 Johannes Born
Johannes Born Felicitas Pfeifer
Felicitas Pfeifer- Microbiology and Archaea, Department of Biology, Technische Universität Darmstadt, Darmstadt, Germany
The study of promoter activities in haloarchaea is carried out exclusively using enzymes as reporters. An alternative reporter is the gene encoding the Green Fluorescent Protein (GFP), a simple and fast tool for investigating promoter strengths. However, the GFP variant smRS-GFP, used to analyze protein stabilities in haloarchaea, is not suitable to quantify weak promoter activities, since the fluorescence signal is too low. We enhanced the fluorescence of smRS-GFP 3.3-fold by introducing ten amino acid substitutions, resulting in mGFP6. Using mGFP6 as reporter, we studied six haloarchaeal promoters exhibiting different promoter strengths. The strongest activity was observed with the housekeeping promoters Pfdx of the ferredoxin gene and P2 of the ribosomal 16S rRNA gene. Much lower activities were determined for the promoters of the p-vac region driving the expression of gas vesicle protein (gvp) genes in Halobacterium salinarum PHH1. The basal promoter strength dropped in the order PpA, PpO > PpF, PpD. All promoters showed a growth-dependent activity pattern. The GvpE-induced activities of PpA and PpD were high, but lower compared to the Pfdx or P2 promoter activities. The mGFP6 reporter was also used to investigate the regulatory effects of 5′-untranslated regions (5′-UTRs) of three different gvp mRNAs. A deletion of the 5′-UTR always resulted in an increased expression, implying a negative effect of the 5′-UTRs on translation. Our experiments confirmed mGFP6 as simple, fast and sensitive reporter to study gene expression in haloarchaea.
Introduction
The expression of many haloarchaeal genes depends on the growth phase and environmental conditions such as oxygen availability, salt concentration or temperature (Pfeifer, 2015). Studies by transcriptome or proteome analyses yield global data on gene expression, but the analysis of single genes and promotors is still required to characterize regulatory elements. So far, enzymatic assays were used to study promoter activities in haloarchaea. Commonly applied reporters are the beta-galactosidase BgaH of Haloferax (Hfx.) alicantei and the dihydrofolate reductase of Hfx. volcanii (Danner and Soppa, 1996; Gregor and Pfeifer, 2001). In Escherichia coli, the green fluorescent protein (GFP) is a suitable alternative reporter (Lissemore et al., 2000). Compared to enzymatic assays, GFP-fluorescence based analyses are less time consuming, require no additional substrates and/or cofactors and also no cell lysis. A fusion of the gfp reading frame to the promoter under investigation leads to fluorescent E. coli cells when the promoter is active. The strength of the GFP signal correlates with the promoter activity and thus with the expression of the gene investigated (Albano et al., 1996, 1998).
Different GFP proteins are used in bacteria and eukaryotes, but in archaea their use is still scarce due to the extreme growth conditions. In the case of haloarchaea the application of GFP is restricted due to the high intracellular salt concentration of 2–5.3 M KCl and a temperature optimum of 42°C (Cormack et al., 1996; Patterson et al., 1997; Nomura and Harada, 1998; Oren, 2002; Enoki et al., 2004; Reuter and Maupin-Furlow, 2004; Robinson et al., 2005; Arpino et al., 2012; Stepanenko et al., 2014). A GFP derivative with an adequate fluorescence in haloarchaea is the short-lived, red-shifted GFP smRS-GFP that has been used to investigate protein degradation in Hfx. volcanii (Reuter and Maupin-Furlow, 2004; Schmidt and Pfeifer, 2013). However, the study of weak promoters is not possible since the fluorescence signal is too low.
SmRS-GFP carries the substitutions S65T, Q80R, P99S, M153T, and V163A, improving its solubility and thermostability in the haloarchaeal intracellular milieu (Reuter and Maupin-Furlow, 2004; Baffour-Awuah et al., 2005). The literature on GFP provides additional mutations to improve its properties. The exchange of alanine 206, leucine 221, and phenylalanine 223 by arginine or lysine reduces the dimerization potential of GFP (Zacharias et al., 2002; Enoki et al., 2004; Jackson et al., 2006). Substitutions improving the folding rate and protein stability are S30R, Y39N, S147P, or N149K (Kimata et al., 1997; Pédelacq et al., 2005; Kremers et al., 2011). A substitution with a dramatic effect on GFP fluorescence is F64L, leading to a better maturation at 37°C (Cormack et al., 1996; Scholz et al., 2000). We already introduced the substitution F64L in smRS-GFP, and the resulting mGFP2 shows a 2.5-fold increase in fluorescence (Winter et al., 2018). The mGFP2 variant was used to establish the split-GFP method for haloarchaea and helped to identify several interactions of gas vesicle proteins of Halobacterium (Hbt.) salinarum PHH1.
Gas vesicle formation involves the 14 gvp genes arranged as oppositely oriented gene clusters p-gvpACNO and p-gvpDEFGHIJKLM in the p-vac region (Figure 1; Horne et al., 1991; Englert et al., 1992a). Transcription of the gvp genes is controlled by the promoters PpA and PpO as well as PpD and PpF (Hofacker et al., 2004). PpA drives the expression of p-gvpACNO encoding the two major gas vesicle structural proteins GvpA and GvpC, whereas the PpO promoter is responsible for the transcription of p-gvpO encoding a protein of unknown function (Offner et al., 1996). In the opposite direction, the PpD promoter drives the expression of p-gvpDE encoding the two regulatory proteins GvpE (activator) and GvpD (repressor). GvpE activates the oppositely oriented PpA and PpD at high levels (Hofacker et al., 2004). In the presence of GvpD, the GvpE-mediated activation is reduced (Englert et al., 1992b; Zimmermann and Pfeifer, 2003; Schmidt and Pfeifer, 2013). PpF is responsible for the transcription of p-gvpFGHIJKLM encoding the accessory proteins GvpF through GvpM that are required in minor amounts during initial stages of gas vesicle formation (Offner et al., 2000; Winter et al., 2018). The mRNAs of p-gvpACNO, p-gvpDE and p-gvpFGHIJKLM all contain 5′-untranslated regions (5′-UTR), and these 20 nt (p-gvpA), 72 nt (p-gvpD), and 169 nt (p-gvpF) 5′-UTRs might increase the mRNA stability and/or regulate translation initiation.
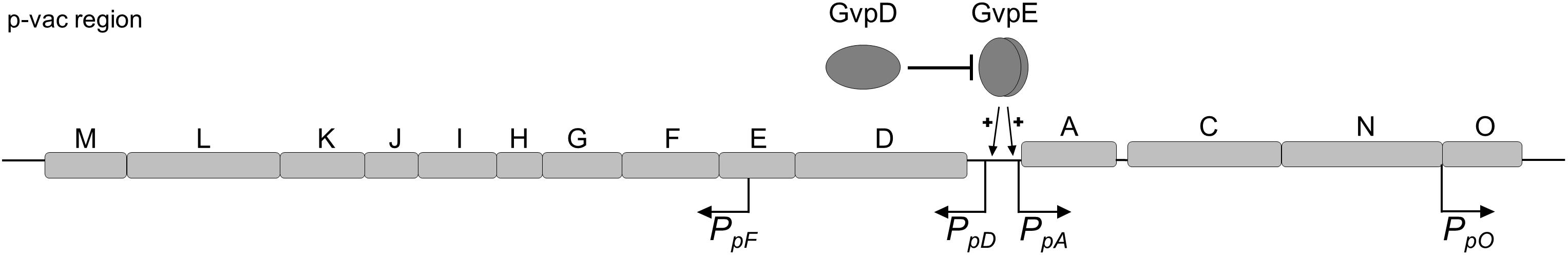
Figure 1. Schematic representation of the p-vac region of Hbt. salinarum PHH1. The 14 gas vesicles genes are arranged in two oppositely oriented gene clusters. The expression is driven by the four promoters PpA, PpD, PpF, and PpO. The activity of PpA and PpD is enhanced by the endogenous activator GvpE. In presence of GvpD the amount of GvpE decreases leading to a reduced expression. The p-gvpACNO, p-gvpDE, and p-gvpF-M transcripts contain a 5′-UTR of 20, 72, and 169 nt, respectively.
In the present report we constructed additional variants of smRS-GFP and tested their application to analyze promoter and translational activities in Hfx. volcanii. This species is a suitable host since it grows faster than Hbt. salinarum and lacks all gvp genes. Both haloarchaeal species produce large amounts of carotenoids leading to an autofluorescence; thus, weak promoter activities are difficult to detect by fluorescence. We introduced additional mutations in smRS-GFP and analyzed the fluorescence properties of these variants in Hfx. volcanii. The highest fluorescence was observed with mGFP6 harboring ten additional alterations. mGFP6 was used to investigate six haloarchaeal promoters exhibiting weak or strong activities during growth. Due to the fast degradation of mGFP6, we were able to observe growth-dependent expressions. mGFP6 was also used to study the regulatory effects of the three 5′-UTRs on the expression of gvp genes.
Materials and Methods
Strains and Cultivation Conditions
The Escherichia coli strain Top10F’ (Invitrogen, Carlsbad, United States) was grown at 37°C overnight in Luria-Bertani broth. For selection of ampicillin-resistant clones, the medium was supplemented with 100 μg/ml ampicillin. The Hfx. volcanii strains used are listed in Supplementary Table S1. Hfx. volcanii WFD11 and WR340 were cultivated in medium containing 3 M NaCl, 150 mM MgSO4, 50 mM KCl, 10 nM MnCl2, 25 mM Tris/HCl pH 7.2, 0.5% (w/v) tryptone, 0.3% (w/v) yeast extract, and 0.02% (w/v) histidine. In case of Hfx. volcanii H1424, Hv-Ca medium (Allers et al., 2004) was used, supplemented with thymidine (40 μg/ml) and uracil (50 μg/ml). Hfx. volcanii transformants were selected by 6 mg/ml lovastatin (Lam and Doolittle, 1989). Plates with solid media (containing 1.8% [w/v] agar) were incubated in plastic bags at 42°C under humid conditions for 4–5 days.
Construction of Plasmids for Promoter Studies
For promotor studies, various shuttle plasmids were generated based on pLacZJB18 or pLacZJB18+E (Supplementary Figure S1). Plasmid pLacZJB18 was produced with the NEBuilder® HiFi DNA Assembly Master Mix (New England Biolabs) and carries the lacZ gene upstream of the smRS-gfp reading frame allowing blue-white selection in E. coli. The lacZ-smRS-gfp fragment is framed by the archaeal L11e rRNA terminator (t.L11e) and a synthetic terminator sequence (Allers et al., 2010) to prevent an unintended read-through of transcripts. This vector enables a replacement of lacZ with the promoter sequence of choice via NcoI and BamHI restriction sites. Additionally, the gfp reporter gene can be exchanged using BamHI and KpnI. For a high copy number in E. coli, pLacZJB18 contains the pMB1 origin derivative of pUC19, whereas the pHK2 origin is used for the replication in haloarchaea, resulting in 7–8 copies per genome equivalent (Holmes et al., 1994). As selectable markers served ampR (E. coli) and the hmgA gene (haloarchaea) (Supplementary Figure S1A). To generate pLacZJB18+E including the activator gene c-gvpE, the reading frame was amplified by PCR. One of the oligonucleotides used contained the sequence of the Pfdx promoter to drive the expression (Table 1). The Pfdx-c-gvpE sequence was inserted behind t.L11e in pLacZJB18 using NheI and XbaI. In order to exclude an inhibition of c-gvpE expression by t.L11e, a 587 nt non-coding sequence was inserted between the terminator and Pfdx (Supplementary Figure S1B). The c-gvpE gene can be replaced using SpeI and XbaI, while substitution of Pfdx by another promoter sequence could be performed via NheI and SpeI.

Table 1. Oligonucleotides used in this study.
To fuse promoter sequences up to 120 bp with the gfp reporter gene, two complementary oligonucleotides covering the entire promoter sequence were synthesized and hybridized to yield a double-stranded fragment with nucleotide overhangs of NcoI and BamHI. After 5′-phosphorylation, the promoter fragment was inserted in pLacZJB18 or one of its derivatives, via BamHI and NcoI. Promoter sequences exceeding 120 bp were amplified by PCR, and the resulting fragments were inserted in pLacZJB18. For the integration of different gfp reporter genes served BamHI and KpnI. In case of plasmids used to quantify promoter activities, an AscI recognition site was present in the oligonucleotide upstream of the NcoI-site and served for analytical digests. All constructs were verified by DNA sequencing (Eurofins genomics). Hfx. volcanii was transformed as described by Pfeifer and Ghahraman (1993) and the transformants were controlled by PCR and DNA sequencing (Eurofins genomics) for uptake of the desired plasmid. All oligonucleotides used for these constructions are listed in Table 1.
Detection and Quantification of Fluorescence in vivo
For the detection of the smRS-GFP fluorescence in colonies of Hfx. volcanii, the cells were grown for 6 days on agar plates and then inspected using a fluorescence binocular. Colonies of the wild type served as negative control. To quantify promoter activities in vivo, the fluorescence was measured in Hfx. volcanii liquid cultures. For that, 20 ml cultures were inoculated to OD600 0.02 and cultivated at 42°C and 180 rpm to an optical density of OD600 0.3, 0.6, 1.2, and 3.0. This corresponded to an incubation time of 16, 24, 36, and 48 h. Cells of a 1 ml sample of each culture were harvested by centrifugation at 5,000 ×g for 5 min at 20°C. The sediment was washed twice with salt water (3 M NaCl, 150 mM MgSO4, 50 mM KCl, 0.02% [w/v] histidine) and then resuspended in 2 ml salt water. Fluorescence measurements were conducted with a Fluorolog FL3-22 (Horiba Jobin Yvon) at 25°C with an excitation wavelength of 488 nm (slit 5 nm) and an emission wavelength of 509 nm (slit 5 nm). As integration time 0.5 s was selected. In case of different fluorescent proteins, the fluorescence was determined at the respective optimal excitation and emission wavelength. The optical density of each culture was determined to normalize the fluorescence intensity. The autofluorescence of the wild type was subtracted from the data. Each experiment was carried out in triplicates with three biological replicates.
Fluorescence Microscopy
Hfx. volcanii cells were visualized by brightfield and fluorescence microscopy using a Leica TCS SP5 II confocal microscope in combination with Leica application suite software. For this, cells of a 2 ml sample of culture were harvested by centrifugation at room temperature at 8,000 rpm for 2 min, the supernatant was removed and the cell sediment resuspended in salt water. The detection of the fluorescence of 20 μl of the suspension was done by an excitation of 488 nm and an emission of 510–580 nm. For image processing the software Fiji was used.
Mutation of smRS-GFP
To improve the fluorescence of smRS-GFP, different substitutions were introduced in the gene by site-directed mutagenesis. The mutagenesis was performed by amplification of the complete vector using two oligonucleotides flanking each other at the 5′ ends. One of the oligonucleotides contained one to three nucleotide substitutions leading to the desired mutations in the smRS-gfp gene. The oligonucleotides used are listed in Table 1. After PCR the template vector was degraded with DpnI at 37°C for 1 h, and the amplified PCR-fragments were purified. The 5′ ends of the linear PCR products were phosphorylated by T4-polynucleotide kinase and ligated by T4-Ligase in one step (1 h, 37°C) to generate the circular plasmid. E. coli Top10 was transformed and the plasmids obtained from the transformants were controlled by DNA sequencing (Eurofins).
Western Analysis
The presence of GvpE in transformants harboring derivatives of pLacZJB18+E was determined by Western analysis. Transformants were grown in 50 ml cultures to OD600 1.2, harvested by centrifugation (2,000 ×g, 45 min, 4°C) and the sediment was resuspended in 2–3 ml lysis buffer (2.5 M KCl, 50 mM MgCl2, 1 mM EDTA, 5% [v/v] glycerol, 50 mM Tris-HCl pH 8.0). The cells were disrupted by sonication on ice (2 × 5 min, Branson sonifier 250, 3 mm disruptor horn) and the suspension centrifuged at 2,000 ×g for 45 min at 4°C. To remove the high amount of salt the suspension was dialyzed against 10 mM Tris-HCl pH 7.2 for 12 h. After dialysis, 20 μg of the total protein were separated by SDS-PAGE (Schägger and von Jagow, 1987) and transferred to a PVDF membrane (Roti®-Fluoro PVDF, Carl Roth) using the PerfectBlueTM “Semi-Dry”-Blotter. The membrane was subsequently incubated for 1 h at 37°C, reactivated in 100% methanol, washed for 4 min with PBS (1.37 M NaCl, 27 mM KCl, 100 mM Na2HPO4, 20 mM KH2PO4, pH 7.4) and blocked for 1 h with Odyssey Blocking Buffer (LI-COR). After incubation with the GvpE antiserum in Odyssey Blocking Buffer overnight, the membrane was washed four times for 5 min with PBS + 0.1% (v/v) Tween® 20 and incubated with the secondary antibody IRDye 800CW (LI-COR) coupled with a fluorophore detectable at 800 nm. The detection was done with the Odyssey Fc Imager (LI-COR).
Results
Fluorescence of smRS-GFP in Three Hfx. volcanii Strains at Various Temperatures
The expression of smRS-gfp was analyzed in the three Hfx. volcanii strains WR340, WFD11, and H1424 available in our laboratory (Cline et al., 1989; Bitan-Banin et al., 2003; Stroud et al., 2012). The lacZ gene of plasmid pLacZJB18 was exchanged for the ferredoxin promoter Pfdx of Hbt. salinarum (Pfeifer et al., 1993). The three strains were transformed with the resulting plasmid pPfdxJB18 and the fluorescence of the colonies was inspected on agar plates illuminated with blue light (Figure 2). The cells were also analyzed by fluorescence microscopy. Only a small fraction of colonies (WFD11) or about half of the colonies (H1424) exhibited a fluorescence, but every colony of WR340 was fluorescent. Hfx. volcanii WR340 appeared to be more stable in the expression of smRS-gfp and was used for all further experiments. To investigate the influence of the temperature on the smRS-gfp expression, pPfdxJB18 WR340 transformants were grown at 37, 42°C, or 45°C to OD600 0.6. The cells grown at 42°C reached a 1.5- to 1.9-fold higher fluorescence (367 ± 34 ×103 AU) than those cultured at 37°C (188 ± 17 ×103AU) or 45°C (242 ± 39 ×103 AU). Thus, all further growth experiments were performed at the optimal growth and folding temperature of 42°C.
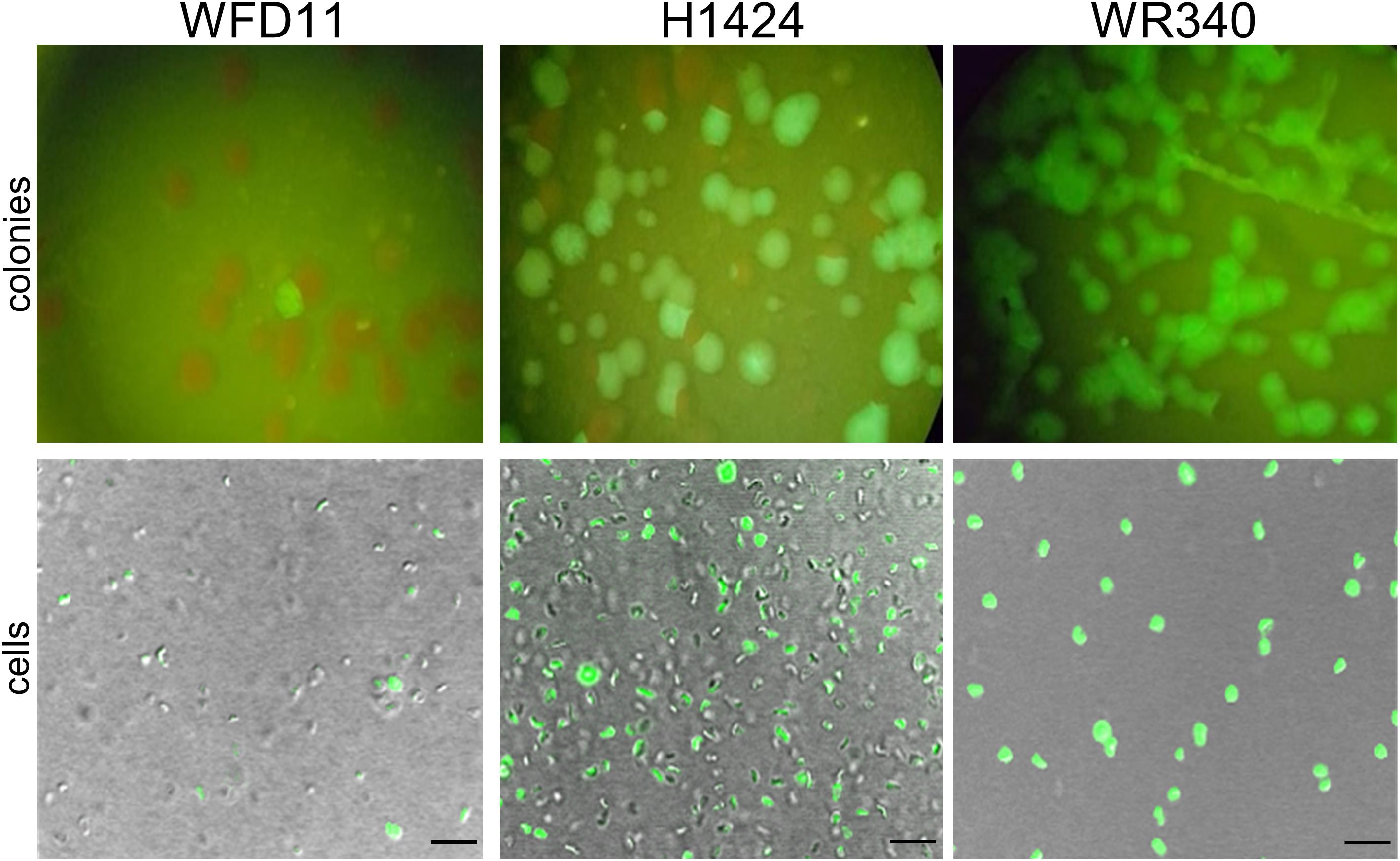
Figure 2. Expression of smRS-gfp in three different Hfx. volcanii strains. The transformants of Hfx. volcanii WFD11, H1224, and WR340 carry pPfdxJB18 harboring the smgRS-gfp reporter under control of Pfdx. The fluorescence of the colonies was visualized under a fluorescence binocular (with blue light) and the fluorescence of cells by confocal laser scanning microscopy. Scale bars equal 10 μm in each case.
Comparison of smRS-GFP With Other Fluorescent Proteins in Hfx. volcanii
To test other available fluorescent proteins, the smRS-gfp reading frame of vector pPfdxJB18 was exchanged with the reading frames encoding mTagBFP (blue), eCFP (cyan), sfGFP (superfolder GFP, green), eYFP, Citrine as well as SYFP2 (yellow), or mCherry (red). The respective Hfx. volcanii WR340 transformants were grown to OD600 0.6 at 42°C and their fluorescence was determined. The highest fluorescence (108,000 AU) was observed with the original smRS-GFP, but the fluorescence of sfGFP, mCherry, SYFP2, and mTagBFP were well detectable (between 16,000 and 60,000 AU) (Figure 3A). Cells were also detectable by fluorescence microscopy (Figure 3B). However, the fluorescence of eCFP, eYFP or Citrine in the transformants was very low and difficult to detect.
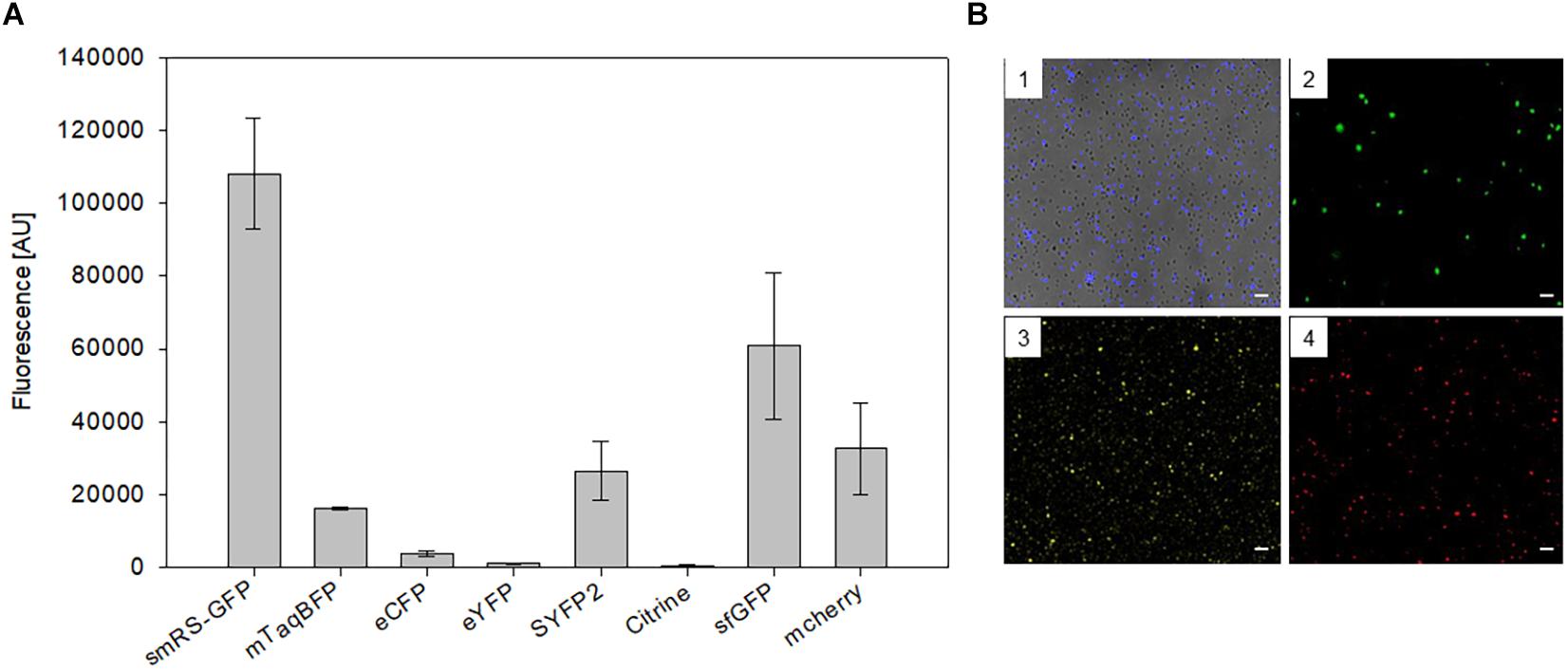
Figure 3. Expression of different fluorescent proteins in Hfx. volcanii WR340. (A) The expression of the fluorescent protein genes was driven by the strong Pfdx promoter. The transformants were grown to OD600 0.6 at 42°C. The fluorescence is given in arbitrary units (AU). The autofluorescence of Hfx. volcanii was subtracted from each value. All experiments were performed in triplicates with three different biological samples. (B) Fluorescence microscopy of Hfx. volcanii WR430 expressing the reading frames encoding mTagBFP (1), mGFP2 (2), SYFP2 (3), and mCherry (4). Scar bars equal 10 μm in each case.
Mutation of smRS-GFP to Enhance the Fluorescence
To investigate whether smRS-GFP allows the analysis of weak promoter activities, the promoter PpD (plus 5′-UTR) of the p-vac region was chosen. The respective plasmid pPDJB18 (smRS-gfp under control of PpD) was constructed and investigated in Hfx. volcanii WR340 transformants. The fluorescence of cells in the early exponential growth phase (OD600 0.3) was hardly above the autofluorescence of Hfx. volcanii (Figure 4B). In order to generate smRS-GFP variants exhibiting a stronger fluorescence, additional mutations (F64L, A206K, L221K, F223R, S30R, Y39N, S147P, N149K, and S72A) known to improve the GFP properties in E. coli (Kimata et al., 1997; Pédelacq et al., 2005; Kremers et al., 2011) were introduced individually or in combination to increase its brightness in haloarchaea (Figure 4A). The fluorescence of the respective Hfx. volcanii transformants harboring these variants was measured in the early exponential growth phase at OD600 0.3 (Figure 4B). The mGFP-F64L mutant (mGFP2) yielded a 2.5-fold higher fluorescence, similar to mGFP3 and mGFP4, whereas variant mGFP5 showed a 2.9-fold higher fluorescence compared to smRS-GFP. The strongest fluorescence (3.3-fold enhanced) was observed with mGFP6 carrying all ten substitutions (Figure 4B).
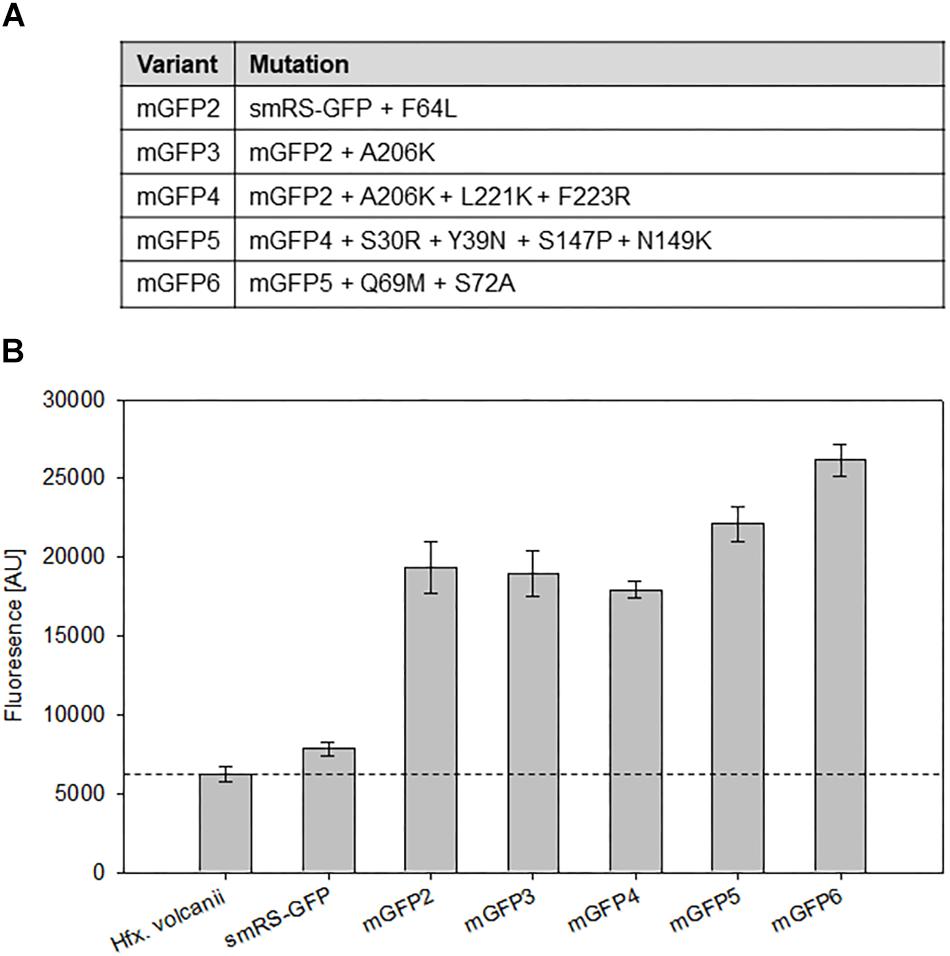
Figure 4. Point mutations in smRS-GFP. (A) Mutations in smRS-GFP to yield the variants mGFP2 through mGFP6. The selected substitutions influence maturation at 37°C, the tendency of self-aggregation, the folding rate, or protein stability. (B) Fluorescence of smRS-GFP variants in vivo. Fluorescence of wild type cells and transformants carrying the reading frame encoding one of the variants under control of the weak PpD promoter. The cells were grown for 24 h to OD600 0.6 at 42°C. The fluorescence is given in arbitrary units (AU). Each experiment was performed in triplicates of three different biological samples.
Quantification of Promoter Activities Using mGFP6
The strength of six different haloarchaeal promoters was determined using mGFP6 as reporter. The two house-keeping gene promoters Pfdx (ferredoxin gene), P2 (ribosomal 16S rRNA gene) and the four promoters of the gas-vesicle encoding p-vac region of Hbt. salinarum PHH1 were investigated. In case of the gas vesicle promoters, the complete promoter sequence up to the first 15 nt (including the ATG start) of the corresponding gvp gene were fused to mgfp6 lacking its AUG start codon. Samples of the respective Hfx. volcanii transformants were taken at OD600 0.3, 0.6, 1.2, and 3.0, i.e., after 16, 24, 36, or 48 h of growth, and analyzed for fluorescence (Table 2; see Figure 5). The Pfdx-mgfp6 transformants yielded the highest fluorescence in the exponential growth phase, and the activity decreased approximately twofold during stationary growth (Table 2). These results implied that the product ferredoxin is mainly required during exponential growth. P2 was constitutively active in the exponential growth phase, but at a slightly lower level compared to Pfdx. In the stationary growth phase, the P2-driven expression was comparable to that of Pfdx (Table 2). The much lower basal activity of PpA was highest in the early exponential growth phase, a reduced fluorescence was observed in late exponential and early stationary growth, and the fluorescence was again higher in the late stationary growth phase (Table 2). PpO yielded a high fluorescence in early exponential growth, which was reduced during exponential growth, increased and dropped down again in the late stationary phase (Table 2). In contrast, PpD and PpF yielded a 3- to 5-fold lower fluorescence compared to PpA and PpO. Both showed their highest value in the early exponential growth phase, followed by a 3- to 4.5-fold reduction up to the stationary growth phase (Table 2). Overall, mGFP6 allowed the detection of all these promoter activities and uncovered different patterns of promoter activity during growth.
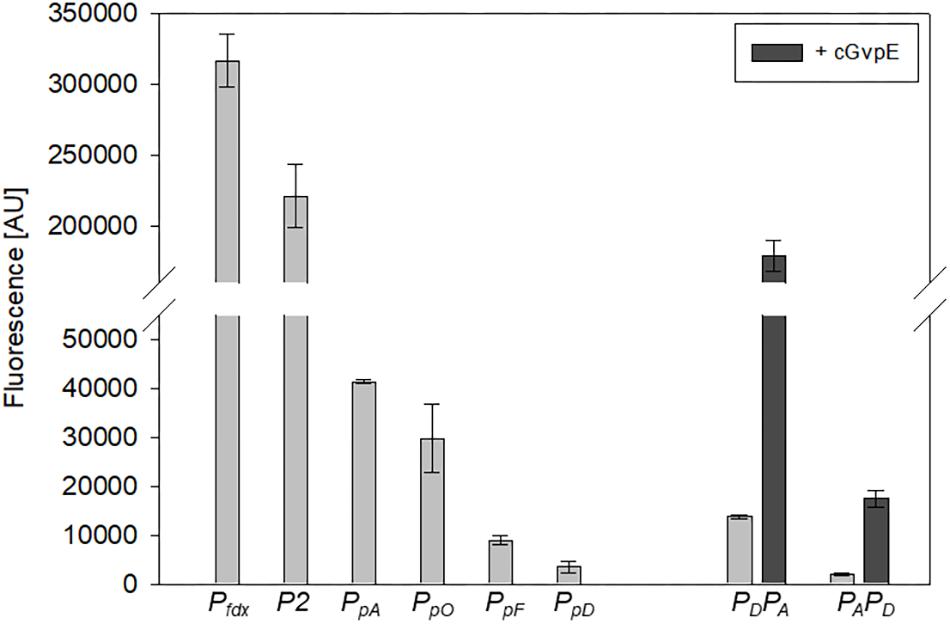
Figure 5. Activities of the six different haloarchaeal promoters. The activity was quantified at OD600 0.6 (42°C). The fluorescence is given in arbitrary units (AU) (see Table 2). The autofluorescence of Hfx. volcanii was subtracted from the values. Each experiment was performed in triplicates of three different biological samples. The PpA and PpD activities induced by cGvpE are shown on the right (OD600 0.6). The PDPA and PAPD constructs used for these experiments include the entire PpA-PpD promoter region. For more information, see text.
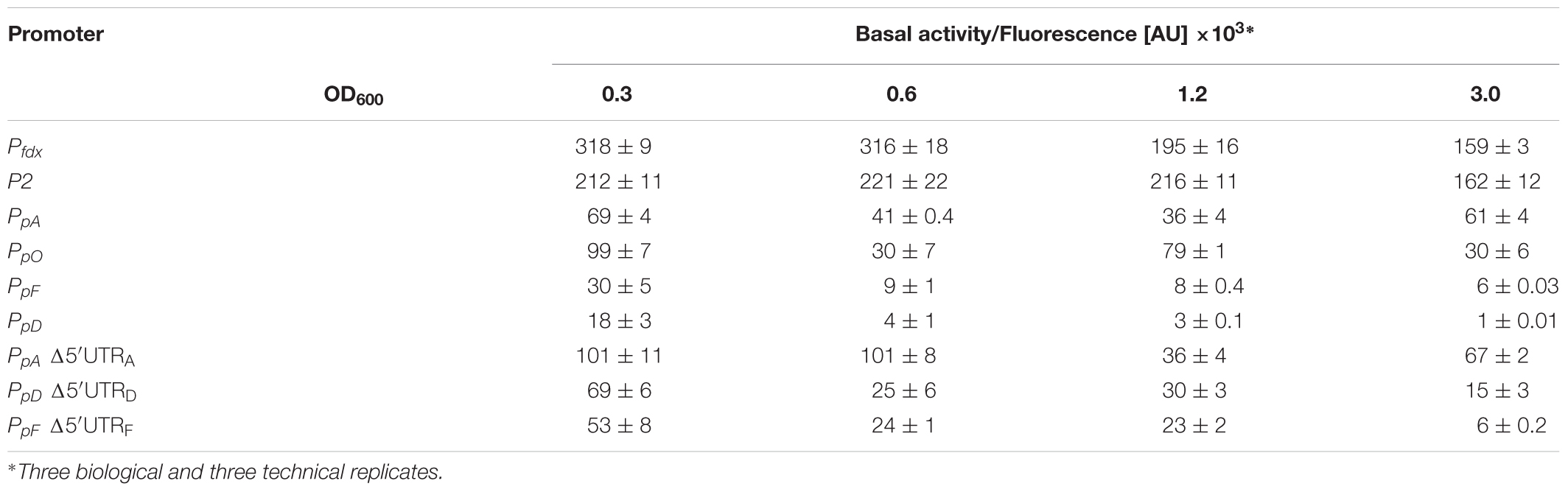
Table 2. Promoter activities throughout the growth.
Activation of PpA and PpD by GvpE
The GvpE-mediated activation of the two promoters PpA and PpD was investigated using the vector construct pLacZJB18+E. The plasmid harbors the c-gvpE reading frame encoding the activator of the c-vac region of Hbt. salinarum PHH1 expressed under the control of the strong Pfdx promoter (Supplementary Figure S1B). The cGvpE protein was used for these studies because of its higher activation capability compared to pGvpE of the p-vac region (Gregor and Pfeifer, 2001). The lacZ reading frame was substituted by the entire PpD-PpA promoter region. The resulting plasmid pPDPAJB18+E (designated PDPA+E) served to investigate the activation of PpA, whereas pPAPDJB18+E (PAPD+E) was used to investigate the GvpE-activation of PpD. Construct PDPA+E contained the first 15 nt of gvpA fused to mgfp6 (i.e., gfp under PA promoter control) and in the opposite direction the entire DNA sequence up to the first 15 nt of the gvpD reading frame. Construct PAPD+E contained the mgfp6 reading frame fused to the first 15 nt of gvpD (gfp under PD promoter control), and in addition all sequences up to the first 15 nt of gvpA. Western analysis performed with total proteins of the PDPA+E or PAPD+E transformants using an antiserum raised against cGvpE yielded the presence of GvpE in both cases (Supplementary Figure S2). To determine the basal activities of PpA or PpD, similar vectors but lacking c-gvpE were used (designated PDPA and PAPD). Based on the high activity of PpA and PpD in the exponential growth phase, the fluorescence of the respective Hfx. volcanii transformants was analyzed in cells grown to OD600 0.6 at 42°C. The fluorescence of the PDPA transformants (basal PpA activity) was almost sevenfold higher than the signal of the PAPD transformants (basal PpD activity), underlining that the PpA promoter is stronger than PpD (Figure 5, right side; Table 3). The GvpE-mediated activation resulted in a much higher fluorescence: PDPA+E transformants were 13-fold enhanced in fluorescence compared to PDPA transformants, and the fluorescence of PAPD+E transformants was 8-fold enhanced compared to PAPD (Figure 5 and Table 3). In summary, the expression derived from the cGvpE-induced PpA promoter was much higher compared to the induced expression derived from PpD.

Table 3. GvpE-induced promoter activities∗.
Effect of 5′-UTRs on gvp Translation
The influence of the 5′-untranslated regions of the p-vac mRNAs p-gvpACNO (5′-UTRA), p-gvpDE (5′-UTRD) and p-gvpFGHIJKLM (5′-UTRF) on translation was investigated using mGFP6 as reporter. The corresponding promoter sequences (including the first 15 nt of the respective reading frame, but lacking the 5′-UTR) were fused to mgfp6 and the fluorescence was determined in Hfx. volcanii transformants at OD600 0.3, 0.6, 1.2, and 3.0 (Figure 6). In each case, lack of the 5′-UTR resulted in a higher fluorescence, especially during the exponential growth phase, suggesting a negative influence of the 5′-untranslated leader regions on translation. The highest fluorescence occurred with Δ5′-UTRA transformants, followed by Δ5′-UTRD and Δ5′-UTRF transformants (Table 2). The strongest effect was observed with Δ5′-UTRD leading up to a sixfold increase in fluorescence in exponential growth. During late exponential growth phase, the fluorescence of the Δ5′-UTRD transformants dropped down, but was still higher than in the presence of the 5′-UTRD (Figure 6 and Table 2). Thus, all three 5′-UTRs significantly reduce the translation of the respective gvp mRNA.
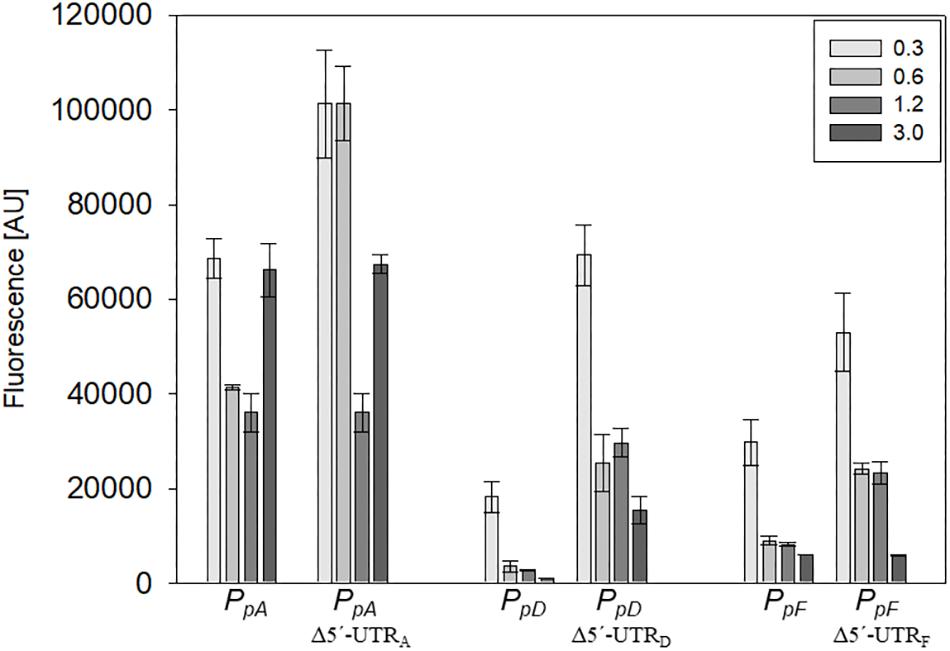
Figure 6. Effect of the 5′-UTR on the expression during growth. The effect of the 5′-UTRA, 5′-UTRD, and 5′-UTRF on the expression of the corresponding mRNA was quantified throughout growth. The fluorescence of the transformants measured at OD600 0.3, 0.6, 1.2, and 3.0 (42°C) is given in arbitrary units (AU). The autofluorescence of Hfx. volcanii was subtracted from each value. Each experiment was performed in triplicates of three different biological samples.
Discussion
To study the activity of weak haloarchaeal promoters, smRS-GFP was altered by additional mutations to improve its brightness. The best variant obtained was mGFP6 containing ten additional mutations and offering a 3.3-fold higher fluorescence signal in Hfx. volcanii compared to smRS-GFP. Using mGFP6 as reporter, six haloarchaeal promoters ranging from weak to high activities were investigated, and the activities were also quantified throughout growth. In addition, mGFP6 was used to study the effect of 5′-untranslated regions (5′-UTRs) on the translation of transcripts derived from the p-vac region.
mGFP6 Is Suitable to Study Weak Promotor Activities
The high salt concentration in the haloarchaeal cytoplasm and growth temperatures above 45°C restrict the GFP fluorescence (Cormack et al., 1996; Nomura and Harada, 1998; Enoki et al., 2004; Stepanenko et al., 2014). The soluble smRS-GFP shows a sufficient signal to investigate protein degradation in haloarchaea, but the signal is too low to quantify weak promoter activities. Several substitutions were introduced in smRS-GFP that are known to improve its maturation at higher temperature, reduce protein self-aggregation and increase folding as well as protein stability (Cormack et al., 1996; Zacharias et al., 2002; Enoki et al., 2004; Jackson et al., 2006). The F64L substitution strongly increases the maturation rate in E. coli (Cormack et al., 1996; Scholz et al., 2000). Indeed, the resulting mGFP2 showed a 2.5-fold enhanced fluorescence compared to the initial signal in Hfx. volcanii (Figure 4B). The high salt concentrations in the haloarchaeal cytoplasm of 2–5 M KCl may limit the fluorescence of GFP by an increased tendency to self-aggregate or alterations of the protein structure (Deschamps et al., 1995; Ishii et al., 2007). The substitutions A206K, F221K, and F223R were inserted individually or in combination in mGFP2 to reduce its dimerization potential (Zacharias et al., 2002; Enoki et al., 2004; Jackson et al., 2006). However, the mutation A206K (mGFP3) as well as A206K, F221K, and F223R (mGFP4) did not result in an increased fluorescence compared to mGFP2 (Figure 4B).
A most widely used GFP variant in E. coli and mammalian cells is sfGFP, whose good folding and stability properties are mainly based on the two substitutions S30R and Y39N (Pédelacq et al., 2005). Other mutations known to improve folding, maturation and protein stability at higher temperatures are the substitutions N149K and S147P (Kimata et al., 1997; Tsien, 1998; Cubitt et al., 1999; Baffour-Awuah et al., 2005; Pédelacq et al., 2005; Choi et al., 2017). The implementation of these four mutations into mGFP4 (to yield mGFP5) led to a 1.2-fold increase in fluorescence. The S72A mutation improves GFP stabilization and folding in E. coli (Cubitt et al., 1999), particularly when combined with F64L, S65T and N149K (Teerawanichpan et al., 2007). The resulting mGFP6 showed the strongest fluorescence (3.3-fold increase) compared to smRS-GFP and a 1.2-fold higher signal than mGFP5 in haloarchaea (Figure 4B). It should be noted that mGFP6 carries at position 69 a methionine instead of a glutamine due to a PCR error. The Q69M mutation is known to increase photostability and reduces the sensitivity to chloride ions and low pH values in yellow fluorescent proteins, YFP (Griesbeck et al., 2001). However, the chromophore structure of YFP is not comparable to GFP and the substitution Q69M might not have any effect (Tsien, 1998). Taken together our results suggest that the stabilizing substitutions play a much larger role in the improvement of smRS-GFP brightness in haloarchaea than mutations that might affect its solubility and self-aggregation. The mGFP6 signal finally obtained was high enough to quantify weak promoter activities in haloarchaea.
Promoter Activities Are Growth-Phase Dependent
To evaluate mGFP6 as a fast and easy alternative to an enzymatic assay we expressed the mgfp6 reading frame under the control of different promoters and quantified the signal in Hfx. volcanii transformants. We selected the house-keeping promoter Pfdx of the ferredoxin gene, the P2 promoter of ribosomal 16S rRNA gene, and for a moderate to very weak expression PpA, PpD, PpF, and PpO of the p-vac region of Hbt. salinarum. The highest fluorescence was obtained with Pfdx, followed by P2 (Figure 5). In the case of Pfdx, a constantly high fluorescence signal was observed throughout growth, and a decrease occurred at the beginning of the stationary growth phase. Ferredoxin serves as major electron carrier in haloarchaea and especially in the decarboxylation of α-keto acids (Kerscher and Oesterhelt, 1977; Pfeifer et al., 1993; Falb et al., 2008). The dominant production of fdx mRNA during the exponential growth phase is due to the use of ferredoxin as electron transport system in the metabolism of haloarchaea. In contrast, the P2 promoter showed a constantly high activity throughout growth (Table 2), suggesting that the 16S rRNA is continuously produced (Reuter and Maupin-Furlow, 2004).
In contrast, the promoters PpA, PpO, PpD, and PpF of the p-vac region exhibited much lower activities and also varied in activity throughout growth (Figure 5). Two of these, PpA and PpD, are induced by the endogenous activator GvpE (Hofacker et al., 2004). The basal activity of PpA yielded the highest fluorescence in the early exponential growth phase, and the fluorescence was reduced up to the early stationary growth phase, but increased again in the late stationary growth phase (Table 2). These results were consistent with previous observations using the haloarchaeal beta-galactosidase BgaH as reporter (Hofacker et al., 2004). PpA drives the transcription of p-gvpACNO, leading to large amounts of p-gvpA mRNA and minor amounts of p-gvpAC, p-gvpACN, and p-gvpACNO transcripts (Offner et al., 1996). The promoter is also induced by the endogenous activator protein GvpE and leads to the formation of the major gas vesicle structural proteins GvpA and GvpC. The activity of PpO resulted in a leaderless p-gvpO transcript. The promoter PpO was characterized for the first time, since an activity was not detectable when measured with BgaH as reporter (Hofacker et al., 2004). The reason could be a loss of function of BgaH due to the N-terminal fusion of the first five amino acids of pGvpO (MADPA). The highest activity of PpO was observed in the early growth phase, the activity decreased during exponential growth, increased at the end of this growth phase and was reduced again in the stationary growth phase (Table 2). The reason for this behavior is not known.
The PpD promoter exhibited the lowest basal activity determined here. Its highest basal activity was observed in the early exponential growth phase, but then the activity dropped continuously down (Table 2). In Hbt. salinarum, the PpD promoter is activated by GvpE, leading to a much higher expression of the two regulatory proteins GvpD and GvpE. In previous Northern analyses performed with total RNA of Hbt. salinarum PHH1, the p-gvpDE mRNA was observed in the stationary growth phase only, suggesting that PpD is active in the late growth phase only. A reason for this difference could be undetectable amounts of transcripts in the early growth phase due to the weak activity of PpD, whereas GvpE-activation of PpD leads to a detectable amount of p-gvpDE mRNA in the stationary phase. Our analysis on the activity of PpA and PpD in the presence of cGvpE in Hfx. volcanii transformants yielded a 13-fold induction of PpA and an eightfold induction of PpD when c-gvpE was expressed under Pfdx control (Figure 5 and Table 3). However, the final activity of the GvpE-induced PpA promoter was still lower than the activity of Pfdx.
The PpF promoter yielded the highest activity in the early exponential growth phase, followed by a continuous reduction up to the stationary growth phase – similar to the growth phase-dependent basal activity of PpD (Figure 5 and Table 2). Both promoters exhibited the weakest activities of all p-vac promoters tested, but in contrast to PpD, the PpF promoter cannot be induced by GvpE. The higher activity of PpF in the early exponential growth phase underlines the early production of the accessory gas vesicle proteins GvpF through GvpM required in minor amounts during the initial stages of gas vesicle formation (Offner et al., 2000; Winter et al., 2018).
In summary, different temporal activity patterns were observed with the six promoters tested here. All promoters decreased their activity in the stationary growth phase as suggested by the reduced fluorescence of the cells. These results implied that the cells did not accumulate GFP over the time. The relatively slow growth rate of haloarchaea with a generation time of approximately 4 h allows GFP folding and also degradation, which is indispensable for promoter studies.
Overall, mGFP6 is useful to detect weak promoter activities in vivo and is even more sensitive compared to the BgaH reporter system. The mGFP6 reporter is also more reliable, since the BgaH activity is influenced by temperature, whereas the fluorescence of GFP is temperature independent. Another advantage of the analysis the mGFP6 fluorescence is that the cells are only washed to remove the media, whereas the BgaH enzyme assay requires cell lysis. Yet, the data obtained with both methods was consistent. The two different haloarchaeal reporter systems now enable the simultaneous and quantitative analysis of two promoters, such as the divergent PA-PD of the gas vesicle protein genes. The BgaH / mRNA analysis applied previously (Marschaus and Pfeifer, 2012) could be substituted by BgaH / GFP to determine both promoter activities in a quantitative way throughout growth in the same cell culture.
5′-UTRs Reduced the Translation of gvp mRNAs
Many 5′-UTRs of transcripts found in bacteria or eukaryotes supply binding sites for regulatory proteins, small regulatory RNAs, or metabolites (riboswitches) and control the translation. Also, the Shine-Dalgarno sequence is located here, providing the positioning of the mRNA for translation initiation at the 30S ribosomal subunit in bacteria. Thus, sequences and secondary structures of the 5′-UTRs play a pivotal role in gene expression regulation (Cho et al., 2017). In haloarchaea, approximately 30% of the mRNAs are leaderless and lack a 5′-untranslated region (Brenneis et al., 2007). Examples for leaderless transcripts are the fdx and the gvpO mRNA (Pfeifer et al., 1993; Offner et al., 1996). The fdx mRNA occurs in large amounts and is efficiently translated. The insertion of reading frames in the expression vector pJAS35 also results in leaderless transcripts that are efficiently expressed. Also, the lack of the 5′-UTR of the p-gvpH mRNA leads to a 15-fold increase in translation (Sartorius-Neef and Pfeifer, 2004). It appears that 5′-UTRs lead to a reduced translation in all these cases.
In this report we used mGFP6 to study the effect of the 5′-UTRs on the expression of p-gvpACNO (5′UTRA), p-gvpDE (5′UTRD), and p-gvpFGHIJKLM (5′UTRF). In all three cases, the deletion of the 5′-UTR resulted in an increase of the GFP fluorescence, especially in early growth stages (Figure 6 and Table 2). Bioinformatic analyses indicate secondary structures in 5′-UTRA and 5′-UTRD that might interfere with the initiation of translation. The higher expression of the leaderless mRNAs could be explained with a higher accessibility of the mRNA to the ribosome. A 1.4- to 2.4-fold increase in fluorescence due to the deletion of 5′-UTRA was observed in the early exponential growth phase, suggesting a growth depending regulation of 5′-UTRA (Figure 6 and Table 2). In contrast, deletion of the 5′-UTRD and 5′-UTRF led to a 3.3- to 4.5-fold higher translation throughout growth when compared to the respective leader-containing transcript.
Conclusion
Overall, mGFP6 appears to be an easy, fast and sensitive alternative to investigate the gene expression at the level of transcription and translation in haloarchaea. Promoter activities can be determined, and the original vector pPLacZJB18 is useful to substitute lacZ with the desired promoter sequences allowing blue/white selection in E. coli. mGFP6 is a highly fluorescent, short-lived protein and can be applied to investigate the time-dependent transcription or translation in haloarchaea. In future, the regulatory effect of 5′-UTRs on the gvp gene expression will be investigated in further detail.
Data Availability
The raw data supporting the conclusions of this manuscript will be made available by the authors, without undue reservation, to any qualified researcher.
Author Contributions
JB and FP planned the study, discussed the results, wrote the manuscript, and approved the final manuscript. JB performed the analysis.
Funding
This work was financially supported by LOEWE Schwerpunkt CompuGene (Project A3) at TU Darmstadt. We also acknowledge support by the German Research Foundation and the Open Access Publishing Fund of Technische Universität Darmstadt.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer MLDS declared a past co-authorship with one of the authors FP to the handling Editor.
Acknowledgments
We thank Arnulf Kletzin for valuable discussions and Kerstin Weitzel for technical assistance. We also wish to thank Martin M. Rudolph, Alisa Jost and Kerstin Winter for carefully proofreading of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01200/full#supplementary-material
References
Albano, C. R., Randers-Eichhorn, L., Bentley, W. E., and Rao, G. (1998). Green fluorescent protein as a real time quantitative reporter of heterologous protein production. Biotechnol. Prog. 14, 351–354. doi: 10.1021/bp970121b
Albano, C. R., RandersEichhorn, L., Chang, Q., Bentley, W. E., and Rao, G. (1996). Quantitative measurement of green fluorescent protein expression. Biotechnol. Tech. 10, 953–958.
Allers, T., Barak, S., Liddell, S., Wardell, K., and Mevarech, M. (2010). Improved strains and plasmid vectors for conditional overexpression of His-tagged proteins in Haloferax volcanii. Appl. Environ. Microbiol. 76, 1759–1769. doi: 10.1128/AEM.02670-09
Allers, T., Ngo, H. P., Mevarech, M., and Lloyd, R. G. (2004). Development of additional selectable markers for the halophilic archaeon Haloferax volcanii based on the leuB and trpA genes. Appl. Environ. Microbiol. 70, 943–953. doi: 10.1128/AEM.70.2.943-953.2004
Arpino, J. A., Rizkallah, P. J., and Jones, D. D. (2012). Crystal structure of enhanced green fluorescent protein to 1.35 A resolution reveals alternative conformations for Glu222. PLoS One 7:e47132. doi: 10.1371/journal.pone.0047132
Baffour-Awuah, N. Y., Fedeles, F., and Zimmer, M. (2005). Structural features responsible for GFPuv and S147P-GFP’s improved fluorescence. Chem. Phys. 310, 25–31. doi: 10.1016/j.chemphys.2004.09.031
Bitan-Banin, G., Ortenberg, R., and Mevarech, M. (2003). Development of a gene knockout system for the halophilic archaeon Haloferax volcanii by use of the pyrE gene. J. Bacteriol. 185, 772–778. doi: 10.1128/JB.185.3.772-778.2003
Brenneis, M., Hering, O., Lange, C., and Soppa, J. (2007). Experimental characterization of Cis-acting elements important for translation and transcription in halophilic archaea. PLoS Genet. 3:e229. doi: 10.1371/journal.pgen.0030229
Cho, S. H., Haning, K., Shen, W., Blome, C., Li, R., Yang, S., et al. (2017). Identification and characterization of 5′ untranslated regions (5′-UTRs) in Zymomonas mobilis as regulatory biological parts. Front. Microbiol. 8:2432. doi: 10.3389/fmicb.2017.02432
Choi, J. Y., Jang, T. H., and Park, H. H. (2017). The mechanism of folding robustness revealed by the crystal structure of extra-superfolder GFP. FEBS Lett. 591, 442–447. doi: 10.1002/1873-3468.12534
Cline, S. W., Schalkwyk, L. C., and Doolittle, W. F. (1989). Transformation of the archaebacterium Halobacterium volcanii with genomic DNA. J. Bacteriol. 171, 4987–4991. doi: 10.1128/jb.171.9.4987-4991.1989
Cormack, B. P., Valdivia, R. H., and Falkow, S. (1996). FACS-optimized mutants of the green fluorescent protein (GFP). Gene 173, 33–38. doi: 10.1016/0378-1119(95)00685-0
Cubitt, A. B., Woollenweber, L. A., and Heim, R. (1999). Understanding structure-function relationships in the Aequorea victoria green fluorescent protein. Methods Cell Biol. 58, 19–30. doi: 10.1016/S0091-679X(08)61946-9
Danner, S., and Soppa, J. (1996). Characterization of the distal promoter element of halobacteria in vivo using saturation mutagenesis and selection. Mol. Microbiol. 19, 1265–1276. doi: 10.1111/j.1365-2958.1996.tb02471.x
Deschamps, J. R., Miller, C. E., and Ward, K. B. (1995). Rapid purification of recombinant green fluorescent protein using the hydrophobic properties of an HPLC size-exclusion column. Protein Expr. Purif. 6, 555–558. doi: 10.1006/prep.1995.1073
Englert, C., Kruger, K., Offner, S., and Pfeifer, F. (1992a). Three different but related gene clusters encoding gas vesicles in halophilic archaea. J. Mol. Biol. 227, 586–592. doi: 10.1016/0022-2836(92)90914-6
Englert, C., Wanner, G., and Pfeifer, F. (1992b). Functional analysis of the gas vesicle gene cluster of the halophilic archaeon Haloferax mediterranei defines the vac-region boundary and suggests a regulatory role for the gvpD gene or its product. Mol. Microbiol. 6, 3543–3550. doi: 10.1111/j.1365-2958.1992.tb01789.x
Enoki, S., Saeki, K., Maki, K., and Kuwajima, K. (2004). Acid denaturation and refolding of green fluorescent protein. Biochemistry 43, 14238–14248. doi: 10.1021/bi048733
Falb, M., Muller, K., Konigsmaier, L., Oberwinkler, T., Horn, P., von Gronau, S., et al. (2008). Metabolism of halophilic archaea. Extremophiles 12, 177–196. doi: 10.1007/s00792-008-0138-x
Gregor, D., and Pfeifer, F. (2001). Use of a halobacterial bgaH reporter gene to analyse the regulation of gene expression in halophilic archaea. Microbiology 147(Pt 7), 1745–1754. doi: 10.1099/00221287-147-7-1745
Griesbeck, O., Baird, G. S., Campbell, R. E., Zacharias, D. A., and Tsien, R. Y. (2001). Reducing the environmental sensitivity of yellow fluorescent protein. Mechanism and applications. J. Biol. Chem. 276, 29188–29194. doi: 10.1074/jbc.M102815200
Hofacker, A., Schmitz, K. M., Cichonczyk, A., Sartorius-Neef, S., and Pfeifer, F. (2004). GvpE- and GvpD-mediated transcription regulation of the p-gvp genes encoding gas vesicles in Halobacterium salinarum. Microbiology 150, 1829–1838. doi: 10.1099/mic.0.27078-0
Holmes, M., Pfeifer, F., and Dyall-Smith, M. (1994). Improved shuttle vectors for Haloferax volcanii including a dual-resistance plasmid. Gene 146, 117–121. doi: 10.1016/0378-1119(94)90844-3
Horne, M., Englert, C., Wimmer, C., and Pfeifer, F. (1991). A DNA region of 9 kbp contains all genes necessary for gas vesicle synthesis in halophilic archaebacteria. Mol. Microbiol. 5, 1159–1174. doi: 10.1111/j.1365-2958.1991.tb01889.x
Ishii, M., Kunimura, J. S., Jeng, H. T., Penna, T. C., and Cholewa, O. (2007). Evaluation of the pH- and thermal stability of the recombinant green fluorescent protein (GFP) in the presence of sodium chloride. Appl. Biochem. Biotechnol. 137-140, 555–571. doi: 10.1007/s12010-007-9079-6
Jackson, S. E., Craggs, T. D., and Huang, J. R. (2006). Understanding the folding of GFP using biophysical techniques. Expert. Rev. Proteomics 3, 545–559. doi: 10.1586/14789450.3.5.545
Kerscher, L., and Oesterhelt, D. (1977). Ferredoxin is the coenzyme of α-ketoacid oxidoreductases in Halobacterium halobium. FEBS Lett. 83, 197–201. doi: 10.1016/0014-5793(77)81004-1
Kimata, Y., Iwaki, M., Lim, C. R., and Kohno, K. (1997). A novel mutation which enhances the fluorescence of green fluorescent protein at high temperatures. Biochem. Biophys. Res. Commun. 232, 69–73. doi: 10.1006/bbrc.1997.6235
Kremers, G. J., Gilbert, S. G., Cranfill, P. J., Davidson, M. W., and Piston, D. W. (2011). Fluorescent proteins at a glance. J. Cell Sci. 124(Pt 2), 157–160. doi: 10.1242/jcs.072744
Lam, W. L., and Doolittle, W. F. (1989). Shuttle vectors for the archaebacterium Halobacterium volcanii. Proc. Natl. Acad. Sci. U.S.A. 86, 5478–5482. doi: 10.1073/pnas.86.14.5478
Lissemore, J. L., Jankowski, J. T., Thomas, C. B., Mascotti, D. P., and deHaseth, P. L. (2000). Green fluorescent protein as a quantitative reporter of relative promoter activity in E. coli. Biotechniques 82-84, 88–89. doi: 10.2144/00281st02
Marschaus, L., and Pfeifer, F. (2012). A dual promoter region with overlapping activator sequences drives the expression of gas vesicle protein genes in haloarchaea. Microbiology 158, 2815–2825. doi: 10.1099/mic.0.060178-0
Nomura, S., and Harada, Y. (1998). Functional expression of green fluorescent protein derivatives in Halobacterium salinarum. FEMS Microbiol. Lett. 167, 287–293. doi: 10.1111/j.1574-6968.1998.tb13241.x
Offner, S., Hofacker, A., Wanner, G., and Pfeifer, F. (2000). Eight of fourteen gvp genes are sufficient for formation of gas vesicles in halophilic archaea. J. Bacteriol. 182, 4328–4336. doi: 10.1128/JB.182.15.4328-4336.2000
Offner, S., Wanner, G., and Pfeifer, F. (1996). Functional studies of the gvpACNO operon of Halobacterium salinarium reveal that the GvpC protein shapes gas vesicles. J. Bacteriol. 178, 2071–2078. doi: 10.1128/jb.178.7.2071-2078.1996
Oren, A. (2002). Diversity of halophilic microorganisms: environments, phylogeny, physiology, and applications. J. Ind. Microbiol. Biotechnol. 28, 56–63. doi: 10.1038/sj/jim/7000176
Patterson, G. H., Knobel, S. M., Sharif, W. D., Kain, S. R., and Piston, D. W. (1997). Use of the green fluorescent protein and its mutants in quantitative fluorescence microscopy. Biophys. J. 73, 2782–2790. doi: 10.1016/S0006-3495(97)78307-3
Pédelacq, J.-D., Cabantous, S., Tran, T., Terwilliger, T. C., and Waldo, G. S. (2005). Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88. doi: 10.1038/nbt1172
Pfeifer, F. (2015). Haloarchaea and the formation of gas vesicles. Life 5, 385–402. doi: 10.3390/life5010385
Pfeifer, F., and Ghahraman, P. (1993). Plasmid pHH1 of Halobacterium salinarium: characterization of the replicon region, the gas vesicle gene cluster and insertion elements. Mol. Gen. Genet. 238, 193–200. doi: 10.1007/BF00279547
Pfeifer, F., Griffig, J., and Oesterhelt, D. (1993). The fdx gene encoding the [2Fe-2S] ferredoxin of Halobacterium salinarium (H. halobium). Mol. Gen. Genet. 239, 66–71. doi: 10.1007/BF00281602
Reuter, C. J., and Maupin-Furlow, J. A. (2004). Analysis of proteasome-dependent proteolysis in Haloferax volcanii cells, using short-lived green fluorescent proteins. Appl. Environ. Microbiol. 70, 7530–7538. doi: 10.1128/AEM.70.12.7530-7538.2004
Robinson, J. L., Pyzyna, B., Atrasz, R. G., Henderson, C. A., Morrill, K. L., Burd, A. M., et al. (2005). Growth kinetics of extremely halophilic archaea (Family Halobacteriaceae) as revealed by arrhenius plots. J. Bacteriol. 187, 923–929. doi: 10.1128/JB.187.3.923-929.2005
Sartorius-Neef, S., and Pfeifer, F. (2004). In vivo studies on putative shine-dalgarno sequences of the halophilic archaeon Halobacterium salinarum. Mol. Microbiol. 51, 579–588. doi: 10.1046/j.1365-2958.2003.03858.x
Schägger, H., and von Jagow, G. (1987). Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166, 368–379. doi: 10.1016/0003-2697(87)90587-2
Schmidt, I., and Pfeifer, F. (2013). Use of GFP-GvpE fusions to quantify the GvpD-mediated reduction of the transcriptional activator GvpE in haloarchaea. Arch. Microbiol. 195, 403–412. doi: 10.1007/s00203-013-0885-7
Scholz, O., Thiel, A., Hillen, W., and Niederweis, M. (2000). Quantitative analysis of gene expression with an improved green fluorescent protein. Eur. J. Biochem. 267, 1565–1570. doi: 10.1046/j.1432-1327.2000.01170.x
Stepanenko, O. V., Stepanenko, O. V., Kuznetsova, I. M., Verkhusha, V. V., and Turoverov, K. K. (2014). Sensitivity of superfolder GFP to ionic agents. PLoS One 9:e110750. doi: 10.1371/journal.pone.0110750
Stroud, A., Liddell, S., and Allers, T. (2012). Genetic and biochemical identification of a novel single-stranded DNA-binding complex in Haloferax volcanii. Front. Microbiol. 3:224. doi: 10.3389/fmicb.2012.00224
Teerawanichpan, P., Hoffman, T., Ashe, P., Datla, R., and Selvaraj, G. (2007). Investigations of combinations of mutations in the jellyfish green fluorescent protein (GFP) that afford brighter fluorescence, and use of a version (VisGreen) in plant, bacterial, and animal cells. Biochim. Biophys. Acta 1770, 1360–1368. doi: 10.1016/j.bbagen.2007.06.005
Tsien, R. Y. (1998). The green fluorescent protein. Annu. Rev. Biochem. 67, 509–544. doi: 10.1146/annurev.biochem.67.1.509
Winter, K., Born, J., and Pfeifer, F. (2018). Interaction of haloarchaeal gas vesicle proteins determined by split-GFP. Front. Microbiol. 9:1897. doi: 10.3389/fmicb.2018.01897
Zacharias, D. A., Violin, J. D., Newton, A. C., and Tsien, R. Y. (2002). Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 296, 913–916. doi: 10.1126/science.1068539
Keywords: promoter studies, 5′-untranslated region, Halobacterium, Haloferax, gas vesicle genes
Citation: Born J and Pfeifer F (2019) Improved GFP Variants to Study Gene Expression in Haloarchaea. Front. Microbiol. 10:1200. doi: 10.3389/fmicb.2019.01200
Received: 08 March 2019; Accepted: 13 May 2019;
Published: 29 May 2019.
Edited by:
Harold J. Schreier, University of Maryland, Baltimore County, United StatesReviewed by:
Mike L. Dyall-Smith, The University of Melbourne, AustraliaKelly Bidle, Rider University, United States
Copyright © 2019 Born and Pfeifer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Felicitas Pfeifer, cGZlaWZlckBiaW8udHUtZGFybXN0YWR0LmRl