 Suresh Panthee
Suresh Panthee Atmika Paudel
Atmika Paudel Jochen Blom
Jochen Blom Hiroshi Hamamoto
Hiroshi Hamamoto Kazuhisa Sekimizu
Kazuhisa Sekimizu- 1Institute of Medical Mycology, Teikyo University, Hachioji, Japan
- 2Bioinformatics and Systems Biology, Justus-Liebig-University Giessen, Giessen, Germany
- 3Genome Pharmaceuticals Institute, Bunkyōku, Japan
Weissella genus from Leuconostocaceae family forms a group of Gram-positive lactic acid bacteria (LAB) that mostly reside in fermented foods and some have been isolated from the environment and vertebrates including humans. Currently there are 23 recognized species, 16 complete and 37 draft genome assemblies for this genus. Weissella hellenica has been found in various sources and is characterized by their probiotic and bacteriocinogenic properties. Despite its widespread importance, little attention has been paid to genomic characterization of this species with the availability of draft assembly of two species in the public database so far. In this manuscript, we identified W. hellenica 0916-4-2 from fermented kimchi and completed its genome sequence. Comparative genomic analysis identified 88 core genes that had interspecies mean amino acid identity of more than 65%. Whole genome phylogenetic analysis showed that three W. hellenica strains clustered together and the strain 0916-4-2 was close to strain WiKim14. In silico analysis for the secondary metabolites biosynthetic gene cluster showed that Weissella are far less producers of secondary metabolites compared to other members of Leuconostocaceae. The availability of the complete genome of W. hellenica 0916-4-2 will facilitate further comparative genomic analysis of Weissella species, including studies of its biotechnological potential and improving the nutritional value of various food products.
Introduction
The non-spore forming lactic acid bacteria (LAB) of Weissella genus within the Leuconostocaceae family contains 23 validly described species. In nature, Weissella spp. have been found in a wide range of habitats (Fusco et al., 2015) such as traditional fermented foods, milk, vegetables, feces, environment and vertebrates including humans. In traditional Korean fermented vegetable food kimchi, Weissella form the dominant genus at the late stage of fermentation, partly due to their high acid-tolerant property (Kim et al., 2016). Recently, a higher level of interest is being paid for the probiotic, biotechnological and bacteriocinogenic potential of Weissella, although some strains are known to act as opportunistic pathogen (Kamboj et al., 2015). Given that most strains regarded as opportunistic pathogens were isolated from the hosts with underlying risk factor, such as immunocompromised condition (Fusco et al., 2015; Kamboj et al., 2015), distinguishing the probiotic and pathogenic Weissella has always been a challenging task. Interestingly, the contemporary research has also identified Weissella as a producer of botulinum-like toxin (Mansfield et al., 2015; Zornetta et al., 2016). Despite the widespread importance, little attention has been paid to genomic characterization of this genus. This is further evident from the number of genome assemblies available in public database. So far, only 16 species of this genus have been sequenced and complete genome sequence is available for seven species only. kimchi, Weissella form the dominant genus at the late stage of fermentation, partly due to their high acid-tolerant property (Kim et al., 2016). Recently, a higher level of interest is being paid for the probiotic, biotechnological and bacteriocinogenic potential of Weissella, although some strains are known to act as opportunistic pathogen (Kamboj et al., 2015). Given that most strains regarded as opportunistic pathogens were isolated from the hosts with underlying risk factor, such as immunocompromised condition (Fusco et al., 2015; Kamboj et al., 2015), distinguishing the probiotic and pathogenic Weissella has always been a challenging task. Interestingly, the contemporary research has also identified Weissella as a producer of botulinum-like toxin (Mansfield et al., 2015; Zornetta et al., 2016). Despite the widespread importance, little attention has been paid to genomic characterization of this genus. This is further evident from the number of genome assemblies available in public database. So far, only 16 species of this genus have been sequenced and complete genome sequence is available for seven species only.
Weissella hellenica 0916-4-2 (highlighted in the figures and tables by bold font) was isolated in our laboratory from Korean fermented pickle kimchi and the strain was identified by 16s rRNA sequencing. Although this species harbors a prominent bacteriocinogenic potential, with the production of various bacteriocins such as 7293A (Woraprayote et al., 2015), Weissellicin L (Leong et al., 2013), Weissellicin D (Chen et al., 2014), Weissellicin M (Masuda et al., 2012), and Weissellicin Y (Masuda et al., 2012), there is a great lack of genomic studies among W. hellenica. In this manuscript, we completed the genome sequence of W. hellenica 0916-4-2 and performed its comparative genomic analysis with publicly available Weissella genomes. We found that W. hellenica 0916-4-2 clustered with W. hellenica Wikim14 based on whole genome phylogeny and harbored two putative genes clusters for the biosynthesis of bacteriocin and non-ribosomal peptide synthetase.
Materials and Methods
Strain and DNA Extraction
The strain used in this study was W. hellenica 0916-4-2 isolated from Korean pickle kimchi using MRS medium. Genomic DNA was isolated as explained (Panthee et al., 2017b).
Whole Genome Sequencing
The library preparation for Oxford Nanopore MinION and Thermo Fisher Ion PGM sequencing was performed as explained previously (Panthee et al., 2017a, c, 2018). Briefly, for ultra-long reads library, 1 μg genomic DNA was end-prepped using the NEBNext End repair/dA-tailing Module (New England Biolabs, Inc., Ipswich, MA, United States) and the DNA was cleaned up using Agencourt AMPure XP (Beckman Coulter Inc., Brea, CA, United States). The DNA was then ligated to the adapter using NEB Blunt/TA Ligase Master Mix (NEB). The library was purified using Agencourt AMPure XP (Beckman Coulter Inc) and reads were obtained using the 48-h protocol and live base-calling after loading the library to a primed FLO-MIN106 R9.4 SpotON Flow Cell. The 400 base-reads was prepared after fragmentation of 100 ng of the DNA using the Ion XpressTM Plus Fragment Library Kit (Thermo Fisher Scientific, Waltham, MA, United States). The libraries were enriched in an Ion TM318 Chip v2 using Ion Chef (Thermo Fisher Scientific), and subsequent sequencing was performed in the Ion PGM System (Thermo Fisher Scientific).
Read Correction and Genome Assembly
Read correction and genome assembly was performed as explained previously (Panthee et al., 2018). We obtained 2M reads (mean length 277 bp) from Ion PGM, and 247K reads from MinION (mean length 7 kb), accounting for approximately 273-fold and 900-fold genome coverage, respectively. The high quality MinION long reads were filtered using Filtlong and self-correction and trimming was performed using canu 1.7 (Koren et al., 2017). The short reads from Ion PGM were corrected using SPADES 3.11 (Bankevich et al., 2012). The hybrid error correction of long reads was then performed using LoRDEC (Salmela and Rivals, 2014). The final genome assembly was performed from the long corrected reads using Flye 2.3.3 assembler (Kolmogorov et al., 2018). Further polishing of the assembly was performed by mapping the short reads to the assembly followed by consensus generation. The assembled genome was annotated using the NCBI Prokaryotic Genome Annotation Pipeline (PGAP) to find 1778 protein coding genes and 101 RNA genes. The genome was further submitted to PathogenFinder (Cosentino et al., 2013) to predict the absence of pathogenic genes. Functional analysis of the protein coding genes was performed in Blast2GO ver 5.1.13 (BioBam Bioinformatics, Valencia, Spain).
Genomic Data of Other Weissella Species and Comparative Genomic Analysis
Genomic data of the additional Weissella species analyzed in this study were obtained from the NCBI. The accession numbers and assembly status are indicated in the Supplementary Table S1. To construct the phylogenetic tree, first the core genes of these genomes were computed, and the alignment of each core gene set was generated using MUSCLE, and the alignments were concatenated to create a single alignment. This alignment was used to generate the phylogenetic tree using neighbor-joining algorithm in EDGAR (Blom et al., 2016). The core- and pan-proteomes were predicted using EDGAR (Blom et al., 2016). The COG analysis of the core proteome was performed using eggNOG-Mapper (Huerta-Cepas et al., 2017). The analysis of secondary metabolite gene clusters was performed as explained (Panthee et al., 2017b) using antiSMASH (Blin et al., 2017).
Weissella Virulence Factors Analysis
For the bioinformatic analysis of virulence factors in Weissella genomes, we downloaded the amino acid sequence of core virulence factors from the virulence factor database (Chen et al., 2005; Liu et al., 2018) and performed a BLAST search against the Weissella proteome with a cut off e-value of e–30 and minimum identity of >70%. To analyze the virulence potential of W. hellenica 0916-4-2 in a mouse model, bacteria was cultivated overnight in MRS at 30°C. The culture was centrifuged and resuspended in phosphate-buffered saline (PBS). Six weeks old female ICR mice (n = 5) were injected with bacteria equivalent to 200 μl of the overnight culture (1.5 × 109 CFU) through intraperitoneal route and survival was observed for 5 days. All mouse experimental protocols were approved by the Animal Use Committee at the Genome Pharmaceuticals Institute.
Results and Discussion
Assembly and Analysis of the W. hellenica 0916-4-2 Genome
The final 0916-4-2 assembly had a genome size of 1.93 Mb with a circular chromosome and two plasmids (Table 1). A total of 4828 Gene Ontology (GO) terms were assigned to 1503 (85%) genes, which included 1703 (35%), 2193 (46%), and 932 (19%) GO annotations for the biological process, molecular function, and cellular component category, respectively (Figures 1A–C). The most abundant GO annotation included oxidation-reduction process, ATP binding, and integral component of the membrane, respectively. InterProScan showed that a total of 3132 families were assigned for 1585 (89%) of the total proteins, where P-loop containing nucleoside triphosphate hydrolase family (IPR027417) was the highest 143 (4.6%) (Figure 1D). Given that some of the Weissella species harbored the genes for drug resistance (Abriouel et al., 2015), none were detected in the genome of the 0916-4-2 strain.

Table 1. General feature(s) of Weissella hellenica 0916-4-2 genome.
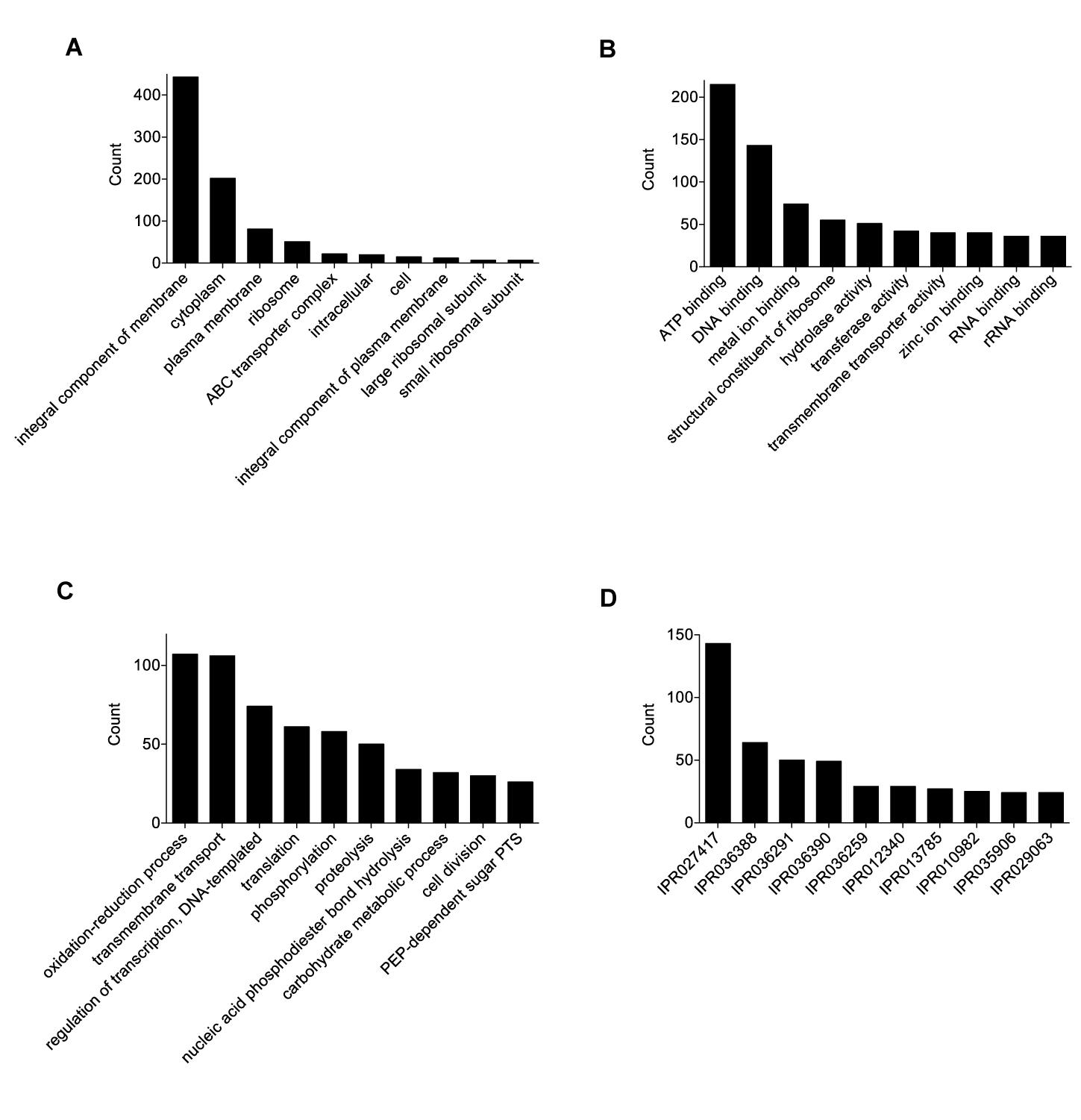
Figure 1. Functional annotation and analysis of W. hellenica 0916-4-2 genome. Gene Ontology (GO) term counts for: (A) cellular component, (B) molecular functions, and (C) biological processes. (D) InterPro scan for gene families.
General Features and Comparative Genomics of Weissella Species
As a first step toward the comparative analysis of the W. hellenica 0916-4-2 genome, we analyzed the gene sets with W. hellenica R-53116 and Wikim14 genomes using pairwise alignment (Figure 2A) and identified the strain-specific and commonly shared genes (Figure 2B). Based on the analysis, we found that 1377 genes were distributed throughout the strains; the 0916-4-2 genome had a larger set of genes shared with Wikim14 genome compared to R-53116 genome and the number of genes unique to 0916-4-2 was 133. Interestingly, nearly half of the unique genes were regarded either hypothetical protein or domain of unknown function harboring proteins. Next, to provide an overview of Weissella genus and perform a comparative genomic analysis, the sequence data of 53 Weissella strains, that included 16 complete genome sequence assemblies, was obtained from NCBI. Of the 23 recognized species as of December 2018, these genomes represent only 16 species and complete genome is available only for seven species: W. ceti, W. cibaria, W. jogaejeotagli, W. koreensis, W. paramesenteroides, and W. soli. The assembly accession numbers and status of assemblies are shown in Supplementary Table S2. To determine the genomic variability between Weissella species, we performed the comparative genomic analysis of all the genome assemblies. The set of commonly shared genes, core genes, in the genus was determined using EDGAR (Blom et al., 2016) and the whole genome phylogenetic tree was constructed. The phylogenetic tree indicated that this analysis was capable of grouping the various strains of a single species into a single cluster (Figure 3). W. hellenica 0916-4-2 clustered with W. hellenica Wikim14, suggesting that these two strains might harbor similar biological properties. Mean AAI analysis of the core proteome showed that interspecies similarity was higher than ∼65% among all the species whereas the value was much higher for any of the W. bombi, W. hellenica, W. paramesenteroides, W. jogaejeotgali, W. thailandensis, W. cibaria, and W. confusa pair. Intraspecies similarity was further higher with more than 97% for all the species (Table 2 and Supplementary Table S2). We found that all the strains of W. ceti, W. soli and W. paramesenteroids and some strains of W. cibaria harbored 100% intraspecies similarity (Supplementary Table S1). Among the interspecies similarity, a high degree of similarity was obtained for W. jogaejeotgali FOL01 and W. thailandensis KCTC3751 with a 99% mean AAI (Table 2).
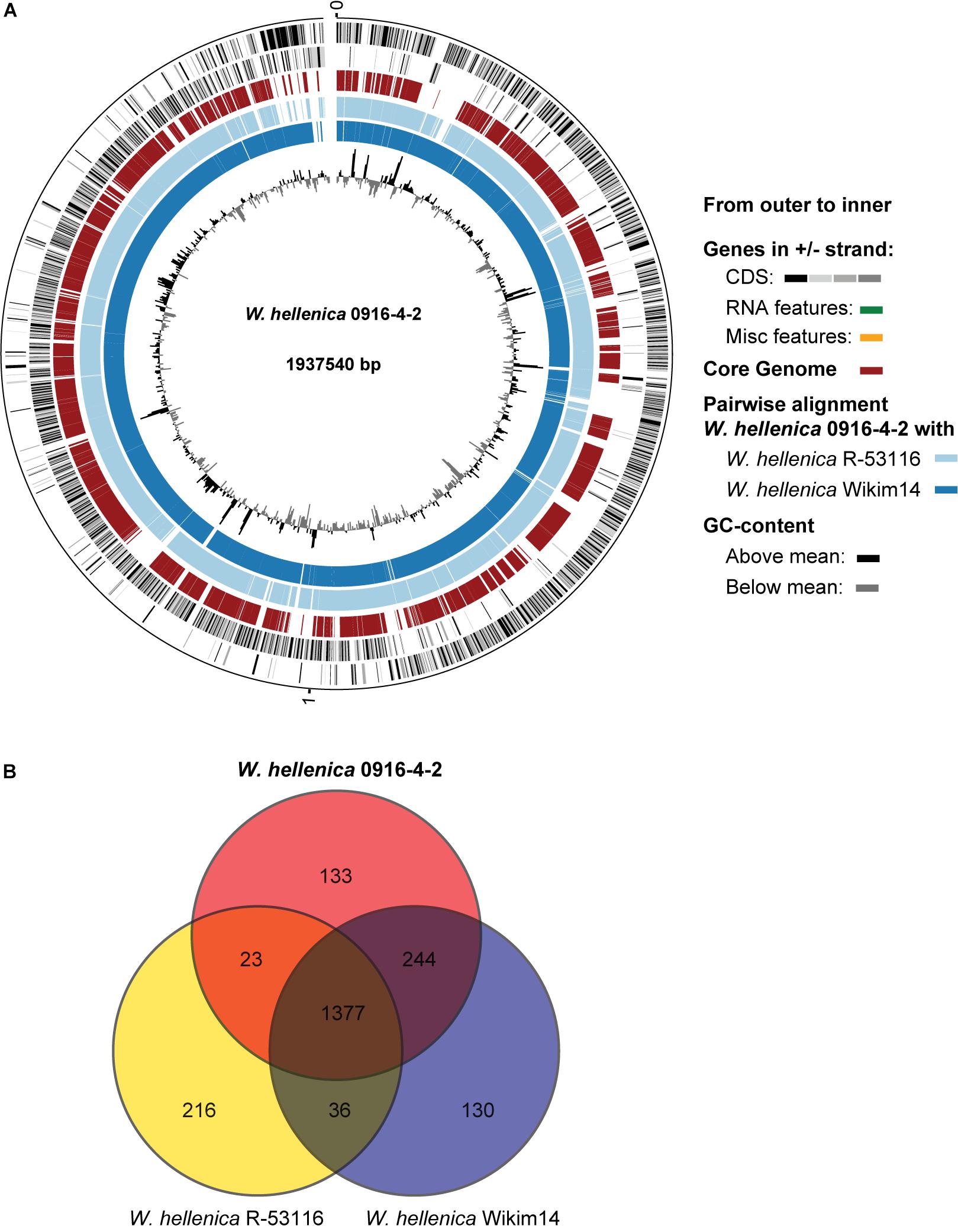
Figure 2. Comparative genomic analysis of W. hellenica. (A) Circular genome comparison showing the core genome, pairwise alignment, and GC content. The meaning of each circle is indicated by the legend in the figure. (B) Venn diagram showing shared and unique genes among W. hellenicas.
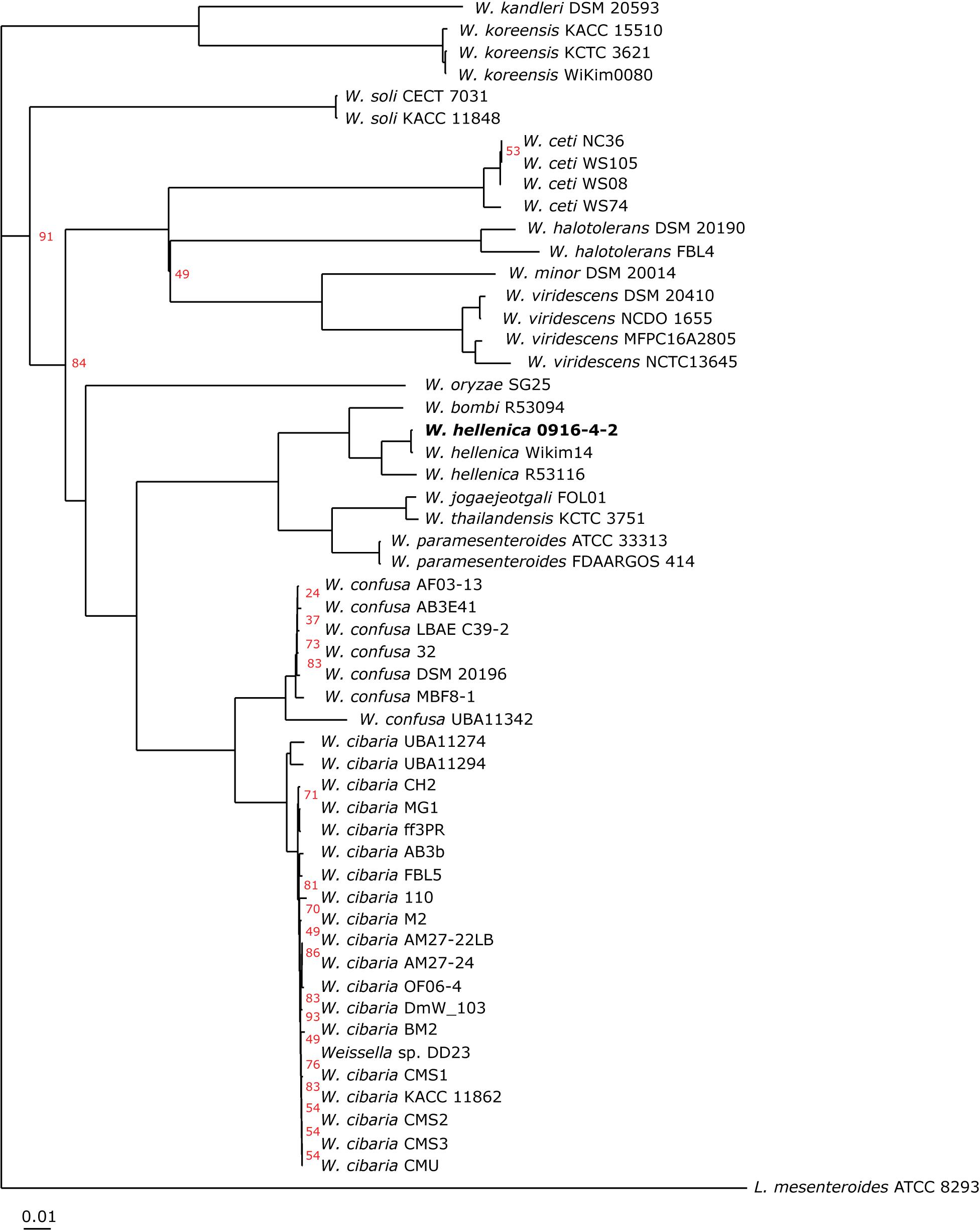
Figure 3. Phylogeny of Weissella species based on single-copy core orthologs. W. hellenica 0916-4-2 proteome, along with 52 Weissella and Leuconostoc mesenteroides ATCC 8293 proteomes obtained from public database and core genome was computed. The alignment of each core gene set was generated using MUSCLE, and the alignments were concatenated to create a single alignment. This alignment was used to generate the phylogenetic tree using neighbor-joining algorithm in EDGAR using L. mesenteroides ATCC 8293 as an outgroup. Bootstrap conservation values are shown in percent out of 200 iterations. Branches without support value showed 100% bootstrap support. Tree for 54 genomes, built out of a core of 67 genes with 20037 AA-residues per genome, 1081998 in total.
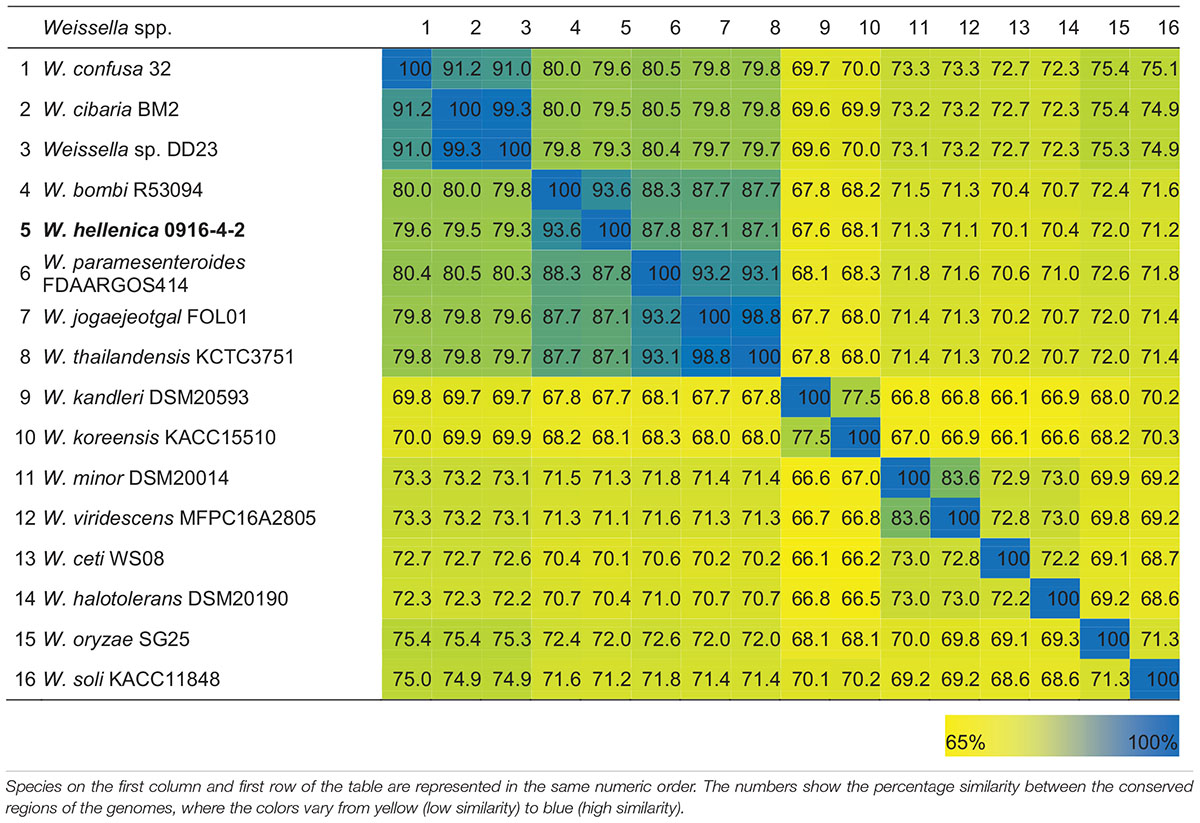
Table 2. Heatmap of the percentage AAI similarity between the conserved regions of the genus Weissella.
The members of Weissella genus are heteroformentative (Collins et al., 1993) attributed to the lack of phosphofructokinase (Sun et al., 2015). We did not find homologs for phosphofructokinase and lactate dehydrogenase in the 0916-4-2 genome. This suggested that 0916-4-2 lacks ability to produce L-lactate and this was further consistent with the observation of multiple copies of D-2-hydroxyacid dehydrogenase gene responsible for metabolism of pyruvate through D-lactate pathway. An analysis of Weissella genomes indicated that majority of Weissella had the genes for D-lactate formation with W. viridescens, W. minor, W. confusa, W. cibaria, and Weissella sp. DD23 harboring the genes for the production of both D/L configuration of lactic acid (Table 3). This might provide a possible explanation for the presence of significant amount of D-lactic acid (Yoon et al., 2005) and increase in lactic acid content at late stage of kimchi fermentation (You et al., 2017).
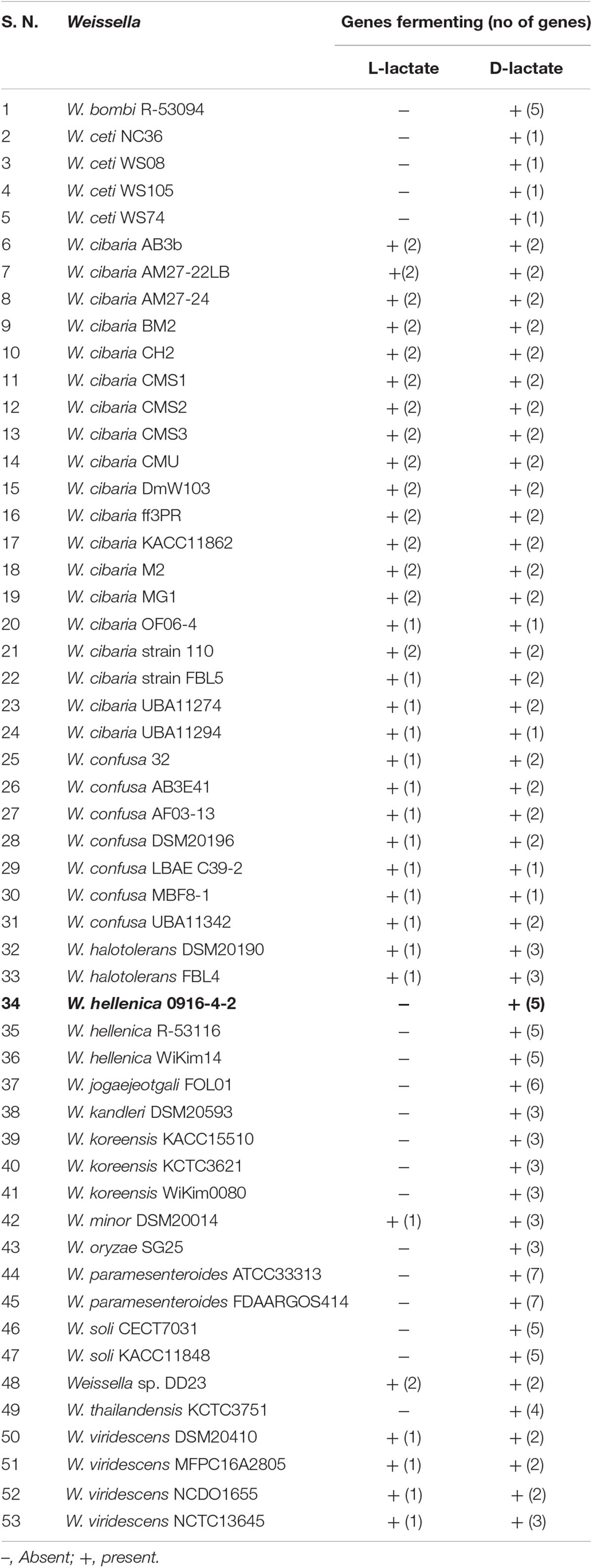
Table 3. Genes involved in D/L-lactate fermentation in Weissella genomes.
W. hellenica was first described in 1993 where Collins et al. (1993) reported the inability of this species to utilize various carbohydrates including D-cellobiose, D-raffinose, and gentiobiose for acid production. We analyzed the carbohydrate utilization pattern and found that W. hellenica 0916-4-2 can utilize D-cellobiose and D-raffinose very well; and gentiobiose partially (Supplementary Figure S1). This indicated the variation in strain specific properties of W. hellenica toward carbohydrate fermentation. Moreover, the metabolism of D-cellobiose and D-raffinose was consistent with the presence of the phosphotransferase system with β-glucosidase and α-galactosidase genes in the genome. Raffinose, present in various cruciferous vegetables including cabbage (Santarius and Milde, 1977), is not metabolized by humans and results in various intestinal disorders like flatulence (Rackis, 1981). Our study indicates that vegetable fermentation using 0916-4-2 strain might enhance the nutritional value of the products such as kimchi.
Core Genome Analysis of Weissella Species
Among the 53 Weissella strains analyzed, the core and pan genome size was 88 and 11,519, respectively (Figure 4). Among the 88 core genes in Weissella, three were found to be categorized as hypothetical and a COG analysis of the remaining 85 genes showed that more than a third of the genes were categorized to be involved in translation and nearly 13% of the genes were categorized to have unknown function (Table 4). Further, with addition of each species, there was a significant change in the number of core and pan genome suggesting a great genomic variation among the species within genus which could be a result of genomic fluidity or significant gene gain/loss during adaptation in natural niche.
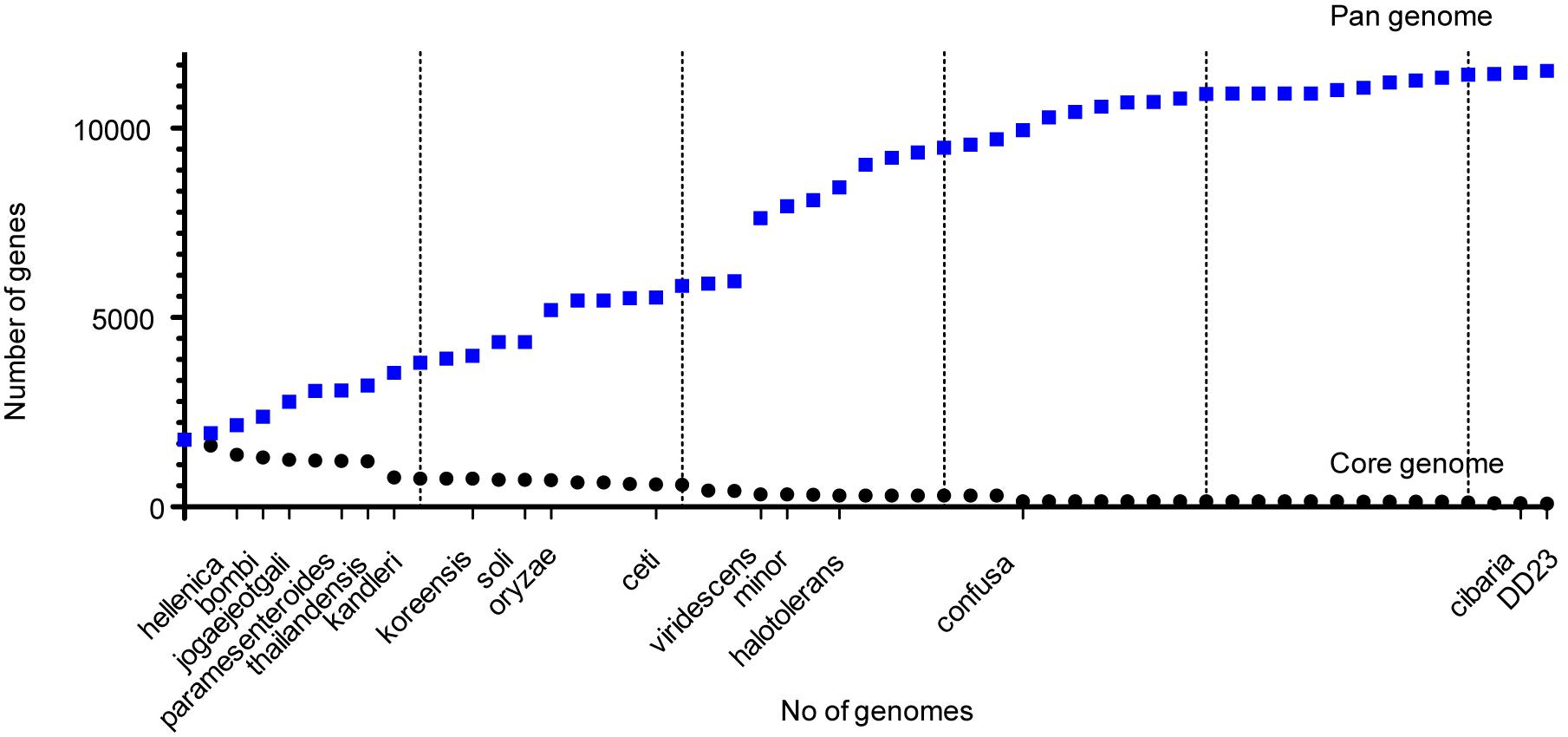
Figure 4. Core and pan proteome analysis of Weissella.
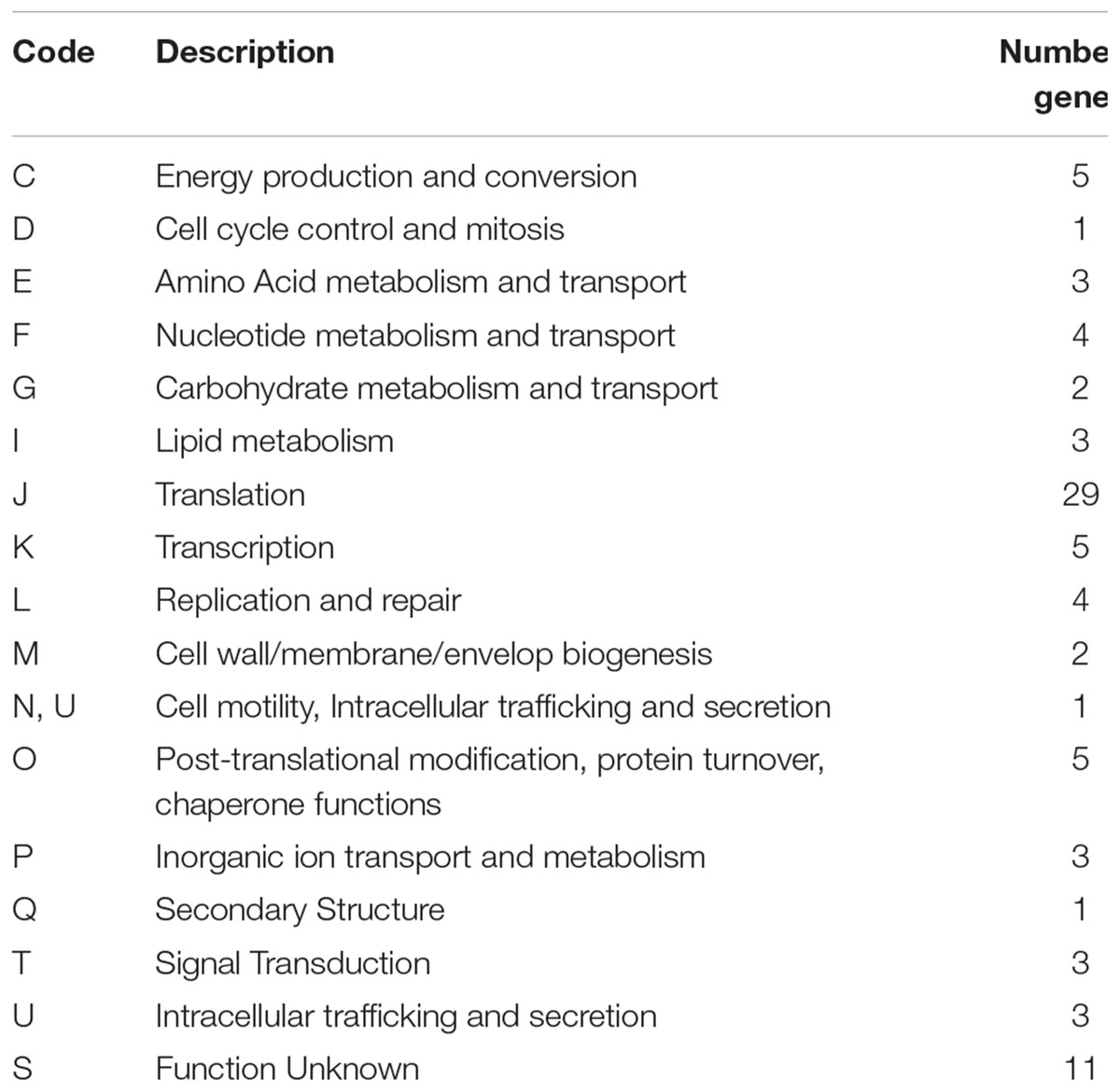
Table 4. COG analysis of core Weissella proteome.
Virulence Factors in Weissella Genome and W. hellenica 0916-4-2
Based on the BLAST search against the core virulence factors from the virulence factor database (Chen et al., 2005; Liu et al., 2018), we did not find homologs associated with toxin production system, including botulinum neurotoxin homolog from W. oryzae SG25 (Mansfield et al., 2015; Zornetta et al., 2016), in the analyzed Weissella genomes. We detected some of the genes, hasC (UDP-glucose pyrophosphorylase); cpsI (UDP-galactopyranose mutase); gnd (6-phosphogluconate dehydrogenase); cpsF and clpE (ATP-dependent protease) involved in virulence in various bacteria. These genes constitute a part of a pathway, indicating that the existence of a single genes is not sufficient to exert virulence. This suggested the need of a detailed investigation into the role of these genes in functional Weissella trait. Interestingly, all but Weissella sp. DD23 harbored hasC and there was no occurrence of species-specific virulence gene. We further expanded our study by the examination of the pathogenicity of W. hellenica 0916-4-2 through intraperitoneal injection to five mice. We found that mice did not die for 5 days post-injection suggesting its non-pathogenicity (data not shown). Although some members of the genus Weissella are opportunistic pathogen, there is a lack of clear demarcation between the probiotic and pathogenic strains among the strains analyzed in this report suggesting a need of a detailed investigation regarding Weissella pathogenicity.
Bacteriophages and Phage Defense
Phages are critical part of bacterial genome that facilitate various beneficial traits for bacteria such as adaptation in new environmental niches and acquisition of bacterial resistance. We used PHASTer (Arndt et al., 2016) to search for putative prophage elements in the complete and draft Weissella genomes to identify a total of 127 phages, of which 44 were considered as intact (Supplementary Table S1) and the number of phages in each strain ranged from 1 to 9. The large number of incomplete phages might be due to the draft nature of the assembly, as the phages generally have the repeated sequences. The intact phages from Weissella, ranged from 14 to 74 kb in length and majority had a length of 20–40 kb (Figure 5A). To identify the presence of potential pathogenic genes in the intact phages, we looked for the possible presence of virulence factors and antibiotic resistance genes. We did not find any virulence factors in the phages and a search in the CARD database (McArthur et al., 2013) using perfect and strict hits did not find any genes responsible for drug resistance. All the intact phages were further analyzed using Victor (Meier-Kolthoff and Göker, 2017) and phylogenetic tree was created. Based on the analysis, we grouped the phages in five groups. Our finding suggested that among the two intact phages present in W. hellenica 0916-4-2, one clustered with the phage from W. jogaejeotgali FOL01 and the next one fell onto relatively distinct cluster (Figure 5B). Interestingly, we found that two separate strains of W. soli (CECT7031 and KACC11848) and W. cibaria (AM27-22LB and AM27-24) shared a common phage, suggesting a very close intraspecies relationship.
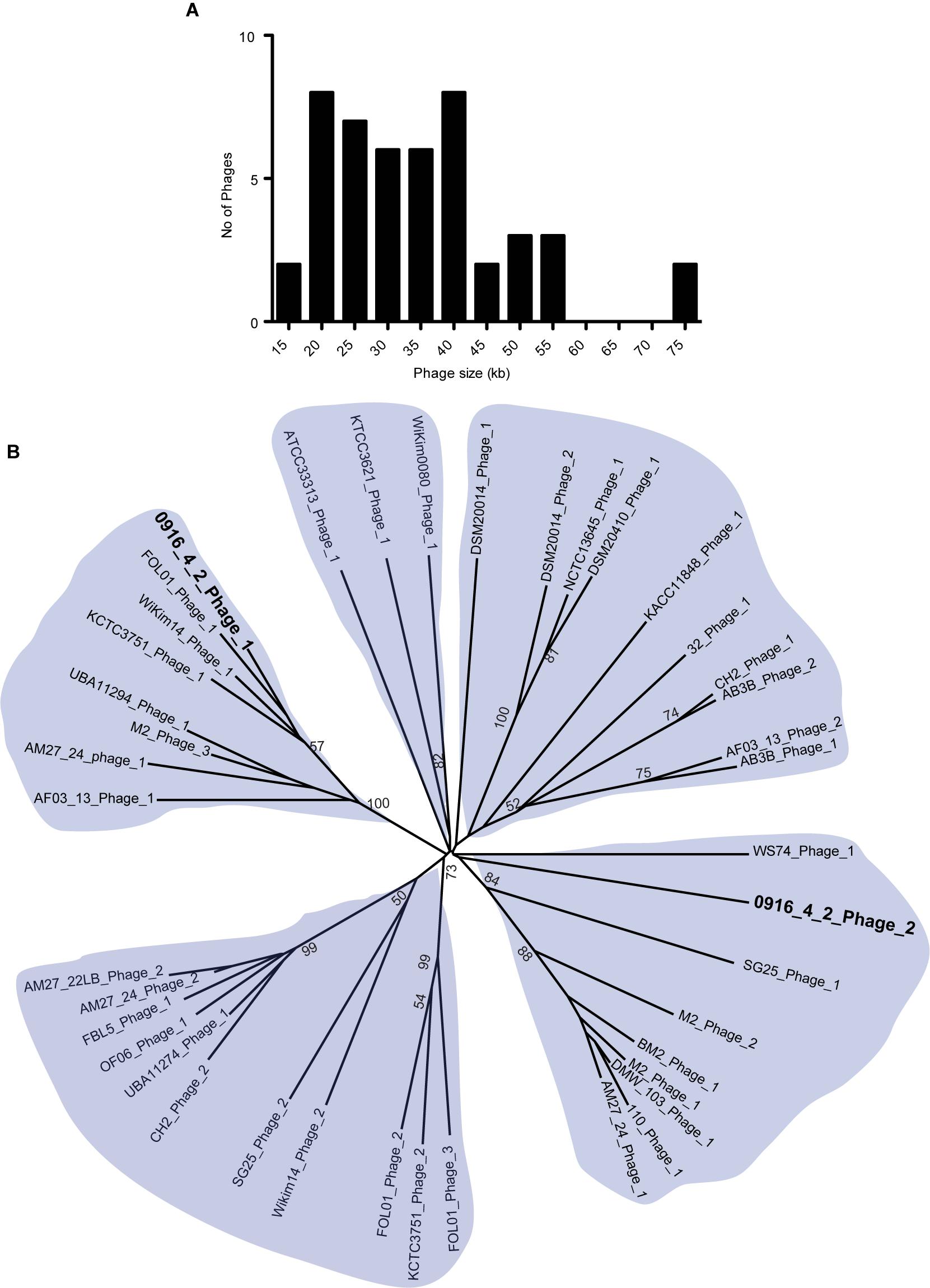
Figure 5. Analysis of Weissella phages. Histogram of length (A) and radial phylogenetic tree (B) of the complete phages identified in Weissella. The phages were analyzed using VICTOR (Meier-Kolthoff and Göker, 2017) and pairwise comparisons of the nucleotide sequences were conducted using the Genome-BLAST Distance Phylogeny method under settings recommended for prokaryotic viruses. The numbers above branches are the bootstrap support values from 100 replications (values ≥50 are shown).
Secondary Metabolic Potential
The genome assemblies were analyzed using antiSMASH (Blin et al., 2017) for the possible presence of gene clusters encoding for secondary metabolites. We found a total of 10 predicted secondary metabolites classified as: arylpolyene (1), bacteriocin (4), lassopeptide (2), NRPS (1), and thiopeptide (2) (Table 5). Given that only 9 out of 53 strains harbored the gene clusters for secondary metabolites, the genus Weissella can be regarded as a low producer of secondary metabolites. Further, this data was compared with other families of the order Lactobacillales. The genome assemblies were downloaded from NCBI and bioinformatic analysis for the presence of secondary metabolite biosynthetic gene clusters was performed. Bacteriocin constituted the major class of metabolite predicted to be present in the genome. Furthermore, in contrast to Weissella, the other members of Leuconostocaceae family were found to harbor about one secondary metabolite gene cluster per genome (Table 6). Among all the Lactobacillales, Lactococcus and Streptococcus were found to be relatively higher producers of metabolites with the presence of about three gene clusters per genome.

Table 5. Secondary metabolic potential of Weissella species.
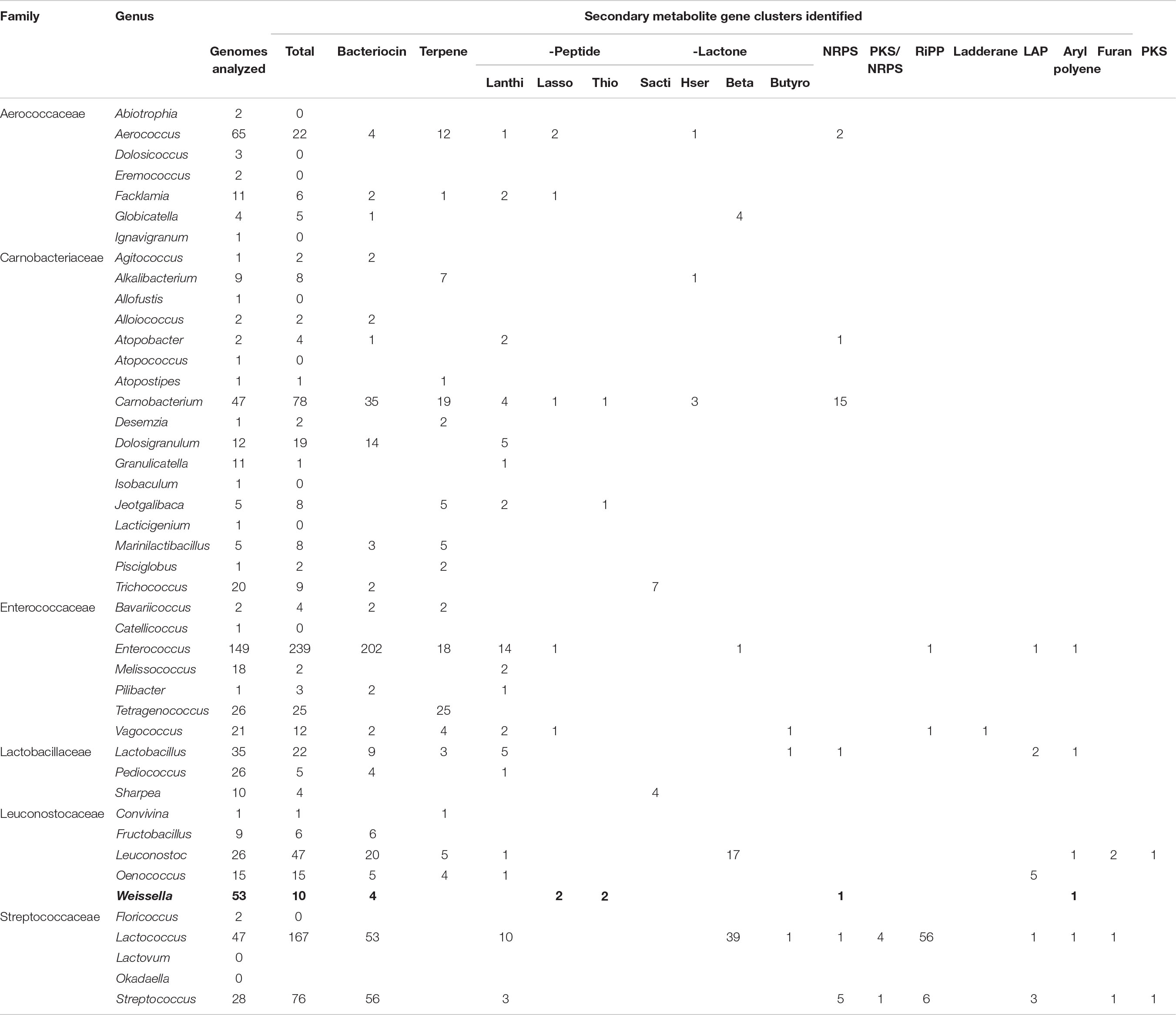
Table 6. Secondary metabolic potential of Lactobacillales.
Summary
The finished genome of W. hellenica 0916-4-2 is 1.93 Mb with a chromosome and two plasmids. We found that this strain can utilize D-cellobiose, a cellulose derivative, and harbored two putative secondary metabolite biosynthetic gene clusters. The ability of 0916-4-2 to utilize raffinose can be exploited to enhance the nutritional value of fermented vegetables. Comparative genomic analysis revealed a high degree of genomic variation among Weissella species. In recent post-genomic era, although the genome sequence data are becoming increasingly available for diverse bacterial species including Weissella, a species which has both probiotic and opportunistic pathogenic potential, the future research should focus on the detailed investigation of the genome and the association to specific gene(s) to the functional traits.
Data Availability
The complete genome assembly of Weissella hellenica 0916-4-2 has been deposited at DDBJ/ENA/GenBank with accession numbers: CP033608, CP033609, and CP033610 for chromosome, pWHSP041, and pWHSP020, respectively. The BioProject accession number for this project is: PRJNA503947.
Author Contributions
SP, HH, and KS designed the study. SP and AP performed the genome sequencing and annotation. SP, AP, and JB performed the comparative genomic analysis. SP and AP wrote the manuscript. HH and KS integrated the research and critically revised the manuscript for important intellectual content. KS approved the final version of the manuscript.
Funding
This work was supported in part by a Grant-in-Aid for Scientific Research (S) (JP15H05783) to KS, Grant-in-Aid for Scientific Research (C) (JP19K07140) to HH, Grant-in-Aid for Early-Career Scientists (JP19K16653) to AP, and Grant-in-Aid for JSPS Fellows (JP17F17421) to SP and KS.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Mr. Takashi Yamashita and Ms. Kiyomi Kyogoku from the Genome Pharmaceuticals Institute for their help with the isolation and animal studies of W. hellenica 0916-4-2.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01619/full#supplementary-material
References
Abriouel, H., Lerma, L. L., Casado Muñoz, M. D. C., Montoro, B. P., Kabisch, J., Pichner, R., et al. (2015). The controversial nature of the Weissella genus: technological and functional aspects versus whole genome analysis-based pathogenic potential for their application in food and health. Front. Microbiol. 6:1197. doi: 10.3389/fmicb.2015.01197
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Blin, K., Weber, T., Kim, H. U., Lee, S. Y., Takano, E., Breitling, R., et al. (2017). antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identification. Nucleic Acids Res. 45, W36–W41.
Blom, J., Kreis, J., Spänig, S., Juhre, T., Bertelli, C., Ernst, C., et al. (2016). EDGAR 2.0: an enhanced software platform for comparative gene content analyses. Nucleic Acids Res. 44, W22–W28. doi: 10.1093/nar/gkw255
Chen, C., Chen, X., Jiang, M., Rui, X., Li, W., and Dong, M. (2014). A newly discovered bacteriocin from Weissella hellenica D1501 associated with Chinese Dong fermented meat (Nanx Wudl). Food Control 42, 116–124. doi: 10.1016/j.foodcont.2014.01.031
Chen, L., Yang, J., Yu, J., Yao, Z., Sun, L., Shen, Y., et al. (2005). VFDB: a reference database for bacterial virulence factors. Nucleic Acids Res. 33, D325–D328.
Collins, M., Samelis, J., Metaxopoulos, J., and Wallbanks, S. (1993). Taxonomic studies on some Leuconostoc-like organisms from fermented sausages: description of a new genus Weissella for the Leuconostoc paramesenteroides group of species. J. Appl. Bacteriol. 75, 595–603. doi: 10.1111/j.1365-2672.1993.tb01600.x
Cosentino, S., Larsen, M. V., Aarestrup, F. M., and Lund, O. (2013). PathogenFinder-distinguishing friend from foe using bacterial whole genome sequence data. PLoS One 8:e77302. doi: 10.1371/journal.pone.0077302
Fusco, V., Quero, G. M., Cho, G.-S., Kabisch, J., Meske, D., Neve, H., et al. (2015). The genus Weissella: taxonomy, ecology and biotechnological potential. Front. Microbiol. 6:155. doi: 10.3389/fmicb.2015.00155
Huerta-Cepas, J., Forslund, K., Coelho, L. P., Bork, P., Von Mering, C., Szklarczyk, D., et al. (2017). Fast genome-wide functional annotation through orthology assignment by eggNOG-Mapper. Mol. Biol. Evol. 34, 2115–2122. doi: 10.1093/molbev/msx148
Kamboj, K., Vasquez, A., and Balada-Llasat, J.-M. (2015). Identification and significance of Weissella species infections. Front. Microbiol. 6:1204. doi: 10.3389/fmicb.2015.01204
Kim, H.-Y., Bong, Y.-J., Jeong, J.-K., Lee, S., Kim, B.-Y., and Park, K.-Y. (2016). Heterofermentative lactic acid bacteria dominate in Korean commercial kimchi. Food Sci. Biotechnol. 25, 541–545. doi: 10.1007/s10068-016-0075-x
Kolmogorov, M., Yuan, J., Lin, Y., and Pevzner, P. (2018). Assembly of long error-prone reads using repeat graphs. bioRxiv 247148.
Koren, S., Walenz, B. P., Berlin, K., Miller, J. R., Bergman, N. H., and Phillippy, A. M. (2017). Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27, 722–736. doi: 10.1101/gr.215087.116
Leong, K.-H., Chen, Y.-S., Lin, Y.-H., Pan, S.-F., Yu, B., Wu, H.-C., et al. (2013). Weissellicin L, a novel bacteriocin from sian-sianzih-isolated Weissella hellenica 4-7. J. Appl. Microbiol. 115, 70–76. doi: 10.1111/jam.12218
Liu, B., Zheng, D., Jin, Q., Chen, L., and Yang, J. (2018). VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 47, D687–D692. doi: 10.1093/nar/gky1080
Mansfield, M. J., Adams, J. B., and Doxey, A. C. (2015). Botulinum neurotoxin homologs in non-Clostridium species. FEBS Lett. 589, 342–348. doi: 10.1016/j.febslet.2014.12.018
Masuda, Y., Zendo, T., Sawa, N., Perez, R. H., Nakayama, J., and Sonomoto, K. (2012). Characterization and identification of weissellicin Y and weissellicin M, novel bacteriocins produced by Weissella hellenica QU 13. J. Appl. Microbiol. 112, 99–108. doi: 10.1111/j.1365-2672.2011.05180.x
McArthur, A. G., Waglechner, N., Nizam, F., Yan, A., Azad, M. A., Baylay, A. J., et al. (2013). The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57, 3348–3357. doi: 10.1128/AAC.00419-13
Meier-Kolthoff, J. P., and Göker, M. (2017). VICTOR: genome-based phylogeny and classification of prokaryotic viruses. Bioinformatics 33, 3396–3404. doi: 10.1093/bioinformatics/btx440
Panthee, S., Hamamoto, H., Ishijima, S. A., Paudel, A., and Sekimizu, K. (2018). Utilization of hybrid assembly approach to determine the genome of an opportunistic pathogenic fungus, Candida albicans TIMM 1768. Genome Biol. Evol. 10, 2017–2022. doi: 10.1093/gbe/evy166
Panthee, S., Hamamoto, H., Paudel, A., and Sekimizu, K. (2017a). Genomic analysis of vancomycin-resistant Staphylococcus aureus VRS3b and its comparison with other VRSA isolates. Drug Discov. Ther. 11, 78–83. doi: 10.5582/ddt.2017.01024
Panthee, S., Hamamoto, H., Suzuki, Y., and Sekimizu, K. (2017b). In silico identification of lysocin biosynthetic gene cluster from Lysobacter sp. RH2180-5. J. Antibiot. 70, 204–207. doi: 10.1038/ja.2016.102
Panthee, S., Paudel, A., Hamamoto, H., and Sekimizu, K. (2017c). Draft genome sequence of the vancomycin-resistant clinical isolate Staphylococcus aureus VRS3b. Genome Announc. 5:e00452-17. doi: 10.1128/genomeA.00452-17
Rackis, J. J. (1981). Flatulence caused by soya and its control through processing. J. Am. Oil Chem. Soc. 58, 503–509. doi: 10.1007/bf02582414
Salmela, L., and Rivals, E. (2014). LoRDEC: accurate and efficient long read error correction. Bioinformatics 30, 3506–3514. doi: 10.1093/bioinformatics/btu538
Santarius, K., and Milde, H. (1977). Sugar compartmentation in frost-hardy and partially dehardened cabbage leaf cells. Planta 136, 163–166. doi: 10.1007/BF00396193
Sun, Z., Harris, H. M. B., Mccann, A., Guo, C., Argimón, S., Zhang, W., et al. (2015). Expanding the biotechnology potential of Lactobacilli through comparative genomics of 213 strains and associated genera. Nat. Commun. 6:8322. doi: 10.1038/ncomms9322
Woraprayote, W., Pumpuang, L., Tosukhowong, A., Roytrakul, S., Perez, R. H., Zendo, T., et al. (2015). Two putatively novel bacteriocins active against Gram-negative food borne pathogens produced by Weissella hellenica BCC 7293. Food Control 55, 176–184. doi: 10.1016/j.foodcont.2015.02.036
Yoon, H.-S., Son, Y.-J., Han, J. S., Lee, J., and Han, N. S. (2005). Comparison of D-and L-lactic acid contents in commercial kimchi and sauerkraut. Food Sci. Biotechnol. 14, 64–67.
You, S.-Y., Yang, J.-S., Kim, S. H., and Hwang, I. M. (2017). Changes in the physicochemical quality characteristics of Cabbage Kimchi with respect to storage conditions. J. Food Qual. 2017, 1–7. doi: 10.1155/2017/9562981
Keywords: Weissella hellenica, comparative genomics, secondary metabolism, lactic acid bacteria, probiotic
Citation: Panthee S, Paudel A, Blom J, Hamamoto H and Sekimizu K (2019) Complete Genome Sequence of Weissella hellenica 0916-4-2 and Its Comparative Genomic Analysis. Front. Microbiol. 10:1619. doi: 10.3389/fmicb.2019.01619
Received: 09 April 2019; Accepted: 01 July 2019;
Published: 24 July 2019.
Edited by:
Daniel Yero, Autonomous University of Barcelona, SpainReviewed by:
Andrew Charles Doxey, University of Waterloo, CanadaValerie Diane Valeriano, Umeå University, Sweden
Copyright © 2019 Panthee, Paudel, Blom, Hamamoto and Sekimizu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhisa Sekimizu, c2VraW1penVAbWFpbi50ZWlreW8tdS5hYy5qcA==