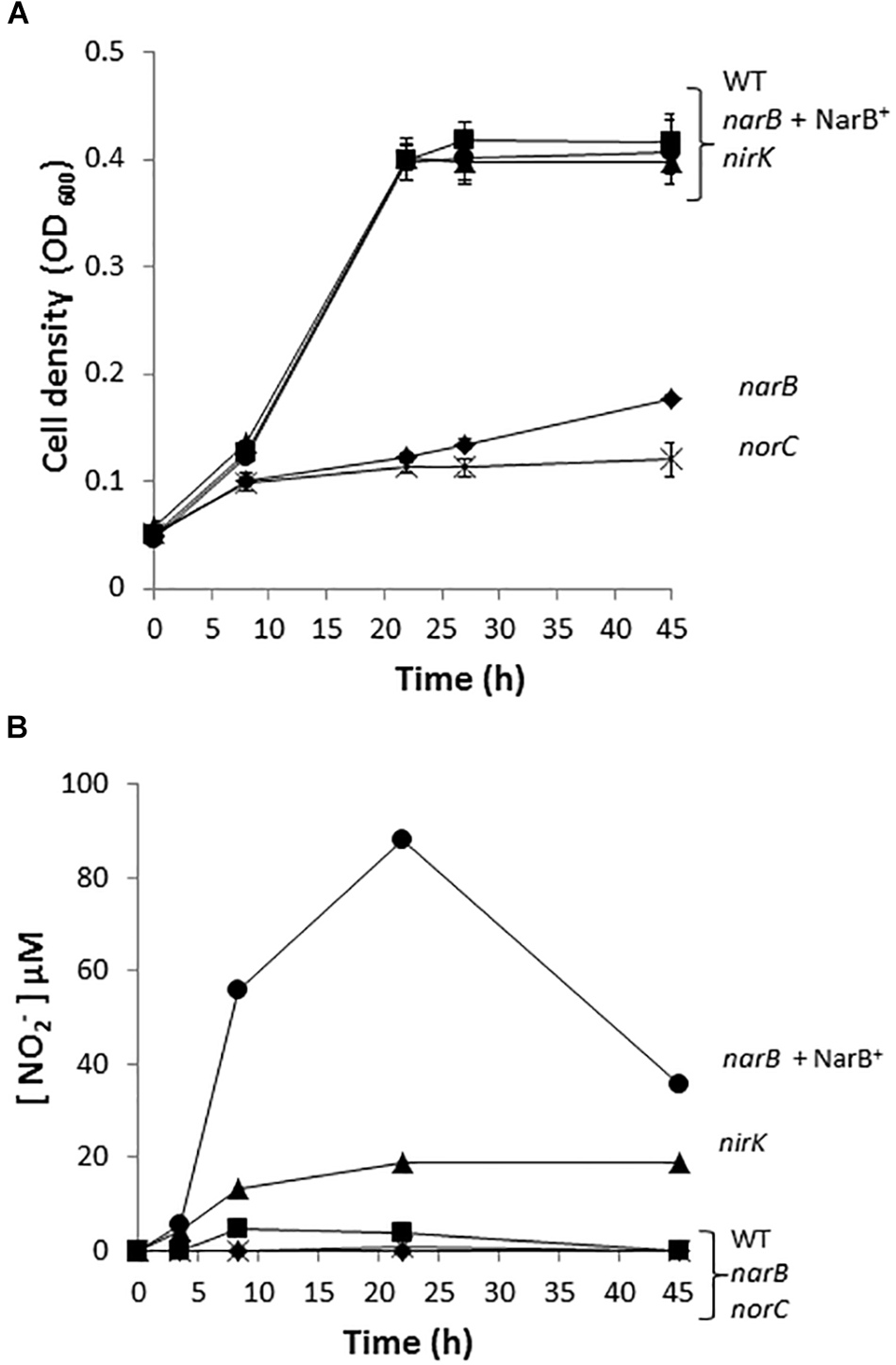
Nitrate dependent micro-oxic growth of R. etli narB, nirK, and norC mutants. (A) Growth curves. (B) Extracellular nitrite concentration in the growth medium. R. etli WT (), narB (), narB+ NarB+ (), norC (X), and nirK () strains were cultured microoxically in minimal medium with NO3− as sole N-source. Data are expressed as the mean value and error bars from two different cultures assayed in triplicate.
In order to investigate the denitrification capacity of the narB, nirK and norC mutants, we determined MV-Nir, NO consumption activity (Nor), NO production capacity and N2O accumulation (Figure 4 and Table 2). As shown in Table 2, MV-Nir activity was about 5-times lower in the nirK mutant compared to that observed in WT, narB or narB containing pDR4002 (narB + NarB+) strains. The residual Nir activity observed in the nirK mutant could be due to the activity of an assimilatory Nir, enzyme that is also encoded in the R. etli CFN42 genome. Concerning Nor activity, it was significantly lower (around ninefold) in cells of the norC mutant compared to the values reached by WT cells (Table 2). These observations indicate that NirK and NorC are the main enzymes responsible of MV-Nir and Nor activities, respectively. Furthermore, a significant reduction (around twofold) of Nor activity was observed in the narB mutant. On the contrary, the constitutive expression of narB in the narB mutant background (narB + NarB+) resulted in an increase of about 3.4-fold of Nor activity compared to that observed in the narB mutant (Table 2). Additionally, we measured NO production capacity of nirK, norC, and narB mutants cultured microoxically with nitrate that were transferred to a reaction chamber with NO2− as substrate (Figure 4A). The norC mutant accumulated about 10-times more NO than the WT being the toxicity of NO the reason that might explain the inability of this mutant to grow under microoxic conditions (Figure 3A, 4A). On the contrary, the nirK mutant did not produce NO. NO accumulation capacity by the narB mutant was 4-times higher than that observed in the WT strain (Figure 4A) probably due to the twofold reduction of NO consumption activity observed in the narB mutant (Table 2) compared to WT cells. Accordingly, NO produced by the narB mutant containing pDR4002 (narB + NarB+) decreased to WT levels (Figure 4A). Interestingly, N2O production was observed in the headspace of WT cultures grown under microoxic conditions with nitrate as the only N source (Figure 4B). By contrary, narB, nirK and norC mutants appeared to be unable to produce N2O (Figure 4B). However, the narB mutant complemented with pDR4002 (narB + NarB+) showed a significant accumulation (about sixfold) of N2O compared to those levels produced by the WT strain in the presence of nitrate in the growth medium. These results clearly demonstrate the involvement of nitrate reduction by NarB on R. etli N2O emission in cells grown with nitrate. However, when cells were grown in the presence of nitrite as the sole nitrogen source, the narB mutant as well as the narB + NarB+ strain reached similar values of N2O accumulation to the WT (Figure 4B). As observed in nitrate-cultured cells, nirK and norC mutants were defective in their capacity to produce N2O in cells with NO2− as N source (Figure 4B). These results indicate that NirK and NorC but not NarB are required for N2O production by R. etli cells cultured with nitrite as N source.
FIGURE 4
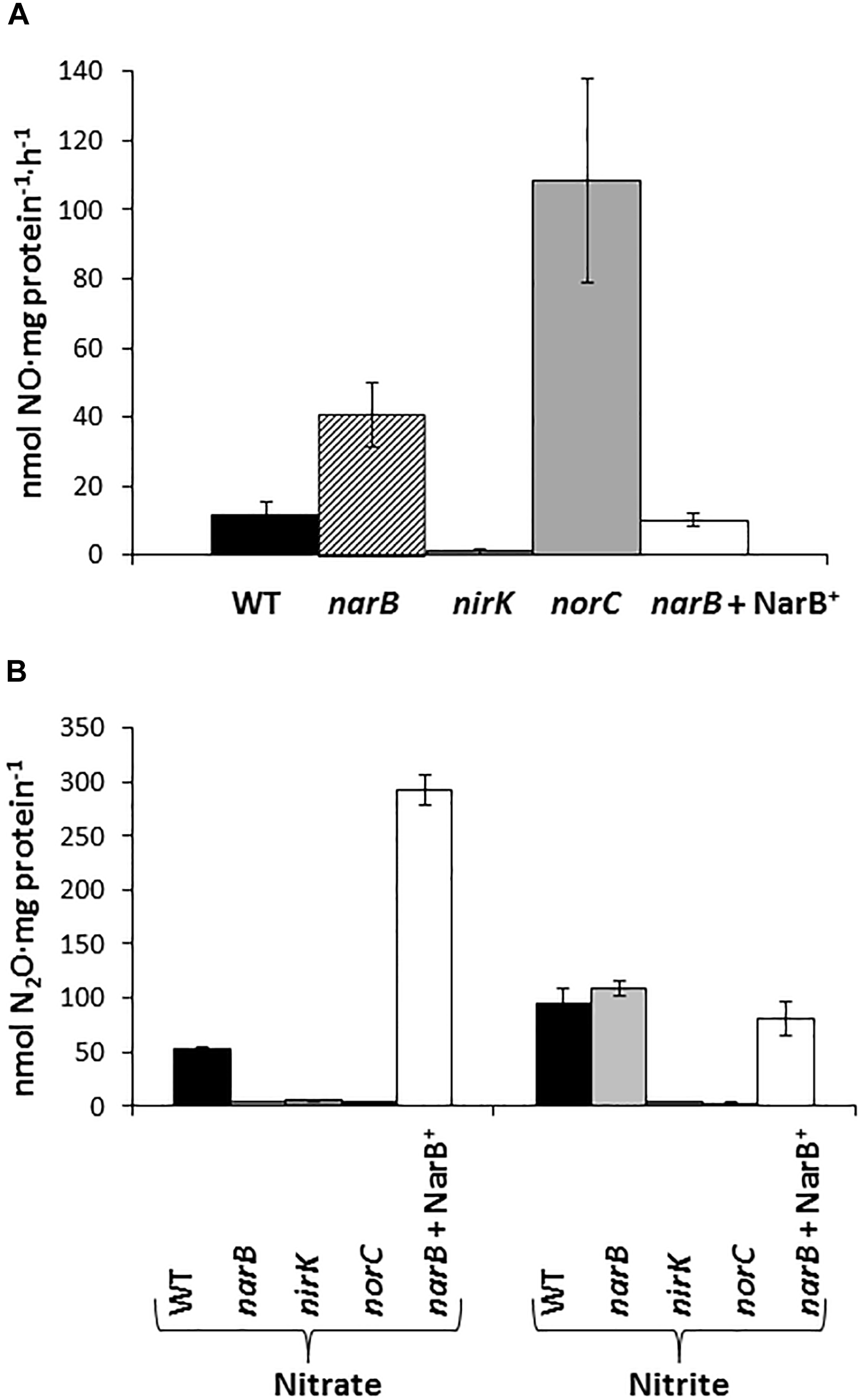
Nitric oxide production capacity (A) and N2O accumulation (B) by R. etli narB, narB + NarB+, nirK, and norC mutants. In (A) cells incubated microoxically with NO3− were transferred to a reaction chamber with NO2−. In (B) cells were cultured microoxically with NO3− or NO2− as the sole N-source. Data are expressed as the mean value and error bars from two different cultures assayed in triplicate.
Table 2
| Activities | |||
|---|---|---|---|
| Strains | Genotype | MV-Nir | Nor |
| CE3 | WT | 195 ± 2.3 | 269 ± 58 |
| DR4000 | narB | 182 ± 1.6 | 155 ± 43 |
| CFNX702 | nirK | 40 ± 2.3 | nd |
| CFNX701 | norC | nd | 31 ± 5.4 |
| DR4000-pDR4002 | narB + NarB+ | 190 ± 4.5 | 532 ± 72 |
MV-Nir and Nor activities of R. etli narB, nirK, norC, or narB complemented with pDR4002 (NarB+).
Cells were cultured microoxically with NO3− as the sole N-source. MV-NiR and Nor activities are expressed as nmol of NO2− or NO consumed⋅h−1⋅mg protein−1, respectively. Data are expressed as the mean value ± SD from at least two different cultures assayed in triplicate; nd, not determined.
Table 3
| β-glucuronidase specific activity1 | |||
|---|---|---|---|
| Strains | Genotype | NH4+ | NO3− |
| CE3-pRK415-pNIC-03 | WT (nirK::uidA) | 1115 ± 72 | 2087 ± 106 |
| CE3-pRK415-pNIC-01 | WT (norC::uidA) | 352 ± 51 | 1451 ± 185 |
| DR4000-pRK415-pNIC-03 | narB (nirK::uidA) | 912 ± 17 | 1617 ± 131 |
| DR4000-pRK415-pNIC-01 | narB (norC::uidA) | 245 ± 6 | 223 ± 8 |
| DR4000-pLG4002-pNIC-03 | narB+NarB+ (nirK::uidA) | 1236 ± 75 | 2356 ± 122 |
| DR4000-pLG4002-pNIC-01 | narB+NarB+ (norC::uidA) | 453 ± 58 | 1493 ± 108 |
| CFNX702- pNIC-01 | nirK (norC::uidA) | 91 ± 33 | 119 ± 23 |
β-glucuronidase specific activity of nirK::uidA and norC::uidA transcriptional fusions in R. etli WT, narB, or narB complemented with pLG4002 (NarB+).
Cells were cultured microoxically with NH4+ or NO3− as the sole N source. 1Values are expressed as nmol min−1mg protein−1. Data are the mean of three replicates from two independent experiments ± SD.
Involvement of Nitrate Reduction by NarB in R. etli nirK and nor Expression
Results from Table 2 suggest the involvement of NarB in Nor activity, but not in Nir activity. Our next goal was to evaluate the participation of NarB in the expression of nirK and nor genes. To achieve this goal, a nirK::uidA and a norC::uidA transcriptional fusions present in plasmids pNIC-03 or pNIC-01, respectively (Gómez-Hernández et al., 2011) were used in this work. R. etli WT cells grown with NO3− showed a slight increase of about twofold of nirK::uidA expression compared to that from NH4+-grown cells (Table 3). However, about fourfold increase of norC::uidA was observed in NO3−-grown cells compared to those grown with NH4+ as N source (Table 3). Interestingly, a differential dependence on NarB for expression of nirK and norC genes was observed when NO3− was present in the culture medium. While nirK showed only a marginal dependence of NarB for expression in this condition (2087 ± 106 in the WT vs. 1617 ± 131 in the narB mutant), the fourfold induction of norC expression by nitrate in the WT was not observed in the narB mutant (Table 3). As shown in Table 3, the expression of norC in the narB mutant strain was restored when it contained plasmid pLG4002 with a constitutive expression of narB (223 ± 8 to 1493 ± 108 activity values). By contrast, the presence of pLG4002 in the narB mutant slightly increased nirK::uidA expression in NO3−-grown cells (1617 ± 131 to 2356 ± 122 activity units). These results clearly show that the induction of the microoxic expression of norC by nitrate is dependent on NarB. We also observed that the induction of the norC::uidA expression in response to nitrate did not occur in a nirK mutant background (Table 3). These results suggest that NO produced from NO3− reduction to NO2− by NarB and from NO2− reduction by NirK is the nitrogen oxide (NOx) required for nor expression.
To confirm the participation of NarB in the induction of nor genes, we examined the expression of NorC by performing haem c staining analyses in proteins from membranes of a narB mutant cultured microoxically with nitrate as N source. To identify NorC protein, we also included in these experiments the R. etli norC mutant. As shown in Figure 5, a 32- and 27-kDa c-type cytochromes, identified previously as the FixP and FixO subunits of the terminal oxygen high-affinity cbb3-type cytochrome oxidase (Soberon et al., 1999) were observed in all strains. A third band of about 17 kDa which was observed in WT cells grown with NO3− could not be detected in membranes from the norC mutant cultured under the same conditions (Figure 5, lanes 2 and 3). These results allowed us to identify by the first time in R. etli the NorC component of cNor. By contrary to WT nitrate-dependent cells, growth with NH4+ as N source did not allowed expression of NorC suggesting the requirement of nitrate in the medium to induce NorC in R. etli (Figure 5, lanes 1 and 2). Interestingly, NorC could not be detected in membranes from the narB mutant grown with nitrate indicating that nitrate reduction by NarB, is required to induce NorC expression in R. etli (Figure 5, lanes 2 and 4). In fact, constitutive expression of NarB in the narB mutant (narB + NarB+) restored the expression of NorC in R. etli narB mutant cultured microoxically with nitrate (Figure 5, lanes 2, 4, and 5). These results clearly demonstrate the role of nitrate reduction by NarB in NorC expression in R. etli. In this bacterium, the reduction products of nitrate under microoxic conditions are NO2− or NO. In order to identify the NOx (NO2− or NO) required for expression of NorC, we included in the haem-staining experiments the nirK mutant which does not produce NO from nitrate (Figure 4A). As shown in Figure 5 (lane 6), NorC was not detected in membranes from the nirK mutant in response to nitrate, suggesting that NO could be the signal molecule implicated in NorC expression.
FIGURE 5
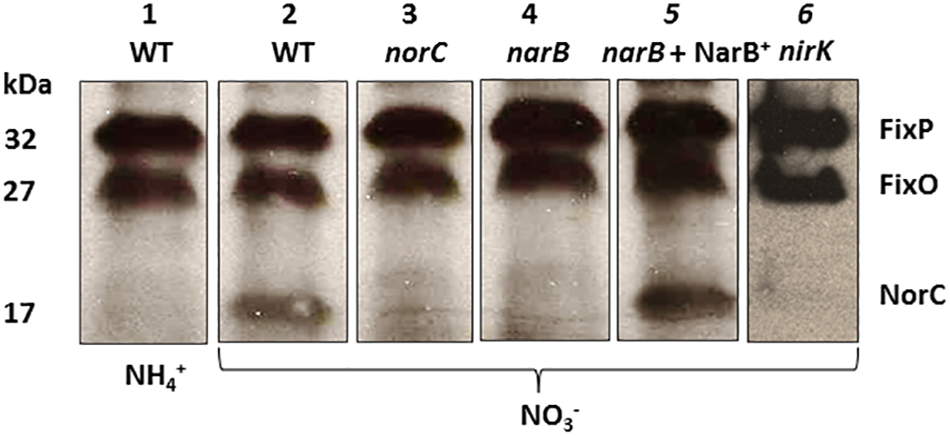
Expression of NorC in R. etli narB mutant. Haem-stained proteins of membranes prepared from R. etli WT (lanes 1 and 2), norC (lane 3), narB (lane 4), narB + NarB+ (lane 5), and nirK (lane 6) mutants cultured microoxically with NH4+ (lane 1) or NO3- (lanes 2, 3, 4, 5, and 6) as the sole N-source. Each lane contains 25 μg of membrane proteins. Haem-stained c-type cytochromes indentified previously (FixP and FixO) and in this work (NorC) are indicated. Apparent protein molecular masses (kDa) are also shown.
Discussion
R. etli NarB Is Required for Nitrate Assimilation and Denitrification
Rhizobium etli is a N2-fixing soil bacterium able to establish endosymbiotic associations with common bean plants. Up to now, the capacity of this bacterium to perform other processes of the N-cycle was unknown. In this work, we have demonstrated for the first time the ability of R. etli to assimilate nitrate as well as to produce NO and N2O from NO3− through denitrification. However, R. etli is unable to grow under anoxic conditions using NO3− as the final electron acceptor since it lacks the respiratory Nap or Nar nitrate reductases. The denitrification enzymes NirK and cNor are not proton pump or electrogenic. In this case, is the electron transfer from UQ pool to bc1 complex that can be used to drive the translocation of protons across the mitochondrial membrane to generate a trans-membrane proton electrochemical gradient or proton motive force (Δp) that can drive the synthesis of ATP. However, in incomplete denitrifiers like R. etli CFN42 that lack Nar or Nap, ATP synthesis from electron transfer to NirK and cNor is limited and it does not allow cells to grow from nitrate respiration anoxically. An inspection of the R. etli CFN42 genome1, allowed us to identify a gene (RHE_CHO1780) annotated as narB, which encodes a putative NarB. Two classes of assimilatory nitrate reductases (Nas) have been described from microorganisms: the NADH-dependent Nas and the ferrodoxin- or flavodoxin-dependent Nas (Fd-Nas, Moreno-Vivian et al., 1999). NADH-Nas proteins are heterodimers consisting of a 45 kDa FAD-containing diaphoarase and the 95 kDa catalytic subunit with a molybdenum bis-molybdopterin dinucleotide (Mo-bis-MGD) cofactor and a N-terminal [4Fe-4S] center (Richardson et al., 2001). Fd-Nas usually present in cyanobacteria are monomers with a molecular weight between 75 and 85 kDa (Moreno-Vivián and Flores, 2007). The in silico analysis of the R. etli NarB sequence (web.expasy.org) found that it has 885 aa, a predicted molecular weight of approximately 94.5 kDa, as well as the typical Mo-bis-MGD binding domain and the consensus motifs for co-ordination of an N-terminal [4Fe-4S] cluster present in NADH-Nas.
In nitrate assimilation, NO3− is incorporated into the cells by its sequential reduction to NO2− and NH4+ by the assimilatory nitrate and nitrite reductases, respectively. The presence of NarB in R. etli led us to hypothesize that this bacterium could use this enzyme to assimilate nitrate together with the two additional ORFs located in the same gene cluster that are predicted to encode the NirB and NirD components of an assimilatory nitrite reductase. The NH4+ produced is further incorporated into carbon skeletons (reviewed by Luque-Almagro et al., 2011). In fact, a R. etli narB mutant was unable to grow aerobically with nitrate as the only N source and was impaired in nitrate reductase activity.
Rhizobium etli CFN42 only possesses in its genome the nirK and nor denitrification genes, encoding NirK and cNor, respectively. Previous results have demonstrated that NirK is required for nitrite reduction to NO and that cNor is needed to detoxify NO (Bueno et al., 2005; Gómez-Hernández et al., 2011). However, up to now, no evidence has been reported about the putative link between NarB and nitrite and NO detoxification in nitrate-dependent microoxically grown cells. In this work, we have demonstrated that NarB is required for N2O formation in cells grown microoxically with nitrate as N source given the inability of the R. etli narB mutant to produce N2O from nitrate. These observations led us to suggest that NO2− produced by NO3− reduction in the cytoplasm through NarB activity might be exported outside the cell where is detoxified by NirK and cNor to produce NO and N2O, respectively, in the periplasmic space. In the same genomic region where NarB is located, there is an ORF (RHE_CHO1783) that is predicted to encode a major facilitator family NO3−/NO2− transporter (NarK) similar to that found in B. diazoefficiens that has been reported to be involved in NO2− extrussion (Cabrera et al., 2016). The implication of this NarK-like protein in transporting NO2− from the cytoplasm to the periplasm is under investigation. The involvement of NarB in nitrate assimilation and denitrification was also demonstrated by complementing the narB mutant with the constitutively expressed narB gene allowing the restoration of the ability to assimilate nitrate and to produce N2O in the narB mutant. Interestingly, the constitutive expression of narB in the narB mutant resulted in a remarkable increase of NR activity, NO2− accumulation in the medium and Nor activity that resulted in higher levels of N2O respect to WT cells under microoxic conditions. However, this increase in nitrate reduction and N2O formation did not result in higher growth rates probably due to the fact that NarB, NirK, and cNor enzymes do not allow the cells to obtain energy through the assimilative NO3− reduction by NarB coupled to denitrification by NirK and cNor. In fact, constitutive expression of NarB in R. etli WT strain did not allow the cells to grow under anoxic conditions with nitrate (data not shown). On the contrary, it has been recently reported that constitutive expression of Nap allowed Ensifer meliloti to increase the production of NO2−, NO, and N2O as well as its capacity to grow anoxically using nitrate as respiratory substrate (Torres et al., 2018). In spite of R. etli narB, nirK, and norC genes do not allow cell growth through denitrification, when cells are cultured microoxically through nitrate assimilation, nirK, and norC have a detoxifying role preventing the accumulation of the cytotoxic molecules nitrite and NO and contributing to the production of the GHG N2O having an environmental impact to climate change. In this context, the role of R. etli NirK and cNor on nitrite and NO detoxification has been previously reported (Gómez-Hernández et al., 2011).
Nitrate-Dependent Induction of R. etli nor Expression Requires NarB
Bradyrhizobium diazoefficiens, the symbiont of soybeans, is considered a model for the study of denitrification in rhizobia given its capacity to grow anoxically from nitrate respiration. In this bacterium, where denitrification has been extensively studied, expression of nap, nirK, nor, and nos genes requires both oxygen limitation and the presence of a NOx (for a recent review see Torres et al., 2016). In R. etli, it has been reported that low-oxygen concentration (1%) induces expression of nirK and norC denitrification genes (Gómez-Hernández et al., 2011). In this work, we demonstrate that in addition to low oxygen conditions, nitrate or a product derived from its reduction generated by NarB activity is also required for induction of nor genes but not for nirK. In fact, Gómez-Hernández et al. (2011) found that R. etli norC and nirK genes display a different level of dependence for the transcriptional regulator NnrR. A null mutation in nnrR causes a drastic drop in the expression of norC, while nirK still exhibits significant expression. In B. diazoefficiens, NnrR is the direct transcriptional regulator of nor genes in response to NO but not of nirK that is controlled directly by FixK2 in response to low oxygen (Bueno et al., 2017). These findings are in agreement with the different dependency on nitrate and NarB of the R. etli nor and nirK genes we show in this work.
In order to identify the NOx (NO2− or NO) derived from nitrate reduction required for induction of R. etli nor genes, we analyzed the expression of the norC::uidA transcriptional fusion as well as the haem c component of cNor (NorC) in a R. etli nirK mutant that is defective in nitrite reduction to NO as it has been demonstrated in this work. The absence of NorC in membranes as well as the very basal levels of β-glucuronidase activity in the nirK mutant cultured with nitrate as sole N source indicates that the signal molecule required for induction of nor genes is NO. Similarly, it has been recently demonstrated in B. diazoefficiens that norCBQD expression requires, in addition to microoxia, the presence of NO (Bueno et al., 2017).
In this work, we propose for the first time a new pathway in bacteria to produce N2O by coupling nitrate assimilation and denitrification under microoxic conditions. In this context, it has been recently identified in B. diazoefficiens, an integrated system for nitrate assimilation and nitric oxide detoxification which is connected to denitrification through the induction of nor genes when a single domain hemoglobin (Bjgb) encoded in this pathway is not functional (Cabrera et al., 2016).
Statements
Author contributions
AH-G and MD conceived and designed the study. AH-G, MT, AS, and LG performed the experiments. AH-G, MT, AS, LG, and MD analyzed the results and wrote the manuscript. EB critically revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by Fondo Europeo de Desarrollo Regional (FEDER)-co-financed grants (AGL2013-45087-R and AGL2017-85676-R) from the Ministerio de Economía y Competitividad (Spain). Grant P12-AGR-1968 from the Junta de Andalucía was also acknowledged.
Acknowledgments
We are grateful to G. Tortosa (EEZ, CSIC, Granada, Spain), M. Rodríguez, and M. P. Salas (CCG, UNAM, Cuernavaca, México) for their excellent technical assistance. We thank D. Francis Lewis for the improvement of the written English. We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.00980/full#supplementary-material
Footnotes
References
1
Al-AttarS.de VriesS. (2015). An electrogenic nitric oxide reductase.FEBS Lett.5892050–2057. 10.1016/j.febslet.2015.06.033
2
BedmarE. J.BuenoE.CorreaD.TorresM. J.DelgadoM. J.MesaS. (2013). “Ecology of denitrification in soils and plant-associated,” in Beneficial Plant-Microbial Interactions: Ecology and Applications, edsRodelasB.Gonzalez-LópezJ. (Boca Ratón, FL: CRC Press), 164–182.
3
BedmarE. J.RoblesE. F.DelgadoM. J. (2005). The complete denitrification pathway of the symbiotic, nitrogen-fixing bacterium Bradyrhizobium japonicum.Biochem. Soc. Trans.33141–144. 10.1042/BST0330141
4
BeringerJ. E. (1974). R factor transfer in Rhizobium leguminosarum.J. Gen. Microbiol.84188–198. 10.1099/00221287-84-1-188
5
BravoA.MoraJ. (1988). Ammonium assimilation in Rhizobium phaseoli by the glutamine synthetase-glutamate synthase pathway.J. Bacteriol.170980–984. 10.1128/jb.170.2.980-984.1988
6
BuenoE.Gomez-HernandezN.GirardL.BedmarE. J.DelgadoM. J. (2005). Function of the Rhizobium etli CFN42 nirK gene in nitrite metabolism.Biochem. Soc. Trans.33162–163. 10.1042/BST0330162
7
BuenoE.RoblesE. F.TorresM. J.KrellT.BedmarE. J.DelgadoM. J.et al (2017). Disparate response to microoxia and nitrogen oxides of the Bradyrhizobium japonicum napEDABC, nirK and norCBQD denitrification genes.Nitric. Oxide68137–149. 10.1016/j.niox.2017.02.002
8
Butterbach-BahlK.BaggsE. M.DannenmannM.KieseR.Zechmeister-BoltensternS. (2014). Nitrous oxide emissions from soils: how well do we understand the processes and their controls?Philos. Trans. R. Soc. B368:20130122. 10.1098/rstb.2013.0122
9
CabreraJ. J.SalasA.TorresM. J.BedmarE. J.RichardsonD. J.GatesA. J.et al (2016). An integrated biochemical system for nitrate assimilation and nitric oxide detoxification in Bradyrhizobium japonicum.Biochem. J.473297–309. 10.1042/BJ20150880
10
Calvo-BegueriaL.RubioM. C.MartinezJ. I.Perez-RontomeC.DelgadoM. J.BedmarE. J.et al (2018). Redefining nitric oxide production in legume nodules through complementary insights from electron paramagnetic resonance spectroscopy and specific fluorescent probes.J. Exp. Bot.693703–3714. 10.1093/jxb/ery159
11
de VriesS.SuhartiS.PouvreauL. A. M. (2007). “Nitric oxide reductase: structural variations and catalytic mechanism,” in Biology of the Nitrogen Cycle, ed.NewtonW. E. (Amsterdam: Elservier Science), 57–67.
12
DelgadoM. J.CasellaS.BedmarE. J. (2007). “Denitrification in rhizobia-legume symbiosis,” in Biology of the Nitrogen Cycle, ed.NewtonW. E. (Amsterdam: Elservier Science).
13
GirardL.BromS.DavalosA.LopezO.SoberonM.RomeroD. (2000). Differential regulation of fixN-reiterated genes in Rhizobium etli by a novel fixL-fixK cascade.Mol. Plant Microbe Interact.131283–1292. 10.1094/MPMI.2000.13.12.1283
14
Gómez-HernándezN.Reyes-GonzalezA.SanchezC.MoraY.DelgadoM. J.GirardL. (2011). Regulation and symbiotic role of nirK and norC expression in Rhizobium etli.Mol. Plant Microbe Interact.24233–245. 10.1094/MPMI-07-10-0173
15
GonzálezV.SantamariaR. I.BustosP.Hernandez-GonzalezI.Medrano-SotoA.Moreno-HagelsiebG.et al (2006). The partitioned Rhizobium etli genome: genetic and metabolic redundancy in seven interacting replicons.Proc. Natl. Acad. Sci. U.S.A.1033834–3839. 10.1073/pnas.0508502103
16
HirayamaJ.EdaS.MitsuiH.MinamisawaK. (2011). Nitrate-dependent N2O emission from intact soybean nodules via denitrification by Bradyrhizobium japonicum bacteroids.Appl. Environ. Microbiol.778787–8790. 10.1128/AEM.06262-11
17
InabaS.IkenishiF.ItakuraM.KikuchiM.EdaS.ChibaN.et al (2012). N2O emission from degraded soybean nodules depends on denitrification by Bradyrhizobium japonicum and other microbes in the rhizosphere.Microbes Environ.27470–476. 10.1264/jsme2.me12100
18
InabaS.TanabeK.EdaS.IkedaS.HigashitaniA.MitsuiH.et al (2009). Nitrous oxide emission and microbial community in the rhizosphere of nodulated soybeans during the late growth period.Microbes Environ.2464–67. 10.1264/jsme2.me08544
19
Intergovernmental Panel on Climate Change [IPCC] (2014). Climate Change 2014: Synthesis Report Contribution of Working Groups I, II and III to the Fifth Assessment Report of the Intergovernmental Panel on Climate Change.Geneva: IPCC. 10.1264/jsme2.me08544
20
KeenN. T.TamakiS.KobayashiD.TrollingerD. (1988). Improved broad-host-range plasmids for DNA cloning in gram-negative bacteria.Gene70191–197. 10.1016/0378-1119(88)90117-5
21
KovachM. E.ElzerP. H.HillD. S.RobertsonG. T.FarrisM. A.RoopR. M.et al (1995). Four new derivatives of the broad-host-range cloning vector pBBR1MCS, carrying different antibiotic-resistance cassettes.Gene166175–176. 10.1016/0378-1119(95)00584-1
22
Luque-AlmagroV. M.GatesA. J.Moreno-VivianC.FergusonS. J.RichardsonD. J.RoldanM. D. (2011). Bacterial nitrate assimilation: gene distribution and regulation.Biochem. Soc. Trans.391838–1843. 10.1042/BST20110688
23
Moreno-VivianC.CabelloP.Martinez-LuqueM.BlascoR.CastilloF. (1999). Prokaryotic nitrate reduction: molecular properties and functional distinction among bacterial nitrate reductases.J. Bacteriol.1816573–6584.
24
Moreno-ViviánC.FloresE. (2007). “Nitrate assimilation in bacteria,” in Biology of the Nitrogen Cycle, edsBotheH.FergusonS. J.NewtonW. E. (Amsterdam: Elsevier), 263–282. 10.1016/b978-044452857-5.50018-7
25
NicholasD. J. D.NasonA. (1957). “Determination of nitrate and nitrite,” in Methods in Enzymolog, ed.KaplanN. O. (London: Academic Press), 974–977.
26
NoelK. D.SanchezA.FernandezL.LeemansJ.CevallosM. A. (1984). Rhizobium phaseoli symbiotic mutants with transposon Tn5 insertions.J. Bacteriol.158148–155.
27
PilegaardK. (2013). Processes regulating nitric oxide emissions from soils.Philos. Trans. R. Soc. Lond. B Biol. Sci.368:20130126. 10.1098/rstb.2013.0126
28
PrentkiP.KrischH. M. (1984). In vitro insertional mutagenesis with a selectable DNA fragment.Gene29303–313. 10.1016/0378-1119(84)90059-3
29
QuintoC.de la VegaH.FloresM.FernándezL.BalladoT.SoberónG.et al (1982). Reiteration of nitrogen fixation gene sequences in Rhizobium phaseoli.Nature299724–726. 10.1038/299724a0
30
RavishankaraA. R.DanielJ. S.PortmannR. W. (2009). Nitrous oxide (N2O): the dominant ozone-depleting substance emitted in the 21st century.Science326123–125. 10.1126/science
31
RichardsonD. J. (2011). “Redox complexes of the nitrogen cycle,” in Nitrogen Cycling in Bacteria, ed.MoirJ. W. B. (Norkfolk: Caister Academic Press), 23–39.
32
RichardsonD. J.BerksB. C.RussellD. A.SpiroS.TaylorC. J. (2001). Functional, biochemical and genetic diversity of prokaryotic nitrate reductases.Cell. Mol. Life Sci.58165–178. 10.1007/pl00000845
33
SambrookJ.FritschE. F.ManiaticsT. (1989). Molecular Cloning: A Laboratory Manual.New York, NY: Cold Spring Harbor Laboratory Press. 10.1007/pl00000845
34
SambrookJ.RussellD. W. (2001). Molecular Cloning : A Laboratory Manual.New York, N.Y: Cold Spring Harbor Laboratory. 10.1007/pl00000845
35
SánchezC.BedmarE. J.DelgadoM. J. (2011). “Denitrification in legume-associated endosymbiotic Bacteria,” in Nitrogen Cycling in Bacteria, ed.MoirJ. W. B., (Norfolk: Caister Academic Press), 197–210.
36
SchäferA.TauchA.JagerW.KalinowskiJ.ThierbachG.PuhlerA. (1994). Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum.Gene14569–73. 10.1016/0378-1119(94)90324-7
37
SimonJ.KlotzM. G. (2013). Diversity and evolution of bioenergetic systems involved in microbial nitrogen compound transformations.Biochim. Biophys. Acta1827114–135. 10.1016/j.bbabio.2012.07.005
38
SimonR.PrieferU.PühlerA. (1983). “Vector plasmids for in vivo and in vitro mainpulation of gram-negative bacteria,” in Molecular Genetics of the Bacteria-Plant Interaction, ed.PühlerA. (Heidelberg: Springer-Verlag), 98–106. 10.1007/978-3-642-69338-0_11
39
SmithK. A.MosierA. R.CrutzenP. J.WiniwarterW. (2012). The role of N2O derived from crop-based biofuels, and from agriculture in general, in Earth’s climate.Philos. Trans. R. Soc. Lond. B Biol. Sci.3671169–1174. 10.1098/rstb.2011.0313
40
SoberonM.LopezO.MoreraC.GirardM. L.TabcheM. L.MirandaJ. (1999). Enhanced nitrogen fixation in a Rhizobium etli ntrC mutant that overproduces the Bradyrhizobium japonicum symbiotic terminal oxidase cbb3.Appl. Environ. Microbiol.652015–2019.
41
ThomsonA. J.GiannopoulosG.PrettyJ.BaggsE. M.RichardsonD. J. (2012). Biological sources and sinks of nitrous oxide and strategies to mitigate emissions.Philos. Trans. R. Soc. Lond. B Biol. Sci.3671157–1168. 10.1098/rstb.2011.0415
42
TorresM. J.AvilaS.BedmarE. J.DelgadoM. J. (2018). Overexpression of the periplasmic nitrate reductase supports anaerobic growth by Ensifer meliloti.FEMS Microbiol. Lett.365:fny041. 10.1093/femsle/fny041
43
TorresM. J.Hidalgo-GarciaA.BedmarE. J.DelgadoM. J. (2013). Functional analysis of the copy 1 of the fixNOQP operon of Ensifer meliloti under free-living micro-oxic and symbiotic conditions.J. Appl. Microbiol.1141772–1781. 10.1111/jam.12168
44
TorresM. J.RubiaI.de la PenaT. C.PueyoJ. J.BedmarE. J.DelgadoM. J. (2014). Genetic basis for denitrification in Ensifer meliloti.BMC Microbiol.14:142. 10.1186/1471-2180-14-142
45
TorresM. J.SimonJ.RowleyG.BedmarE. J.RichardsonD. J.GatesA. J.et al (2016). Nitrous oxide metabolism in nitrate-reducing bacteria: physiology and regulatory mechanisms.Adv. Microb. Physiol.68353–432. 10.1016/bs.ampbs.2016.02.007
46
van SpanningR. J. (2011). “Structure, function, regulation and evolution of the nitrite and nitrous oxide reductase: denitrification enzymes with a β-propeller fold,” in Nitrogen Cycling in Bacteria, ed.MoirJ. W. B. (Norkfolk: Caister Academic Press), 135–161.
47
VargasC.McEwanA. G.DownieJ. A. (1993). Detection of c-type cytochromes using enhanced chemiluminescence.Anal. Biochem.209323–326. 10.1006/abio.1993.1127
48
ZumftW. G.KroneckP. M. (2007). Respiratory transformation of nitrous oxide (N2O) to dinitrogen by Bacteria and archaea.Adv. Microb. Physiol.52107–227. 10.1016/S0065-2911(06)52003-X
Summary
Keywords
assimilatory nitrate reductase, denitrification, gene expression, soil bacteria, nitrous oxide
Citation
Hidalgo-García A, Torres MJ, Salas A, Bedmar EJ, Girard L and Delgado MJ (2019) Rhizobium etli Produces Nitrous Oxide by Coupling the Assimilatory and Denitrification Pathways. Front. Microbiol. 10:980. doi: 10.3389/fmicb.2019.00980
Received
30 November 2018
Accepted
18 April 2019
Published
07 May 2019
Volume
10 - 2019
Edited by
Marcus A. Horn, Leibniz University Hannover, Germany
Reviewed by
Stephen Spiro, The University of Texas at Dallas, United States; Katharina Kujala, University of Oulu, Finland
Updates
Copyright
© 2019 Hidalgo-García, Torres, Salas, Bedmar, Girard and Delgado.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maria J. Delgado, mdelgado@eez.csic.es
This article was submitted to Terrestrial Microbiology, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.