Abstract
Syphilis, caused by Treponema pallidum subsp. pallidum (TPA), remains an important public health problem with an increasing worldwide prevalence. Despite recent advances in in vitro cultivation, genetic variability of this pathogen during infection is poorly understood. Here, we present contemporary and geographically diverse complete treponemal genome sequences isolated directly from patients using a methyl-directed enrichment prior to sequencing. This approach reveals that approximately 50% of the genetic diversity found in TPA is driven by inter- and/or intra-strain recombination events, particularly in strains belonging to one of the defined genetic groups of syphilis treponemes: Nichols-like strains. Recombinant loci were found to encode putative outer-membrane proteins and the recombination variability was almost exclusively found in regions predicted to be at the host-pathogen interface. Genetic recombination has been considered to be a rare event in treponemes, yet our study unexpectedly showed that it occurs at a significant level and may have important impacts in the biology of this pathogen, especially as these events occur primarily in the outer membrane proteins. This study reveals the existence of strains with different repertoires of surface-exposed antigens circulating in the current human population, which should be taken into account during syphilis vaccine development.
Introduction
Treponema pallidum subsp. pallidum (TPA) is the causative agent of syphilis, a globally occurring disease. Although the worldwide number of syphilis cases dramatically decreased after the introduction of penicillin therapy in the 1940s, the estimated number of new syphilis cases per year remains over 5.6 million. Especially alarming is the number of congenital syphilis cases, which is approaching one million cases per year (Newman et al., 2012; Peeling et al., 2017). In developed countries, syphilis is often transmitted among MSM patients (men who have sex with men). Moreover, MSM patients with syphilis are often co-infected with HIV (42% in Western Europe) (Dubourg et al., 2015). It is believed that syphilis facilitates the HIV infection, since syphilitic genital ulcers are infiltrated with lymphocytes (the primary target cells for HIV-infection) and provide a portal of entry for HIV acquisition. The rising prevalence of syphilis among MSM patients has coincided with the introduction of highly active anti-retroviral drugs leading to decreased HIV-associated mortality and the re-emergence of unsafe sexual behavior among MSM (Stolte et al., 2001). TPA infections are characterized by early and fast dissemination, immune evasion and long persistence in untreated patients. However, the underlying molecular mechanisms remain poorly understood (Radolf et al., 2016).
In spite of recent advances in in vitro cultivation of TPA (Edmondson et al., 2018), routine laboratory cultivation of this pathogen directly from patient samples is not yet possible. Therefore, most of the information on TPA genetics comes from genome sequencing studies, where DNA was isolated from bacteria propagated in experimentally infected rabbits (Fraser et al., 1998; Matĕjková et al., 2008; Giacani et al., 2010, 2014; Pětrošová et al., 2012, 2013; Zobaníková et al., 2012; Tong et al., 2017). The research community uses culture-independent enrichment techniques prior to whole genome sequencing of TPA clinical samples due to the overwhelming levels of human DNA and very low amounts of TPA DNA (1000:1 ratio of human to TPA DNA) found in clinical samples. However, available enrichment techniques demonstrate low efficiency (e.g., Anti-treponemal antibody enrichment, ATAE) (Grillová et al., 2018b) or are based on sequence-specific protocols (e.g., DNA-capture microarray and “in solution” capture techniques) (Arora et al., 2016; Pinto et al., 2016; Knauf et al., 2018; Marks et al., 2018), thus preventing the recovery of unique sequences not present in the reference genomes.
Genetically, TPA can be divided into two separate groups – SS14-like and Nichols-like strains (Nechvátal et al., 2014; Arora et al., 2016). As revealed by molecular typing studies of TPA isolates, most of the examined patients were infected with SS14-like strains (94.1%) (Woznicová et al., 2007; Flasarová et al., 2012; Grillová et al., 2014, 2018c; Arora et al., 2016; Gallo Vaulet et al., 2017; Mikalová et al., 2017a; Pospíšilová et al., 2018). The reason for the predominance of one genetic group is widely discussed, but was not clarified yet (Arora et al., 2016; Šmajs et al., 2016).
In this study, we performed direct whole genome sequencing of 25 TPA clinical samples isolated from different geographical areas using methyl-directed enrichment prior to next generation sequencing (NGS) (Barnes et al., 2014). Using this approach, we obtained 11 complete genome sequences, which represents the vast majority (92%) of complete TPA genomes sequenced directly from clinical samples. The subsequent detailed comparative genomic analyses revealed unexpected variability among Nichols-like genomes driven by inter-clade and/or intra-strain recombination events, which were accumulated mainly in the genes encoding predicted outer membrane proteins. This discovery, beyond being relevant to the understanding of basic biology of treponemes, highlights the presence of different repertoires of alleles coding for potential virulence factors, which circulate in the current human population.
Results
Clinical Samples
We selected 25 TPA samples recently isolated from 24 patients diagnosed with syphilis for whole genome sequencing. The sample set was selected to contain (i) samples with the highest possible genetic diversity, (ii) samples from different geographical areas and (iii) samples representing contemporary TPA infections. The samples were collected in four countries on three different continents (Australia, Cuba, Czechia, and France), mostly from males (92%) from which 74% were MSM (Table 1). Samples were taken as genital (n = 16), anal (n = 4), buccal (n = 3), or skin smears (n = 1), with one sample of lung tissue from a fatal case of congenital syphilis. As a result of the non-random sample selection, Nichols-like strains were overrepresented in our sample set (31%) compared to their prevalence in the infected population (5.9%) (Šmajs et al., 2018). Samples belonged to 9 different sequencing types (STs) and most (72%) carried the A2058G mutations in both rrn operons leading to resistance to macrolide antibiotics (Table 1).
TABLE 1
| ID | Origin | Year of isolation | Material | Sexual orientation | Stage | Allelic profile (ST) | Genetic group | Macrolide resistance/sensitivity (mutation) |
| CW30* | Czechia | 2014 | genital smear | MSM | Primary | 1.3.1 (1) | SS14-like | sensitive |
| CW84 | France | 2015 | genital smear | MSW | Primary | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW85 | France | 2016 | genital smear | MSM | Primary | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW87 | France | 2016 | bucal smear | Unknown | Primary | 1.23.1 (35) | SS14-like | resistant (A2058G) |
| CW88 | Czechia | 2017 | genital smear | Unknown | Primary | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW56 | Cuba | 2013 | genital smear | MSM | Unknown | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW82 | Cuba | 2016 | genital smear | MSM | Unknown | 15.7.3 (37) | Nichols-like | sensitive |
| CW65 | Australia | 2014 | anal smear | Unknown | Secondary | 9.14.3 (47) | Nichols-like | sensitive |
| CW83 | Cuba | 2015 | genital smear | MSM | Unknown | 9.24.8 (38) | Nichols-like | sensitive |
| CW86 | France | 2013 | genital smear | MSM | Secondary | 9.20.3 (31) | Nichols-like | sensitive |
| CW59 | France | 2012 | anal smear | MSM | Secondary | 9.7.3 (26) | Nichols-like | sensitive |
| CW57 | Cuba | 2014 | genital smear | MSM | Unknown | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW51 | Cuba | 2016 | anal smear | MSM | Unknown | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW53 | Cuba | 2015 | genital smear | MSM | Unknown | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW29 | Czechia | 2013 | genital smear | MSW | Primary | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW45∗∗ | Czechia | 2013 | genital smear | WSM | Secondary | 1.26.1 (25) | SS14-like | resistant (A2058G) |
| CW89 | Czechia | 2017 | genital smear | MSW | Primary | 1.26.1 (25) | SS14-like | resistant (A2058G) |
| CW33 | Czechia | 2012 | lung | Unknown | Congenital | 1.1.8 (3) | SS14-like | sensitive |
| CW35 | Czechia | 2013 | genital smear | MSM | Primary/Secondary | 9.7.3 (26) | Nichols-like | resistant (A2058G) |
| CW31 | Czechia | 2013 | bucal smear | WSM | Secondary | 1.26.1 (25) | SS14-like | resistant (A2058G) |
| CW44∗∗ | Czechia | 2013 | bucal smear | WSM | Secondary | 1.26.1 (25) | SS14-like | resistant (A2058G) |
| CW52 | Cuba | 2016 | genital smear | MSM | Unknown | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW55 | Cuba | 2015 | skin smear | MSM | Unknown | 1.3.1 (1) | SS14-like | resistant (A2058G) |
| CW58 | France | 2013 | genital smear | unknown | Unknown | 9.7.3 (26) | Nichols-like | resistant (A2058G) |
| CW61 | Czechia | 2014 | anal smear | MSM | Primary | 1.3.1 (1) | SS14-like | resistant (A2058G) |
Clinical characteristics of samples and their genotyping data.
*Sample enriched by ATAE (Grillová et al., 2018b) prior to the DpnI enrichment. ∗∗Parallel samples taken from the same patient but isolated from different clinical material. MSM, men who have sex with men; MSW, men who have sex with women; WSM, women who have sex with men.
The number of TPA DNA copies as well as human DNA copies were determined by qPCR in all examined samples. The number of TPA DNA varied from 1 to 105 copies per μl with the TPA DNA/human DNA ratio ranging from 0.01 to 3.69 (Supplementary Table S1). Given the fact that the human genome is approximately 3000 times larger than the TPA genome, the samples contained 103–105 times more human DNA than treponemal DNA requiring TPA DNA enrichment prior to sequencing.
Methyl-Directed Enrichment Using Restriction Endonuclease DpnI
The method we are presenting in this paper is based on binding activity of the DpnI endonuclease. DpnI is a restriction endonuclease that recognizes DNA methylated on adenine residue within the GATC sequence. This methylated DNA motif occurs in all bacteria that have deoxyadenosine methyltransferase (DAM) and is not present in higher eukaryotes. Immobilized DpnI proteins on magnetic beads were used for specific capture of prokaryotic DNA. In the absence of Mg2+ ions, DpnI binds its recognition sequence without cutting it. A previous study by Stamm et al. (1997) identified methylated adenine residues in GATC sequences in the TPA genome. To ensure the appropriate binding of DpnI and enrichment effectivity of the DpnI enrichment, a pilot experiment on one sample (CW88; Table 1) was performed. Before enrichment, the sample was sequenced with 384,489,027 reads and 25,168 of them were mapped to the treponemal genome. This resulted in 92% broad genome coverage and median sequencing depth of 6.6× and more than 100 sequencing gaps. After enrichment, the same sample was sequenced with a similar number of reads (332, 564, 761) revealing 296, 978 treponemal reads, which resulted in a near complete genome (98%) with a median sequencing depth of 68×. Except for the paralogous and repetitive regions (listed in the Supplementary Table S2), which were excluded from the reference-guided approach, the draft genome of CW88 had even sequencing coverage depth without any detectable biases. This indicated that Gm6ATC DNA motifs are evenly distributed across the TPA genomes and that the DpnI method is suitable for the TPA DNA enrichment.
Direct Whole Genome Sequencing
Methyl-directed enrichment was applied to all examined clinical samples (n = 25) prior to NGS (details in section “Materials and Methods”). We used the preliminary sequencing to calculate the appropriate number of reads needed for the best possible coverage in the next sequencing runs. The NGS statistics are given in the Supplementary Table S3. In order to determine if the new sequencing approach was comparable to the traditional pooled segment genome sequencing (PSGS) approach, we sequenced isolates from the same organism (strain Phi-1 and Grady) following each method and did not find any discrepancies between the genomes (data not shown). According to the preliminary sequencing results, we selected samples containing treponemal DNA showing the highest breadth coverage (>97%, n = 11) as candidates for complete genome sequencing and samples with lower coverage (69–97%, n = 8) as candidates for genome-wide analyses (with a depth of coverage of 3 or greater). The samples with the lowest coverage (<10%, n = 7) were excluded from further analyses (Supplementary Table S3). For the whole genome determination, we amplified and Sanger sequenced regions with low coverage (less than 3 good-quality reads; in average 10 regions per genome) as well as regions, which were excluded from the bioinformatic pipeline including paralogous regions (tpr genes and rrn operons) and repetitive regions (e.g., arp and TP0470 genes) (details in section “Materials and Methods”).
All samples from SS14-like and Nichols-like clades revealed complete gene synteny. However, we identified significant differences between the genetics of these two TPA clades (Figure 1 and Supplementary Figure S1). The variability observed in Nichols-like strains was about one order of magnitude higher compared to that of the SS14-like strains. Moreover, in contrast to SS14-like strains, Nichols-like strains showed accumulation of a high number of single nucleotide variants (SNVs) in several genes. The manual inspection of the genes with a high SNV density (defined as more than 4 SNVs per gene) revealed that the genetic diversity was a result of inter-clade and/or intra-strain recombination events (altogether covering 147 SNVs, which represents 49.5% of variability found within Nichols-like strains). The intra-strain recombination events included rearrangements of genes coding for lipoproteins (TP0856 and TP0858), which possessed modular structures (Strouhal et al., 2018), rrn spacers and predicted virulence factors tprG and tprJ (described below, Figure 2). The inter-clade recombination events were identified as sequences resembling both different syphilis genetic groups (i.e., SS14-like and Nichols-like groups) in the TP0136 gene encoding an outer membrane protein. In addition, the inter-clade recombination included sequences resembling both syphilis and bejel sequences (in TP0117 coding for TprC; in TP0317 coding for TprG; in TP0462 encoding probable lipoprotein; in TP0483 coding for hypothetical protein; in TP0621 coding for TprJ; in TP0865 encoding a putative outer membrane protein) (Table 2 and Figure 3).
FIGURE 1

Phylogeny of all TPA complete genome sequences determined to date. Maximum likelihood bootstrapping method was used to generate the phylogenetic tree based on 2273 variable positions found exclusively in the whole genome sequences available to date (Table 2). The bootstrap values, when above 60, are given next to the branches in red. SS14-like strains are represented by red squares, Nichols-like strains by blue squares. The year of isolation is given next to the branches. Strains designated CW30-CW88 are the whole genome sequences determined in this study by DpnI enrichment while Phi-1 and Grady were established in this study by PSGS. These represent 65% of all whole genome sequences of TPA to date. tpr genes, repetitive regions and inter- and intra-recombinant loci were excluded from this analysis (Supplementary Table S2 and Figures 2, 5). We used the genome of Samoa D (T. pallidum subsp. pertenue) as an outgroup.
FIGURE 2
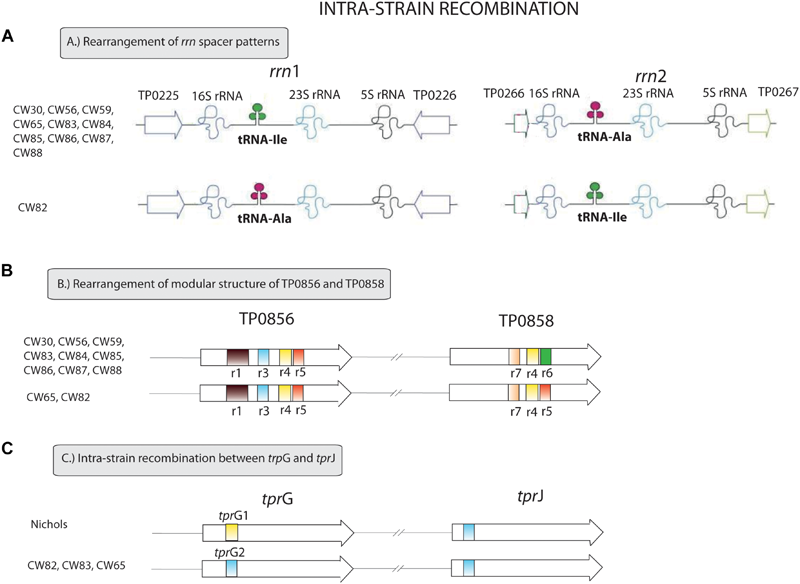
Intra-strain recombination events found among examined clinical samples. (A) Rearrangement of rrn spacer patterns. The ribosomal operon with Ile/Ala tRNA patterns differ from the ribosomal operon with Ala/Ile tRNA patterns in 34 nucleotide positions. The figure was modified according to Čejková et al. (2013). (B) Rearrangement of the modular structure of TP0856 and TP0858 genes. We identified two variants of the modular structure of the TP0856 and TP0858 genes – r1r3r4r5/r7r4r5 and r1r3r4r5/r7r4r6. These structures differ in 30 nucleotide sites. The figure was modified according to Strouhal et al. (2018). (C) Intra-strain recombination between tprG and tprJ. tprG1 and tprG2 allele variants differ in 18 variable positions.
TABLE 2
| Intra-strain recombination | Inter-clade recombination | ||||||||||||||
| Strain | Year of isolation | rrn1/rrn21 | TP08582 | tprC/D3 | tprG4 | TP01365 | TP03266 | TP04627 | TP04838 | TP04889 | TP086510 | tprC11 | tprG12 | tprJ13 | |
| Nichols-like | Nichols (CP004010.2) | 1912 | Ile/Ala | R7R4R6 | D | G1 | – | – | – | – | – | – | C | – | – |
| strains | Chicago (CP001752.1) | 1951 | Ile/Ala | R7R4R6 | D | G1 | – | – | – | – | – | – | C | – | – |
| previously | DAL-1 (CP003115.1) | 1991 | Ile/Ala | R7R4R6 | D | G1 | – | – | – | – | – | – | C | – | – |
| described | SEA81-4 (CP003679.1) | 2014 | ? | R3R4R5 | D2 | G2 | – | – | – | – | – | Nichols/TEN | C | – | Nichols/TEN or TPE |
| Nichols-like | CW82 (CP34972) | 2016 | Ala/Ile | R7R4R5 | D2 | G2 | Nichols/SS14 | – | – | – | – | – | C3 | – | – |
| strains in this | CW65 (CP34918) | 2014 | Ile/Ala | R7R4R5 | D2 | G2 | – | – | – | – | – | – | C4 | – | – |
| study | CW83 (CP34917) | 2015 | Ile/Ala | R7R4R6 | D2 | G2 | – | – | – | Nichols/TEN | – | – | C3 | – | – |
| CW59 (CP34919) | 2012 | Ile/Ala | R7R4R6 | D2 | G2 | – | – | Nichols/TEN or TPE | – | – | Nichols/TEN | C3 | – | Nichols/TEN or TPE | |
| CW86 (CP34914) | 2013 | Ile/Ala | R7R4R6 | D2 | G2 | – | – | Nichols/TEN or TPE | – | – | Nichols/TEN | C3 | – | – | |
| SS14-like | SS14 (CP004011.1) | 1977 | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – |
| strains | Mexico A (CP003064.1) | 1953 | Ala/Ile | R7R4R6 | D2 | G1 | – | SS14/TEN | – | – | SS14/TEN | – | C2 | – | – |
| previously | Phi-1 (CP035193) | 1988 | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – |
| described | Grady (CP035104) | 1980s | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – |
| UZ1974 (CP028438.1) | 2012 | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – | |
| SS14-like | CW30 (CP034921) | 2014 | Ile/Ala | R7R4R6 | D2 | – | – | – | – | – | – | – | C2 | SS14/Nichols | – |
| strains in this | CW84 (CP34916) | 2015 | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – |
| study | CW85 (CP34915) | 2016 | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – |
| CW87 (CP34913) | 2016 | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – | |
| CW88 (CP34912) | 2017 | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – | |
| CW56 (CP34920) | 2013 | Ile/Ala | R7R4R6 | D2 | G1 | – | – | – | – | – | – | C2 | – | – | |
Inter-clade and intra-strain recombination in examined samples and in previously determined whole genome sequences of TPA.
1Intra-genomic rearrangements of rrn spacer patterns (Ile/Ala) (Čejková et al., 2013; Figure 3). 2Intra-genomic rearrangements of modular structure of TP0856 and TP0858 (Strouhal et al., 2018; Figure 3). 3Intra-genomic recombination of tprC and tprD. Also described in Kumar et al. (2018). 4Intra-genomic recombination of tprG and tprJ (Figure 3). 5The inter-clade recombination of the TP0136 gene (Figure 4), reported also in Grillová et al. (2018c) and predicted in Arora et al. (2016). 6The inter-clade recombination of the TP0326 gene, as described in Pětrošová et al. (2012) and predicted in Arora et al. (2016). 7,8The inter-clade recombination of the TP0462 and TP0483 genes (Figure 4). 9The inter-clade recombination of the TP0488 gene, as described in Pětrošová et al. (2012). 10–13The inter-clade recombination of the TP0865, as predicted in Arora et al. (2016), tprC, tprG, and tprJ genes (Figure 4). Inter-clade and intra-strain recombination events are highlighted in gray.
FIGURE 3
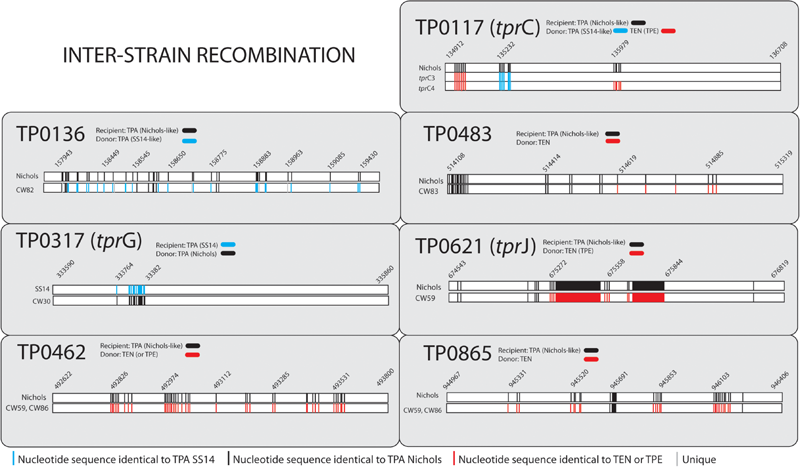
Inter-clade recombination events found among examined clinical samples. Coordinates according to the reference Nichols (CP04010.1) and reference SS14 (CP004011.2) for the Nichols-like and SS14-like clinical samples, respectively. Only positions with nucleotide differences are shown. The detailed description of the Figure 3 is given in the Supplementary Text S2.
The detailed overview concerning different genome dynamics of SS14-like and Nichols-like strains are described in the Supplementary Text S1.
Analyses of tpr Genes
When analyzing tpr genes among the SS14-like samples we did not find any significant variability with exception of the sample CW30, where we found a Nichols-like allele in the tprG gene (differing in 29 SNVs from the SS14 sequence) probably as the result of an inter-clade recombination event (Figure 3). Otherwise, the SS14-like samples differed only in a few SNVs in the tprC and tprI genes compared to the SS14 reference (1 and 3, respectively, Figure 4 and Supplementary Figure S2).
FIGURE 4
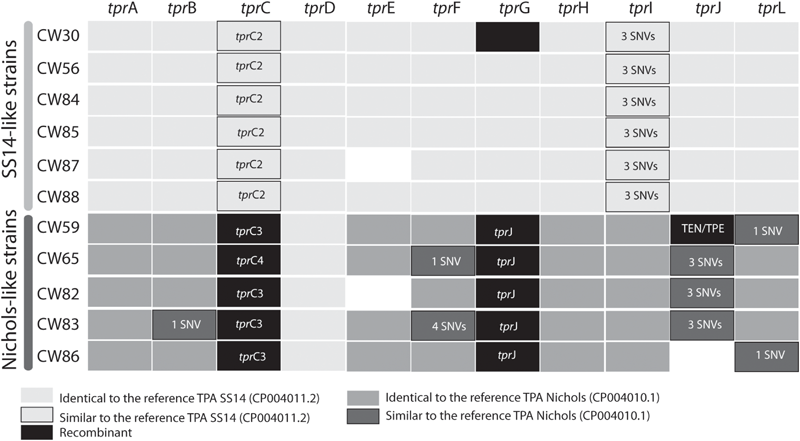
Allele variants of tpr genes found among examined clinical samples. To overcome the paralogous character of tpr genes, these loci were PCR amplified using primers with unique binding sites upstream and downstream the genes. The details are given in the section “Materials and Methods”. The allele variants are presented as a different color code (see legend under the figure). The blank spaces (e.g., tprE in CW87 and CW82; tprJ in CW86) represent genome regions which were not amplifiable in the given samples. TprK variants are missing due to their hyper-variable sequences based on different subpopulations of the same strain. The tprC allelic variants are shown in Supplementary Figure S2.
In Nichols-like samples, the analyses of tpr genes revealed higher sequence variability compared to the group of SS14-like samples. We have identified gene conversion of a partial sequence of the tprJ gene into the tprG gene (intra-strain recombination, Figure 2). Moreover, in sample CW59, we identified inter-clade recombination in the tprJ allele (with a putative TEN – T. pallidum subsp. endemicum or TPE – T. pallidum subsp. pertenue donor sequence, Figure 3). And finally, we have identified new alleles in the tprC locus (tprC3, tprC4, Figure 3) represented by different branches in maximum likelihood phylogeny (Supplementary Figure S2). After manual inspection of the tprC3 and tprC4 alleles, we have identified the putative donor sequences of these alleles as SS14-like, bejel (TEN), or yaws (TPE) strains. The remaining tpr genes among Nichols-like strains were quite uniform, except for a few SNVs found in alleles present in the tprB, tprI, tprJ, tprL loci (Figure 4).
Recombination
When analyzing all published complete TPA genomes (including genomes from this study), we observed inter-clade or intra-strain recombination events frequently among Nichols-like strains, and only sporadically among SS14-like strains (Figure 1 and Table 2). More specifically, intra-strain recombination was found in all complete genomes of Nichols-like clade. Interestingly, inter-clade recombination was found more frequently in Nichols-like strains that were recently isolated (2013–2016) (Figure 1). To determine if the Nichols-like strains show differences in putative genes involved in recombination, we analyzed the rec genes (recA, recB, recF, recG, recJ, recN, recO, recQ, recR, recX) and genes encoding recombinases (XerD1 and XerD2). Except for one SNV in the recB gene distinguishing Nichols-like and SS14-like strains (and encoding non-synonymous amino acid substitution), no other differences were found.
Analyses of the Strains Based on the Conserved Genomic Regions
To analyze the phylogeny of TPA strains, we excluded all identified inter-clade and intra-strain recombinant loci identified in this study and in the previous studies (Figure 5) and variable genes such as tpr genes, and reconstructed Network phylogenetic tree of these two clades based solely on the conserved genomic regions. Except for Mexico A strain, SS14-like strains created a star-like topology as described previously (Arora et al., 2016) and the highest genetic distance (18 SNVs) was found between sample CW87 and the SS14 reference genome (Supplementary Figure S3). Nichols-like strains were found to be more genetically diverse than SS14-like strains with the minimum genetic distance represented by 12 SNVs in the case of DAL-1 (1991) and Nichols (1912), and the highest genetic distance represented by 160 SNVs in the case of SEA81-4 (2014) and DAL-1 (1991).
FIGURE 5
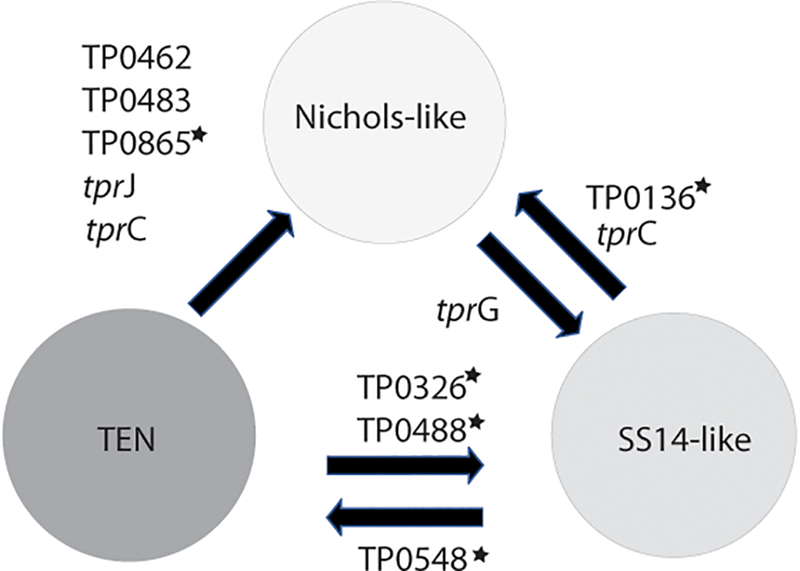
Inter-clade recombinations identified to date. The genes with stars represent loci, which were previously identified as recombinant (Pětrošová et al., 2012; Arora et al., 2016; Mikalová et al., 2017b; Grillová et al., 2018c). Five (out of 10) loci were identified in this study and were found among contemporary clinical samples enriched by a sequence-independent enrichment method.
Interestingly, SNVs distinguishing Nichols-like and SS14-like strains code for a significantly higher proportion of synonymous substitutions of amino acids (45.3%) compared to the SNVs found inside the SS14-like strains (17%) and within the Nichols-like strains (18%), suggesting that separate evolutionary forces operate inside and between each clade. Since we observed the same recombination events in the different phylogenetic branches (e.g., inter-clade recombination of TP0865 and tprJ in SEA-81-4 and CW59; Table 2 and Figure 1), these recombination events may have emerged several times independently.
Recombination Loci Encode Putative Surface-Exposed Antigens
Predicted protein structures encoded by recombinant loci provide important insights into structural and functional implications of recombination-driven variability. From the 8 recombinant protein-coding regions identified in this study, 6 were predicted to code for outer membrane proteins (TP0136, TP0858, TP0865, tprC, tprG and tprJ) and all of them contained predicted antigenic peptides (15–29). The prediction of treponemal protein structures is quite limited due to the lack of the protein homologues. However, a protein structure for tprC was predicted by Centurion-Lara et al. (2013) and more recently by Kumar et al. (2018). The recombinant regions identified in the new tprC3 and tprC4 alleles found in this study (Supplementary Figure S2) correspond to the identified extracellular loops (L3, L4, L5) of the β-barrel outer membrane protein which were predicted to serve as B-cells epitopes2 (Kumar et al., 2018; Figure 6). Moreover, the newly predicted 3D structures of proteins TP0858 and TP0865 (Figure 6) revealed that the observed recombination-driven diversity almost strictly corresponds to the residues located at the host-pathogen interface.
FIGURE 6
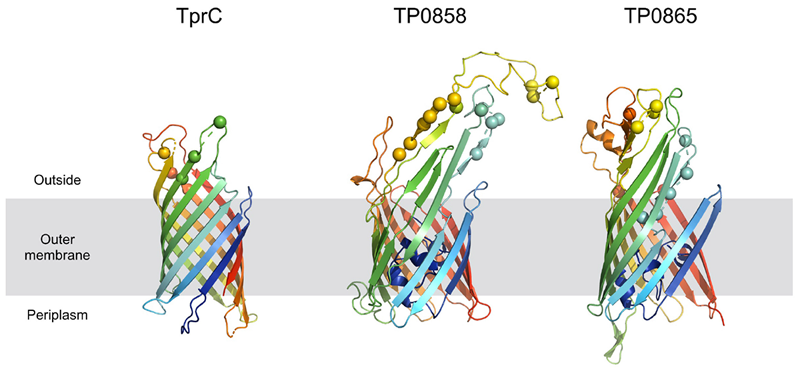
Homology models of TprC, TP0858, and TP0865. Homology models are shown in cartoon representation. The mutated residues are depicted with spheres. The models are colored based on a rainbow coloring scheme (with N-terminal of protein colored blue and C-terminal colored red).
Discussion
Efforts to understand the pathogenesis of TPA have been hindered by the inability to routinely propagate the bacterium in vitro and the lack of an efficient method for obtaining genomes directly from clinical samples. TPA isolates form two separate clusters, i.e., SS14-like and Nichols-like clades (Pětrošová et al., 2013; Nechvátal et al., 2014; Arora et al., 2016; Šmajs et al., 2016, 2018; Figure 1 and Supplementary Figure S1). We have found striking genetic diversity of the contemporary Nichols-like strains when compared to SS14-like strains. Nichols-like strains represent only a minority (about 6%) of contemporary strains circulating in the syphilis-infected population (Woznicová et al., 2007; Flasarová et al., 2012; Grillová et al., 2014, 2018c; Arora et al., 2016; Gallo Vaulet et al., 2017; Mikalová et al., 2017a; Pospíšilová et al., 2018), as revealed by molecular typing studies of TPA isolates. Although there are several possible explanations available, the ultimate reasons for this contemporary worldwide predominance of SS14-like isolates in the human population remain unknown (Šmajs et al., 2016). However, the high prevalence of SS14-like strains circulating in the contemporary syphilis-infected population could be due to the recent expansion of these strains. Therefore, it would not be surprising that most of the SS14-like strains are in fact of more clonal character than the Nichols-like strains. The observed genetic diversity of the contemporary Nichols-like strains could, therefore, be a result of sampling bias. This possibility is supported by the existence of the sequentially diverse SS14-like strain, TPA Mexico A, isolated in 1953 (Pětrošová et al., 2012). To further address this question, additional molecular typing studies, accompanied by whole genome sequencing of genetically diverse MLST types, would be needed.
The methyl-directed enrichment used in this study allowed us to discover that about 50% of this genetic diversity was a result of inter-clade and/or intra-strain recombination events. Although molecular mechanisms of inter-clade and/or intra-strain recombination events could differ, both these processes can provide new alleles to TPA strains that are positively selected by the immune host system. In fact, most of the detected variability within both SS14- and Nichols-like clades predominantly led to non-synonymous amino acid changes which is consistent with positive selection of the corresponding genetic loci.
Intra-Strain Recombinant Events
As described previously, the mechanisms resulting in intra-strain recombinant events include gene conversion in regions with modular character, e.g., tpr (T. pallidum repeat) genes (predicted to code for the potential virulence factors) (Gray et al., 2006; Strouhal et al., 2018), duplication or deletion of repetitive sequences (arp, TP0470) (Harper et al., 2008; Šmajs et al., 2018) and reciprocal translocation (rrn operons, tprCD loci) (Čejková et al., 2013; Centurion-Lara et al., 2013).
The tprD2 allele, but not tprD (differing in 328 nucleotide positions), was previously predicted to be an outer membrane protein (Centurion-Lara et al., 2000), which suggests a different functional role for each allele during the course of infection. In our study, tprD2 alleles were found among all completely sequenced TPA isolates (belonging to both SS14- and Nichols-like clades) despite the fact, that tprD2 was previously believed to occur exclusively among SS14-like strains and tprD allele among Nichols-like strains (Fraser et al., 1998; Zobaníková et al., 2012; Centurion-Lara et al., 2013). This suggests that in the ancestor of these Nichols-like strains, “tprD allele” arose by duplication (gene conversion) of the tprC gene. A similar finding was recently published by Kumar et al. (2018). Interestingly, although no such recombination was found among SS14-like strains, in the SS14 genome, a minor tprD allele has been found in the tprD locus (Pětrošová et al., 2013).
Similar recombination of predicted virulence factors identified among the clinical samples in this study was observed between the tprG and tprJ genes resulting in a new tprG2 allele in Nichols-like clinical samples. The same pattern of recombination was already predicted as possible in the work of Strouhal et al. (2018).
Another recombination found in this study resulted in new patterns in the TP0856 and TP0858 genes, following a pattern previously recognized among TPE strain Kampung-Dalan K363, TPA SEA81-4 (Strouhal et al., 2018) and TPE strains from the Solomon Islands (Marks et al., 2018). Both these proteins showed structural similarity to FadL, a long-chain fatty acid transporter (Kumar et al., 2018) required for the specific binding and transport of exogenous long-chain fatty acids prior to metabolic utilization. Moreover, the predicted 3D structures of TP0858 revealed that the recombination-driven diversity found almost entirely corresponds to residues located at the host-pathogen interface (Figure 6).
Finally, as a result of reciprocal translocation, we identified an inverse rrn spacer pattern in one of the Nichols-like samples. However, these rrn spacer patterns (Ile/Ala or Ala/Ile) appeared to be distributed randomly across species/subspecies classification, time and the geographical source of the treponemal strains (Čejková et al., 2013) and the impact of this intra-strain recombination remains unknown.
Inter-Clade Recombinant Events
Treponema pallidum subsp. pallidum is not considered a competent bacterium and does not possess gene transfer mechanisms. In addition, no plasmid or phages have been described as of yet. Despite this, several apparent recombinant loci appear to result from inter-clade genetic recombinations. Previous studies identified such recombinations in TP0136 (Arora et al., 2016; Grillová et al., 2018c), TP0548 (Mikalová et al., 2017b), TP0326, TP0488 (Pětrošová et al., 2012), and TP0865 (Arora et al., 2016). In this study, we have identified five new recombinant loci representing one half of all inter-clade recombinant genes identified to date. We have identified recombination events between Nichols-like and SS14-like strains (in TP0117 [tprC], TP0136, TP0317 [tprG]); and Nichols-like strains and bejel treponemes (or possibly in some cases also TPE) (TP0117 [tprC], TP0462, TP0483, TP0621 [tprJ], TP0865) (Figure 5). In the case of tprG, we have found both intra-strain recombinations (recombination between tprG and tprJ genes resulting in a new tprG2 allele) and inter-clade recombination (in the Nichols-like strain CW30 with sequence originating from SS14-like strains, Figure 3). In addition, the recombinant regions identified in the new tprC3 and tprC4 alleles found in this study (Supplementary Figure S2) correspond to the identified extracellular loops (L3, L4, L5) of the β-barrel outer membrane protein which were predicted to serve as B-cells epitopes (Kumar et al., 2018). Similarly, the newly predicted 3D structures of protein TP0865 in this study (Figure 6) showed the accumulation of recombination-driven diversity in the residues located at the host-pathogen interface.
While the intra-strain recombinant events are relatively easy to explain, the presence of inter-clade recombinations would require a DNA transfer to the recipient bacterium from the outside, likely during co-infection of patients with treponemes either belonging to two different syphilis clades (Nichols, SS14) or two different subspecies of Treponema (causing syphilis and bejel). Cross-immunity experiments (Turner and Hollander, 1957) revealed that there is no protective immunity between different TPA strains and between TPA and TEN strains enabling co-infections or overlapping infections of different treponemal strains. The subsequent homologous recombination of DNA taken up by recipient cells could provide alleles encoding protein sequences allowing persistence and escape from the immune response of the host.
In addition to inter-clade and intra-strain recombinations identified in this study, we have analyzed all publicly available TPA genome sequences (Supplementary Table S4) for the presence of such recombinant events. Among whole genome sequences (n = 20), we identified a SEA81-4 strain (Nichols-like strain; CP003679.1) that carries the tprG2 allele and the r3r4r6 modular structure of TP0858 gene as results of intra-strain recombination events and we have identified TP0865 and tprJ as recombinant loci. Among draft genomes (n = 74), we have identified strains UW189B as inter-clade recombinant in both the TP0462 and TP0865 loci.
Given the adaptive evolution operating within both clades of syphilis treponemes, recombinant loci appear to be important in the treponemal pathogenesis and bacterium-host interactions. Despite the fact that TPA contains a low abundance of surface-exposed antigens, most of the Tpr proteins and recombinant proteins including TP0136, TP0326, TP0462, TP0483, TP0488, TP0548, and TP0865 encode for outer membrane proteins which are either targets for interactions with the immune system or structures enabling binding to host tissues (Brinkman et al., 2008; Arora et al., 2016; Kumar et al., 2018). In general, these proteins should be an important candidates for vaccine development. This is opportune, since several research teams are currently working on the development of a vaccine against syphilis. A comprehensive syphilis vaccine needs to react with antigens present in the reference strains but also on variants among contemporary TPA strains circulating in the human population. The current research identifying molecular types of TPA strains and their subsequent genomic analyses should be able to provide the required inventory of treponemal antigens and their variants.
Materials and Methods
Collection of Clinical Samples
Clinical samples were collected between 2013 and 2016 from several clinical departments in the Czechia (Department of Dermatovenerology, University Hospital Brno, Czechia); France (Institut Cochin U1016 Equipe Batteux, Laboratoire de Dermatologie–CNR Syphilis, Faculté de Médecine, Université Sorbonne Paris Descartes, Paris, France); Cuba (Instituto de Medicina Tropical “Pedro Kourí”, Havana, Cuba) and Australia (Melbourne Sexual Health Centre, Australia) (Table 1). Patients were considered as syphilis-positive when clinical symptoms were combined with positive syphilis serology or with positive PCR detection of treponemal DNA. All clinical samples were received after patients signed an informed-consent form and the written informed consent was obtained. The design of the study was approved by the ethics committee of the Faculty of Medicine, Masaryk University and the study was conducted in compliance with the Declaration of Helsinki.
Isolation of DNA, MLST, and Quantification of Treponemal and Human DNA Present in the Clinical Material
Swab extracts (prepared by submersion of swabs into 1.5 ml of PBS and agitation for 5 min at room temperature) and tissue sample (25 mg) were used for isolation of DNA using a QIAamp DNA Blood Mini kit and a DNeasy Blood & Tissue Kit (QIAGEN, Hilden, Germany) according to manufacturer’s recommendations. Multi-locus sequence typing was performed as described previously (Grillová et al., 2018a). The sequences were submitted to BIGSdb of T. pallidum subsp. pallidum available at pubMLST (Grillová et al., 2019), and the allelic profiles, STs and clonal complexes were determined (Table 1).
The number of copies of treponemal and human DNA in samples subjected to the whole genome sequencing were determined by using Applied Biosystems® 7500 Real-Time PCR System (United States). Treponemal DNA was detected by primers Tpa_polA_F (5′-GAGTGTGCAGTCCGCTATGC-3′) and Tpa_polA_R (5′-AGGCAAAAGCGGCATTTCTA-3′) amplifying 381 bp partial sequence of the polA gene and the probe Tpa_polA_P (5′-FAM-TCCGCTTGGAAAGAGCA-BHQ1-3′) (Dubourg et al., 2015). Human DNA was detected by primers FP (5′-CCAAGTGTGAGGGCTGAAAAG-3′) and RP (5′-TGTTGTGGCTGATGAACTATAAAAGG-3′) targeting 80 bp partial sequence of the RNase P gene and the probe (5′-FAM-CCCCAGTCTCTGTCAGCACTCCCTTC-BHQ1-3′) (Chi et al., 2015). Real-time PCR mix was composed of 13 μl of QuantiFast® Probe PCR Kit (QIAGEN, Hilden, Germany) with ROX dye passive reference, 0.1 μl of each primer and 0.05 μl of the probe of 100 μM (final concentration 0.4 and 0.2 μM, respectively). Total volume of each PCR reaction was 25 μl. Cycling conditions were 95°C (3 min); and 95°C (10 s), and 65°C (31 s) for 40 cycles.
Methyl-Directed Enrichment Using Restriction Endonuclease DpnI
The endonuclease DpnI cleaves the tetramer GATC when methylated at the N6 position of adenine. When used under conditions which prevent digestion, DpnI binds the methylated tetramer which is distributed approximately every 256 bases on average in DAM positive bacteria such as TPA but is absent in mammalian genomes. This enables selective bacterial DNA enrichment from the excess of human DNA found in syphilis samples. To accomplish methyl-directed enrichment, clinical DNA samples (10–40 μl) were added to DpnI-coated beads in 1.7 mL Eppendorf tubes in a final volume of 50 μl, as described previously (Barnes et al., 2014). The beads were mixed by end-over-end rotation for 30 min and the DpnI-magnetic beads were separated using a magnetic stand. The beads were washed once with Wash Buffer (10 mM Tris pH 7.9, 500 mM NaCl, 10 mM CaCl2, 0.1% Tween 20) followed by a single Binding Buffer wash (10 mM Tris pH 7.9, 50 mM NaCl, 10 mM CaCl2, 0.01% Tween 20). DNA was eluted from beads by incubation with 20 μl of 5 M guanidinium thiocyanate at room temperature for 5 min and subsequently desalted via dialysis for 45 min using 20,000 MWCO Slide-A-Lyzer MINI dialysis cups (Thermo Scientific, Waltham, MA, United States).
Next Generation Sequencing
The Nextera XT DNA library Preparation Kit (Illumina, San Diego, CA, United States) was used to produce barcoded libraries for all DpnI enriched fractions. Library products were amplified using 19 cycles and DNA was purified with AMPure XP beads (New England BioLabs, Ipswich, MA, United States) with an elution volume of 20 μl. Library quality and size distributions were determined with the Lab Chip GX Touch-HT (Perkin Elmer, Waltham, MA, United States) and High Sensitivity DNA Analysis Kit (Perkin Elmer, Waltham, MA, United States). Libraries were diluted for sequencing and pooled as appropriate for the targeted sequencing depth on a single flow cell. NextSeq runs were prepared using NextSeq 500/550 300 Cycle High Output v2 (FC-404-2004), loaded at approximately 3.9 pM. All runs were configured to obtain 149 nucleotide paired-end read lengths.
The bioinformatic analysis was performed according to the pipeline described previously (Grillová et al., 2018b). Briefly, the quality check of the raw reads was performed using FastQC (v0.11.5, Andrews, 2010) and the raw reads were pre-processed using Cutadapt (v1.15, Martin, 2011) and Fastx-toolkit (v0.0.14, Gordon, 2014). The total set of pre-processed reads was mapped to the human genome reference (hg38) and the human-matching reads were removed using BBMap (v37.25, Bushnell, 2017). Subsequently, the remaining reads were mapped to the TPA reference genomes using BWA MEM (v0.7.15, Li, 2014). SS14-like strains were mapped to the SS14 TPA reference genome (GenBank Acc. No. CP004011.1) and Nichols-like strains were mapped to the Nichols TPA reference genome (GenBank Acc. No. CP004010.2). The post-processing of the mapping was performed using Samtools (v1.4, Li et al., 2009), Picard (v2.8.1, Broad Institute, 2015), GATK (v3.7, McKenna et al., 2010), and NGSUtils/bamutils (v0.5.9, commit a7f08f5, Breese and Liu, 2013). Low-quality mappings were omitted from the analyses (mapping quality; MAPQ < 40) as well as genome regions biased by mapping of short reads generated by NGS (e.g., repetitive and paralogous regions; Supplementary Table S2). Possible cross-mapping artifacts from other prokaryotes were removed by filtering of minimal alignment length (35 bp), maximum allowed mismatches (5 mismatches or 5% of the read length), and maximum soft-clipping (5% of the read length). The broad coverage was established based on at least three good-quality aligned reads. In parallel, de novo assemblies were performed by SPAdes assembler (v 3.11.1, Bankevich et al., 2012) using unfiltered reads aligned to the TPA reference (k-mer = 15, 21, 27, 33, 55, 77, 99, 127; minimal contig coverage 5). QUAST (v4.5, Gurevich et al., 2013) was used to evaluate the completeness of the assembly. Consensus sequences from reference-guided mapping and the de novo assemblies of TPA genomes were manually combined to create a final WGS for each sample. Jupyter Notebook containing workflow for methyl-directed enrichment sequencing analyses can be found at https://github.com/opplatek/bacterial_genome_analysis.
SNV and Indel Identification
Single nucleotide variants and insertions and deletions (indels) were identified using FreeBayes (v0.9.21-19-gc003c1e, Garrison and Marth, 2012) and filtered by vcflib (v 0.1.15, Garrison, 2016). Prior to the variant call, mappings were indel realigned (GATK) and PCR duplicates were removed (v3.7, Broad Institute, 2015). FreeBayes was run with recommended settings for bacterial samples by the tool authors (ploidy 1; minimal coverage depth 3; minimal variant quality 50). Filtered variants were annotated using SnpEff (v4.2, Cingolani et al., 2012) with corresponding GenBank annotations.
Completion of Whole Genomes
In the samples with the highest NGS broad coverage (>97%, n = 11), the Sanger sequencing of regions with low coverage was performed (approximately 10 regions for every genome). Moreover, paralogous tpr genes (tprC, tprD, tprE, tprF, tprG, tprI, tprJ) were amplified with Long-range PCR (Supplementary Table S2) under conditions described in the Quality of treponemal DNA paragraph and Sanger sequenced using sequencing primers presented in the Supplementary Table S5. The number of 60 bp-long repetitions in the arp gene and the number of 24 bp-long repetition in the TP0470 gene were determined by Sanger sequencing using primers listed in the Supplementary Table S2. The number of repetitions was verified by gel electrophoresis. To determine the intergenic spacers between rRNA encoding rrn loci, we designed primers for nested PCR using one unique primer in outer step for distinguishing Ala/Ile and Ile/Ala patterns (Supplementary Table S6). The Sanger sequencing reads were combined with the Illumina sequencing reads using Lasergene software (DNASTAR v. 7.1.0.; DNASTAR, Madison, WI, United States). The workflow to obtain the whole genome sequences of TPA is given in the Supplementary Figure S4.
Sequences of treponemal rRNA operons (rrn1, rrn2) were searched against the de novo assemblies (BLAST) (v2.2.31+, Camacho et al., 2009) and all the hits with more than 90% identity, and alignment length of 100 bp were extracted in fasta and bed format (Bedtools) (v2.27.0, Quinlan and Hall, 2010). We have used BLAST to discover all potential sequences of rRNA regions or their fragments. We manually inspected all the BLAST hits and selected those that most likely represented the real rRNA regions.
Phylogenetic Analyses
Maximum likelihood phylogeny based on whole genomes was done using MEGA (v6.0, Tamura et al., 2011) using the Tamura-Nei model and 1000 bootstrap replications. The visualization of the phylogenetic tree was done using iTOL (v4, Letunic and Bork, 2007). Median-joining (MJ) networks were generated with Network version 4 (Bandelt et al., 1999).
Detection of Recombination Events
Recombination patterns were identified by manual inspection of gene sequences that had a high number of SNVs (identified by SNV call) and displayed phylogeny incongruent with the one derived from whole genome sequences. The high number of SNVs was defined as the presence of at least 4 SNVs per gene, which is about 10-times higher number of polymorphic sites than expected between TPA clades (Pětrošová et al., 2013; Šmajs et al., 2016), and this threshold was calculated from the previously published recombination events found in other treponemal genomes (Pětrošová et al., 2012; Štaudová et al., 2014). The gene tree topology was tested against the tree topology derived from the whole genome sequences.
Pooled Segment Genome Sequencing (PSGS) of TPA Phi-1 and Grady Strains
Philadelphia 1 (Phi-1) strain was isolated in Philadelphia, United States in 1988 (Harper et al., 2008) and Grady strain was isolated in Atlanta United States in 1980s. Both strains were provided by David L. Cox (Centers for Disease Control and Prevention, Atlanta, GA, United States) as a rabbit testicular tissue containing treponemal cells. Whole genomic DNA was amplified from rabbit testicular tissue using QIAGEN REPLI-g kit (QIAGEN, Hilden, Germany) according to manufacturer’s instructions. Amplified DNA served as a template for T. pallidum intervals (TP intervals) amplification during the PSGS phase as described previously (Weinstock et al., 2000; Čejková et al., 2012; Strouhal et al., 2017).
The amplified TP intervals (n = 279 and 272) of the Phi-1 and Grady samples, respectively, were sequenced using the Illumina platform (NextSeq 500) at CEITEC (Brno, Czechia). To separate paralogous regions, the amplified TP intervals were labeled with multiplex identifier adapters and sequenced as four different samples (NexteraTM XT DNA Sample Preparation Kit, Illumina Inc., Madison, WI, United States). The sequencing reads were trimmed (Trimmomatic) (v0.32, Bolger et al., 2014), and low-quality bases were removed with a sliding window (window length of 4 nt; average quality of at least Phred 17). The sequencing reads shorter than 50 bp were omitted from the analyses. Reads were analyzed with respect to four distinct pools and were de novo assembled using SeqMan NGen software (v4.1.0, DNASTAR, Madison, WI, United States) as well as mapped to the TPA reference genome (GenBank Acc. No. CP004011.1).
Annotation of Complete Genomes
For gene annotation, Geneious software (v5.6.5, Biomatters ApS, Aarhus, Denmark) was used. The tprK gene showed intra-strain variability in all samples and the corresponding nucleotides positions were denoted as “N.” Raw data were deposited with NCBI under BioProject number: PRJNA508872 and the complete genome sequences can be found under the following Accession Numbers: CP034921, CP034920, CP034919, CP034918, CP034972, CP034917, CP034916, CP034915, CP034914, CP034913, CP034912, CP035104, and CP035193.
3D Structure Prediction
The 3D structures of TP0858 and TP0865 were generated using the SWISS-MODEL server (Waterhouse et al., 2018) using as templates the Protein Data Bank entries 3DWO and 3BS0, respectively. HHblits was used to find suitable template models (Remmert et al., 2011). The orientation of proteins with respect to the outer membrane corresponded to that predicted in the OPM database1 (Lomize et al., 2012). The TprC model was built using the TMBpro server (Randall et al., 2008) according to Kumar et al. (2018). The antigenic peptides were predicted by “Predicted Antigenic Peptides tool2.” Predictions were based on a table that reflects the occurrence of amino acid residues in experimentally known segmental epitopes. Segments were only reported if they had a minimum size of 8 residues.
Statements
Data availability statement
The datasets generated for this study can be found in NCBI, BioProject number: PRJNA508872.
Ethics statement
All patients signed the informed consent. The study protocol was approved by the Ethics Committee of all institutions involved in this study and was conducted in compliance with the Declaration of Helsinki.
Author contributions
LGr, RF, and DŠ designed the experiments. LGr, LM, MN, AN, PP, and CW performed the experiments. LGr, JO, AM, and DČ analyzed data. LGr, AAN, PG, ND, RS, MC, ID, and NA collected clinical samples. LGr, MP, and DŠ wrote the manuscript. All authors provided critical feedback.
Funding
This research was supported by funds from the Faculty of Medicine, Masaryk University to junior researchers (LGr, MN, and PP), the Grant Agency of the Czechia (GA17-25455S) and by the Ministry of Health of the Czechia (17-31333A) to DŠ. Core Facility Bioinformatics of CEITEC Masaryk University is gratefully acknowledged for the obtaining of the scientific data presented in this manuscript. Computational resources were provided by the CESNET LM2015042 and the CERIT Scientific Cloud LM2015085, provided under the program “Projects of Large Research, Development, and Innovations Infrastructures”.
Acknowledgments
We would like to thank Prof. Nicholas Robert Thomson (Wellcome Sanger Institute) for his valuable insights during the preparation of the manuscript. We also thank Robert Anthony Gaultney for his assistance with the English revision of the manuscript.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01691/full#supplementary-material
References
1
AndrewsS. (2010). FastQC A Quality Control tool for High Throughput Sequence Data.Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed June 09, 2018).
2
AroraN.SchuenemannV. J.JägerG.PeltzerA.SeitzA.HerbigA.et al (2016). Origin of modern syphilis and emergence of a pandemic Treponema pallidum cluster.Nat. Microbiol.2:16245. 10.1038/nmicrobiol.2016.245
3
BandeltH. J.ForsterP.RöhlA. (1999). Median-joining networks for inferring intraspecific phylogenies.Mol. Biol. Evol.1637–48. 10.1093/oxfordjournals.molbev.a026036
4
BankevichA.NurkS.AntipovD.GurevichA. A.DvorkinM.KulikovA. S.et al (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing.J. Comput. Biol.19455–477. 10.1089/cmb.2012.0021
5
BarnesH. E.LiuG.WestonC. Q.KingP.PhamL. K.WaltzS.et al (2014). Selective microbial genomic DNA isolation using restriction endonucleases.PLoS One9:e109061. 10.1371/journal.pone.0109061
6
BolgerA. M.LohseM.UsadelB. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data.Bioinformatics302114–2120. 10.1093/bioinformatics/btu170
7
BreeseM. R.LiuY. (2013). NGSUtils: a software suite for analyzing and manipulating next-generation sequencing datasets.Bioinformatics29494–496. 10.1093/bioinformatics/bts731
8
BrinkmanM. B.McGillM. A.PetterssonJ.RogersA.MatejkovaP.ŠmajsD.et al (2008). A novel Treponema pallidum antigen, TP0136, is an outer membrane protein that binds human fibronectin.Infect. Immun.761848–1857. 10.1128/IAI.01424-07
9
Broad Institute (2015). Picard Toolkit.Available at: http://broadinstitute.github.io/picard/ (accessed October 24, 2018).
10
BushnellB. (2017). BBMap.Available at: sourceforge.net/projects/bbmap/ (accessed May 25, 2017).
11
CamachoC.CoulourisG.AvagyanV.MaN.PapadopoulosJ.BealerK.et al (2009). BLAST+: architecture and applications.BMC Bioinformatics10:421. 10.1186/1471-2105-10-421
12
ČejkováD.ZobaníkováM.ChenL.PospíšilováP.StrouhalM.QinX.et al (2012). Whole genome sequences of three Treponema pallidum ssp. Pertenue strains: yaws and syphilis treponemes differ in less than 0.2% of the genome sequence.PLoS Negl. Trop. Dis.6:e1471. 10.1371/journal.pntd.0001471
13
ČejkováD.ZobaníkováM.PospísilováP.StrouhalM.MikalováL.WeinstockG. M.et al (2013). Structure of rrn operons in pathogenic non-cultivable treponemes: sequence but not genomic position of intergenic spacers correlates with classification of Treponema pallidum and Treponema paraluiscuniculi strains.J. Med. Microbiol.62(Pt 2)196–207. 10.1099/jmm.0.050658-0
14
Centurion-LaraA.GiacaniL.GodornesC.MoliniB. J.Brinck ReidT.LukehartS. A. (2013). Fine analysis of genetic diversity of the tpr gene family among treponemal species, subspecies and strains.PLoS Negl. Trop. Dis.7:e2222. 10.1371/journal.pntd.0002222
15
Centurion-LaraA.SunE. S.BarrettL. K.CastroC.LukehartS. A.Van VoorhisW. C. (2000). Multiple alleles of Treponema pallidum repeat gene D in Treponema pallidum isolates.J. Bacteriol.1822332–2335. 10.1128/jb.182.8.2332-2335.2000
16
ChiK.-H.DanavallD.TaleoF.PillayA.YeT.NachamkinE.et al (2015). Molecular differentiation of Treponema pallidum subspecies in skin ulceration clinically suspected as yaws in Vanuatu using real-time multiplex PCR and serological methods.Am. J. Trop. Med. Hyg.92134–138. 10.4269/ajtmh.14-0459
17
CingolaniP.PlattsA.WangL. L.CoonM.NguyenT.WangL.et al (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3.Fly680–92. 10.4161/fly.19695
18
DubourgG.EdouardS.PrudentE.FournierP.-E.RaoultD. (2015). Incidental syphilis diagnosed by real-time PCR screening of urine samples.J. Clin. Microbiol.533707–3708. 10.1128/jcm.01026-15
19
EdmondsonD. G.HuB.NorrisS. J. (2018). Long-Term In Vitro culture of the syphilis spirochete Treponema pallidum subsp. pallidum.mBio9:e01153-18. 10.1128/mBio.01153-18
20
FlasarováM.PospíšilováP.MikalováL.VališováZ.DastychováE.StrnadelR.et al (2012). Sequencing-based molecular typing of treponema pallidum strains in the Czech Republic: all identified genotypes are related to the sequence of the SS14 strain.Acta. Derm. Venereol.92669–674. 10.2340/00015555-1335
21
FraserC. M.NorrisS. J.WeinstockG. M.WhiteO.SuttonG. G.DodsonR.et al (1998). Complete genome sequence of Treponema pallidum, the syphilis spirochete.Science281375–388. 10.1126/science.281.5375.375
22
Gallo VauletL.GrillováL.MikalováL.CascoR.Rodríguez FermepinM.PandoM. A.et al (2017). Molecular typing of Treponema pallidum isolates from Buenos Aires, Argentina: frequent nichols-like isolates and low levels of macrolide resistance.PLoS One12:e0172905. 10.1371/journal.pone.0172905
23
GarrisonE. (2016). Vcflib, A Simple C++ Library for Parsing and Manipulating VCF files.Available at: https://github.com/vcflib/vcflib (accessed July 28, 2016).
24
GarrisonE.MarthG. (2012). Haplotype-Based Variant Detection from Short-Read Sequencing.Available from: http://arxiv.org/abs/1207.3907 (accessed October 24, 2018).
25
GiacaniL.Iverson-CabralS. L.KingJ. C. K.MoliniB. J.LukehartS. A.Centurion-LaraA. (2014). Complete genome sequence of the Treponema pallidum subsp. pallidum Sea81-4 Strain.Genome Announc.2:e0333-14. 10.1128/genomeA.00333-14
26
GiacaniL.JeffreyB. M.MoliniB. J.LeH. T.LukehartS. A.Centurion-LaraA.et al (2010). Complete genome sequence and annotation of the Treponema pallidum subsp. pallidum Chicago strain.J. Bacteriol.1922645–2646. 10.1128/JB.00159-10
27
GordonA. (2014). FASTX-Toolkit: FASTQ/A Short-Reads Pre-Processing Tools.Available at: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed November 29, 2016).
28
GrayR. R.MulliganC. J.MoliniB. J.SunE. S.GiacaniL.GodornesC.et al (2006). Molecular evolution of the tprC, D, I, K, G, and J genes in the pathogenic genus Treponema.Mol. Biol. Evol.232220–2233. 10.1093/molbev/msl092
29
GrillováL.BawaT.MikalováL.Gayet-AgeronA.NieseltK.StrouhalM.et al (2018a). Molecular characterization of Treponema pallidum subsp. pallidum in Switzerland and France with a new multilocus sequence typing scheme.PLoS One13:e0200773. 10.1371/journal.pone.0200773
30
GrillováL.GiacaniL.MikalováL.StrouhalM.StrnadelR.MarraC.et al (2018b). Sequencing of Treponema pallidum subsp. pallidum from isolate UZ1974 using anti-treponemal antibodies enrichment: first complete whole genome sequence obtained directly from human clinical material.PLoS One13:e0202619. 10.1371/journal.pone.0202619
31
GrillováL.NodaA. A.LienhardR.BlancoO.RodríguezI.ŠmajsD. (2018c). Multilocus sequence typing of Treponema pallidum subsp. pallidum in Cuba from 2012 to 2017.J. Infect. Dis.2191138–1145. 10.1093/infdis/jiy604
32
GrillováL.JolleyK.ŠmajsD.PicardeauM. (2019). A public database for the new MLST scheme for Treponema pallidum subsp. Pallidum: surveillance and epidemiology of the causative agent of syphilis.PeerJ6:e6182. 10.7717/peerj.6182
33
GrillováL.PetrošováH.MikalováL.StrnadelR.DastychováE.KuklováI.et al (2014). Molecular typing of Treponema pallidum in the Czech Republic during 2011 to 2013: increased prevalence of identified genotypes and of isolates with macrolide resistance.J. Clin. Microbiol.523693–3700. 10.1128/JCM.01292-14
34
GurevichA.SavelievV.VyahhiN.TeslerG. (2013). QUAST: quality assessment tool for genome assemblies.Bioinformatics291072–1075. 10.1093/bioinformatics/btt086
35
HarperK. N.OcampoP. S.SteinerB. M.GeorgeR. W.SilvermanM. S.BolotinS.et al (2008). On the origin of the treponematoses: a phylogenetic approach.PLoS Negl. Trop. Dis.2:e148. 10.1371/journal.pntd.0000148
36
KnaufS.GogartenJ. F.SchuenemannV. J.De NysH. M.DüxA.StrouhalM.et al (2018). Nonhuman primates across sub-Saharan Africa are infected with the yaws bacterium Treponema pallidum subsp. pertenue.Emerg. Microbes Infect.7:157.
37
KumarS.CaimanoM. J.AnandA.DeyA.HawleyK. L.LeDoytM. E.et al (2018). Sequence variation of rare outer membrane protein β-barrel domains in clinical strains provides insights into the evolution of Treponema pallidum subsp. pallidum, the Syphilis Spirochete.mBio9:e01006-18. 10.1128/mBio.01006-18
38
LetunicI.BorkP. (2007). Interactive tree of life (iTOL): an online tool for phylogenetic tree display and annotation.Bioinformatics23127–128. 10.1093/bioinformatics/btl529
39
LiH. (2014). Toward better understanding of artifacts in variant calling from high-coverage samples.Bioinformatics302843–2851. 10.1093/bioinformatics/btu356
40
LiH.HandsakerB.WysokerA.FennellT.RuanJ.HomerN.et al (2009). The sequence alignment/map format and SAMtools.Bioinformatics252078–2079. 10.1093/bioinformatics/btp352
41
LomizeM. A.PogozhevaI. D.JooH.MosbergH. I.LomizeA. L. (2012). OPM database and PPM web server: resources for positioning of proteins in membranes.Nucleic Acids Res.40D370–D376. 10.1093/nar/gkr703
42
MarksM.FookesM.WagnerJ.ButcherR.GhinaiR.SokanaO.et al (2018). Diagnostics for yaws eradication: insights from direct next-generation sequencing of cutaneous strains of Treponema pallidum.Clin. Infect. Dis.66818–824. 10.1093/cid/cix892
43
MartinM. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads.EMBnet.J.17:10. 10.14806/ej.17.1.200
44
MatĕjkováP.StrouhalM.ŠmajsD.NorrisS. J.PalzkillT.PetrosinoJ. F.et al (2008). Complete genome sequence of Treponema pallidum ssp. pallidum strain SS14 determined with oligonucleotide arrays.BMC Microbiol.8:76. 10.1186/1471-2180-8-76
45
McKennaA.HannaM.BanksE.SivachenkoA.CibulskisK.KernytskyA.et al (2010). The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data.Genome Res.201297–1303. 10.1101/gr.107524.110
46
MikalováL.GrillováL.OsbakK.StrouhalM.KenyonC.CrucittiT.et al (2017a). Molecular typing of syphilis-causing strains among human immunodeficiency virus-positive patients in antwerp, Belgium.Sex Transm. Dis.44376–379. 10.1097/OLQ.0000000000000600
47
MikalováL.StrouhalM.OppeltJ.GrangeP. A.JanierM.BenhaddouN.et al (2017b). Human Treponema pallidum 11q/j isolate belongs to subsp. Endemicum but contains two loci with a sequence in TP0548 and TP0488 similar to subsp. pertenue and subsp. pallidum, respectively.PLoS Negl. Trop. Dis.11:e0005434. 10.1371/journal.pntd.0005434
48
NechvátalL.PětrošováH.GrillováL.PospíšilováP.MikalováL.StrnadelR.et al (2014). Syphilis-causing strains belong to separate SS14-like or Nichols-like groups as defined by multilocus analysis of 19 Treponema pallidum strains.Int. J. Med. Microbiol.304645–653. 10.1016/j.ijmm.2014.04.007
49
NewmanL.RowleyJ.Vander HoornS.WijesooriyaN. S.UnemoM.LowN.et al (2012). Global estimates of the prevalence and incidence of four curable sexually transmitted infections in based on systematic review and global reporting.PLoS One10:e0143304. 10.1371/journal.pone.0143304
50
PeelingR. W.MabeyD.KambM. L.ChenX.-S.RadolfJ. D.BenzakenA. S. (2017). Syphilis.Nat. Rev. Dis. Primers.3:17073. 10.1038/nrdp.2017.73
51
PětrošováH.PospíšilováP.StrouhalM.ČejkováD.ZobaníkováM.MikalováL.et al (2013). Resequencing of Treponema pallidum ssp. pallidum strains Nichols and SS14: correction of sequencing errors resulted in increased separation of syphilis treponeme subclusters.PLoS One8:e74319. 10.1371/journal.pone.0074319
52
PětrošováH.ZobaníkováM.ČejkováD.MikalováL.PospíšilováP.StrouhalM.et al (2012). Whole genome sequence of Treponema pallidum ssp. pallidum, strain Mexico A, suggests recombination between yaws and syphilis strains.PLoS Negl. Trop. Dis.6:e1832. 10.1371/journal.pntd.0001832
53
PintoM.BorgesV.AnteloM.PinheiroM.NunesA.AzevedoJ.et al (2016). Genome-scale analysis of the non-cultivable Treponema pallidum reveals extensive within-patient genetic variation.Nat. Microbiol.2:16190. 10.1038/nmicrobiol.2016.190
54
PospíšilováP.GrangeP. A.GrillováL.MikalováL.MartinetP.JanierM.et al (2018). Multi-locus sequence typing of Treponema pallidum subsp. Pallidum present in clinical samples from France: infecting treponemes are genetically diverse and belong to 18 allelic profiles.PLoS One13:e0201068. 10.1371/journal.pone.0201068
55
QuinlanA. R.HallI. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features.Bioinformatics26841–842. 10.1093/bioinformatics/btq033
56
RadolfJ. D.DekaR. K.AnandA.ŠmajsD.NorgardM. V.YangX. F. (2016). Treponema pallidum, the syphilis spirochete: making a living as a stealth pathogen.Nat. Rev. Microbiol.14744–759. 10.1038/nrmicro.2016.141
57
RandallA.ChengJ.SweredoskiM.BaldiP. (2008). TMBpro: secondary structure, beta-contact and tertiary structure prediction of transmembrane beta-barrel proteins.Bioinformatics24513–520. 10.1093/bioinformatics/btm548
58
RemmertM.BiegertA.HauserA.SödingJ. (2011). HHblits: lightning-fast iterative protein sequence searching by HMM-HMM alignment.Nat. Methods9173–175. 10.1038/nmeth.1818
59
ŠmajsD.MikalovaL.StrouhalM.GrillováL. (2016). Why are there two genetically distinct syphilis-causing strains?Forum Immunopathol. Dis. Ther.7181–190. 10.1615/forumimmundisther.2017020184
60
ŠmajsD.StrouhalM.KnaufS. (2018). Genetics of human and animal uncultivable treponemal pathogens.Infect. Genet. Evol.6192–107. 10.1016/j.meegid.2018.03.015
61
StammL. V.GreeneS. R.BarnesN. Y.BergenH. L.HardhamJ. M. (1997). Identification and characterization of a Treponema pallidum subsp. Pallidum gene encoding a DNA adenine methyltransferase.FEMS Microbiol. Lett.155115–119. 10.1016/s0378-1097(97)00375-3
62
ŠtaudováB.StrouhalM.ZobaníkováM.ČejkováD.FultonL. L.ChenL.et al (2014). Whole genome sequence of the Treponema pallidum subsp. Endemicum strain BosniaA: the genome is related to yaws treponemes but contains few loci similar to syphilis treponemes.PLoS Negl. Trop. Dis.8:e3261. 10.1371/journal.pntd.0003261
63
StolteI. G.DukersN. H.de WitJ. B.FennemaJ. S.CoutinhoR. A. (2001). Increase in sexually transmitted infections among homosexual men in Amsterdam in relation to HAART.Sex Transm. Infect.77184–186. 10.1136/sti.77.3.184
64
StrouhalM.MikalováL.HaviernikJ.KnaufS.BruistenS.NoordhoekG. T.et al (2018). Complete genome sequences of two strains of Treponema pallidum subsp. Pertenue from Indonesia: modular structure of several treponemal genes.PLoS Negl. Trop. Dis.12:e0006867. 10.1371/journal.pntd.0006867
65
StrouhalM.MikalováL.HavlíčkováP.TentiP.ČejkováD.RychlíkI.et al (2017). Complete genome sequences of two strains of Treponema pallidum subsp. Pertenue from Ghana, Africa: identical genome sequences in samples isolated more than 7 years apart.PLoS Negl. Trop. Dis.11:e0005894. 10.1371/journal.pntd.0005894
66
TamuraK.PetersonD.PetersonN.StecherG.NeiM.KumarS. (2011). MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods.Mol. Biol. Evol.282731–2739. 10.1093/molbev/msr121
67
TongM.-L.ZhaoQ.LiuL.-L.ZhuX.-Z.GaoK.ZhangH.-L.et al (2017). Whole genome sequence of the Treponema pallidum subsp. Pallidum strain amoy: an asian isolate highly similar to SS14.PLoS One12:e0182768. 10.1371/journal.pone.0182768
68
TurnerT. B.HollanderD. H. (1957). Biology of the treponematoses based on studies carried out at the international treponematosis Laboratory Center of the Johns Hopkins University under the auspices of the World Health Organization.Monogr. Ser. World Health Organ.353–266.
69
WaterhouseA.BertoniM.BienertS.StuderG.TaurielloG.GumiennyR.et al (2018). SWISS-MODEL: homology modelling of protein structures and complexes.Nucleic Acids Res.46W296–W303. 10.1093/nar/gky427
70
WeinstockG. M.SmajsD.HardhamJ.NorrisS. J. (2000). From microbial genome sequence to applications.Res. Microbiol.151151–158. 10.1016/s0923-2508(00)00115-7
71
WoznicováV.SmajsD.WechslerD.MatĕjkováP.FlasarováM. (2007). Detection of Treponema pallidum subsp. pallidum from skin lesions, serum, and cerebrospinal fluid in an infant with congenital syphilis after clindamycin treatment of the mother during pregnancy.J. Clin. Microbiol.45659–661. 10.1128/jcm.02209-06
72
ZobaníkováM.MikolkaP.ČejkováD.PospíšilováP.ChenL.StrouhalM.et al (2012). Complete genome sequence of Treponema pallidum strain DAL-1.Stand. Genomic Sci.712–21. 10.4056/sigs.2615838
Summary
Keywords
Treponema pallidum subsp. pallidum, syphilis, direct whole genome sequencing, recombination-driven diversity, culture-independent bacterial enrichment
Citation
Grillová L, Oppelt J, Mikalová L, Nováková M, Giacani L, Niesnerová A, Noda AA, Mechaly AE, Pospíšilová P, Čejková D, Grange PA, Dupin N, Strnadel R, Chen M, Denham I, Arora N, Picardeau M, Weston C, Forsyth RA and Šmajs D (2019) Directly Sequenced Genomes of Contemporary Strains of Syphilis Reveal Recombination-Driven Diversity in Genes Encoding Predicted Surface-Exposed Antigens. Front. Microbiol. 10:1691. doi: 10.3389/fmicb.2019.01691
Received
28 February 2019
Accepted
09 July 2019
Published
31 July 2019
Volume
10 - 2019
Edited by
Vasco Ariston De Carvalho Azevedo, Federal University of Minas Gerais, Brazil
Reviewed by
Jason Sahl, Northern Arizona University, United States; Sandeep Tiwari, Federal University of Minas Gerais, Brazil; Guilherme Campos Tavares, Universidade Nilton Lins, Brazil; Alice Rebecca Wattam, University of Virginia, United States; Ulisses Padua Pereira, State University of Londrina, Brazil
Updates
Copyright
© 2019 Grillová, Oppelt, Mikalová, Nováková, Giacani, Niesnerová, Noda, Mechaly, Pospíšilová, Čejková, Grange, Dupin, Strnadel, Chen, Denham, Arora, Picardeau, Weston, Forsyth and Šmajs.
This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Šmajs, dsmajs@med.muni.cz
†These authors have contributed equally to this work
This article was submitted to Evolutionary and Genomic Microbiology, a section of the journal Frontiers in Microbiology
Disclaimer
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.