 Junrong Liang1†Zengqiang Kou2†Shuai Qin1Yuhuang Chen3
Junrong Liang1†Zengqiang Kou2†Shuai Qin1Yuhuang Chen3 Zhenpeng Li1Chuchu Li1,4Ran Duan1Huijing Hao5Tao Zha6
Zhenpeng Li1Chuchu Li1,4Ran Duan1Huijing Hao5Tao Zha6 Wenpeng Gu7
Wenpeng Gu7 Yuanming Huang1Meng Xiao1
Yuanming Huang1Meng Xiao1 Huaiqi Jing1
Huaiqi Jing1 Xin Wang1*
Xin Wang1*- 1State Key Laboratory of Infectious Disease Prevention and Control, Collaborative Innovation Center for Diagnosis and Treatment of Infectious Diseases – Chinese Center for Disease Control and Prevention, National Institute for Communicable Disease Control and Prevention, Beijing, China
- 2Shandong Provincial Centre for Disease Control and Prevention, Jinan, China
- 3Shenzhen Nanshan Maternity and Child Heath Care Hospital, Shenzhen, China
- 4Department of Pathogenic Biology, School of Medical Science, Jiangsu University, Zhenjiang, China
- 5Chang Ping Women and Children Health Care Hospital, Beijing, China
- 6Wuhu Municipal Centre for Disease Control and Prevention, Wuhu, China
- 7Yunnan Provincial Centre for Disease Control and Prevention, Kunming, China
Yersinia enterocolitica is a major agent of foodborne diseases worldwide. Prophage plays an important role in the genetic evolution of the bacterial genome. Little is known about the genetic information about prophages in the genome of Y. enterocolitica, and no pathogenic Y. enterocolitica prophages have been described. In this study, we induced and described the genomes of six prophages from pathogenic Y. enterocolitica for the first time. Phylogenetic analysis based on whole genome sequencing revealed that these novel Yersinia phages are genetically distinct from the previously reported phages, showing considerable genetic diversity. Interestingly, the prophages induced from O:3 and O:9 Y. enterocolitica showed different genomic sequences and morphology but highly conserved among the same serotype strains, which classified into two diverse clusters. The three long-tailed Myoviridae prophages induced from serotype O:3 Y. enterocolitica were highly conserved, shared ≥99.99% identity and forming genotypic cluster A; the three Podoviridae prophages induced from the serotype O:9 strains formed cluster B, also shared more than 99.90% identity with one another. Cluster A was most closely related to O:5 non-pathogenic Y. enterocolitica prophage PY54 (61.72% identity). The genetic polymorphism of these two kinds of prophages and highly conserved among the same serotype strains, suggested a possible shared evolutionary past for these phages: originated from distinct ancestors, and entered pathogenic Y. enterocolitica as extrachromosomal genetic components during evolution when facing selective pressure. These results are critically important for further understanding of phage roles in host physiology and the pathology of disease.
Introduction
As the most abundant microorganisms on the planet (Brussow and Hendrix, 2002; Olszak et al., 2017), phages can be divided into two kinds. The lytic phages contain a copy of the phage genome packaged in its capsid, which is built into its quaternary structure prior to lysing the host cell and its subsequent release. In contrast, the temperate phages integrate into bacterial genomes as prophages and represent an important source of genetic variation on bacterial evolution, such as producing toxins responsible for the virulence of major pathogens, producing cell adhesion molecules, nutrient uptake, immune response evasion, fimbriae, and others (Ikeda and Tomizawa, 1968; Briani et al., 2001; Chen and Lu, 2002; Abedon and Lejeune, 2007; St-Pierre and Endy, 2008; Ariff et al., 2015). These prophage-bacterial combinations are described as lysogenic bacteria or lysogens. With the development of high-throughput sequencing technology, many genomes have been sequenced, and numerous prophages have been identified. These results can further our understanding of roles in phage host physiology and the pathology of disease.
Yersinia enterocolitica is an important zoonotic pathogen leading to human and animal enteric infections and includes three lineages: avirulent strains belonging to biotype 1A, highly pathogenic strains of biotype 1B, and weakly pathogenic strains of biotypes 2–5 (Cornelis et al., 1998; Revell and Miller, 2001; Reuter et al., 2014; McNally et al., 2016). Compared with other countries, there is no pathogenic O:8 strains in China, only serotypes O:3 and O:9 Y. enterocolitica strains, which carry virulence determinants that can cause human infections (Wang et al., 2008, 2009). In many years of Y. enterocolitica surveillance in China, we found that the O:3 serotype strains have replaced O:9 strains and become the main pathogenic Y. enterocolitica (Wang et al., 2009; Liang et al., 2015). Currently, there are 15 Y. enterocolitica and 27 Yersinia phages of full-length genomic sequences in the National Center for Biotechnology Information (NCBI) database. Genomic sequencing of Y. enterocolitica has made it possible to identify prophages and perform comparative genomic analysis of phage sequences. To the best of our knowledge, several O:3 and O:9 Yersinia lytic bacteriophages have been described and some can subtype Y. enterocolitica (Leon-Velarde et al., 2016; Jun et al., 2018; Salem and Skurnik, 2018). The lytic phages phiYe-F10 and φYeO3-12 displayed specificity for Y. enterocolitica O:3 (Pajunen et al., 2003; Liang et al., 2016), phage φR1-37 has a broader host range within Y. enterocolitica also it can infect some Y. intermedia and Y. similis strains (Kiljunen et al., 2005), and phage vB_YenP_AP5 can form plaques only on Y. enterocolitica serotypes O:3, O:2, and O:1 (Leon-Velarde et al., 2014). However, there have not been any reports describing the prophages of main pathogenic O:3 and O:9 Y. enterocolitica, and only one Y. enterocolitica prophage has been isolated. The temperate phage PY54 isolated from non-pathogenic Y. enterocolitica O:5 strain has a lambda-like morphology and infects the O:5 strains and pathogenic O:5,27 strains (Hertwig et al., 2003a,b). In this study, we first induced 6 novel prophages from serotype O:3 and O:9 pathogenic Y. enterocolitica and performed comparative genomic and phylogenetic analyses with other phages.
Materials and Methods
Bacterial Strains and Induction of Prophages
The lysogenic phages were induced using mitomycin C (Ivánovics et al., 1976; Faelen et al., 1993). Strains were grown overnight on brain heart infusion [BHI] Agar (Oxoid) at 27°C. The colonies were transferred to BHI culture medium and shaken to OD600 = 0.4∼0.8 at 27°C, followed by four additions to BHI culture medium for further replication. The experimental groups were induced with 0.5 μg/ml mitomycin C (Sigma) and the control group received nothing and was incubated for 8–14 h at 27°C with gentle shaking. If the difference of OD600 between the experimental group and control group was equal or greater than 0.5, it was suggested that bacteriophage particles may be induced. We confirmed the prophages with a double-layer plaque assay at 25°C. The supernatant was filtered through a sterile disposable filter of 0.45 μm pore size (Thermo Fisher Scientific, Mississauga, ON, Canada) and standard double agar overlay plaque assays were used to identify plaques.
Electron Microscopy
The presence of induced phages were confirmed by transmission electron microscopy. Filtered phage lysates were pelleted at 25,000 ×g for 1 h at 4°C, using a Beckman high-speed centrifuge and a JA-18.1 fixed-angle rotor (Beckman, Palo Alto, CA, United States). The phage pellets were washed twice under the same conditions in neutral 0.1 M ammonium acetate. The final phage sediment was re-suspended in 150 μL of SM-buffer supplemented with 5 mM CaCl2. Samples were then deposited onto carbon-coated Formvar films on copper grids and stained with 20 μl of 2% potassium phosphotungstate (PT, pH 7.2). The dye was removed with filter paper and the sample was air dried and examined under a Tecnai G2 F20 transmission electron microscope (FEI, Hillsboro, OR, United States), operating at 120 keV. Images were collected and analyzed using Digital MicrographTM Software (Gatan, Pleasanton, CA, United States).
Isolation of Phage DNA
To separate the phage, the phage inducing lysate was centrifuged at 10,000 ×g for 15 min at 4°C and the supernatant was filtered through a 0.22 μm low protein binding filter (Millipore, United States). Contaminating nucleic acids in the supernatant were digested with pancreatic DNase I and RNase A, and each was added to obtain a final concentration of 10 μg/mL (Sigma-Aldrich Canada Ltd., Oakville, ON, United States) and incubated for 15 min at 37°C. DNA isolation was then performed according to Molecular Cloning: A Laboratory Manual, Third Edition (Sambrook J., Russell D.W. Molecular Cloning: A Laboratory Manual. 3rd ed. Volme 1 Cold Spring Harbor Press; Cold Spring Harbor, NY, United States: 2001), with minor modifications. For phage DNA purification the suspension was extracted twice with an equal volume of phenol-chloroform, once with chloroform and precipitated with ethanol. The pellets were washed in 70% ethanol, vacuum dried, and resuspended in 20 ml distilled water. PCR verification was performed on 1 ml of the phage DNA preparation.
Genome Sequencing and Assembly
We tested the quality of the whole genomes of the phages with Qubit3.0 (Life Technology, United States) followed by NEB Next Ultra DNA Library Prep Kit for library construction. Whole genome sequencing was carried out using the Illumina HiSeq2500 (PE25) genome analyser. Generated reads were assembled using SPAdes. The assembled contigs were annotated using Rapid Annotation using Subsystem Technology (RAST, version 2.0) and PHASTER web server (PHAge Search Tool Enhanced Release1) (Fancello et al., 2011; Arndt et al., 2016, 2017).
Prophage Sequence Detection
The complete genomes of Y. enterocolitica were downloaded from NCBI database, which were analyzed by PHASTER to identify the presence of prophages. The prophages were identified as intact, questionable or incomplete by PHASTER. Intact phages were estimated as complete functional phages, and functionality needs to be tested with plaque formation analyses. Questionable and incomplete phages do not contain sufficient prophage genes considered as unfunctional phages (Arndt et al., 2017).
We analyzed the strains of O:9 serotype Y. enterocolitica with the designed O:9 serotype phage-specific primers (F: TCAGGTAGTCTGACTTGACCGA, R: TCACGCTGGATGTGCCTATTGT).
Comparative Genomic Analyses
Data from 21 available whole Yersinia (pro)phages genomic sequences and 85 of the other Enterobacteriaceae phages were downloaded from the NCBI website and analyzed with the 6 Yersinia enterocolitica prophage sequences in this study (Supplementary Table S2). The comparison heat map of the phage genome structure was drawn as a flow diagram: first the non-redundant genes were obtained for all the genomes using cd-hit; second, an m∗n matrix (m indicated the number of pangenes, n indicated the number of phages) was constructed. If the individual gene existed on the specific phage, the corresponding position of the matrix was tagged as 1; otherwise, it was tagged as 0. Euclidean distance was computed between the different phages based on the matrix, and a new matrix n∗n was constructed. The item of the matrix indicated the distance of the corresponding phage. Last, the heat map was constructed using the pheatmap package. Average nucleotide identity (ANI) of the phage genomes were calculated using the BLASTn algorithm in JSpecies 1.2.1 (Richter and Rossello-Mora, 2009).
Comparative genomic analyses were carried out using the BLAST software with an e-value of 1e-2, and the alignments of >1 kb were retained. PRODIGIAL v2.6 was used to predict the CDS for all of the phage genomes. A Perl script was used to draw the phage genome structure.
Results
Induction of Lysogenic Bacteriophages and Electron Micrographs of Phage Particles
Altogether, 6 bacteriophages were induced from Y. enterocolitica with mitomycin C (Table 1). The induced prophages are able to form turbid plaques on Y. enterocolitica isolates that do not harbor the cognate prophage, but they do not infect the Yersinia pestis, Yersinia pseudotuberculosis, Escherichia coli, and Salmonella isolates we tested. The bacteriophages were negatively stained and examined using transmission electron microscopy. These phages showed hexagonal outlines, indicating their icosahedral nature. However, approximately 95% of the O:3 phage particles released and directly analyzed in the crude lysate were defective, consisting of DNA-filled heads without tails. The phages YeP1, YeP2 and YeP3 were induced from the 3/O:3 Y. enterocolitica serotype and were characterized by icosahedral symmetry head (ranged from 56.0 nm to 64.0 nm) and long (ca. 140–160 nm), flexible, contractile tails which belonged to Myoviridae family. YeP4, YeP5, and YeP6 were induced from the O:9 Y. enterocolitica serotype, with icosahedral head (ranged from 52.0 nm to 58.0 nm) and very short non-contractile tail, and the morphological group corresponds to the Podoviridae family (Figure 1).
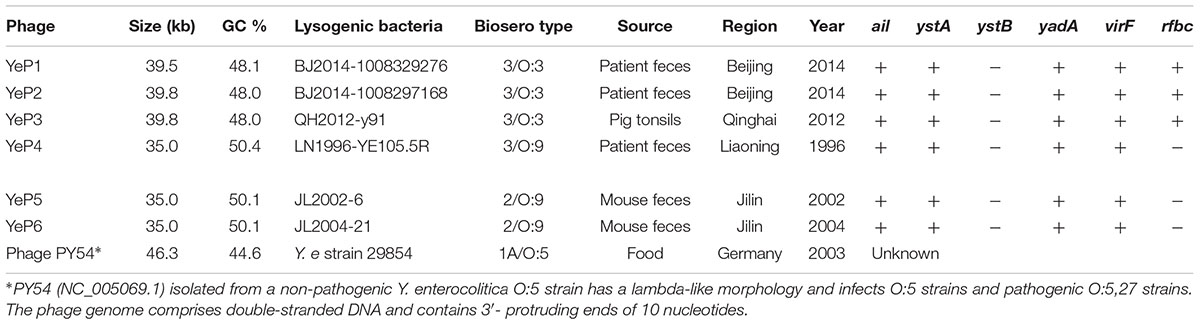
Table 1. The information of lysogenic bacteria from which Y. enterocolitica temperate bacteriophages were induced.
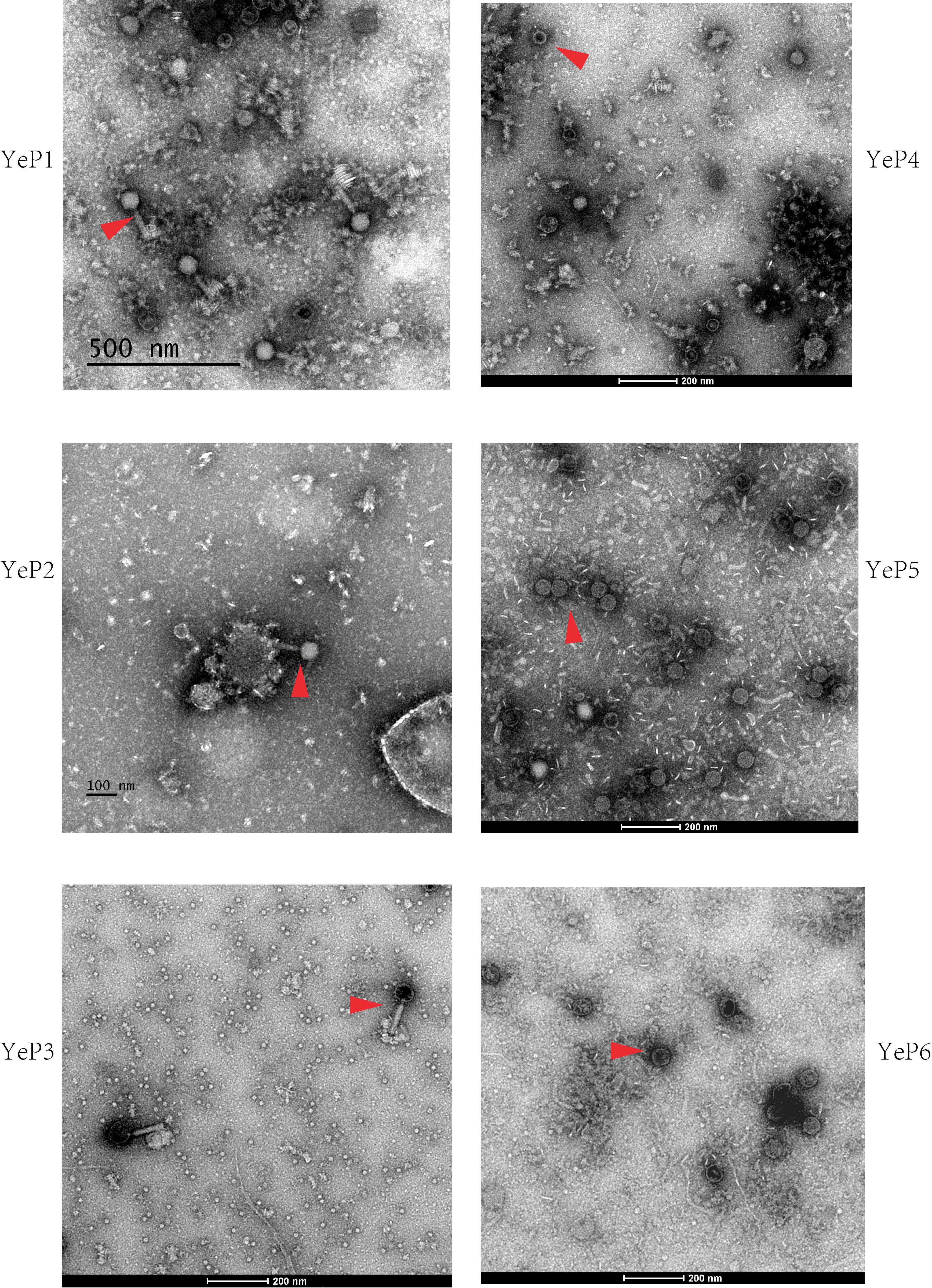
Figure 1. The electron micrographs of the six prophages. The phages are negatively stained with 2% potassium phosphotungstate and showed with red arrows. Scale bar indicates size in nm.
Prophages Genome Analysis
The phages that resulted from induction of the prophages in the lysogens were purified and their DNA was sequenced. Altogether, 1.5G raw data was available for each phage with a sequencing depth of >8000×. The cleaned raw data obtained had Q30 >80% after quality checks for sequencing. We assembled the raw data using SPAdes genome assembler version 3.10.1. Each of the phage genomes was assembled as a circular molecule when the sequencing was completed. The phage genome sizes ranged from 34,969 bp to 39,751 bp, and the G+C contents varied between 48.0 and 50.4% (Table 1), similar to the 48.5 ± 1.5% reported for their Y. enterocolitica hosts. Phage YeP1 induced from strain BJ20141008329276 had a total genome length of 39,492 bp; YeP2 induced from BJ20141008297168 had a total genome size of 39,751 bp; YeP3 induced from strain QH2012-Y91 with 39,751 bp. Features of the open reading frames (ORFs) of each prophage were listed on Supplementary Table S1. YeP1 phage group from serotype O:3 strains with 61 proposed ORFs and three tRNA were predicted, and predicted functions were attributed to 38 of the ORFs based on similarities of the predicted products to the known proteins. Phage YeP4 induced from strain YE105.5R had a genome length of 35,029 bp; YeP5 and YeP6 induced from strain JL2002-6 and JL2004-21 had genome length of 34,969 bp, both with 39 ORFS predicted, of which 16 had predicted functions. The genomic structure of O:3 and O:9 Y. enterocolitica prophages were shown as Figure 2. PY54 was isolated from a non-pathogenic Y. enterocolitica O:5 strain and was the first temperate Yersinia phage that had been sequenced and characterized by Hertwig et al. (2003a,b).
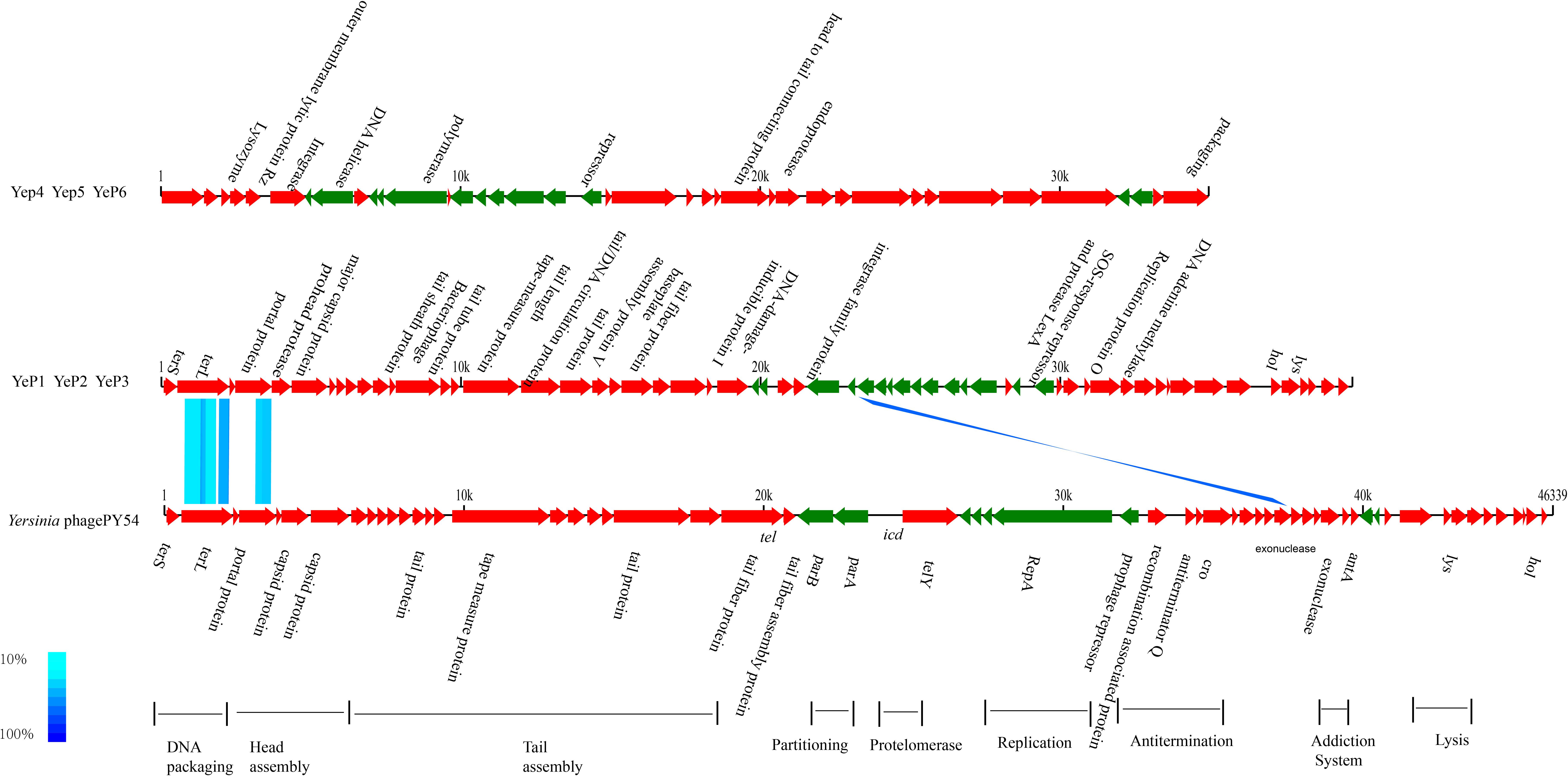
Figure 2. Genomic comparison between prophages from O:3/O:9 Yersinia enterocolitica strain and Phage PY54. Each arrow identifies an open reading frame. Color shading was used to show the amino acid identity between the two ORFs. The absence of shading indicates no significant similarity. The phage genome structure were classified into two groups: prophage YeP1, YeP2 and YeP3 were from O:3 Y. enterocolitica; and YeP4, YeP5 and YeP6 were from O:9 Y. enterocolitica. The genome sequence of Y. enterocolitica bacteriophage PY54 was from GenBank (Accession number NC_005069).
Comparative Genomic Analyses
The six Y. enterocolitica prophage genome sequences showed little similarities with the previously reported phages (Figures 3, 4 and Supplementary Figure S1). Phages YeP1, YeP2, and YeP3 induced from the O:3 Y. enterocolitica serotype clustered together with an identity of more than 99.99%; the three O:9 Y. enterocolitica serotype lysogenic phages (YeP4, YeP5, and YeP6) were identity up to 99.93% (Figure 3). Little similarity existed between the lysogenic phages from O:3 and those from the O:9 serotype of Y. enterocolitica (Figures 2, 3). The comparative genomic analyses showed that the Yersinia phages were sorted into five large clusters (cluster A, cluster B, cluster C, cluster D, and cluster E) and 10 single clusters based on genomic similarities (Figure 3 and Supplementary Figure S1). Cluster A contained three O:3 prophages: YeP1, YeP2, and YeP3; cluster B contained three O:9 lysogenic phages YeP4, YeP5 and YeP6; and cluster C contained three O:3 serotype-specific virulent phages: phiYeO3-12, Yersinia phage vB_YenP_AP5 and Yersinia Phage phiYe-F10. Y. enterocolitica lysogenic phage PY54 showed 61.72% similarity to the O:3 Y. enterocolitica lysogenic phages (Figure 3). However, the Y. enterocolitica lysogenic phage PY54 formed a separate cluster. Y. pestis phages were divided into two large clusters: cluster D contained YepΦ, Berlin, Yepe2, and YpP-G, cluster E contained YpP-Y, YpP-R, YpsP-G, and ΦA1122, and the rest were all sepereted (Figure 4). The phage genome sequence average nucleotide identity showed the similarity results (Figure 3).
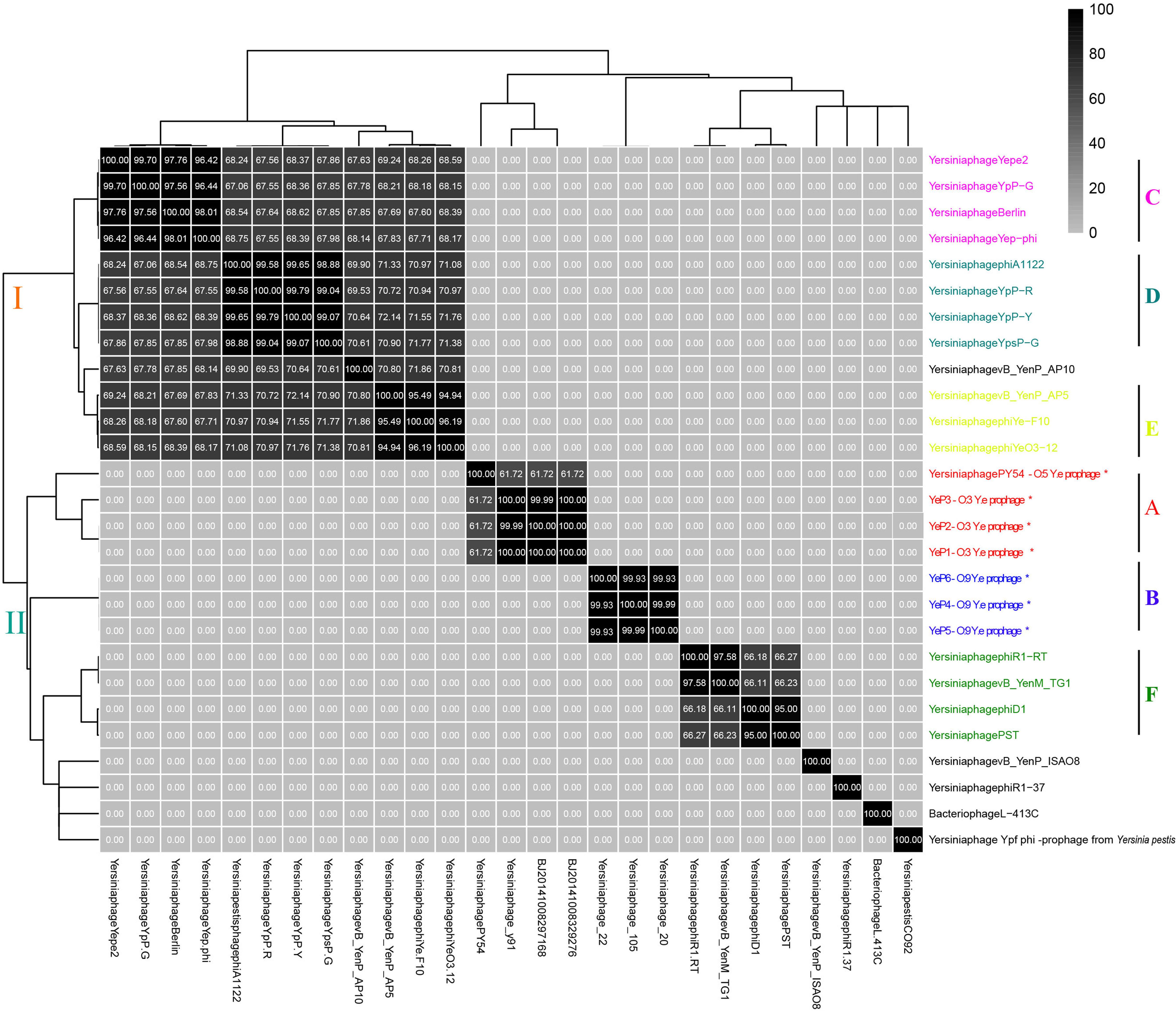
Figure 3. The sequence average nucleotide identity of phage genomes. ∗Indicates the temperate phage of Yersinia.
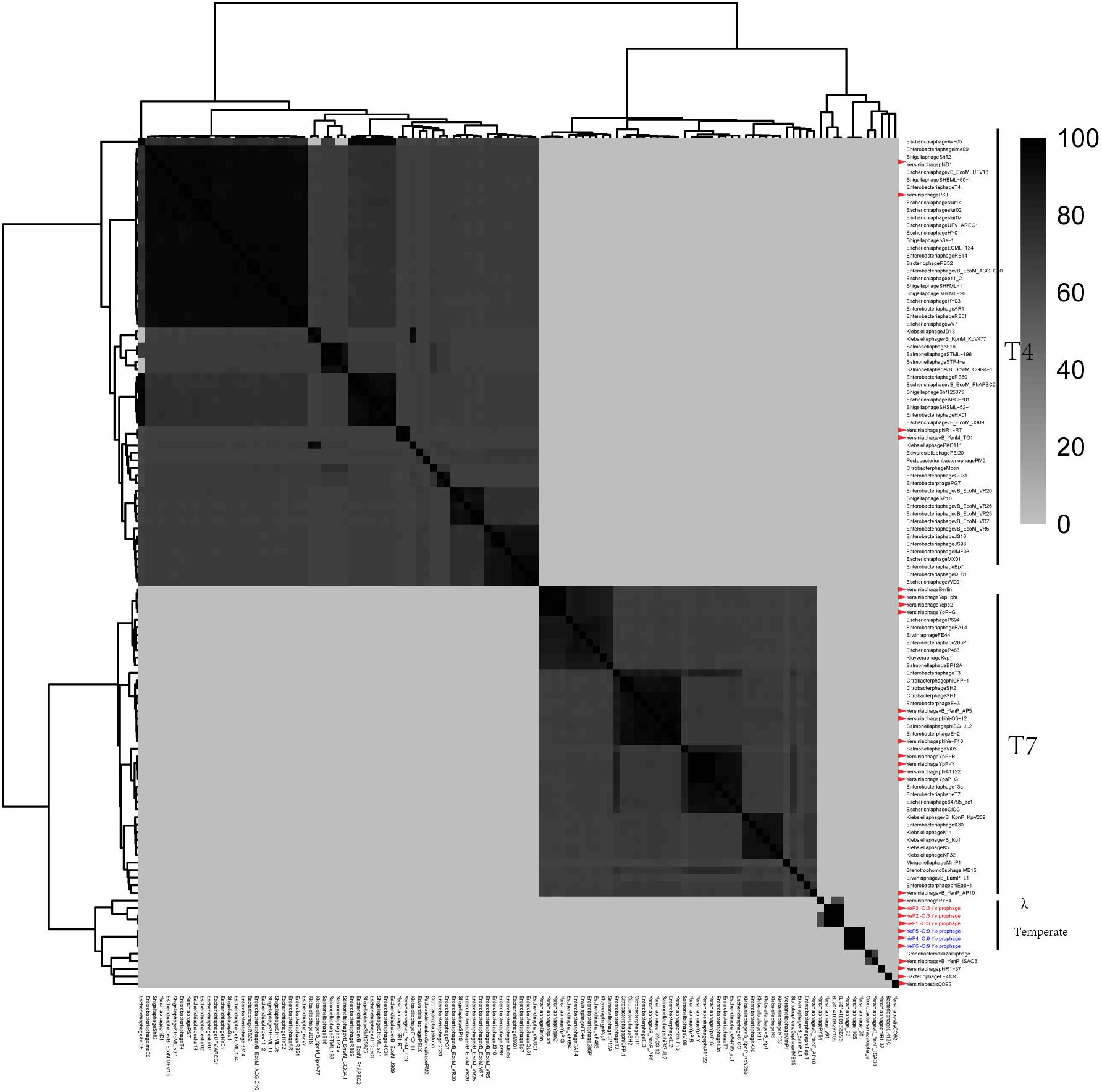
Figure 4. The comparative genomic structure analyses of all Yersinia phages and some of the Enterobacteriaceae phages.
The closely related phage of O:3 Y. enterocolitica prophage was prophage PY54 (ANI: 61.72%) and shared the same new branch in the phylogenetic tree (Figure 3 and Supplementary Figure S2). We identified homology with only very short protein fragments of Yersinia phages PY54 compared to YeP2 prophage, and mauve alignment showed 1,326 SNPs between them. The peg.2 (533–2230 bp) of YeP2 was similar to the Phage terminase of PY54 (PY54p02, protein_id: NP_892047.1), with identity 63%; the peg.3 (23208–23787 bp) of YeP2 was similar to portal protein of PY54 (PY54p03, protein_id: NP_892048.1) with identity 71%; and the peg.33 (23229–23780 bp) of YeP2 was similar to exonuclease (PY54p50, protein_id: NP_892096.1) with identity 82%. The proteins encoded by the remaining ORFs had no homology with other known phages proteins.
Identified the Prophage-Like Elements of Yersinia enterocolitica
The prophages were found among all of the 15 genomes of Y. enterocolitica from GenBank with PHASTER. Altogether, 117 prophage-like elements were identified with 55 intact prophages and 62 defective prophages. Their size ranged from 6 kb to 102.9 kb. The average of prophages per genome for Y. enterocolitica was 7.8.
Six prophage-like elements were identified within the whole genomes of Yersinia enterocolitica subsp. palearctica 105.5R(r) (CP002246.1) (Table 2). The genome sizes of prophages ranged from approximately 11.2–54.9 kb, and the GC content varied between 41.17 and 48.87%. Only two prophage regions were predicted intact with the region’s total scores 150 and 116, and the remaining four were incomplete or questionable. The lysogenic bacteriophage YeP4 was induced from YE105.5R genome (CP002246.1), the coordinates was located from 1584464 to 1619508, which determined by BLASTN with sequences alignment. The sequence of YeP4 (from 3500 to 35029) was highly homologous to YE105.5R genome (from 1584464 to 1619508) with nucleotide identities higher than 99.99%. And the sequence of YeP4 (from 1 to 3518) was identical to YE105.5R genome (from 1615991 to 1619508).
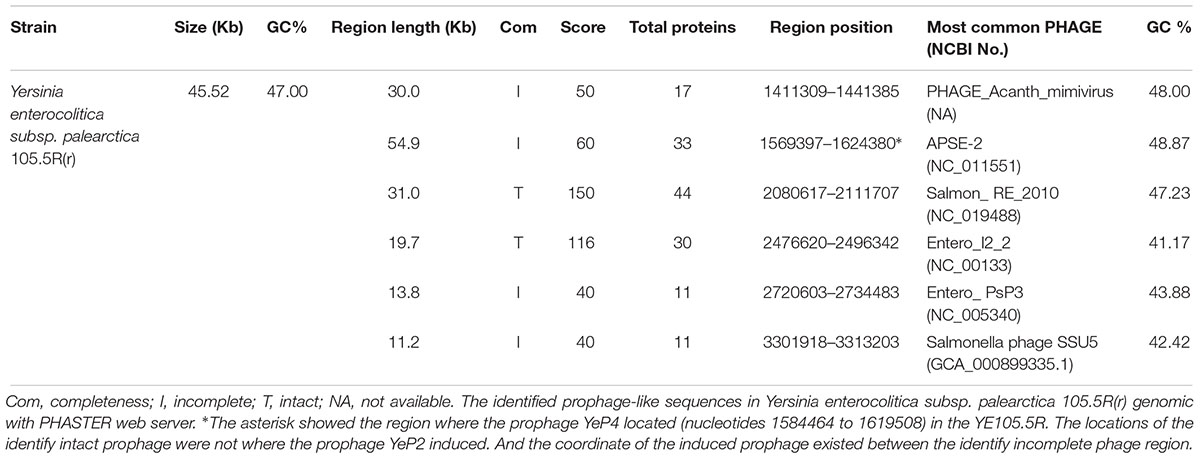
Table 2. Yersinia enterocolitica subsp. palearctica 105.5R(r) genomic prophage prediction results.
Phage Genome Sequences and Accession Numbers
The complete sequences of YeP1, YeP2, YeP3, YeP4, YeP5, and YeP6 were submitted to the NCBI databank under the Accession numbers as follows: MK733259, MK733260, MK733261, MK733262, MK733263, and MK733264, respectively. All of the sequencing data assembled were linear double strand DNA.
Discussion
As we all know, prophages can play particularly important roles in bacterial evolution since phage-mediated mobility can result in faster gene evolution through increased rates of mutation, recombination, or adaptation (Casjens, 2003; Labrie et al., 2010). In addition, prophages also have an intimate association with the novel phenotypic properties of bacterial hosts, such as pathogenicity and genomic variation (Abedon and Lejeune, 2007; Fortier and Sekulovic, 2013; Pleska et al., 2018). Little is known about the genetic information of prophages of Y. enterocolitica, a major pathogen of human intestinal disease. In this study, we induced and isolated 6 prophages from Y. enterocolitica, which had few similarities compared to the genomic sequences of previously reported phages, suggesting that they were novel Yersinia phages and have considerable genetic diversity. Comparative genomic and phylogenetic analyses of prophage sequences revealed that the 27 Yersinia prophages could be sorted into 5 large clusters and 8 separate clusters. The three O:3 Y. enterocolitica prophages are from a common ancestor, with a hexagonal capsid, a long non-contractile tail and a sequence identity more than 99%, and as such, were denoted group A (Figure 3 and Supplementary Figure S1). The three Podoviridae O:9 Y. enterocolitica prophages, clustered as group B. However, there was no sequence similarities between group A and B, indicating that during the evolution of the O:3 and O:9 serotypes of Y. enterocolitica, they obtained different prophages that persistently presented in the genome as extrachromosomal genetic elements under selective pressure. In this study, the prophage sequence diversity in different Y. enterocolitica serovars, the findings presented suggest a possible different lysogenesis evolutionary past for these strains. Salmonella enterica prophage sequence profiles can reflected genome diversity and can be used for high discrimination subtyping (Mottawea et al., 2018). Prophages have also been used for bacterial typing in E. coli, Streptococcus pneumonia, and Bacillus anthracis based on their ubiquitous and unique features (Sozhamannan et al., 2006; Romero et al., 2009; Kwon et al., 2013).
Prophage PY54 shared little nucleotide identity (61.72%) with prophage YeP1 and YeP2 (Figure 3) and could not be grouped into a cluster according to genomic similarity (Supplementary Figure S1). Yersinia phages and other T4 and T7 Enterobacteriaceae phages clustered together in comparative genomic analyses (Grose and Casjens, 2014; Figure 4), which implied that the Y. enterocolitica phage had a genetic relationship, to a certain extent, compared to other Enterobacteriaceae phages. The virulent phages of Y. enterocolitica O:3 serotypes, Yersinia phage phiYeO3-12, Yersinia phage phiYe-F10, and Yersinia phage vB_YenP_AP5, had a high genomic similarity and clustered together (Liang et al., 2016).
Among the 8 sequenced strains of Y. pestis virulent phages, Berlin, ΦA1122, Yepe2, YpP-G, Yep-phi, YpP-Y, YpP-R and YpsP-G all belonged to the T7 phage family, Podoviridae and Caudovirales, with the same size of the hexagonal structure head and a short non-stretching conical tail (Garcia et al., 2003; Schwudke et al., 2008; Zhao et al., 2011; Rashid et al., 2012). Although they are all double-stranded DNA viruses with the same genome size, alignments showed that the eight phages clustered into two groups: YepΦ group (YepΦ, Berlin, Yepe2, and YpP-G) and ΦA1122 group (YpP-Y, YpP-R, YpsP-G, and ΦA1122) (Figure 3). Interestingly, they exhibit a different host range (Filippov et al., 2012): ΦA1122 and YpP-G cannot infect some strains of Y. pseudotuberculosis, while YpP-Y, YpP-R, YpsP-G, and YpsP-PST can. In addition, YpP-Y, YpP-R, YpsP-G, YpsP-PST and ΦA1122 can also infect E. coli ATCC35401, Klebsiella pneumoniae env17, and Shigella sonnei S43-46 (probably as the common receptors for Y. pestis and Shigella) (Garcia et al., 2003). To date, the molecular interactions between bacteriophages and Y. pestis have been studied. The LPS and two OMPs (Ail and OmpF) are currently recognized as the receptor for bacteriophages of Yersinia pestis (Filippov et al., 2011; Kiljunen et al., 2011).
The genome of the lysogenic bacteria YE105.5R was completely sequenced by our laboratory (GenBank: CP002246.1) (Wang et al., 2011). The locations of the identify intact prophage were not where the prophage YeP2 induced. And the coordinate of the induced prophage existed between the identify incomplete phage region. The lysogenic bacteriophage YeP4 was induced from YE105.5R genome (CP002246.1), the coordinates was located from 1584464 to 1619508, which determined by BLASTN with sequences alignment. The sequence of YeP4 (from 3500 to 35029) was highly homologous to YE105.5R genome (from 1584464 to 1619508) with nucleotide identities higher than 99.99%. And the sequence of YeP4 (from 1 to 3518) was identical to YE105.5R genome (from 1615991 to 1619508). The YeP4 genome integrated into the YE105.5R chromosomal sites of YE105_RS07025 (protein_id = “WP_013649519.1”) and YE105_RS07220 (protein_id = “WP_013649558.1”). Blast screen of the NCBI database showed that, much to our surprise, YeP4 had similarities of up to 99% with sequences of Y. enterocolitica KNG22703 (GenBank: CP011286.1) (Range: 1374407–1403830), Y. enterocolitica 2516–2587 (GenBank: CP009838.1) (Range: 3262942–3292365) and Y. enterocolitica W22703 biovar 2, serovar O:9 (GenBank: FR718687.1) (Range: 1–29424). It is noteworthy that these strains were all O:9 serotypes, which indicated that prophage YeP4 can be found in the genomes of many O:9 serotype Y. enterocolitica strains from diverse localities. We analyzed 24 strains of O:9 serotype Y. enterocolitica with the designed O:9 serotype phage-specific primers, with 14 being positive. After that, we sequenced the genomes of two of these lysogenic bacteriophages, YeP5, and YeP6. The sequencing results showed that the identity of phage YeP5, YeP6, and YeP4 sequences was as high as 99.9%, implying a high similarity and prevalence of the O:9 serotype prophage. It is possible that, during the evolution of Y. enterocolitica O:9 serotype strains, they obtained the same phage due to selection pressure. However, YeP1 phage genomes shared low nucleotide identity with other available Yersinia genome sequences in GenBank.
In the 15 genomes analyzed 55 complete prophages were identified (data not show). YE105.5R contained 6 prophage regions, with 2 complete phages with intact genomes (2080617–2111707 and 2476620–2496342), and the remaining 4 prophages were incomplete or questionable (1411309–1441385, 1569397–1624380, 2720603–2734483, and 3301918–3313203). PHASTER web server results showed that prophage-like elements were identified in every complete Y. enterocolitica genomic sequence from the NCBI database. Interestingly, the induced lysogenic phage genome of YeP4 was located at 1584464–1619508 in YE105.5R. This position corresponds to an incomplete prophage region in the phaster prediction, with a score of 50 points; however, no prophages were induced in the two intact regions (scores 150 and 116) (Table 2). Thus, we concluded that the predicted regions of the prophages on phaster were inconsistent with the actual lysogenic phage regions, and the predicted intact phage regions were not actually the functional prophage regions.
The primary determinant in the infection of a bacterial host by a bacteriophage is the adsorption of the phage receptor binding proteins, e.g., the tail fiber, to the host receptor (Morita et al., 2002). Conversely, bacteriophages are capable of rapid adaptive responses to evolutionary changes in their hosts. As a defensive measure, evolution can modify the phage’s receptors binding proteins to achieve infection and kill the resistant bacterium. Its specificity depends on the complementarity between the phage receptor binding protein and the surface structure of the host bacterial receptor (Heilpern and Waldor, 2000). There may be a strong selection for fixing adaptive mutations, and tail fiber proteins can recognize a variety of host bacterial receptors (such as outer membrane proteins or lipopolysaccharide, LPS). Our previous studies have found that there is a correlation between tail fiber sequence and target serotype: the phage phiYe-F10, phiYeO3-12 and vB_YenP_AP5 display specificity for Y. enterocolitica O:3 were clustered together (Liang et al., 2016). The alignment showed high similarity among the prophage tail fiber proteins from the same serotype, which were consistent with earlier results. However, there are few similarities between different serotypes.
To the best of our knowledge, this is the first systematic analysis of temperate phages from pathogenic O:3 and O:9 Y. enterocolitica. The genomes and genetic information of Y. enterocolitica prophages play an important role in the analysis of the genetic evolution of the bacterial genome. This study showed that these novel genome of Y. enterocolitica prophages had diversity and highly conserved among the same serotype strains. The specificity of the phage tail fiber protein played a key role in the identification of surface receptors of different host bacteria. We suggested that the different tail fiber proteins from the O:3, O:9 serotype Y. enterocolitica lysogenic bacteriophages result in a difference in the specificity of the two phage hosts. These phages could also be used for specific and efficient detection of pathogenic Y. enterocolitica.
Author Contributions
JL, ZK, ZL, and XW conceived and designed the experiments, sequenced, assembled, and annotated the genomes of prophages. XW and HJ supervised the study and contributed to manuscript writing. YC, WG, SQ, and YH performed the data analysis and assisted in the preparation of figures and text. CL and MX participated in the data interpretation. JL and ZL contributed to bioinformatic analysis. JL, RD, HH, TZ, and YC contributed to manuscript writing. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation of China (General Project Nos. 31500117 and 81470092).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Liuying Tang and American Journal Experts for their critical reading and helpful comments on our manuscript (Sub ID N8L3RSVD).
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01184/full#supplementary-material
FIGURE S1 | Similarity matrix of 27 Yersinia phages and prophages based on the presence/absence of genes. The heatmap was generated based on the number of proteins shared by phages. Deeper shade of blue indicated a closer relationship.
FIGURE S2 | The phylogenetic tree of the 27 Yersinia phages genome sequences using the program VICTOR (https://ggdc.dsmz.de/victor.php).
TABLE S1 | Features of the open reading frames (ORFs) of six prophages.
TABLE S2 | The (pro)phages used in this study.
Footnotes
References
Abedon, S. T., and Lejeune, J. T. (2007). Why bacteriophage encode exotoxins and other virulence factors. Evol. Bioinform. Online 1, 97–110.
Ariff, A., Wise, M. J., Kahler, C. M., Tay, C. Y., Peters, F., Perkins, T. T., et al. (2015). Novel Moraxella catarrhalis prophages display hyperconserved non-structural genes despite their genomic diversity. BMC Genomics 16:860. doi: 10.1186/s12864-015-2104-2101
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). Phaster: a better, faster version of the phast phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Arndt, D., Marcu, A., Liang, Y., and Wishart, D. S. (2017). Phast, Phaster and Phastest: tools for finding prophage in bacterial genomes. Brief Bioinform. [Epub ahead of print]
Briani, F., Deho, G., Forti, F., and Ghisotti, D. (2001). The plasmid status of satellite bacteriophage P4. Plasmid 45, 1–17. doi: 10.1006/plas.2000.1497
Casjens, S. (2003). Prophages and bacterial genomics: what have we learned so far? Mol. Microbiol. 49, 277–300. doi: 10.1046/j.1365-2958.2003.03580.x
Chen, F., and Lu, J. (2002). Genomic sequence and evolution of marine cyanophage P60: a new insight on lytic and lysogenic phages. Appl. Environ. Microbiol. 68, 2589–2594. doi: 10.1128/aem.68.5.2589-2594.2002
Cornelis, G. R., Boland, A., Boyd, A. P., Geuijen, C., Iriarte, M., Neyt, C., et al. (1998). The virulence plasmid of yersinia, an antihost genome. Microbiol. Mol. Biol. Rev. 62, 1315–1352.
Faelen, M., Merlin, C., Geuskens, M., Mergeay, M., and Toussaint, A. (1993). Characterization of a temperate phage hosted by Alcaligenes eutrophus strain A5. Res. Microbiol. 144, 627–631. doi: 10.1016/0923-2508(93)90065-a
Fancello, L., Desnues, C., Raoult, D., and Rolain, J. M. (2011). Bacteriophages and diffusion of genes encoding antimicrobial resistance in cystic fibrosis sputum microbiota. J. Antimicrob. Chemother. 66, 2448–2454. doi: 10.1093/jac/dkr315
Filippov, A. A., Sergueev, K. V., He, Y., Huang, X. Z., Gnade, B. T., Mueller, A. J., et al. (2011). Bacteriophage-resistant mutants in Yersinia pestis: identification of phage receptors and attenuation for mice. PLoS One 6:e25486. doi: 10.1371/journal.pone.0025486
Filippov, A. A., Sergueev, K. V., He, Y., and Nikolich, M. P. (2012). Bacteriophages capable of lysing Yersinia pestis and Yersinia pseudotuberculosis: efficiency of plating tests and identification of receptors in Escherichia coli K-12. Adv. Exp. Med. Biol. 954, 123–134. doi: 10.1007/978-1-4614-3561-7_16
Fortier, L. C., and Sekulovic, O. (2013). Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 4, 354–365. doi: 10.4161/viru.24498
Garcia, E., Elliott, J. M., Ramanculov, E., Chain, P. S., Chu, M. C., and Molineux, I. J. (2003). The genome sequence of Yersinia pestis bacteriophage phiA1122 reveals an intimate history with the coliphage T3 and T7 genomes. J. Bacteriol. 185, 5248–5262. doi: 10.1128/jb.185.17.5248-5262.2003
Grose, J. H., and Casjens, S. R. (2014). Understanding the enormous diversity of bacteriophages: the tailed phages that infect the bacterial family Enterobacteriaceae. Virology 46, 421–443. doi: 10.1016/j.virol.2014.08.024
Heilpern, A. J., and Waldor, M. K. (2000). CTXphi infection of Vibrio cholerae requires the tolQRA gene products. J. Bacteriol. 182, 1739–1747. doi: 10.1128/jb.182.6.1739-1747.2000
Hertwig, S., Klein, I., and Appel, B. (2003a). Properties of the temperate Yersinia enterocolitica bacteriophage PY54. Adv. Exp. Med. Biol. 529, 241–243. doi: 10.1007/0-306-48416-1_46
Hertwig, S., Klein, I., Schmidt, V., Beck, S., Hammerl, J. A., and Appel, B. (2003b). Sequence analysis of the genome of the temperate Yersinia enterocolitica phage PY54. J. Mol. Biol. 331, 605–622. doi: 10.1016/s0022-2836(03)00763-0
Ikeda, H., and Tomizawa, J. (1968). Prophage P1, and extrachromosomal replication unit. Cold Spring Harb. Symp. Quant. Biol. 33, 791–798. doi: 10.1101/sqb.1968.033.01.091
Ivánovics, G., Gaál, V., Nagy, E., Prágai, B., and Simon, M. Jr. (1976). Studies on megacinogeny in Bacillus cereus. II. Bacillus cereus isolates characterized by prophage-controlled production of megacin A (phospholipase A). Acta Microbiol. Acad. Sci. Hung. 23, 283–291.
Jun, J. W., Park, S. C., Wicklund, A., and Skurnik, M. (2018). Bacteriophages reduce Yersinia enterocolitica contamination of food and kitchenware. Int. J. Food Microbiol. 271, 33–47. doi: 10.1016/j.ijfoodmicro.2018.02.007
Kiljunen, S., Datta, N., Dentovskaya, S. V., Anisimov, A. P., Knirel, Y. A., Bengoechea, J. A., et al. (2011). Identification of the lipopolysaccharide core of Yersinia pestis and Yersinia pseudotuberculosis as the receptor for bacteriophage phiA1122. J. Bacteriol. 193, 4963–4972. doi: 10.1128/JB.00339-11
Kiljunen, S., Hakala, K., Pinta, E., Huttunen, S., Pluta, P., Gador, A., et al. (2005). Yersiniophage phiR1-37 is a tailed bacteriophage having a 270 kb DNA genome with thymidine replaced by deoxyuridine. Microbiology 151(Pt 12), 4093–4102. doi: 10.1099/mic.0.28265-0
Kwon, H. J., Seong, W. J., and Kim, J. H. (2013). Molecular prophage typing of avian pathogenic Escherichia coli. Vet. Microbiol. 162, 785–792. doi: 10.1016/j.vetmic.2012.10.005
Labrie, S. J., Samson, J. E., and Moineau, S. (2010). Bacteriophage resistance mechanisms. Nat. Rev. Microbiol. 8, 317–327. doi: 10.1038/nrmicro2315
Leon-Velarde, C. G., Happonen, L., Pajunen, M., Leskinen, K., Kropinski, A. M., Mattinen, L., et al. (2016). Yersinia enterocolitica-specific infection by bacteriophages TG1 and varphiR1-RT is dependent on temperature-regulated expression of the phage host receptor OmpF. Appl. Environ. Microbiol. 82, 5340–5353. doi: 10.1128/AEM.01594-16
Leon-Velarde, C. G., Kropinski, A. M., Chen, S., Abbasifar, A., Griffiths, M. W., and Odumeru, J. A. (2014). Complete genome sequence of bacteriophage vB_YenP_AP5 which infects Yersinia enterocolitica of serotype O:3. Virol. J. 11:188. doi: 10.1186/1743-422X-11-188
Liang, J., Duan, R., Xia, S., Hao, Q., Yang, J., Xiao, Y., et al. (2015). Ecology and geographic distribution of Yersinia enterocolitica among livestock and wildlife in China. Vet. Microbiol. 178, 125–131. doi: 10.1016/j.vetmic.2015.05.006
Liang, J., Li, X., Zha, T., Chen, Y., Hao, H., Liu, C., et al. (2016). DTDP-rhamnosyl transferase RfbF, is a newfound receptor-related regulatory protein for phage phiYe-F10 specific for Yersinia enterocolitica serotype O:3. Sci. Rep. 6:22905. doi: 10.1038/srep22905
McNally, A., Thomson, N. R., Reuter, S., and Wren, B. W. (2016). ‘Add, stir and reduce’: Yersinia spp. as model bacteria for pathogen evolution. Nat. Rev. Microbiol. 14, 177–190. doi: 10.1038/nrmicro.2015.29
Morita, M., Tanji, Y., Mizoguchi, K., Akitsu, T., Kijima, N., and Unno, H. (2002). Characterization of a virulent bacteriophage specific for Escherichia coli O157:H7 and analysis of its cellular receptor and two tail fiber genes. FEMS Microbiol. Lett. 211, 77–83. doi: 10.1016/s0378-1097(02)00656-0
Mottawea, W., Duceppe, M. O., Dupras, A. A., Usongo, V., Jeukens, J., Freschi, L., et al. (2018). Salmonella enterica prophage sequence profiles reflect genome diversity and can be used for high discrimination subtyping. Front. Microbiol. 9:836. doi: 10.3389/fmicb.2018.00836
Olszak, T., Latka, A., Roszniowski, B., Valvano, M. A., and Drulis-Kawa, Z. (2017). Phage life cycles behind bacterial biodiversity. Curr. Med. Chem. 24, 3987–4001. doi: 10.2174/0929867324666170413100136
Pajunen, M. I., Molineux, I. J., and Skurnik, M. (2003). Yersiniophages. special reference to phi YeO3-12. Adv. Exp. Med. Biol. 529, 233–240. doi: 10.1007/0-306-48416-1_45
Pleska, M., Lang, M., Refardt, D., Levin, B. R., and Guet, C. C. (2018). Phage-host population dynamics promotes prophage acquisition in bacteria with innate immunity. Nat. Ecol. Evol. 2, 359–366. doi: 10.1038/s41559-017-0424-z
Rashid, M. H., Revazishvili, T., Dean, T., Butani, A., Verratti, K., Bishop-Lilly, K. A., et al. (2012). A Yersinia pestis-specific, lytic phage preparation significantly reduces viable Y. pestis on various hard surfaces experimentally contaminated with the bacterium. Bacteriophage 2, 168–177. doi: 10.4161/bact.22240
Reuter, S., Connor, T. R., Barquist, L., Walker, D., Feltwell, T., Harris, S. R., et al. (2014). Parallel independent evolution of pathogenicity within the genus Yersinia. Proc. Natl. Acad. Sci. U.S.A. 111, 6768–6773. doi: 10.1073/pnas.1317161111
Revell, P. A., and Miller, V. L. (2001). Yersinia virulence: more than a plasmid. FEMS Microbiol. Lett. 205, 159–164. doi: 10.1016/s0378-1097(01)00476-1
Richter, M., and Rossello-Mora, R. (2009). Shifting the genomic gold standard for the prokaryotic species definition. Proc. Natl. Acad. Sci. U.S.A. 106, 19126–19131. doi: 10.1073/pnas.0906412106
Romero, P., Garcia, E., and Mitchell, T. J. (2009). Development of a prophage typing system and analysis of prophage carriage in Streptococcus pneumoniae. Appl. Environ. Microbiol. 75, 1642–1649. doi: 10.1128/AEM.02155-08
Salem, M., and Skurnik, M. (2018). Genomic characterization of sixteen Yersinia enterocolitica-infecting podoviruses of pig origin. Viruses 10:E174. doi: 10.3390/v10040174
Schwudke, D., Ergin, A., Michael, K., Volkmar, S., Appel, B., Knabner, D., et al. (2008). Broad-host-range yersinia phage PY100: genome sequence, proteome analysis of virions, and DNA packaging strategy. J. Bacteriol. 190, 332–342. doi: 10.1128/JB.01402-07
Sozhamannan, S., Chute, M. D., McAfee, F. D., Fouts, D. E., Akmal, A., Galloway, D. R., et al. (2006). The Bacillus anthracis chromosome contains four conserved, excision-proficient, putative prophages. BMC Microbiol. 6:34. doi: 10.1186/1471-2180-6-34
St-Pierre, F., and Endy, D. (2008). Determination of cell fate selection during phage lambda infection. Proc. Natl. Acad. Sci. U.S.A. 105, 20705–20710. doi: 10.1073/pnas.0808831105
Wang, X., Cui, Z., Jin, D., Tang, L., Xia, S., Wang, H., et al. (2009). Distribution of pathogenic Yersinia enterocolitica in China. Eur. J. Clin. Microbiol. Infect. Dis. 28, 1237–1244. doi: 10.1007/s10096-009-0773-x
Wang, X., Li, Y., Jing, H., Ren, Y., Zhou, Z., Wang, S., et al. (2011). Complete genome sequence of a Yersinia enterocolitica “Old World” (3/O:9) strain and comparison with the “New World” (1B/O:8) strain. J. Clin. Microbiol. 49, 1251–1259. doi: 10.1128/JCM.01921-10
Wang, X., Qiu, H., Jin, D., Cui, Z., Kan, B., Xiao, Y., et al. (2008). O:8 serotype Yersinia enterocolitica strains in China. Int. J. Food Microbiol. 125, 259–266. doi: 10.1016/j.ijfoodmicro.2008.04.016
Keywords: Yersinia enterocolitica, prophage, Podoviridae, Myoviridae, comparative genomic analysis
Citation: Liang J, Kou Z, Qin S, Chen Y, Li Z, Li C, Duan R, Hao H, Zha T, Gu W, Huang Y, Xiao M, Jing H and Wang X (2019) Novel Yersinia enterocolitica Prophages and a Comparative Analysis of Genomic Diversity. Front. Microbiol. 10:1184. doi: 10.3389/fmicb.2019.01184
Received: 06 February 2019; Accepted: 09 May 2019;
Published: 29 May 2019.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Jens Andre Hammerl, Federal Institute for Risk Assessment (BfR), GermanySherwood Reid Casjens, University of Utah, United States
Copyright © 2019 Liang, Kou, Qin, Chen, Li, Li, Duan, Hao, Zha, Gu, Huang, Xiao, Jing and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xin Wang, d2FuZ3hpbkBpY2RjLmNu
†These authors have contributed equally to this work