 Hui Wang
Hui Wang Nan Wu
Nan Wu Yan Liu
Yan Liu Jiban Kumar Kundu
Jiban Kumar Kundu Wenwen Liu
Wenwen Liu Xifeng Wang
Xifeng Wang- 1State Key Laboratory for Biology of Plant Diseases and Insect Pests, Institute of Plant Protection, Chinese Academy of Agricultural Sciences, Beijing, China
- 2Division of Crop Protection and Plant Health, Crop Research Institute, Prague, Czechia
The bacterial communities in the gut of an insect have important ecological and functional effects on the insect. However, the community composition and diversity of the gut microbiota in insects that vector plant viruses are poorly understood. As an important insect vector, Psammotettix alienus transmits various viruses including wheat dwarf virus (WDV). Here, we used the combination of leafhopper and WDV as model to survey the influence of gut microbiota on virus transmission characteristic of insect vector and vice versa. We have characterized 22 phyla and 249 genera of all gut bacterial communities in the leafhopper populations collected from six geographic regions in China. Community composition and diversity varied across different geographic populations. However, WDV transmission efficiencies of these six field populations were all greater than 80% with no significant difference. Interestingly, the transmission efficiency of WDV by laboratory reared insects with decreased gut bacterial diversity was similar to that of field populations. Furthermore, we found that the composition of the leafhopper gut bacteria was dynamic and could reversibly respond to WDV acquisition. Higher bacterial diversity and abundance of gut microbiota in different leafhopper populations did not influence their WDV transmission efficiency, while the acquisition of WDV changes gut microbiota by a dynamic and reversible manner. This report provides insight into the complex relationship between the gut microbiota, insect vector and virus.
Introduction
Almost all insects harbor gut microbial communities that play important functions for their hosts (Dillon and Dillon, 2004). In general, such microbial partners can provide nutrition, contribute to host reproduction and survival (Broderick et al., 2006; Sharon et al., 2010), mediate detoxification of insect diets (Genta et al., 2006; Cejanavarro et al., 2015), or confer resistance to insecticides (Kikuchi et al., 2012; Cheng et al., 2017). In addition, some microbes can help or inhibit pathogen transmission (Douglas, 2015). Wolbachia inhibits replication of dengue virus in the vector mosquito (Aedes aegypti; Moreira et al., 2009). On the contrary, the gut microbiome of the vector sand fly (Lutzomyia longipalpis) is essential for survival of Leishmania infantum, because successive daily antibiotic treatments inhibited growth and development of the parasite into its infectious metacyclic forms (Kelly et al., 2017). In addition, the composition and diversity of insect gut microbes are influenced by the external factors, such as climate change (Sheik et al., 2011), soil attributes (da C Jesus et al., 2009; Young et al., 2018), pathogens and ingested food (Knief et al., 2010; Ben Guerrero et al., 2016).
Hemipteran insects have a needle-like sucking stylet, and some are notorious agricultural pests that cause serious economic loss not only by directly sucking the plant sap but also by transmitting plant viruses (Wang et al., 2015; Liu W. et al., 2018; Qin et al., 2018). Hemipterans that are phytophagous usually feed on nutritionally deficient xylem or phloem diets, but endosymbionts in these insects can provide essential amino acids and other nutrients (Redak et al., 2004). Most leafhoppers (Hemiptera: Cicadellidae) harbor the bacterium Candidatus Sulcia muelleri, which coexists with other bacteria such as Candidatus Baumannia cicadellinicola, Candidatus Zinderia insecticola, Candidatus Nasuia or Hodgkinia cicadicola (Koga et al., 2013). In addition to obligate endosymbionts, leafhoppers also host various facultative endosymbionts such as Wolbachia, Rickettsia or Cardinium (Zheng et al., 2017).
The European grass feeding leafhopper (Psammotettix alienus) can lead to great yield losses by transmitting viruses such as Russian mosaic virus (Vacke, 1961), wheat yellow striate virus (Liu Y. et al., 2018) and wheat dwarf virus (WDV) in a persistent circulative manner (Wang et al., 2014). Periodic outbreaks of wheat dwarf disease outbreaks have caused economic losses in European (Benkovics et al., 2010), African (Najar et al., 2000) and Asian countries (Zhang et al., 2010), with the incidence of wheat dwarf disease in Swedish wheat fields reaching 90% in severe cases (Lindblad and Waern, 2002). In China, the first WDV disease outbreak reached up to 80% incidence, and yield was reduced by 50–80% in Hancheng, Shaanxi Province in 2007 (Wang et al., 2008). High population numbers and expanding distribution of P. alienus are important factors contributing to these epidemics.
Similar to viruses transmitted in a persistent circulative manner (Mar et al., 2014), WDV invades the midgut via receptor-mediated endocytosis and spreads into the salivary glands through the hemolymph, but it also rapidly moves to the hemocoel through the filter chamber (Wang et al., 2014). Interestingly, some endosymbionts are involved in the spread of viruses within the insect vectors (Kliot and Ghanim, 2013). For example, the GroEL protein of the endosymbiont Buchnera is crucial for determining the persistent nature of potato leafroll virus in Myzus persicae (Hogenhout et al., 2000). Similarly, the GroEL protein produced by Arsenophonus in the Asia II genetic group of Bemisia tabaci can interact with the viral coat protein encoded by cotton leaf curl virus (CLCuV) and is involved in virus transmission (Rana et al., 2012). Also, in B. tabaci, Hamiltonella produces a GroEL protein that is involved in transmission of tomato yellow leaf curl virus (TYLCV; Gottlieb et al., 2010; Ghanim, 2014). Recently, a symbiotic bacterium, Sulcia, in a leafhopper was found to directly mediate transovarial transmission of rice dwarf virus (Jia et al., 2017).
Although microbes in insect vectors might be involved in virus transmission, little is known about the change/function of insect vector gut microbiota in virus transmission. The present study wants to comprehensively characterize the bacterial communities in the gut of P. alienus from six locations in China by high-throughput sequencing. We also focused on the gut bacterial community changes during WDV acquisition (Supplementary Figure S1). In general, the gut bacterial communities of the field-collected leafhoppers represented 22 phyla and 249 genera, meanwhile the difference of gut bacterial community composition and diversity of the six field populations do not influence the virus transmission. Interestingly, the WDV transmission efficiency of laboratory reared leafhoppers with decreased gut bacterial diversity is similar to field populations. Moreover, composition of the leafhopper gut bacterial communities during the acquisition period was dynamic and reversible over time.
Materials and Methods
Leafhopper Collection
In China, wheat dwarf disease occurs frequently in Tianshui (Gansu Province), Hancheng (Shaanxi Province), Taiyuan (Shanxi Province) and Linfen (Shanxi Province), but occasionally in Baoding (Hebei Province), Shijiazhuang (Hebei Province) and Tianjin (Zhang et al., 2017). Based on the epidemic pattern of the disease, vector leafhoppers (P. alienus) were collected from six regions in China (Tianshui, Hancheng, Linfen, Taiyuan, Baoding and Tianjin) during April 2017 (Table 1 and Supplementary Figure S2), and taken immediately on healthy wheat seedlings in different tubes to the laboratory for transmission assay and 16S rDNA sequencing.

Table 1. Sampling locations and dates in 2017 for Psammotettix alienus and number of reads obtained from high-throughput Illumina sequencing of gut microbiome.
Similarly, we selected more than 200 various instars laboratory-maintained leafhoppers (collected from Linfen in 2011) and then placed them on WDV-infected wheat to feed. After an acquisition access period (AAP) of 5, 10, and 20 d, 15 adult leafhoppers as one replicate (three repetitions for each time) were collected, dissected and extracted total DNA from guts for sequencing, respectively. Nonviruliferous adult leafhoppers (1 to 3 days old) were fed on healthy wheat as controls. WDV transmission efficiency was measured as above.
DNA Extraction From Excised Guts
Adults of P. alienus were first surface-sterilized with 70% ethanol for 1 min, then washed three times for 1 min each with ddH2O. Fifteen guts from the same population were collected in one tube as one replicate (three repetitions for every field population). DNA was extracted from all samples using a Wizard genomic DNA isolation kit (Promega, Madison, WI, United States) according to the manufacturer’s instruction.
PCR Amplification of 16S rDNA
Genomic DNA of the gut microbiome of leafhoppers was amplified by primers V3-V4F (5′-ACTCCTACGGGAGGCA GCA-3′) and V3-V4R (5′-GGACTACHVGGGTWTCTAAT-3′) specific for the 16S rDNA hypervariable V3-V4 region. The PCR mix contained 1.5 μl PrimerF, 1.5 μl PrimerR, 0.5 μl Q5 High-Fidelity DNA Polymerase (TaKaRa, Dalian, China), 10 μl High GC Enhancer (TaKaRa), 10 μl 5 × PCR Buffer (TaKaRa), 1 μl dNTP (TaKaRa) and 40ng DNA and ddH2O. The thermal cycling conditions for the indexing PCR consisted of an initial denaturation at 95°C for 5 min, followed by 30 cycles of 95°C for 1 min, 55°C for 1 min and 72°C for 1 min. The PCR products were purified, quantified and amplified again as template by Solexa PCR with an initial denaturation of 98°C for 30 s, followed by 40 cycles of 98°C for 10 s, 65°C for 30 s and 65°C for 30 s. Solexa PCR products were purified and quantified for high-throughput sequencing (Illumina) by Biomarker Technologies (Beijing, China).
Sequence Data Analyses
After sequencing, sequences were trimmed and assembled by Flash (version 1.2.7, http://ccb.jhu.edu/software/FLASH/; Magoc and Salzberg, 2011), and reads that could not be assembled were discarded. Chimeras were identified and removed using Uchime (Mothur; version 1.31.2, http://www.mothur.org/; Edgar et al., 2011). The cleaned Fastq data were aligned into operational taxonomic units (OTUs) by UCLUST (Edgar, 2010; QIIME; Caporaso et al., 2010) based on a similarity of 97%. OTUs were assigned taxons using the RDP CLASSIFIER (version 2.2, http://sourceforge.net/projects/rdp-CLASSIFIER) against the Silva database1. Relative OTU abundances were summarized across taxonomic levels from phylum to genus. The raw data were available under SRA accession number PRJNA495407.
Diversity Analyses
ACE and Chao 1 indices for alpha diversity, which reflects the diversity and richness of individual samples, were plotted using the Mother package (QIIME)2. Beta diversity was determined to evaluate the degree of similarity of gut bacterial communities from different samples using QIIME. Principal coordinate analysis (PCoA; Sakaki et al., 1994), heat maps, dendrograms based on unweighted pair-group method with arithmetic mean (UPGMA; Kim et al., 2017) and nonmetric multidimensional scaling (NMDS; Looft et al., 2012) were used to analyze the beta diversity.
Analysis of Significant Differences in Relative Abundance of Gut Microbes
To discover biomarkers that differed significantly among populations, we used a linear discriminant analysis (LDA) of effect size to determine OTUs that discriminate among the leafhopper populations with an LDA score is more than 4.0. Colors were used to indicate the different populations of the phylogenetic component contributing to group uniqueness. A cladogram was also constructed to show the LDA results. Levels of the cladogram represented, from the inner to outer rings, phylum, class, order, family, and genus. Color codes indicated the condition, and letters indicated the taxa that contribute to the uniqueness of the corresponding populations at an LDA score greater than 4.0.
Function Analysis
To predict putative KEGG functions for the gut microbiome of leafhoppers from different geographic locations, we compared the predicted relative abundances of KEGG orthologs based on evenly rarefied 16S rDNA gene amplicon sequences using PICRUST version 1.0.0 (Parks et al., 2014). Fisher tests were used to compare the KEGG function predictions for the six populations.
WDV Transmission Assay
WDV transmission efficiency of the different leafhopper populations was determined using 50 leafhoppers that were allowed to feed on WDV-infected wheat for 3 days. They were then transferred to wheat seedlings (1 insect/plant) for a 72-hour inoculation access period, and the seedlings were then grown in a greenhouse. At 21 days post-inoculation, wheat plants were analyzed for virus symptoms and tested using specific primers for WDV infection by PCR with primer pairs 5′-ATGGTGACCAACAAGGAC-3′ and 5′-TAACACGCGTGCGTATAGGC-3′ (Zhang et al., 2010). The experiments were performed three times.
In addition, we compared the WDV transmission efficiency of the laboratory-maintained leafhoppers (from Linfen) and field population.
Results
Sequencing Quality and Operational Taxonomic Unit (OTU) Analysis
To study the composition and diversity of the gut bacterial communities in the leafhopper field population, three DNA pools of leafhoppers were extracted from each of the six geographic locations (Table 1), and then subjected to Illumina sequencing of 16S rDNA. The all samples generated 1,436,533 raw reads in total. Following demultiplexing, quality filtering and chimera removal, 1,221,573 clean tags were retained for all samples, and every sample had average 67,865 high-quality sequences. In all, 499 distinct OTUs were recognized with 97% similarity cutoff (Figure 1C).
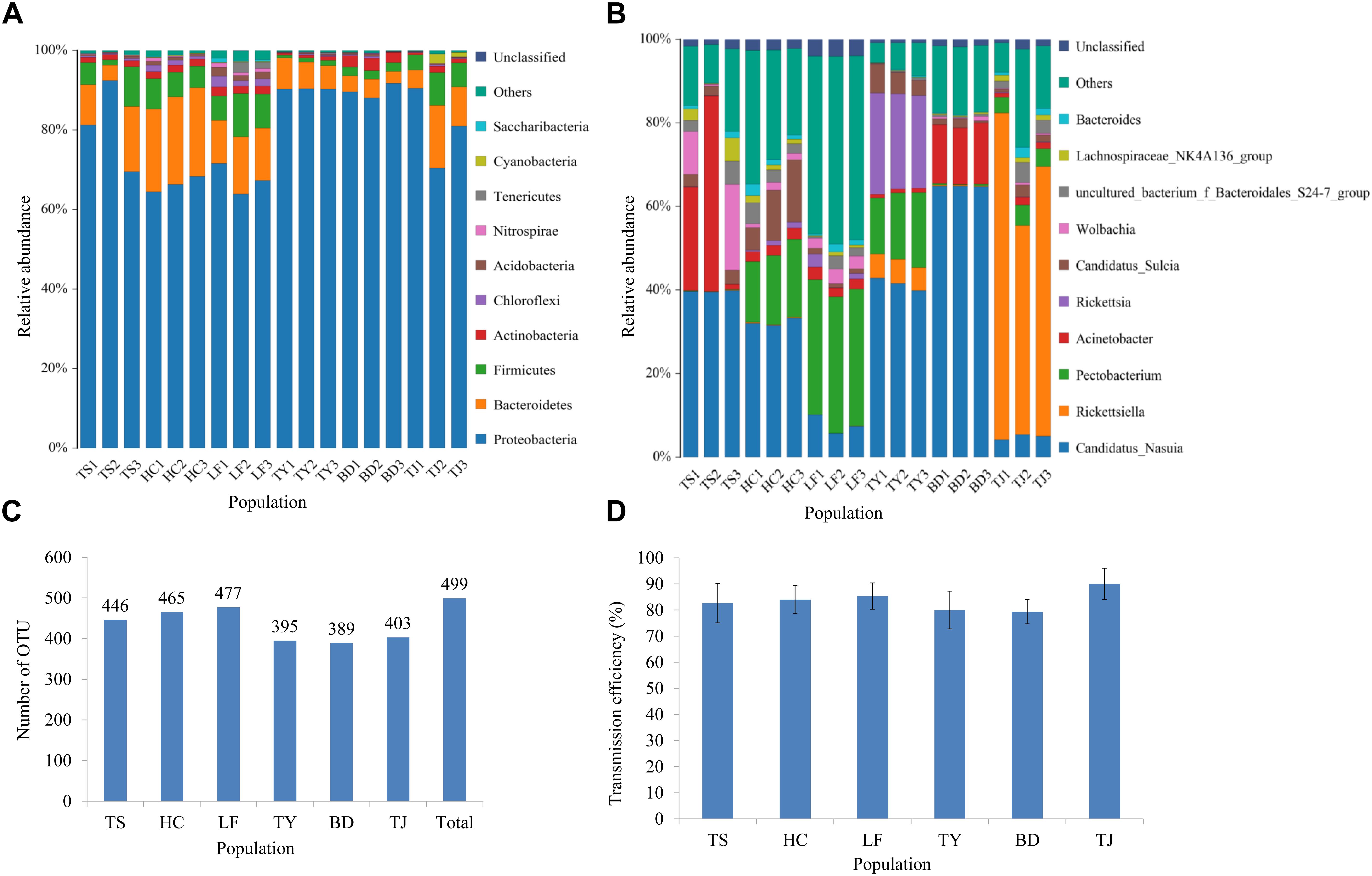
Figure 1. Analysis of gut microbiota and transmission efficiency of wheat dwarf virus for the field populations of leafhoppers from six locations in China. Taxonomic composition at the phylum level (A) and genus level (B); The number of OTUs (C); WDV transmission efficiency (D). Only the top 10 most abundant taxa are shown for each. Results are shown for three bulked samples (N = 15 adults/sample) for each geographic population. The bacterial community composition varied among the locations. TS: Tianshui; HC: Hancheng; LF: Linfen; TY: Tianjin; BD: Baoding; TJ: Tianjin.
The OTUs were assigned to taxonomic groups using the BLAST algorithm in a search against the SILVA ribosomal RNA gene database3. The identified sequences were distributed across 22 assigned bacterial phyla. Members of Proteobacteria, Bacteroidetes, Firmicutes and Actinobacteria had a cumulative relative abundance of more than 80% (Figure 1A). The remaining phylum all had a very low abundance. Overall, 249 genera were represented (Figure 1B and Supplementary Table S1). At the genus level, Acinetobacter, Candidatus_Nasuia and Wolbachia were the three main genera in the Tianshui population, but Pectobacterium, Candidatus_Nasuia and Candidatus_Sulcia were three most abundant genera in the Hancheng population (Figure 1B). Similarly, the dominant genus in the Linfen population was Pectobacterium, followed in order by Candidatus_Nasuia, Wolbachia, Rickettsia and Acinetobacter (Figure 1B). In addition, two genera Ruminobacter and Prevotellaceae_UCG-004 predominated in the Linfen population. Candidatus_Nasuia, Pectobacterium and Rickettsia were the three most abundant in order in the Taiyuan population. The relative abundance of Candidatus_Nasuia surpassed 60% in the Baoding population, followed by Acinetobacter (Figure 1B). The dominant genus in the Tianjin population was Rickettsiella, accounting for more than 70%, followed by Candidatus_Nasuia and Pectobacterium (Figure 1B). When we determined the WDV transmission efficiency of these six populations, all populations were greater than 80% and did not differ significantly among the populations (Figure 1D).
Differences in the Gut Bacterial Communities in Geographic Populations of P. alienus
We used the Chao 1 and ACE indices to evaluate alpha diversity across the different populations. The results showed a saturating number of OTUs by rarefaction curves (Supplementary Figure S3), indicating adequate sampling of 16S rDNA sequences for all the samples. On the basis of the Chao 1 (Supplementary Figure S4A) and ACE index (Supplementary Figure S4B), bacterial diversity in the Linfen population was the highest in all populations, the lowest in the Taiyuan, Baoding and Tianjin population, intermediate in the Tianshui and Hancheng populations, and highest in the Linfen population (Supplementary Figures S4A,B).
In an analysis of the beta diversity of the gut bacterial communities based on PCoA (Figure 2A) NMDS (Figure 2B), the results showed a similar tendency in the difference between the Tianjin population (blue circle) and the other five populations. In addition, the Hancheng and Linfen populations (red circle) always formed a tight cluster, as did the Tianshui and Taiyuan populations (green circle), and the Baoding population was near the cluster with Hancheng and Linfen. The heat map (Figure 2C) and UPGMA (Figure 2D) indicated that the distance between different regions was consistent with the above results.
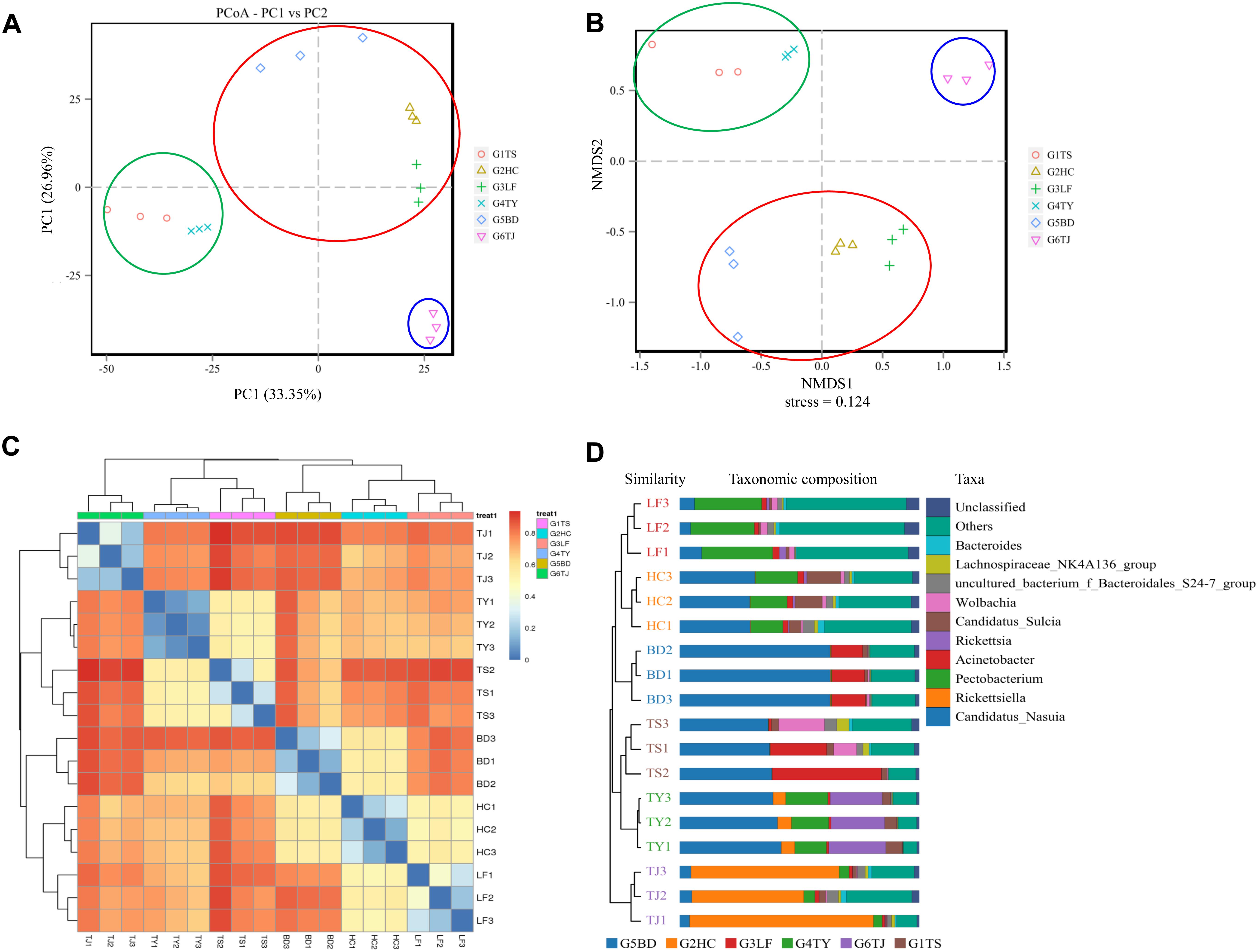
Figure 2. Similarity of bacterial communities of leafhoppers among six locations in China. PCoA analysis (A), NMDS analysis (B), heatmap analysis (C), and UPGMA (D). G1TS: Tianshui; G2HC: Hancheng; G3LF: Linfen; G4TY: Tianjin; G5BD: Baoding; G6TJ: Tianjin.
In a LDA to determine the bacteria that differed significantly among the six geographic populations, Wolbachia (Figures 3A,C) and Actinobacteria (Figures 3A,D) accounted significantly for the divergence of the bacterial community in the Tianshui population from the other five populations. Candidatus_Sulcia (Figures 3A,E) was an important biomark for the Hancheng population. The genera Neisseria (Figures 3A,F), Streptococcus (Figures 3A,G) and Pectobacterium (Figures 3A,H) distinguished the Linfen population from the other groups. The presence of Rickettsia (Figures 3A,I) in the Taiyuan population was a distinct difference from the other five populations. The most distinctive difference in bacterial abundance in the Baoding population was Candidatus_Nasuia (Figures 3A,J), whereas Rickettsiella differentiated the Tianjin population from the other populations (Figures 3A,K). The OTUs represented in each population are illustrated by phylogenetic levels from phylum to genus in a cladogram (Figure 3B).
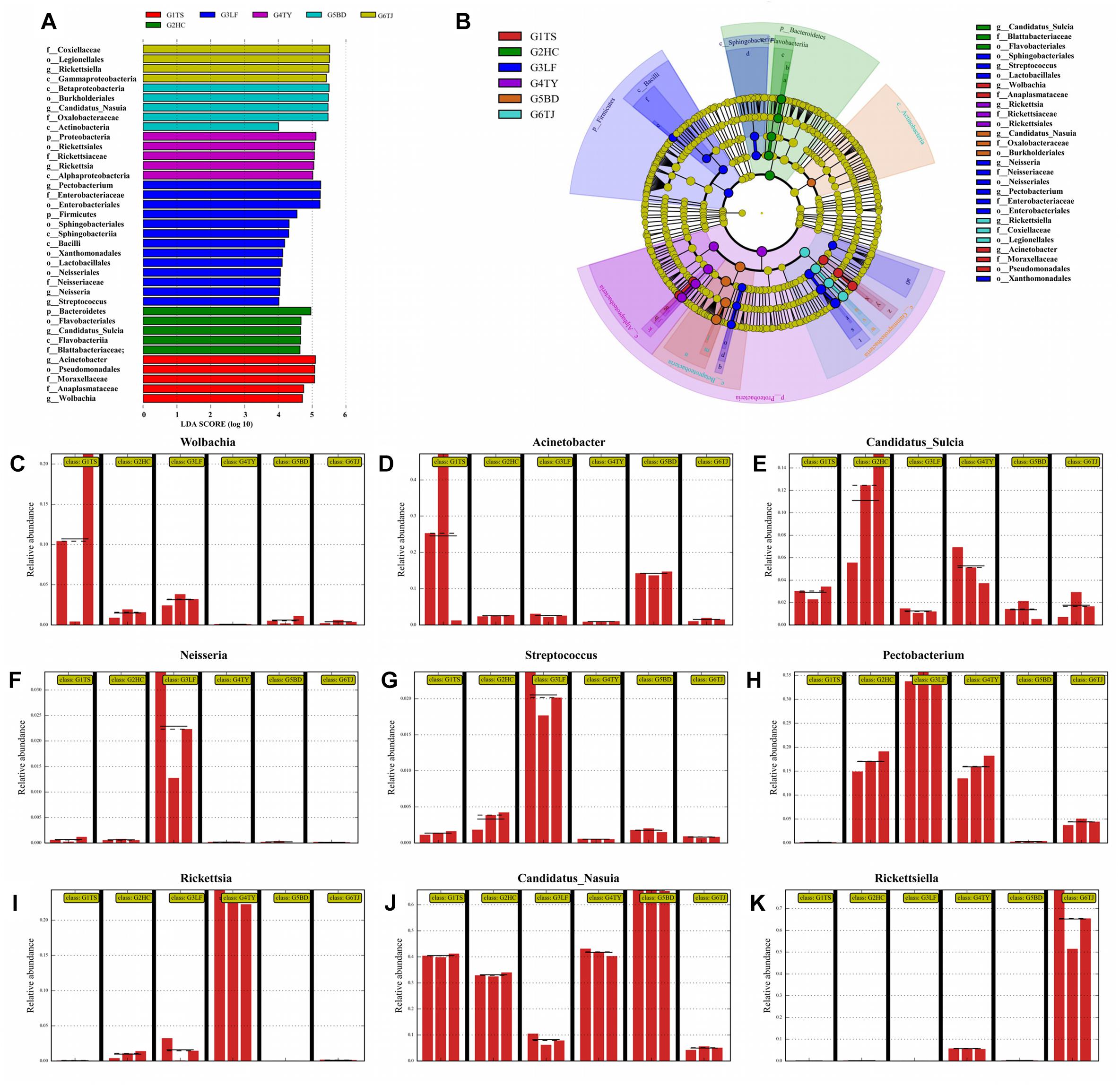
Figure 3. Distinct biomarkers of the bacterial community from different leafhopper populations revealed by linear discriminant analysis (LDA). Multiple regression results are shown for LDA of microbiomes from different populations with LDA > 4. Colors indicate the different populations of the phylogenetic component contributing to group uniqueness (A). Cladogram of the LDA results in panel A. Levels of the cladogram represent, from the inner to outer rings, phylum, class, order, family, and genus. Color codes indicate the six populations, and lower-case letters indicate the taxa that contribute to the uniqueness of the corresponding leafhopper populations (B). (C–K) Relative abundance of distinct bacteria in the different populations: Wolbachia (C), Acinetobacter (D), Candidatus_Sulcia (E), Neisseria (F), Streptococcus (G), Pectobacterium (H), Rickettsia (I), Candidatus_Nasuia (J), Rickettsiella (K). G1TS: Tianshui; G2HC: Hancheng; G3LF: Linfen; G4TY: Tianjin; G5BD: Baoding; G6TJ: Tianjin.
Functional Prediction of the Gut Bacterial Community
In a pathway analysis of the microbes using the Kyoto Encyclopedia of Genes and Genomes (KEGG) database based on their presence within the genomes, bacterial communities in the six natural populations were involved in 43 pathways including amino acid metabolism, carbohydrate metabolism, cofactor and vitamin metabolism (Supplementary Figure S5A). However, the relative abundance of these functions varied among the six groups (Supplementary Figures S5B–E). The membrane transport pathway was predicted to be significantly lower in the microbiota of Tianjin population than in that of the Hancheng, Linfen, Taiyuan and Baoding populations (Supplementary Figures S5B–E). Fewer microbes were involved in signal transduction in Tianjin populations than Hancheng, Taiyuan and Baoding populations (Supplementary Figures S5B,D,E). Significantly more microbes with energy metabolism function were present in Taiyuan, than in Tianjin, followed by the Hancheng and Baoding populations (Supplementary Figures S5B,D,E).
Composition and Abundance of the Gut Bacterial Community Change Dynamically During WDV Acquisition
In a comparison of the gut microbe composition in the nonviruliferous laboratory reared leafhoppers (derive from Linfen) to that in the field-collected leafhoppers from Linfen, the laboratory-maintained leafhoppers had fewer microbes and lower diversity (Figure 4A). However, the virus transmission efficiencies do not have significant difference (Figure 4B).
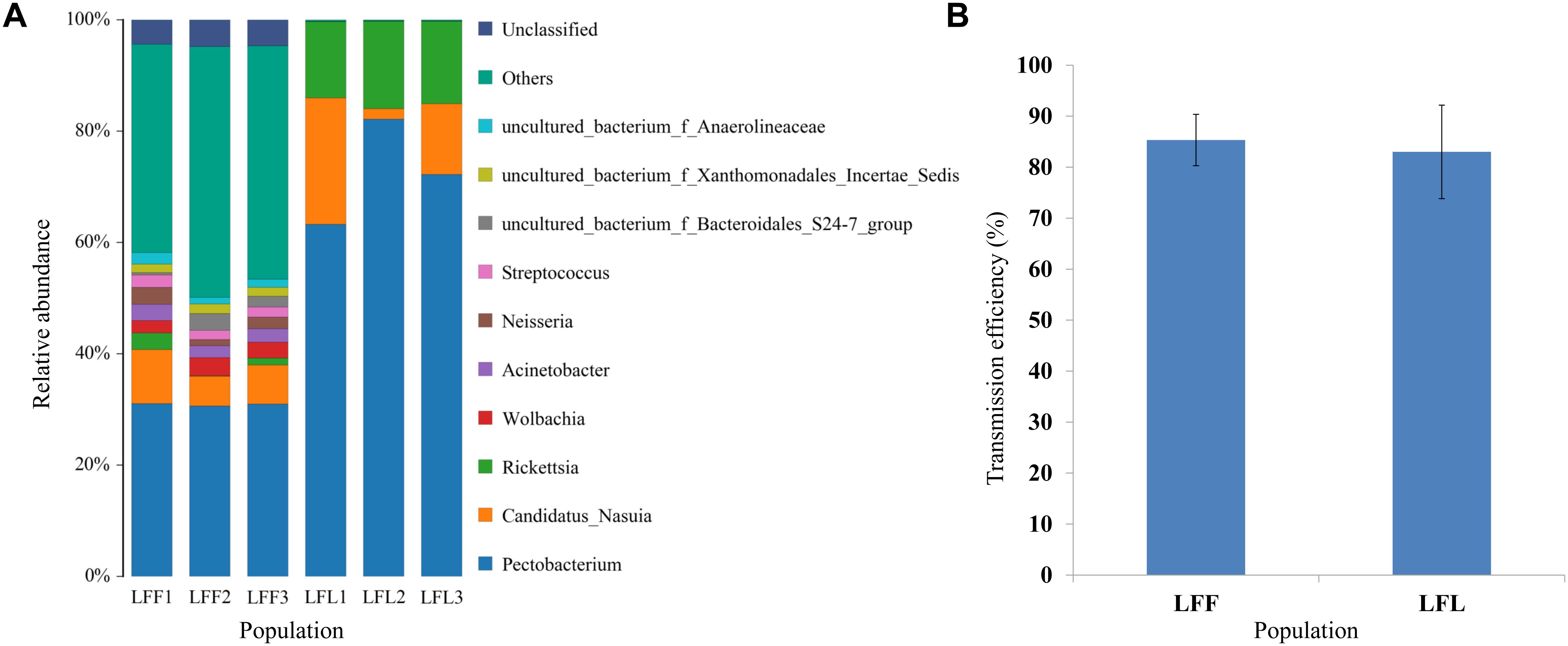
Figure 4. Comparison of gut bacterial community composition (A) and virus transmission efficiencies (B) between field populations (LFF) from China and laboratory-maintained leafhoppers (LFL). Two nonviruliferous populations (NV) were originally collected from Linfen in 2010. Only the top 10 most abundant are shown.
We further used the laboratory reared leafhoppers to study gut bacterial community composition in various times (acquisition access period [AAP] of 0, 5, 10, and 20 d) during WDV acquisition. At all four sampling times of laboratory-maintained leafhoppers, Pectobacterium, Rickettsia and Candidatus_Nasuia accounted for over 90% of the microbes, but Pectobacterium has similar relative abundance, whereas Rickettsia and Candidatus_Nasuia changed in relative abundance with increasing duration of access to the WDV-infected plants (Figure 5A). After 5 d AAP, the relative abundance of Rickettsia was upregulated and accounted for more than 40% of the bacterial microbiome compared to 15% in the controls. By 10 d AAP, the percentage of Rickettsia fell to about 15%, and by 20 d AAP, the Rickettsia had decreased to less than 10% (Figure 5C). Conversely, the proportion of Candidatus_Nasuia among the total bacteria had severely dropped in the leafhopper by 5 d AAP compared to the control, but had begun rising by 10 d AAP and continued to rise in abundance through 20 d AAP, indicating that WDV impacted the composition of the gut bacterial microbiota in a dynamic and reversible fashion (Figure 5D). Several minor species such as Acinetobacter, Pseudomonas, Candidatus_Sulcia and Wolbachia were also identified, but they did not change in relative abundance, i.e., consistent among different samples. The relative abundances of all genera are listed in Supplementary Table S2. Beta diversity was used to evaluate the degree of similarity of bacterial microbiota associated with different times. The results based on a heat map (Figure 5B) showed that the composition of the gut bacterial microbiota from healthy leafhopper, viruliferous leafhoppers after 10 d AAP and 20 d AAP clustered together. In addition, viruliferous leafhoppers after 5 d AAP did not cluster together with the other three groups (healthy leafhopper, viruliferous leafhoppers after 10 d AAP and 20 d AAP). These results further confirm that the composition and abundance of the gut bacterial community recovered during WDV acquisition.
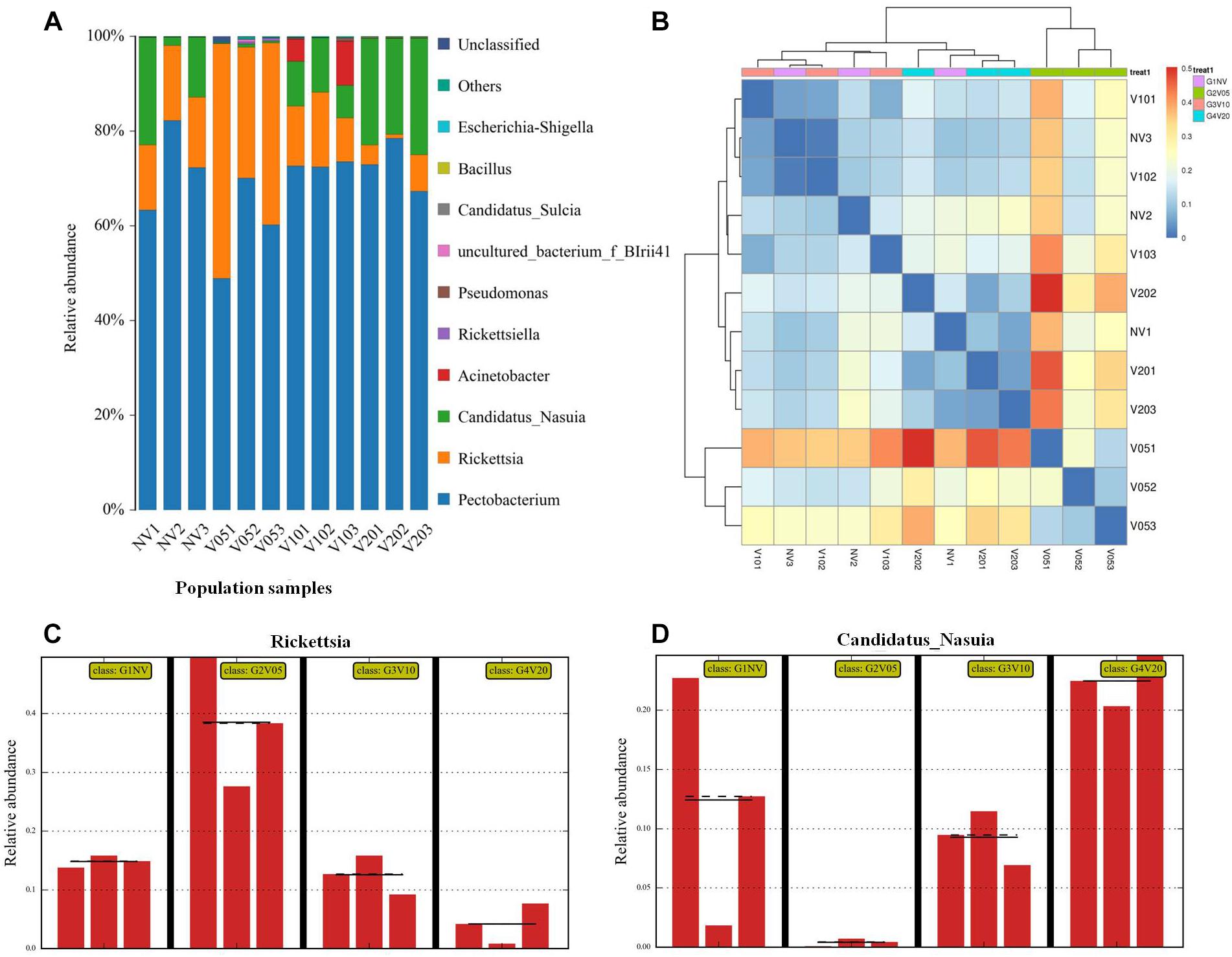
Figure 5. Composition and abundance of the gut bacterial community during the WDV acquisition period. Composition of gut bacterial community at the genus level (A). Only the top 10 most abundant are shown. Comparison of similarities of leafhopper gut bacterial communities at various times during the WDV acquisition period based on Heat map analysis (B). Relative abundance of Rickettsia (C) and Candidatus_Nasuia (D) during WDV acquisition. NV: nonviruliferous leafhopper; V05: viruliferous leafhopper after 5 d AAP; V10: viruliferous leafhopper after 10 d AAP; V20: viruliferous leafhopper after 20 d AAP.
Discussion
High-throughput sequencing of insect gut microbial community has unveiled vital microbial functions such as cellulose degradation and essential amino acids synthesis that complement metabolic pathways of the host (Lee et al., 2017; Smith et al., 2017). However, there were few studies on the gut bacterial community of sap-sucking insects which transmit various viruses (Jupatanakul et al., 2014). Thus, we focused here on the gut bacterial community of P. alienus, which widely distributes in Europe, Asia and Africa and also transmits several cereal-infecting viruses including WDV. Previous studies on microbes in leafhoppers from subfamily Cicadellidae using traditional methods have identified members of the bacteria Sulcia, Sodalis-like and Pectobacterium as resident in the green leafhopper Cicadella viridis (Michalik et al., 2014) and Rickettsia in the green rice leafhopper Nephotettix cincticep (Noda et al., 2012). Recently, Kobialka et al. (2018) examined symbiotic microbes associated with 13 species of Deltocephalinae using traditional methods and found several highly abundant microorganisms. Here, we found 22 phyla and 249 genera bacteria in the gut of P. alienus from six regions in China using high-throughput sequencing. The bacteria composition and abundance varied among the geographical locations.
The gut bacterial community of P. alienus also clustered into different phylogenetic groups depending on the geographical location, suggesting that environment influences the diversity of the gut bacterial communities in the leafhoppers. Tianjin is a coastal city on the Bohai Sea with a typical semi-humid monsoon climate and less temperature and humidity changes than in inland areas. In contrast, the other five regions are far from the sea and have a continental monsoon climate. This means that climate may contribute to the differences in the gut bacterial communities between Tianjin and the other five populations, because the Tianjin population is located on an independent branch in the PCoA and UPGMA analysis. According to climatic data from the China National Meteorological Information Center,4 average annual rainfall was about 400–500 mm in the other five regions, but more than 650 mm in Tianjin. Further, the food sources of P. alienus in these five regions are similar, i.e., cereal crops and gramineous weeds. Mean annual temperature in Tianshui and Taiyuan is around 10°C, without any extreme high or low temperature during the year. However, mean annual temperature in Hancheng, Linfen and Baoding is between 12.6 and 13.5°C, with the extreme high around 41°C and low about -20°C, indicating a large annual temperature difference. The difference in temperature may explain the high similarity in the gut microbiota among populations of Hancheng, Linfen and Baoding. Previous studies have also shown that the gut microbiome of the scarab beetle (Holotrichia parallela) from 10 locations in China was also determined by environmental heterogeneity (Huang and Zhang, 2013). High temperature would also affect normal growth and survival of green stinkbug (Nezara viridula) by suppressing obligate gut bacterial symbionts (Kikuchi et al., 2016). Therefore, climate had a greater impact than geographic latitude on the composition and diversity of the gut bacterial community of P. alienus. Large differences in annual temperature and humidity could alter the bacterial composition and diversity, and assist insect survival in these local environments.
Psammotettix alienus may survive better in different regions through the contribution of various gut bacteria, but whether these bacteria influence virus transmission is not known. Various microbes in insect vector of animal or plant viruses are known to affect viral infection capacity, such as virus entry into host cells. (Moreira et al., 2009; Kliot et al., 2014). In Aedes albopictus, the symbiont Wolbachia did not affect replication of dengue virus, but it reduced the titre of viruses that entered host cells, thus leading to lower virus transmission efficiency (Mousson et al., 2012). Another symbiont Chromobacterium in A. aegypti can also reduce mosquito susceptibility to dengue virus infection, resulting in lower virus titre in the mosquito midgut cells (Jupatanakul et al., 2014). Other bacteria such as Rickettsia apparently enable B. tabaci to acquire more TYLCV from infected plants and promote the virus transmission (Kliot et al., 2014). Because some symbionts can alter virus titre in the insect vector and subsequent transmission efficiency to a new host, therefore we examined WDV transmission efficiency of six geographic P. alienus populations. The composition and abundance of bacterial communities differed among the six populations, but WDV transmission efficiency was similar among all populations. Although a much higher level of Rickettsia existed in the Taiyuan population than in the others, the virus transmission efficiency showed no significant difference among these populations. Comparing with the field populations, the gut bacterial diversity in laboratory reared leafhoppers decreased obviously, but the WDV transmission efficiency was not affected. Thus, the composition and diversity of the gut bacterial community in P. alienus apparently did not affect transmission characteristic as those in other insects.
A previous study showed that infection of the human pathogen Leishmania infantum could decrease the bacterial richness in the gut of the vector Lutzomyia longipalpis (Kelly et al., 2017). Another study indicated that Marek’s disease virus modifies the core gut microbiome of chickens during the early and late phases of viral replication by enriching several specific genera that might influence inflammation and immunosuppression of T and B cells (Perumbakkam et al., 2014). Interestingly, in the present study, WDV changed the composition and abundance of some gut bacteria which eventually recovered as the AAP lengthened. Relative abundance of Rickettsia had increased but Candidatus_Nasuia decreased by five AAP, then Rickettsia decreased and Candidatus_Nasuia increased until the levels resembled those of the nonviruliferous group by 10 AAP, indicating that WDV only impacted some bacteria for a short period. We propose that, unlike the effects of replicating pathogens, a persistent, non-propagative virus might only influence bacterial abundance in the early acquisition phase to enable their entry into the insect vector. Finally, these bacterial changes would progressively recover in the later stage.
In conclusion, resident bacterial community in the gut of P. alienus were distributed among 22 phyla and 249 genera; the main genera were Pectobacterium, Acinetobacter, Candidatus_Nasuia, Rickettsiella, Candidatus_Sulcia, Rickettsia and Wolbachia. The composition and abundance of the bacterial community in P. alienus varied among the six geographic regions apparently due to local conditions, whereas the diversity in bacterial functions was similar except for differences in the relative abundance of some functions. However, diversity in the gut bacterial composition did not affect transmission efficiency of the virus as those in other insect vectors. As a persistent, circulative, non-propagative virus, WDV only influenced the abundance of some gut bacteria during early acquisition; the change was dynamic and recovered later in the acquisition period. Our results help to elucidate the complex bacterial communities in leafhopper populations and provide important information for further studies on the complex interactions among the insect vector, microbial symbionts, and the vectored virus and on their coevolution.
Data Availability
All the data were already available under SRA accession number PRJNA495407.
Author Contributions
XW designed the experiments and reviewed the manuscript. XW, HW, and WL analyzed the data and wrote the manuscript. HW, WL and JK did preliminary data processing, analysis and manuscript correction. HW, YL and NW processed samples, isolated and sequenced DNA. XW, HW, NW and WL collected samples and edited the manuscript. All authors read and approved the final manuscript.
Funding
This work was funded by the National Key Research and Development Program of China: Inter-Governmental S&T Cooperation Proposal (2016YFE0131000), the National Natural Science Foundation of China (31871938) and the Research Program of the Ministry of Education, Youth and Sports of the Czechia (LTACH-17010) provided support.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Dr. B. E. Hazen (Willows End scientific editing and writing, United States) for critical reading and revising of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01144/full#supplementary-material
FIGURE S1 | Scheme of experiment design. (A) The composition of gut bacterial community and WDV transmission efficiency in the leafhopper populations collected from six geographic regions. (B) Composition and abundance of gut bacterial community in laboratory reared leafhoppers during WDV acquisition. (C) Comparison of the gut bacterial community and WDV transmission efficiency between laboratory reared and field collected leafhoppers.
FIGURE S2 | Location information of the sampling of Psammotettix alienus conducted in this study. TS: Tianshui; HC: Hancheng; LF: Linfen; TY: Tianjin; BD: Baoding; TJ: Tianjin.
FIGURE S3 | Rarefaction curves for ‘observed OTUs’ for six field populations. For all gut samples, the number of observed species plateaued as sequence counts increased. G1TS: Tianshui; G2HC: Hancheng; G3LF: Linfen; G4TY: Tianjin; G5BD: Baoding; G6TJ: Tianjin.
FIGURE S4 | Diversity indices of the bacterial communities in leafhoppers from the six locations in China. Differences in the Chao 1 index (A) and ACE index (B) for different geographic locations. G1TS: Tianshui; G2HC: Hancheng; G3LF: Linfen; G4TY: Tianjin; G5BD: Baoding; G6TJ: Tianjin.
FIGURE S5 | Comparison of predicted KEGG pathways of gut microbiome from six field leafhopper populations from China. Classification of gene functions (A). Significant difference analysis in relative abundance of metabolic pathways from microbiota functional genes between two populations (B–E). All statistically significant pathways were determined using Fisher test. p ≤ 0.01. G1TS: Tianshui; G2HC: Hancheng; G3LF: Linfen; G4TY: Tianjin; G5BD: Baoding; G6TJ: Tianjin.
Footnotes
References
Ben Guerrero, E., Soria, M., Salvador, R., Ceja-Navarro, J. A., Campos, E., Brodie, E. L., et al. (2016). Effect of different lignocellulosic diets on bacterial microbiota and hydrolytic enzyme activities in the gut of the cotton boll weevil (Anthonomus grandis). Front. Microbiol. 7:2093. doi: 10.3389/fmicb.2016.02093
Benkovics, A. H., Vida, G., Nelson, D., Veisz, O., Bedford, I., Silhavy, D., et al. (2010). Partial resistance to wheat dwarf virus in winter wheat cultivars. Plant Pathol. 59, 1144–1151. doi: 10.1111/j.1365-3059.2010.02318.x
Broderick, N. A., Raffa, K. F., and Handelsman, J. (2006). Midgut bacteria required for Bacillus thuringiensis insecticidal activity. Proc. Natl. Acad. Sci. U.S.A. 103, 15196–15199. doi: 10.1073/pnas.0604865103
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Cejanavarro, J. A., Vega, F. E., Karaoz, U., Zhao, H., Jenkins, S., Lim, H. C., et al. (2015). Gut microbiota mediate caffeine detoxification in the primary insect pest of coffee. Nat. Commun. 6:7618. doi: 10.1038/ncomms8618
Cheng, D., Guo, Z., Riegler, M., Xi, Z., Liang, G., and Xu, Y. (2017). Gut symbiont enhances insecticide resistance in a significant pest, the oriental fruit fly Bactrocera dorsalis (Hendel). Microbiome 5:13. doi: 10.1186/s40168-017-0236-z
da C Jesus, E., Marsh, T. L., Tiedje, J. M., de S Moreira, F. M. (2009). Changes in land use alter the structure of bacterial communities in Western Amazon soils. ISME J. 3, 1004–1011. doi: 10.1038/ismej.2009.47
Dillon, R. J., and Dillon, V. M. (2004). The gut bacteria of insects: nonpathogenic interactions. Annu. Rev. Entomol. 49, 71–92. doi: 10.1146/annurev.ento.49.061802.123416
Douglas, A. E. (2015). Multiorganismal insects: diversity and function of resident microorganisms. Annu. Rev. Entomol. 60, 17–34. doi: 10.1146/annurev-ento-010814-020822
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Edgar, R. C., Haas, B. J., Clemente, J. C., Quince, C., and Knight, R. (2011). UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 27, 2194–2200. doi: 10.1093/bioinformatics/btr381
Genta, F. A., Dillon, R. J., Terra, W. R., and Ferreira, C. (2006). Potential role for gut microbiota in cell wall digestion and glucoside detoxification in Tenebrio molitor larvae. J. Insect Physiol. 52, 593–601. doi: 10.1016/j.jinsphys.2006.02.007
Ghanim, M. (2014). A review of the mechanisms and components that determine the transmission efficiency of tomato yellow leaf curl virus (Geminiviridae; Begomovirus) by its whitefly vector. Virus Res. 186, 47–54. doi: 10.1016/j.virusres.2014.01.022
Gottlieb, Y., Zchori-Fein, E., Mozes-Daube, N., Kontsedalov, S., Skaljac, M., Brumin, M., et al. (2010). The transmission efficiency of tomato yellow leaf curl virus by the whitefly Bemisia tabaci is correlated with the presence of a specific symbiotic bacterium species. J. Virol. 84, 9310–9317. doi: 10.1128/Jvi.00423-10
Hogenhout, S. A., van der Wilk, F., Verbeek, M., Goldbach, R. W., and van den Heuvel, J. F. (2000). Identifying the determinants in the equatorial domain of Buchnera GroEL implicated in binding potato leafroll virus. J. Virol. 74, 4541–4548. doi: 10.1128/jvi.74.10.4541-4548.2000
Huang, S., and Zhang, H. (2013). The impact of environmental heterogeneity and life stage on the hindgut microbiota of Holotrichia parallela larvae (Coleoptera: Scarabaeidae). PLoS One 8:e57169. doi: 10.1371/journal.pone.0057169
Jia, D., Mao, Q., Chen, Y., Liu, Y., Chen, Q., Wu, W., et al. (2017). Insect symbiotic bacteria harbour viral pathogens for transovarial transmission. Nat. Microbiol. 2:17025. doi: 10.1038/nmicrobiol.2017.25
Jupatanakul, N., Sim, S., and Dimopoulos, G. (2014). The insect microbiome modulates vector competence for arboviruses. Viruses 6, 4294–4313. doi: 10.3390/v6114294
Kelly, P. H., Bahr, S. M., Serafim, T. D., Ajami, N. J., Petrosino, J. F., Meneses, C., et al. (2017). The gut microbiome of the vector Lutzomyia longipalpis is essential for survival of Leishmania infantum. mBio 8:e1121-16. doi: 10.1128/mBio.01121-16
Kikuchi, Y., Hayatsu, M., Hosokawa, T., Nagayama, A., Tago, K., and Fukatsu, T. (2012). Symbiont-mediated insecticide resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 8618–8622. doi: 10.1073/pnas.1200231109
Kikuchi, Y., Tada, A., Musolin, D. L., Hari, N., Hosokawa, T., Fujisaki, K., et al. (2016). Collapse of insect gut symbiosis under simulated climate change. mBio 7:e01578. doi: 10.1128/mBio.01578-16
Kim, M., Kim, Y., Qian, L., and Song, J. S. (2017). TeachEnG: a teaching engine for genomics. Bioinformatics 33, 3296–3298. doi: 10.1093/bioinformatics/btx447
Kliot, A., Cilia, M., Czosnek, H., and Ghanim, M. (2014). Implication of the bacterial endosymbiont Rickettsia spp. in interactions of the whitefly Bemisia tabaci with tomato yellow leaf curl virus. J. Virol. 88, 5652–5660. doi: 10.1128/JVI.00071-14
Kliot, A., and Ghanim, M. (2013). The role of bacterial chaperones in the circulative transmission of plant viruses by insect vectors. Viruses 5, 1516–1535. doi: 10.3390/v5061516
Knief, C., Ramette, A., Frances, L., Alonso-Blanco, C., and Vorholt, J. A. (2010). Site and plant species are important determinants of the Methylobacterium community composition in the plant phyllosphere. ISME J. 4, 719–728. doi: 10.1038/ismej.2010.9
Kobialka, M., Michalik, A., Szwedo, J., and Szklarzewicz, T. (2018). Diversity of symbiotic microbiota in deltocephalinae leafhoppers (insecta, Hemiptera, Cicadellidae). Arthropod Struct. Dev. 47, 268–278. doi: 10.1016/j.asd.2018.03.005
Koga, R., Bennett, G. M., Cryan, J. R., and Moran, N. A. (2013). Evolutionary replacement of obligate symbionts in an ancient and diverse insect lineage. Environ. Microbiol. 15, 2073–2081. doi: 10.1111/1462-2920.12121
Lee, J. H., Lee, K. A., and Lee, W. J. (2017). Microbiota, gut physiology, and insect immunity. Insect Immun. 52, 111–138. doi: 10.1016/bs.aiip.2016.11.001
Lindblad, M., and Waern, P. (2002). Correlation of wheat dwarf incidence to winter wheat cultivation practices. Agric. Ecosyst. Environ. 92, 115–122. doi: 10.1016/s0167-8809(01)00302-4
Liu, W., Hajano, J. D., and Wang, X. (2018). New insights on the transmission mechanism of tenuiviruses by their vector insects. Curr. Opin. Virol. 33, 13–17. doi: 10.1016/j.coviro.2018.07.004
Liu, Y., Du, Z., Wang, H., Zhang, S., Cao, M., and Wang, X. (2018). Identification and characterization of wheat yellow striate virus, a novel leafhopper-transmitted nucleorhabdovirus infecting wheat. Front. Microbiol. 9:468. doi: 10.3389/fmicb.2018.00468
Looft, T., Johnson, T. A., Allen, H. K., Bayles, D. O., Alt, D. P., Stedtfeld, R. D., et al. (2012). In-feed antibiotic effects on the swine intestinal microbiome. Proc. Natl. Acad. Sci. U.S.A. 109, 1691–1696. doi: 10.1073/pnas.1120238109
Magoc, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mar, T., Liu, W. W., and Wang, X. F. (2014). Proteomic analysis of interaction between P7-1 of Southern rice black-streaked dwarf virus and the insect vector reveals diverse insect proteins involved in successful transmission. J. Proteom. 102, 83–97. doi: 10.1016/j.jprot.2014.03.004
Michalik, A., Jankowska, W., Kot, M., Golas, A., and Szklarzewicz, T. (2014). Symbiosis in the green leafhopper, Cicadella viridis (Hemiptera, Cicadellidae). association in statu nascendi?. Arthropod Struct. Dev. 43, 579–587. doi: 10.1016/j.asd.2014.07.005
Moreira, L. A., Iturbe-Ormaetxe, I., Jeffery, J. A., Lu, G., Pyke, A. T., Hedges, L. M., et al. (2009). A Wolbachia symbiont in Aedes aegypti limits infection with dengue. chikungunya, and plasmodium. Cell 139, 1268–1278. doi: 10.1016/j.cell.2009.11.042
Mousson, L., Zouache, K., Arias-Goeta, C., Raquin, V., Mavingui, P., and Failloux, A. B. (2012). The native Wolbachia symbionts limit transmission of dengue virus in Aedes albopictus. PLoS Negl. Trop. Dis. 6:e1989. doi: 10.1371/journal.pntd.0001989
Najar, A., Khaled, M., Boudhir, H., and Safaa, G. K. (2000). Viral diseases of cultivated legume and cereal crops in tunisia. Phytopathol. Mediterr. 29,423–432.
Noda, H., Watanabe, K., Kawai, S., Yukuhiro, F., Miyoshi, T., Tomizawa, M., et al. (2012). Bacteriome-associated endosymbionts of the green rice leafhopper Nephotettix cincticeps (Hemiptera: Cicadellidae). Appl. Entomol. Zool. 47,217–225. doi: 10.1007/s13355-012-0110-1
Parks, D. H., Tyson, G. W., Hugenholtz, P., and Beiko, R. G. (2014). STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics 30, 3123–3124. doi: 10.1093/bioinformatics/btu494
Perumbakkam, S., Hunt, H. D., and Cheng, H. H. (2014). Marek’s disease virus influences the core gut microbiome of the chicken during the early and late phases of viral replication. FEMS Microbiol. Ecol. 90, 300–312. doi: 10.1111/1574-6941.12392
Qin, F., Liu, W., Wu, N., Zhang, L., Zhang, Z., Zhou, X., et al. (2018). Invasion of midgut epithelial cells by a persistently transmitted virus is mediated by sugar transporter 6 in its insect vector. PLoS Pathogen 14:e1007201. doi: 10.1371/journal.ppat.1007201
Rana, V. S., Singh, S. T., Priya, N. G., Kumar, J., and Rajagopal, R. (2012). Arsenophonus GroEL interacts with CLCuV and is localized in midgut and salivary gland of whitefly B. tabaci. PLoS One 7:e42168. doi: 10.1371/journal.pone.0042168
Redak, R. A., Purcell, A. H., Lopes, J. R. S., Blua, M. J., Mizell, R. F., and Andersen, P. C. (2004). The biology of xylem fluid-feeding insect vectors of Xylella fastidiosa and their relation to disease epidemiology. Annu. Rev. Entomol. 49, 243–270. doi: 10.1146/annurev.ento.49.061802.123403
Sakaki, T., Takeshima, T., Tominaga, M., Hashimoto, H., and Kawaguchi, S. (1994). Recurrence of ICA-PCoA aneurysms after neck clipping. J. Neurosurg. 80, 58–63. doi: 10.3171/jns.1994.80.1.0058
Sharon, G., Segal, D., Ringo, J. M., Hefetz, A., Zilber-Rosenberg, I., and Rosenberg, E. (2010). Commensal bacteria play a role in mating preference of Drosophila melanogaster. Proc. Natl. Acad. Sci. U.S.A. 107, 20051–20056. doi: 10.1073/pnas.1009906107
Sheik, C. S., Beasley, W. H., Elshahed, M. S., Zhou, X., Luo, Y., and Krumholz, L. R. (2011). Effect of warming and drought on grassland microbial communities. ISME J. 5, 1692–1700. doi: 10.1038/ismej.2011.32
Smith, C. C., Srygley, R. B., Healy, F., Swaminath, K., and Mueller, U. G. (2017). Spatial structure of the mormon cricket gut microbiome and its predicted contribution to nutrition and immune function. Front. Microbiol. 8:801. doi: 10.3389/fmicb.2017.00801
Wang, H., Wu, K., Liu, Y., Wu, Y., and Wang, X. (2015). Integrative proteomics to understand the transmission mechanism of Barley yellow dwarf virus-GPV by its insect vector Rhopalosiphum padi. Sci. Rep. 5:10971. doi: 10.1038/srep10971
Wang, J. F., Liu, Y. B., Wu, B. L., Xie, J. J., and Wang, X. F. (2008). Identification and analyses of the pathogen causing the wheat dwarf virus in hancheng of shaanxi province, China. Plant Prot. 37, 17–21.
Wang, Y., Mao, Q., Liu, W., Mar, T., Wei, T., Liu, Y., et al. (2014). Localization and distribution of wheat dwarf virus in its vector leafhopper, Psammotettix alienus. Phytopathology 104, 897–904. doi: 10.1094/PHYTO-09-13-0251-R
Young, E., Carey, M., Meharg, A. A., and Meharg, C. (2018). Microbiome and ecotypic adaption of Holcus lanatus (L.) to extremes of its soil pH range, investigated through transcriptome sequencing. Microbiome 6:48. doi: 10.1186/s40168-018-0434-3
Zhang, P., Liu, Y., Liu, W., Massart, S., and Wang, X. (2017). Simultaneous detection of wheat dwarf virus, northern cereal mosaic virus, barley yellow striate mosaic virus and rice black-streaked dwarf virus in wheat by multiplex RT-PCR. J. Virol. Methods 249, 170–174. doi: 10.1016/j.jviromet.2017.09.010
Zhang, X., Zhou, G., and Wang, X. (2010). Detection of wheat dwarf virus (WDV) in wheat and vector leafhopper (Psammotettix alienus Dahlb.) by real-time PCR. J. Virol. Methods 169, 416–419. doi: 10.1016/j.jviromet.2010.07.029
Keywords: European grass feeding leafhopper (Psammotettix alienus), 16S rDNA high-throughput sequencing, gut bacterial community, wheat dwarf virus (WDV), geographic location
Citation: Wang H, Wu N, Liu Y, Kundu JK, Liu W and Wang X (2019) Higher Bacterial Diversity of Gut Microbiota in Different Natural Populations of Leafhopper Vector Does Not Influence WDV Transmission. Front. Microbiol. 10:1144. doi: 10.3389/fmicb.2019.01144
Received: 06 March 2019; Accepted: 06 May 2019;
Published: 29 May 2019.
Edited by:
George Tsiamis, University of Patras, GreeceReviewed by:
Jia Dongsheng, Fujian Agriculture and Forestry University, ChinaElena Gonella, University of Turin, Italy
Copyright © 2019 Wang, Wu, Liu, Kundu, Liu and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenwen Liu, bGl1d2Vud2VuOThAMTYzLmNvbQ==; Xifeng Wang, eGZ3YW5nQGlwcGNhYXMuY24=