Abstract
Pyrococcus furiosus DSM 3638 is a model organism for hyperthermophilic archaea with an optimal growth temperature near 100°C. The genome was sequenced about 18 years ago. However, some publications suggest that in contrast to other Pyrococcus species, the genome of P. furiosus DSM 3638 is prone to genomic rearrangements. Therefore, we re-sequenced the genome using third generation sequencing techniques. The new de novo assembled genome is 1,889,914 bp in size and exhibits high sequence identity to the published sequence. However, two major deviations were detected: (1) The genome is 18,342 bp smaller than the NCBI reference genome due to a recently described deletion. (2) The region between PF0349 and PF0388 is inverted most likely due an assembly problem for the original sequence. In addition, numerous minor variations, ranging from single nucleotide exchanges, deletions or insertions were identified. The total number of insertion sequence (IS) elements is also reduced from 30 to 24 in the new sequence. Re-sequencing of a 2-year-old “lab culture” using Nanopore sequencing confirmed the overall stability of the P. furiosus DSM 3638 genome even under normal lab conditions without taking any special care. To improve genome annotation, the updated DNA sequence was combined with an RNA sequencing approach. Here, RNAs from eight different growth conditions were pooled to increase the number of detected transcripts. Furthermore, a differential RNA-Seq approach was employed for the identification of transcription start sites (TSSs). In total, 2515 TSSs were detected and classified into 834 primary (pTSS), 797 antisense (aTSS), 739 internal and 145 secondary TSSs. Our analysis of the upstream regions revealed a well conserved archaeal promoter structure. Interrogation of the distances between pTSSs and aTSSs revealed a significant number of antisense transcripts, which are a result of bidirectional transcription from the same TATA box. This mechanism of antisense transcript production could be further confirmed by in vitro transcription experiments. We assume that bidirectional transcription gives rise to non-functional antisense RNAs and that this is a widespread phenomenon in archaea due to the architecture of the TATA element and the symmetric structure of the TATA-binding protein.
Introduction
Pyrococcus furiosus was isolated from geothermally heated marine sediments taken from the beach of Porto di Levante, Vulcano Island, Italy (Fiala and Stetter, 1986). It is a strictly anaerobic heterotroph, growing on maltose, starch, pyruvate, peptone and complex organic substrates. When carbohydrates are used as energy source, acetate, carbon dioxide and hydrogen are the major fermentation products (Schäfer and Schönheit, 1992; Kengen et al., 1994). In the presence of peptides, elemental sulfur is required for efficient growth and hydrogen sulfide is generated as end product. With a doubling time of only 37 min at the optimal growth temperature of 100°C, P. furiosus has developed to one of the best studied hyperthermophilic organisms. The first published genome sequence of P. furiosus DSM 3638 revealed a GC content of 40.8% and a genome size of 1.91 Mb encoding 2,225 genes and 2,122 proteins (Robb et al., 2001).
However, some publications suggest that the P. furiosus genome is susceptible to genomic rearrangements in comparison to the related Pyrococcus species Pyrococcus abyssi and Pyrococcus horikoshii (DiRuggiero et al., 2000; Brügger et al., 2002; Zivanovic et al., 2002). Genome comparison suggests that transposition events, most frequently induced by insertion sequence (IS) elements, are the major driving force for such genome variations in the P. furiosus genome. No full-length IS elements were identified in the genomes of the other two Pyrococcus species (Zivanovic et al., 2002). IS elements are short DNA sequences with a typical length between 700 and 2500 bp (Siguier et al., 2014). They contain an open reading frame (ORF) encoding a transposase, which is usually flanked by inverted repeats and promote translocation of DNA segments within and between genomes. A study analyzing the IS elements of a collection of Pyrococcus strains isolated from the original habitat, Vulcano Island, suggested that these elements play an important role for genetic drift in the diversification of a geographically isolated population of P. furiosus (Escobar-Páramo et al., 2005). Furthermore, the identification of an almost identical 16 kb region transposable region between P. furiosus and Thermococcus litoralis with only 153 nucleotide differences, indicates that these mobile elements are also involved in horizontal gene transfer (DiRuggiero et al., 2000). This region belongs to one of six highly variable chromosomal regions, which were previously identified by comparative genome hybridization using P. furiosus and seven Pyrococcus isolates from Vulcano Island (White et al., 2008). The 16 kb transposable region harbors genes encoding an ABC transport system for maltose and trehalose and is only present in P. furiosus, but absent from all other Vulcano isolates. This is also true for Pyrococcus woesei, which was isolated 1 year later from the same habitat (Zillig et al., 1987). The physiology of P. woesei seems to be very similar to that of P. furiosus and the rRNA operons of these strains have identical sequences (Kanoksilapatham et al., 2004). Although the complete genome sequence of P. woesei is not available, hybridization of genomic sequence from P. woesei to a DNA microarray of P. furiosus revealed the presence of additional genes in two clusters in P. furiosus (Hamilton-Brehm et al., 2005). One of these clusters is the 16 kb transposable region involved in the maltose metabolism. It is interesting to note that a ChIP-Seq approach from our group recently revealed the deletion of this 16 kb fragment also in P. furiosus and an additional southern blot analysis with a new strain from the German Collection of Microorganisms and Cell Cultures (DSMZ, Braunschweig, Germany) confirmed a rapid deletion of the fragment even with growth on maltose (Reichelt et al., 2016). Altogether, these findings support the previous suggestion to rename P. woesei as P. furiosus subsp. woesei (Kanoksilapatham et al., 2004).
Genome variability of P. furiosus strains can be observed in the natural habitat but also in strains cultivated in the laboratory. Several years of re-cultivation from stocks stored at 4°C resulted in the emergence of at least two additional P. furiosus strains, LS and BBR, in the Archaea Center at the University of Regensburg (Näther-Schindler et al., 2014). Both strains differ in comparison to the deposited type strain P. furiosus DSM 3638 in the degree of flagellation and cell morphology (Daum et al., 2017). A similar observation concerning the occurrence of a lab strain was made in Michael Adams group (University of Georgia, Athens, Georgia). This lab strain exhibits an extended exponential growth phase and atypical cell aggregation behavior (Lewis et al., 2015). The genome sequence of the mutant showed 145 genes with one or more insertions, deletions or substitutions. The data clearly demonstrate that P. furiosus has most likely due to the presence of IS elements an inherent property for efficient genome rearrangements. This facilitates the selection of special mutants under a proper selection pressure, but also stimulates the non-specific accumulation of different mutations within the genome. The best example for this process is the development of the genetically tractable P. furiosus strain COM1 (Bridger et al., 2012). The genome sequence of this strain is 1,571 bp longer than the type strain and contains numerous chromosomal rearrangements, deletions, and single base changes, which lead to the inactivation of 20 genes and to amino acid sequence variations of another 102 gene products. These changes impact various cellular functions including a riboflavin requirement for growth. The alignment of the COM1 genome sequence with the published P. furiosus genome revealed major inversions, but an additional analysis of the chromosomal orientations of the original DSMZ strain (ordered in October 2010) by PCR showed that some of this major inversions are also present in the original DSMZ strain (Bridger et al., 2012).
The P. furiosus genome sequence was published 18 years ago, but sequencing technologies and bioinformatic analysis have been revolutionized during the last 10 years. The introduction of massively parallel sequencing led to a significant reduction of sequencing costs. However, these so-called second-generation sequencing techniques produce only short reads, which impedes the assembly of the complete genome as repetitive regions cannot be resolved (Verma et al., 2017). Meanwhile, third-generation sequencing techniques have also entered the market. These systems act directly on the native DNA without the requirement for PCR amplification and show a significant increase in read length, which facilitates complete genome assemblies.
The discrepancy between the published P. furiosus genome sequence (Robb et al., 2001) and the detected deviations in the genome from a recently ordered P. furiosus strain DSM 3638 from the DSMZ (Bridger et al., 2012; Reichelt et al., 2016) and the fact that many groups make use of the originally described P. furiosus strain, encouraged us to re-sequence the type strain P. furiosus. To address the problem concerning the observed genome rearrangements and to allow for complete genome assembly we used a hybrid approach of third generation long-read PacBio sequencing complemented with highly accurate short-read Illumina sequencing (Rhoads and Au, 2015). We amended the DNA-sequencing approach with differential RNA sequencing data, to generate a high-resolution annotation using the ANNOgesic pipeline (Sharma et al., 2010; Yu et al., 2018). Last but not least, to gain insight into the genomic variability of continuously cultivated lab strains and to investigate, if it is possible to maintain genome stability by avoiding strong selection pressure during cultivation, we re-sequenced P. furiosus again after 2 years of cultivation employing the recently developed Nanopore sequencing technology (Loman et al., 2015). Our results indicate a quite stable genome even with the strain cultivated for 2 years in the lab and differential RNA-Seq data revealed that bidirectional transcription is a significant source for archaeal antisense transcripts.
Materials and Methods
Strains and Growth Conditions
Pyrococcus furiosus strain DSM 3638 was stored as freeze-dried culture at 12°C in the dark at the Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ) in Braunschweig, Germany. For the isolation of DNA for combined PacBio and Illumina sequencing, cells were grown anaerobically in 20 ml SME medium supplemented with 0.1% yeast extract and 0.1% starch at 95°C to late-exponential phase (Fiala and Stetter, 1986).
For Nanopore sequencing, a culture was obtained from the DSMZ in 2015 and after growth in SME media the strain was stored in the gas phase of liquid nitrogen at the archaea center in Regensburg for 1 year. After that, the strain was recultivated and handled in the lab for about 2 years with numerous inoculations, to simulate storage and daily life usage conditions of many labs with a focus on microbiology. We assume that during these 2 years the culture was about -roughly estimated- thirty times transferred into fresh medium. In between, liquid cultures were stored at room temperature or 4°C. For the isolation of DNA cells were grown anaerobically in 40 ml SME medium supplemented with 40 mM pyruvate, 0.1% peptone and 0.1% yeast extract at 85°C to mid-exponential phase.
For RNA sequencing P. furiosus cells were grown under eight different conditions to maximize the number of different transcripts in the genome: Cells were grown anaerobically in 20 ml SME medium supplemented with 0.1% starch, 0.1% peptone and 0.1% yeast extract at 95°C to early- (1 × 107 cells/ml), mid-exponential (5 × 107 cells/ml) or late-exponential (1 × 108 cells/ml) phase (conditions 1, 2, and 3). In addition, cells were grown in 20 ml SME medium supplemented with 0.1% starch, 0.1% peptone and 0.1% yeast extract at 95°C to late-exponential phase, further incubated at 4°C for 1 h (condition 4; cold shock) or 110°C for 15 min (condition 5; heat shock). Moreover, cells were grown anaerobically in 20 ml SME medium supplemented with 0.1% starch, 0.1% peptone and 0.1% yeast extract at 75°C to late-exponential phase (condition 6; cold adaption). Furthermore, cells were grown anaerobically in 20 ml SME medium supplemented with 0.1% yeast extract and 0.1% starch (condition 7; glycolytic growth) or 40 mM pyruvate (condition 8; gluconeogenic growth) at 95°C to late-exponential phase. Cells were harvested by centrifugation at 3,939 × g for 45 min at 4°C, cell pellets were frozen in liquid nitrogen and stored at −80°C until used for the isolation of DNA or RNA.
DNA Isolation
Genomic DNA was isolated using ReliaPrepTM gDNA Tissue Miniprep System (Promega) according to the instructions of the manufacturer. Quantity and quality were analyzed using Nanodrop One, Qubit dsDNA HS assay kit (both from Thermo Fisher Scientific) and agarose gel electrophoresis. For Nanopore sequencing size distribution was checked using pulsed field gel electrophoresis on a CHEF-DR®III system (Bio-rad).
RNA Extraction
P. furiosus total RNA was purified using the peqGOLD TriFastTM reagent (VWR). 20 ml cell culture was pelleted, and cells were lysed by addition of 1 ml TriFast followed by rigorous shaking for 15 min. After adding 0.2 ml 2 M sodium acetate pH 4.0 RNA was isolated according to the manufacturer instructions. Contaminating DNA was removed via the TURBO DNA-freeTM Kit (Thermo Fisher Scientific, Waltham, MA, United States). The integrity of the total RNA was assessed via a Bioanalyzer (Agilent) run using the RNA 6000 Pico Kit (Agilent) and purified RNA was stored at −80.
PacBio Library Preparation and Sequencing
SMRTbellTM template library was prepared according to the instructions from Pacific Biosciences, Menlo Park, CA, United States, following the Procedure and Checklist – Greater Than 10 kb Template Preparation. Briefly, for preparation of 15 kb libraries 8 μg genomic DNA was sheared using g-tubesTM from Covaris, Woburn, MA, United States according to the manufacturer’s instructions. DNA was end-repaired and ligated overnight to hairpin adapters applying components from the DNA/Polymerase Binding Kit P6 from Pacific Biosciences, Menlo Park, CA, United States. Reactions were carried out according to the manufacturer’s instructions. BluePippinTM Size-Selection to greater than 7 kb was performed according to the manufacturer’s instructions (Sage Science, Beverly, MA, United States). Conditions for annealing of sequencing primers and binding of polymerase to purified SMRTbellTM template were assessed with the Calculator in RS Remote, Pacific Biosciences, Menlo Park, CA, United States. SMRT sequencing was carried out on the PacBio RSII (Pacific Biosciences, Menlo Park, CA, United States) taking one 240-min movie on two SMRT Cells.
Genome Assembly, Error Correction, and Annotation
SMRT Cell data was assembled using the “RS_HGAP_Assembly.3” protocol included in SMRT Portal version 2.3.0 using default parameters. The assembly revealed a single circular chromosome. The chromosome was circularized, particularly artificial redundancies at the ends of the assembled contig were removed and adjusted to cdc6 as the first gene. Error-correction was performed by a mapping of paired-end reads of 2 × 100 bp generated on an Illumina HiSeq 2500 onto finished genomes using BWA (Li and Durbin, 2010) with subsequent variant and consensus calling using VarScan (Koboldt et al., 2012). A consensus concordance of QV60 could be confirmed. Finally, an annotation was carried out using NCBI prokaryotic genome annotation pipeline (Tatusova et al., 2016). The genome sequence was deposited in NCBI GenBank under Accession Number CP023154.
Nanopore Sequencing (MinION)
Library Preparation and Sequencing
Library preparation was performed according to Oxford Nanopore Technologies (ONT, Oxford, United Kingdom) protocol for multiplexing samples (1D native barcoding genomic DNA with EXP-NBD103 and SQK-LSK108). Sequencing was performed on ONT’s MinION MK1B device, connected to a laptop computer via USB3, using MinKNOW software (v.1.4.3). After confirmation of a sufficient number of active pores on the flow cell (SpotON Flow cell Mk I R9 version), the prepared DNA library was loaded onto the flow cell and sequencing was started choosing the 48 h sequencing protocol suggested by MinKNOW.
Basecalling, de novo Assembly, Polishing and Evaluation
For MinION data analysis raw reads in fast5 data format were base-called and de-multiplexed using Albacore 2.3.4. In a first step a de novo genome assembly was done using canu 1.8 (genomeSize = 1.9 m, minReadLength = 500, minOverlapLength = 100) (Koren et al., 2016), before improving the consensus sequence in a second step with nanopolish 0.11 (min-candidate-frequency = 0.1) (Simpson et al., 2017). The chromosome was circularized, artificial redundancies at the ends of the assembled contig removed and adjusted to cdc6 as the first gene (compare 2.5). To determine the identity of the de novo assembly to the hybrid PacBio-Illumina approach, statistics from dnadiff (MUMmer version 3) were calculated and visualized using R package genoPlotR (Kurtz et al., 2004; Guy et al., 2011). Read length and nucleotide frequencies were analyzed using the statistical program R with ggplot2 (R Development Core Team, 2011; Wickham, 2016). Code is available at: https://github.com/felixgrunberger/pyrococcus_reannotation.
Illumina Sequencing (RNA-Seq)
RNAs purified from cells grown under eight different growth conditions were pooled equally and submitted for library preparation and sequencing to the Core Unit Systems Medicine (SysMed) at the University of Würzburg, Germany. Three different libraries were prepared to fulfill the requirements for usage in the ANNOgesic pipeline: fragmented, unfragmented with terminator exonuclease treatment (+TEX) and unfragmented without TEX-treatment (−TEX). For the fragmented sample, RNA was fragmented for 2 min at 94°C using the NEBNext®Magnesium RNA Fragmentation Module. Afterward RNA was treated with T4 Polynucleotide Kinase (PNK) without ATP for 6 h at 37°C and 1 h at 37°C with 2 mM ATP and fresh T4 PNK. After overnight ethanol precipitation, 5′ triphosphates were removed using RNA 5′ Pyrophosphohydrolase (RppH) for 1 h at 37°C. RNA was again precipitated and resuspended in 6 μl H2O. The two samples (+/− TEX) for the transcription start site detection were either treated with TEX (+ TEX) or with H2O as a mock control (− TEX) for 30 min at 37°C. Afterward both samples were treated with RppH for 1 h at 37°C before they were precipitated, and the RNA was resuspended in 6 μl H2O. After the pre-treatment, all three libraries were prepared using the NEBNext®Multiplex Small RNA Library Prep Kit for Illumina according to the manufacturer’s protocol with small modifications. The first linker ligation was performed for 18 h at 16°C and libraries were amplified with 12 PCR cycles with an extended elongation time of 75 s. Libraries were pooled in a 2:1:1 ratio (fragmented: + TEX : − TEX) and sequenced on an Illumina NextSeq 500 high-output single-end 75 nt run.
Trimming and Mapping of RNA-Seq Reads
Illumina reads in FASTQ format were quality/length/adapter trimmed using trimmomatic (v.0.36) in single-end-mode allowing for a minimum length of 12 nt and a cut-off Phred score of 20, calculated in a sliding window of 4 bases (Bolger et al., 2014). Next, reads were mapped using STAR aligner (v.2.5.3) to the new assembled genome of P. furiosus (Dobin et al., 2013). Mapping statistics (input, filtered, uniquely aligned reads) can be found in the Supplementary Table 3. To use ANNOgesic for RNA-based annotation of P. furiosus, strand-specific coverage files in wiggle format of all three sequencing data sets were generated (Yu et al., 2018). As recommended, reads were additionally mapped with segemehl 0.2.0 to detect circular RNAs within the ANNOgesic pipeline (Otto et al., 2014).
Reference Genome Annotation Using ANNOgesic
ANNOgesic is a recently published pipeline that predicts transcriptome-wide features based on a combination of differential and fragmented RNA sequencing (Yu et al., 2018). Amongst others, it is built on TSSpredator, using adaptive parameter optimization, which simplifies and improves detection of transcription start sites (TSS) (Dugar et al., 2013). Following subcommands were executed in the provided Docker image of ANNOgesic to improve annotation of P. furiosus (basic parameters if not stated otherwise): optimize_tss_ps (with 50 manually detected TSS as a reference, 4000 iterations), tss_ps (with optimized parameters from previous step), transcript, terminator, utr, operon, srna, sorf, circrna (cut-off supported reads: 200), promoter, crispr. Features in gff file format were combined with the merge_features command and added to the gff file from DNA sequencing and assembly (Supplementary Table 5).
RNA-Seq Data Analysis
Data analysis of output files from ANNOgesic was done using the R/Bioconductor environment (R Development Core Team, 2011). Scripts for analysis were uploaded to https://github.com/felixgrunberger/pyrococcus_reannotation.
Detection of Promoter Elements
For the detection of common archaeal promoter elements, a position weight matrix (PWM) was calculated from sequences 50 bases upstream to 10 bases downstream of all available TSS. The resulting motif was visualized in R using ggseqlogo (Wagih, 2017).
The sequences 51 bases upstream of every TSS were extracted to identify the best ranking promoter motif for each TSS category using MEME with default options except “search given strand only” (Bailey et al., 2009). Motifs and position tables were further analyzed using ggseqlogo and ggplot2. For internal TSS a repetitive sequence coming from CRISPR regions gave the best motif but was excluded from further analysis.
Length of 5′ UTRs of pTSS and sTSS was already calculated in the ANNOgesic pipeline and visualized using ggplot2. Internal and antisense TSS positions relative to a gene were sorted in three windows: 300 bp upstream, 300 bp downstream and in between annotated genes. Positions between start and stop site were scaled according to gene length.
To find a motif for possible bidirectional promoters we filtered all primary TSS that had strong TEX signal on the antisense strand (more than 40% of the reads from −400 to +400 in the region 100 bp upstream of pTSS) and calculated a motif using MEME (default options).
Coverage Plots
We generated average coverage profile plots to check for the enrichment of TSS in the TEX data set and to validate the TSSpredator classification. The R package CoverageView was used to calculate the coverage for each sequencing data set from 400 bp upstream and downstream relative to a TSS in a window of 10 bp (Lowy, 2017). Every position for a single TSS was scaled proportionally, before calculating mean values for plotting.
Coverage plots were also generated for the analysis of putative bidirectional promoters. A similar protocol as mentioned above was used. We split the data set into two groups (head-to-head and head-to-tail) considering the orientation of the upstream gene. From this data set we also calculated the intergene distance for annotated genes.
Antisense Enrichment Around IS Elements
Insertion sequence elements for the available Pyrococcus assemblies were identified using ISEScan 1.6 (Xie and Tang, 2017). Genomic positions of these elements were extracted to scan for antisense TSS nearby. The relative position was calculated in a window 100 bp upstream from the start of an IS element, 100 bp downstream from the end and in between scaled according to length. The aTSS and IS elements used for the analysis to create Figure 8B are listed in Supplementary Table 6.
In vitro Analysis of Bidirectional Transcription
For the analysis of bidirectional transcription in vitro transcription reactions were assembled in 100 μl reaction volumes (40 mM Na-HEPES, pH 7.3, 0.1 mM EDTA, 0.1 mg/ml BSA, 2.5 mM MgCl2, 250 mM KCl): 8.8 nM DNA was combined with 190 nM TBP, 105 nM TFB, 108 nM TFE, and 10.5 nM RNA polymerase. The 349 bp DNA fragment was prepared by PCR with the primers 5′-gaaagggcgaaccagttagattgaacg and 5′-tgttgggcttctccccaagctgag using genomic DNA as template. Transcription was initiated by the addition of NTPs to a final concentration of 100 μM and reactions were incubated at 80°C for 10 min. Reactions were stopped by extraction with one volume phenol/chloroform/isoamyl alcohol, RNA was precipitated with ethanol and resuspended in 20 μl H2O. For the analysis of the transcripts primer extension experiments were carried out using labeled primers in sense or antisense direction. 10 μl in vitro RNA were combined with 125 nM of the corresponding primer in a total volume of 15 μl. After RNA denaturing at 70°C for 5 min, primer annealing was performed at 0°C for 5 min. Primer extension was started by the addition of 5 μl reverse transcription mixture containing 50 units of M-MLV RNase H minus (Promega) and 1 mM dNTPs. After incubation at 48°C for 30 min, cDNA products were purified by ethanol precipitation, resuspended in 10 μl formamide buffer and analyzed on a 20% denaturing polyacrylamide gel. The DNA fragments were visualized with a ChemiDoc MP imaging system (Bio-rad).
Results and Discussion
Strategy for Genome Re-annotation of Pyrococcus furiosus
In order to address the described questions concerning the stability of the Pyrococcus genome, we employed a combination of DNA and RNA sequencing techniques to generate an updated version of the P. furiosus genome (Figure 1). We used the current gold standard in genome assembly approaches, a combination of long read PacBio sequencing and short read Illumina sequencing, to obtain a highly accurate reference genome of QV60 (<1 error per Mbp) for further analysis. Differential RNA sequencing was used to map primary transcription start sites (TSS) and to improve genome annotation with the recently developed ANNOgesic pipeline (Yu et al., 2018). In order to test whether P. furiosus’ genome is subject to genome instability upon long-term cultivation of the strain, we re-sequenced a 2-year-old lab culture of P. furiosus employing the Nanopore sequencing technique.
FIGURE 1
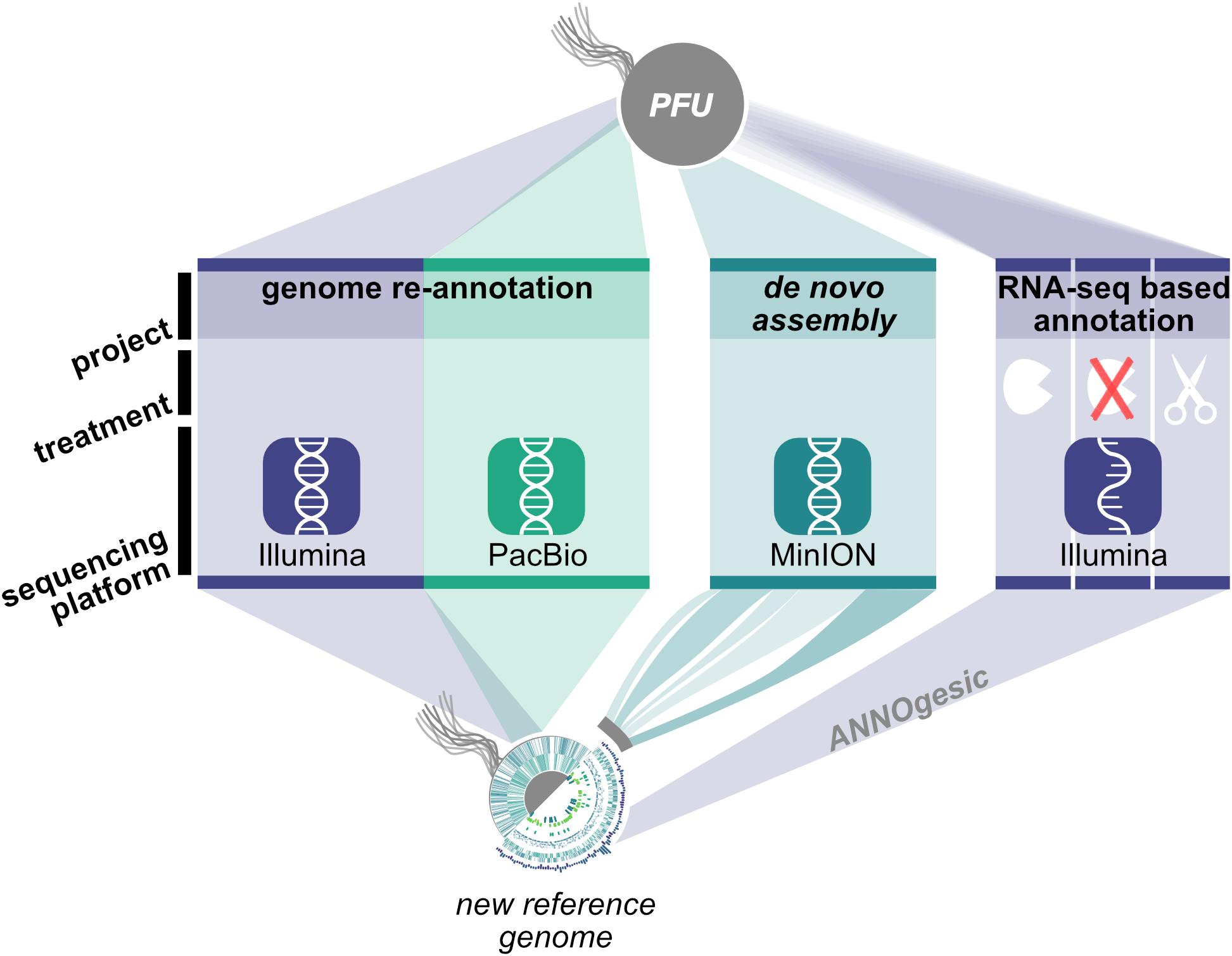
Workflow for genome re-annotation of P. furiosus. To build the new reference genome of P. furiosus DSM 3638 we used a hybrid PacBio-Illumina approach. After 2 years of subcultivation genome stability of the same strain was tested using Nanopore MinION sequencing and de novo assembly. Genome annotation was improved with an RNA-Seq based approach of eight mixed growth conditions to cover a broad range of transcripts. Three different RNA treatments (terminator-exonuclease treated, not-treated, fragmented) were used to map transcription start sites and additional features using the ANNOgesic pipeline (Yu et al., 2018).
Genome Comparison
A New Reference Genome With Two Major Deviations
Based on PacBio sequencing data that provided a 194-fold coverage of the genome, the P. furiosus type strain DSM 3638 genome was assembled de novo to a single contig sequence, which was error-corrected by Illumina data. The comparison with the current NCBI reference sequence (NC_003413) revealed that the new genome sequence (CP023154) is strongly syntenous to the published sequence (Figure 2, upper part). However, we found two major variations:
FIGURE 2
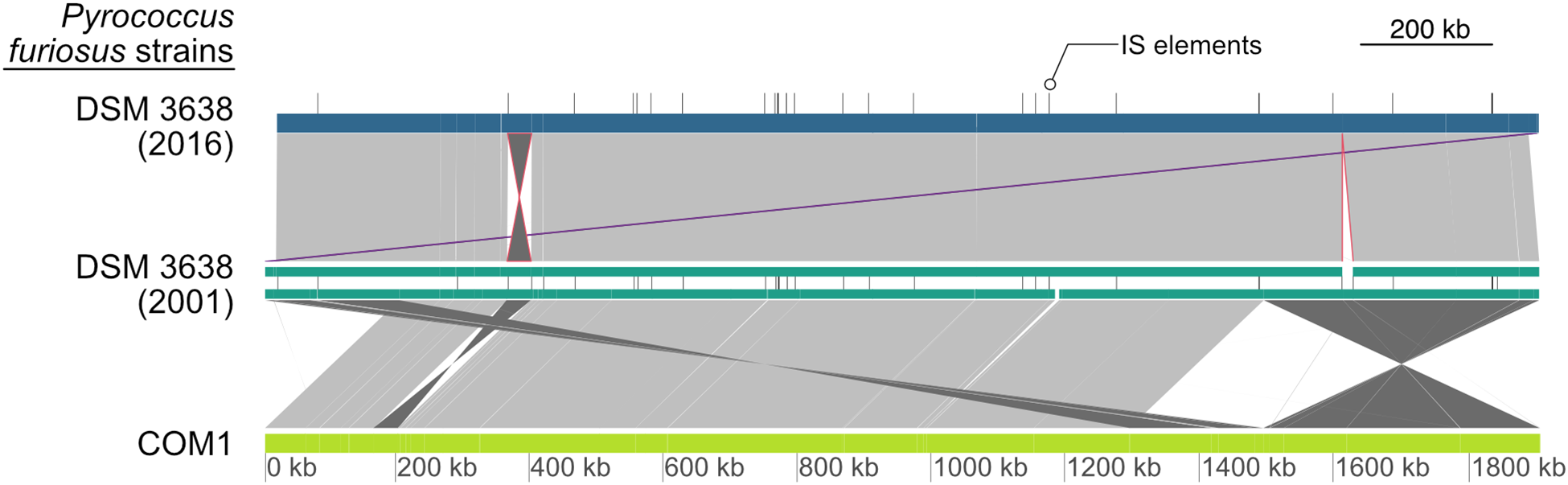
Global pairwise comparison of the genome organization of the new and the original P. furiosus DNA sequence together with the COM1 strain (Bridger et al., 2012). Whole genome alignments were calculated with dnadiff and visualized using GenoPlotR (Kurtz et al., 2004; Guy et al., 2011). Each genome is shown as colored blocks (blue, green, light green), whereas IS elements of DSM 3638 are indicated by vertical lines. Direct matches with high similarity between genomes are colored in light gray and inversions in dark gray. The inversion of the fragment from PFDSM3638_01715 to PFDSM3638_01910 and the deletion of the 17,075 bp fragment are indicated in red and the adjusted annotation start to cdc6 as the first gene is highlighted in purple. Additional minor variations are below the resolution of the presented map. In contrast to the large genomic rearrangements between the COM1 strain and the type strain of P. furiosus, there are only minor differences between the old and the new DNA sequence of P. furiosus DSM 3638.
- (1)
The fragment encoding the genes PFDSM3638_01715 to PFDSM3638_01910 is inverted in comparison to the corresponding region PF_RS01790 (PF0349) to PF_RS01990 (PF0388) in the original reference sequence NC_003413. This inversion represents one of numerous described chromosomal rearrangements identified for the P. furiosus strain COM1 (Figure 2, lower part) (Bridger et al., 2012). But in contrast to the other differences this deviation was also found in the sequencing data of a newly ordered type strain from the DSMZ by the same group (Bridger et al., 2012). This indicates that this inversion is most likely caused by an assembly problem of the original sequence (Robb et al., 2001).
- (2)
The region from 1,613,139 to 1,630,214 is deleted in the re-sequenced genome. The deletion of this region has also been identified previously by a ChIP-Seq approach (Reichelt et al., 2016). It belongs to a highly variable chromosomal region, which is flanked by two IS elements and proposed as an example for a recent transposon mediated gene transfer between P. furiosus and Thermococcus litoralis (DiRuggiero et al., 2000). The fragment encodes a trehalose/maltose-specific-ABC-transporter. Even growth on maltose could not prevent this deletion (Reichelt et al., 2016).
Due to the above-mentioned deletion and a reduced number of IS elements, the complete sequence consists of 1,889,914 bp, which is 18,342 bp smaller than the NCBI reference genome (Figure 1, upper part, and Table 1). The GC content and the total number of genes are also slightly reduced due to the deletion events. Using the NCBI prokaryotic genome annotation pipeline (Tatusova et al., 2016), the new genome sequence harbors 2,035 genes of which 1,982 encode proteins, whereas the residual 68 genes transcribe four rRNA genes (one 23S rRNA, one 16S rRNA and two 5S rRNAs), 46 tRNAs and 18 additional ncRNAs. The values are very similar in comparison to the annotations from the other two assemblies (Table 1). Moreover, the number of pseudogenes decreased in the new assembly from 74 to 53, which indicates that the re-sequencing allowed for correction of frameshifted genes now being present correctly annotated in full-length. All three genomes harbor seven CRISPR arrays, but the number of spacers in the CRISPR array 6 differs between the new and old P. furiosus type strain DSM 3638 assemblies (36 vs. 21). This might be due to assembly problems of repetitive sequences in the initial sequencing in 2002. In addition, numerous minor variations were identified including single nucleotide exchanges (causing silent or missense mutations), frameshift insertions or deletions and deletions or insertions of complete genes (summarized in Supplementary Table 1). Some of these variations were already reported by previous studies. For example, the flaB0 gene was discovered in an earlier study, which encodes the major flagellin of the P. furiosus archaellum apparatus (Näther-Schindler et al., 2014). A comparison of all coding sequences from the annotation based on the new genome assembly (CP023154) with the annotation based on the genome assembly (NC_003413) from 2002 is shown in the Supplementary Table 2.
Table 1
| NCBI GenBank Accession | DSM 3638_2016 CP023154 | DSM 3638_2001 NC_003413 | COM_1 NC_018092 |
|---|---|---|---|
| Genome length (bp)1 | 1889914 | 1908256 | 1909827 |
| GC content [%] | 40.75 | 40.77 | 40.79 |
| Genes (total)1 | 2,035 | 2,053 | 2,066 |
| Genes (coding)1 | 1,982 | 1,979 | 2,001 |
| Genes (RNA)1 | 68 | 68 | 67 |
| complete rRNAs1 | 2, 1, 1 | 2, 1, 1 | 2, 1, 1 |
| (5S, 16S, 23S) | (5S, 16S, 23S) | (5S, 16S, 23S) | |
| tRNAs1 | 46 | 46 | 46 |
| ncRNAs1 | 18 | 18 | 17 |
| Pseudo Genes (total)1 | 53 | 74 | 65 |
| CRISPR Arrays1,2 | 7 | 7 | 7 |
| CRISPR1 Spacer2 | 51 | 51 | 51 |
| CRISPR2 Spacer2 | 21 | 20 | 21 |
| CRISPR3 Spacer2 | 23 | 22 | 23 |
| CRISPR4 Spacer2 | 30 | 30 | 30 |
| CRISPR5 Spacer2 | 45 | 45 | 44 |
| CRISPR6 Spacer2 | 36 | 21 | 36 |
| CRISPR7 Spacer2 | 11 | 11 | 11 |
| Total no. of IS elements3 | 24 | 30 | 40 |
| IS200/IS6053 | 1 | 1 | 1 |
| IS63 | 17 | 23 | 33 |
| IS9823 | 5 | 5 | 5 |
| new3 | 1 | 1 | 1 |
Genome comparison of the re-sequenced Pyrococcus furiosus DSM 3638 together with the first published NCBI reference sequence (NC_003413) and P. furiosus COM1 (NC_018092).
1NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al., 2016). 2CRISPRFinder (Grissa et al., 2007). 3ISEScan (Xie and Tang, 2017).
Nanopore Sequencing Confirms Genome Stability
Re-sequencing of the type strain, which was stored for more than 15 years under optimal conditions at the DSMZ, indicated indeed a very stable genome over the years. But is this also true for a strain handled in the lab which is repeatedly inoculated and stored over time in liquid cultures? To answer this question, we re-sequenced the “lab culture” about 2 years after we performed the Illumina/PacBio sequencing employing this time the recently developed Nanopore sequencing technique. During these 2 years the culture was about -roughly estimated- thirty times transferred into fresh medium. A total of 397,582 reads were accumulated of which 328,862 (82.7%) had a mean Phred-based quality score (qscore albacore) equal or better than 7.0 representing ∼308-fold genome coverage. The median Phred quality score for all reads used for further assembly steps was 8.82.
We performed a de novo assembly of the genome based on the Nanopore sequencing data. First, we generated a draft genome using Canu. Subsequently, we improved the consensus sequence with Nanopolish (Koren et al., 2016; Simpson et al., 2017). In the first step, we were able to reach a closed assembly with 1 contig (1,891,829 bp) with no genomic rearrangements observed compared to the PacBio/Illumina reference sequence. In general, the genome was only 0.1% assembled larger with an identity of 99.42% compared to the reference genome (Table 2). After polishing, the sequence identity further improved to 99.92%. Most of the additional base pairs (+0.35%) can be explained by insertions throughout the genome with a slight preference for additional As and Ts (A: 27%, T: 27% of all insertions).
Table 2
| Assembly | Total Bases | No. of contigs | GC content | % Identity (1-to-1 dnadiff mummer) |
|---|---|---|---|---|
| Illumina/PacBio | 1889914 | 1 | 40.75 | 100 |
| Nanopore raw (Canu) | 1891829 | 1 | 40.69 | 99.42 |
| Nanopore polished (Nanopolish) | 1896610 | 1 | 40.75 | 99.92 |
Nanopore sequencing is suitable for generating a high identity genome de novo in comparison with hybrid Illumina/PacBio data.
A closed genome (No. of contigs = 1) from Oxford Nanopore data was reached with Canu. One round of Nanopolish was used to further increase sequence identity.
One major advantage of the Nanopore technique is the significantly increased read length in comparison to Illumina sequencing, which facilitates genome assembly and helps to identify genome rearrangements. The usage of a column-based DNA purification protocol led to a fragmentation of DNA, which can be observed in the length distribution of sequenced reads (median length: 1,160 bp; longest read: 31,965 bp) (Figure 3A). Most of the errors in the non-polished assembly were in fact not random but can be explained when the counts of sixmers (all combinations of 6 nucleotides) in both assemblies are analyzed (Figure 3B). Bioinformatical polishing successfully reduced the differences in sixmer usage and lead to a very high sequence identity. All in all, we could not observe any large differences in genome organization after 2 years on/off cultivation and storage. Therefore, we conclude that the Pyrococcus genome is more stable than previously expected.
FIGURE 3
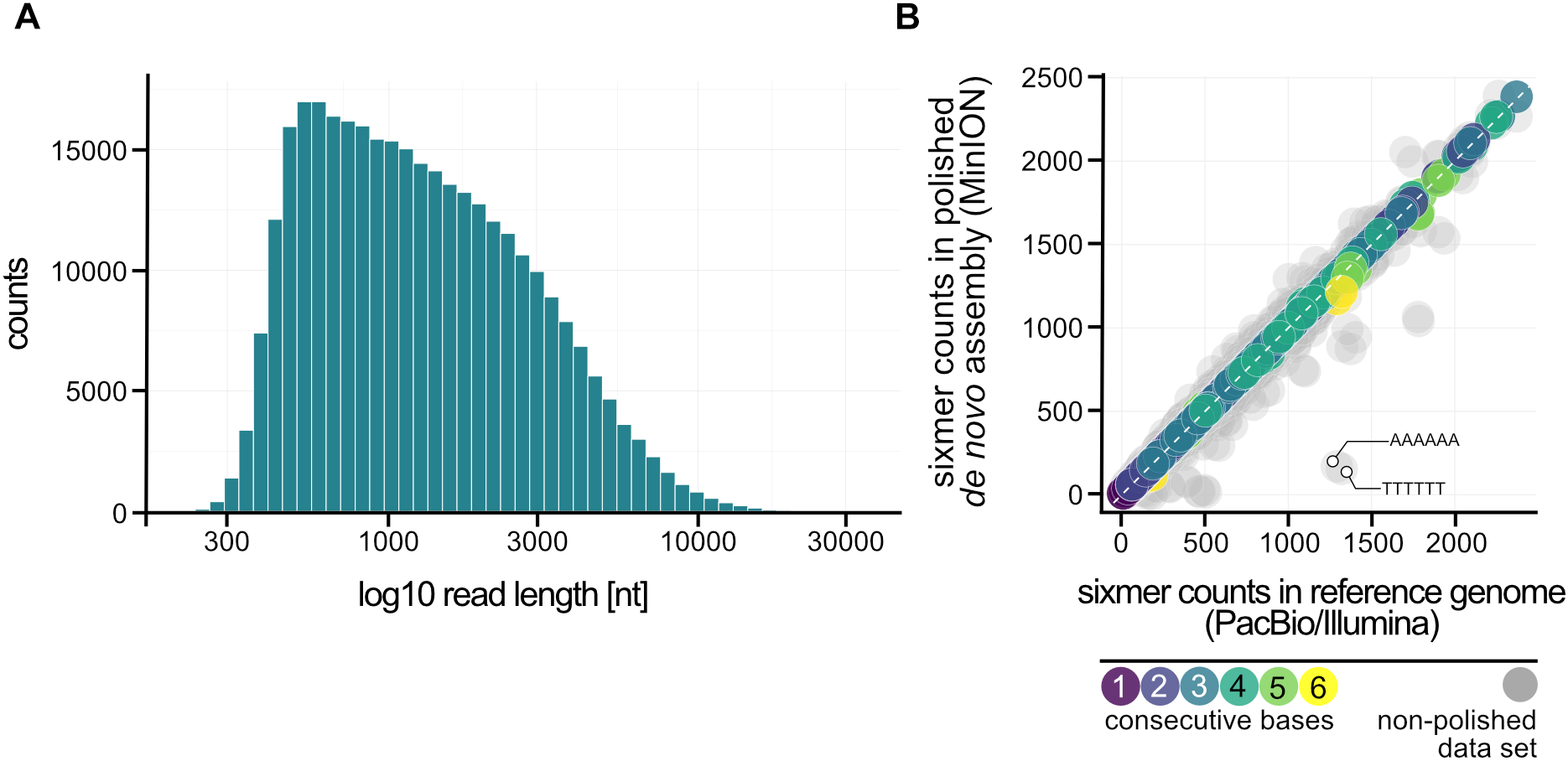
Nanopore sequencing of P. furiosus. (A) Fragmentation of the DNA due to the DNA purification protocol used can be ascertained from the read length distribution after sequencing (median: 1,160 bp). (B) Sixmer-comparison of PacBio/Illumina assembly to Nanopore de novo assembly (polished: colored-, not-polished: gray) shows known drawbacks of Nanopore-sequencing with limited resolution of homopolymers that requires bioinformatic polishing (Simpson et al., 2017).
Analysis of the Primary Transcriptome
We combined the updated DNA sequence with an RNA sequencing approach to improve genome annotation with the recently developed ANNOgesic pipeline (Yu et al., 2018). To maximize the number of detected transcripts in the P. furiosus genome we pooled RNA preparations from eight different growth conditions. In one dataset we fragmented the RNA before generation of cDNA libraries to increase RNA coverage. For the enrichment of primary transcription start sites (TSS), we employed the differential RNA-Seq (dRNA-Seq) approach, which uses a terminator exonuclease (TEX) treatment to degrade RNAs that exhibit a 5′ monophosphate that arise from nucleolytic degradation of primary transcripts but not RNAs with a 5′ triphosphate (Sharma et al., 2010). A TEX-untreated cDNA library served as a control, which includes in addition the 5′ ends of processed or degraded RNAs. Sequencing and mapping statistics can be found in Supplementary Table 3.
Transcription start sites were identified within the ANNOgesic pipeline using TSSpredator and classified into 834 primary (P), 797 antisense (A), 739 internal (I), and 145 secondary (S) transcripts according to their position relative to the next gene (Figure 4A,B; Dugar et al., 2013; Yu et al., 2018). After using an iterative optimization process in the parameter selection module of the newly developed TSSpredator (Yu et al., 2018), the total number of TSS is similar to previously published archaeal primary transcriptome sets considering different genome sizes (Jäger et al., 2009, 2014; Babski et al., 2016; Cho et al., 2017; Smollett et al., 2017). As a result of the densely packed genome, some of these identified transcripts belong to more than one category. For example, 212 TSS were categorized both as pTSS and aTSS that arise from head-to-head oriented genes.
FIGURE 4

Transcription start site (TSS) classification. (A) Primary (P), Secondary (S), Internal (I), and Antisense (A) TSSs are classified by TSSpredator according to their position relative to the next gene (Dugar et al., 2013). (B) Number of TSS identified in each category after using adaptive parameter optimization from ANNOgesic (Yu et al., 2018). (C) Known archaeal promoter elements can be detected by visualizing a PWM calculated from all TSS from −50 to +10 bases to a start site. (D) Promoter motifs for the individual TSS categories identified by MEME search of all upstream sequences (−50, +1). The e-values and the number of sequences contributing to each motif are shown on top of each panel. The panels at the bottom of each category shows a histogram aligned to the third T of the corresponding TATA boxes. (E) Relative coverage of all reads in three sequencing datasets fragmented (FRAG, purple), terminator-exonuclease treated (TEX, blue) and TEX-non-treated-control (NOTEX, green) are plotted in relative position to a TSS in a window of −400 to +400 to confirm the output of the classification algorithm. (F) Position of all TSS according to next gene with normalized length in a window of −300 to annotation-start and +300 to annotation-end (on gene level) are plotted. Primary TSS have predominantly short 5′ UTR lengths with a median of 13 nt, secondary TSS have larger 5′ UTRs and internal TSS are equally distributed across the corresponding gene length. Antisense TSS seem to be enriched in gene-flanking regions.
Using Operon-mapper we analyzed in more detail how transcription of the 2035 identified genes is organized (Taboada et al., 2018). The program recognized 953 transcription units, which consist of 501 single genes and 452 operon structures, which contain the residual 1534 genes. One half (760) is organized in operons with two or three genes and the other half (774) is located in more complex operons with four or more genes (Supplementary Table 4). A comparison of the pTSS with the 953 transcription units revealed that almost 70% (571) of the identified primary transcripts match perfectly with the identified transcription units. It has to be emphasized that the in silico based operon prediction is purely based on intergenic distances and functional relationships between the genes (Taboada et al., 2018). Using a multiple conditions approach, we were able to use high-resolution transcriptomic data to improve current annotation by predicting operons this time based on TSSs, transcripts and genes within the ANNOgesic pipeline (Yu et al., 2018). The total number of transcription units decreased to 693 with the great majority (473, 68.25%) being identical to the Operon-mapper predicted units. The transcriptomic-based detection of transcription units reflects the true biological organization rather than an artifact from annotation, because we were able to detect every single gene in the fragmented RNA-Seq dataset (minimum number of reads per gene: 69).
We also analyzed in more detail the promoter structure of these transcription units. In general, most archaeal promoters consist of three conserved parts, TFB recognition element (BRE), TATA box and a pyrimidine/purine dinucleotide (INR) at the −1/+1 position of the TSS (Hausner et al., 1991; Soppa, 1999; Van De Werken et al., 2006). A position weight matrix (PWM) calculated in a region −50 to +10 from all TSS confirmed the presence of a highly conserved promoter structure with consensus sequences for the BRE −36(RRAAA)−32, the TATA box −30(WTTTAAAW)−23, and the INR −1(YR)+1 (Figure 4C). It is also possible to identify the initially melted region from −11 to −2 which facilitates open complex formation due to accumulation of AT sequences. The identified promoter for P. furiosus fits well with published data of related organisms, e.g., P. abyssi, Thermococcus kodakarensis or Thermococcus onnurineus NA1 (Toffano-Nioche et al., 2013; Jäger et al., 2014; Cho et al., 2017). This is also an additional indication that the TEX treatment was successful and all the identified TSS indeed represent initiation start points of the RNA polymerase. To answer the question if different promoter structures are used in the case of secondary, internal or antisense transcripts, the sequences from −50 to +1 of all identified TSS were also individually analyzed using MEME for identification of the best fitting motif (Figure 4D; Bailey et al., 2009). All motifs exhibit typical BRE and TATA box sequences, but with reduced conservation for secondary, internal and antisense TSS. Furthermore, the location of the TATA box in relation to the TSS is slightly different. In the case of pTSS the last conserved adenine nucleotide of the TATA box is 23 bp upstream of the TSS, which is in perfect agreement with the consensus sequence of all TSS (Figure 4C) and with published data (Toffano-Nioche et al., 2013; Jäger et al., 2014;Cho et al., 2017). In contrast, the position for sTSS is at −27, for iTSS at −22 and for aTSS at −25. From previous in vitro transcription experiments it is known, that these distances still enable transcription, but most likely with a reduced efficiency at least for the sTSS and aTSS (Hausner et al., 1991). To further validate the identification of different TSS classes, we analyzed the distribution of reads in a window from −400 to +400 bp of all annotated TSSs (Figure 4E). As expected, we observed the highest enrichment for the TEX dataset in all TSS classes, confirming a successful enzymatical treatment and downstream bioinformatical analysis. To exclude any further bias and gain insights into possible different regulation mechanisms of the four TSS groups, we plotted the positions of all TSS 300 bp upstream and downstream of the corresponding coding sequences (Figure 4F). Most of the transcripts initiate in close proximity to the coding sequence (pTSS, median: 13 nt, mean: 49.17 nt), indicated by the strong peak of the pTSS near the start codon in contrast to sTSS that exhibit significantly longer 5′ UTR sequences. iTSS are equally distributed over the whole gene length with one prominent peak at the end of the coding sequence. Due to the high gene density in P. furiosus it is possible that some of these iTSS represent pTSS of downstream genes. This is also true for the strong peak upstream of the coding sequence within the aTSS. In general, the distribution of 5′ untranslated regions (5′ UTR) is similar to the data published in the Thermococcales (Jäger et al., 2014; Cho et al., 2017), but quite different from predominantly leaderless transcripts in Haloferax volcanii (Babski et al., 2016) and long untranslated leader regions in Methanocaldococcus jannaschii (Smollett et al., 2017; Supplementary Figure 1).
Characterization of Bidirectional Transcription in the Context of aTSS
It is interesting to note the high number of aTSS which is in agreement with data from other archaeal organisms (Babski et al., 2016). The function of these transcripts is not known, but we assume that at least some of them are most likely nonsense transcripts, which arise from symmetric promoter sequences. A closer look to the consensus sequence (Figure 4C) revealed an almost symmetric TATA box with TTTAAA as the most conserved structure and 3 bp upstream AAA and three bp downstream TTT, although less conserved. TBP is known to bind symmetrically to the TATA box and the orientation of transcription is determined by the binding of TFB (Cox et al., 1997; Bell et al., 1999; Werner and Grohmann, 2011). Therefore, it is possible that some of these antisense transcripts are initiated by opposed TBP binding, which in turn results in two transcripts on opposite strands. An inspection of individual pTSSs using the IGV browser (Robinson et al., 2011) reveals strong signal counts on the antisense strand in short distance upstream of promoter elements for many transcripts. Most striking examples with head-to-tail orientation of the neighboring genes are shown in Figure 5.
FIGURE 5
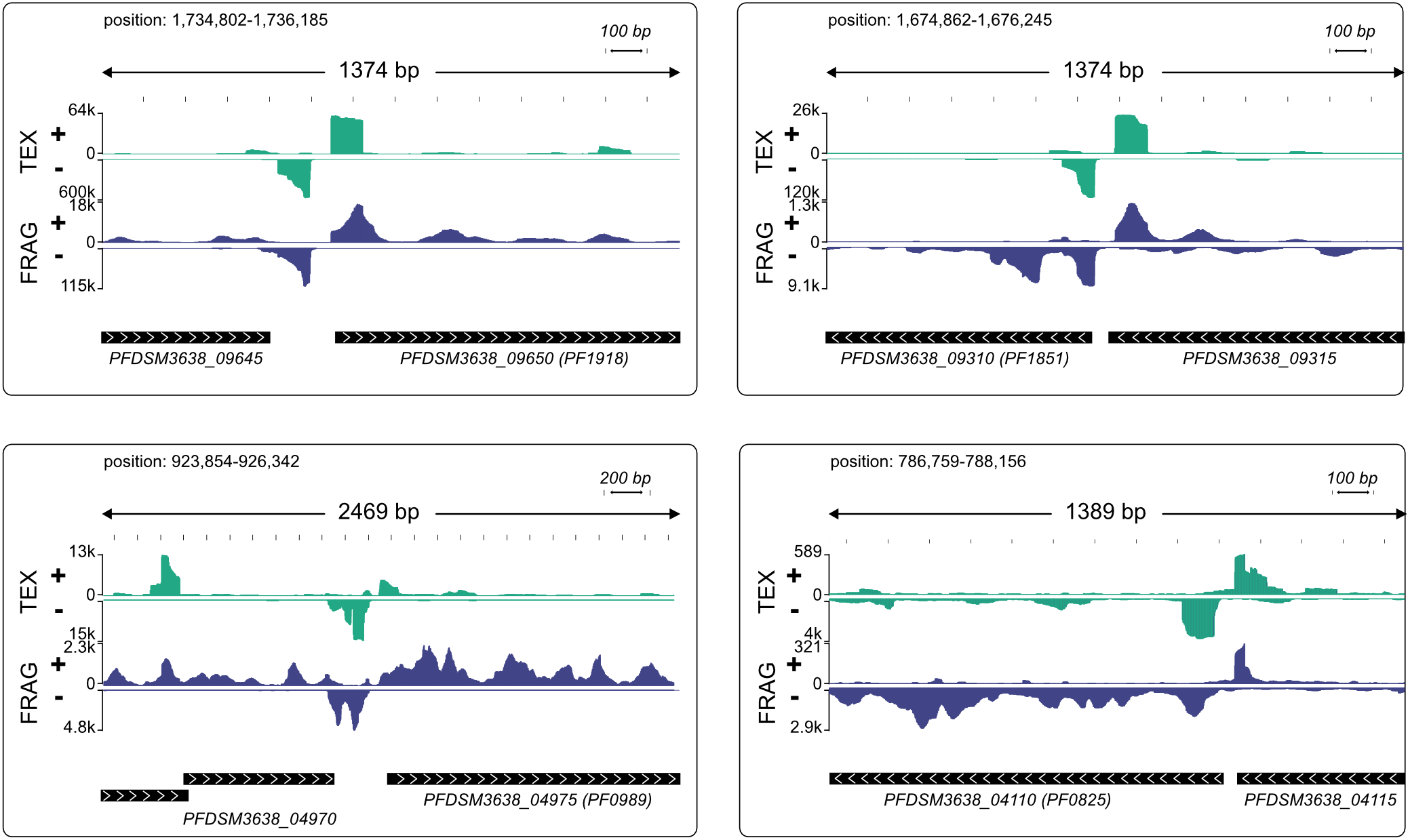
Modified IGV snapshots from head-to-tail genes with an antisense TSS in close proximity to a pTSS. The exact positions of each fragment within the genome are indicated. The genes are annotated with the new genome locus tags and with old locus tags in brackets. RNA coverage on both strands is autoscaled to fit the window, with sequencing depth indicated on the y-axis, TEX-treated RNA in green and fragmented RNA in blue.
To investigate the occurrence of these bidirectional transcription reads in more detail we plotted the read density for all genes with a detected pTSS from TSSpredator on sense and antisense strand (n = 834). To exclude bias from genes in head-to-head orientation with actual pTSS on negative strand we split our dataset according to gene orientation (head-to-head: 388, head-to-tail: 442). This analysis showed a strong antisense peak for head-to-tail orientated genes starting about 50 bp upstream of pTSS (Figure 6A). More than 10% of the 442 genes (49) with this orientation had very strong TEX signals in the region up to 100 bp upstream of the pTSS (more than 40% of the reads from −400 to 400 region). In the case of head-to-head orientated genes, we also observed an enrichment of TEX signals on the antisense strand 50 bp upstream of a pTSS (Figure 6B). The expanded signal distribution is most likely caused by overlapping signals of aTTS and corresponding pTTS of upstream located genes. A more detailed analysis of the intergenic region for head-to-head genes confirmed the short intergene distances (median 117 bp) which impedes any possibility to discriminate between both signals (Figure 6C). The strong accumulation of these antisense transcripts in a distance of approximately 50 bp upstream of the pTSS most likely indicates a shared TATA element for the primary and the corresponding antisense transcript. In this case, we expect an additional BRE element downstream of the TATA element for TFB recruitment in antisense direction. To circumvent the problem with head-to-head orientated genes we only analyzed promoter sequences with head-to-tail orientation. In fact, about 78% of these promoter regions (38) exhibited a bidirectional BRE-TATA-BRE motif located in the middle between a pTSS (position 0) and an aTSS (position −50). The BRE on the antisense strand is less prominent than on the sense strand but can still be detected (Figure 6D).
FIGURE 6
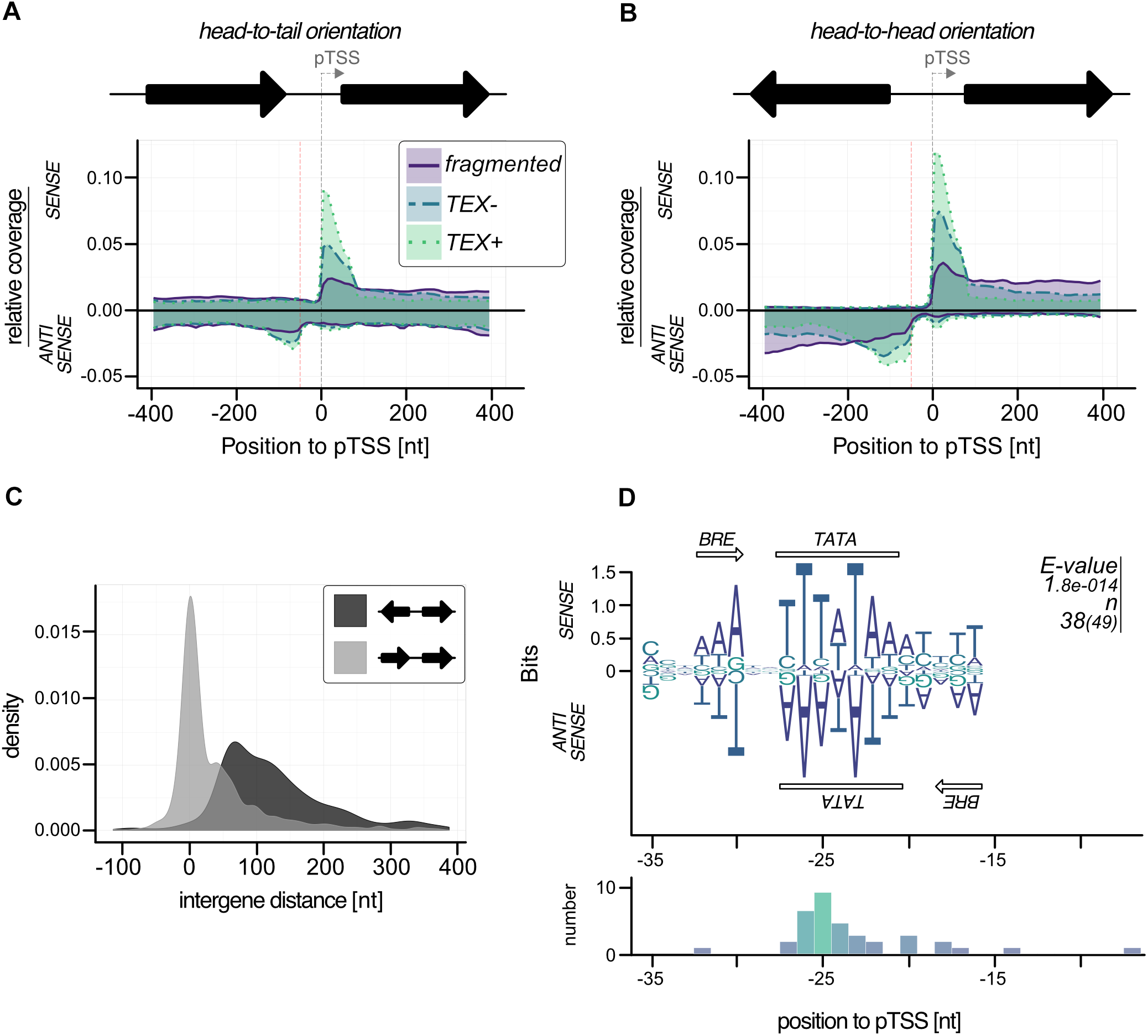
Bidirectional transcription in P. furiosus. Genes were sorted in (A) head-to-tail or (B) head-to-head oriented groups and relative coverage plots (compare Figure 4E) were calculated for both sense and antisense strand. The distance of 50 b upstream of the pTSS is indicated by a red dotted line. (C) The intergene distance for both groups is based on gene annotation, head-to-tail orientation are shown in light gray, head-to-head orientation in dark gray. (D) MEME motif search for promoter regions with strong antisense signals resulted in a bidirectional BRE-TATA-BRE motif, that is shown on both strands. The e-values and the number of sequences contributing to the motif are shown on the right of the panel. This distance to pTSS is shown in the lower panel.
To get additional evidence for antisense transcription induced by bidirectional promoter sequences, we analyzed the promoter of PF1918 (PFDSM3638_09650, Figure 5 upper left) in more detail using in vitro transcription. A detailed sequence analysis of the upstream region confirmed the presence of a bidirectional promoter (Figure 7A) and the gene upstream of PF1918 is located in head-to-tail orientation (Figure 7B). To distinguish between sense and antisense transcripts in vitro, the RNA was analyzed by primer extension experiments (Figure 7C). A comparison of the 40-nucleotide sense and the 39-nucleotide antisense signal revealed that this bidirectional promoter produces the main transcript as well as the antisense transcript in almost similar amount. The distance between both TSS is 49 bp, which clearly indicates that both transcripts originate from the same TATA element. This is the first in vitro evidence in archaea that some of the numerous antisense transcripts can be induced by bidirectional transcription. It is possible that the AT-rich promoter sequence in combination with the low GC content of P. furiosus increases the frequency of bidirectional promoters, but we assume that the symmetrical binding of the archaeal TBP to the TATA element (Cox et al., 1997) is especially prone to antisense transcription from bidirectional promoter sequences. This is in line with recent findings in eukaryotes indicating the promoter regions are intrinsically bidirectional and are shaped by evolution to bias transcription toward coding versus non-coding RNAs (Xu et al., 2009; Jin et al., 2017). Furthermore, divergent transcription is a mechanistic feature that does not imply a function for these transcripts. Transcriptional noise as a main result of antisense transcription seems to be also common in bacteria in particular in combination with a high AT content (Lloréns-Rico et al., 2016).
FIGURE 7
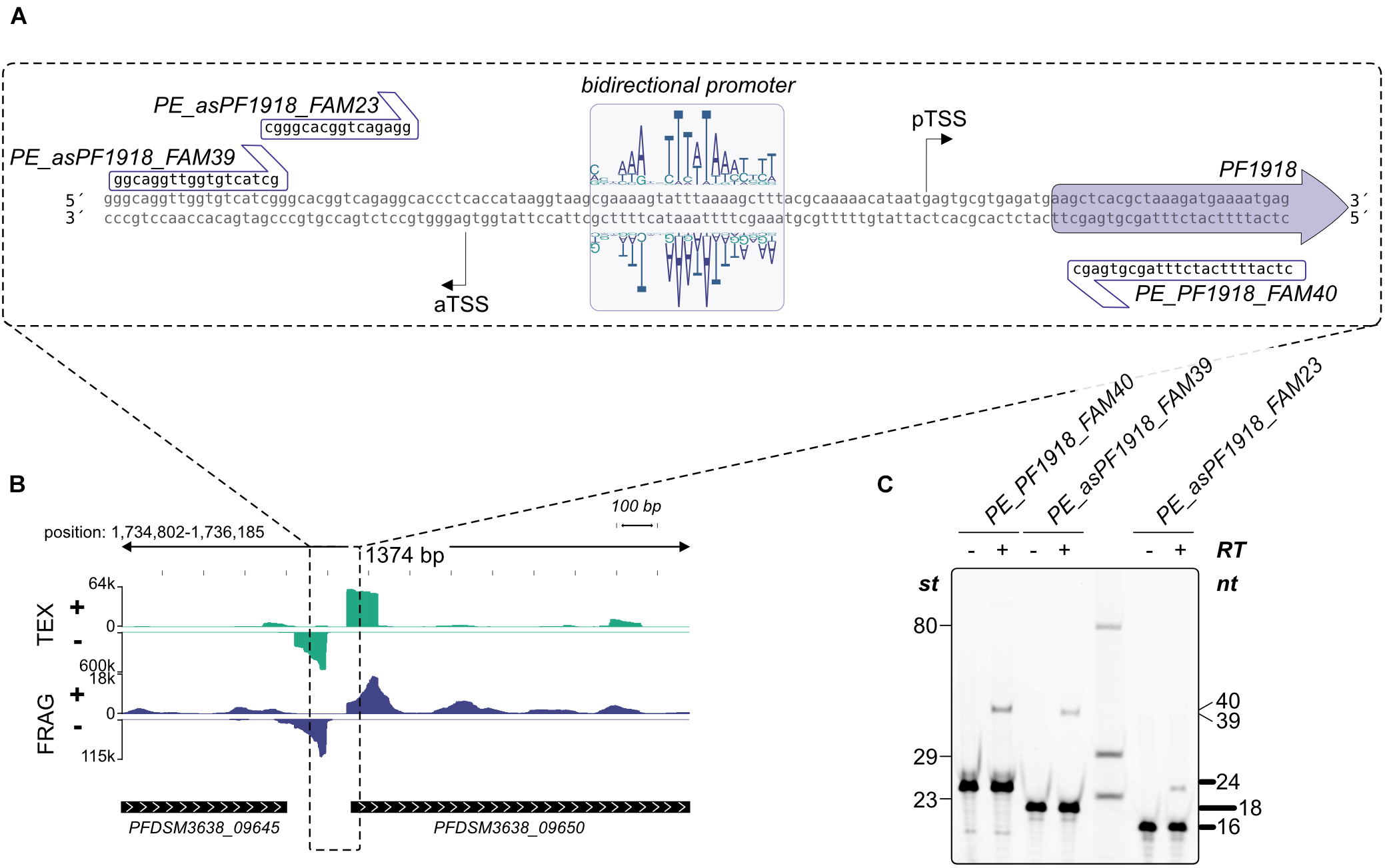
Validation of bidirectional transcripts (A) The promoter region of PFDSM3638_09650 (old locus tag PF1918) with the bidirectional motif and the corresponding sequences used for primer extension are specified. (B) Zoom out of the promoter region. RNA coverage on both strands is indicated. (C) Primer extension analysis of in vitro synthesized RNA. The presence (+) or absence (−) of Reverse Transcriptase and the used primers are shown on top of each lane. Lengths of marker fragments (st) are shown on the left, primer signals in bold on the right and expected length of primer extension signals also on the right side.
IS Elements and a Potential Regulation by Antisense Transcripts
As already mentioned in the introduction, P. furiosus seems to prone to genomic rearrangements most likely due to an increased number of IS elements, which has been known for a long time as driving force for genomic reorganizations (Sapienza et al., 1982; DiRuggiero et al., 2000; Brügger et al., 2002; Zivanovic et al., 2002). Using ISEScan 1.6 we identified 24 IS elements in the new assembled genome (Figure 8A), whereas the number of IS elements in the “old” strain is 30 and 40 in the COM1 strain. In detail, the number of the IS6 family type of transposable elements is reduced (Table 1). IS6-mediated gene rearrangements have been already described in the early 2002’s as the first genome sequences of different Pyrococcus species became available (Zivanovic et al., 2002). Therefore, we assume that the decreased number of IS elements in the new sequenced DSM 3638 strain is a decisive point that ensures genome integrity.
FIGURE 8
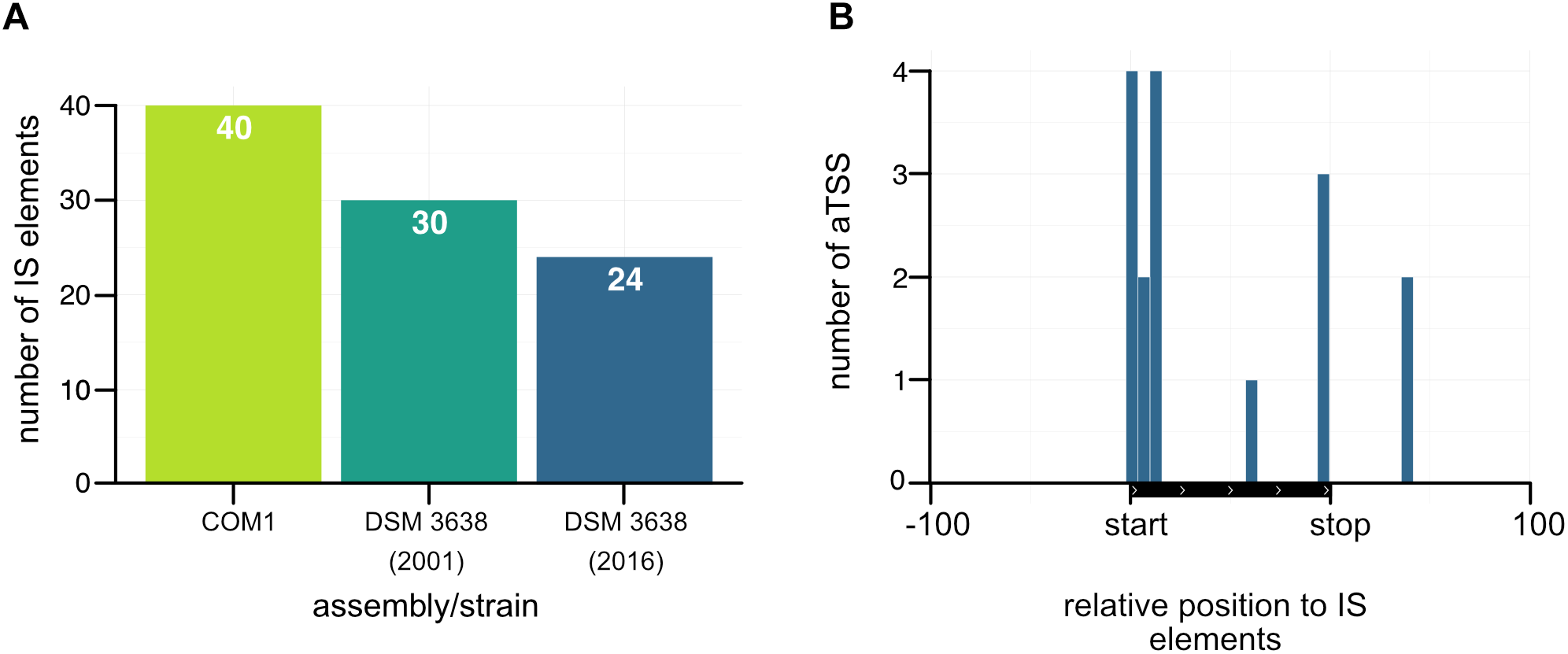
Accumulation of antisense transcripts in the neighborhood of IS elements. (A) Comparison of the number of IS elements in the COM1 strain (Bridger et al., 2012) together with the two sequences of the P. furiosus type strain. (B) Position of aTSSs relative to the transposon coding sequence of IS elements.
Furthermore, there is increasing evidence that the activity of IS elements could be suppressed by corresponding antisense RNAs in particular under stress conditions (Ellis and Haniford, 2016). As our RNA library pool also contained stress conditions like heat or cold shock and the accumulation of antisense transcripts associated with transposase-encoding genes has also been observed in other archaea (Tang et al., 2002, 2005; Jäger et al., 2009; Straub et al., 2009; Wurtzel et al., 2010; Yoon et al., 2011; Bernick et al., 2012; Heyer et al., 2012; Su et al., 2013; Toffano-Nioche et al., 2013) we mapped aTSS to the relative position of IS elements (Figure 8B). This analysis revealed an increased number of antisense transcripts, which in most cases overlap with the start of the open reading frame of the transposase. Therefore, it seems feasible that antisense transcripts of IS elements might also play a role in gene silencing in P. furiosus to avoid genome instability.
Conclusion
This study provides an updated genome assembly of P. furiosus using a combination of long-read PacBio sequencing and short-read Illumina sequencing. The new genome is 18,342 bp smaller than the NCBI reference from 2001 mainly due to a recently described deletion (Reichelt et al., 2016), but the overall structure is still almost identical to the published sequence of P. furiosus. The stability of the P. furiosus genome was confirmed by re-sequencing of a “lab culture” 2 years after initial sequencing of the strain. Our data demonstrate that it is possible to ensure genome stability in “lab cultures” by avoiding strong selection pressure, even with a strain which was assumed highly susceptible for genome rearrangements (DiRuggiero et al., 2000; Brügger et al., 2002; Zivanovic et al., 2002).
The updated DNA sequence in combination with RNA sequencing enabled us to improve genome annotation using the recently developed pipeline ANNOgesic. We included additional features, such as operon structures, TSSs and terminator sequences as well as non-coding or circRNAs to provide a comprehensive dataset of the genome features of the P. furiosus type strain DSM 3638 for future research (Supplementary Figure 2 and Supplementary Table 5).
Dedication
This paper is dedicated to the memory of our colleague and friend, Prof. Dr. Reinhard Wirth, who recently passed away. Reinhard has identified the LS and the BBR strain of Pyrococcus furiosus which exhibit different amounts of flagella and unusual cell morphology.
Statements
Data availability statement
Raw sequencing data has been submitted to the NCBI Sequence Read Archive (BioProject: PRJNA382684, BioSample: SAMN06711904). Code, raw figures, and data used during the bioinformatical analysis were uploaded to https://github.com/felixgrunberger/pyrococcus_reannotation.
Author contributions
RoR prepared the RNA from Pyrococcus. BB, CS, and JO performed the PacBio and Illumina sequencing and FG the nanopore sequencing. The bioinformatical analysis was carried out by FG and RoR. FG, RoR, DG, and WH wrote the manuscript. WH, RR, and DG coordinated and supervised the work. All authors approved the final version of the manuscript.
Funding
This work was supported by the Institute of Microbiology and Archaea Center of the University of Regensburg, the SFB960, and by the German Research Foundation (DFG) with the funding program Open Access Publishing. The CU SysMed was supported by the IZKF at the University of Würzburg (project Z-6).
Acknowledgments
The authors thank Renate Richau from the University of Regensburg and Simone Severitt and Nicole Heyer from the DSMZ for excellent technical assistance. The authors thank Esther Schüler (DSMZ, Department Microorganisms) for providing biomass and Annett Bellack for providing genomic DNA from Pyrococcus furiosus.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01603/full#supplementary-material
References
1
BabskiJ.HaasK. A.Näther-SchindlerD.PfeifferF.FörstnerK. U.HammelmannM.et al (2016). Genome-wide identification of transcriptional start sites in the haloarchaeon Haloferax volcanii based on differential RNA-Seq (dRNA-Seq).BMC Genomics17:629. 10.1186/s12864-016-2920-y
2
BaileyT. L.BodenM.BuskeF. A.FrithM.GrantC. E.ClementiL.et al (2009). MEME suite: tools for motif discovery and searching.Nucleic Acids Res.37W202–W208. 10.1093/nar/gkp335
3
BellS. D.KosaP. L.SiglerP. B.JacksonS. P. (1999). Orientation of the transcription preinitiation complex in Archaea.Proc. Natl. Acad. Sci. U.S.A.9613662–13667. 10.1073/PNAS.96.24.13662
4
BernickD. L.DennisP. P.LuiL. M.LoweT. M. (2012). Diversity of antisense and other non-coding RNAs in archaea revealed by comparative small RNA sequencing in four Pyrobaculum species.Front. Microbiol.3:231. 10.3389/fmicb.2012.00231
5
BolgerA. M.LohseM.UsadelB. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data.Bioinformatics302114–2120. 10.1093/bioinformatics/btu170
6
BridgerS. L.Andrew LancasterW.PooleF. L.SchutG. J.AdamsM. W. W. (2012). Genome sequencing of a genetically tractable Pyrococcus furiosus strain reveals a highly dynamic genome.J. Bacteriol.1944097–4106. 10.1128/JB.00439-12
7
BrüggerK.RedderP.SheQ.ConfalonieriF.ZivanovicY.GarrettR. A. (2002). Mobile elements in archaeal genomes.FEMS Microbiol. Lett.206131–141. 10.1016/S0378-1097(01)00504-3
8
ChoS.KimM.-S.JeongY.LeeB.-R.LeeJ.-H.KangS. G.et al (2017). Genome-wide primary transcriptome analysis of H2-producing archaeon Thermococcus onnurineus NA1.Sci. Rep.7:43044. 10.1038/srep43044
9
CoxJ. M.HaywardM. M.SanchezJ. F.GegnasL. D.van der ZeeS.DennisJ. H.et al (1997). Bidirectional binding of the TATA box binding protein to the TATA box.Proc. Natl. Acad. Sci. U.S.A.9413475–13480. 10.1073/pnas.94.25.13475
10
DaumB.VonckJ.BellackA.ChaudhuryP.ReicheltR.AlbersS.-V.et al (2017). Structure and in situ organisation of the Pyrococcus furiosus archaellum machinery.eLife6:e27470. 10.7554/elife.27470
11
DiRuggieroJ.DunnD.MaederD. L.Holley-ShanksR.ChatardJ.HorlacherR.et al (2000). Evidence of recent lateral gene transfer among hyperthermophilic archaea.Mol. Microbiol.38684–693.
12
DobinA.DavisC. A.SchlesingerF.DrenkowJ.ZaleskiC.JhaS.et al (2013). STAR: ultrafast universal RNA-seq aligner.Bioinformatics2915–21. 10.1093/bioinformatics/bts635
13
DugarG.HerbigA.FörstnerK. U.HeidrichN.ReinhardtR.NieseltK.et al (2013). High-resolution transcriptome maps reveal strain-specific regulatory features of multiple Campylobacter jejuni isolates.PLoS Genet.9:e1003495. 10.1371/journal.pgen.1003495
14
EllisM. J.HanifordD. B. (2016). Riboregulation of bacterial and archaeal transposition.Wiley Interdiscip. Rev. RNA.7382–398. 10.1002/wrna.1341
15
Escobar-PáramoP.GhoshS.DiRuggieroJ. (2005). Evidence for genetic drift in the diversification of a geographically isolated population of the hyperthermophilic archaeon Pyrococcus.Mol. Biol. Evol.222297–2303. 10.1093/molbev/msi227
16
FialaG.StetterK. O. (1986). Pyrococcus furiosus sp. nov. represents a novel genus of marine heterotrophic archaebacteria growing optimally at 100°C.Arch. Microbiol.14556–61.
17
GrissaI.VergnaudG.PourcelC. (2007). CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats.Nucleic Acids Res.35W52–W57. 10.1093/nar/gkm360
18
GuyL.KultimaJ. R.AnderssonS. G. E.QuackenbushJ. (2011). GenoPlotR: comparative gene and genome visualization in R.Bioinformatics262334–2335. 10.1093/bioinformatics/btq413
19
Hamilton-BrehmS. D.SchutG. J.AdamsM. W. W. (2005). Metabolic and evolutionary relationships among Pyrococcus species: genetic exchange within a hydrothermal vent environment.J. Bacteriol.1877492–7499. 10.1128/JB.187.21.7492-7499.2005
20
HausnerW.FreyG.ThommM. (1991). Control regions of an archaeal gene. A TATA box and an initiator element promote cell-free transcription of the tRNAValgene of Methanococcus vannielii.J. Mol. Biol.222495–508. 10.1016/0022-2836(91)90492-O
21
HeyerR.DörrM.Jellen-RitterA.SpäthB.BabskiJ.JaschinskiK.et al (2012). High throughput sequencing reveals a plethora of small RNAs including tRNA derived fragments in Haloferax volcanii.RNA Biol.91011–1018. 10.4161/rna.20826
22
JägerD.FörstnerK. U.SharmaC. M.SantangeloT. J.ReeveJ. N. (2014). Primary transcriptome map of the hyperthermophilic archaeon Thermococcus kodakarensis.BMC Genomics15:684. 10.1186/1471-2164-15-684
23
JägerD.SharmaC. M.ThomsenJ.EhlersC.VogelJ.SchmitzR. A. (2009). Deep sequencing analysis of the Methanosarcina mazei Go1 transcriptome in response to nitrogen availability.Proc. Natl. Acad. Sci. U.S.A.10621878–21882. 10.1073/pnas.0909051106
24
JinY.EserU.StruhlK.ChurchmanL. S. (2017). The ground state and evolution of promoter region directionality.Cell170889.e–898.e. 10.1016/j.cell.2017.07.006
25
KanoksilapathamW.GonzálezJ. M.MaederD. L.DiruggieroJ.RobbF. T. (2004). A proposal to rename the hyperthermophile Pyrococcus woesei as Pyrococcus furiosus subsp. woesei.Archaea1277–283. 10.1155/2004/513563
26
KengenS. W. M.De BokF. A. M.Van LooN. D.DijkemaC.StamsA. J. M.De VosW. M. (1994). Evidence for the operation of a novel Embden-Meyerhof pathway that involves ADP-dependent kinases during sugar fermentation by Pyrococcus furiosus.J. Biol. Chem.26917537–17541.
27
KoboldtD. C.ZhangQ.LarsonD. E.ShenD.McLellanM. D.LinL.et al (2012). VarScan 2: somatic mutation and copy number alteration discovery in cancer by exome sequencing.Genome Res.22568–576. 10.1101/gr.129684.111
28
KorenS.WalenzB. P.BerlinK.MillerJ. R.BergmanN. H.PhillippyA. M. (2016). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation.Genome Res.27722–736. 10.1101/gr.215087.116
29
KurtzS.PhillippyA.DelcherA. L.SmootM.ShumwayM.AntonescuC.et al (2004). Versatile and open software for comparing large genomes.Genome Biol.5:R12. 10.1186/gb-2004-5-2-r12
30
LewisD. L.NoteyJ. S.ChandrayanS. K.LoderA. J.LipscombG. L.AdamsM. W. W.et al (2015). A mutant (‘lab strain’) of the hyperthermophilic archaeon Pyrococcus furiosus, lacking flagella, has unusual growth physiology.Extremophiles19269–281. 10.1007/s00792-014-0712-3
31
LiH.DurbinR. (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform.Bioinformatics26589–595. 10.1093/bioinformatics/btp698
32
Lloréns-RicoV.CanoJ.KammingaT.GilR.LatorreA.ChenW.-H.et al (2016). Bacterial antisense RNAs are mainly the product of transcriptional noise.Sci. Adv.2:e1501363. 10.1126/sciadv.1501363
33
LomanN. J.QuickJ.SimpsonJ. T. (2015). A complete bacterial genome assembled de novo using only nanopore sequencing data.Nat. Methods12733–735. 10.1038/nmeth.3444
34
LowyE. (2017). CoverageView: Coverage Visualization Package for R. R Package Version 1.16.0.
35
Näther-SchindlerD. J.SchopfS.BellackA.RachelR.WirthR. (2014). Pyrococcus furiosus flagella: biochemical and transcriptional analyses identify the newly detected flaB0 gene to encode the major flagellin.Front. Microbiol.5:695. 10.3389/fmicb.2014.00695
36
OttoC.StadlerP. F.HoffmannS. (2014). Lacking alignments? The next-generation sequencing mapper segemehl revisited.Bioinformatics301837–1843. 10.1093/bioinformatics/btu146
37
R Development Core Team. (2011). R: A Language and Environment for Statistical Computing.Vienna: R Foundation for Statistical Computing.
38
ReicheltR.GindnerA.ThommM.HausnerW. (2016). Genome-wide binding analysis of the transcriptional regulator TrmBL1 in Pyrococcus furiosus.BMC Genomics17:40. 10.1186/s12864-015-2360-0
39
RhoadsA.AuK. F. (2015). PacBio sequencing and its applications.Genomics Proteomics Bioinforma13278–289. 10.1016/j.gpb.2015.08.002
40
RobbF. T.MaederD. L.BrownJ. R.DiRuggieroJ.StumpM. D.YehR. K.et al (2001). Genomic sequence of hyperthermophile, Pyrococcus furiosus: implications for physiology and enzymology.Methods Enzymol.330134–157. 10.1016/s0076-6879(01)30372-5
41
RobinsonJ. T.ThorvaldsdóttirH.WincklerW.GuttmanM.LanderE. S.GetzG.et al (2011). Integrative genomics viewer.Nat. Biotechnol.2924–26. 10.1038/nbt.1754
42
SapienzaC.RoseM. R.DoolittleW. F. (1982). High-frequency genomic rearrangements involving archaebacterial repeat sequence elements.Nature.299182–185. 10.1038/299182a0
43
SchäferT.SchönheitP. (1992). Maltose fermentation to acetate, CO2and H2in the anaerobic hyperthermophilic archaeon Pyrococcus furiosus: evidence for the operation of a novel sugar fermentation pathway.Arch. Microbiol.158188–202. 10.1007/BF00290815
44
SharmaC. M.HoffmannS.DarfeuilleF.ReignierJ.FindeißS.SittkaA.et al (2010). The primary transcriptome of the major human pathogen Helicobacter pylori.Nature.464250–255. 10.1038/nature08756
45
SiguierP.GourbeyreE.ChandlerM. (2014). Bacterial insertion sequences: their genomic impact and diversity.FEMS Microbiol. Rev.38865–891. 10.1111/1574-6976.12067
46
SimpsonJ. T.WorkmanR. E.ZuzarteP. C.DavidM.DursiL. J.TimpW. (2017). Detecting DNA cytosine methylation using nanopore sequencing.Nat. Methods14407–410. 10.1038/nmeth.4184
47
SmollettK.BlombachF.ReicheltR.ThommM.WernerF. (2017). A global analysis of transcription reveals two modes of Spt4/5 recruitment to archaeal RNA polymerase.Nat. Microbiol.2:17021. 10.1038/nmicrobiol.2017.21
48
SoppaJ. (1999). Transcription initiation in Archaea: facts, factors and future aspects.Mol. Microbiol.311295–1305. 10.1046/j.1365-2958.1999.01273.x
49
StraubJ.BrenneisM.Jellen-RitterA.HeyerR.SoppaJ.MarchfelderA. (2009). Small RNAs in Haloarchaea: identification, differential expression and biological function.RNA Biol.6281–292. 10.4161/rna.6.3.8357
50
SuA. A. H.TrippV.RandauL. (2013). RNA-Seq analyses reveal the order of tRNA processing events and the maturation of C/D box and CRISPR RNAs in the hyperthermophile Methanopyrus kandleri.Nucleic Acids Res.416250–6258. 10.1093/nar/gkt317
51
TaboadaB.EstradaK.CiriaR.MerinoE. (2018). Operon-mapper: a web server for precise operon identification in bacterial and archaeal genomes.Bioinformatics.44118–4120. 10.1093/bioinformatics/bty496
52
TangT.-H.BachellerieJ.-P.RozhdestvenskyT.BortolinM.-L.HuberH.DrungowskiM.et al (2002). Identification of 86 candidates for small non-messenger RNAs from the archaeon Archaeoglobus fulgidus.Proc. Natl. Acad. Sci. U.S.A.997536–7541. 10.1073/pnas.112047299
53
TangT. H.PolacekN.ZywickiM.HuberH.BruggerK.GarrettR.et al (2005). Identification of novel non-coding RNAs as potential antisense regulators in the archaeon Sulfolobus solfataricus.Mol. Microbiol.55469–481. 10.1111/j.1365-2958.2004.04428.x
54
TatusovaT.DiCuccioM.BadretdinA.ChetverninV.NawrockiE. P.ZaslavskyL.et al (2016). NCBI prokaryotic genome annotation pipeline.Nucleic Acids Res.446614–6624. 10.1093/nar/gkw569
55
Toffano-NiocheC.OttA.CrozatE.NguyenA. N.ZytnickiM.LeclercF.et al (2013). RNA at 92 °C: the non-coding transcriptome of the hyperthermophilic archaeon Pyrococcus abyssi.RNA Biol.101211–1220. 10.4161/rna.25567
56
Van De WerkenH. J. G.VerheesC. H.AkerboomJ.De VosW. M.Van Der OostJ. (2006). Identification of a glycolytic regulon in the archaea Pyrococcus and Thermococcus.FEMS Microbiol. Lett.26069–76. 10.1111/j.1574-6968.2006.00292.x
57
VermaM.KulshresthaS.PuriA. (2017). “Genome Sequencing,” in Bioinformatics: Volume I: Data, Sequence Analysis, and Evolution, ed.KeithJ. M. (New York, NY: Springer), 3–33. 10.1007/978-1-4939-6622-6_1
58
WagihO. (2017). Ggseqlogo: a versatile R package for drawing sequence logos.Bioinformatics333645–3647. 10.1093/bioinformatics/btx469
59
WernerF.GrohmannD. (2011). Evolution of multisubunit RNA polymerases in the three domains of life.Nat. Rev. Microbiol.985–98. 10.1038/nrmicro2507
60
WhiteJ. R.Escobar-ParamoP.MongodinE. F.NelsonK. E.DiRuggieroJ. (2008). Extensive genome rearrangements and multiple horizontal gene transfers in a population of Pyrococcus isolates from Vulcano Island, Italy.Appl. Environ. Microbiol.746447–6451. 10.1128/AEM.01024-08
61
WickhamH. (2016). ggplot2: Elegant Graphics for Data Analysis.New York, NY: Springer-Verlag, 10.1007/978-0-387-98141-3
62
WurtzelO.SapraR.ChenF.ZhuY.SimmonsB. A.SorekR. (2010). A single-base resolution map of an archaeal transcriptome.Genome Res.20133–141. 10.1101/gr.100396.109
63
XieZ.TangH. (2017). ISEScan: automated identification of insertion sequence elements in prokaryotic genomes.Bioinformatics333340–3347. 10.1093/bioinformatics/btx433
64
XuZ.WeiW.GagneurJ.PerocchiF.Clauder-MünsterS.CamblongJ.et al (2009). Bidirectional promoters generate pervasive transcription in yeast.Nature4571033–1037. 10.1038/nature07728
65
YoonS. H.ReissD. J.BareJ. C.TenenbaumD.PanM.SlagelJ.et al (2011). Parallel evolution of transcriptome architecture during genome reorganization.Genome Res.211892–1904. 10.1101/gr.122218.111
66
YuS.-H.VogelJ.FörstnerK. U. (2018). ANNOgesic: a Swiss army knife for the RNA-Seq based annotation of bacterial/archaeal genomes.Gigascience7:giy096. 10.1101/143081
67
ZilligW.HolzI.KlenkH. P.TrentJ.WunderlS.JanekovicD.et al (1987). Pyrococcus woesei, sp. nov., an ultra-thermophilic marine archaebacterium, representing a novel order, Thermococcales.Syst. Appl. Microbiol.962–70. 10.1016/S0723-2020(87)80057-7
68
ZivanovicY.LopezP.PhilippeH.ForterreP. (2002). Pyrococcus genome comparison evidences chromosome shuffling-driven evolution.Nucleic Acids Res.301902–1910. 10.1093/nar/30.9.1902
Summary
Keywords
archaea, Pyrococcus, RNA sequencing, Nanopore sequencing, PacBio sequencing, bidirectional transcription, antisense transcription
Citation
Grünberger F, Reichelt R, Bunk B, Spröer C, Overmann J, Rachel R, Grohmann D and Hausner W (2019) Next Generation DNA-Seq and Differential RNA-Seq Allow Re-annotation of the Pyrococcus furiosus DSM 3638 Genome and Provide Insights Into Archaeal Antisense Transcription. Front. Microbiol. 10:1603. doi: 10.3389/fmicb.2019.01603
Received
22 March 2019
Accepted
26 June 2019
Published
12 July 2019
Volume
10 - 2019
Edited by
Philippe M. Oger, UMR5240 Microbiologie, Adaptation et Pathogenie (MAP), France
Reviewed by
Michael W. Adams, University of Georgia, United States; Lennart Randau, Max Planck Institute for Terrestrial Microbiology, Germany; Gerrit Jan Schut, University of Georgia, United States
Updates
Copyright
© 2019 Grünberger, Reichelt, Bunk, Spröer, Overmann, Rachel, Grohmann and Hausner.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Winfried Hausner, Winfried.Hausner@ur.de
This article was submitted to Extreme Microbiology, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.