 Lei Wei1,2
Lei Wei1,2 Qingping Wu2*
Qingping Wu2* Jumei Zhang2
Jumei Zhang2 Weipeng Guo2
Weipeng Guo2 Qihui Gu2
Qihui Gu2 Huiqing Wu2
Huiqing Wu2 Juan Wang3
Juan Wang3 Tao Lei2
Tao Lei2 Moutong Chen2
Moutong Chen2 Musheng Wu4Aimei Li4
Musheng Wu4Aimei Li4- 1School of Bioscience and Bioengineering, South China University of Technology, Guangzhou, China
- 2State Key Laboratory of Applied Microbiology Southern China, Guangdong Provincial Key Laboratory of Microbial Culture Collection and Application, Guangdong Open Laboratory of Applied Microbiology, Guangdong Institute of Microbiology, Guangdong Academy of Science, Guangzhou, China
- 3College of Food Science, South China Agricultural University, Guangzhou, China
- 4Guangdong Dinghu Mountain Spring Company Limited, Zhaoqing, China
The aim of this study was to gain insight into the bacterial composition and dynamics in a mineral water treatment system (MWTS). The bacterial community of a full-scale mineral water treatment plant in the Maofeng Mountain, South China, was studied using high-throughput sequencing combined with cultivation-based techniques in both the dry and wet season. Overall, adenosine tri-phosphate (ATP) concentration (6.47 × 10-11 – 3.32 × 10-8 M) and heterotrophic plate counts (HPC) (3 – 1.29 × 103 CFU/mL) of water samples in the wet season were lower than those (ATP concentration 5.10 × 10-11 – 6.96 × 10-8 M, HPC 2 – 1.97 × 103 CFU/mL) in the dry season throughout the whole MWTS. The microbial activity and biomass of water samples obviously changed along with treatment process. All 300 isolates obtained using cultivation-based techniques were distributed in 5 phyla, 7 classes, and 19 genera. Proteobacteria accounted for 55.7% (167) of the total isolates, among which predominant genus was Pseudomonas (19.3%). Illumina sequencing analysis of 16s rRNA genes revealed 15 bacterial phyla (relative abundance >0.1%) as being identified in all water samples. Among these, Proteobacteria constituted the dominant bacteria microbiota in all water samples. A large shift in the proportion of Bacteroidetes, Actinobacteria, and Firmicutes was obtained during the treatment process, with the proportion of Bacteroidetes, Actinobacteria decreasing sharply, whereas that of Firmicutes increased and predominated in the final water product. The core microbiome, which was still present in whole MWTS comprised several genera including Pseudomonas, Acinetobacter, Clostridium, and Mycobacterium, that contain species that are opportunistic pathogens, suggesting a potential threat for mineral water microbiology safety. This study is the first to investigate the bacterial community of a full-scale mineral water treatment plant in China. The results provided data regarding the bacteria composition and dynamics in an MWTS, which will contribute to the beneficial manipulation of the mineral water microbiome.
Introduction
Large-scale outbreaks of infectious diseases via waterborne pathogens constitute a public serious health problem (Faria et al., 2009; Liu et al., 2018). It is cautiously estimated that approximately 350 million people worldwide are infected with waterborne pathogens and 25 million children annually die from diseases caused by drinking contaminated water (Eichler et al., 2006; Deere et al., 2017). Although numerous strictly regulated microbial indicators exist for mineral water, the production of mineral water that meets quality standards does not necessarily guarantee microbial safety (Varga, 2011; Zhang et al., 2015). In particular, several recent studies have found that mineral water contains pathogens and opportunistic pathogens, which can adversely affect the health of consumers (Herath et al., 2012; Wei et al., 2017). The bacterial community is ubiquitous throughout the entire mineral water treatment system (MWTS) (Bharath et al., 2003; Pontara et al., 2011). Therefore, a comprehensive understanding of the composition and dynamics of bacterial communities in the MWTS is essential to guarantee the microbial safety of mineral water.
The spatiotemporal heterogeneity of microbial communities in drinking water treatment system (DWTS) has been investigated in several previous studies. For example, Pinto et al. (2014) found that the microbial community structure exhibits a large temporal change in DWTS. However, some studies have also shown that the primary dynamic pattern of variations in bacterial communities occur during the treatment process rather than via temporal fluctuations (El-Chakhtoura et al., 2015; Li et al., 2017). Hou et al. (2018) reported that microbial community structure had temporal change in DWTS and the treatment processes exerted profound influence on the microbial community composition. However, compared to DWTS, little information is available regarding the spatiotemporal variations of multi-step treatment processes in MWTS, which cannot guarantee the microbial safety of the final mineral water product.
The production process of mineral water mainly includes three levels of filter (quartz sand, activated carbon, and fine filter), ozone sterilization, filling and capping, and light inspection of the finished product (Zhang et al., 2011; Wei et al., 2017). Although some studies have focused on the raw water and the final water product in MWTS, the information is still lacking regarding on the spatiotemporal changes of the bacterial communities in the whole treatment process. Moreover, how each treatment process and season affects the bacterial community structure of mineral water remains unclear, and it is unknown whether common microbial patterns exist in the MWTS. This information was very important for ensuring the microbial safety of mineral water production. In addition, most studies regarding the bacterial communities have been conducted using a single method, such as cultivation-based techniques or molecular biological methods. However, as 90% of microorganisms are uncultured, the results of cultivation-based techniques cannot accurately reflect the actual information of bacterial communities (Griebler and Lueders, 2009; Albertsen et al., 2013). Molecular biology technology which avoids the technical difficulty of obtaining uncultured microorganisms, has fundamental disadvantages. Molecular biology technology does not allow an accurate understanding of the physiological and metabolic characteristics of microorganisms, which is unable to be effectively applied for the control and utilization of microorganisms (Hahn et al., 2016; Pulschen et al., 2017). Therefore, the actual composition and characteristics of the mineral water microbial community needs to be explored in more detail through the use of high-throughput sequencing analysis combined with cultivation techniques.
To our knowledge, a systematic survey of the bacterial community in MWTS has not previously been conducted in China. In the present study, the bacterial community of a full-scale mineral water treatment plant in the Maofeng Mountain, South China was studied using Illumina HiSeq sequencing analyses combined with cultivation-based techniques in both dry and wet season. The aims of this study were to understand the composition and dynamics of the bacterial community in whole MWTS and investigate whether potential pathogens and opportunistic pathogens exist across the mineral water. The information generated in this study will provide strong basis for ensuring the microbial safety of mineral water.
Materials and Methods
Study Site and Sample Collection
The mineral water treatment plant is located in the Maofeng Mountain, south China. Its raw water originates from the groundwater at a depth of 200 meter. The production process of mineral water is shown in Figure 1. Water samples were collected in sterile containers from raw water (A), quartz sand filtered water (B), activated carbon filtered water (C), fine filtered water (D), and final water product (E) in the wet season and dry season in 2017. For each sampling point, three replicate samples were collected. All water samples were maintained at 4°C during transportation and were transported to laboratory within 4 h after sample collection. After all the samples arrived in the laboratory, 2 L of raw water, 5 L of quartz sand filtered water, 5 L of activated carbon filtered water, 10 L of fine filtered water and 10 L of final water product were filtered through sterile 0.22 μm pore-sized polycarbonate membranes (Millipore, Billerica, MA, United States). Membranes were transferred to sterile tubes and stored at -80°C. Each water sample was retested three times. The remainders of the sample were stored at 4°C, until all assayed parameters were confirmed.
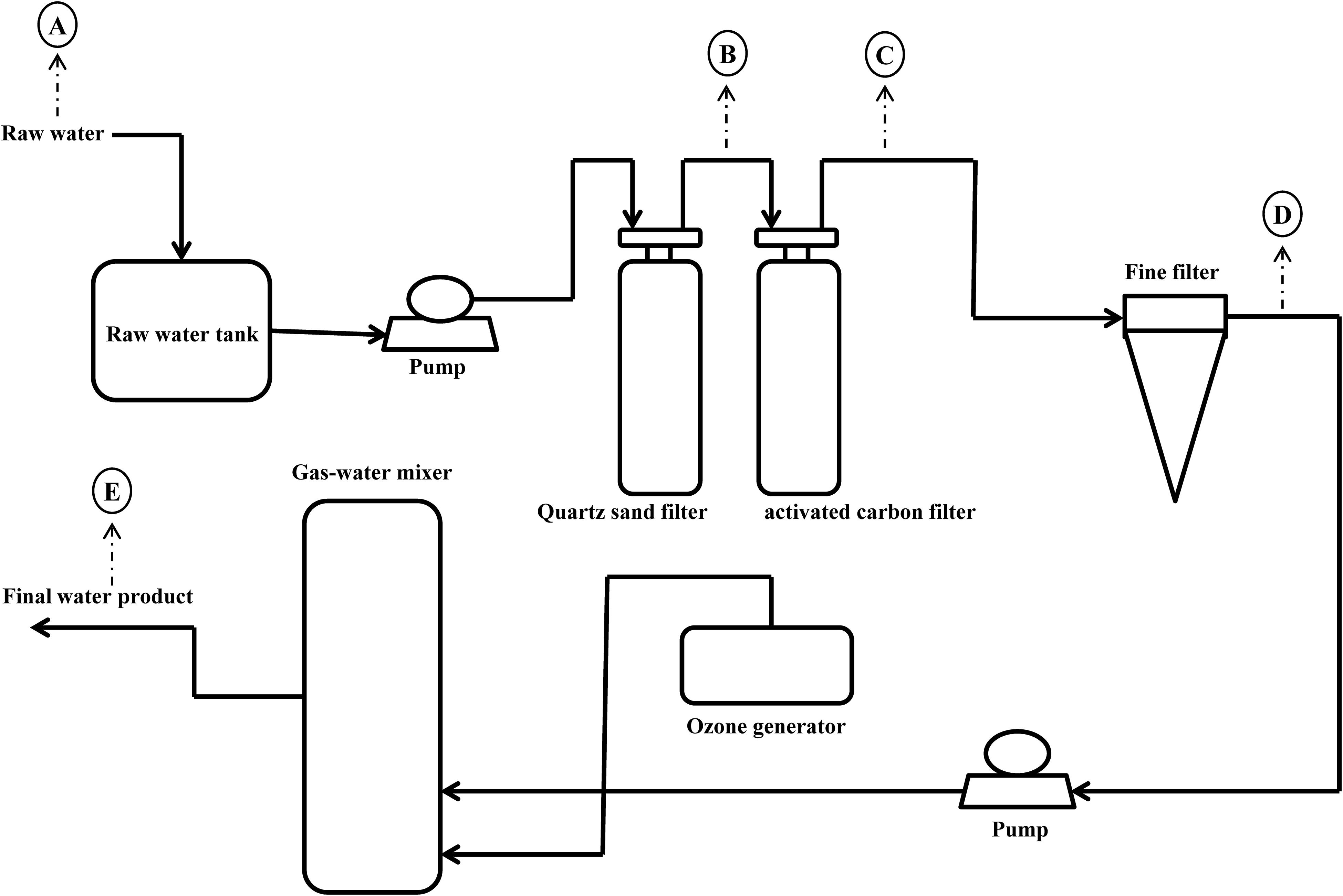
Figure 1. Production flow chart of mineral water in China.
Water Quality Analysis
To understand the water quality of all water samples, several standard physicochemical parameters were measured. Turbidity was measured using a turbidimeter (Mettler Toledo, Zurich, Switzerland). Temperature and pH was measured in situ using a multi-parameter water quality monitoring sonde (INESA Scientific, Shanghai, China). Other physicochemical parameters, including ammonia nitrogen, chemical oxygen demand (COD), total organic carbon (TOC) were measured according to standard examination methods for drinking water (GB/T 5750-2006, China). Ammonia nitrogen was determined by spectrophotometry. COD was measured by titration. TOC was measured using a total machine carbon analyzer (Shimadzu, Kyoto, Japan). All determinations were performed in triplicate. As groundwater was confined water and the dissolved oxygen content is very low, the parameter of dissolved oxygen (DO) was not measured.
Microbial Biomass and Activity
Heterotrophic plate counts (HPC) of all water samples were determined using a spread plating method on Reasoner’s 2A (R2A) agar (Haibo Co., Qingdao, China) (Lehtola et al., 2004). Briefly, all water samples were diluted to the appropriate concentration and 0.2 mL diluent was spread on R2A agar plate. The plates were cultured at 20°C for 7 days before colony-forming units (CFU) counting. The microbial activity was characterized by measuring adenosine tri-phosphate (ATP). ATP concentration was detected using a bioluminescence assay kit (Huankai Co., Guangzhou, China) described by Zhang et al. (2011). Briefly, membranes that filter water samples were fully washed with 0.5 mL ddH2O, and were reacted with 0.5 mL ATP-releasing reagent for 2 min. 0.1 mL reaction solution, 0.2 mL buffer solution, 0.1 mL luminescent reagent were added into a cuvette in sequence and luminous pulse value was detected by Glomax 20/20 luminometer (Promega Co., Madison, WI, United States). The ATP concentration of each sample was calculated according to a calibration curve generated using standards of known ATP. All determinations were performed in triplicate.
Isolation and Identification of Bacteria From Raw Water
Three kinds of medium, nutrient agar (NA) (Huankai Co., Guangzhou, China), trypticase soy agar (TSA) (Huankai Co., Guangzhou, China), and R2A (Haibo Co., Qingdao, China) were used as separate medium for the isolation of bacteria from raw water. Following incubation in 20°C culture for 7 days, colonies with different morphologies (color, size, edge, and transparency) were purified using the streaking method. After purification, all the isolates were cultured with R2A liquid medium to preserve and extract the genomic DNA. The genomic DNA of the isolate was extracted according to the specifications of the Bacterial Genomic DNA Purification Kit (Dongsheng Biotech, Guangzhou, China). Primers 27F (5′-GTGCTGCAGAGAGTTTGATCCTGGCTCAG-3′) and 1492R (5′-CACGGATCCTACGGGTACCTTGTTACGACTT-3′) were used to amplify the 16S rRNA gene of each isolate (Dymock et al., 1996). The polymerase chain reaction (PCR) mixture contained 2 μL of template, 12.5 μL PCR Taq-mix, 0.5 μL of each primer, and 8.5 μL of ddH2O. Amplifications were performed using a DNA thermocycler under the following temperature profiles: 5 min of denaturation at 95°C and 35 cycles of 30 s at 95°C, 45 s at 56°C, and 1.5 min at 72°C, followed by an additional 10 min at 72°C at the end of the 35th cycle for repair and extension. The PCR products were electrophoresed using 1.0% (W/V) agarose gel, and the positive PCR products were selected and sent to Huada Gene Co., (Guangzhou, China) for sequencing using the PCR primers. According to a previously described method (Kim et al., 2012), 16S rDNA sequences were divided into different Operational Taxonomic Units (OTUs) and representative 16S rDNA sequences were selected in each OTU. Representative 16S rDNA sequences were submitted to the National Center for Biotechnology Information (NCBI) and GenBank databases for comparison and identification using BLAST software.
Microbial Community Analysis
The polycarbonate membranes that had been used to analyze the microbial community were cut into pieces with sterilized scissors. Total genomic DNA of all the samples was extracted according to the specifications of the Powersoil DNA extraction kit (Mobio Laboratories, Carlsbad, CA, United States). PCR amplicon libraries were constructed for Illumina HiSeq sequencing using bacterial primers 341F (5′- CCTAYGGGRBGCASCAG-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) targeting the V3+V4 hypervariable regions of the 16S rRNA genes (Wu et al., 2015). The PCR mixture (20 μL) contained 10 ng of template DNA, 4 mL of 5× FastPfu Buffer, 2.5 mM dNTPs, 0.4 mL FastPfu Polymerase, 5 M of each primer, and ddH2O. Amplifications were performed as follows: an initial denaturation at 95°C for 5 min; followed by 27 cycles at 95°C for 30 s, 55°C for 30 s, and 72°C for 45 s; and a final extension at 72°C for 10 min. The obtained PCR products were purified and joined using a sequencing linker. After constructing the gene library, the modified products were subjected to high-throughput sequencing using Illumina Hiseq2500 (San Diego, CA, United States). The reads from the original DNA fragments were merged using FLASH1 (Magoč and Salzberg, 2011). Quality filtering was carried out on the initial data obtained by sequencing to obtain more accurate and high-quality DNA sequence. To obtain high-quality sequences (primer mismatch base number <1%, sequence length 200–500 bp, no chimeras), the original sequences were processed using the protocol described by Caporaso et al. (2010). Sequences were grouped into OTUs at 97% sequence similarity using Mothur version 1.34.02. To obtain the species classification information of each OTU, the representative sequences for each OTU were selected and submitted for taxonomic identification using the Ribosomal Database Project (RDP) classifier (see text footnote 2) (Wang et al., 2007). Mothur version 1.34.0 was used as the processing pipeline for calculation of alpha-diversity indices including the Chao1, Simpson, and Shannon indices. Based on the relative abundance of bacterial phyla, unweighted UniFrac with QIIME3 was used for the unweighted pair-group method with arithmetic mean clustering. Principal component analysis (PCA) and canonical correspondence analysis (CCA) were conducted using Canoco 4.5 software4.
Data Analysis
All the data were processed using Microsoft Excel 2010 software (Redmond, WA, United States) and statistical analysis was performed using IBM SPSSV20.0 software (Armonk, NY, United States). The differences between groups were analyzed using one-way analysis of variance (ANOVA) and the statistical significance level was set at p = 0.05.
Results
Water Quality
As shown in Table 1, a series of specific physicochemical parameters were determined for all water samples. The raw water temperature in the wet season and the dry season were approximately 20.0°C and 20.9°C, respectively, and has no obvious change in the follow-up process treatment. The pH of all the water samples was stable in the range of 7.3 to 7.5 across the whole MWTS in both seasons. The concentrations of ammonia in whole MWTS were <0.02 mg/L in both seasons. The turbidity, TOC, and CODMn of raw water samples were 0.60 nephelometric turbidity units (NTU), 0.61 and 0.41 mg/L in the wet season, and 0.70 NTU, 0.74, and 0.45 mg/L in the dry season, respectively. As the seasons changed, the turbidity, TOC, and CODMn of the water samples showed no significant difference in whole MWTS (p > 0.05). However, the turbidity, TOC, and CODMn of the water samples were changed by most treatment processes (p < 0.05).
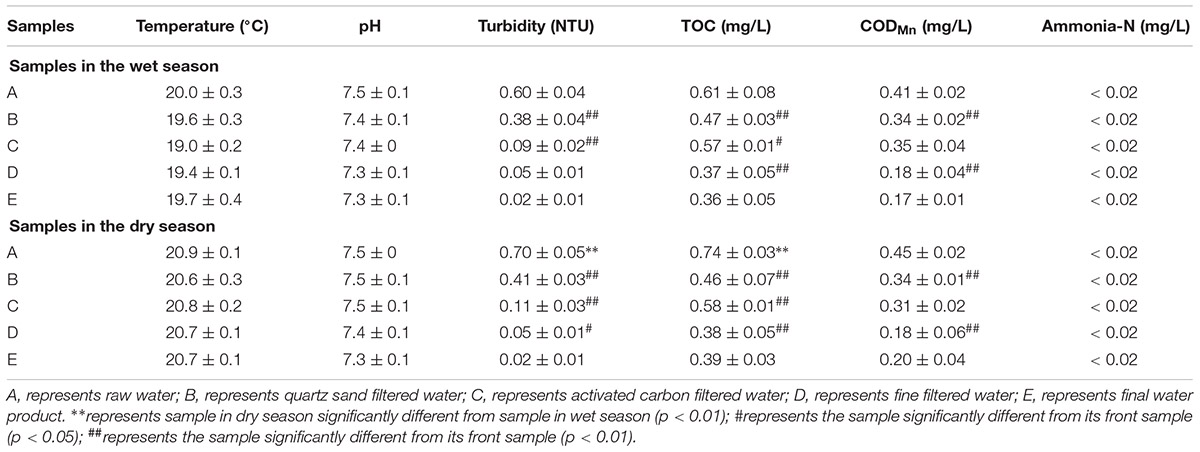
Table 1. Physicochemical parameters of 5 water samples in whole MWTS.
Microbial Biomass and Microbial Activity
The HPC from raw water in the wet and dry season was 1.29 × 103 and 1.97 × 103 CFU/mL, respectively (Table 2). Along with the treatment process in whole MWTS, the HPC showed a downward trend in both seasons (Figure 2A). After filtration by quartz sand and fine filter, the microbial biomass decreased sharply (p < 0.01). However, after activated carbon filtration, the HPC showed no significant change. The HPC of all finished water in both seasons was <4 CFU/mL. The microbial activity of water samples in the wet season was lower than that in the dry season in whole MWTS, especially in the raw water and activated carbon filtered water (p < 0.01). ATP concentration during the treatment process ranged from 3.32 × 10-8 to 6.47 × 10-11 M in the wet season and 6.96 × 10-8 to 5.10 × 10-11 M in the dry season (Figure 2B). Along with treatment process, the ATP concentration was obviously reduced after quartz sand filter, and then exhibited a significant increase after activated carbon filter in both season (p < 0.01). The ATP concentration of activated carbon filtered water was 4.20 × 10-9 M in the wet season and 6.81 × 10-9 M in the dry season.
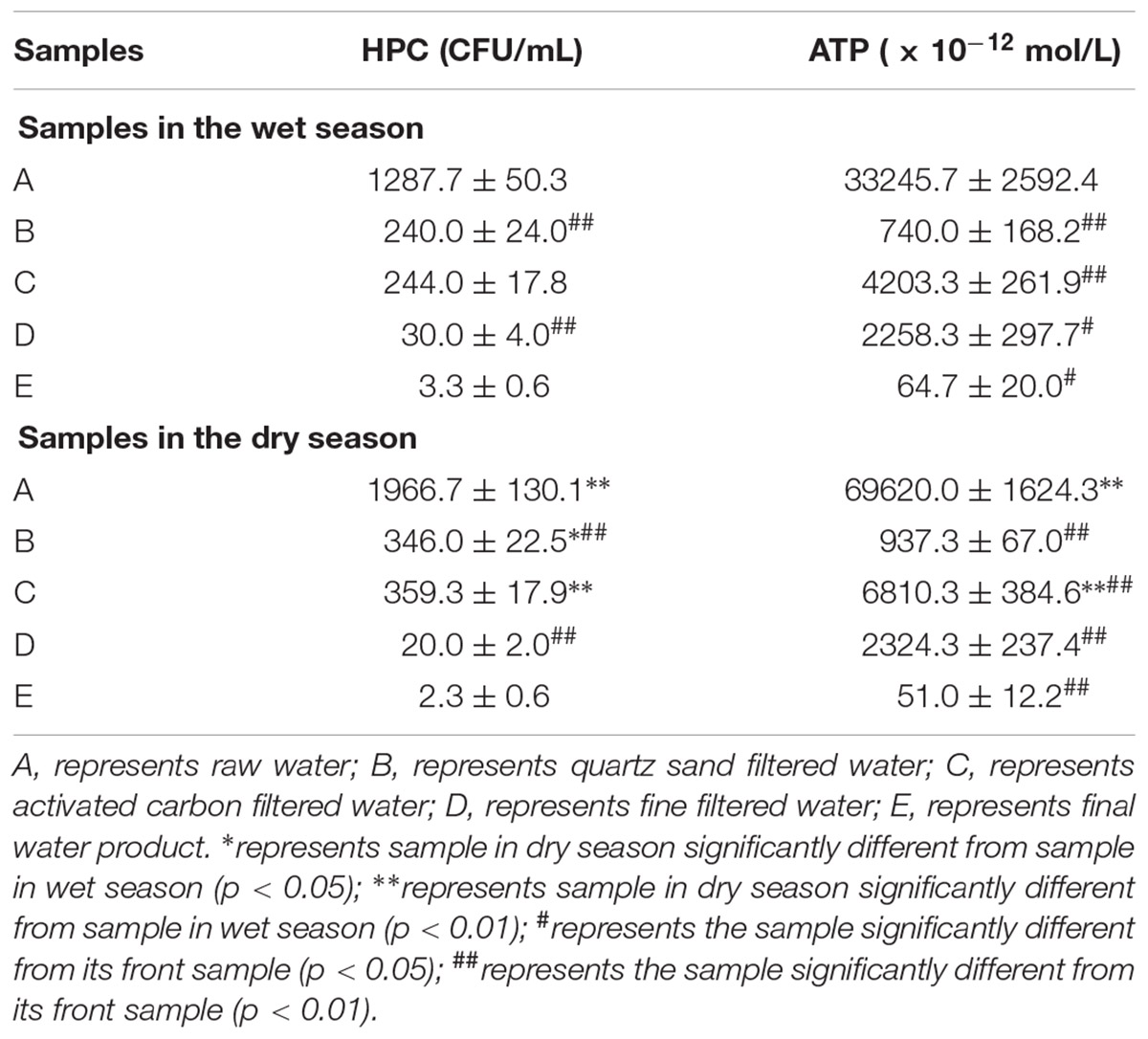
Table 2. Heterotrophic plate counts (HPC) and bacterial adenosine tri-phosphate (ATP) concentrations of 5 water samples in whole MWTS.
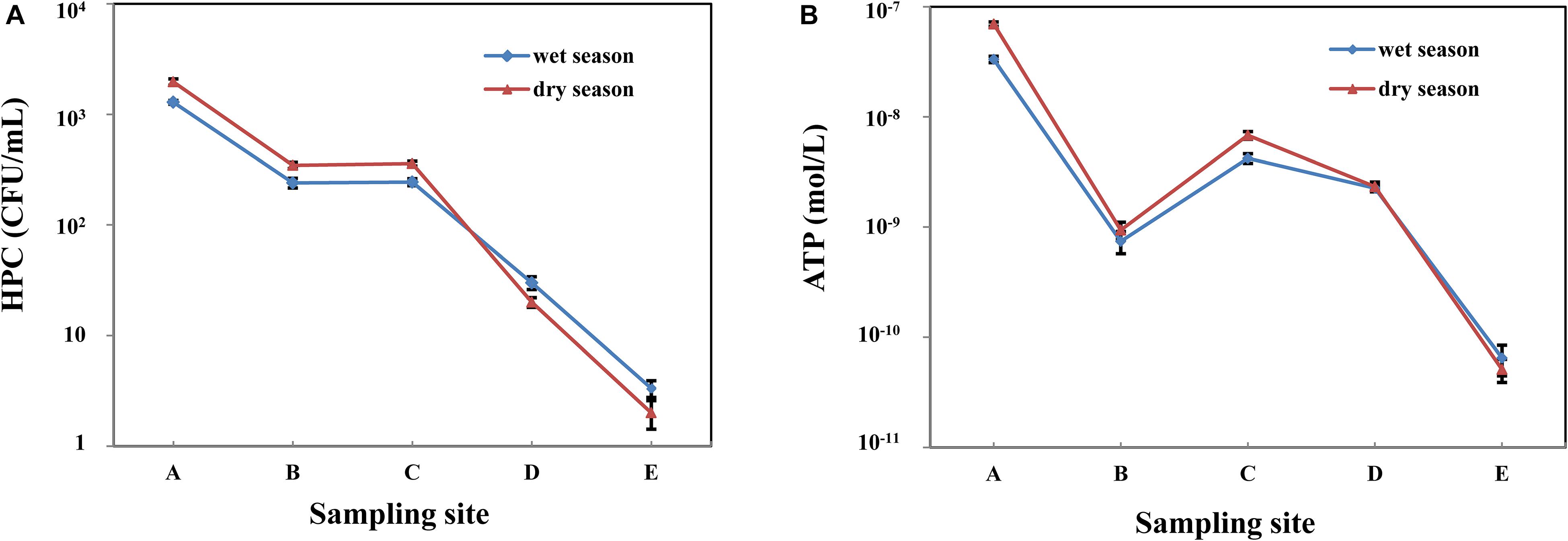
Figure 2. Variations of heterotrophic plate counts (HPC) (A) and bacterial adenosine tri-phosphate (ATP) concentrations (B) in whole MWTS. A, represents raw water; B, represents quartz sand filtered water; C, represents activated carbon filtered water; D, represents fine filtered water; E, represents final water product.
Isolation and Identification of Bacteria From Groundwater
In total, 141 isolates from the wet season and 159 isolates from the dry season were isolated in raw water samples (Supplementary Table S1). A total of 139 isolates were cultured from R2A medium, 93 isolates from NA medium, and 68 isolates from TSA medium. According to 16s rRNA gene sequence, all the isolates could be divided into 23 OTUs (Table 3). All 300 isolates were distributed in 5 phyla (Proteobacteria, Chloroflexi, Firmicutes, Bacteroides, and Actinobacteria), 7 classes (α-proteobacteria, β-proteobacteria, γ-proteobacteria, Chloroflexi, Bacilli, Flavobacteriia, and Actinomycete) and 19 genera (Pseudomonas, Chryseobacterium, Porphyrobacter, Xanthomonas, Brevundimonas, Caulobacter, Acinetobacter, Sphingomonas, Bacillus, Brevibacterium, Arthrobacter, Aquimonas, Chloroflexus, Mycobacterium, Roseomonas, Chromobacterium, Pimelobacter, Microbacterium, and Hydrogenophaga). Proteobacteria accounted for 55.7% (167) of the total isolates, followed by Actinobacteria accounting for 23.7% (71). Among all the isolates, the predominant genera were Pseudomonas (19.3%) and Sphingomonas (7.7%) at genus level. All the isolates cultured from R2A medium were distributed in 18 different genera, and Porphyrobacter, Bacillus, Chromobacterium, Roseomonas, and Pimelobacter were isolated only on R2A medium. Mycobacterium and Pimelobacter were isolated only from the sample in the wet season.
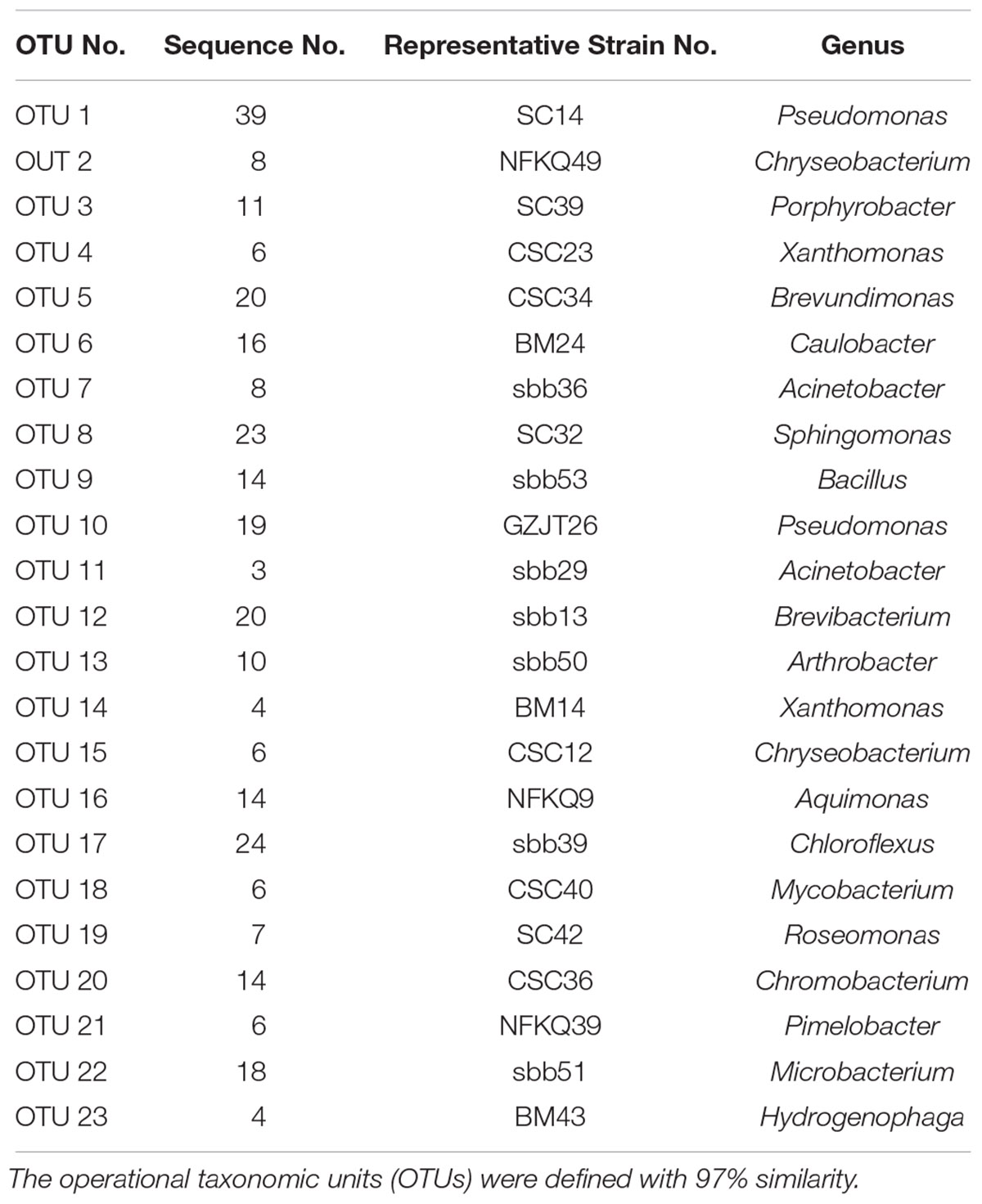
Table 3. Blast results and classify information of representative isolates cultured from raw water sample.
Diversity of Bacterial Community
A range of 20,693 to 39,643 original bacterial 16S rRNA gene sequences were extracted from each sample using Illumina HiSeq sequencing analysis (Table 4). The OTUs of the water samples in the wet season ranged from 2411 to 6607, and ranged 2524 to 7391 for sample in the dry season. As shown in Figure 3, In general, the diversity indices of water samples in the dry season were comparable to those in the wet season. Among all the water samples in both seasons, the diversity indices of raw water samples were the highest. Chao1 richness index ranged from 15421 to 38316 in the wet season, and ranged 15567 to 33647 in the dry season. Shannon index ranged from 3.19 to 5.55 in the wet season, and ranged 3.46 to 6.49 in the dry season. Along with treatment process in whole MWTS, the richness index of Chao1 and the Shannon index gradually decreased to the lowest level and were significantly changed by the fine filtration and ozone sterilization treatments. Simpson diversity index of finished water was 0.1872 in the wet season and 0.1819 in the dry season. However, those of raw water reached 0.0245 and 0.0158 in the wet and dry seasons, respectively.
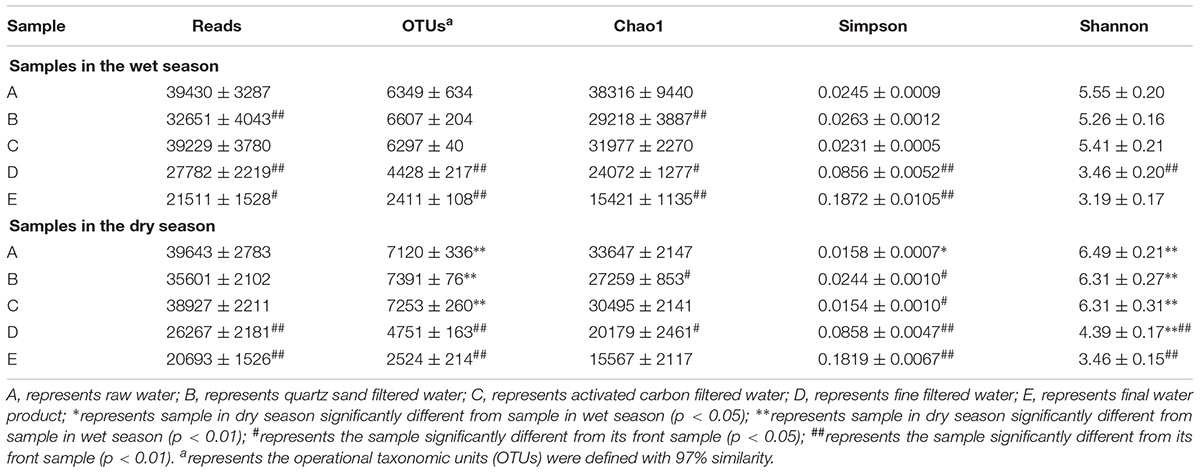
Table 4. Bacterial community richness and diversity indices for 5 water samples in whole MWTS.
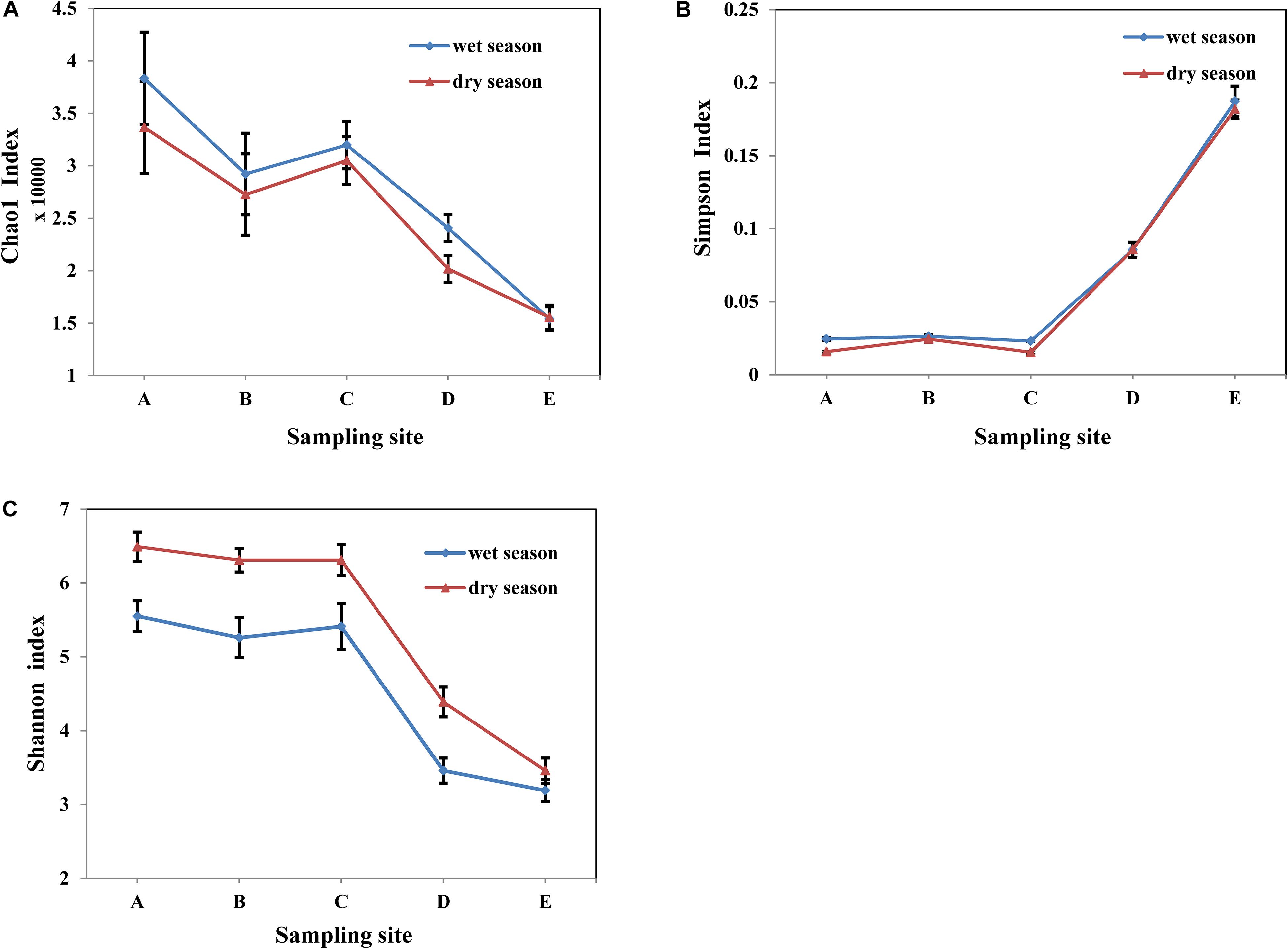
Figure 3. Variations of Diversity indices, Chao1 index (A), Simpson index (B), and Shannon index (C) in whole MWTS. A, represents raw water; B, represents quartz sand filtered water; C, represents activated carbon filtered water; D, represents fine filtered water; E, represents final water product.
Composition of Bacterial Community
The bacterial community composition of all the water samples was identified at the phylum level (Figure 4A). Overall, 15 bacterial phyla (relative abundance >0.1%) including Proteobacteria, Verrucomicrobia, Planctomycetes, Nitrospirae, Gemmatimonadetes, Firmicutes, Cyanobacteria, Chloroflexi, Bacteroidetes, Actinobacteria, Dependentiae (Tm6), Deinococcus–Thermus, Saccharibacteria, Chlamydiae, and Acidobacteria were identified in all the water samples. No obvious difference was observed upon comparison of the bacterial microbiota of water samples in different seasons. Proteobacteria comprised the dominant bacteria microbiota in all the water samples, accounting for approximately 60% of all the samples, especially in quartz filtered water (69.7%) and activated carbon filtered water (67.5%). The relative abundance of Proteobacteria from samples in the wet season was higher than those in the dry season. Along with the treatment processes, significant change in the proportion of Bacteroidetes, Actinobacteria, and Firmicutes was obtained in the MWTS. The relative abundances of Bacteroidetes and Actinobacteria in raw water were 22.3 and 16.1%, respectively. The proportion of Bacteroidetes decreased initially in quartz sand filtered samples, followed by an increase upon activated carbon filtration; however, Bacteroidetes from final water product were present a very low levels (<1%) in both seasons. The proportion of Actinobacteria gradually decreased in both seasons, being only <1% in final water product. Among all bacterial microbiota, the largest change of relative abundance was observed for Firmicutes. The proportion of Firmicutes in raw water was 1.4% in the wet season and 3.4% in the dry season, but was highly abundant in finished water in both seasons (41.8 and 42.7%, respectively). The final water product in both seasons mainly comprised Proteobacteria (50.8%), Firmicutes (42.2%), and Verrucomicrobia (1.4%).
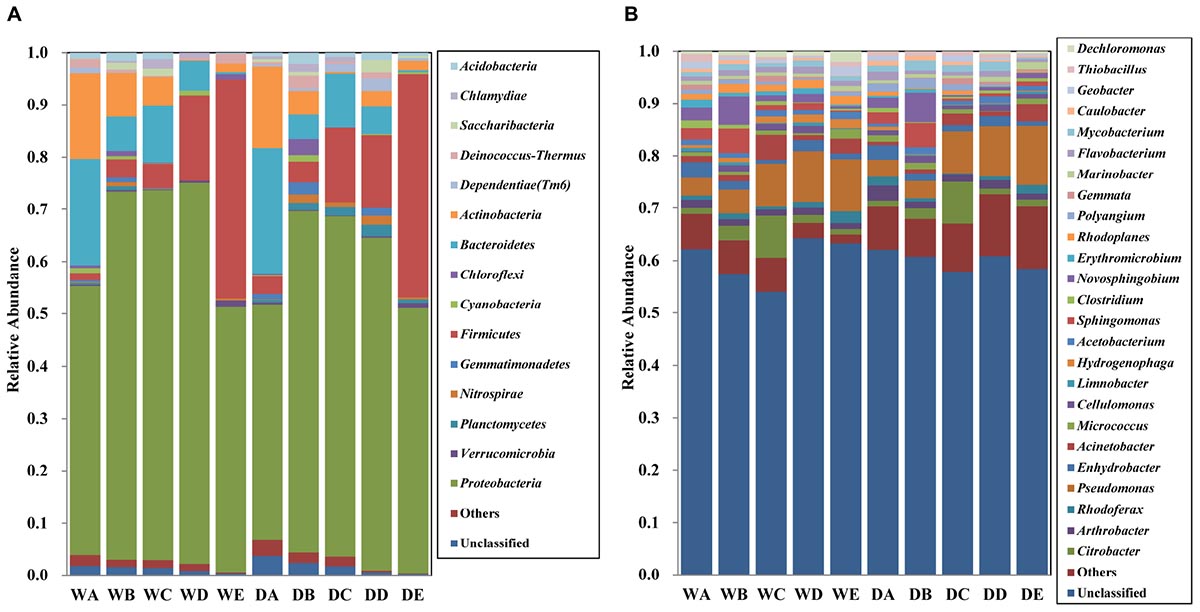
Figure 4. Relative abundances of bacterial community composition at the phylum level (A) and genus level (B) in all water samples. The rare species with relative abundance less than 0.1% are included as others. A, represents raw water; B, represents quartz sand filtered water; C, represents activated carbon filtered water; D, represents fine filtered water; E, represents final water product, Sample names staring with “W” represents samples collected in the wet season, Sample names staring with “D” represents samples collected in the dry season.
The bacterial community composition of all the water samples was identified at the genus level (Figure 4B). Overall, 25 bacterial genera (relative abundance >0.1%), including Citrobacter, Pseudomonas, Enhydrobacter, Acinetobacter, Sphingomonas, and Novosphingobium were detected in all the water samples. The proportion of unclassified genera was range from 60 to 70% in all the samples. The relative abundance of each genus was <3.5% in raw water. Quartz filtered water in both seasons mainly comprised Novosphingobium (5.5%) and Sphingomonas (4.6%), whereas activated carbon water was dominated by Pseudomonas (7.9%), Citrobacter (7.8%), and Acinetobacter (4.7%). The final water product in both seasons mainly comprised Pseudomonas (8.6%), followed by Acinetobacter (2.4%).
Core Microbiome
The bacteria that were still present in whole MWTS were defined as the core microbiome (Shade and Handelsman, 2012). Venn diagrams were constructed to investigate the core microbiome in whole MWTS. As shown in Figure 5, 178 (1.9%) OTUs were shared in all the samples from the wet season and 265 (2.9%) from the dry season. Among these shared OTUs, 94 were shared in both seasons and were identified as Pseudomonadaceae (n = 20), Sphingomonadaceae (n = 9), Enterococcaceae (n = 8), unclassified AB024 (n = 7), AJ234 (n = 6), Moraxellaceae (n = 6), Xanthomonadaceae (n = 6), Comamonadaceae (n = 6), Citrobacteriaceae (n = 5), Caulobacteraceae (n = 5), Bacillaceae (n = 5), Micrococcaceae (n = 4), Microbacteriaceae (n = 3), Mycobacteriaceae (n = 3), and Nocardiaceae (n = 1). The number of shared OTUs between adjacent treatment steps ranged from 370 to 2127 in samples collected during the wet season and from 397 to 2601 in those collected during the dry season, accounting for 4.1 to 23.4% and 4.4 to 29.2% of overall OTUs, respectively.
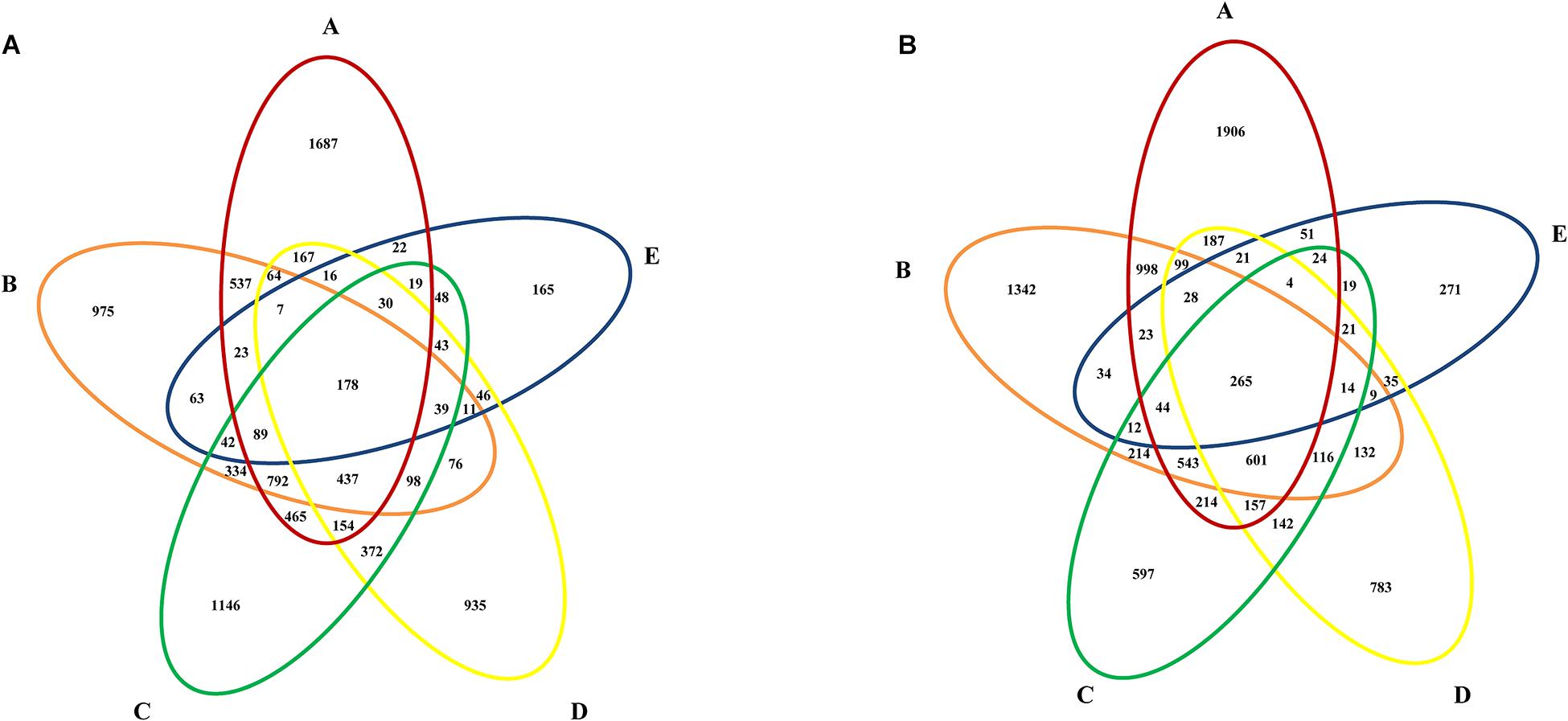
Figure 5. Venn diagrams showing the number of shared OTUs among water samples from different sampling sites during the (A) wet season, (B) dry season. A, represents raw water; B, represents quartz sand filtered water; C, represents activated carbon filtered water; D, represents fine filtered water; E, represents final water product.
Discussion
In this study, we identified that the microbial activity (Figure 2B) (ATP concentration 5.10 × 10-11 – 6.96 × 10-8 M) and biomass (Figure 2A) (HPC 2 – 1.97 × 103 CFU/mL) of mineral water samples from an MWTS in the dry season were higher than those (ATP concentration 6.47 × 10-11 – 3.32 × 10-8 M, HPC 3 – 1.29 × 103 CFU/mL) in the wet season. The seasonal fluctuation of microbial activity and biomass may be attributed to environmental factors (Table 1); specifically, the concentration of TOC. As TOC represents a carbon source, the changing of seasons might alter the nutrient composition of the water environment in the MWTS. The rise of TOC potentially promoting microbial activity and biomass shifts in the dry season in whole MWTS, which is consistent with the description of Li in DWTS (Li et al., 2016). The seasonal fluctuation of microbial activity and biomass had a positive correlation with turbidity and COD. Turbidity is related to suspended, which can adsorb and enrich bacteria. COD is an indicator of organic content and has direct relationship with bacterial biomass. In addition, previous studies in DWTS have shown that environmental parameters such as phosphate, sulfate, pH and water depth were the major factors controlling the temporal variation of bacterial activity and biomass (Lautenschlager et al., 2013; Ji et al., 2015). The temporal pattern of bacteria diversity in DWTS in particular has been thoroughly studied, whereas comparatively little research has been conducted on MWTS. Henne et al. (2013) found obvious seasonal variations of bacterial community composition in surface water reservoirs and Kelly et al. (2014) reported that the diversity of bacteria community in biofilms in the summer months was higher than that observed in the winter. Compared with previous studies in DWTS, the results of the present study showed that the bacterial diversity of water samples have no obvious seasonal variations and bacterial microbiota of water samples in the different season exhibited no obvious difference (Figure 3). The differences in seasonal variations of microbial diversity in the DWTS and MWTS may have been driven by the strong seasonal changes in the DWTS. Previous study also suggested that physicochemical parameters, temperature, and pH, were correlated with bacterial diversity (Chen et al., 2017). In comparison, the stability of the groundwater environment from which the MWTS samples were derived resulted in no obvious seasonal difference of microbial diversity (Fovet et al., 2018). In addition, the groundwater that serves as the raw water of mineral water is not influenced by other external factors as is surface water. For example, after, rainfall or pollution, the surface water bacterial diversity demonstrates significant change (França et al., 2015). In our study, the kinds of bacterial phylum are significantly different from the results of studies in DWTS, which is due to different raw water in DWTS and MWTS.
Distinct differences were identified in the microbial activity, biomass, and diversity at each step of the MWTS. Along with treatment process in whole MWTS, the microbial activity, biomass, and diversity exhibited an obviously downward trend, especially, following the fine filtration and ozone sterilization treatments, which indicated that the water quality was gradually improved. However, after the activated carbon filter treatment process, the activity and diversity of bacteria showed a clear increase in both seasons. Activated carbon filter systems are commonly used for mineral water treatment to ensure equipment life, improve water quality, and prevent pollution (Feng et al., 2013). Activated carbon filters, which contain numerous pores and a large surface area, possess a strong physical adsorption capacity to absorb organic pollutants and microbes (Liao et al., 2013). Our previous study had shown that activated carbon filters could serve as a gathering place for opportunistic pathogens and is the most serious microbial contamination site throughout the whole production process of the MWTS (Wei et al., 2017). The results of the present study thus showed that there was dynamic bacterial community change among the samples of the different treatment processes and that specific treatment processes in the MWTS could affect the bacterial communities in unique ways (Figure 4). In comparison, Poitelon et al. (2010) reported that the influence of water treatment by coagulation-flocculation and sedimentation on bacterial communities is relatively small, whereas Li et al. (2017) observed that bacterial communities significant changed during these steps. Consistent with previous report, the present analysis revealed that Proteobacteria predominated in the water samples, which could mainly be related to the capacity of Proteobacteria with regard to the biodegradation or biotransformation of various organic compounds (Galvão et al., 2005). In addition, significant change in the proportions of the Bacteroidetes, Actinobacteria, and Firmicutes was also obtained in the MWTS. The activated carbon filtered water was mainly composed of Proteobacteria and Citrobacter, which differed from the result of DWTS (Lin et al., 2014). Furthermore, cluster analysis indicated that quartz sand filtration apparently changed the bacterial community structure, with Sphingomonas and Novosphingobium increasing to a higher level in the quartz sand filtered water, which demonstrated that quartz sand filter promoted the proportion of Sphingomonas and Novosphingobium. However, after ozone sterilization treatment, the proportion of Firmicutes exhibited a sharp increase. This result is consistent with the study of Mi et al. (2015) which indicated that disinfection strategies could have a profound impact on the proportion of Firmicutes in microbial communities in MWTS. The disinfection resistance mechanism of Firmicutes remains unresolved, which may be connected with higher spore resistance. Therefore, additional studies are necessary to validate the effect of disinfection on Firmicutes content in mineral water treatment plant bacterial communities.
Despite the spatial-temporal variations of bacterial diversity was detected in MWTS, shared OTUs were found along the treatment processes in different seasons. The bacteria that were present throughout whole MWTS were defined as the core microbiome (Shade and Handelsman, 2012). In the present study, the core microbiome comprised 15 families, which indicated that the core microbiome in MWTS is generally difficult to remove completely and may gradually become a healthy threat (Figure 5). Therefore it is critical to comprehensively understand the core microbiome in MWTS. In particular, some genera, such as Pseudomonas and Acinetobacter, which are highly likely to contain pathogens and opportunistic pathogens, were retrieved from Illumina amplicons of final water product. The occurrence of these genera in mineral water may increase the risks of waterborne diseases and health problems. For example, some Acinetobacter species were important opportunistic pathogens that underlie hospital infections and can cause respiratory tract infection, septicemia, meningitis, endocarditis, and urogenital tract infection (Wong et al., 2017). Some Mycobacterium species were the pathogen that causes tuberculosis (Pfyffer, 2015). Among all 300 isolates obtained using cultivation-based techniques in raw water, Pseudomonas was predominant species (19.3%). Additionally, Illumina sequencing analysis of 16s rRNA genes showed that the final water product in both seasons mainly comprised Pseudomonas (8.6%). Pseudomonas aeruginosa serves as an important opportunistic pathogen that is frequently detected in mineral water for human consumption and causes human urinary tract infections (Lu et al., 2016). Our previous research showed that the Pseudomonas aeruginosa contamination rate of final water product was 2.3%, which can adversely affect the health of consumers (Wei et al., 2014). Moreover, many waterborne bacteria can form biofilms, which are difficult to remove (Flemming and Wingender, 2010). Although it is possible that PCR amplification might amplify DNA fragment from dead cells, there remains a possibility that some pathogens are able to survive the water treatments considering their resistances to disinfectants. Some pathogen may also cause diseases even at relatively low concentrations (Kirk et al., 2015). Illumina sequencing analysis of 16s rRNA genes in final water product showed that some opportunistic pathogens exist in the final water product that was disinfected by ozone. Hence, manufacturers of mineral water need to established measures for monitoring the MWTS.
This study is the first to investigate the composition and dynamics of bacterial community in a full-scale mineral water treatment plant in China during both the wet and dry seasons. In general, the microbial activity (ATP concentration 5.10 × 10-11 – 6.96 × 10-8 M) and biomass (HPC 2 – 1.97 × 103 CFU/mL) of mineral water samples from an MWTS in the dry season were higher than those (ATP concentration 6.47 × 10-11 – 3.32 × 10-8 M, HPC 3 – 1.29 × 103 CFU/mL) in the wet season. All 300 isolates obtained using cultivation-based techniques were distributed in 5 phyla, 7 classes, and 19 genera. Proteobacteria accounted for 55.7% (167) of the total isolates and the predominant species comprised Pseudomonas (19.3%) at the genus level. Illumina sequencing analysis of 16s rRNA genes revealed that 15 bacterial phyla (relative abundance >0.1%) were detected in all the water samples and that Proteobacteria constituted the dominant bacteria microbiota. The bacterial diversity of water samples has no obvious seasonal variations and bacterial microbiota of water samples in the different season exhibited no obvious difference. Distinct differences were identified in the microbial activity, biomass, and diversity at each step of the MWTS. Along with treatment process in whole MWTS, the microbial activity, biomass, and diversity exhibited an obviously downward trend, especially, following the fine filtration and ozone sterilization treatments. In particular, some core microbiome that persisted throughout the treatment process requires special consideration. Notably, opportunistic pathogens including Pseudomonas, Acinetobacter, Clostridium, and Mycobacterium, which can adversely affect the health of consumers, were detected throughout the treatment process. These data may therefore provide useful information for the development of public health policies and effective strategies to ensure the safety of mineral water products.
Author Contributions
QW, JZ, and LW conceived and designed the experiments. LW and WG performed the experiments. LW, QG, and JW analyzed the data. MC, MW, and AL contributed reagents, materials, and analysis tools. LW, HW, and TL contributed to the writing of the manuscript.
Funding
We would like to acknowledge the financial support of the Science and Technology Planning Project of Guangdong Province (2017A07070218) and the GDAS’ Project of Science and Technology Development (2017GDASCX-0201).
Conflict of Interest Statement
MW and AL were employed by Guangdong Dinghu Mountain Spring Company Limited.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01542/full#supplementary-material
Footnotes
- ^http://ccb.jhu.edu/software/FLASH/
- ^https://mothur.org
- ^http://qiime.org/scripts/make_phylogeny.html
- ^http://www.canoco5.com/
References
Albertsen, M., Hugenholtz, P., Skarshewski, A., Nielsen, K. L., Tyson, G. W., and Nielsen, P. H. (2013). Genome sequences of rare, uncultured bacteria obtained by differential coverage binning of multiple metagenomes. Nat. Biotechnol. 31:533. doi: 10.1038/nbt.2579
Bharath, J., Mosodeen, M., Motilal, S., Sandy, S., Sharma, S., Tessaro, T., et al. (2003). Microbial quality of domestic and imported brands of bottled water in Trinidad. Int. J. Food Microbiol. 81,53–62.
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7:335.
Chen, Y., Liu, Y., and Wang, X. (2017). Spatiotemporal variation of bacterial and archaeal com- munities in sediments of a drinking reservoir. Beijing, China. Appl. Microbiol. Biotechnol. 101, 3379–3391. doi: 10.1007/s00253-016-8019-1
Deere, D., Leusch, F. D. L., Humpage, A., Cunliffe, D., and Khan, S. J. (2017). Hypothetical scenario exercises to improve planning and readiness for drinking water quality management during extreme weather events. Water Res. 111, 100–108. doi: 10.1016/j.watres.2016.12.028
Dymock, D., Weightman, A. J., Scully, C., and Wade, W. G. (1996). Molecular analysis of microflora associated with dentoalveolar abscesses. J. Clin. Microbiol. 34:537–542.
Eichler, S., Christen, R., Höltje, C., Westphal, P., Bötel, J., Brettar, I., et al. (2006). Composition and dynamics of bacterial communities of a drinking water supply system as assessed by rna- and dna-based 16s rrna gene fingerprinting. Appl. Environ. Microbiol. 72:1858–1872.
El-Chakhtoura, J., Prest, E., Saikaly, P., Van Loosdrecht, M., Hammes, F., and Vrouwenvelder, H. (2015). Dynamics of bacterial communities before and after distribution in a full-scale drinking water network. Water Res. 74, 180–190. doi: 10.1016/j.watres.2015.02.015
Faria, C., Vaz-Moreira, I., Serapicos, E., Nunes, O. C., and Manaia, C. M. (2009). Antibiotic resistance in coagulase negative staphylococci isolated from wastewater and drinking water. Sci. Total Environ. 407, 3876–3882. doi: 10.1016/j.scitotenv.2009.02.034
Feng, S., Chen, C., Wang, Q. F., Zhang, X. J., Yang, Z. Y., and Xie, S. G. (2013). Characterization of microbial communities in a granular activated carbon–sand dual media filter for drinking water treatment. Int. J. Environ. Sci. Technol. 10, 917–922.
Flemming, H.-C., and Wingender, J. (2010). The biofilm matrix. Nat. Rev. Microbiol. 8:623. doi: 10.1038/nrmicro2415
Fovet, O., Dupas, R., Gruau, G., Jeanneau, L., Jaffrézic, A., Ruiz, L., et al. (2018). “Groundwater storage as a major control of seasonal stream water quality dynamics during both base and storm flows,” in Proceedings of the EGU General Assembly Conference Abstracts, Vienne.
França, L., Lopéz-Lopéz, A., Rosselló-Móra, R., and Da Costa, M. S. (2015). Microbial diversity and dynamics of a groundwater and a still bottled natural mineral water. Environ. Microbiol. 17, 577–593. doi: 10.1111/1462-2920.12430
Galvão, T. C., Mohn, W. W., and De Lorenzo, V. (2005). Exploring the microbial biodegradation and biotransformation gene pool. Trends Biotechnol. 23,497–506.
Griebler, C., and Lueders, T. (2009). Microbial biodiversity in groundwater ecosystems. Freshw. Biol. 54, 649–677.
Hahn, M. W., Jezberová, J., Koll, U., Saueressig-Beck, T., and Schmidt, J. (2016). Complete ecological isolation and cryptic diversity in Polynucleobacter bacteria not resolved by 16S rRNA gene sequences. ISME J. 10, 1642–1655. doi: 10.1038/ismej.2015.237
Henne, K., Kahlisch, L., Höfle, M. G., and Brettar, I. (2013). Seasonal dynamics of bacterial community structure and composition in cold and hot drinking water derived from surface water reservoirs. Water Res. 47, 5614–5630. doi: 10.1016/j.watres.2013.06.034
Herath, A. T., Abayasekara, C. L., Chandrajith, R., and Adikaram, N. K. B. (2012). Temporal variation of microbiological and chemical quality of noncarbonated bottled drinking water sold in sri lanka. J. Food Sci. 77, M160–M164. doi: 10.1111/j.1750-3841.2011.02588.x
Hou, L., Zhou, Q., Wu, Q., Gu, Q., Sun, M., and Zhang, J. (2018). Spatiotemporal changes in bacterial community and microbial activity in a full-scale drinking water treatment plant. Sci. Total Environ. 625, 449–459. doi: 10.1016/j.scitotenv.2017.12.301
Ji, P., Parks, J., Edwards, M. A., and Pruden, A. (2015). Impact of water chemistry, pipe material and stagnation on the building plumbing microbiome. PLoS One 10:e0141087. doi: 10.1371/journal.pone.0141087
Kelly, J. J., Minalt, N., Culotti, A., Pryor, M., and Packman, A. (2014). Temporal variations in the abundance and composition of biofilm communities colonizing drinking water distribution pipes. PLoS One 9:e98542. doi: 10.1371/journal.pone.0098542
Kim, O.-S., Cho, Y.-J., Lee, K., Yoon, S.-H., Kim, M., Na, H., et al. (2012). Introducing eztaxon-e: a prokaryotic 16s rrna gene sequence database with phylotypes that represent uncultured species. Int. J. Syst. Evol. Microbiol. 62, 716–721. doi: 10.1099/ijs.0.038075-0
Kirk, M. D., Pires, S. M., Black, R. E., Caipo, M., Crump, J. A., Devleesschauwer, B., et al. (2015). World health organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: a data synthesis. PLoS Med. 12:e1001921. doi: 10.1371/journal.pmed.1001921
Lautenschlager, K., Hwang, C., Liu, W.-T., Boon, N., Köster, O., Vrouwenvelder, H., et al. (2013). A microbiology-based multi-parametric approach towards assessing biological stability in drinking water distribution networks. Water Res. 47, 3015–3025. doi: 10.1016/j.watres.2013.03.002
Lehtola, M. J., Miettinen, I. T., Keinänen, M. M., Kekki, T. K., Laine, O., Hirvonen, A., et al. (2004). Microbiology, chemistry and biofilm development in a pilot drinking water distribution system with copper and plastic pipes. Water Res. 38, 3769–3779.
Li, C., Ling, F., Zhang, M., Liu, W.-T., Li, Y., and Liu, W. (2017). Characterization of bacterial community dynamics in a full-scale drinking water treatment plant. J. Environ. Sci. 51, 21–30. doi: 10.1016/j.jes.2016.05.042
Li, W., Wang, F., Zhang, J., Qiao, Y., Xu, C., Liu, Y., et al. (2016). Community shift of biofilms developed in a full-scale drinking water distribution system switching from different water sources. Sci. Total Environ. 544, 499–506. doi: 10.1016/j.scitotenv.2015.11.121
Liao, X., Chen, C., Wang, Z., Wan, R., Chang, C.-H., Zhang, X., et al. (2013). Changes of biomass and bacterial communities in biological activated carbon filters for drinking water treatment. Process Biochem. 48, 312–316.
Lin, W., Yu, Z., Zhang, H., and Thompson, I. P. (2014). Diversity and dynamics of microbial communities at each step of treatment plant for potable water generation. Water Res. 52, 218–230. doi: 10.1016/j.watres.2013.10.071
Liu, G., Zhang, Y., Van Der Mark, E., Magic-Knezev, A., Pinto, A., Van Den Bogert, B., et al. (2018). Assessing the origin of bacteria in tap water and distribution system in an unchlorinated drinking water system by SourceTracker using microbial community fingerprints. Water Res. 138, 86–96. doi: 10.1016/j.watres.2018.03.043
Lu, J., Struewing, I., Vereen, E., Kirby, A. E., Levy, K., Moe, C., et al. (2016). Molecular detection of legionella spp. and their associations with mycobacterium spp., pseudomonas aeruginosa and amoeba hosts in a drinking water distribution system. J Appl. Microbiol. 120, 509–521. doi: 10.1111/jam.12996
Magoč, T., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Mi, Z., Dai, Y., Xie, S., Chen, C., and Zhang, X. (2015). Impact of disinfection on drinking water biofilm bacterial community. J. Environ. Sci. 37, 200–205.
Pfyffer, G. E. (2015). “Mycobacterium: general characteristics, laboratory detection, and staining procedures,” in Manual of Clinical Microbiology, 11th Edn, eds J. Jorgensen, M. Pfaller, K. Carroll, G. Funke, M. Landry, S. Richter, et al. (Washington, DC: American Society of Microbiology).
Pinto, A. J., Schroeder, J., Lunn, M., Sloan, W., and Raskin, L. (2014). Spatial-temporal survey and occupancy-abundance modeling to predict bacterial community dynamics in the drinking water microbiome. mBio 5:e01135-14. doi: 10.1128/mBio.01135-14
Poitelon, J.-B., Joyeux, M., Welté, B., Duguet, J.-P., Prestel, E., and Dubow, M. S. (2010). Variations of bacterial 16S rDNA phylotypes prior to and after chlorination for drinking water production from two surface water treatment plants. J. Ind. Microbiol. Biotechnol. 37, 117–128. doi: 10.1007/s10295-009-0653-5
Pontara, A. V., Oliveira, C. D. D. D., Barbosa, A. H., Santos, R. A. D., Pires, R. H., and Martins, C. H. G. (2011). Microbiological monitoring mf mineral water commercialized in Brazil. Braz. J. Microbiol. 42, 554–559. doi: 10.1590/S1517-838220110002000020
Pulschen, A. A., Bendia, A. G., Fricker, A. D., Pellizari, V. H., Galante, D., and Rodrigues, F. (2017). Isolation of uncultured bacteria from antarctica using long incubation periods and low nutritional media. Front. Microbiol. 8:1346. doi: 10.3389/fmicb.2017.01346
Shade, A., and Handelsman, J. (2012). Beyond the venn diagram: the hunt for a core microbiome. Environ. Microbiol. 14, 4–12. doi: 10.1111/j.1462-2920.2011.02585.x
Varga, L. (2011). Bacteriological quality of bottled natural mineral waters commercialized in hungary. Food Control 22, 591–595.
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naïve bayesian classifier for rapid assignment of rrna sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73:5261.
Wei, L., Wu, Q., Zhang, J., Guo, W., Chen, M., Xue, L., et al. (2017). Prevalence and genetic diversity of enterococcus faecalis isolates from mineral water and spring water in china. Front. Microbiol. 8:1099. doi: 10.3389/fmicb.2017.01109
Wei, L., Wu, Q., Zhang, J., Wu, K., Guo, W., and Que, S. (2014). The pollution survey of Pseudomonas aeruginosa in mineral water and spring water and the analyses of virulence genes and antibiotic resistance of the isolates. Microbiol. China 42, 125–132.
Wong, D., Nielsen, T. B., Bonomo, R. A., Pantapalangkoor, P., Luna, B., and Spellberg, B. (2017). Clinical and pathophysiological overview of acinetobacter Infections: a Century of Challenges. Clin. Microbiol. Rev. 30, 409–447.
Wu, H., Zhang, J., Mi, Z., Xie, S., Chen, C., and Zhang, X. (2015). Biofilm bacterial communities in urban drinking water distribution systems transporting waters with different purification strategies. Appl. Microbiol. Biotechnol. 99, 1947–1955. doi: 10.1007/s00253-014-6095-7
Zhang, Y. Q., Wu, Q. P., Zhang, J. M., and Yang, X. H. (2011). Effects of ozone on membrane permeability and ultrastructure in Pseudomonas aeruginosa. J. Appl. Microbiol. 111, 1006–1015. doi: 10.1111/j.1365-2672.2011.05113.x
Keywords: mineral water treatment system, bacterial community, high-throughput sequencing, core microbiome, opportunistic pathogens
Citation: Wei L, Wu Q, Zhang J, Guo W, Gu Q, Wu H, Wang J, Lei T, Chen M, Wu M and Li A (2019) Composition and Dynamics of Bacterial Communities in a Full-Scale Mineral Water Treatment Plant. Front. Microbiol. 10:1542. doi: 10.3389/fmicb.2019.01542
Received: 07 January 2019; Accepted: 20 June 2019;
Published: 24 July 2019.
Edited by:
Learn-Han Lee, Monash University Malaysia, MalaysiaReviewed by:
Margarita Kambourova, Institute of Microbiology (BAS), BulgariaJames Scott Maki, Marquette University, United States
Carmen Wacher, National Autonomous University of Mexico, Mexico
Copyright © 2019 Wei, Wu, Zhang, Guo, Gu, Wu, Wang, Lei, Chen, Wu and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingping Wu, d3VxcDIwM0AxNjMuY29t