 César Gago-Córdoba1
César Gago-Córdoba1 Jorge Val-Calvo1
Jorge Val-Calvo1 Andrés Miguel-Arribas1
Andrés Miguel-Arribas1 Ester Serrano1†
Ester Serrano1† Praveen K. Singh1†
Praveen K. Singh1† David Abia1
David Abia1 Ling Juan Wu2
Ling Juan Wu2 Wilfried J. J. Meijer1*
Wilfried J. J. Meijer1*- 1Laboratory 402, Centro de Biología Molecular “Severo Ochoa” (CSIC-UAM), Department of Virology and Microbiology, Autonomous University of Madrid, Madrid, Spain
- 2Centre for Bacterial Cell Biology, Institute for Cell and Molecular Biosciences, Newcastle University, Newcastle upon Tyne, United Kingdom
During conjugation a genetic element is transferred from a bacterial donor to a recipient cell via a connecting channel. It is the major route responsible for the spread of antibiotic resistance. Conjugative elements can contain exclusion system(s) that inhibit its transfer to a cell already harboring the element. Our limited knowledge on exclusion systems is mainly based on plasmids of Gram-negative bacteria. Here we studied the conjugative plasmid pLS20 of the Gram-positive Bacillus subtilis. We demonstrate that pLS20 contains an exclusion system and identified the single gene responsible for exclusion, named sespLS20, which is embedded in the conjugation operon. SespLS20 is the founding member of a novel family of surface exclusion proteins encoded by conjugative elements of Gram-positive origin. We show that the extent of surface exclusion correlates with the level of sespLS20 expression, and that sespLS20 is expressed at basal low-levels in all donor cells but becomes highly expressed in conjugating cells. Accordingly, the transfer of pLS20 from a conjugation-primed donor cell to an un-primed or conjugation-primed donor is inhibited moderately and very efficiently, respectively. The consequences of this differential regulation, which appears to be a conserved feature of surface exclusion systems of Gram-positive and Gram-negative origin, are discussed.
Introduction
Horizontal gene transfer (HGT) refers to the transfer of DNA regions from one cell to another which happens at large scale in bacteria (for review see, Ochman et al., 2000; Frost et al., 2005; Thomas and Nielsen, 2005). HGT plays a major role in the evolution of bacteria, but unfortunately is also responsible for the emergence and dissemination of antibiotic resistance (Mazel and Davies, 1999). The World Health Organization has highlighted the problem with antibiotic resistance and warned that the problem is getting worse. Of the various HGT mechanisms, conjugation is the principal route that is responsible for the emergence and dissemination of antibiotic resistance (Mazel and Davies, 1999). Conjugation is the process by which a conjugative element is transferred from a donor to a recipient cell via a connecting channel. Conjugative elements can be embedded in the bacterial genome (named integrative and conjugative elements; ICE) or present on plasmids, which are named conjugative plasmids. Most conjugation studies are based on a limited number of plasmids of Gram-negative (G−) origin. Antibiotic resistance is a problem though in both G− and Gram-positive (G+) bacteria, particularly in bacteria belonging to the phylum of Firmicutes (Sommer et al., 2009). We have been studying the native conjugative plasmid pLS20 from the G+ Firmicutes Bacillus subtilis. Bacillus strains form part of the microbiome of humans and animals and Bacilli are widely used as probiotics in animals. pLS20cat, a derivative of pLS20 containing a chloramphenicol-resistance gene, contains a large conjugation operon encompassing genes 28–74 (according to our gene annotation) that is under the control of the strong promoter Pc. Expression of the conjugation genes is regulated by genes 25–27. Gene 27 encodes the master regulator of conjugation, RcopLS20, which binds to the operator sites located near the divergently oriented promoters Pc and Pr, the latter driving expression of rcopLS20. This binding results in repression of Pc promoter and activation of the Pr promoter thereby keeping conjugation in the default “OFF” state (Singh et al., 2013; Ramachandran et al., 2014). Gene 25 encodes an anti-repressor, RappLS20, whose activity is regulated by the signaling molecule Phr∗pLS20, that is encoded by gene 26 (Singh et al., 2013; Ramachandran et al., 2014). In this study we addressed the question whether pLS20cat contains an exclusion system and what role(s) it plays in conjugation.
Exclusion systems inhibit the transfer of a conjugative element to a cell already harboring the element. They were first described more than half a century ago (Lederberg et al., 1952). Despite many studies, their mechanisms of action remain poorly understood. Escherichia coli plasmid F and related plasmids have been studied in most depth. The F plasmid contains two genes, traS and traT, each encoding for a protein responsible for a distinct type of exclusion, named entry exclusion (EE; traS) and surface exclusion (SE; traT). Other conjugative plasmids of G− origin contain either a surface or an EE system, or both. EE systems appear to be more effective than SE systems (Garcillan-Barcia and De la Cruz, 2008). The TraS EE system involves interaction of two inner membrane proteins present on the two mating cells: TraS on the recipient cell and TraG on the conjugation-primed donor cell (Achtman et al., 1979; Anthony et al., 1999; Audette et al., 2007), which causes abortion of the conjugation process after stable mating pairs have been formed (Ou, 1975; Hartskeerl and Hoekstra, 1984). The traT SE gene, on the other hand, encodes an outer membrane protein and its presence apparently reduces the ability of the cell to form a stable mating aggregate (Achtman et al., 1977).
Far less is known about exclusion systems present on conjugative plasmids of G+ origin. Here we show that pLS20cat contains an exclusion system. We identified gene 29 as the only gene responsible for exclusion, and name it sespLS20. We show that SespLS20 is a surface-located protein and that it is the prototype of a new family of SE proteins encoded by multiple conjugative elements present in G+ bacteria, including pathogens. The principal function of exclusion systems is generally believed to prevent redundant transfer of conjugative elements. Interestingly, we found that sespLS20, which is located within the conjugation operon, is not only controlled by the strong conjugation Pc promoter, but that it is also expressed from a weak constitutive promoter, P29. Consequently, sespLS20 is weakly expressed in all donor cells and strongly expressed in those donor cells having switched on the conjugation process. We also show that the level of exclusion correlates with the level of sespLS20 expression. This means that the main function of the exclusion system is not to prevent redundant transfer per se but that this is restricted to the minor population of conjugation-primed donor cells. Probably this type of regulation applies to many or perhaps all other SE systems known and hence our findings shed a new light on the role of the exclusion systems in general.
Materials and Methods
Bacterial Strains, Plasmids, Media and Oligonucleotides
Bacillus subtilis and Escherichia coli strains were grown in Luria-Bertani (LB) liquid medium or on 1.5% LB agar plates. When appropriate, media were supplemented with the following antibiotics: ampicillin (100 μg/ml), spectinomycin (100 μg/ml), chloramphenicol (5 μg/ml), erythromycin (1 and 150 μg/ml in B. subtilis and E. coli, respectively), and kanamycin (10 and 30 μg/ml in B. subtilis and E. coli, respectively). All B. subtilis strains used were isogenic with B. subtilis strain 168 (see Supplementary Table S1). Plasmids and oligonucleotides (Isogen Life Science, Netherlands) used are listed in Supplementary Tables S2, S3, respectively.
Transformation
Escherichia coli cells were transformed using standard methods (Sambrook et al., 1989). Generation of competent B. subtilis cells and transformation were done as before (Bron et al., 1989).
Construction of Plasmids and Strains
All cloned PCR fragments were checked for their correctness by sequence analysis. Total DNA extracted from pLS20cat harboring strain PKS11 was used as template to amplify pLS20cat regions by PCR.
Plasmid pLS20 was labeled with a spectinomycin resistance gene as follows. Plasmid pCm::Sp, designed to replace a chloramphenicol resistance (cat) gene by a spectinomycin resistance (spec) gene (Steinmetz and Richter, 1994), was used to transform competent B. subtilis cells of strain PKS56, which harbors pLS20cat. Spectinomycin-resistant transformants were checked for sensitivity to chloramphenicol. The total DNAs isolated from several SpecR/CmS clones were used as templates in PCR reactions to confirm replacement of the cat by the spec gene. Next, the total DNA of a representative clone, named PKS76, was used to transform competent cells of B. subtilis strain 168. The presence of pLS20spec in a spectinomycin-resistant transformant was demonstrated by PCR and conjugation experiments. The resulting strain was named PKS91.
The promoter strength screening vector pKSsfGFP is a derivative of pJS104, which is an amyE integration vector containing a gfp reporter gene that has been modified in multiple ways to enhance its expression and stability: it has an extension of eight codons of the B. subtilis comGA gene at the N-terminus (Veening et al., 2004); it has a changed codon bias compromised to obtain optimum expression in both E. coli and B. subtilis; it contains the mut3 gfp mutations (S2R, S65G, S72A) (Andersen et al., 1998) combined with the superfolder mutations (S30R, Y39N, F64L, G65T, F99S, N105T, Y145F, M153T, V163A, I171V, A206V) (Pedelacq et al., 2006), and is monomeric in solution (Pedelacq et al., 2006). For simplicity we refer to this gene as gfp. The gfp gene in pJS104 is preceded by a 124 bp EcoRI fragment containing the constitutive promoter of bacteriophage SPO1 gene 26, and two unique restriction sites (BamHI, HindIII). The following strategy was used to replace the promoter region by a 25 bp region including additional unique restriction sites (EcoRI, NheI and SpeI). A 328 bp DNA fragment overlapping with the N-terminus of the gfp gene was amplified by PCR with primer set [oPKS45-oSeqpKSGFP_Dn] and using pJS104 as template DNA. This fragment was digested with EcoRI and KpnI and the 224 bp was purified and cloned into pJS104 digested with the same enzymes. The resulting pKSsfGFP vector contains a promoter less gfp gene that is preceded by the unique restriction sites BamHI, HindIII, EcoRI, NheI, and SpeI.
The pKSsfGFP vector was used to construct B. subtilis strains containing a cassette at the chromosomal amyE containing either a promoter-less gfp fusion or gfp fused to promoter Pc or P29. In the case of the P29 promoter, the 358 bp region upstream of pLS20cat gene 29 was amplified with primer set [29_UpHindIII - 29DnHIII]. This PCR fragment was digested with HindIII and cloned in the HindIII-linearized vector pKSsfGFP. Colony PCR using primer set [oSeqpJS104_Dn – oSeqpKSGFP_Dn] was used to identify pKSsfGFP derivatives containing the insert, and sequencing of positive clones was performed to confirm the absence of mutations and to select the vector having the insert cloned in the desired orientation. The resulting plasmid was named pCG1 (P29-gfp). In the case of the Pc promoter, the 583 bp region containing promoter Pc (Ramachandran et al., 2014) was amplified using primer set [Prom28UP_Hind - Prom28Dn_Bam]. After purification, the 3′-ends of the fragment were extended with an ATP nucleotide by incubating the fragment with Taq DNA polymerase in the presence of dATP at 73°C for 20 min. Next, the purified fragment was cloned into the TA cloning vector pTZ57R/T (Thermo Fisher Scientific, United States). The absence of mutations in the plasmid isolated from a white transformant growing on LB agar plate supplemented with ampicillin, isopropyl β-D-1-thiogalactopyranoside (IPTG, 1 mM) and 5-bromo-4-chloro-indolyl-β-D-galactopyranoside (Xgal; 40 μg/ml) was confirmed by sequencing. The resulting plasmid was named pTAP28Hind_B. Next, the HindIII fragment containing promoter Pc of pTAP28Hind_B was cloned into the HindIII linearized vector pKSsGFP. Colony PCR using primer set [oSeqpJS104_Dn – oSeqpKSGFP_Dn] was used to identify the pKSsGFP derivative containing the insert, and sequencing of positive clones was performed to select the vector having the insert cloned in the desired orientation. The resulting vector was named pAND2A (Pc-gfp). Plasmids pKSsfGFP, pCG1 and pAND2A were isolated from the corresponding E. coli strains and used to transform competent B. subtilis 168 cells. Transformants were selected on LB plates containing spectinomycin. Double-crossover integration into the chromosome was checked by the loss of amylase activity.
The gfp reporter gene and the pLS20cat genes 29 and/or 30 were cloned behind the IPTG-inducible Pspank or Physpank promoter in the B. subtilis amyE integration vector pDR110 (Pspank promoter) or pDR111 (Physpank promoter) using a similar strategy. The genes were amplified by PCR using appropriate primer sets (oCG11-oCG12, oEST13-oEST17, oEST18-oEST14, and oEST13-oEST14 for cloning of gene(s) gfp, 29, 30, and [29–30], respectively). Plasmid pKSsfGFP was used as template to amplify the gfp gene. After purification, the PCR fragments were digested with [HindIII and SalI] (in the case of the gfp containing fragment), or with [SalI and SphI] (in the case of the other fragments) and then ligated with the vector pDR110 or pDR111 cut with the same enzymes. Plasmid DNA of the constructed vectors pCG35 (Pspank-gfp), pCG36 (Physpank-gfp), pCG2 (Pspank-29), pCG3 (Pspank-30), pEST19 (Pspank-29-30), and pCG106 (Physpank-29) was isolated from E. coli cells and used to transform competent B. subtilis 168 cells. Transformants were selected on LB plates containing spectinomycin. Double-crossover integration into the chromosome was checked by the loss of amylase activity.
Strain CG129, which contains an IPTG-inducible sespLS20-cMyc fusion at the amyE locus, was constructed as follows. In a first PCR reaction the pLS20cat gene 29 was amplified and extended in frame with a “GGGGS” linker region. For this PCR, pLS20cat was used as template DNA in combination with primer set oEST13-oCG64. The cMyc encoding tag was then added in frame to the linker region in two additional PCR reactions. In the first one, the amplified product of the foregoing PCR was used as template in combination with primer set oEST13-oCG67. In the final PCR, the product of the foregoing PCR was used as template in combination with primer set oEST13-oCG68. The final PCR product was purified, digested with SalI and SphI and used to clone into vector pDR111 digested with the same enzymes. The resulting vector, pCG129, was then used to transform competent B. subtilis 168 cells. Spectinomycin-resistant transformants were checked for double cross-over of the cassette at the chromosomal amyE locus by loss of amylase activity. Strain CG133 (amyE::Physpank-sespLS20-cMyc, lacA::Pxyl-gfp) was constructed by transforming competent CG129 cells with plasmid pAX01-sfGFP (see below construction of pAX01-sfGFP). Several erythromycin resistant transformants were screened for double cross-over of the Pxyl-gfp cassette at lacA by PCR. Thus, total DNAs of transformants were used as templates in PCR reactions with primer sets [opAX1up-opAX1Dn] and [oPAX2Up-oPAX2Dn]. Control strain CG47 (lacA::Pxyl-gfp) was constructed using the same strategy as for constructing CG133 but using competent cells of wild type strain 168, instead of CG129.
Plasmid pAX01-sfGFP was constructed as follows. The gfp gene was amplified by PCR from the pKSsfGFP plasmid with primer set oCG26-oCG27. The resulting PCR fragment was digested with BamHI and cloned into the BamHI site of pAX01. Transformants were checked for the presence of the insert by PCR using primer set pAXseqDn-pAXseqUp, and by screening cells grown in the presence of xylose for fluorescence.
Marker less in-frame deletions of either gene 29 or 30, or both [29–30] on pLS20cat were constructed making use of the pMiniMAD2 vector (Patrick and Kearns, 2008). The method is based on single cross-over integration of a temperature-sensitive replicon via homologous recombination at restrictive temperature followed by permitting replication of the integrated replicon by growth at the permissive temperature to provoke deletion of replicon thereby generating the desired deletion (Arnaud et al., 2004). The pMiniMAD2 vector is based on the E. coli vector pUC19, and contains a fragment of the pE194 rolling-circle plasmid encompassing its erythromycin resistance gene and its replication functions. Hence, it can replicate in B. subtilis and is selected by erythromycin. However, the pE194 replication gene contains a mutation causing the encoded replication protein to be temperature sensitive. The deletion strategy is based on the following procedure. Regions of ∼600 bp flanking the up- and downstream region to be deleted were amplified and fused by overlapping PCR, and subsequently cloned into the pMiniMAD2 vector. The resulting plasmid was isolated from the RecA-proficient E. coli strain JM101 and used to transform freshly prepared competent B. subtilis cells containing pLS20cat (strain PKS11). After transformation, cells were plated on LB plates supplemented with erythromycin and incubated overnight at 37°C forcing the pMiniMAD2-derivative to integrate into pLS20cat via a single cross-over event. Several transformants were randomly selected and grown in liquid medium without antibiotic pressure during 14–16 h at 22°C. Under these latter conditions the replicons of both pLS20cat and pE194 are functional creating stress that can be relieved by homologous recombination which results in either the deletion of the entire pMiniMAD2-derivative plasmid or by deletion of the desired region. Thus, after growth at 22°C appropriate dilutions of the cultures were plated on LB plates supplemented with chloramphenicol. After overnight growth at 37°C at least 100 colonies were tooth picked on plates containing erythromycin to select clones that had lost the pE194 replicon including the erythromycin resistance gene. Next, the total DNA of several erythromycin-sensitive colonies was isolated and used as template in PCR reactions to check for deletion of the desired fragment. The following primer sets were used to generate the different in frame deletions. Gene 29: [Up fragment, D29_1 and oCG28] [down fragment, oCG29 and oCG30]. Gene 30: [Up fragment, D30_p8 and Connect_C29_C30_P9] [down fragment, D29_30_p6 and D29_30_p7]. Gene 29-30: [Up fragment, D29_1 and connectN29_C30_p5] [down fragment, D29_30_p6 and D29_30_p7]. For each deletion, first the “UP” and “Down” regions were generated by PCR. Next, equal amounts (generally corresponding to 100 ng) of each purified PCR fragment were used for preparing a PCR reaction mixture lacking primers. This mixture was then used to fuse both fragments by performing extension reactions (50 μl) using the following settings [(2 min 94°C); 13 rounds (15 s 94°C; 1 min 55°C; 1.5 min 73°C)]. Next, 2 μl of this elongation reaction was used as template in a subsequent conventional PCR reaction (100 μl) containing the appropriate outer primers using the following settings [(4 min 94°C); 30 rounds (30 s 94°C; 30 s 55°C; 1.5 min 73°C)]. The resulting fused PCR product was purified and digested with HindIII and SalI and cloned into the pMiniMAD2 vector digested with the same enzymes. The following primer sets were used to check deletion of the desired regions by PCR using as template total DNA of erythromycin-sensitive clones. Gene 29: oCG14-oCG15; gene 30: oCG18-oCG19; gene 29-30: oCG16-oCG17. Consequently, the following in frame deletions were created. Gene 29: codon eight fused to codon 370; Gene 30: codon three fused to codon 79; Gene 29–30: codon three of gene 29 fused to codon 79 of gene 30.
In silico Analyses
Identification of Membrane and/or Surface Proteins
Deduced pLS20cat protein sequences of all ORFs larger than 40 residues were screened online for the presence of transmembrane spanning domains and their transmembrane topology using the hidden Markov model based TMHMM server version 2.0 of the center for biological sequence analyses of the Technical University of Denmark (DTU1) (Sonnhammer et al., 1998; Krogh et al., 2001). The presence of potential signal peptidase cleavage sites 1 were predicted using the SignalP 4.1 server (DTU2) using default settings for proteins of Gram-positive bacteria.
Identification of Genes Encoding Proteins Showing Significant Similarity to SespLS20
SespLS20 and Sesp576 were used separately as a query sequence to execute blastp searches3 against the NCBI nr protein database (version 2.5.0+, February 2017) (Altschul et al., 1997, 2005; Schaffer et al., 2001). This search resulted in the detection of 46 sequences sharing significant similarity with SespLS20 and Sesp576. The program “USEARCH”4 (version 8.0.1517_i86linux32) was then used to identify and remove redundant sequences showing 100% identity (Edgar, 2010), resulting in 30 unique hits.
Crossing SespLS20 Homologs Against Constructed Plasmid Database
Plasmids deposited in the NCBI nr database were retrieved by screening the annotations for the keywords “plasmid” and “circular DNA.” The 10,904 plasmids retrieved at 6 Nov 2016 were used to build a blast database. Next, each SespLS20 homolog identified was run against the constructed plasmid database using tblastn. A SespLS20 homolog member was considered to be located on a plasmid if an identity of more than 95% was identified over more than 80% of the entire protein sequence.
RNA Isolation, RNA Sequencing and Bioinformatic Analyses of RNAseq Data
RNAseq experiments were performed as described before (Singh et al., 2013). In short, total RNA was isolated from late exponentially growing PKS11 (pLS20cat) or PKS14 (amyE::Pspank-rcopLS20, pLS20cat) cells, and possible traces of DNA were removed with a DNase treatment step. Next, after removal of rRNA, 150–250 ng of RNA was used to prepare cDNA libraries using a procedure that preserves the information about the direction of transcription. Bar-coded fragmented samples were titrated, bound at a final concentration of 10 pM to an Ilumina SR-flowcell. Libraries were then run according to the Illumina “sequencing by synthesis” (SBS) technology under a single-read 1x75 protocol. Quality filtering was performed according Illumina specification and fastq files generated. The data sets analyzed were constituted by two B. subtilis subsp subtilis strain 168 and plasmid pLS20cat samples with totals of 56⋅106 and 48⋅106 single end reads of 50 nt in FASTQ format, respectively. Quality control of the reads, alignments and calculations of expression levels were performed as described before (Singh et al., 2013).
Conjugation Assays
Conjugation was carried out in liquid medium as described previously (Singh et al., 2013).
Flow Cytometry
Overnight grown cultures were diluted 100-fold in pre-warmed LB medium. Two ml of the culture was centrifuged (1 min 14,000 g) when the OD600 was between 0.8 and 1.0. After a washing step (2 ml 0.2 μM filtered 1xPBS), the pellet was resuspended in 1 ml 0.2 μM filtered 1xPBS. Next, cells were directly measured on a FacsCalibur cytometer (Becton Dickinson, United States) equipped with an argon laser (488 nm). The fluorescence of at least 100,000 cells was analyzed using a 530/30 nm band pass filter using arbitrary units (AU). Sample data was collected using CellQuest Pro (Becton Dickinson, United States) software and afterward analyzed using FlowJo 6.4.1 mac (TreeStar, United States) software. B. subtilis strain 168 was included in each flow cytometry experiment as negative control. Values showed and represented in graphs, corresponds with Geomean estimated by flow jo.
Results
Terminology Used
In most conjugation systems, including pLS20, the conjugation genes are activated only in a subpopulation of cells (Chatterjee et al., 2011; Singh and Meijer, 2014; Rosch and Graumann, 2015). To distinguish different cells in mating experiments we use the following nomenclature. Donor: cell containing pLS20 or a derivative, regardless whether or not it has activated the conjugation genes. Conjugation-primed donor: cell containing pLS20 (or derivative) that has activated the conjugation process. Recipient: cell amenable to receive a plasmid when it mates with a conjugation-primed donor cell. Receptive-donor: cell containing pLS20 (or derivative) that acts as a recipient cell when mating with a conjugation-primed donor cell.
pLS20 Contains an Exclusion System
The presence of an exclusion system on a conjugative element would result in lower conjugation efficiencies (CEs) for matings between two donor strains as compared to standard matings (i.e., between donor and plasmid-free recipient strains). Using a standard protocol (Singh et al., 2013), the maximum CE of pLS20cat, a pLS20-derivative labeled with a chloramphenicol marker, at the end of the exponential growth phase is in the order of 10−3 transconjugants per donor (Singh et al., 2013, see Table 1). To test the CE between two donor strains we constructed pLS20spec, which carries a spectinomycin resistance marker instead of a chloramphenicol marker. Whereas the CE obtained for pLS20spec in matings with plasmid-free recipient cells was very similar to that obtained with pLS20cat, the CE using two donor strains was about 10-fold lower (see Table 1). This strongly indicated that pLS20 contains an exclusion system.
To facilitate the comparison of the extent of exclusion, we index the exclusion level as done for other exclusion systems (Marrero and Waldor, 2005; Garcillan-Barcia and De la Cruz, 2008). The surface exclusion index (SEI) is defined as the ratio of conjugation efficiency (CE) of matings between [donor cells and plasmid-free recipient cells] and [donor cells and recipient cells of interest]. Thus, stronger exclusion effects are reflected by higher SEIs.
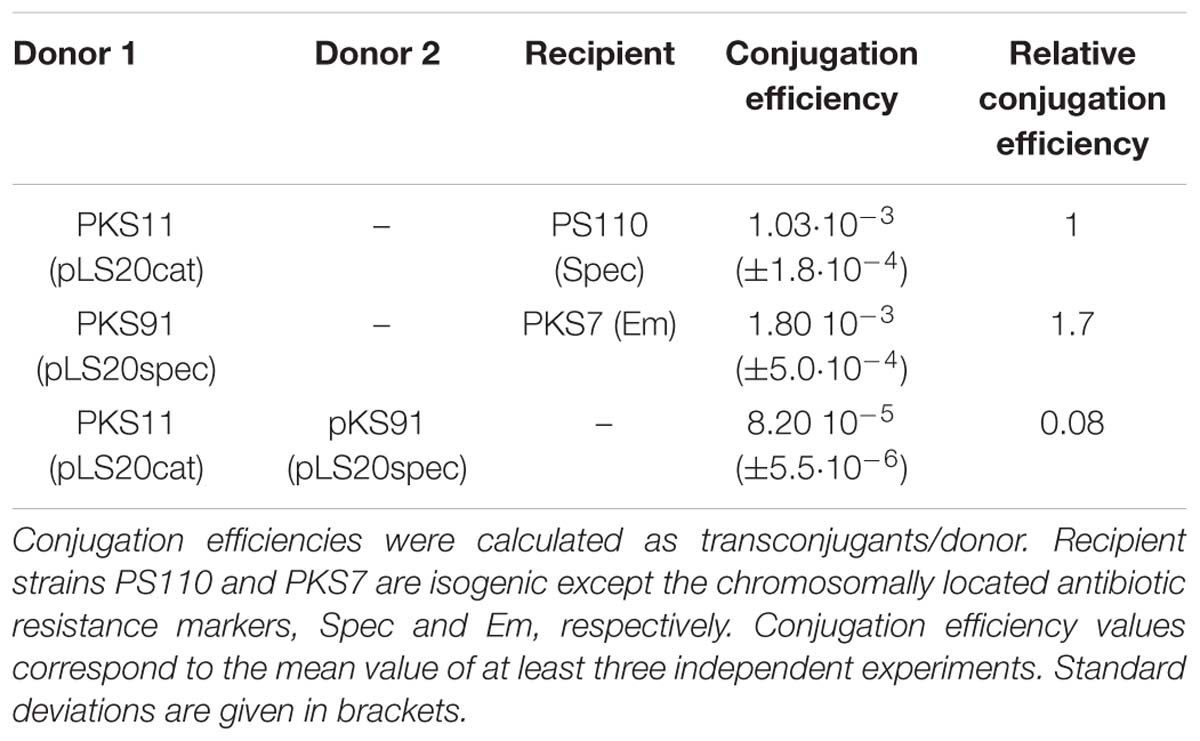
Table 1. Evidence that pLS20 contains an exclusion system.
Gene 29 Is Crucial for pLS20cat Exclusion
To identify the protein(s) responsible for pLS20cat exclusion we performed pBlast and tBlastN searches to find possible similarities between pLS20 (protein) sequences and those of known exclusion system(s). These analyses did not result in the identification of any significant similarity, indicating that pLS20cat contains a hitherto unknown exclusion system. To make the searches more specific we screened all (putative) pLS20cat genes for candidates encoding putative surface proteins, and identified gene 29 (390 codons) and the flanking gene 30 (99 codons) as potential exclusion genes because they were predicted to encode proteins having an N-terminal transmembrane spanning domain but lacking a predicted cleavage site for a signal peptidase. The majority of the p29 protein was predicted to be located at the outside of the cell membrane (see Supplementary Figure S1A), while the majority of the p30 protein was predicted to be located at the inside of the cell membrane (see Supplementary Figure 1B). Based on the genetic organization we considered that both gene 29 and 30 might play a role in exclusion (see Supplementary Figure 1C).
To test whether gene 29 and/or 30 are involved in SE we constructed derivatives of pLS20cat lacking either gene 29 (pLS20catΔ29) or both genes (pLS20catΔ29-30), and used strains harboring either plasmid as donor in matings with another donor strain harboring a wild type version of the plasmid. CEs between donor strains CG52 (pLS20catΔ29) and PKS91 (pLS20spec) were about 50-fold higher than those obtained between PKS11 (pLS20cat) and PKS91 (pLS20spec) (see Figure 1). This strongly indicated that gene 29 played a crucial role in SE. In addition, these results demonstrated that the decreased conjugation efficiencies observed in matings between two donor strains harboring a wild type plasmid (pLS20cat/spec) were not due to incompatibility of the transferred plasmid with the resident plasmid in the cell. Finally, it is worth noting that matings between two donor cells allows bi-directional transfer of the plasmid. This probably explains why the CE between donor strains CG52 and PKS91 was about 5-fold higher than the CE obtained in standard matings between a donor and plasmid-free cell.
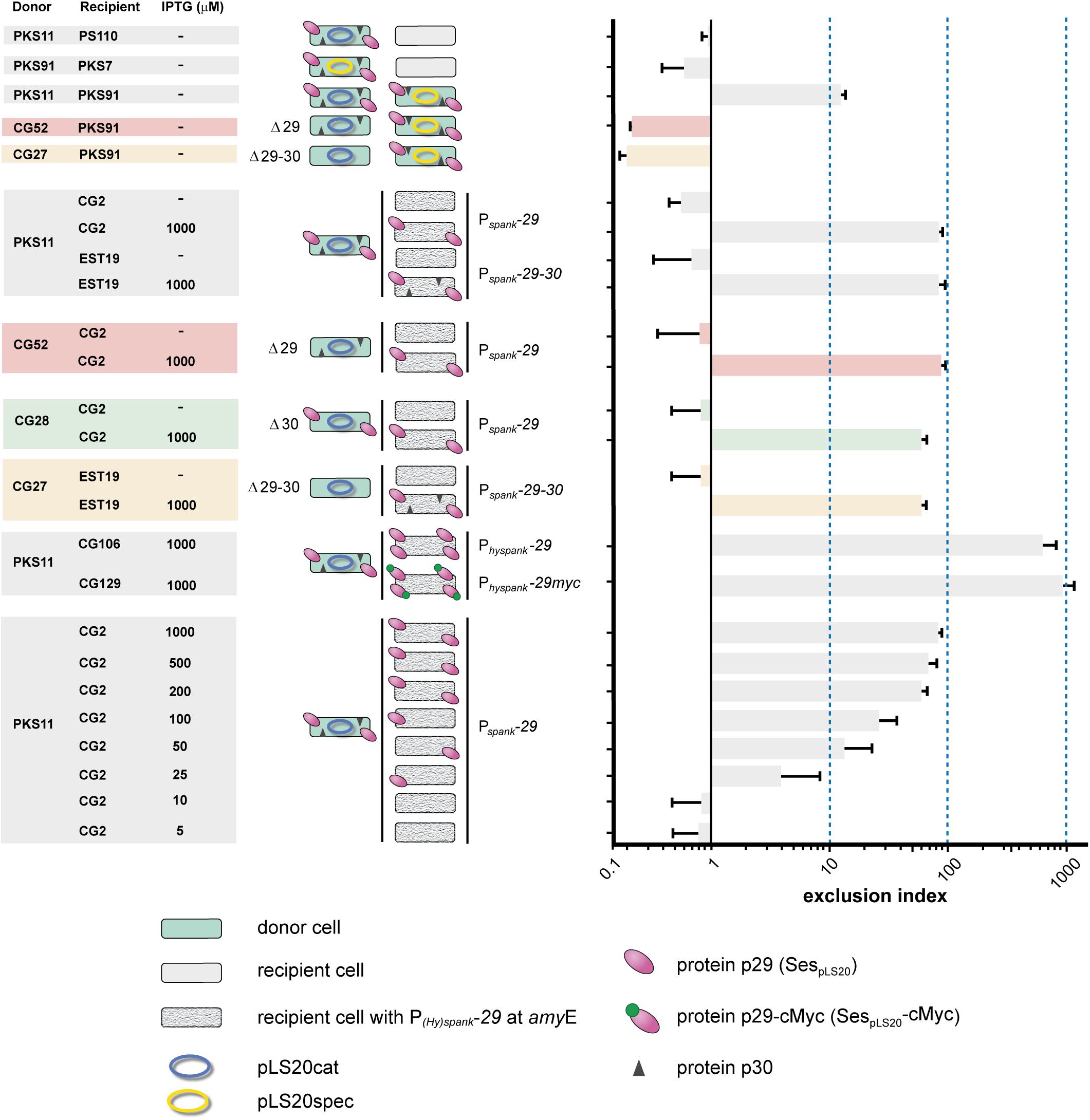
Figure 1. Surface exclusion indexes of matings. Names of donor and recipient strains and induction levels are given on the left. Cartoons of each mating are shown in the middle panel. Donor cells are shown in green, and the plasmid is indicated with an ellipse; blue and yellow representing pLS20cat and pLS20spec, respectively. Deletion derivatives of pLS20cat lacking gene 29, 30 or both are indicated (Δ29, Δ30, Δ29-30, respectively). Color codes are used to indicate the genotype of the plasmid: Δ29, red; Δ30, green; Δ29-30, orange. Plasmid-free recipient cells are shown in gray, and with a granite pattern if they harbor a chromosomal cassette containing a copy of pLS20cat gene 29, 30 or both under the control of the IPTG-inducible P(hy)spank promoter. pLS20cat protein p29 and p30 are schematically indicated with pink ovals and black triangles, respectively. The cMyc tag is indicated with small green oval on top of the protein p29 symbol. Each mating was performed at least three times and the bars correspond to the mean value of these experiments with error bars indicating standard deviations.
Conjugation experiments using the double deletion strain CG27 (pLS20catΔ29-30) gave very similar conjugation efficiencies to that of strain CG52 (pLS20catΔ29) when mated with PKS91 (pLS20spec) (see Figure 1), showing that the absence of protein p30 did not inflict an additional defect on the SE system.
Expression of Gene 29 in Recipient Cells Is Sufficient for Exclusion
To study whether gene 29 is sufficient for SE we constructed strain CG2 in which gene 29 was placed at the chromosomal amyE locus under the control of the isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible Pspank promoter. We also constructed strain EST19 containing both genes 29 and 30 under the control of the Pspank promoter. Next, each of these two strains was employed as the recipient in conjugation experiments using PKS11 (pLS20cat) as donor. As expected, in the absence of IPTG, low SEIs were obtained for matings using CG2 (Pspank-29) or EST19 (Pspank-29-30) as recipient. Importantly, high SEIs were obtained when either recipient strain was grown in the presence of 1 mM IPTG (see Figure 1). These results demonstrate that (i) expression of only gene 29 is sufficient for SE, and (ii) protein p29 is functional in the absence of protein p30.
We next tested whether SE required protein p29 to be present on both mating cells by comparing matings between recipient CG2 cells (Pspank-29) ectopically expressing gene 29, and CG52 donor cells harboring pLS20catΔ29. As shown in Figure 1, high SEIs were obtained for these matings when gene 29 was induced in recipient cells, indicating that SE was not due to interaction between protein p29 molecules on both mating cells.
The results presented in this section and those presented above demonstrate that gene 29 encodes the SE protein of pLS20cat, which henceforth we name sespLS20 (surface exclusion system pLS20).
SespLS20 Is Located on the Cell Surface
To investigate whether SespLS20 is a surface protein as predicted we determined the cellular location of a cMyc-tagged version of SespLS20, named sespLS20-cMyc. Thus, we first constructed strain CG129 which contained a copy of sespLS20-cMyc (the only copy of sespLS20) at the amyE locus under the control of the IPTG-inducible Physpank promoter. High SEI levels were obtained when the expression of sespLS20-cMyc was induced in recipient cells, showing that the fusion protein is functional (Figure 1). We then introduced into strain CG129 a cassette containing the gfp gene under the control of the xylose-inducible promoter Pxyl at the lacA locus, resulting in strain CG133. The intracellular soluble GFP protein served as an internal control in the experiments described below. To study if SespLS20 is a membrane protein we prepared total (T), cytosolic (C) and membrane (M) fractions of CG133 cells grown in the presence of IPTG and xylose, and subjected these to SDS-PAGE followed by immunoblotting using antibodies against cMyc. Figure 2A shows that a clear signal corresponding to a protein having an estimated size of ∼48 kDa, which is slightly more than the calculated weight of 43.3 kDa for SespLS20-cMyc, was obtained for the total protein sample (upper panel, lane 4) of the CG133 cells but not in identically treated samples of the negative control strain CG47 (upper panel, lane 1). A strong signal migrating to the same position was also detected in the membrane fraction of CG133 cells, but only a very minor signal was observed in the cytosolic fraction (upper panel, lanes 6 and 5, respectively). As a control, a duplicate immunoblot was performed with antibodies against GFP. As expected, the vast majority of the GFP signal was obtained in the cytosolic fractions (Figure 2A, lower panel). Together, these results provide compelling evidence that SespLS20-cMyc is a membrane protein.
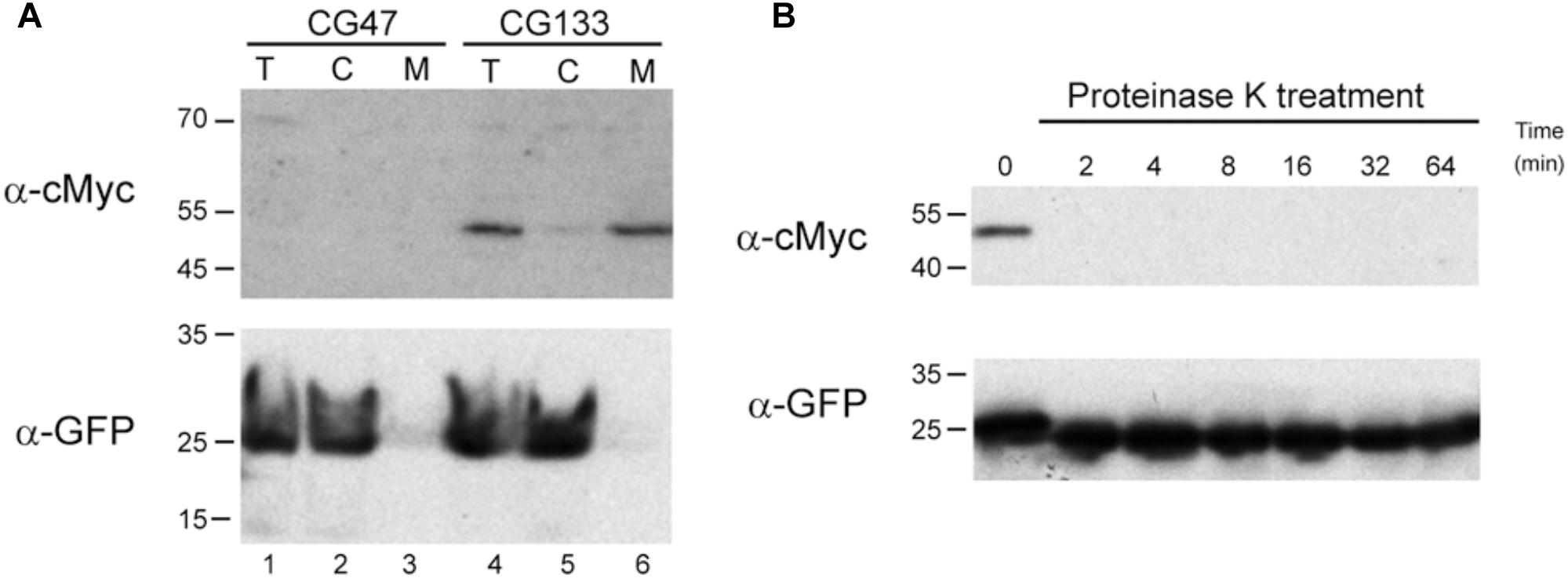
Figure 2. SespLS20 is located on the cell surface. (A) Western blots of fractionated cell samples incubated with antibodies against either the cMyc epitope tag or GFP. Samples of CG47 (lacA::Pxyl-gfp, lanes 1–3) and CG133 (amyE:: Physpank-sespLS20-cMyc, lacA::Pxyl-gfp, lanes 4–6) cultures growing in the presence of IPTG (1 mM) and xylose (2%) were processed resulting in total (T, lanes 1 and 4), cytosolic (C, lanes 2 and 5), and membrane (M, lanes 3 and 6) fractions. These samples were used for two identically loaded SDS-PAA gels, which were subsequently subjected to immunoblotting using antibodies against either the cMyc epitope tag or GFP. Positions of molecular weight markers (kDa) are given on the left. (B) Proteinase K treatment of intact cells expressing sespLS20-cMyc. CG133 (amyE:: Physpank-sespLS20-cMyc, lacA::Pxyl-gfp) cells grown in the presence of IPTG and xylose were harvested, resuspended in a proteinase K containing buffer, and incubated at 37°C. One aliquot of this sample was taken immediately (t = 0) and other aliquots were withdrawn at the indicated times (min). After processing, these samples were analyzed on two identically loaded SDS-PAA gels, which were subsequently subjected to immunoblotting using antibodies against the cMyc epitope tag or GFP. Positions of molecular weight markers (kDa) are given on the left.
To determine whether SespLS20-cMyc is located at the cell surface we treated intact CG133 cells, grown in the presence of both IPTG and xylose, with proteinase K (protK), and then assessed whether this affected the detection of SespLS20-cMyc or GFP. The aliquots of the cells treated with protK for different periods of time were lysed and subjected to SDS-PAGE, followed by immunobloting using antibodies against cMyc or GFP. As expected, a clear signal corresponding to SespLS20-cMyc was detected in the protK-untreated sample (t = 0) (Figure 2B, upper panel), but the signal was no longer detected when the samples had been treated for two min or more with protK. On the contrary, strong signals were still observed for GFP even in samples treated for 64 min with protK (Figure 2B, lower panel). These results show that protK degraded the cMyc epitope fused to SespLS20 but not the cytosolic GFP protein, strongly indicating that SespLS20-cMyc is located at the surface of the cells.
Genes sespLS20 and 30 Are Under the Control of Two Promoters
To carry out their function, exclusion proteins are expected to be expressed constitutively. However, sespLS20 is located within the conjugation operon of pLS20cat that is controlled by the main conjugation promoter Pc (Singh et al., 2013, see Figure 3A). This promoter is strictly repressed except during a transient period of time when an anti-repressor relieves the repression (Singh et al., 2013; Ramachandran et al., 2014). We therefore re-evaluated our previous RNAseq data, obtained from pLS20cat-containing cell samples with/out ectopic overexpression of the repressor of promoter Pc, RcopLS20 (Singh et al., 2013), which showed that all the conjugation genes were repressed most of the time. Previously, a heatmap presentation was used in which color codes indicate differential gene expression under both conditions. Since this might obscure possible expression from a promoter different from Pc we plotted the levels of expression along the plasmid genome for either condition, using the RNAseq data of a newly executed experiment (Figure 3) as well as those of the experiment published before (Singh et al., 2013, Supplementary Figure S2). As expected, genes in the conjugation operon were expressed at high and low levels without and with ectopic rcopLS20 induction, respectively. However, substantial levels of expression were still observed for genes sespLS20 and 30 when rcopLS20 was induced, raising the possibility that genes sespLS20 and 30 were expressed from another promoter that is not controlled by RcopLS20. To test this, we cloned the sespLS20 upstream region in front of a gfp reporter gene (see Materials and Methods; Rudge et al., 2013) and placed it at the chromosomal amyE locus (strain CG1). Using the same strategy, we also constructed strain AND2A, in which the gfp gene is controlled by the strong Pc promoter (Ramachandran et al., 2014). FACS analysis of late exponential cells (OD600 = 1) was then used to determine fluorescence levels (Figure 4). As expected, a high level of fluorescence was observed for strain AND2A (Pc-gfp). Importantly, although lower than that observed for AND2A, CG1 cells were also fluorescent, demonstrating that the cloned region upstream of the sespLS20 gene contains a promoter, which we name P29. Fluorescence levels of strains CG35 and CG36 are explained below.
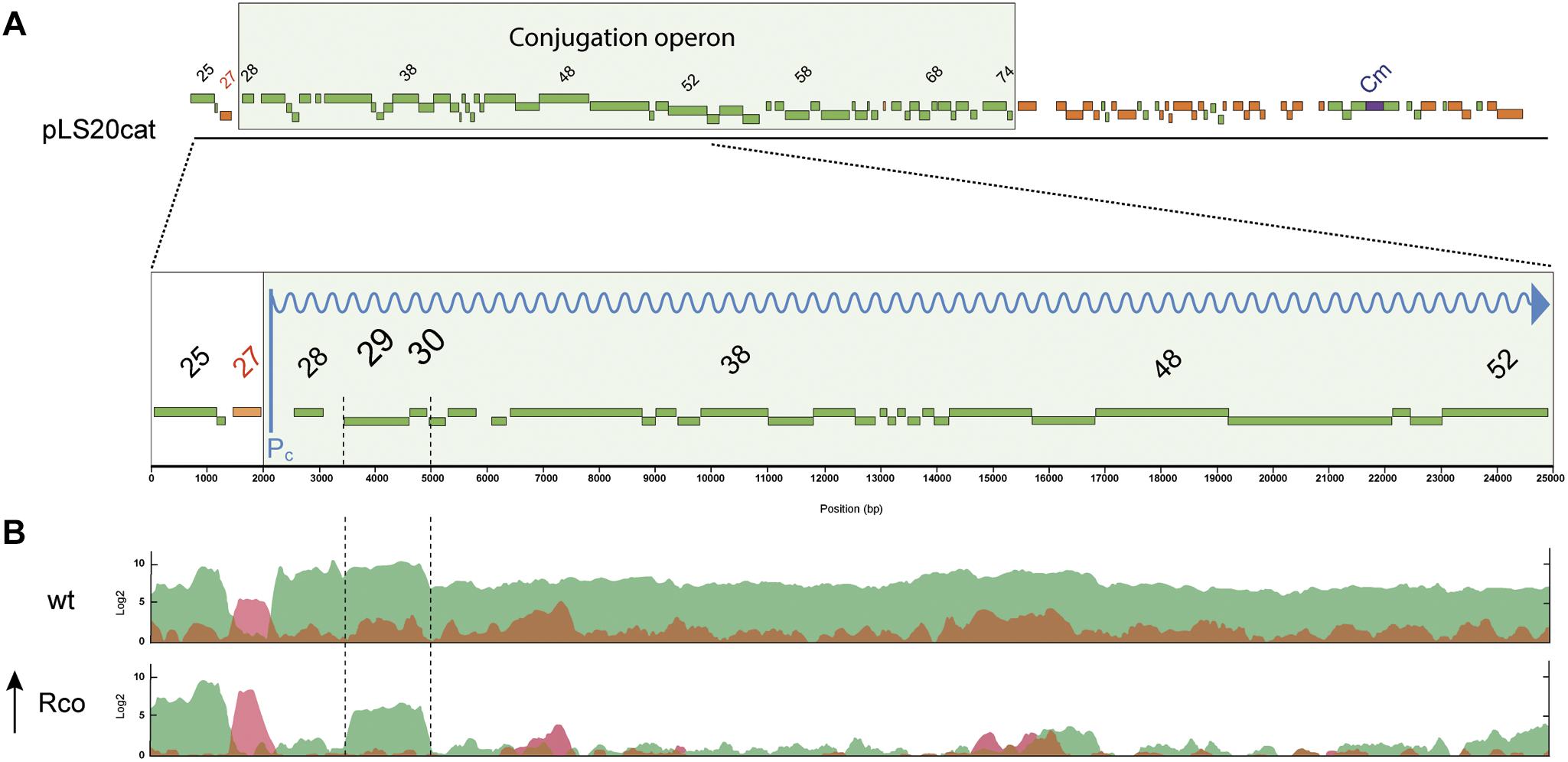
Figure 3. pLS20cat genes sespLS20 and 30 are expressed independently of the main conjugation promoter Pc. (A) Schematic presentation of the genetic organization of pLS20cat. Position and size of genes are indicated with rectangular bars; green and red colors present forward and reverse orientation of the genes, respectively. The conjugation operon is boxed. The chloramphenicol resistance gene (Cm) is indicated in blue. A blow-up of the region spanning genes 25 to 52 is shown in the lower part. The reversely oriented gene 27 encodes the transcriptional regulator RcopLS20. (B) Expression profiles of pLS20cat genes determined by RNAseq. Total RNA was isolated from late exponentially growing cells of strains PKS11 (pLS20cat; upper panel) and PKS14 (Pspank-rcopLS20, pLS20cat, lower panel) grown in the presence of 1 mM IPTG. Expression levels are presented on a log2 scale. Forward and reversely oriented transcripts are indicated in green and red, respectively. Note that rcopLS20 (gene 27) was expressed at a higher level in PKS14 than in PKS11 cells due to the additional induced expression of this gene from the ectopic locus at amyE. Results corresponding to pLS20cat genes 25 till 52 are shown. Data presented here are from a newly executed RNAseq experiment. Supplementary Figure S2 shows that a very similar expression profile was obtained using the data of the previously published RNAseq experiment (Singh et al., 2013).
Gene 30 Is Not Involved in the Exclusion System of pLS20cat
Although gene 30 is co-transcribed with sespLS20 from the P29 promoter, results presented above did not indicate that it had a role in exclusion. To study a possible role of gene 30 in more detail we constructed strain CG28 that harbored a derivative of pLS20cat lacking gene 30 (pLS20catΔ30, see Materials and Methods) and used it as donor in matings with a plasmid-free recipient strain expressing sespLS20 (i.e., CG2). The absence of gene 30 did not significantly affect CE (Figure 1). This, together with the results presented above, demonstrates that gene 30 does not play a crucial role in SE.
Correlation Between the Level of Surface Exclusion and sespLS20 Induction
Induction of sespLS20 in recipient cells from the Pspank promoter in the presence of 1 mM IPTG provoked SE (Figure 1). Interestingly, in this experimental set up the SEI was about 10-fold higher than that observed for matings between two donor strains. This discrepancy might be due to different sespLS20 expression levels in the two experimental set-ups. To test this, we performed matings in which sespLS20 was induced to different levels in the recipient strain CG2. The results presented in Figure 1 show a direct correlation between the extent of SE and the level of gene sespLS20 induction. Next, we expressed sespLS20 in recipient cells from a promoter stronger than Pspank, the IPTG-inducible Physpank promoter. The strain, CG106, was used as a recipient strain in matings with PKS11 (pLS20cat) as donor strain. When grown in the presence of 1 mM IPTG, the SEI value obtained for strain CG106 (Physpank-sespLS20) was about 10-fold higher than that obtained for strain CG2 (Pspank-sespLS20) and about 100-fold higher than that obtained for matings between two donor strains (see Figure 1).
To compare the (maximum) strengths of promoters P29, Pc, Pspank, and Physpank we constructed strains CG35 and CG36 containing a cassette in which gfp was placed under the control of the Pspank and the Physpank promoter, respectively, and determined the fluorescence levels of these cells growing in the presence of 1 mM IPTG in parallel with those of strains CG1 (P29-gfp) and AND2A (Pc-gfp) (Figure 4). The results obtained confirmed that the maximum strength of Physpank promoter was considerably stronger (by at least 6-fold) than that of the Pspank promoter. In addition, the results showed that P29 was the weakest of these promoters, and that the strength of Pc was similar to that of the Physpank promoter induced to maximum levels. Taken together, these results provide strong evidence that the amount of protein SespLS20 determines the strength of the SE system.
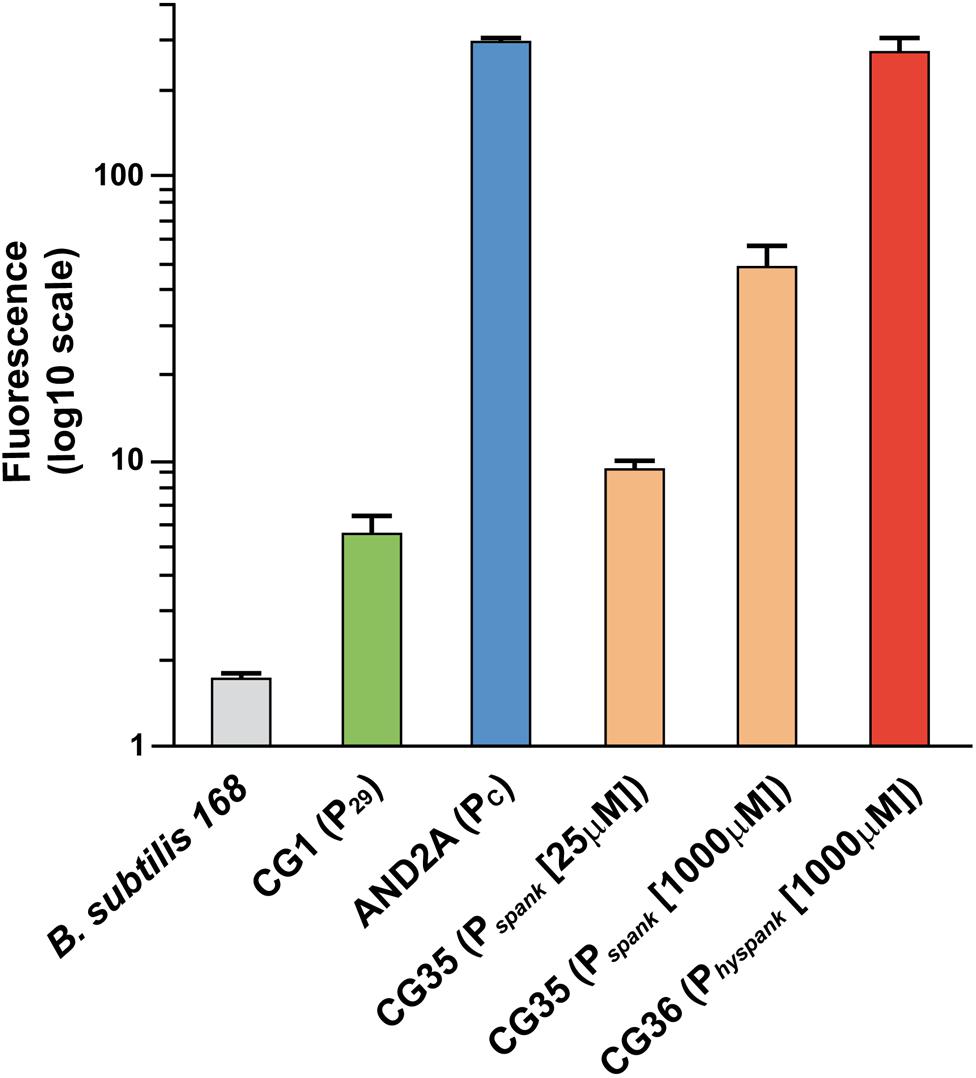
Figure 4. Relative promoter strength determined by FACS analysis of strains containing a transcriptional gfp fusion. Strains having at the amyE locus a cassette containing a single copy of the gfp reporter gene fused to a promoter of interest, and a negative control strain were used for FACS analysis at an OD600 = 1. At least 100,000 cells were analyzed for each sample. Strains: 168, gray; CG1 (P29-gfp), green; AND2A (Pc-gfp), blue; CG35 (Pspank-gfp) grown in the presence of 25 or 1,000 μM IPTG, orange; CG36 (Physpank-gfp) grown in the presence of 1 mM IPTG, red. For each strain the mean values of geomean determinations of at least three independent FACS analyses are given together with their standard deviations.
Although the maximum strength of Physpank was similar to that of the Pc promoter (Figure 4), the SEI of matings between two donor strains was much lower than that in matings using recipient cells expressing sespLS20 from the Physpank promoter at 1 mM IPTG. These results could be explained bearing in mind that the Pc promoter became activated only in a subpopulation of the donor cells (Rosch and Graumann, 2015). The modest SEI levels observed for matings between two donor strains would therefore be the consequence of the low-level sespLS20 expression from the weak P29 promoter in receptive donor cells that have not activated the conjugation promoter Pc. This conclusion was supported by the observations that the SEI obtained between two donor strains was similar to that between a donor strain and recipient strain CG2 (Pspank-sespLS20) grown in the presence of 25 μM IPTG (Figure 1), and that the strength of promoter P29 was similar to that of the Pspank promoter induced at 25 μM IPTG (Figure 4).
SespLS20 Is the Founding Member of a New Family of Surface Exclusion Proteins Whose Homologs Are Present in a Number of Firmicutes Bacteria Including Pathogens
Bacillus pumilus strain NRS576 harbors a plasmid, p576, which is related to pLS20: they share a similar origin of replication and plasmid segregation module, and they both have a conjugation operon (Singh et al., 2010; Val-Calvo et al., 2019). Based on this we analyzed whether p576 contained a homologue of sespLS20. This analysis revealed that the second gene of its putative conjugation operon, gene 38, a predicted membrane protein that shares significant similarity with SespLS20 (51% similarity; see Supplementary Figure S3). Based on the similarity and its conserved genetic location within the conjugation operon it was likely that p576 gene 38 encoded a homologue of SespLS20 which we tentatively named sesp576. To study whether public databases contained genes encoding proteins similar to SespLS20/Sesp576 we used these protein sequences to perform ‘blastp’ searches. After removing redundant sequences these searches resulted in the identification of 30 hits showing significant similarity to SespLS20 (see Supplementary Table S4). All the identified genes, which were annotated as unknown function, could encode a putative protein of similar size to SespLS20. Interestingly, all these hits corresponded to putative genes present in the phylum Firmicutes genera Bacilli, Listeria or Clostridia. Results of additional analyses supported the view that the identified genes encoded homologs of SespLS20. First, all except one were predicted with more than 95% confidence to encode a protein containing an N-terminal membrane anchor. In the case of the single exception, an N-terminal transmembrane spanning domain was predicted with 60% confidence. In some cases, an analysis of its surrounding context was limited because the identified gene was located at the beginning of a (small) contig. Nevertheless, in 24 out of the 30 cases it could be established that the identified gene formed part of a likely conjugation operon, and in most of these cases the putative conjugation operons was located on a plasmid. Strikingly, the sespLS20 homologs were one of the first genes of their corresponding putative conjugation operon, as in pLS20 and p576. In summary, our data provide compelling evidence that SespLS20 constitutes the founding member of a family of SE proteins that forms part of a conjugative element that is present in a subset of bacteria belonging to the Gram+ phylum Firmicutes.
Finally, we also used an in silico approach to analyze whether the genes downstream of the identified sespLS20 homologs shared similarity with pLS20cat gene 30. As shown in Supplementary Table S5, only the protein encoded by the gene downstream of the sespLS20 homolog, present on an unnamed putative conjugative Bacillus atrophaeus plasmid, shared more than 20% identity with pLS20cat encoded protein p30. This further supports the view that the identified SE system is composed of the single sespLS20-like gene.
Discussion
Probably all conjugative plasmids contain at least one exclusion gene (Garcillan-Barcia and De la Cruz, 2008). Improving our knowledge about exclusion systems may open ways to impede or block conjugation-mediated spread of antibiotic resistance. Most information available on exclusion systems is related to conjugative plasmids replicating in G− bacteria, prompting us to study the SE system of the Gram+ B. subtilis conjugative plasmid pLS20. Taking into account the large differences in the cell wall organization between G− and G+ bacteria, the SE proteins encoded by conjugative elements replicating in G+ bacteria may be very different from those replicating in G− bacteria. The little information that is available indicates that this is indeed the case. As far as we know, the only genuine G+ exclusion systems described are those encoded by the so-called enterococcal conjugative sex-pheromone plasmids, particularly those present on pCF10 and pAD1 (Dunny et al., 1985; Kao et al., 1991; Weidlich et al., 1992). These plasmids contain a conserved gene, sec10 (a.k.a. prgA) or sea1 encoding a surface-located protein that is responsible for SE (Clewell and Brown, 1980; Dunny et al., 1985).
Here we have demonstrated that pLS20 contains a SE system that is different from all exclusion systems described so far, and that homologues of sespLS20 are present on other (likely) conjugative plasmids of Firmicutes bacteria including (opportunistic) pathogens like Listeria or Clostridia.
It is generally believed that the primary function of exclusion systems is to prevent redundant transfer of conjugative elements. However, matings between two pLS20-containing donor strains revealed that, although redundant transfer was inhibited, it was far from being blocked. As outlined in more detail below, this is also the case for most if not all SE systems studied so far and indicates that the function of exclusion systems is more complex than “simply” preventing redundant transfer of a conjugative element.
The major findings of these studies can be summarized as follows: (i) sespLS20 is the only necessary gene to exert exclusion, (ii) there is a direct relation between the level of sespLS20 expression and the level of exclusion, (iii) sespLS20 is differentially expressed in donor cells having or not activated the conjugation pathway; i.e., sespLS20 is expressed at low basal levels in all donor cells from the weak promoter P29 that is located immediately upstream of sespLS20, but it is expressed to much higher levels in conjugation-primed cells due to the additional expression from the strong conjugation promoter Pc. Whereas the basal expression level of sespLS20 in all donor cells inhibits redundant plasmid transfer only moderately (∼10-fold), its high-level expression in conjugation-primed cells inhibits redundant plasmid transfer very efficiently (∼1,000-fold). Thus, as schematically shown in Figure 5, our results revealed that the SE system of pLS20 has a dual effect: it inhibits moderately the transfer of the plasmid between donor cells in general, but efficiently prevents the transfer of pLS20 between two conjugation-primed cells.
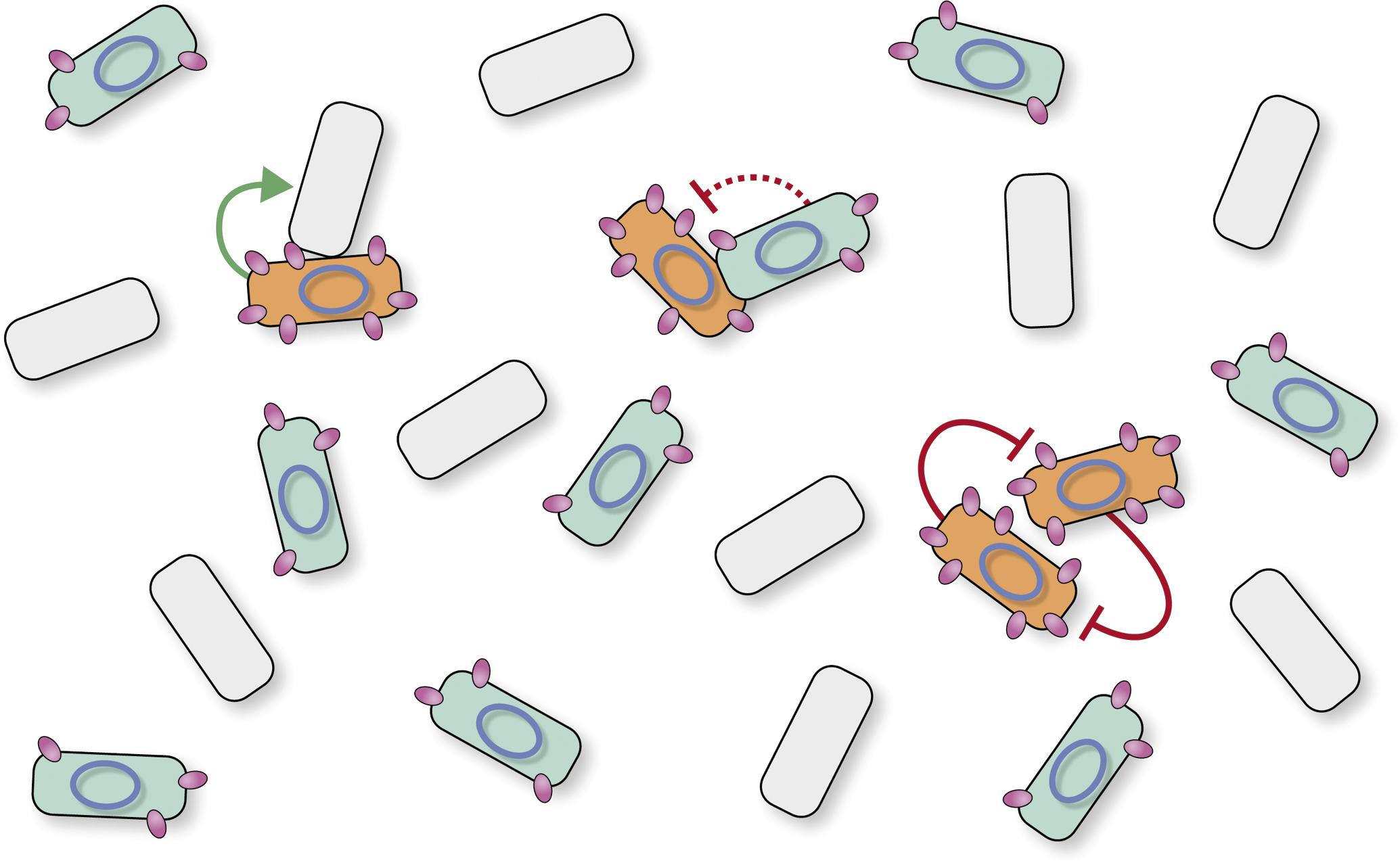
Figure 5. Model depicting different surface exclusion levels for conjugative transfers between two conjugation-primed donor cells, and between conjugation-primed donor cells with non-primed donor cells. Cartoon of a mixed population of cells consisting of plasmid-free recipients (shown in gray) and pLS20-containing donor cells (cells with a blue ellipse). Donor cells express low levels of SespLS20 (shown as small purple ovals) due to the weak constitutive P29 promoter (cells shown in blue). However, because sespLS20 is also under the control of the strong conjugation promoter Pc, its expression becomes up-regulated in the subpopulation of donor cells that have activated the conjugation genes (shown as orange cells). The extent of surface exclusion is directly related to the level of sespLS20 expression. Conjugation is not inhibited between conjugation-primed donor and plasmid-free recipient cells that do not express SespLS20 (reflected with green solid line arrow), resulting therefore in optimal conjugation efficiencies and maximum spread of conjugative plasmid in the population. Low-level expression of SespLS20 in non-conjugation primed donor cells moderately inhibits conjugative transfer between [conjugation-primed and non-conjugation-primed donor cells] (indicated with interrupted red line ending in a “T” shape) favoring efficient spread of the plasmid in the population, as well as genetic exchange (i.e., evolutionary adaptation) between genes and/or modules associated with the conjugation module. Finally, high levels of SespLS20 expression efficiently inhibit conjugation between two conjugation-primed donor cells (indicated with solid red lines ending in a “T”-shape).
The fact that a sophisticated transcriptional circuitry has evolved to differentially express sespLS20 in primed and non-primed donor cells suggests that the resulting differences in SE levels in these cells underlie evolutionary advantage(s). For instance, inhibition of plasmid transfer between two donor cells will contribute to more efficient spreading of the plasmid to cells not harboring the plasmid. However, by preventing redundant transfer only partially it also may contribute to genetic flexibility of the plasmid and the host. For this it has to be taken into account that plasmids have a modular organization; the essential backbone of a plasmid, -comprising the replication and maintenance functions-, is generally combined with one or more non-essential modules, including a conjugation module, which can be interchanged with other (extra)chromosomal DNA residing in the bacterium (for review see, Frost et al., 2005; Norman et al., 2009). Therefore, moderate inhibition of redundant plasmid transfer is beneficial for the plasmid and their hosts in evolutionary terms: it favors efficient spreading of the plasmid in a population of cells, without losing the ability to exchange modules or genes that are associated with another conjugative element sharing the same SE system. Probably this is also the reason why the SE gene is embedded within the conjugation operon. If located outside of the conjugation operon, it can become uncoupled from the conjugation operon if such a plasmid recombines with another (conjugative) DNA element. A conjugation module lacking a SE system will be compromised in efficient spreading of the plasmid and be outcompeted by an element containing a SE system.
Transfer of plasmids between two conjugation-primed donor cells, on the other hand, is efficiently inhibited. In addition to preventing futile plasmid exchanges, the other reason for the strong inhibition could be that simultaneous bidirectional transfer is not compatible. The transfer process involves the establishment of a mating pair, local lysis of the cell wall, and the formation of a sophisticated membrane-associated translocation machinery that connects the mating cells and transfers – besides some proteins – many kbp of DNA (∼65 kb in the case of pLS20) in its single-stranded form. It is conceivable that simultaneous bidirectional transfer of the plasmid is not feasible due to, for instance, incompatibility of the translocation machineries with opposing directionalities and/or incompatibility of the incoming ssDNA with the simultaneous production of ssDNA to be transferred.
Similar to what we observed for pLS20, all SE systems studied so far, regardless whether present on ICEs or conjugative plasmids of G− or G+ origin, do not efficiently inhibit conjugation when analyzed at population level, with a inhibition range of 10 to 20-fold (Clewell and Brown, 1980; Dunny et al., 1985) (for review see Garcillan-Barcia and De la Cruz, 2008). Furthermore, it appears that most if not all SE genes are located within the conjugation operon. This applies to exclusion genes present in ICEs as well as conjugative plasmids of G− or G+ origin. Therefore, the particular structural organization in which the SE gene is embedded in the conjugation operon, and the differential transcriptional expression of the SE gene in non-conjugation-primed and conjugation-primed donor cells appears to be a common feature of SE systems.
In summary, besides unraveling a novel SE system present on pLS20, our results permitted the identification of putative similar systems on other conjugative elements present in Gram+ bacteria and adds a new dimension to the function of SE systems in general.
Data Availability
RNAseq data are deposited in the SRA repository (submission SUB5726602).
Author Contributions
All authors have made substantial, direct experimental and/or intellectual contribution to the work. CG-C, JV-C, AM-A, ES, and PS generated all plasmids and strains, purified proteins, and conducted all the experiments. DA and JV-C carried out in silico analyses. DA also contributed to the general design and analyses of the results. LW and WM designed the experimental plan and were principally responsible for analyzing the results and writing the manuscript. WM supervised the contributions of CG-C, JV-C, AM-A, ES, and PS.
Funding
Work in the Meijer lab was funded by grants Bio2013-41489-P and BIO2016-77883-C2-1-P of the Ministry of Economy and Competitiveness of the Spanish Government to WM, which also funded CG-C, JV-C, and AM-A. Part of the economic support of the two aforementioned grants was provided by the “Agencia Estatal de Investigación (AEI)” and “Fondo Europeo de Desarrollo Regional (FEDER).” This research was also supported by institutional grants from the “Fundación Ramón Areces” and “Banco de Santander” to the Centro de Biología Molecular “Severo Ochoa.” LW’s work was supported by a Wellcome Trust grant awarded to Jeff Errington (209500). The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We acknowledge support of the publication fee by the CSIC Open Access Publication Support Initiative through its Unit of Information Resources for Research (URICI). We thank Paul J. Steiner for sending us plasmid pJS104, José Belio for help with preparing the figures, Sara Pico for technical help with Western blotting, and Margarita Salas and Jeff Errington for their support on our work. We also want to acknowledge helpful discussion with other lab members.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01502/full#supplementary-material
Footnotes
- ^ www.cbs.dtu.dk/servises/TMHMM
- ^ http://www.cbs.dtu.dk/services/SignalP/
- ^ https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE=Proteins
- ^ https://www.drive5.com/usearch/
References
Achtman, M., Kennedy, N., and Skurray, R. (1977). Cell–cell interactions in conjugating Escherichia coli: role of trat protein in surface exclusion. Proc. Natl. Acad. Sci. U.S.A. 74, 5104–5108. doi: 10.1073/pnas.74.11.5104
Achtman, M., Manning, P. A., Edelbluth, C., and Herrlich, P. (1979). Export without proteolytic processing of inner and outer membrane proteins encoded by F sex factor tra cistrons in Escherichia coli minicells. Proc. Natl. Acad. Sci. U.S.A. 76, 4837–4841. doi: 10.1073/pnas.76.10.4837
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids. Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Altschul, S. F., Wootton, J. C., Gertz, E. M., Agarwala, R., Morgulis, A., Schaffer, A. A., et al. (2005). Protein database searches using compositionally adjusted substitution matrices. FEBS J. 272, 5101–5109. doi: 10.1111/j.1742-4658.2005.04945.x
Andersen, J. B., Sternberg, C., Poulsen, L. K., Bjorn, S. P., Givskov, M., and Molin, S. (1998). New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl. Environ. Microbiol. 64, 2240–2246.
Anthony, K. G., Klimke, W. A., Manchak, J., and Frost, L. S. (1999). Comparison of proteins involved in pilus synthesis and mating pair stabilization from the related plasmids F and R100-1: insights into the mechanism of conjugation. J. Bacteriol. 181, 5149–5159.
Arnaud, M., Chastanet, A., and Debarbouille, M. (2004). New vector for efficient allelic replacement in naturally nontransformable, low-GC-content, gram-positive bacteria. Appl. Environ. Microbiol. 70, 6887–6891. doi: 10.1128/aem.70.11.6887-6891.2004
Audette, G. F., Manchak, J., Beatty, P., Klimke, W. A., and Frost, L. S. (2007). Entry exclusion in F-like plasmids requires intact TraG in the donor that recognizes its cognate TraS in the recipient. Microbiology 153(Pt 2), 442–451. doi: 10.1099/mic.0.2006/001917-0
Bron, S., Peijnenburg, A., Peeters, B. P. H., Haima, P., and Venema, G. (1989). “Cloning and plasmid (in)stability in Bacillus subtilis,” in Genetic Transformation And Expression, eds L. O. Butler, C. R. Harwood, and B. E. B. Moseley (Andover: Intercept Ltd.), 205–219.
Chatterjee, A., Johnson, C. M., Shu, C. C., Kaznessis, Y. N., Ramkrishna, D., Dunny, G. M., et al. (2011). Convergent transcription confers a bistable switch in Enterococcus faecalis conjugation. Proc. Natl. Acad. Sci. U.S.A. 108, 9721–9726. doi: 10.1073/pnas.1101569108
Clewell, D. B., and Brown, B. L. (1980). Sex pheromone cAD1 in Streptococcus faecalis: induction of a function related to plasmid transfer. J. Bacteriol. 143, 1063–1065.
Dunny, G. M., Zimmerman, D. L., and Tortorello, M. L. (1985). Induction of surface exclusion (entry exclusion) by Streptococcus faecalis sex pheromones: use of monoclonal antibodies to identify an inducible surface antigen involved in the exclusion process. Proc. Natl. Acad. Sci. U.S.A. 82, 8582–8586. doi: 10.1073/pnas.82.24.8582
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Frost, L. S., Leplae, R., Summers, A. O., and Toussaint, A. (2005). Mobile genetic elements: the agents of open source evolution. Nat. Rev. Micobiol. 3, 722–732. doi: 10.1038/nrmicro1235
Garcillan-Barcia, M. P., and De la Cruz, F. (2008). Why is entry exclusion an essential feature of conjugative plasmids? Plasmid 60, 1–18. doi: 10.1016/j.plasmid.2008.03.002
Hartskeerl, R. A., and Hoekstra, W. P. (1984). Exclusion in IncI-type Escherichia coli conjugations: the stage of conjugation at which exclusion operates. Antonie van Leeuwenhoek 50, 113–124. doi: 10.1007/bf00400171
Kao, S. M., Olmsted, S. B., Viksnins, A. S., Gallo, J. C., and Dunny, G. M. (1991). Molecular and genetic analysis of a region of plasmid pCF10 containing positive control genes and structural genes encoding surface proteins involved in pheromone-inducible conjugation in Enterococcus faecalis. J. Bacteriol. 173, 7650–7664. doi: 10.1128/jb.173.23.7650-7664.1991
Krogh, A., Larsson, B., von, H. G., and Sonnhammer, E. L. (2001). Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J. Mol. Biol. 305, 567–580. doi: 10.1006/jmbi.2000.4315
Lederberg, J., Cavalli, L. L., and Lederberg, E. M. (1952). Sex compatibility in Escherichia coli. Genetics 37, 720–730.
Marrero, J., and Waldor, M. K. (2005). Interactions between inner membrane proteins in donor and recipient cells limit conjugal DNA transfer. Dev. Cell 8, 963–970. doi: 10.1016/j.devcel.2005.05.004
Mazel, D., and Davies, J. (1999). Antibiotic resistance in microbes. Cell Mol. Life Sci. 56, 742–754.
Norman, A., Hansen, L. H., and Sorensen, S. J. (2009). Conjugative plasmids: vessels of the communal gene pool. Philos. Trans. R. Soc. Lond. B Biol. Sci. 364, 2275–2289. doi: 10.1098/rstb.2009.0037
Ochman, H., Lawrence, J. G., and Groisman, E. A. (2000). Lateral gene transfer and the nature of bacterial innovation. Nature 405, 299–304. doi: 10.1038/35012500
Ou, J. T. (1975). Mating signal and DNA penetration deficiency in conjugation between male Escherichia coli and minicells. Proc. Natl. Acad. Sci. U.S.A. 72, 3721–3725. doi: 10.1073/pnas.72.9.3721
Patrick, J. E., and Kearns, D. B. (2008). MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol. Microbiol. 70, 1166–1179. doi: 10.1111/j.1365-2958.2008.06469.x
Pedelacq, J. D., Cabantous, S., Tran, T., Terwilliger, T. C., and Waldo, G. S. (2006). Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24, 79–88. doi: 10.1038/nbt1172
Ramachandran, G., Singh, P. K., Luque-Ortega, J. R., Yuste, L., Alfonso, C., Rojo, F., et al. (2014). A complex genetic switch involving overlapping eivergent promoters and DNA looping regulates expression of conjugation genes of a Gram-positive plasmid. PLoS Genet. 10:e1004733. doi: 10.1371/journal.pgen.1004733
Rosch, T. C., and Graumann, P. L. (2015). Induction of plasmid conjugation in Bacillus subtilis es bistable and driven by a direct interaction of a Rap/Phr quorum-sensing system with a master repressor. J. Biol. Chem. 290, 20221–20232. doi: 10.1074/jbc.m115.664110
Rudge, T. J., Federici, F., Steiner, P. J., Kan, A., and Haseloff, J. (2013). Cell polarity-driven instability generates self-organized, fractal patterning of cell layers. ACS Synth. Biol. 2, 705–714. doi: 10.1021/sb400030p
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: a Laboratory Manual. New York, NY: Cold Spring Harbor Laboratory Press.
Schaffer, A. A., Aravind, L., Madden, T. L., Shavirin, S., Spouge, J. L., Wolf, Y. I., et al. (2001). Improving the accuracy of PSI-BLAST protein database searches with composition-based statistics and other refinements. Nucleic Acids Res. 29, 2994–3005. doi: 10.1093/nar/29.14.2994
Singh, P. K., Ballestero-Beltran, S., Ramachandran, G., and Meijer, W. J. (2010). Complete nucleotide sequence and determination of the replication region of the sporulation inhibiting plasmid p576 from Bacillus pumilus NRS576. Res. Microbiol 161, 772–782. doi: 10.1016/j.resmic.2010.07.007
Singh, P. K., and Meijer, W. J. (2014). Diverse regulatory circuits for transfer of conjugative elements. FEMS Microbiol. Lett. 358, 119–128. doi: 10.1111/1574-6968.12526
Singh, P. K., Ramachandran, G., Ramos-Ruiz, R., Peiro-Pastor, R., Abia, D., Wu, L. J., et al. (2013). Mobility of the native Bacillus subtilis conjugative plasmid pLS20 is regulated by intercellular signaling. PLoS Genet. 9:e1003892. doi: 10.1371/journal.pgen.1003892
Sommer, M. O., Dantas, G., and Church, G. M. (2009). Functional characterization of the antibiotic resistance reservoir in the human microflora. Science 325, 1128–1131. doi: 10.1126/science.1176950
Sonnhammer, E. L., von Heijne, G., and Krogh, A. (1998). A hidden Markow model for predicting transmembrane helices in protein sequences. Proc. Int. Conf. Intell. Syst. Mol. Biol. 6, 175–182.
Steinmetz, M., and Richter, R. (1994). Plasmids designed to alter the antibiotic resistance expressed by insertion mutations in Bacillus subtilis, through in vivo recombination. Gene 142, 79–83. doi: 10.1016/0378-1119(94)90358-1
Thomas, C. M., and Nielsen, K. M. (2005). Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat. Rev. Microbiol. 3, 711–721. doi: 10.1038/nrmicro1234
Val-Calvo, J., Miguel-Arribas, A., Gago-Cordoba, C., Lopez-Perez, A., Ramachandran, G., Singh, P. K., et al. (2019). Draft genome sequences of sporulation-impaired Bacillus pumilus strain NRS576 and its native plasmid p576. Microbiol. Resour. Announc. 8:e00089-19. doi: 10.1128/MRA.00089-19
Veening, J. W., Smits, W. K., Hamoen, L. W., Jongbloed, J. D., and Kuipers, O. P. (2004). Visualization of differential gene expression by improved cyan fluorescent protein and yellow fluorescent protein production in Bacillus subtilis. Appl. Environ. Microbiol. 70, 6809–6815. doi: 10.1128/aem.70.11.6809-6815.2004
Keywords: surface exclusion, antibiotic resistance, Firmicutes, horizontal gene transfer, conjugation, Gram-positive bacteria, gene expression, surface protein
Citation: Gago-Córdoba C, Val-Calvo J, Miguel-Arribas A, Serrano E, Singh PK, Abia D, Wu LJ and Meijer WJJ (2019) Surface Exclusion Revisited: Function Related to Differential Expression of the Surface Exclusion System of Bacillus subtilis Plasmid pLS20. Front. Microbiol. 10:1502. doi: 10.3389/fmicb.2019.01502
Received: 07 May 2019; Accepted: 14 June 2019;
Published: 10 July 2019.
Edited by:
Rustam Aminov, University of Aberdeen, United KingdomReviewed by:
Daniel B. Kearns, Indiana University Bloomington, United StatesElisabeth Grohmann, Beuth Hochschule für Technik Berlin, Germany
Copyright © 2019 Gago-Córdoba, Val-Calvo, Miguel-Arribas, Serrano, Singh, Abia, Wu and Meijer. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wilfried J. J. Meijer, d21laWplckBjYm0uY3NpYy5lcw==
†Present address: Ester Serrano, Laboratorio 214, Centro Nacional de Biotecnología, Consejo Superior de Investigaciones Científicas, Madrid, Spain Praveen K. Singh, Max Planck Institute for Terrestrial Microbiology, Marburg, Germany