 Yongsheng Xie1†
Yongsheng Xie1† Xiaoru Wang2†
Xiaoru Wang2† Danping Su1†
Danping Su1† Junsen Feng1
Junsen Feng1 Liuming Wei1
Liuming Wei1 Weiyou Cai1
Weiyou Cai1 Jinhui Li1
Jinhui Li1 Shaorong Lin1
Shaorong Lin1 He Yan2*
He Yan2* Dongsheng He1*
Dongsheng He1*- 1College of Veterinary Medicine, South China Agricultural University, Guangzhou, China
- 2School of Food Science and Engineering, South China University of Technology, Guangzhou, China
Atypical porcine pestivirus (APPV) is an RNA virus newly discovered from swine in Asia, Europe, and the Americas. This novel virus has been confirmed as the cause of congenital tremor (CT) in piglets, which causes extensive economic losses to the swine industry. To investigate the genetic diversity and evolutionary relationship of APPV in China, 83 piglet samples with severe CT clinical signs were obtained from 12 commercial swine farms in 3 provinces of Southern China. RT-PCR revealed that the positive rates of APPV were as high as 100% (12/12) for the swine farms and 90.4% (75/83) for the samples. Subsequently, 21 positive samples and 3 positive samples were selected for partial E2 gene and complete polyprotein gene sequencing, respectively. Phylogenetic analysis showed that 62.5% of the sequences belonged to a novel APPV clade provisionally named genotype 3, which showed 81.0–82.1% sequence identity to genotypes 1 and 2. Amino acid sequence alignment showed that E2 protein of genotype 3 has three specific mutation sites, namely I19V, Y82F, and N107G. The results of the present study demonstrate that a novel APPV subgenotype, which is widely distributed in severe CT clinical samples in Southern China, was genetically diverse. We advocate for the inclusion of genotype 3 during revision of the APPV typing method.
Introduction
Pestiviruses are highly mutable RNA viruses comprising bovine viral diarrhea viruses 1 and 2, border disease virus, and classical swine fever virus (CSFV), which are responsible for huge economic losses in the swine industry worldwide (Postel et al., 2016, 2017; Smith et al., 2017; Zhou et al., 2019). In 2015, Hause et al. (2015) using metagenomic sequencing, found a novel pestivirus, named atypical porcine pestivirus (APPV), in swine serum samples that were positive for porcine reproductive and respiratory syndrome virus. Soon thereafter, this novel virus, which has caused extensive economic losses to the pig industry, was confirmed as the cause of congenital tremor (CT) in piglets in Netherlands, United States, Germany, Austria, China, Spain, Hungary, Brazil, and other countries (Arruda et al., 2016; de Groof et al., 2016; Munoz-Gonzalez et al., 2017; Postel et al., 2017; Schwarz et al., 2017; Yuan et al., 2017; Zhang et al., 2017; Denes et al., 2018; Mosena et al., 2018; Possatti et al., 2018). Recent studies have shown that APPV also circulates in boars in Germany and US; this finding merits further research in this regard (Cagatay et al., 2018; Gatto et al., 2018a).
Pigs affected by CT are commonly known as “shaker pigs” or “dancing pigs” (de Groof et al., 2016; Gatto et al., 2018b; Zhang et al., 2018b; Pan et al., 2019). The main clinical symptoms are rhythmic tremors of the head, abdomen, and limbs. These tremors gradually become more debilitating, causing difficulty in standing or a complete inability to walk, ultimately leading to starvation of affected piglets as they cannot stand to feed (de Groof et al., 2016; Schwarz et al., 2017; Yuan et al., 2017; Gatto et al., 2018b).
APPV is a highly variable single-stranded RNA virus with a genome of approximately 12 kb in length; it contains a long open reading frame (ORF) encoding a polyprotein, composed of four structural proteins (C, Erns, E1, and E2) and eight non-structural proteins (Npro, P7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B) (Hause et al., 2015; Pan et al., 2019). de Groof et al. (2016) provisionally divided APPV into three clusters based on genome analyses. However, as more sequences have been discovered, the phylogenetic tree of APPV has become more complicated (de Groof et al., 2016; Zhang et al., 2018b; Pan et al., 2019). Latest research divided APPV into five different clusters (A-E) (Zhou et al., 2019).
So far, although some novel APPV sequences have been identified (Postel et al., 2017; Zhang et al., 2018b, 2018c; Pan et al., 2019; Zhou et al., 2019), strains available for biological, origin, and evolution analyses are scarce, rendering studies for understanding APPV genetic diversity and evolutionary relationships inadequate. This study aimed to investigate the diversity of APPV sequences and their evolutionary relationships in Southern China to improve understanding of the epidemiological characteristics, evolution, and genetic divergence of APPV.
Materials and Methods
Collection of CT Clinical Samples
From May 2017 to March 2018, 83 pathology samples of newborn piglets which died from CT were collected from 12 commercial swine farms in Southern China (Guangdong, Jiangxi, and Anhui provinces), and submitted for immediate diagnostic investigation. Each sample tested included the brain, lung, heart, kidney, spleen, and lymph nodes of the piglet. Seventy samples were from 10 swine farms in Guangdong province, six samples were from a swine farm in Jiangxi province, and seven samples were from a swine farm in Anhui province. Each of the twelve commercial swine farms housed more than 2,000 breeding sows with no pigs being introduced from other countries in the last 2 years. Detailed clinical sample information is shown in Table 1. This work complied with the Laboratory Animals-Guideline of Welfare and Ethics published by the General Administration of Quality Supervision, Inspection, and Quarantine of the People’s Republic of China. Studies were carried out in accordance with animal ethics guidelines and approved by the Animal Care and Use Committee of South China Agriculture University, China.
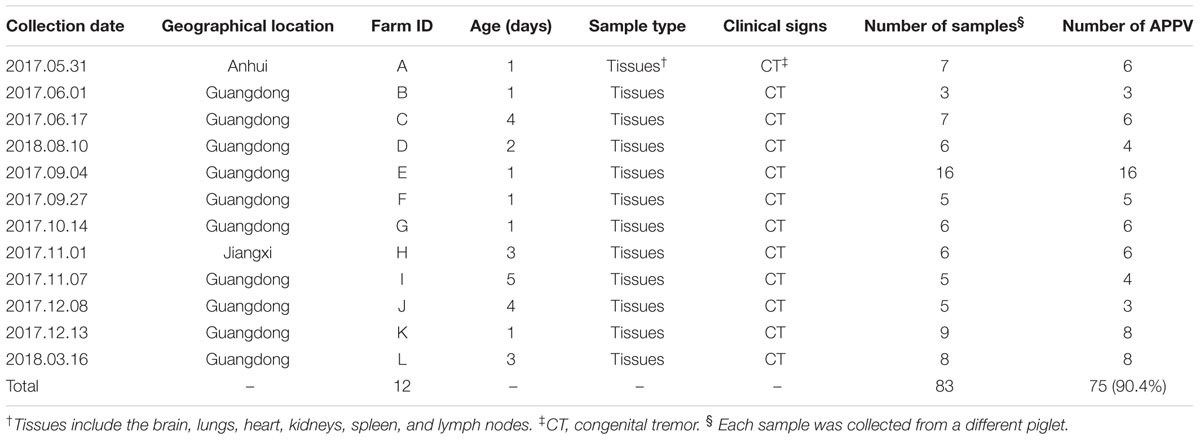
Table 1. Clinical samples collected between 2017 and 2018 in Southern China and results of APPV detection using RT-PCR.
Viral RNA Extraction
Pathology samples were homogenized using TissueLyser II (Qiagen, Hilden, Germany) and diluted 10-fold with 0.1 M phosphate-buffered saline (pH 7.4), frozen and thawed three times, then centrifuged at 8,000 × g for 30 min at 4°C. Viral RNA was isolated from the supernatants using the QIAamp Viral RNA Mini Kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions and immediately stored at -80°C until use.
Primer Design and RT-PCR Detection of APPV
Based on published APPV sequences, primers specific to conserved regions of the genome (primers F0 and R0; Table 2) were designed for RT-PCR detection of APPV using Primer 6.0 software. RT-PCR amplification was performed using a one-step RT-PCR kit (TaKaRa, Dalian, China) according to the manufacturer’s instructions. The amplification volume was 25 μL, and the amplification conditions were as follows: 50°C for 30 min and 94°C for 2 min for the RT reaction, followed by 35 cycles of amplification at 94°C for 30 s, 56°C for 30 s, and 72 C for 30 s, with a final extension at 72 C for 7 min.

Table 2. Primers used for the detection of APPV and amplification of the complete polyprotein gene.
Amplification and Sequencing of the E2 Gene and Complete APPV Genome
Based on the number of APPV positive samples in each farm (Table 1), we selected 21 positive samples and 3 positive samples from 75 positive samples for partial E2 gene, and complete ORF gene sequencing, respectively. Ten pairs of specific primers (F0–F9 and R0–R9, Table 2) were used to amplify the complete APPV polyprotein gene, of which primers F2 and R2 (Table 2) were used to amplify the E2 gene. The PCR products were purified and then cloned into the pMD19-T vector (TaKaRa, Dalian, China). Ten positive plasmids of each amplicons were selected and sequenced in both directions by Guangzhou BGI. Finally, we obtained 24 APPV sequences (including 3 complete ORF sequences and 21 partial E2 gene sequences) from 12 different swine farms, and these sequences have been deposited in the GenBank database under accession numbers MK347474 to MK347497 (Supplementary Table S2).
Sequence Analysis
Complete genomic sequences and E2 gene sequences of APPV available in the National Center of Biotechnology Information database (accessed January 2019) were downloaded for phylogenetic and nucleotide alignment analyses, including 30 complete ORF sequences and 46 E2 gene sequences. The MegAlign software was used for multiple sequence alignment analysis. Phylogenetic analysis was performed by the neighbor-joining (NJ) method with 1,000 bootstrap replicates in MEGA 6.06 software.
Results
Brief Description of CT at Swine Farms and Detection of APPV by RT-PCR
The clinical samples utilized in this study were all obtained from piglets within the first week of birth. Affected piglets experienced whole-body tremors shortly after birth, leading to difficulty in walking and inability to feed. Many cachexic piglets died within a week of CT onset. The incidence of CT in new-born piglets in different litters varied from 10 to 60%. Piglet mortality rates from CT at swine farms D, E, K, and L (Table 1) averaged at approximately 20%, whereas the mortality rate at the remaining eight farms was significantly lower at 10%.
RT-PCR analysis of the 83 CT samples collected from the 12 commercial swine farms showed that 75 samples (90.4%) were positive for APPV, and swine farm positive rates were as high as 100%, indicating that APPV was directly correlated with CT. In addition, we found APPV infections at a swine farm within Anhui Province of China, which has not been reported before.
Alignment of Complete Genome and E2 Gene Sequences
Three complete ORF sequences, obtained from two different swine farms in Guangdong province, were named GD-LDCT1, GD-YJHSEY2N, and GD-YJHSEY3N. The ORF sequence alignments revealed that average nucleotide identity (ANI) among these three sequences was 81.2–99.9%, and ANI with reference sequences from China was between 81.0 and 99.6% (Table 3). GD-LDCT1 had the lowest ANI of 81.0% with sequences KY475593 and KY612413 found in Guangdong province by Shen et al. (2018) and Zhang et al. (2017), respectively, whereas GD-YJHSEY2N showed the highest homology at 99.6% to sequence KX950762 found in Guangdong province (Zhang et al., 2017; Table 3). Furthermore, the three complete ORF sequences shared only 81.6–83.7% ANI with complete APPV sequences identified from other countries (Table 3). GD-LDCT1 showed the highest homology of 82.1% with 2 strains (LT594521, KX929062) from Germany and Netherlands and had lowest nucleotide similarity of 81.6% with the strain KU041639 from Germany and strain KU194229 from United States (Arruda et al., 2016; de Groof et al., 2016; Postel et al., 2016; Beer et al., 2017; Table 3). GD-YJHSEY3N shared the highest homology of 83.7% with MF979135 identified in Korea and displayed lowest nucleotide similarity of 83.2% with KU194229 from US (Arruda et al., 2016; Table 3). Taken together, ORF sequence alignment results demonstrated that the three complete ORF sequences possessed low nucleotide identity to reference sequences from other countries but showed higher homology with sequences isolated in China.
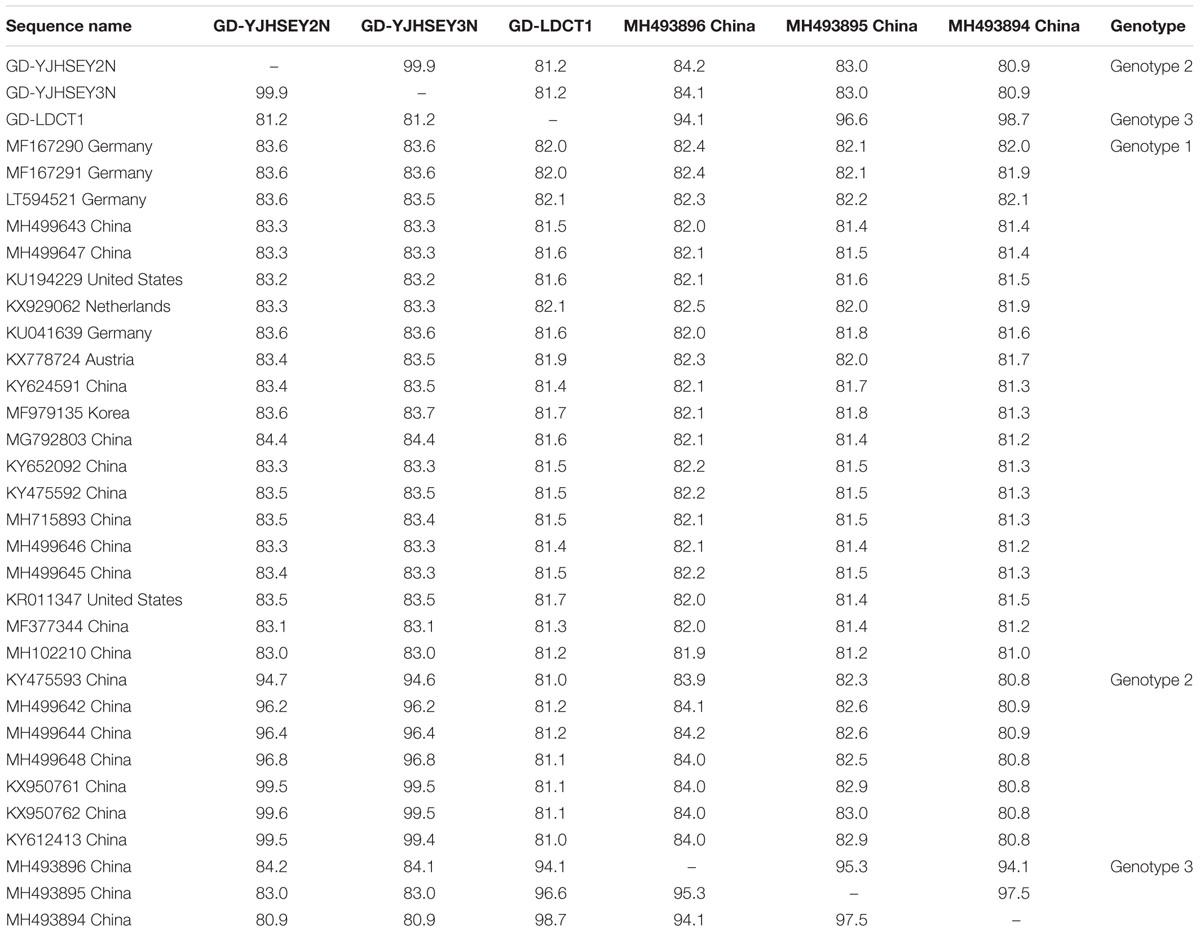
Table 3. Nucleotide identity (%) of three complete ORF sequences with 30 published reference sequences available in GenBank.
Similar to most published reference sequences, the ORF gene for these three complete ORF sequences had a sequence length of 10,908 bp, without deletions and insertions, with the polyprotein comprising 3,635 amino acids. However, amino acid alignment between the three new APPV sequences and the reference sequences from GenBank showed that a total of four APPV reference sequences had amino acid deletions or insertions in the polyprotein. The ORF of reference sequences MH499648 (genotype 2) and MG792803 (genotype 1, 1c cluster) was 10,911 bp long and translated into 3,636 amino acids (Wu et al., 2018; Zhou et al., 2019). MH499648 has a lysine inserted at position 653 of the polyprotein (Figure 1A), while MG792803 has a tyrosine inserted at position 3533 (Figure 1B). In addition, the sequence MH499645 (genotype 1, 1c cluster) ORF is 10,923 bp long and is translated into 3,640 amino acids (Zhou et al., 2019); it has five amino acids inserted at position 1810 of the polyprotein compared to other APPV sequences (Figure 1C). Furthermore, the MH499647 ORF (genotype 1, 1a cluster) has a length of 10,905 bp and is translated into 3,634 amino acids, with a deletion of glycine at position 945 (Figure 1D; Zhou et al., 2019). Currently, it is unclear whether amino acid deletions or insertions in the polyprotein of APPV can affect the pathogenicity of this virus, and further research is required to determine this.

Figure 1. Polyprotein amino acid of APPV alignment by Megalign software available in DNASTAR (http://www.dnastar.com/). Amino acid deletion and insertion regions of the four reference sequences are indicated using rectangular boxes. (A) MH499648 (genotype 2) has a lysine inserted at position 653 of the polyprotein. (B) MG792803 (genotype 1) has a tyrosine inserted at position 3533. (C) MH499645 (genotype 1) has five amino acids (serine, leucine, valine, arginine, and threonine) inserted at position 1810. (D) MH499647 (genotype 1) with a deletion of glycine at position 945.
In addition, partial E2 gene sequence alignments were performed with 46 E2 gene reference sequences. We found that E2 sequences identified in this study shared 79.5–100% ANI with each other (Supplementary Table S1). However, the homology with reference sequences from other countries was between 80.6 and 86.6%, and the homology with reference sequences published in China was 79.3–99.8% (Supplementary Table S1), indicating that the E2 gene sequences in this study showed higher homology to the domestically identified sequences.
Phylogenetic Analysis and Molecular Features of APPV
The phylogenetic tree based on complete ORF gene was constructed using the 30 published APPV full-length strains. This analysis showed that all APPV strains from different countries were distributed in different clusters (Figure 2). Strains identified from China were distributed in almost every cluster of the phylogenetic tree, demonstrating that the origin of Chinese APPV strains is very complex. Detailed analysis of the phylogenetic tree showed that all sequences could be divided into three clades. Based on the genotype naming convention for Pestivirus, these three clades were provisionally named genotypes 1, 2, and 3 (Blome et al., 2017; Postel et al., 2018). Genotype 1 included two subclades (1.1 and 1.2) based on evolutionary relationship (Figure 2). In addition, the 1.1 subclade covered most sequences, which can be further divided into three small clusters, 1.1a, 1.1b, and 1.1c; and the only four complete APPV sequences identified in Germany were located in 1.1a and 1.1b clusters (Figure 2). Strains in 1.1c cluster were all isolated from China, and the only complete APPV sequence isolated from Korea belonged to 1.1b (Figure 2). In this study, strains GD-YJHSEY2N and GD-YJHSEY3N belonged to genotype 2, but strains GD-LDCT1 belonged to genotype 3. Unexpectedly, three mutant APPV sequences discovered by Zhang et al. (2018c) also belonged to genotype 3. This is a newly discovered subgenotype with less than 85% ANI compared to genotypes 1 and 2 (Table 3).
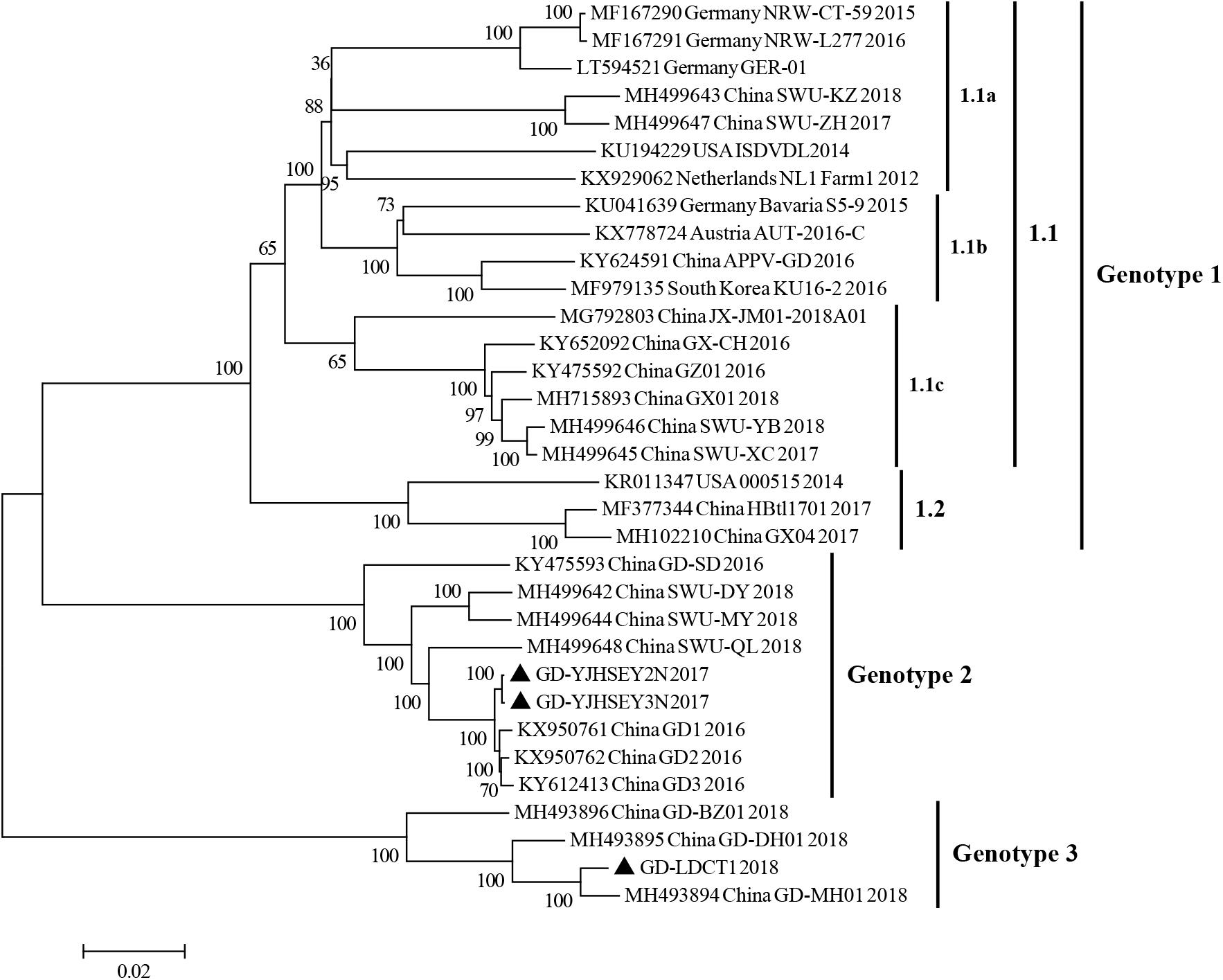
Figure 2. Phylogenetic trees based on the sequences encoding the polyprotein of APPV. Three complete ORF sequences obtained in this study are marked with black triangles. Published sequences, including 30 complete APPV sequences, were downloaded from the GenBank database. MEGA 6.0 software was used to construct the neighbor-joining (NJ) phylogenetic trees using a p-distance method with a bootstrap of 1,000 replicates.
To further explore the evolutionary relationship between E2 sequences identified in this study and previously published APPV E2 sequences, a phylogenetic tree analysis based on partial E2 gene was also performed. Similar to the results obtained using the polyprotein gene phylogenetic tree, all E2 sequences analyzed could be divided into three clades (genotypes 1, 2, and 3) (Figure 3). Further analysis indicated that, in this study, 62.5% of the sequences belonged to genotype 3, 37.5% belonged to genotype 2, and while no sequence belonged to genotype 1.
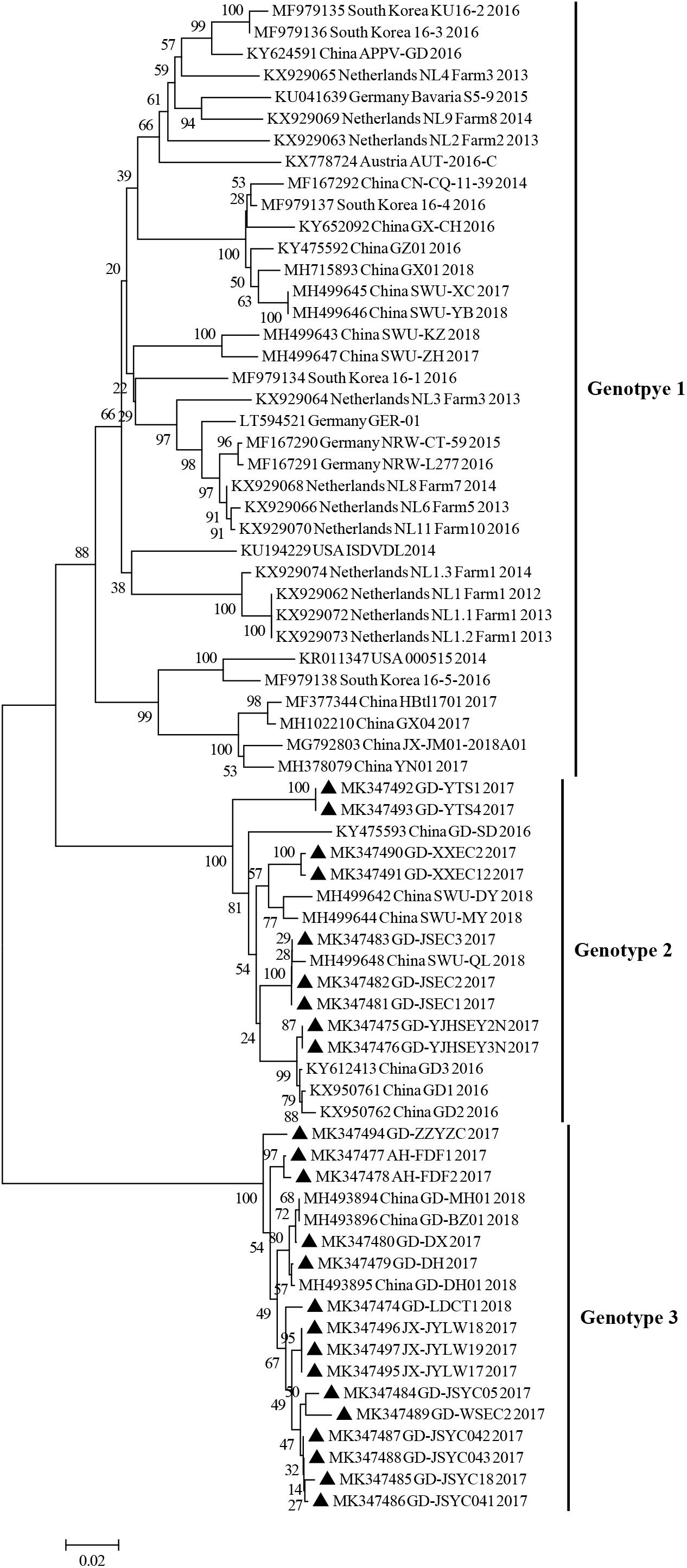
Figure 3. Phylogenetic trees based on the partial sequences (633 bp) of the E2 gene. E2 gene sequences obtained in this study are marked with black triangles. Published sequences, including 46 E2 gene sequences, were downloaded from the GenBank database. MEGA 6.0 software was used to construct the NJ phylogenetic trees using a p-distance method with a bootstrap of 1,000 replicates.
Amino acid alignment of the E2 protein (partial, 211 amino acids) revealed that there were four specific amino acid mutation sites in these three genotypes, which may affect the genotypes of different strains (Figures 4A,B). Genotype 3 contained three amino acid mutations at positions 19, 82, and 107, which were valine, phenylalanine, and glycine, respectively; while genotype 2 contained the S39T mutation; and genotype 1 contained isoleucine, serine, tyrosine, and asparagine at positions 19, 39, 82, and 107, respectively (Figures 4A,B). E2 gene is one of the structural protein genes of APPV and contains several B and T cell epitopes; therefore, E2 protein is subjected to greater immune selection pressure, and we speculated that these mutation sites may be related to some antigenic epitopes of the virus.
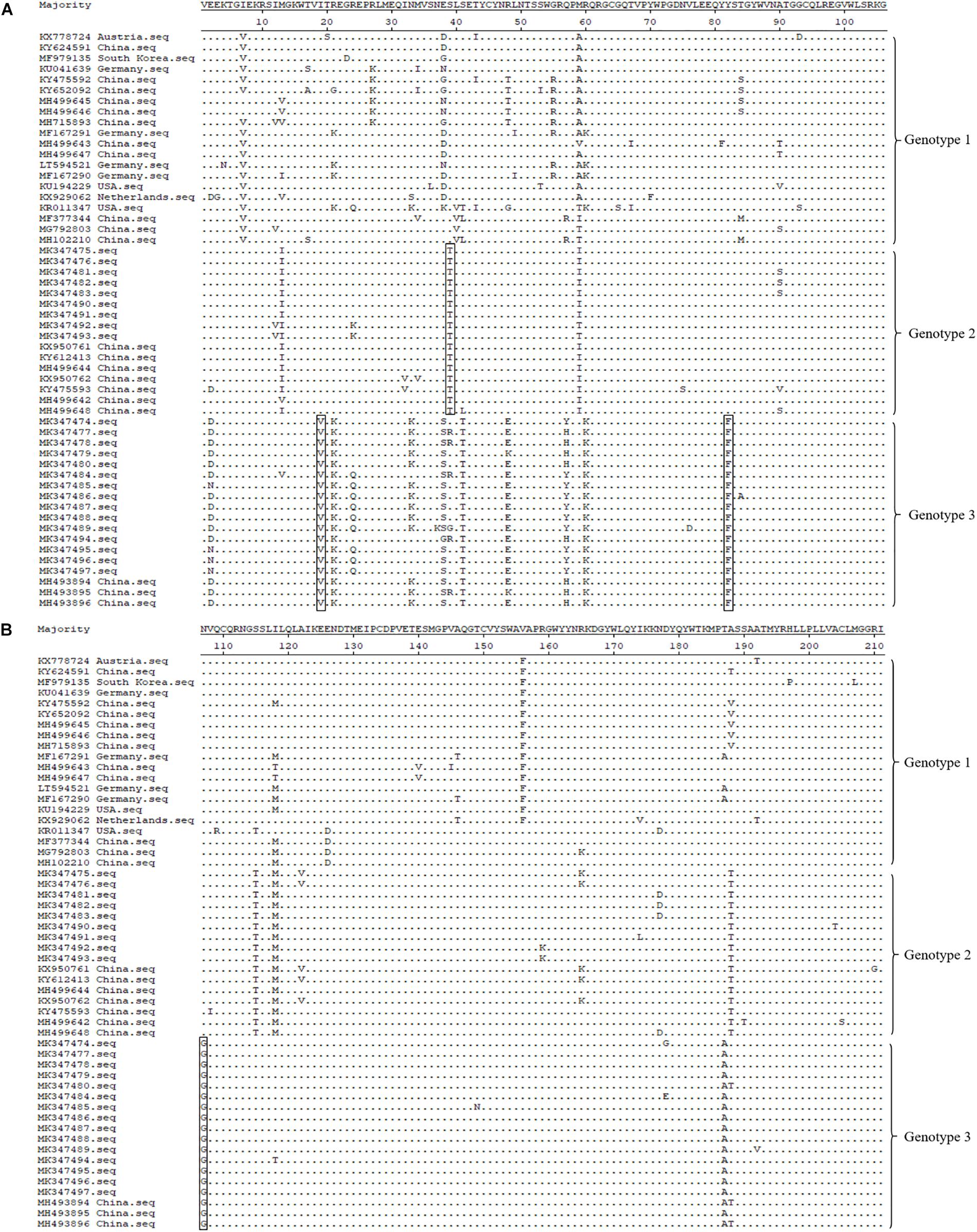
Figure 4. Molecular feature analysis of APPV E2 protein (partial, 211 amino acids). Four specific amino acid mutation sites (I19V, S39T, Y82F, and N107G), which are used for different APPV genotype divisions, are depicted using rectangular boxes. Panel (A) displayed is the alignment of the 106 amino acid sequence at the front end of E2 gene. Panel (B) displayed is the alignment of the 105 amino acid sequences at the end of E2 gene.
Discussion
Congenital tremor is a common clinical disease in piglets in the swine industry, the clinical symptoms of which were first described nearly a century ago (de Groof et al., 2016; Yuan et al., 2017). After that, studies have shown that factors such as malnutrition, genetics, and viral infections can cause CT clinical signs in piglets (Knox et al., 1978; Vannier et al., 1981; Bradley et al., 1983). In China, the earliest studies on CT can be traced back to the 1960s (Yuan et al., 2017). Through a series of experiments conducted in the following decades, some researchers determined that CT was caused by viral infection, but were not able to find the true pathogen (Yuan et al., 2017). de Groof et al. (2016) confirmed through experiments in pigs that CT was associated with APPV. Then, many studies further confirmed the correlation between APPV and CT. In Austria, APPV infection directly led to a 10% increase in piglet mortality, and APPV antibodies could be detected in both sows and affected piglets; quantitative RT-PCR results showed that adult pigs had a high viral load in the saliva and semen (Schwarz et al., 2017). In Spain, APPV infection can be traced back to 1997, and researchers detected APPV infection in 2-day-old piglets with CT symptoms (Munoz-Gonzalez et al., 2017). In China, Yuan et al. (2017) detected the first APPV sequence in Asia in new-born piglets with CT symptoms, and found the highest viral load in the submaxillary lymph nodes. Two APPV sequences discovered in Guangdong province by Zhang et al. (2017) formed an independent cluster in the phylogenetic tree. Shen et al. (2018) found that APPV infection can cause 60% of new-born piglets to die or be eliminated. In this study, we detected APPV in piglets that died from CT in Southern China; the positive rate of the sample was 90.4% and that of farms was as 100%. Among the piglets infected with CT, the morbidity at different farms ranged from 10 to 60%, and four farms had a mortality rate of 20% because of CT, which resulted in huge economic losses to these farms. Most importantly, two strains of APPV (MK347477 and MK347478) were identified from the Anhui province of China for the first time. This research further confirmed that APPV had a direct correlation with CT, which was consistent with the published studies (Arruda et al., 2016; de Groof et al., 2016; Munoz-Gonzalez et al., 2017; Schwarz et al., 2017; Yuan et al., 2017; Zhang et al., 2017; Shen et al., 2018).
E2 gene is one of the major structural protein genes of APPV. In this study, to analyze the evolutionary relationship of APPV sequences, phylogenetic tree of E2 gene sequences were constructed. Results showed that the phylogenetic trees of the complete ORF genes and E2 genes were similar, and all APPV sequences could be divided into 3 clades (Figures 3, 4). As established, the APPV genome is long and susceptible to mutations, and thus the complete genomic sequence cannot be easily obtained (Postel et al., 2017; Yuan et al., 2017). Therefore, in epidemiological studies, researchers have used different genes (such as E2, NS2-3, NS5B, and Npro) instead of complete genes to construct phylogenetic trees for APPV genotype determination (Beer et al., 2017; Cagatay et al., 2018; Gatto et al., 2018b; Pan et al., 2019; Zhou et al., 2019). However, in this study, the results from phylogenetic analysis of complete ORF genes and E2 genes were quite similar, indicating that the E2 gene could be utilized for phylogenetic analysis in future researches to determine the correct genotypes of different sequences.
It should be noted that many researchers have found sequences of genotype 1 in Chinese swine herds, but sequence alignment and phylogenetic analysis showed that these sequences have high homology and phylogenetic relationship with genotype 1 sequences identified from other countries (Yuan et al., 2017; Zhou et al., 2019). Zhang et al. (2018c) have recently discovered three novel APPV sequences, and ORF sequence alignments revealed that GD-LDCT1 showed the highest ANI of 94.1–98.7% with these three sequences (Table 3); however, the authors did not investigate the prevalence of APPV in China. In this study, phylogenetic analysis showed that these sequences also belonged to genotype 3, and we found many genotype 3 APPV sequences in CT-affected piglets in the southern province of China. Further, our results showed that APPV sequences identified from eight swine farms belonged to genotype 3, indicating a high prevalence of genotype 3. So far, only China has reported sequences of genotype 3 of APPV, and the 12 commercial swine farms in this study had no breeding pigs introduced from other countries in the past 2 years. Sequence alignment results showed that genotype 3 shared less than 85% nucleotide identities with genotypes 1 and 2 (Table 3); thus, this is a novel sequence, and it can be speculated that the genotype 3 APPV strain originated in China rather than being imported from another country.
It is well known that new-born piglets have poor resistance to disease. After APPV infection, CT can cause piglets to walk with difficulty and be unable to feed on colostrum, thereby starving or being crushed to death (Yuan et al., 2017; Gatto et al., 2018b). Vaccines reducing the effects of APPV on new-born piglets may be a highly effective treatment. The E2 protein of CSFV can induce a strong immune response as well as resistance to viral infection; based on this principle, Zhang et al. (2018a) have successfully expressed the E2 protein of APPV in vitro in a baculovirus system. Results showed that the E2 protein, combined with a different adjuvant, may induce the production of APPV-specific antibodies in mice, indicating that the E2 protein could potentially be utilized as an effective subunit vaccine to prevent APPV infection (Zhang et al., 2018a).
Conclusion
This study found APPV in CT samples from 12 commercial pig farms in Southern China. Sequencing and phylogenetic analysis were performed on positive samples. Results clearly showed that 62.5% of the sequences belonged to genotype 3. Thus, further research into modes of transmission and vaccine development is paramount in order to reduce future negative impact to the swine industry.
Data Availability
The datasets generated for this study can be found in GenBank database, MK 347474 to MK347497.
Author Contributions
YX conceived and designed the experiments and drafted the manuscript. XW analyzed the data and tools. DS, LW, and WC completed the collection of samples. JF, JL, and SL performed the experiments. HY and DH conceived the study and critically revised the manuscript. All authors read and approved the final version of the manuscript.
Funding
This work was supported by the Guangdong Province Pig Industrial System Innovation Team (Grant No. 2018LM1103), the National Key Basic Research Program (Grant No. 2016YFD0500606), the Construction of the First Class Universities (Subject) and Special Development Guidance Special Fund (Grant No. K5174960), and the Fundamental Research Funds for the Central Universities, SCUT (Grant No. D2170320).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01406/full#supplementary-material
References
Arruda, B. L., Arruda, P. H., Magstadt, D. R., Schwartz, K. J., Dohlman, T., Schleining, J. A., et al. (2016). Identification of a divergent lineage porcine pestivirus in nursing piglets with congenital tremors and reproduction of disease following experimental inoculation. PLoS One 11:e0150104. doi: 10.1371/journal.pone.0150104
Beer, M., Dräger, C., Höper, D., Pohlmann, A., Bergermann, C., et al. (2017). High prevalence of highly variable atypical porcine pestiviruses found in Germany. Transbound. Emerg. Dis. 64, e22–e26. doi: 10.1111/tbed.12532
Blome, S., Staubach, C., Henke, J., Carlson, J., and Beer, M. (2017). Classical swine fever-an updated review. Viruses 9:E86. doi: 10.3390/v9040086
Bradley, R., Done, J. T., Hebert, C. N., Overby, E., Askaa, J., Basse, A., et al. (1983). Congenital tremor type AI: light and electron microscopical observations on the spinal cords of affected piglets. J. Comp. Pathol. 93, 43–59. doi: 10.1016/0021-9975(83)90042-7
Cagatay, G. N., Antos, A., Meyer, D., Maistrelli, C., Keuling, O., Becher, P., et al. (2018). Frequent infection of wild boar with atypical porcine pestivirus (APPV). Transbound. Emerg. Dis. 65, 1087–1093. doi: 10.1111/tbed.12854
de Groof, A., Deijs, M., Guelen, L., van Grinsven, L., van Os-Galdos, L., Vogels, W., et al. (2016). Atypical porcine pestivirus: a possible cause of congenital tremor type a-II in newborn piglets. Viruses 8, 271. doi: 10.3390/v8100271
Denes, L., Biksi, I., Albert, M., Szeredi, L., Knapp, D. G., Szilasi, A., et al. (2018). Detection and phylogenetic characterization of atypical porcine pestivirus strains in Hungary. Transbound. Emerg. Dis. 65, 2039–2042. doi: 10.1111/tbed.12981
Gatto, I. R. H., Arruda, P. H., Visek, C. A., Victoria, J. G., Patterson, A. R., Krull, A. C., et al. (2018a). Detection of atypical porcine pestivirus in semen from commercial boar studs in the United States. Transbound. Emerg. Dis. 65, e339–e343. doi: 10.1111/tbed.12759
Gatto, I. R. H., Harmon, K., Bradner, L., Silva, P., Linhares, D. C. L., Arruda, P. H., et al. (2018b). Detection of atypical porcine pestivirus in Brazil in the central nervous system of suckling piglets with congenital tremor. Transbound. Emerg. Dis. 65, 375–380. doi: 10.1111/tbed.12824
Hause, B. M., Collin, E. A., Peddireddi, L., Yuan, F., Chen, Z., Hesse, R. A., et al. (2015). Discovery of a novel putative atypical porcine pestivirus in pigs in the USA. J. Gen. Virol. 96, 2994–2998. doi: 10.1099/jgv.0.000251
Knox, B., Askaa, J., Basse, A., Bitsch, V., Eskildsen, M., Mandrup, M., et al. (1978). Congenital ataxia and tremor with cerebellar hypoplasia in piglets borne by sows treated with Neguvon vet. (metrifonate, trichlorfon) during pregnancy. Nord Vet Med. 30, 538–545.
Mosena, A. C. S., Weber, M. N., da Cruz, R. A. S., Cibulski, S. P., da Silva, M. S., Puhl, D. E., et al. (2018). Presence of atypical porcine pestivirus (APPV) in Brazilian pigs. Transbound. Emerg. Dis. 65, 22–26. doi: 10.1111/tbed.12753
Munoz-Gonzalez, S., Canturri, A., Perez-Simo, M., Bohorquez, J. A., Rosell, R., Cabezon, O., et al. (2017). First report of the novel atypical porcine pestivirus in Spain and a retrospective study. Transbound. Emerg. Dis. 64, 1645–1649. doi: 10.1111/tbed.12699
Pan, S., Yan, Y., Shi, K., Wang, M., Mou, C., and Chen, Z. (2019). Molecular characterization of two novel atypical porcine pestivirus (APPV) strains from piglets with congenital tremor in China. Transbound. Emerg. Dis. 66, 35–42. doi: 10.1111/tbed.13029
Possatti, F., Headley, S. A., Leme, R. A., Dall Agnol, A. M., Zotti, E., de Oliveira, T. E. S., et al. (2018). Viruses associated with congenital tremor and high lethality in piglets. Transbound. Emerg. Dis. 65, 331–337. doi: 10.1111/tbed.12807
Postel, A., Austermann-Busch, S., Petrov, A., Moennig, V., and Becher, P. (2018). Epidemiology, diagnosis and control of classical swine fever: recent developments and future challenges. Transbound. Emerg. Dis. 65(Suppl. 1), 248–261. doi: 10.1111/tbed.12676
Postel, A., Hansmann, F., Baechlein, C., Fischer, N., Alawi, M., Grundhoff, A., et al. (2016). Presence of atypical porcine pestivirus (APPV) genomes in newborn piglets correlates with congenital tremor. Sci. Rep. 6, 27735. doi: 10.1038/srep27735
Postel, A., Meyer, D., Cagatay, G. N., Feliziani, F., De Mia, G. M., Fischer, N., et al. (2017). High abundance and genetic variability of atypical porcine pestivirus in pigs from Europe and Asia. Emerg. Infect. Dis. 23, 2104–2107. doi: 10.3201/eid2312.170951
Schwarz, L., Riedel, C., Hogler, S., Sinn, L. J., Voglmayr, T., Wochtl, B., et al. (2017). Congenital infection with atypical porcine pestivirus (APPV) is associated with disease and viral persistence. Vet. Res. 48, 1. doi: 10.1186/s13567-016-0406-1
Shen, H., Liu, X., Zhang, P., Wang, L., Liu, Y., Zhang, L., et al. (2018). Identification and characterization of atypical porcine pestivirus genomes in newborn piglets with congenital tremor in China. J. Vet. Sci. 19, 468–471. doi: 10.4142/jvs.2018.19.3.468
Smith, D. B., Meyers, G., Bukh, J., Gould, E. A., Monath, T., Scott Muerhoff, A., et al. (2017). Proposed revision to the taxonomy of the genus Pestivirus, family Flaviviridae. J. Gen. Virol. 98, 2106–2112. doi: 10.1099/jgv.0.000873
Vannier, P., Plateau, E., and Tillon, J. P. (1981). Congenital tremor in pigs farrowed from sows given hog cholera virus during pregnancy. Am. J. Vet. Res. 42, 135–137.
Wu, S., Wang, Z., Zhang, W., and Deng, S. (2018). Complete genome sequence of an atypical porcine pestivirus isolated from Jiangxi province, China. Genome Announc. 6, e439–18. doi: 10.1128/genomeA.00439-18
Yuan, J., Han, Z., Li, J., Huang, Y., Yang, J., Ding, H., et al. (2017). Atypical porcine pestivirus as a novel type of pestivirus in pigs in China. Front. Microbiol. 8:862. doi: 10.3389/fmicb.2017.00862
Zhang, H., Wen, W., Hao, G., Chen, H., Qian, P., and Li, X. (2018a). A subunit vaccine based on E2 protein of atypical porcine pestivirus induces Th2-type immune response in mice. Viruses 10:673. doi: 10.3390/v10120673
Zhang, H., Wen, W., Hao, G., Hu, Y., Chen, H., Qian, P., et al. (2018b). Phylogenetic and genomic characterization of a novel atypical porcine pestivirus in China. Transbound. Emerg. Dis. 65, e202–e204. doi: 10.1111/tbed.12675
Zhang, X., Dai, R., Li, Q., Zhou, Q., Luo, Y., Lin, L., et al. (2018c). Detection of three novel atypical porcine pestivirus strains in newborn piglets with congenital tremor in southern China. Infect. Genet. Evol. 68, 54–57. doi: 10.1016/j.meegid.2018.12.008
Zhang, K., Wu, K., Liu, J., Ge, S., Xiao, Y., Shang, Y., et al. (2017). Identification of atypical porcine pestivirus infection in swine herds in China. Transbound. Emerg. Dis. 64, 1020–1023. doi: 10.1111/tbed.12659
Keywords: atypical porcine pestivirus, APPV, congenital tremor, piglets, phylogenetic analysis, genotype 3, Southern China
Citation: Xie Y, Wang X, Su D, Feng J, Wei L, Cai W, Li J, Lin S, Yan H and He D (2019) Detection and Genetic Characterization of Atypical Porcine Pestivirus in Piglets With Congenital Tremors in Southern China. Front. Microbiol. 10:1406. doi: 10.3389/fmicb.2019.01406
Received: 23 March 2019; Accepted: 04 June 2019;
Published: 20 June 2019.
Edited by:
Douglas Paul Gladue, United States Department of Agriculture, United StatesReviewed by:
Wen Zhang, Jiangsu University, ChinaJulia Francis Ridpath, National Animal Disease Center (USDA – ARS), United States
Copyright © 2019 Xie, Wang, Su, Feng, Wei, Cai, Li, Lin, Yan and He. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: He Yan, eWFuaGVAc2N1dC5lZHUuY24=; Dongsheng He, ZGhlQHNjYXUuZWR1LmNu
†These authors have contributed equally to this work