 Magdalena Kotowska
Magdalena Kotowska Michał Świat
Michał Świat Justyna Zarȩba-Pasławska
Justyna Zarȩba-Pasławska Paweł Jaworski
Paweł Jaworski Krzysztof Pawlik
Krzysztof Pawlik- Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences, Wrocław, Poland
Bacteria from the genus Streptomyces have been long exploited as the most prolific producers of antibiotics, other secondary metabolites and enzymes. They are important members of soil microbial communities that can adapt to changing conditions thank to the fine regulation of gene expression in response to environmental signals. Streptomyces coelicolor A3(2) is a model organism for molecular studies with the most deeply recognized interactions within the complex metabolic and regulatory network. However, details about molecular signals recognized by specialized regulatory proteins as well as their direct targets are often missing. We describe here a zinc-binding protein HypR (SCO6294) which belongs to FadR subfamily of GntR-like regulators. The DNA sequence 5′-TACAATGTCAC-3′ recognized by the HypR protein in its own promoter region was identified by DNase I footprinting. Binding of six DNA fragments containing similar sequences located in other promoter regions were confirmed by the electrophoretic mobility shift assay (EMSA). The sequences of 7 in vitro-determined binding sites were assembled to generate a logo of the HypR binding motif, 5′-CTNTGC(A/C)ATGTCAC-3′. Comparison of luciferase reporter genes expression under the control of cloned promoter regions in S. coelicolor A3(2) wild type and deletion mutant strains revealed, that the HypR protein acts as a repressor of its target genes. Genes belonging to the regulon of HypR code for enzymes putatively involved in collagen degradation and utilization of L-hydroxyproline (L-Hyp) as concluded from predicted structure and conserved domains. Their transcription is induced in the wild type strain by the addition of L-Hyp to the culture medium. Moreover, knockout of one of the genes from the predicted L-Hyp utilization operon abolished the ability of the strain to grow on L-Hyp as a sole source of carbon. To our knowledge, this work is the first indication of the existence of the pathway of L-hydroxyproline catabolism in Streptomycetes.
Introduction
Morphological differentiation and secondary metabolism of Streptomycetes is controlled by a complex regulatory network (Liu et al., 2013). A variety of transcription factors respond to changing environmental conditions by adjusting gene expression. The chemical signals which influence gene expression include available nutrients (or lack of them), metal ions, toxic compounds and signaling molecules produced by other cells of the same organism or by other organisms (van der Heul et al., 2018). One of the driving forces of the studies of the regulatory mechanisms is the hope to increase titers of known useful compounds and to find ways to “wake up” cryptic genes for unknown natural products which may potentially become new drugs (Zarins-Tutt et al., 2016). Streptomyces coelicolor A3(2) has been a model for genetic studies of the genus Streptomyces for over five decades (Hopwood, 1999). It produces four pigmented compounds of polyketide origin: blue actinorhodin, red undecylprodigiosin, gray pigment of spores and yellow coelimycin. A number of regulatory proteins governing the molecular mechanisms of secondary metabolism and differentiation of this bacterium have been identified (Flärdh and Buttner, 2009; Van Keulen and Dyson, 2014). Nevertheless, the characterized regulators compose only a fraction of nearly a thousand of potential regulatory proteins coded by the S. coelicolor A3(2) genome (Bentley et al., 2002).
Bacterial transcription factors from GntR family regulate gene expression in response to environmental signals such as availability of different carbon sources including complex food sources such as chitin. Some of them are pleiotropic regulators, which may act as both repressors and activators of diverse metabolic pathways. They control primary metabolic processes maintaining the balance of specific compounds and are involved in interconnecting the primary and secondary metabolic pathways. They bind DNA by N-terminal winged helix-turn-helix (WHTH) domains and their activity is modulated by binding ligands. The diversity of C-terminal ligand binding domains is the basis for classification of GntR-like proteins into seven subfamilies (FadR, HutC, MocR, YtrA, AraR, DevA, and PlmA) (Hoskisson and Rigali, 2009).
FadR subfamily is the most abundant group of bacterial GntR-like regulators. FadR from Escherichia coli acts as a switch coordinating fatty acid biosynthesis and β-oxidation. When bound to DNA it represses fatty acid degradation (fad) genes and promotes transcription of fabA gene required for biosynthesis of unsaturated fatty acids. Binding of a long chain acyl-CoA effector molecule releases FadR protein from DNA, leading to de-repression of fad genes and inactivation of fabA (Xu et al., 2001). Other members of this subfamily often control transport and catabolism of amino acids and other organic acids.
S. coelicolor A3(2) genome contains 56 genes for GntR-like proteins representing five subfamilies. Eleven of them were described experimentally (Hoskisson et al., 2006; Rigali et al., 2008; Horbal et al., 2013; Persson et al., 2013; Cen et al., 2016; Yu et al., 2015, 2016; Tsypik et al., 2016, 2017), but the low molecular ligands have been identified in only two cases. Glucosamine 6-phosphate is the effector molecule bound by a master regulator DasR which links nutrient stress to antibiotic production (Rigali et al., 2008). The repressor of gluconoate operon, GntR (SCO1678), was recently shown to respond to both gluconoate and glucono-1,5-lactone (Tsypik et al., 2017). In the current work we characterized a GntR-like protein HypR (SCO6294) from S. coelicolor A3(2) and identified its regulatory targets forming putative L-hydroxyproline degradation pathway.
Materials and Methods
DNA Manipulation and Bacterial Strains Growth Conditions
DNA manipulations were carried out by standard protocols (Sambrook and Russell, 2001). All the PCR amplified fragments were first cloned into p-GEM-T Easy vector (Promega) or pTZ57R/T (Thermo Fisher Scientific), verified by DNA sequencing and cloned into appropriate plasmids. Primers, as well as bacterial strains and plasmids, are listed in Supplementary Tables S1, S2, respectively. Culture conditions, transformation and conjugation methods followed the general procedures for E. coli (Sambrook and Russell, 2001) and Streptomyces (Kieser et al., 2000). S. coelicolor was cultivated on the following media: modified 79 medium (without glucose) (Pawlik et al., 2010) and MS (Kieser et al., 2000). For positive selection in bacterial one-hybrid system NM medium was used (Meng et al., 2005). The ability to utilize L-Hyp (50 mM) as a sole source of carbon was tested in the liquid Minimal Medium with (NH4)2SO4 as a nitrogen source (Kieser et al., 2000).
Bacterial One-Hybrid System
A one-step selection procedure of Meng et al. (2005) was applied to isolate clones containing the sequences recognized by HypR protein. The hypR gene was PCR amplified using the primer pair B1 and B2 and cloned into NotI and BamHI restriction sites of pB1H1 plasmid, giving pB1H1-SCO6294, so that HypR was expressed as a fusion protein with the α-subunit of RNA polymerase. Selection strain E. coli US0 (ΔhisBΔpyrF) (Meng et al., 2006) containing plasmid pB1H1-SCO6294 was transformed with a purified library pH3U3-18random (Wolański et al., 2011). Positive clones were selected on plates with NM medium with 5 mM 3-AT (3-amino-triazole, a competitive inhibitor of HIS3 that provides selection for active promoter), kanamycin (30 μg/ml) and chloramphenicol (34 μg/ml). Fragments of pH3U3 plasmid covering the randomized 18 bp sequences from selected clones were amplified using the primer pair B3 and B4 and sequenced. The sequences were analyzed using the MEME algorithm (Bailey and Elkan, 1994)1.
Construction of hypR and SCO6293 Deletion Strains
The hypR gene was deleted by two rounds of homologous recombination (Kieser et al., 2000). Flanking arms were amplified (Supplementary Figure S4) and cloned into pOJ260 plasmid. In the first round the gene was replaced with hygromycin resistance cassette (Ωhyg) using the plasmid pJZ14 resulting in strain P130. In the second round Ωhyg cassette was removed using the plasmid pJZ15 to obtain strain P138. Deletion was verified by PCR (Supplementary Figure S4). SCO6293 gene was deleted using homologous recombination facilitated by SceI meganuclease (Fernández-Martínez and Bibb, 2014). Left and right flanking arms were amplified and sequentially cloned into pIJ12738 vector resulting in pMK44 plasmid (Supplementary Table S2). pIJ12742 plasmid was used to deliver I-SceI meganuclease. Exconjugants after single and double crossing over events were verified by PCR with D12 and D13 primer pair. The SCO6393 deletion mutant strain was named P201.
Construction of Expression Plasmids
hypR gene was amplified using the primer pair G1 and G2 and cloned into NdeI and HindIII sites of pET28a(+) vector to give plasmid pJZE1 for expression of His-tagged protein and into BamHI and HindIII sites of pET28NStrep vector to give plasmid pMS08 for expression of Strep-tagged protein.
Purification of His-Tagged HypR Protein
E. coli BL21(DE3)pLysS strain containing pJZE1 was grown in LB medium with kanamycin (30 μg/ml) and chloramphenicol (34 μg/ml) to an OD600 of 0.4–0.5 at 30°C. Expression was induced with 0.1 mM isopropyl-b-D-thiogalactopyranoside (IPTG) at 30°C for 4 h. After harvesting by centrifugation, the cells from 1 l culture were lysed in 40 ml of BWS-10 buffer (100 mM Tris-HCl pH 8.0, 400 mM NaCl, 10 mM imidazole) by sonication and centrifuged at 4°C, 26,890 × g, 30 min. The clarified lysate was supplemented with 2% Tween 20, mixed with 0.5 ml of Co2+ loaded Chelating Sepharose Fast Flow resin (Amersham Biosciences) and gently agitated for 30 min at room temperature. The resin was collected by centrifugation (4°C, 500 g, 5 min) washed with BWS-10 and transferred to a column. His-tagged HypR protein was eluted by increasing concentration of imidazole in BWS buffer (40 and 250 mM). The eluted fractions were examined using SDS-PAGE. Equal volume of glycerol was added to the fractions (to the final concentration of 50%) and the protein was stored at −20°C.
Purification of Strep-Tagged HypR Protein
E. coli BL21(DE3) strain containing pMS08 was grown in 1 l LB medium with kanamycin (30 μg/ml) to an OD600 of 0.7 at 37°C. Expression was induced with 0.1 mM IPTG at 30°C for 4 h. Cells harvested by centrifugation were disrupted by sonication in 10 ml of buffer W (100 mM Tris-HCl pH 8.0, 150 mM NaCl) and centrifuged at 4°C, 26,890 × g, 30 min. The clarified lysate was mixed with 1 ml of Strep-Tactin Superflow high capacity resin (IBA). The resin was transferred to an empty column and washed with buffer W (7 ml), buffer W supplemented with 200 mM MgCl2 (4 ml) and buffer W (8 ml). Strep-tagged HypR protein was eluted with buffer E (buffer W supplemented with 2.5 mM desthiobiotin). Eluted protein was stored at 4°C until use.
Electrophoretic Mobility Shift Assay (EMSA)
Amplified promoter fragments (see Supplementary Table S1) were cloned directly into the pTZ57R/T vector and used as templates for amplification with IRDye labeled primers P10 and P11 complementary to the sequences flanking the cloning site of pTZ57R plasmid. Each EMSA sample contained 0.03 pmoles of labeled DNA in 16 μl binding buffer (10 mM Tris-HCl pH 7.5, 50 mM KCl, 2.6 mM DTT, 0.16% Tween20, 6 ng/μl herring sperm DNA). His-tagged or Strep-tagged HypR protein was added and samples were incubated for 20 min at room temperature. Four microliter of 40% sucrose was added and samples were loaded on nondenaturing 4% polyacrylamide–Tris–borate–EDTA gels. The results were visualized on Typhoon FLA 9500 apparatus. To test the effect of metal ions on the binding of DNA respective metal chlorides solutions were added at the concentration 5 mM.
DNase I Footprint
For footprinting experiment the fragment p6294 was uniquely radiolabeled at one end during amplification with the primer pair P3 and P4. One of the primers was labeled on its 5′ end with [32P]-ATP using T4 polynucleotide kinase. A binding mixture contained the radiolabeled fragment (approximately 200) in 20 μl binding buffer (10 mM Tris-HCl pH 7.5, 50 mM KCl, 1 mM DTT) and a variable amount of His-tagged HypR protein. The samples were incubated for 20 min at 25°C and 10 min at 30°C. For digestion, 2.5 μl of 10× reaction buffer with MgCl2 for DNase I (Fermentas, Thermo Fisher Scientific) and 2.5 μl of DNase I diluted in 1× reaction buffer (0.005 u/μl) were added and samples were incubated for exactly 5 min at 30°C. The reaction was stopped by addition of 25 μl of Stop buffer (200 mM NaCl, 100 mM EDTA, 1% SDS) and incubation for 10 min at 75°C. DNA from each sample was extracted with phenol/chloroform/isoamyl alcohol (25:24:1) and ethanol-precipitated. The precipitated DNA was dissolved in loading buffer (95% formamide, 20 mM EDTA, 0.05% bromophenol blue, 0.05% xylene cyanol FF), loaded onto a denaturing 8% polyacrylamide –Tris–borate–EDTA gel and visualized with a Typhoon FLA9500 Variable Mode Imager (GE Healthcare). Dideoxy sequencing ladders were generated using Thermo Sequenase Cycle Sequencing Kit (Affymetrix) and the same labeled primers as those used to prepare the probes.
Tryptophan Fluorescence Measurements
Metal ion binding was assayed by monitoring of tryptophan fluorescence (λex = 278 nm, λem = 335 nm) in a 96-well black polystyrene plate on a microplate reader Synergy H4 Hybrid Reader (BioTek). One hundred and fifty microliter samples containing 4 μM Strep-tagged HypR protein in a buffer (100 mM Tris-HCl pH 7.5, 150 mM NaCl) were titrated with 37.5 μM solutions of metal chlorides in 2 μl increments up to the concentration 13 μM in the sample. The data for Zn2+ binding were fitted by DynaFit programme (Kuzmič, 1996) to the appropriate chemical model (2:1; metal to protein) with metal–buffer interactions included (logKZnTris = 2.27; logKNiTris = 2.67) (Zheng et al., 2009).
Luciferase Reporter Gene Activity Assay
DNA fragments containing potential promoter regions were PCR amplified using primers listed in Supplementary Table S1, and cloned into the reporter plasmid pFLUXH. The fragments p5911/p5912 and p7176/p7177 were cloned into NdeI site and clones in both orientations were identified by PCR with P9 starter complementary to the sequence outside of the cloning site in pFLUXH. Due to the use of NdeI site (CATATG), the native sequence of three nucleotides immediately upstream of the translational start codon was replaced with CAT and start codons were replaced with ATG. pFLUXH derivatives were introduced into M145 and P138 Streptomyces strains by conjugation. Negative control strain (MG01) was M145 with an empty pFLUXH plasmid. Two hundred microliter of solid modified 79 medium supplemented with higromycin (50 μg/ml) and 10 μM ZnCl2 if needed, was placed in wells of a white 96-well clear bottom plate and inoculated with 10 μl of spore suspension diluted in water to OD600 = 0.3 to give confluent growth. Cultures were incubated at 30°C directly in the ClarioStar microplate reader. Luminescence and OD600 were measured every hour.
Results
HypR Protein Binds Its Own Promoter Region
Looking for regulators of cpk gene cluster (genes SCO6269 – SCO6288) (Pawlik et al., 2007), coding coelimycin polyketide synthase (Pawlik et al., 2010; Gomez-Escribano et al., 2012), we started to investigate the role of a potential regulator coded by a neighboring gene SCO6294 which we later named hypR (for L-hydroxyproline utilization regulator, as explained below). The protein was expressed in E. coli with N-terminal His-tag and Electrophoretic Mobility Shift Assay (EMSA) was used to test if it binds to promoters of cpk genes, but it was not the case (data not shown). Many GntR like proteins are autoregulators (Hoskisson and Rigali, 2009), therefore hypR promoter region was also included in the assay. Binding of the HypR protein was observed to the DNA fragment p6294/p6295 encompassing its own promoter region and that of the neighboring gene SCO6295 (Figures 1A,B). EMSA experiments with two shorter fragments (not shown) indicated that the binding site is located immediately upstream of hypR.
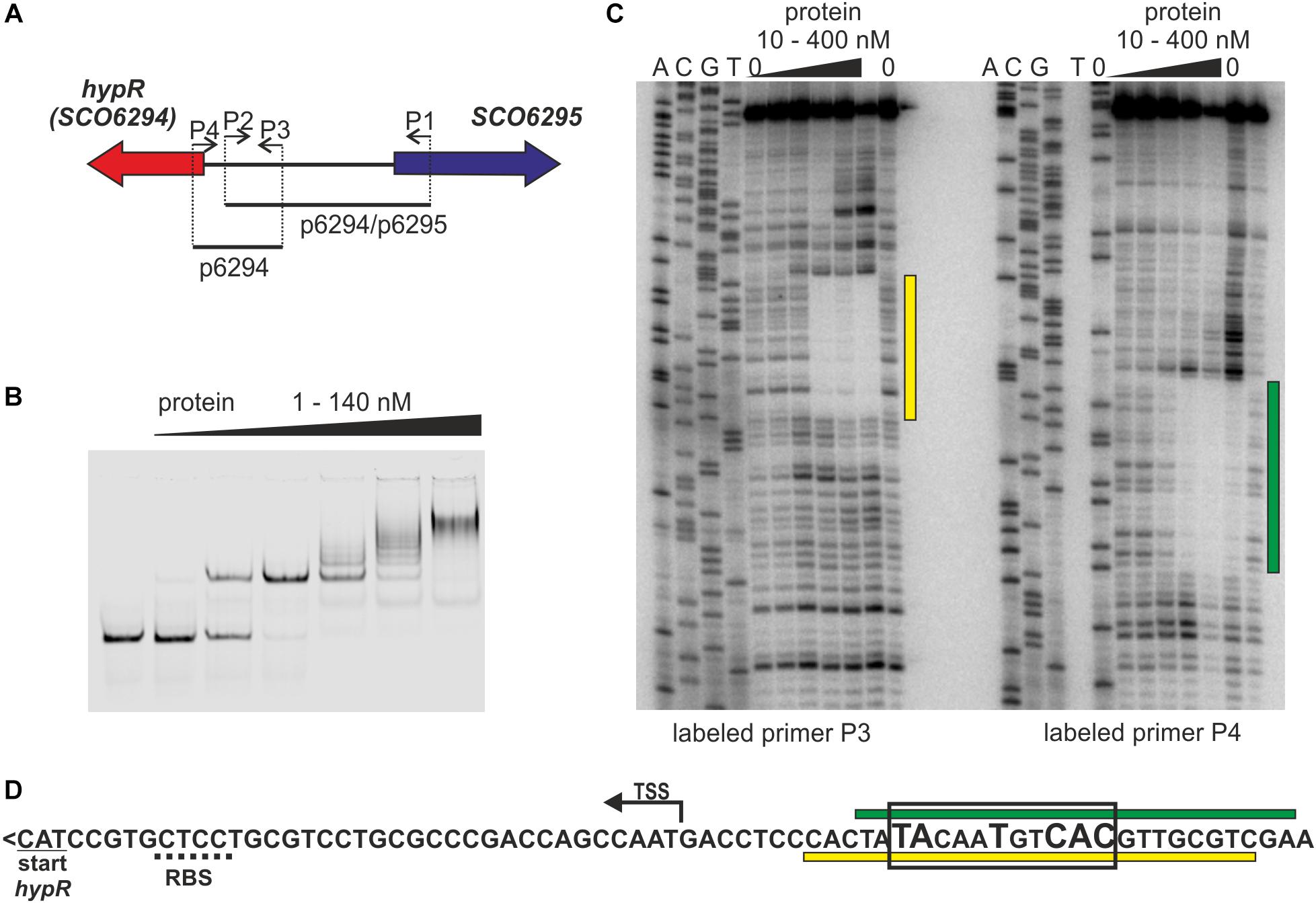
Figure 1. Identification of the DNA sequence recognized by HypR protein in its own promoter region. (A) Schematic representation of the location of primers and fragments used for EMSA and footprint experiments. (B) Binding of HypR protein to IR Dye 800 labeled fragment p6294/p6295 (EMSA). (C) Protection of the fragment p6294 from digestion by HypR protein (DNase I footprint). The radiolabeled primer is indicated below the lanes. Protected regions are marked with yellow and green bars. (D) Nucleotide sequence upstream of hypR (SCO6294) gene. Start codon and predicted ribosome binding site (RBS) are underlined with solid and dotted lines, respectively. TSS – transcription start site (Jeong et al., 2016). Protected regions from panel (C) are marked with yellow and green bars. Sequence similar to consensus motif identified in bacterial one hybrid system (see Supplementary Figure S2) is boxed and conserved nucleotides are bigger.
The DNA sequence bound by HypR protein was found in the DNase I footprint experiment. The 32P labeled p6294 was incubated with increasing amount of His-tagged HypR protein and digested with DNase I (Figure 1C). The 21 bp sequence protected by HypR from DNase I digestion was found upstream of the hypR start codon covering the -10 region of the promoter (Figure 1D; see Supplementary Text S1 and Supplementary Figure S1 for new annotation of hypR start). No other similar sequences were found on S. coelicolor A3(2) chromosome.
To further characterize HypR binding sequence a bacterial one-hybrid system (B1H) was used (Meng et al., 2005). The studied DNA binding regulator was expressed as a fusion protein with the α-subunit of RNA polymerase in the selection strain E. coli US0 (ΔhisBΔpyrF) (Meng et al., 2006) which is unable to grow on the minimal medium lacking histidine. The strain was transformed with a library of 18 bp randomized oligonucleotides cloned upstream of the reporter genes (the yeast HIS3 and URA3). Clones in which interaction between the fusion protein and the promoter region of reporter genes took place were identified by their ability to grow in the absence of histidine. Weak interactions were eliminated by the addition of 5 mM 3-AT (3-amino-triazole, a competitive inhibitor of HIS3). After screening the library, 35 individual clones were selected. Their randomized regions were sequenced and analyzed by a motif finding software MEME (Bailey and Elkan, 1994; Supplementary Figure S2). A consensus sequence 5′-TATNNTNNC(AC)(TC)-3′ recognized by HypR protein was found. A sequence similar to this motif (TACAATGTCAC) with one mismatch (underlined) is present within the region protected by bound HypR protein from digestion by DNase I (Figure 1D). The narrowed down sequence allowed us to identify other promoters recognized by HypR.
HypR Protein Binds Zinc Ions
Predicted structure of HypR protein (details below) resembles that of TM0439 protein from Thermotoga maritima (Zheng et al., 2009), which represents a group of metal binding proteins from FadR subfamily of GntR-like regulators. The physiological role of TM0439 and its interaction with metal ions is unknown.
We tested the influence of several divalent metal ions on DNA binding by HypR. To avoid possible interactions of His-tag with the metal, we used in this experiment Strep-tagged HypR expressed in E. coli from pMS08 plasmid. As observed in EMSA analysis, DNA binding was abolished by the addition of divalent metal ions (Ni2+, Cu2+, and Zn2+) (Figure 2A). Magnesium ions had a weaker effect and the effect of calcium and manganese was negligible.
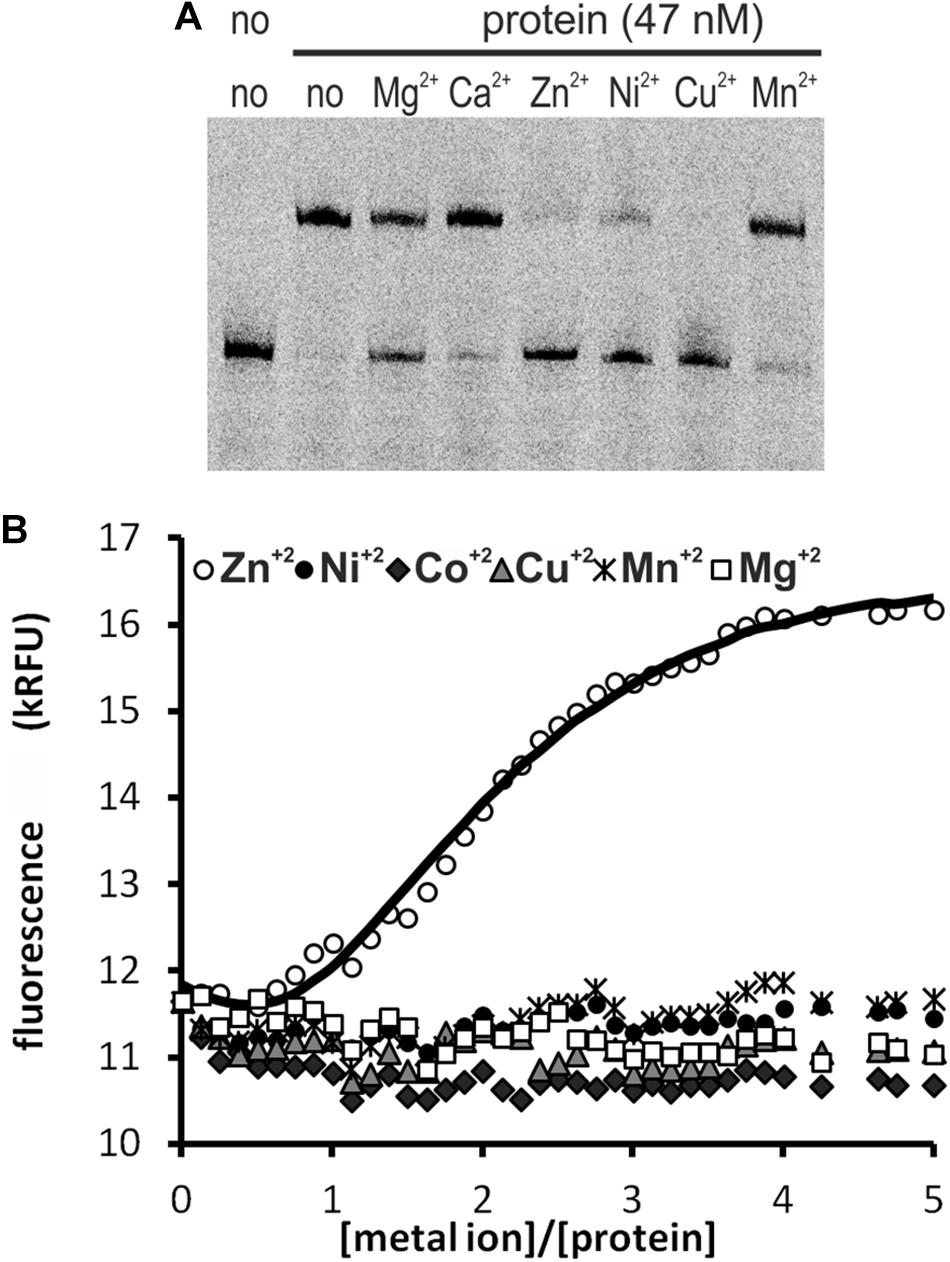
Figure 2. Interactions of HypR protein with metal ions. (A) Influence of divalent metal ions on binding of HypR protein to its own promoter region. Fragment p6294 was labeled with IR Dye 800, 5 mM metal ions were added to the samples, protein:DNA molar ratio was 25:1. (B) Tryptophan fluorescence of HypR protein in the presence of divalent metal ions. Solid line represents the best fit, found by DynaFit programme (Kuzmič, 1996) accounting for appropriate metal–buffer interactions (Zheng et al., 2009).
To assess binding of metal ions by HypR protein we measured its intrinsic tryptophan fluorescence. Samples containing Strep-tagged protein solution were titrated with zinc, nickel, cobalt, copper, manganese and magnesium chlorides. Zinc was the only metal which altered the intensity of fluorescence of the protein (Figure 2B). Zn2+ binding fitted the 2:1 (metal to protein) stoichiometric model with binding constants: K1 = (3 ± 1.5) × 107 M−1, K2 = (6.5 ± 0.33) × 105 M−1, comparable to those reported for TM0439 protein: K1 = (1.4 ± 0.1) × 107 M−1, K2 = (4.5 ± 0.4) × 105 M−1 (Zheng et al., 2009). Data fitting and model selection were done with DynaFit programme (Kuzmič, 1996). Obtained results indicate that zinc is the metal specifically bound by HypR.
Identification of Genes Controlled by the HypR Protein
We searched for other sequences bound by HypR on S. coelicolor A3(2) chromosome with FUZZNUC program2. We used the TACAATGTCAC sequence from hypR promoter allowing for one mismatch. Three locations in potential promoter regions were found (Table 1, positions 2–4). Two of them (promoter of SCO6289 and intergenic region of SCO5911 and SCO5912) were confirmed by EMSA to be recognized by HypR protein (Figure 3 and Supplementary Figure S3). In the second round of search the sequence TGCAATGTCAC common for both p6289 and p5911-12 fragments was taken, again allowing for one mismatch. Three sequences located in five putative promoter regions were found (Table 1, positions 5–9). Binding of four of them was confirmed by EMSA. Similarly as in case of hypR promoter, all the fragments were not bound in the presence of Zn2+ ions (Figure 3). DNA sequences of seven fragments bound by HypR protein were subjected to MEME analysis and a 14-nucleotide consensus sequence CTNTGC(A/C)ATGTCAC was found (Figure 4). Small palindromic elements and, in two cases, direct repeats can be noticed.
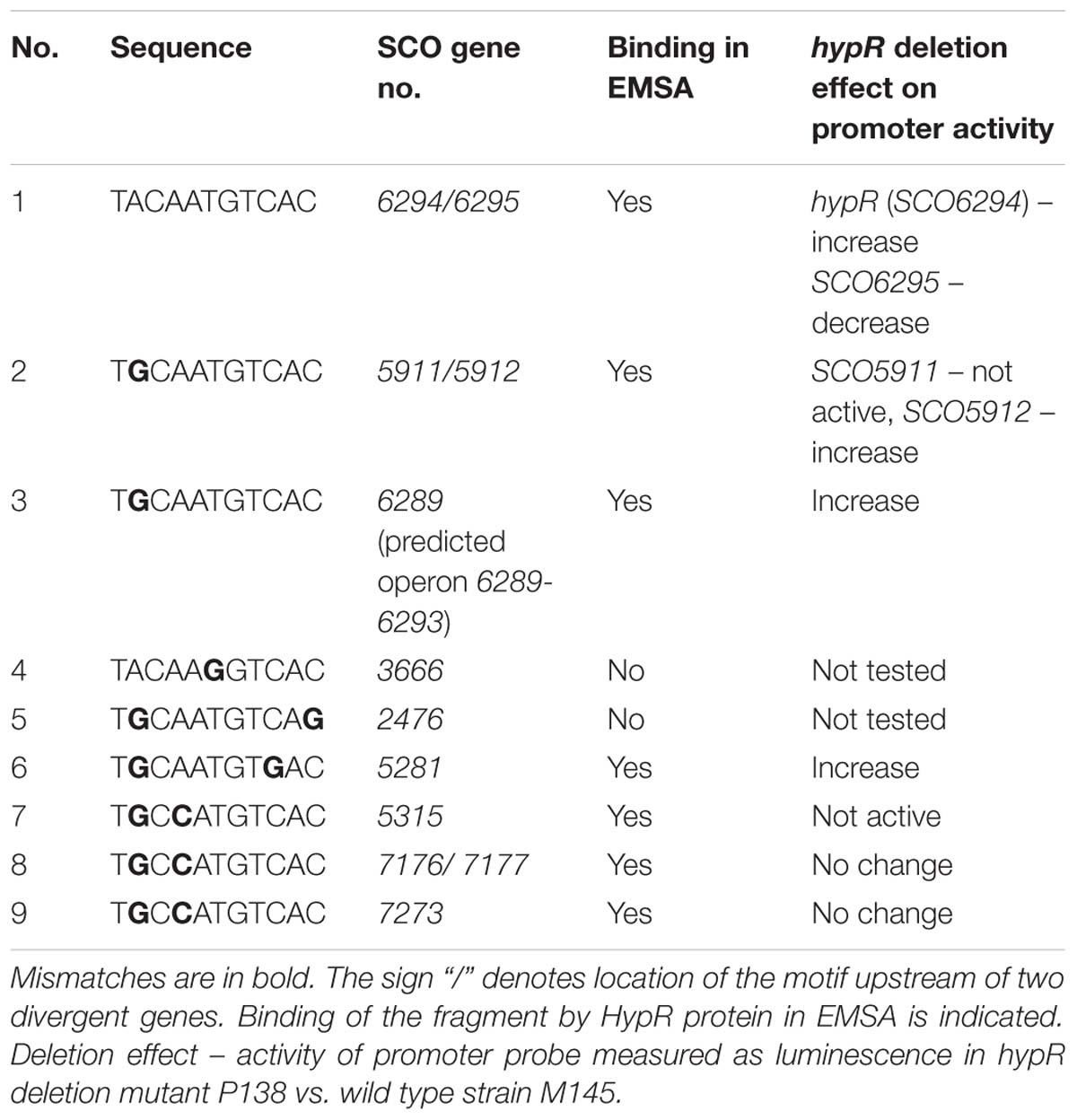
Table 1. Sequences similar to the motif from hypR promoter region found in potential promoter regions on Streptomyces coelicolor A3(2) chromosome.
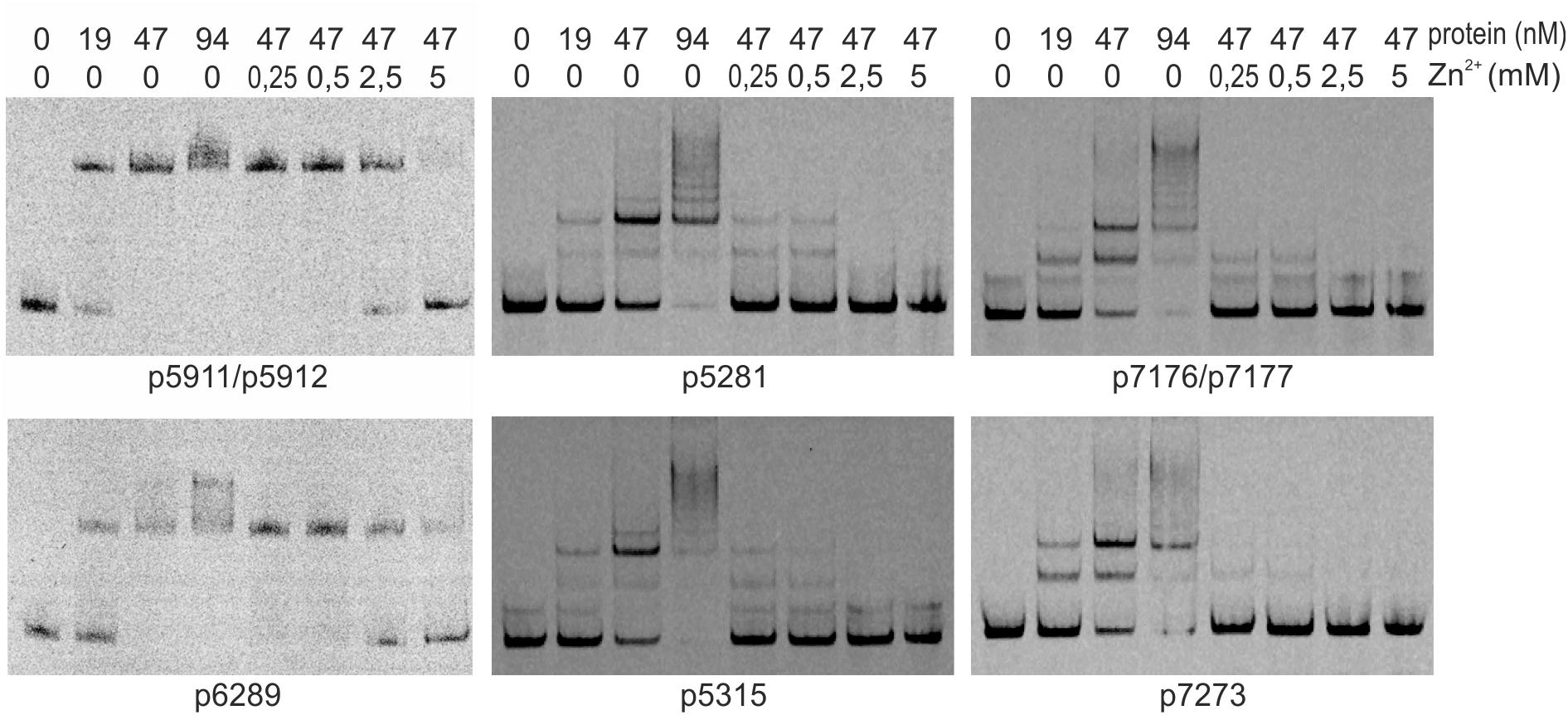
Figure 3. Binding of HypR protein to different promoter regions (EMSA). Fragments p5911/p5912 and p6289 were labeled with IRD-800 and incubated with Strep-tagged protein, remaining fragments were labeled with IRD-700. Protein and zinc chloride were added as indicated. Fragments for EMSA were between 104 and 129 bp long with the identified motifs (listed in Table 1) in the center.
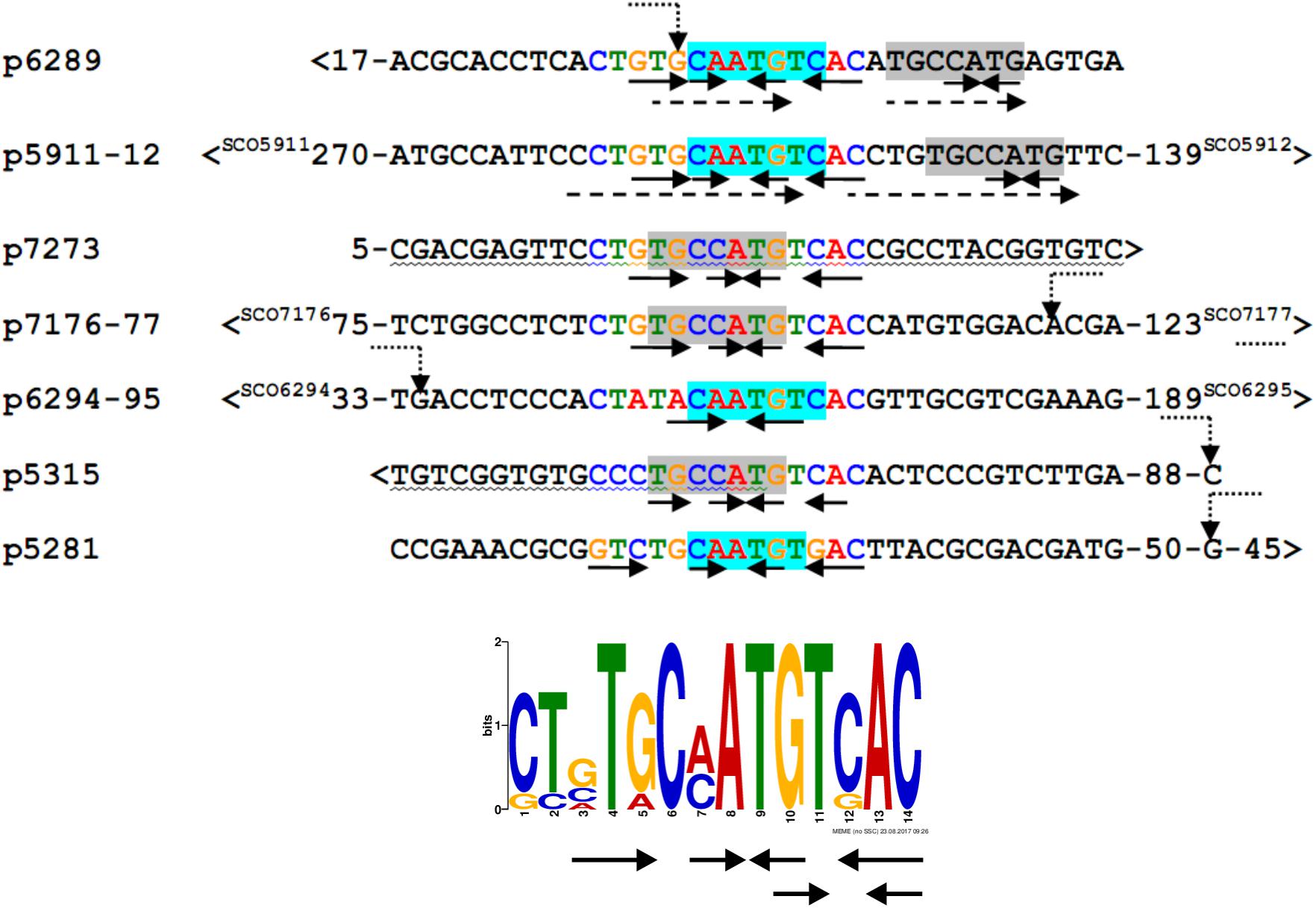
Figure 4. Alignment of DNA sequences of fragments bound by HypR protein and a 14 nt consensus sequence found by MEME program (E-value: 5.8e-012). Letters in the alignment are colored as in the consensus sequence logo. Gray or cyan background indicates identical sequences. Solid arrows – palindromic elements; dashed arrows – direct repeats with 1 mismatch; wavy underlining – coding sequence; >, <– gene direction; numbers – distance to start codons. If known, transcription start sites (TSS) are marked by bent dotted arrows (Jeong et al., 2016).
HypR Is a Repressor of SCO6289-SCO6293 Genes
In order to study the physiological role of the regulator, hypR gene was deleted from S. coelicolor A3(2) chromosome (Supplementary Figure S4). The obtained mutant strain, S. coelicolor P138, was viable and we did not note differences in growth and colored metabolites production between the wild type M145 strain and P138 mutant.
To investigate the in vivo activity of promoters in the presence and absence of HypR protein, promoter probes based on pFLUXH vector (Supplementary Table S1) were introduced into M145 and P138 strains. The plasmid carries a promoterless luciferase operon luxCDAEB encoding luciferase and enzymes necessary for luciferase substrate (tetradecanal) biosynthesis (Craney et al., 2007). It allows direct monitoring of the luminescence in bacterial biomass. Luminescence of strains grown for 45 h is shown in Figure 5 (for time-course see Supplementary Figure S5). The same strains were grown on the medium supplemented with 10 μM ZnCl2 to check if the addition of zinc would have an effect on the activity of promoters in vivo, but the level of luminescence was not changed (Supplementary Figure S5).
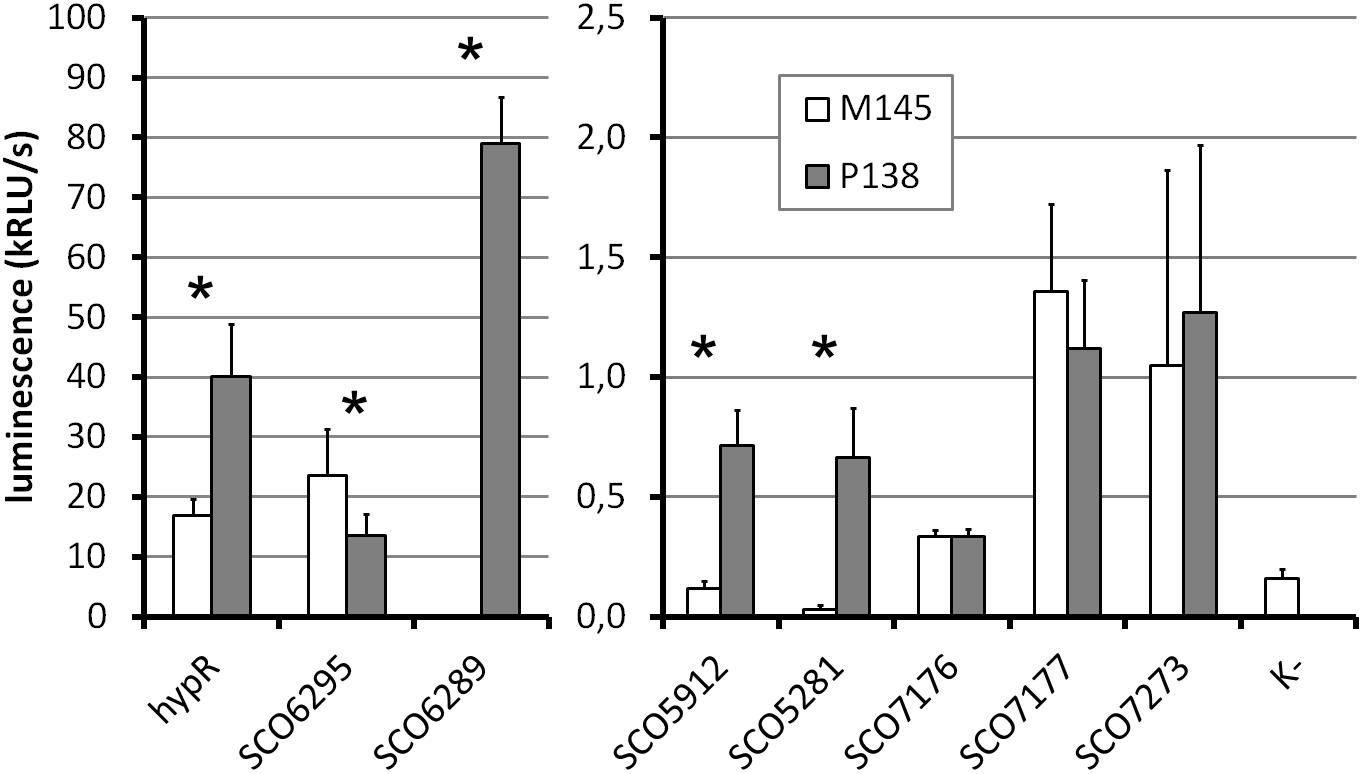
Figure 5. Promoter activity assay. Luminescence intensity after 45 h of growth of wild type (M145) and hypR deletion mutant (P138) strains carrying integrated promoter probe plasmids (pFLUXH derivatives). Negative control (K-) was M145 with an empty pFLUXH plasmid. Average values of data from at least three experiments are shown. Error bars represent standard deviation. Asterisks denote promoters with statistically significant differences of activity between M145 and P138 (P < 0.05 in t-test). The vertical axis description and legend refer to both parts of the graph.
The most striking differences between M145 and P138 were observed in case of SCO6289, SCO5912, and SCO5281 promoters which showed activity only in P138. These results suggest that HypR is a repressor of probably co-transcribed genes SCO6289-SCO6293 as well as SCO5912 and SCO5281. Notably, promoter of SCO6289 is much stronger than the remaining ones tested. Activity of hypR promoter was twice as high in P138 as in M145 strain. This indicates that HypR is an autorepressor. Wild type luminescence intensity in M145 was much higher for hypR promoter than for other identified target genes promoters, for which it was at the level of negative control, which means that the repression of its own promoter is weaker. The lower level of luciferase production from SCO6295 promoter in the deletion strain (57% of that in wild type) suggests that the regulator may activate the expression of the transporter. Activity of SCO7176, SCO7177, and SCO7273 promoters was not influenced by the deletion of hypR and promoters of SCO5911 and SCO5315 were not active in any of the strains.
Annotation of Genes From HypR Regulon
Amino acid sequences and predicted structures of proteins coded by genes controlled by HypR transcription factor were subjected to similarity searches (Table 2). Genes SCO6289-SCO6293 are probably co-transcribed, which indicates the involvement of the respective enzymes in a common pathway. The genes are conserved and share the same order on the chromosomes of Streptomyces, Kitasatospora and, to a lesser extent, Pseudomonas species, as shown by synteny analysis (Supplementary Figure S6; Oberto, 2013). They are predicted to code enzymes required for L-hydroxyproline (L-Hyp) utilization (Figure 6).
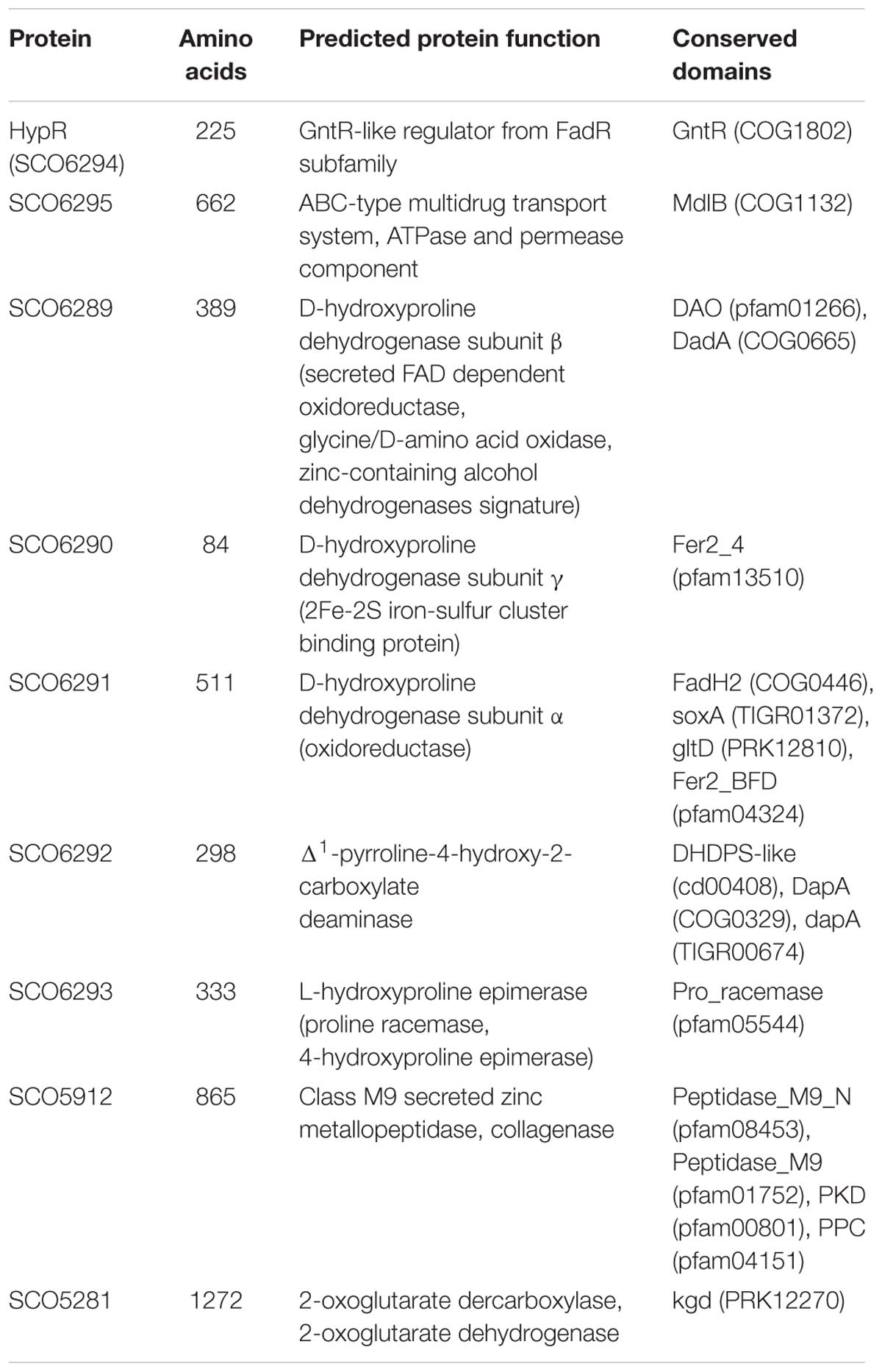
Table 2. Functions of the proteins from HypR regulon predicted on the basis of Blast (Altschul et al., 1990), Phyre2 (Kelley and Sternberg, 2009), and NCBI Conserved Domains search (Marchler-Bauer et al., 2017).
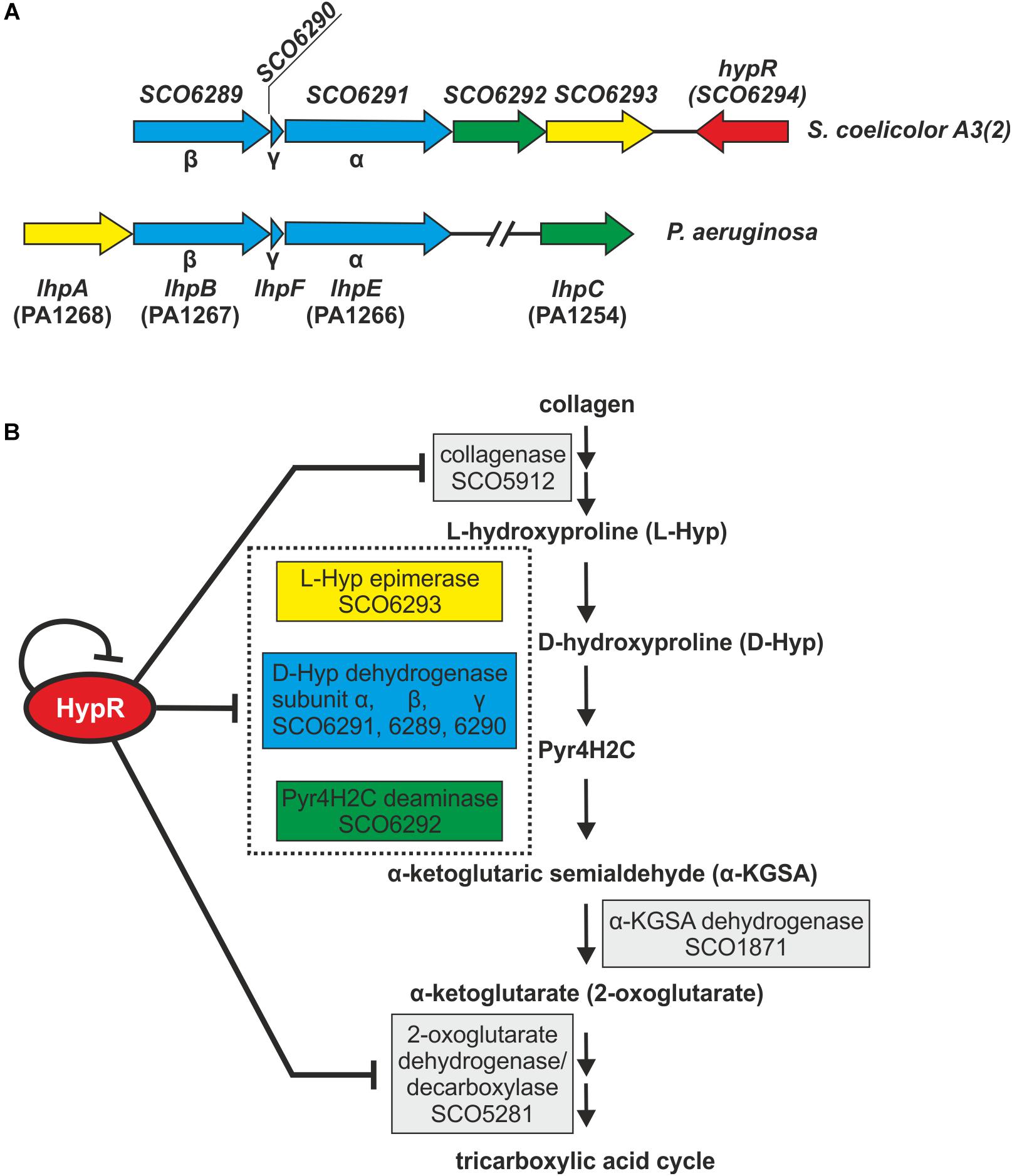
Figure 6. Schematic gene clusters for L-hydroxyproline metabolism in Streptomyces coelicolor A3(2) and Pseudomonas aeruginosa (A). Interactions between HypR repressor and its regulatory target genes related to the predicted L-Hyp utilization pathway in S. coelicolor A3(2) (Watanabe et al., 2012; Chen et al., 2016) (B). Pyr4H2C – Δ1-pyrroline-4-hydroxy-2-carboxylate, yellow – L-Hyp epimerase, blue – D-hydroxyproline dehydrogenase, green – Pyr4H2C deaminase. Dotted box represents enzymes coded in an operon. Influence of HypR protein on the activity of SCO1871 promoter was not tested.
L-Hyp is one of the main components of collagen. Its utilization by bacteria (Figure 6B) starts with conversion to D-hydroxyproline (D-Hyp) by L-Hyp epimerase, followed by oxidation to Δ1-pyrroline-4-hydroxy-2-carboxylate (Pyr4H2C) by D-Hyp dehydrogenase (D-HypDH), spontaneous hydrolysis and enzymatic deamination by Pyr4H2C deaminase to α-ketoglutaric semialdehyde (α-KGSA) and final conversion to α-ketoglutarate by α-KGSA dehydrogenase (Watanabe et al., 2012; Chen et al., 2016).
Conserved domains and molecular masses of proteins SCO6291, SCO6289, and SCO6290 correspond to α, β and γ subunits of D-HypDH from Pseudomonas aeruginosa, with amino acid sequence identity 34, 34, and 41% to respective proteins LhpE, LhpB, and LhpF (Watanabe et al., 2012; Figure 6A). SCO6293 and SCO6292 are homologs of L-Hyp epimerase and Pyr4H2C deaminase, respectively, with amino acid sequence identity 37 and 33% to respective proteins LhpA and LhpC from P. aeruginosa. L-Hyp catabolic pathway has also been described in Sinorhizobium meliloti (White et al., 2012). SCO6293 and SCO6292 have 44 and 32% identical amino acids, respectively, as S. meliloti L-Hyp epimerase (HypRE) and Pyr4H2C deaminase (HypD). However, the putative three subunit D-HypDH of S. coelicolor (SCO6289-91) has no significant homology with its functional counterpart in S. meliloti (HypO). D-HypDH enzymes may have different subunit compositions, while other enzymes of the pathway are conserved (Watanabe et al., 2012; Satomura et al., 2015).
Gene for α-KGSA dehydrogenase is not present in the proximity of the SCO6289-SCO6293 operon, however, SCO1871 was found as a best hit (43–45% amino acid identity) when S. coelicolor A3(2) genome was searched for homologs of α-KGSA dehydrogenases of P. aeruginosa, Pseudomonas putida, Azospirillum brasilense, and Sinorhizobium meliloti.
The remaining genes repressed by the regulator include SCO5912 coding a putative collagenase from class M9 metallopeptidases, as suggested by the order of conserved domains in SCO5912 protein and a presence of a zinc-binding motif HEXXH-E (Duarte et al., 2016), and SCO5281 coding α-ketoglutarate (2-oxoglutarate) dehydrogenase or decarboxylase (Tian et al., 2005; Huergo and Dixon, 2015).
We found that both M145 and P138 strains were able to grow in the liquid Minimal Medium (MM) with L-Hyp as a sole source of carbon. Growth testing on MM agar plates was unsuitable due to the ability of S. coelicolor A3(2) to utilize agar as a source of carbon. The luminescence reporter test showed that transcription of genes from the regulon of HypR was induced by L-Hyp in the wild type strain, while in the deletion mutant, promoter activity was independent from the amino acid addition (Figure 7A). Binding of DNA as tested by EMSA did not change in the presence of L-Hyp, D-Hyp, and L-proline with and without zinc ions (Figure 7B).
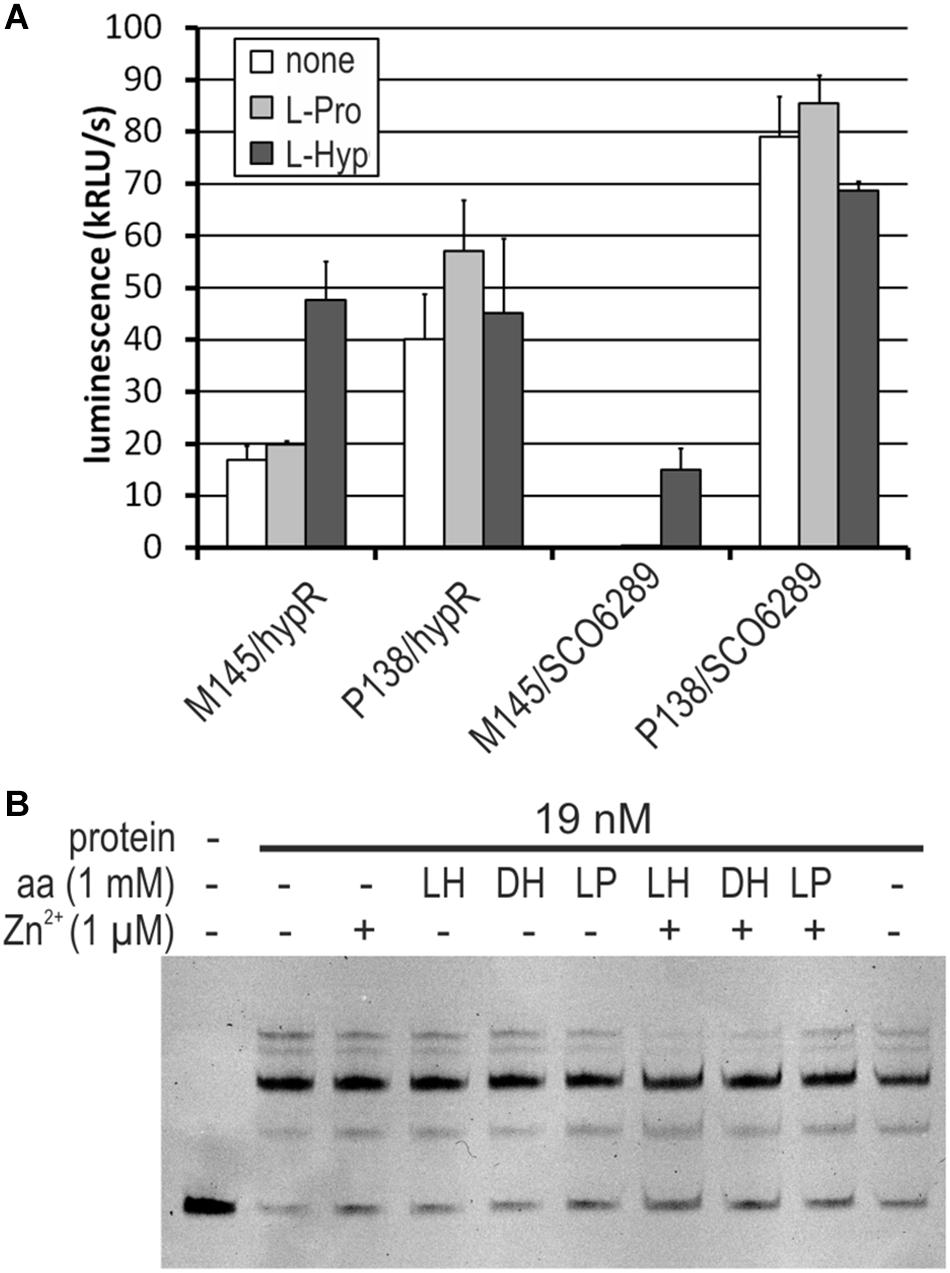
Figure 7. Influence of L-hydroxyproline (LH, L-Hyp), D-hydroxyproline (DH), and L-proline (LP, L-Pro) on the activity of HypR protein in vitro (DNA binding) and on the activity of promoters. (A) Activity of hypR and SCO6289 promoters in M145 and P138 strains measured as luminescence after 45 h of growth on modified 79 medium with L-Hyp or L-Pro at the concentration 15 mM. (B) EMSA assay with IR Dye labeled fragment p6289. HypR protein, amino acids and zinc chloride were added as indicated.
To verify if the operon SCO6289-SCO6293 is responsible for L-Hyp utilization, SCO6293 gene, predicted to code L-Hyp epimerase was deleted. The obtained mutant strain S. coelicolor P201 lost the ability to grow in liquid MM with L-Hyp as a sole source of carbon, while it was still able to grow in the medium containing L-proline.
HypR Protein Predicted Structure
HypR protein, as annotated in NCBI Protein Database under accession number NP_630392.1, has 263 amino acids. However, according to our results (see Supplementary Figure S1 and Supplementary Text S1) the annotation is based on an erroneous start codon and the actual protein is shorter – it consists of 225 aa and its calculated molecular mass is 24.6 kDa. In this work amino acids were numbered according to the corrected start. Similarity search using Blast (Altschul et al., 1990) revealed that HypR belongs to FadR subfamily of HTH GntR proteins and is highly conserved (28 hits with 80–94% identity covering 97–100% of the sequence). A model of three dimensional structure was obtained with Phyre2 program (Kelly et al., 2015) (confidence 100.0) based on the structure of TM0439 protein from Thermotoga maritima (Zheng et al., 2009) for which amino acid sequence identity and similarity are 29 and 53%, respectively (Supplementary Figures S7, S8). TM0439 is a dimer which is typical for FadR subfamily.
The winged helix-turn-helix (WHTH) N-terminal domain is formed by two helices α2 and α3 and two β strands. Based on comparison with crystal structures of FadR (Xu et al., 2001) and TM0439 (Zheng et al., 2009) the following amino acids of HypR are implicated in interactions with DNA: Ser50, Thr52, Arg55, Arg71, and Asn72 (Supplementary Figure S8).
C-terminal domains of GntR proteins from FadR subfamily are composed of six or seven α helices (Hoskisson and Rigali, 2009). Pfam database (Bateman et al., 2002) distinguishes a small FadR_C family (comprising FadR itself with seven α helices) (Pfam07840) and a large FCD family (Pfam007729) which includes C-terminal domains with both six and seven α helices. HypR protein has a six-helical ligand binding FCD domain. It has a potential metal ion binding site formed by three histidine residues His142, His186, and His208 which correspond to His134, His174, and His196 in TM0439 protein. In TM0439 two of the metal-binding histidines are stabilized by hydrogen bonds with Glu90, Asp130, and Glu173 (Zheng et al., 2009). Corresponding amino acids can be also found in HypR sequence (Glu96, Asp138, and Glu185). Mutational analysis of FCD domain of CitO protein, activator of citrate utilization pathway in Enterococcus faecalis, whose predicted structure is similar to HypR, revealed amino acids important for citrate binding and transcription activation in vivo (Arg97, His191, Phe143, Leu214) (Blancato et al., 2016). All the corresponding residues are conserved in HypR sequence (Arg92, His186, Phe141, Leu209) (Supplementary Figure S8).
Discussion
HypR Is a Repressor
Similarity search with amino acid sequence of HypR protein and analysis of its predicted structure shows that it belongs to FadR subfamily of GntR-like proteins. It has a typical N-terminal DNA binding WHTH domain and a metal binding C-terminal FCD domain composed of six α helices.
Recombinant protein HypR bound the DNA in the hypR promoter region as shown by EMSA and DNase I footprint experiments. The sequence protected from DNase I cleavage by binding of HypR to its own promoter corresponded to a motif identified in bacterial one-hybrid system (Figure 1). Binding of the protein was shown for six other DNA fragments with similar sequences located in potential promoter regions (Table 1 and Figure 3) and a consensus sequence motif CTNTGC(A/C)ATGTCAC shared by the fragments was found (Figure 4). Finally, HypR protein was shown by the luciferase reporter assay to be a repressor of SCO6289-SCO6293 operon, SCO5912, and SCO5281 genes and an autorepressor (Figure 5). The repression of its own promoter is weaker than that of other target genes.
Most of the studied GntR-like proteins bind as dimers to twofold symmetric DNA operator sequences, with each monomer recognizing a half-site (Suvorova et al., 2015). Oligomeric state of HypR was not studied. We expect it to be a dimer based on the predicted structure similarity to the dimeric protein TM0439. Symmetry of the consensus binding site sequence of HypR protein is not clear, but short palindromic elements can be noticed (Figure 4) including the GT/AC pair resembling the central part of the conserved motif TKGT/ACMA found for FadR subfamily (Suvorova et al., 2015).
The HypR binding DNA consensus sequence motif is located in diverse positions relative to transcription start sites (TSSs) of respective genes (Figure 4). In case of FadR, it is the exact location of its recognized sequence that determines whether the bound regulator represses or activates target genes (Xu et al., 2001). The binding site of HypR in its own promoter covers the -10 region, which is consistent with its role as a repressor. Similarly, binding of the repressor next to the TSS of SCO6289 should impair access of RNA polymerase. On the other hand, the consensus sequence motif is located more than 60 bp upstream of TSS of SCO5281, a location resembling that of an activator CitO, which binds upstream of -35 regions (Blancato et al., 2008). Observed decrease of activity of SCO6295 promoter in hypR deletion strain suggests that the regulator may activate the expression of the transporter. However, HypR binding site in its own promoter is located as much as 200 bp upstream of the start codon of the divergent gene SCO6295, therefore we consider an indirect effect more likely than direct activation.
Zinc Ion Is a Cofactor of HypR
Majority of FCD domains have conserved histidine residues for coordinating a divalent metal ion (Zheng et al., 2009). Unlike in metal-sensing transcription factors regulating metal uptake/efflux systems, which have metal binding sites at or near dimer interface (Giedroc and Arunkumar, 2007), in FCD domains the site is buried at the bottom of a cavity formed by α helices. The size of the cavity is suitable to accommodate a small organic compound (Zheng et al., 2009). Although it is not known whether acetate presence in the FCD domain of TM0439 is physiologically relevant, its location in the crystal structure helped to identify amino acids important for formation of the ligand binding pocket in CitO from Enterococcus faecalis (Blancato et al., 2016). The same amino acids can be found in respective positions in HypR (Supplementary Figure S8).
CitO activates citrate utilization pathway by binding cit promoters in response to sensing citrate (Blancato et al., 2008). It was shown, that coordinating the metal ion is crucial for binding citrate molecule and for effective DNA binding. The authors proposed that the ligand interacts with the buried Ni2+ or Zn2+ ion in the FCD domain leading to conformational changes of the N-terminal domain that improve recognition of DNA target. DNA binding by CitO-citrate complex in vitro is not disturbed by the addition of metal ions. In the absence of citrate, binding of DNA by the recombinant CitO is much weaker and sensitive to nickel and zinc (Blancato et al., 2016).
Similarly, DNA binding by HypR protein was prevented by several divalent metal ions (Figure 2A). However, changes of tryptophan fluorescence were observed only in case of zinc (Figure 2B). If zinc ion had a signaling function and its binding caused dissociation of the protein from its target sequences in the promoter regions, transcription of respective genes should increase in its presence. However, we found that the addition of ZnCl2 to the medium did not change promoter activity (Supplementary Figure S5). We suspect that zinc ion is a cofactor involved in binding of an organic compound in a way similar to citrate binding by CitO.
Predicted L-Hydroxyproline Utilization Pathway
None of the proteins from HypR regulon have been previously studied experimentally. On the basis of sequence and predicted structure similarity searches (Table 2) we propose that they are involved in the degradation of collagen, metabolism of one of its components (L-hydroxyproline) and directing the product (α-ketoglutarate) to the TCA cycle for production of energy and biosynthetic precursors (Figure 6). We have found here that deletion of putative L-Hyp epimerase gene (SCO6293) leads to the loss of the ability of S. coelicolor A3(2) to grow in the minimal medium containing L-Hyp as a sole source of carbon. This confirms the involvement of SCO689-93 genes in L-Hyp catabolism. Transcription of the operon SCO689-93 and hypR gene is induced by L-Hyp in wild type strain, and not in hypR deletion mutant (Figure 7). Interestingly, SCO6289 promoter activity reached approximately 20% of the value obtained in the absence of the repressor. However, neither L-Hyp nor D-Hyp had any effect on DNA binding by HypR in vitro. This suggests a different effector compound interacting with the repressor (likely a downstream metabolite). It is also possible that transcription induction by L-Hyp does not lead to HypR protein dissociation from the DNA but to a conformational change and/or involves additional factors. Known L-Hyp uptake and utilization pathways of other bacteria are induced by both L-Hyp and D-Hyp, but the molecular mechanisms of transcription regulation are different. In Pseudomonas aeruginosa the expression of lhp genes is activated by LhpR protein of the AraC family as a result of binding L-Hyp (Li et al., 2016). In Sinorhizobium meliloti the hyp genes are controlled by a GntR (FadR)-like repressor HypR. We chose the same name for SCO6294 protein. Analysis of transcription induction in a series of deletion mutants indicates D-Hyp (or a downstream metabolite) as the most likely effector molecule interacting with S. meliloti HypR, but DNA binding tests were not performed (White et al., 2012).
L-Hyp is one of the main components of collagen. It is also present in plant and algae cell walls and in root nodules. Therefore, it is an abundant carbon and nitrogen source for soil-dwelling bacteria (Moe, 2013). However, the ability to hydrolyse collagen and to metabolize L-Hyp is not a common feature of bacteria. It is utilized by some pathogens, such as Pseudomonads, which degrade collagen as a part of invasion of host tissues (Duarte et al., 2016). Molecular mechanism of L-Hyp utilization was also described for nitrogen-fixing plant endosymbiont S. meliloti (White et al., 2012). To our knowledge, the current work is the first indication of the existence of the same pathway of L-Hyp metabolism in Streptomycetes, although collagenases from this genus were reported previously (Sakurai et al., 2009) and gene clusters homologous to SCO6289-93 can be found in many Streptomyces genomes (Supplementary Figure S6). It gives a new insight into the way of adaptation of the model organism S. coelicolor A3(2) to nutrients from plant and animal tissues available in the soil. Moreover, enzymes from HypR regulon may find biotechnological applications. Collagenases are used, among others, in food technology and medical industry (Duarte et al., 2016). Putative L-Hyp dehydrogenase from S. coelicolor A3(2) belongs to Dye-linked D-amino acid dehydrogenases which can be applied to construct electrochemical biosensors (Satomura et al., 2015).
Author Contributions
MK and KP designed the study and evaluated results. MK, MŚ, JZ-P, and PJ conducted the experiments. MK wrote the manuscript.
Funding
This project was supported by the Polish National Science Centre (Narodowe Centrum Nauki) Grant N N405 355639 and by Wrocław Centre of Biotechnology, programme. The Leading National Research Centre (KNOW) for years 2014–2018. Open access publication was supported by the Hirszfeld Institute of Immunology and Experimental Therapy, Polish Academy of Sciences.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Nickolas E. Grossoehme for advice on the use of the DynaFit program and Anna Zawilak-Pawlik for critical reading of the manuscript.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01451/full#supplementary-material
Footnotes
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Bailey, T. L., and Elkan, C. (1994). Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proceedings. Int. Conf. Intell. Syst. Mol. Biol. 2, 28–36.
Bateman, A., Birney, E., Cerruti, L., Durbin, R., Etwiller, L., Eddy, S. R., et al. (2002). The Pfam protein families database. Nucleic Acids Res. 30, 276–280. doi: 10.1093/nar/gkp985
Bentley, S., Chater, K., Cerdeño-Tárraga, A.-M., Challis, G. L., Thomson, N. R., James, K. D., et al. (2002). Complete genome sequence of the model actinomycete Streptomyces coelicolor A3(2). Nature 417, 141–147. doi: 10.1038/417141a
Blancato, V. S., Pagliai, F. A., Magni, C., Gonzalez, C. F., and Lorca, G. L. (2016). Functional analysis of the citrate activator CitO from Enterococcus faecalis implicates a divalent metal in ligand binding. Front. Microbiol. 7:101. doi: 10.3389/fmicb.2016.00101
Blancato, V. S., Repizo, G. D., Suárez, C. A., and Magni, C. (2008). Transcriptional regulation of the citrate gene cluster of Enterococcus faecalis involves the GntR family transcriptional activator CitO. J. Bacteriol. 190, 7419–7430. doi: 10.1128/JB.01704-07
Cen, X. F., Wang, J. Z., Zhao, G. P., Wang, Y., and Wang, J. (2016). Molecular evidence for the coordination of nitrogen and carbon metabolisms, revealed by a study on the transcriptional regulation of the agl3EFG operon that encodes a putative carbohydrate transporter in Streptomyces coelicolor. Biochem. Biophys. Res. Commun. 471, 510–514. doi: 10.1016/j.bbrc.2016.02.044
Chen, S., White, C. E., George, C., Zhang, Y., Stogios, P. J., Savchenko, A., et al. (2016). L-Hydroxyproline and D -proline catabolism in Sinorhizobium meliloti. J. Bacteriol. 198, 1171–1181. doi: 10.1128/JB.00961-15.Editor
Craney, A., Hohenauer, T., Xu, Y., Navani, N. K., Li, Y., and Nodwell, J. (2007). A synthetic luxCDABE gene cluster optimized for expression in high-GC bacteria. Nucleic Acids Res. 35, 1–10. doi: 10.1093/nar/gkm086
Duarte, A. S., Correia, A., and Esteves, A. C. (2016). Bacterial collagenases–a review. Crit. Rev. Microbiol. 42, 106–126. doi: 10.3109/1040841X.2014.904270
Fernández-Martínez, L. T., and Bibb, M. J. (2014). Use of the meganuclease I-SceI of Saccharomyces cerevisiae to select for gene deletions in actinomycetes. Sci. Rep. 4:7100. doi: 10.1038/srep07100
Flärdh, K., and Buttner, M. J. (2009). Streptomyces morphogenetics: dissecting differentiation in a filamentous bacterium. Nat. Rev. Microbiol. 7, 36–49. doi: 10.1038/nrmicro1968
Giedroc, D. P., and Arunkumar, A. I. (2007). Metal sensor proteins: nature’s metalloregulated allosteric switches. Dalt. Trans. 29, 3107–3120. doi: 10.1039/b706769k
Gomez-Escribano, J. P., Song, L., Fox, D. J., Yeo, V., Bibb, M. J., and Challis, G. L. (2012). Structure and biosynthesis of the unusual polyketide alkaloid coelimycin P1, a metabolic product of the cpk gene cluster of Streptomyces coelicolor M145. Chem. Sci. 3:2716. doi: 10.1039/c2sc20410j
Hopwood, D. A. (1999). Forty years of genetics with Streptomyces: from in vivo through in vitro to in silico. Microbiology 145, 2183–2202. doi: 10.1515/ijsl.1999.140.149
Horbal, L., Fedorenko, V., Bechthold, A., and Luzhetskyy, A. (2013). A transposon-based strategy to identify the regulatory gene network responsible for landomycin e biosynthesis. FEMS Microbiol. Lett. 342, 138–146. doi: 10.1111/1574-6968.12117
Hoskisson, P. A., and Rigali, S. (2009). Chapter 1 variation in form and function. The helix-turn-helix regulators of the GntR superfamily. Adv. Appl. Microbiol. 69, 1–22. doi: 10.1016/S0065-2164(09)69001-8
Hoskisson, P. A., Rigali, S., Fowler, K., Findlay, K. C., and Buttner, M. J. (2006). DevA, a GntR-like transcriptional regulator required for development in Streptomyces coelicolor. J. Bacteriol. 188, 5014–5023. doi: 10.1128/JB.00307-06
Huergo, L. F., and Dixon, R. (2015). The emergence of 2-oxoglutarate as a master regulator metabolite. Microbiol. Mol. Biol. Rev. 79, 419–435. doi: 10.1128/MMBR.00038-15
Jeong, Y., Kim, J. N., Kim, M. W., Bucca, G., Cho, S., Yoon, Y. J., et al. (2016). The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3(2). Nat. Commun. 7, 1–11. doi: 10.1038/ncomms11605
Kelley, L. A., and Sternberg, M. J. E. (2009). Protein structure prediction on the web: a case study using the phyre server. Nat. Protoc. 4, 363–373. doi: 10.1038/nprot.2009.2
Kelly, L. A., Mezulis, S., Yates, C., Wass, M., and Sternberg, M. (2015). The Phyre2 web portal for protein modelling, prediction, and analysis. Nat. Protoc. 10, 845–858. doi: 10.1038/nprot.2015-053
Kieser, T., Bibb, M. J., Buttner, M. J., Chater, K. F., and Hopwood, D. A. (2000). Practical Streptomyces Genetics. Norwich: John Innes Cent. Ltd.
Kuzmič, P. (1996). Program DYNAFIT for the analysis of enzyme kinetic data: application to HIV proteinase. Anal. Biochem. 237, 260–273. doi: 10.1006/abio.1996.0238
Li, G., Lu, C., and Lu, C. (2016). Molecular characterization of LhpR in control of hydroxyproline catabolism and transport in Pseudomonas aeruginosa PAO1. Microbiology 162, 1232–1242. doi: 10.1099/mic.0.000300
Liu, G., Chater, K. F., Chandra, G., Niu, G., and Tan, H. (2013). Molecular regulation of antibiotic biosynthesis in streptomyces. Microbiol. Mol. Biol. Rev. 77, 112–143. doi: 10.1128/MMBR.00054-12
Marchler-Bauer, A., Bo, Y., Han, L., He, J., Lanczycki, C. J., Lu, S., et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203. doi: 10.1093/nar/gkw1129
Meng, X., Brodsky, M. H., and Wolfe, S. A. (2005). A bacterial one-hybrid system for determining the DNA-binding specificity of transcription factors. Nat. Biotechnol. 23, 988–994. doi: 10.1038/nbt1120
Meng, X., Smith, R. M., Giesecke, A. V., Joung, J. K., and Wolfe, S. A. (2006). Counter-selectable marker for bacterial-based interaction trap systems. Biotechniques 40, 179–184. doi: 10.2144/000112049
Moe, L. A. (2013). Amino acids in the rhizosphere: from plants to microbes. Am. J. Bot. 100, 1692–1705. doi: 10.3732/ajb.1300033
Oberto, J. (2013). SyntTax: a web server linking synteny to prokaryotic taxonomy. BMC Bioinformatics 14:4. doi: 10.1186/1471-2105-14-4
Pawlik, K., Kotowska, M., Chater, K. F., Kuczek, K., and Takano, E. (2007). A cryptic type I polyketide synthase (cpk) gene cluster in Streptomyces coelicolor A3(2). Arch. Microbiol. 187, 87–99. doi: 10.1007/s00203-006-0176-7
Pawlik, K., Kotowska, M., and Kolesiñski, P. (2010). Streptomyces coelicolor A3(2) produces a new yellow pigment associated with the polyketide synthase Cpk. J. Mol. Microbiol. Biotechnol. 19, 147–151. doi: 10.1159/000321501
Persson, J., Chater, K. F., and Flärdh, K. (2013). Molecular and cytological analysis of the expression of Streptomyces sporulation regulatory gene whiH. FEMS Microbiol. Lett. 341, 96–105. doi: 10.1111/1574-6968.12099
Rigali, S., Titgemeyer, F., Barends, S., Mulder, S., Thomae, A. W., Hopwood, D. A., et al. (2008). Feast or famine: the global regulator DasR links nutrient stress to antibiotic production by Streptomyces. EMBO Rep. 9, 670–675. doi: 10.1038/embor.2008.83
Sakurai, Y., Inoue, H., Nishii, W., Takahashi, T., Iino, Y., Yamamoto, M., et al. (2009). Purification and characterization of a major collagenase from Streptomyces parvulus. Biosci. Biotechnol. Biochem. 73, 21–28. doi: 10.1271/bbb.80357
Sambrook, J., and Russell, D. W. (2001). Molecular Cloning: A Laboratory Manual. Cold Spring Harbor. NY: Cold Spring Harbor Laboratory Press.
Satomura, T., Sakuraba, H., Suye, S., and Ohshima, T. (2015). Dye-linked D-amino acid dehydrogenases: biochemical characteristics and applications in biotechnology. Appl. Microbiol. Biotechnol. 99, 9337–9347. doi: 10.1007/s00253-015-6944-z
Suvorova, I. A., Korostelev, Y. D., and Gelfand, M. S. (2015). GntR family of bacterial transcription factors and their DNA binding motifs: structure, positioning and co-evolution. PLoS One 10:e0132618. doi: 10.1371/journal.pone.0132618
Tian, J., Bryk, R., Itoh, M., Suematsu, M., and Nathan, C. (2005). Variant tricarboxylic acid cycle in Mycobacterium tuberculosis: identification of alpha-ketoglutarate decarboxylase. Proc. Natl. Acad. Sci. U.S.A. 102, 10670–10675. doi: 10.1073/pnas.0501605102
Tsypik, O., Makitrynskyy, R., Bera, A., Song, L., Wohlleben, W., Fedorenko, V., et al. (2017). Role of GntR family regulatory Gene SCO1678 in gluconate metabolism in Streptomyces coelicolor M145. Biomed. Res. Int. 2017:9529501. doi: 10.1155/2017/9529501
Tsypik, O., Yushchuk, O., Zaburannyi, N., Flärdh, K., Walker, S., Fedorenko, V., et al. (2016). Transcriptional regulators of GntR family in Streptomyces coelicolor A3(2): analysis in silico and in vivo of YtrA subfamily. Folia Microbiol. 61, 209–220. doi: 10.1007/s12223-015-0426-7
van der Heul, H. U., Bilyk, B. L., McDowall, K. J., Seipke, R. F., and van Wezel, G. P. (2018). Regulation of antibiotic production in actinobacteria: new perspectives from the post-genomic era. Nat. Prod. Rep. 35, 575–604. doi: 10.1039/C8NP00012C
Van Keulen, G., and Dyson, P. J. (2014). Production of Specialized Metabolites by Streptomyces Coelicolor A3(2), 1st Edn. Amsterdam: Elsevier.
Watanabe, S., Morimoto, D., Fukumori, F., Shinomiya, H., Nishiwaki, H., Kawano-Kawada, M., et al. (2012). Identification and characterization of D-hydroxyproline dehydrogenase and δ1-pyrroline-4-hydroxy-2-carboxylate deaminase involved in novel L-hydroxyproline metabolism of bacteria: metabolic convergent evolution. J. Biol. Chem. 287, 32674–32688. doi: 10.1074/jbc.M112.374272
White, C. E., Gavina, J. M. A., Morton, R., Britz-Mckibbin, P., and Finan, T. M. (2012). Control of hydroxyproline catabolism in Sinorhizobium meliloti. Mol. Microbiol. 85, 1133–1147. doi: 10.1111/j.1365-2958.2012.08164.x
Wolański, M., Donczew, R., Kois-Ostrowska, A., Masiewicz, P., Jakimowicz, D., and Zakrzewska-Czerwiñska, J. (2011). The level of AdpA directly affects expression of developmental genes in Streptomyces coelicolor. J. Bacteriol. 193, 6358–6365. doi: 10.1128/JB.05734-11
Xu, Y., Heath, R. J., Li, Z., Rock, C. O., and White, S. W. (2001). The FadR⋅DNA complex. J. Biol. Chem. 276, 17373–17379. doi: 10.1074/jbc.M100195200
Yu, L., Gao, W., Li, S., Pan, Y., and Liu, G. (2016). GntR family regulator SCO6256 is involved in antibiotic production and conditionally regulates the transcription of myo-inositol catabolic genes in Streptomyces coelicolor A3(2). Microbiol 162, 537–551. doi: 10.1099/mic.0.000235
Yu, L., Li, S., Gao, W., Pan, Y., Tan, H., and Liu, G. (2015). Regulation of myo-inositol catabolism by a GntR-type repressor SCO6974 in Streptomyces coelicolor. Appl. Microbiol. Biotechnol. 99, 3141–3153. doi: 10.1007/s00253-014-6368-1
Zarins-Tutt, J. S., Barberi, T. T., Gao, H., Mearns-Spragg, A., Zhang, L., Newman, D. J., et al. (2016). Prospecting for new bacterial metabolites: a glossary of approaches for inducing, activating and upregulating the biosynthesis of bacterial cryptic or silent natural products. Nat. Prod. Rep. 33, 54–72. doi: 10.1039/c5np00111k
Zheng, M., Cooper, D. R., Grossoehme, N. E., Yu, M., Hung, L. W., Cieslik, M., et al. (2009). Structure of Thermotoga maritima TM0439: implications for the mechanism of bacterial GntR transcription regulators with Zn2+-binding FCD domains. Acta Crystallogr. D Biol. Crystallogr. 65, 356–365. doi: 10.1107/S0907444909004727
Keywords: Streptomyces coelicolor, regulation of gene expression, L-hydroxyproline, GntR-like protein, FadR subfamily, zinc-binding protein
Citation: Kotowska M, Świat M, Zarȩba-Pasławska J, Jaworski P and Pawlik K (2019) A GntR-Like Transcription Factor HypR Regulates Expression of Genes Associated With L-Hydroxyproline Utilization in Streptomyces coelicolor A3(2). Front. Microbiol. 10:1451. doi: 10.3389/fmicb.2019.01451
Received: 23 January 2019; Accepted: 11 June 2019;
Published: 26 June 2019.
Edited by:
Christian Sohlenkamp, National Autonomous University of Mexico, MexicoReviewed by:
Pablo Cruz-Morales, Lawrence Berkeley National Laboratory, United StatesHaike Antelmann, Freie Universität Berlin, Germany
Copyright © 2019 Kotowska, Świat, Zarȩba-Pasławska, Jaworski and Pawlik. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Magdalena Kotowska, bWFnZGFsZW5hLmtvdG93c2thQGhpcnN6ZmVsZC5wbA==; Krzysztof Pawlik, a3J6eXN6dG9mLnBhd2xpa0BoaXJzemZlbGQucGw=
†Present address: Micha łŚwiat, Pure Biologics S.A. Wrocław, Poland Justyna Zarȩba-Pasławska, Department of Clinical Neuroscience (CNS), Karolinska Institutet, Stockholm, Sweden