 Roxanne A. Beinart1*
Roxanne A. Beinart1* Chengwei Luo2†
Chengwei Luo2† Konstantinos T. Konstantinidis2,3
Konstantinos T. Konstantinidis2,3 Frank J. Stewart3
Frank J. Stewart3 Peter R. Girguis4
Peter R. Girguis4- 1Graduate School of Oceanography, University of Rhode Island, Narragansett, RI, United States
- 2School of Civil and Environmental Engineering, Georgia Institute of Technology, Atlanta, GA, United States
- 3School of Biological Sciences, Georgia Institute of Technology, Atlanta, GA, United States
- 4Department of Organismic and Evolutionary Biology, Harvard University, Cambridge, MA, United States
Symbiosis has evolved between a diversity of invertebrate taxa and chemosynthetic bacterial lineages. At the broadest level, these symbioses share primary function: the bacterial symbionts use the energy harnessed from the oxidation of reduced chemicals to power the fixation of inorganic carbon and/or other nutrients, providing the bulk of host nutrition. However, it is unclear to what extent the ecological niche of the host species is influenced by differences in symbiont traits, particularly those involved in chemoautotrophic function and interaction with the geochemical environment. Hydrothermal vents in the Lau Basin (Tonga) are home to four morphologically and physiologically similar snail species from the sister genera Alviniconcha and Ifremeria. Here, we assembled nearly complete genomes from their symbionts to determine whether differences in chemoautotrophic capacity exist among these symbionts that could explain the observed distribution of these snail species into distinct geochemical habitats. Phylogenomic analyses confirmed that the symbionts have evolved from four distinct lineages in the classes γ-proteobacteria or Campylobacteria. The genomes differed with respect to genes related to motility, adhesion, secretion, and amino acid uptake or excretion, though were quite similar in chemoautotrophic function, with all four containing genes for carbon fixation, sulfur and hydrogen oxidation, and oxygen and nitrate respiration. This indicates that differences in the presence or absence of symbiont chemoautotrophic functions does not likely explain the observed geochemical habitat partitioning. Rather, differences in gene expression and regulation, biochemical differences among these chemoautotrophic pathways, and/or differences in host physiology could all influence the observed patterns of habitat partitioning.
Introduction
Symbiosis with chemosynthetic bacteria has independently evolved in many hydrothermal vent, cold seep, and shallow-water invertebrate taxa. Broadly speaking, the animal hosts rely on the fixed carbon produced by their obligate symbionts for the majority of their nutrition (Dubilier et al., 2008). The bacterial partners of such associations are phylogenetically diverse, representing multiple bacterial lineages across two phyla. Nevertheless, with respect to energy metabolism, they perform similar metabolic functions: these symbionts use reduced chemicals like hydrogen sulfide as electron donors, respire oxygen and/or nitrate, and fix inorganic carbon into organic carbon (Dubilier et al., 2008; Childress and Girguis, 2011).
Chemoautotrophic symbionts can, at times, exhibit marked differences in gene content, which can subsequently confer differences in metabolism that may have ecological implications for their host species. For example, a comparison of the genomic content among some of the γ-proteobacterial symbionts of vent animals has shown that they can differ in a diversity of genes and gene networks, from energy metabolism to nitrogen acquisition (Kleiner et al., 2012). While it is reasonable to assert that genomic content that confers symbiont functional traits could affect the distribution of host species into distinct physicochemical habitats, it is still unknown whether this occurs. Interpretation of the similarities and differences in the genomes of chemosynthetic symbionts has been confounded by the fact that the comparisons to date have been made among symbionts from highly divergent host taxa and/or from different geographical locations with distinct environmental characteristics and biogeographic histories. Thus, it is difficult to untangle whether observed differences in symbiont gene content are driven by co-evolution with physiologically and ecologically distinct host taxa, or adaptation to dissimilar habitats. Comparisons among the symbionts of closely related host species can help resolve whether differences in symbiont genomic traits impact their distribution into distinct geochemical habitats.
The hydrothermal vent snail genera Alviniconcha and Ifremeria provide a unique opportunity to examine gene content differences among the symbionts of closely related and regionally sympatric host species that occupy distinct geochemical habitats. Alviniconcha and Ifremeria are sister genera of provannid gastropod molluscs (snails) that dominate hydrothermal vent communities in the southwestern Pacific (Desbruyères et al., 1994). At vents along the Eastern Lau Spreading Center (Tonga), three species of Alviniconcha (A. boucheti, A. kojimai, and A. strummeri), co-occur with each other and with Ifremeria nautilei (Beinart et al., 2012; Johnson et al., 2015). These snail species associate with a total of four distinct phylotypes from the phylum Proteobacteria (class γ-proteobacteria) and one from the phylum Campylobacterota (class Campylobacteria), which are all hosted as intracellular gill endosymbionts (Suzuki et al., 2005a, b, 2006a, b; Urakawa et al., 2005; Beinart et al., 2012). The highly reduced gut and carbon stable isotopic composition of host tissue indicates that these symbionts provide nutrition for their hosts through chemoautotrophy (Waren and Bouchet, 1993; Suzuki et al., 2005a, b, 2006a, b; Beinart et al., 2012). Since they represent different bacterial lineages and do not show a pattern of co-divergence with host species (Suzuki et al., 2006a; Beinart et al., 2012), the symbionts are thought to be acquired horizontally from the environment. However, there is strong specificity between each snail species and its symbiont phylotypes(s): surveys of hundreds of individuals across a range of habitats have demonstrated that each snail species associates with only one or two of the symbiont phylotypes, with individual snails usually dominated by just one symbiont phylotype at a time (Table 1; Beinart et al., 2012; Seston et al., 2016).

Table 1. Specificity between host species and possible symbiont phylotypes, with symbiont taxonomy to the level of class.
As such, it is plausible that differences in gene content among these symbionts could influence the geochemical niche of the host snails at the Eastern Lau Spreading Center. Indeed, each snail species is restricted to associations with only one or two of the symbiont phylotypes, so differences in symbiont physiological traits could partition host species into distinct habitats based on these traits. The four snail species have similar anatomy in terms of overall body size, as well as gill and circulatory structure (Waren and Bouchet, 1993; Johnson et al., 2015). Despite these shared features, Alviniconcha and I. nautilei occupy distinct geochemical habitats (Waite et al., 2008; Podowski et al., 2009, 2010; Beinart et al., 2012), and this segregation is thought to primarily be driven by the distribution of reducing compounds that could be used by their particular symbionts for chemoautotrophy (Henry et al., 2008; Beinart et al., 2012, 2015; Sanders et al., 2013). Within a single vent field, Alviniconcha and I. nautilei show consistent patterns of zonation, with Alviniconcha species found nearest to vent orifices and I. nautilei on the edges of Alviniconcha patches (Podowski et al., 2009, 2010). Thus, compared to I. nautilei, Alviniconcha species occur where concentrations of vent-derived reductants and temperatures are higher, and oxygen and thiosulfate concentrations are lower (Waite et al., 2008; Podowski et al., 2009, 2010). However, at a regional scale, the dominant Alvinconcha species at each vent field varies according to the particular geochemistry of that site (Beinart et al., 2012). Specifically, the Alviniconcha species (A. boucheti) hosting the Campylobacterial symbiont dominates at vent fields with higher concentrations of hydrogen and hydrogen sulfide, whereas the Alviniconcha species (A. kojimai and A. strummeri) hosting γ-proteobacterial symbiont phylotypes dominate at vents with lower concentrations of these two reductants (Beinart et al., 2012). These vent fields are separated by 10 s to 100 s of kilometers, but are part of one biogeographic region with no known barriers to dispersal among sites (Speer and Thurnherr, 2012; Mitarai et al., 2016), suggesting that the distribution of Alviniconcha species could be tied to how their specific symbionts interact with the varying concentrations of chemical reductants at each site. Physiological experiments have directly demonstrated that some of these species can oxidize hydrogen sulfide and thiosulfate to support high rates of carbon fixation (Henry et al., 2008; Beinart et al., 2015). In addition, studies of gene transcription in situ and in laboratory experiments have demonstrated differences in expression of genes for sulfur oxidation, hydrogen oxidation, and both assimilatory and dissimilatory nitrate reduction (Sanders et al., 2013; Seston et al., 2016). These studies provide an understanding of metabolic pathway use and rates under conditions at the time of sampling and treatment, but do not allow for a comparison of the full metabolic and physiological potential of each symbiont.
To investigate whether variation in chemoautotrophic capacity (i.e., the genomic content of each specific symbiont) confers differences in energy and carbon metabolism that play a role in the distribution of the host species, we assembled the genomes of the dominant four symbiont phylotypes associated with Alviniconcha species and I. nautilei from the Eastern Lau Spreading Center (Tonga). We specifically concentrate on comparisons of the genes for chemoautotrophic metabolism of these symbionts, since the host species segregate into distinct niches that differ primarily in the chemicals used by the symbionts for chemoautotrophy (hydrogen, sulfur) (Waite et al., 2008; Podowski et al., 2009, 2010; Beinart et al., 2012), suggesting that their symbionts could differ in their capacity to use these chemicals. Subsequent phylogenomic and comparative analyses with these nearly complete genomes demonstrated that the symbionts, although representing multiple lineages, exhibit broad similarity in the representation of genes associated with chemoautotrophic energy generation and biosynthesis. These data underscore that gene content alone is not robust as a causal factor for the observed habitat partitioning among these symbioses. Rather, these data raise alternative hypotheses about the factors controlling habitat partitioning, for example the extent to which differential gene expression and regulation, biochemical differences in the particular pathways employed by the symbionts, and the possibility of uncharacterized differences in host or symbiont physiology, play a role in governing the distribution of Alviniconcha species and I. nautilei.
Materials and Methods
Sample Collection
Snails were collected from the ABE, Tu’i Malila, and Kilo Moana vent fields at the Eastern Lau Spreading Center (Table 1) by the remotely operated vehicle JASON II during the R/V Thomas G. Thompson expedition TM-235 in 2009. Alviniconcha were recovered in insulated containers and, once on board, were briefly kept in 4°C seawater until dissection of symbiont-containing gill tissue. Gill tissues were stored at −80°C until DNA extraction. DNA was extracted from 25 mg subsamples of frozen gill tissue using the AutoGenprep 965 automatic extraction system with the AutoGenprep 965/960 Tissue DNA Extraction kit (AutoGen, Inc.), as previously described (Beinart et al., 2012). The I. nautilei individual used for sequencing was part of a high-pressure physiological experiment (see Beinart et al., 2015). A the conclusion of the experiment, symbiont-containing gill tissue was dissected, homogenized in TrizolTM (Thermo Fisher Scientific, Inc.) preservative, and stored at −80°C until DNA extraction as described in Seston et al. (2016).
Sequencing, Genomic Bin Assembly, and Annotation
Gill DNA consists of both host and symbiont genomic material. Thus, the resulting metagenomic sequences are a metagenomic mixture of both host and symbiont genomes. Two separate approaches were used to assemble the symbiont genomes from these metagenomes. For the symbionts of Alviniconcha, a novel method was developed to separate symbiont reads from host reads prior to assembly (described herein). For the I. nautilei symbionts, all reads were assembled, and then binning of contigs based on an Expectation-Maximization algorithm was used to separate symbiont contigs from each other and the host genome (Wu et al., 2014).
Three Alviniconcha and one I. nautilei individuals were selected for sequencing (Table 1). Each Alviniconcha individual was dominated by only one of the three phylotypes of bacterial symbiont, as previously assessed via symbiont-specific quantitative PCR assays (see Beinart et al., 2012). The I. nautilei individual (Table 1) had been previously shown, via 16S rRNA gene amplicon sequencing, to be dominated by one major symbiont phylotype from the order Chromatiales (Ifr1), with a very small sub-population of a second phylotype from the family Methylococcaceae in some individuals (Ifr2) (Seston et al., 2016). Symbiont 16S rRNA gene sequence similarity is >98% among host individuals sharing the same symbiont phylotype (Beinart et al., 2012, 2015; Seston et al., 2016). This low level of intra-phylotype diversity is at approximately the threshold typically used to differentiate bacterial species (Konstantinidis et al., 2017). Therefore, each sequenced individual (host) in our study acts as a representative for its phylotype, with each phylotype constituting a sub-species-level population. In addition, the Alviniconcha individuals were genotyped via amplification and sequencing of the host mitochondrial CO1 gene (to date Alviniconcha species cannot be distinguished morphologically, see (Beinart et al., 2012; Johnson et al., 2015; Table 1).
Gill DNA from the three Alviniconcha individuals was used for Illumina HiSeq sequencing (read length: 2 × 150 bp). Reads were trimmed using the protocol described in Luo et al. (2011), followed by a K-mer-based algorithm to separate the Alviniconcha reads and the bacterial reads. The algorithm leverages the fact that the Alviniconcha snail genome is significantly larger than the bacterial genome, and hence, random reads generation would result in a bivariate normal distribution in read coverage. Such a bivariate normal distribution would be reflected in K-mer frequency, which can be used as described below as a method for read separation:
Assuming the Alviniconcha genome S size is s, and the symbiont bacterial genome Q size is q, for a given K-mer, ki, its occurrence in the Alviniconcha genome is Ns(ki) and its occurrence in the bacterial genome is Ng1(ki). Also, assuming that there are n reads of l –bp long each in the sample, and denoting the Alviniconcha reads’ fraction, R, then the number of Alviniconcha base pairs in reads is nlR, and the number of bacterial base pairs in reads is nl(1-R). Assuming DNA sequencing covers the genomes uniformly, for a given K-mer, ki, the expected observance, Nm(ki), is:
We denote the K-mers a read contains as K = {k1, k2, …, km} with corresponding occurrences O = {o1, o2, …, om}. We can transform occurrences into ranks, denoted as R = {r1, r2, …, rm}. With these, the likelihood that a paired-end read, r, is from the symbiotic bacterial genome is:
P(r ∈ q) and P(r ∈ q) represent the probability of a read originating from the bacterial genome and from the Alviniconcha genome, respectively, and they can be initialized by using the average genome sizes of the closest relative available. In our practice, we have found that since the genome sizes are significantly different, the accuracy of the genome size estimation has little effect on the end results.
P(O|r ∈ q) is the conditional probability of observing such K-mer occurrences as in O if a read was originated from the symbiotic bacterial genome q, and it was calculated as:
where P(oj|r ∈ q) can be approximated using the discrete ranks as:
where K is the set of all ranks of K-mers in the metagenome.
As such, for each read, we counted the K-mer occurrences using Jellyfish (Marçais and Kingsford, 2011), and calculated the probability of it originating from Alviniconcha. A K-means (K = 2) clustering was performed on to separate the reads. The Velvet assembler (Zerbino and Birney, 2008) was then used with K = 41 and default settings to assemble the reads into a cluster identified as the symbiotic bacterium.
Gill DNA from I. nautilei was sequenced on an Illumina MiSeq (2 × 300 bp). Resulting sequences were quality filtered and trimmed with TrimGalore!1 using a read quality cutoff of Q25, read length cutoff of 100 and Nextera flag for adapter detection. Approximately 21 million read pairs were assembled using IDBA-UD with default parameters (Peng et al., 2012). Using MaxBin v1.4.5 (Wu et al., 2014), two I. nautilei symbiont genome bins were recovered from the 294 Mb assembly that included both host and symbiont contigs.
In all cases, only contigs >500 bp were used for downstream analyses of the putative symbiont genome bins. Symbiont genome assembly characteristics, completeness, contamination, quality, and taxonomy assignment were assessed via the Microbial Genomes Atlas (MiGA) webserver (Rodriguez-R et al., 2018). Gene prediction and annotation were done with RASTtk (Brettin et al., 2015) using the default workflow. Sequences annotated as hydrogenases were further classified with HydDB (Søndergaard et al., 2016).
Phylogenomic Analysis
All Chromatiales (NCBI Tax. ID: 135613), Sulfurimonas (NCBI Tax. ID 202746), and sulfur-oxidizing symbiont (NCBI Tax. ID: 32036) genomes from NCBI were placed into a bacterial phylogeny with the Alviniconcha and I. nautilei symbiont genomes using a 6,989 position amino acid alignment of 43 concatenated phylogenetically informative marker genes with PPlacer (Matsen et al., 2010) in CheckM (Parks et al., 2015). A subset of the genomes most closely related to each of the Alviniconcha and I. nautilei symbiont genomes were selected for further phylogenetic analysis, and the marker gene alignment of each subset was exported from CheckM (Parks et al., 2015). The best model for amino acid evolution was determined for each alignment using ModelGenerator v0.851 (Keane et al., 2006): LG + G + F for the Alviniconcha ε and γ-1 alignments, and LG + I + G + F for the alignment containing the two other symbionts (γ-Lau and Ifr1). Maximum likelihood phylogenies were constructed with RAxML v.8.2.10 (Stamatakis, 2014) via the CIPRES web server (Miller et al., 2010). Clade support was estimated using bootstrapping halted automatically by RAxML. Average amino acid identities (AAI) were calculated for each symbiont and its closest relative using the AAI tool that is part of the enveomics collection (Rodriguez-R and Konstantinidis, 2016).
Results and Discussion
Genome Assemblies
A single symbiont genome, representing the previously identified dominant symbiont, was assembled from each Alviniconcha individual, while two symbiont genomes were assembled from the I. nautilei individual (herein, called Ifr1 and Ifr2). Each symbiont genome contained a single-copy 16S rRNA gene that had 99% identity with the published 16S rRNA genes for the symbionts of Alviniconcha and I. nautilei in the Lau Basin (Beinart et al., 2012; Seston et al., 2016). The 16S rRNA genes from each genome also matched the dominant symbiont phylotype expected for each individual based on previous assessment of the symbionts associated with each snail by qPCR or 16S rRNA gene amplicon sequencing (Beinart et al., 2012; Seston et al., 2016). The Alviniconcha symbionts had previously been called the ε, γ-Lau, and γ-1 symbiont phylotypes (Beinart et al., 2012; Johnson et al., 2015). Taxonomy assignment based on marker gene sequences was possible to the level of family for the A. ε symbiont (Helicobacteraceae) and the Ifr2 symbiont (Methylococcaceae). The A. γ-Lau, Ifr1, and A. γ-1 symbiont were classified to the level of the class γ-proteobacteria (Table 2). Because the Ifr2 symbiont genome bin, which represents the minority symbiont in I. nautilei (Seston et al., 2016), was large (∼8 Mbp), highly fragmented, had a lower estimated completion (62%), relatively high level of contamination (6%), and intermediate quality (31%) (Table 2), it was not included in further analysis. In contrast, the other four symbiont genomes were near complete (87–96%), with low estimated contamination (<4%), high to excellent quality (69–90%), sizes from 2.0 to 3.7 Mb, and GC content from 33 to 59% (Table 2).
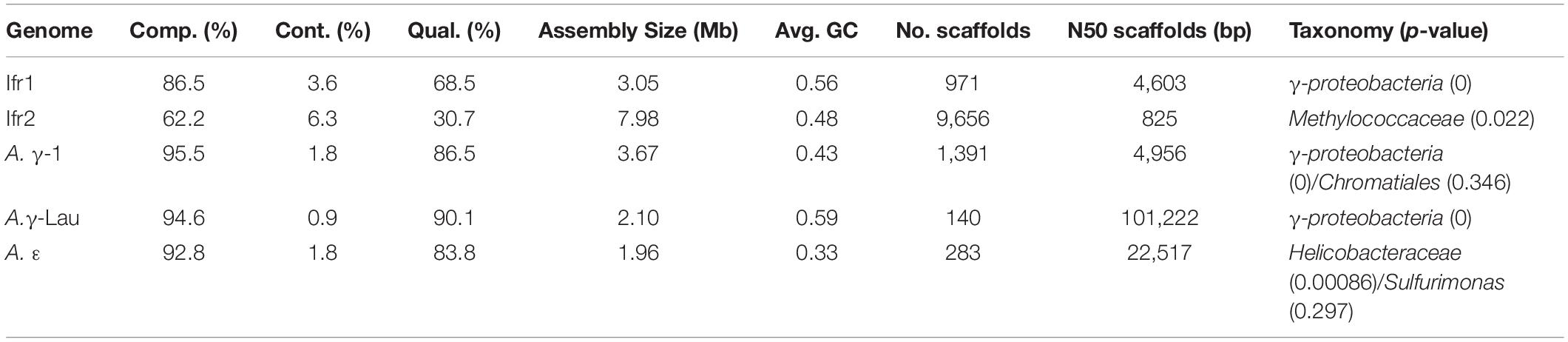
Table 2. Characteristics of symbiont genomes including the percent completeness (Comp.) and contamination (Cont.), the total size, average GC content, number of contigs, N50 of the contigs, and taxonomy assigned to lowest possible rank.
Phylogenomic Analysis
Phylogenomic analyses of the four symbiont genomes based on 43 marker genes showed that each was affiliated with a different clade of γ-proteobacteria or Campylobacteria (Figures 1–3), confirming previous 16S rRNA gene phylogenies and indicating that the Alviniconcha and I. nautilei symbionts were acquired separately by each host species (Urakawa et al., 2005; Suzuki et al., 2005a, b, 2006a, b; Beinart et al., 2012). The A. γ-Lau and Ifr1 symbionts clustered in a clade that is entirely comprised of genomes assembled from host-associated, chemoautotrophic symbionts, with the exception of that from Thiolapillus brandeum (NCBI Assembly: GCA_000828615), a free-living close relative of the Ifr1 symbiont that was isolated from a western Pacific hydrothermal vent (Nunoura et al., 2014; Figure 1). T. brandeum has 97% identity with the Ifr1 16S rRNA gene and a two-way average amino acid identity (AAI) of 76% (1998 proteins). A. γ-Lau is very closely related to the symbiont of Chrysomallon squamiferum, a snail from hydrothermal vents in the Indian Ocean (Nakagawa et al., 2014), showing 97% 16S rRNA gene identity, and 88% two-way AAI (1547 proteins). These 16S rRNA gene identities and AAIs represent values that are typically seen between bacterial species from the same genus (Rodriguez-R and Konstantinidis, 2014).
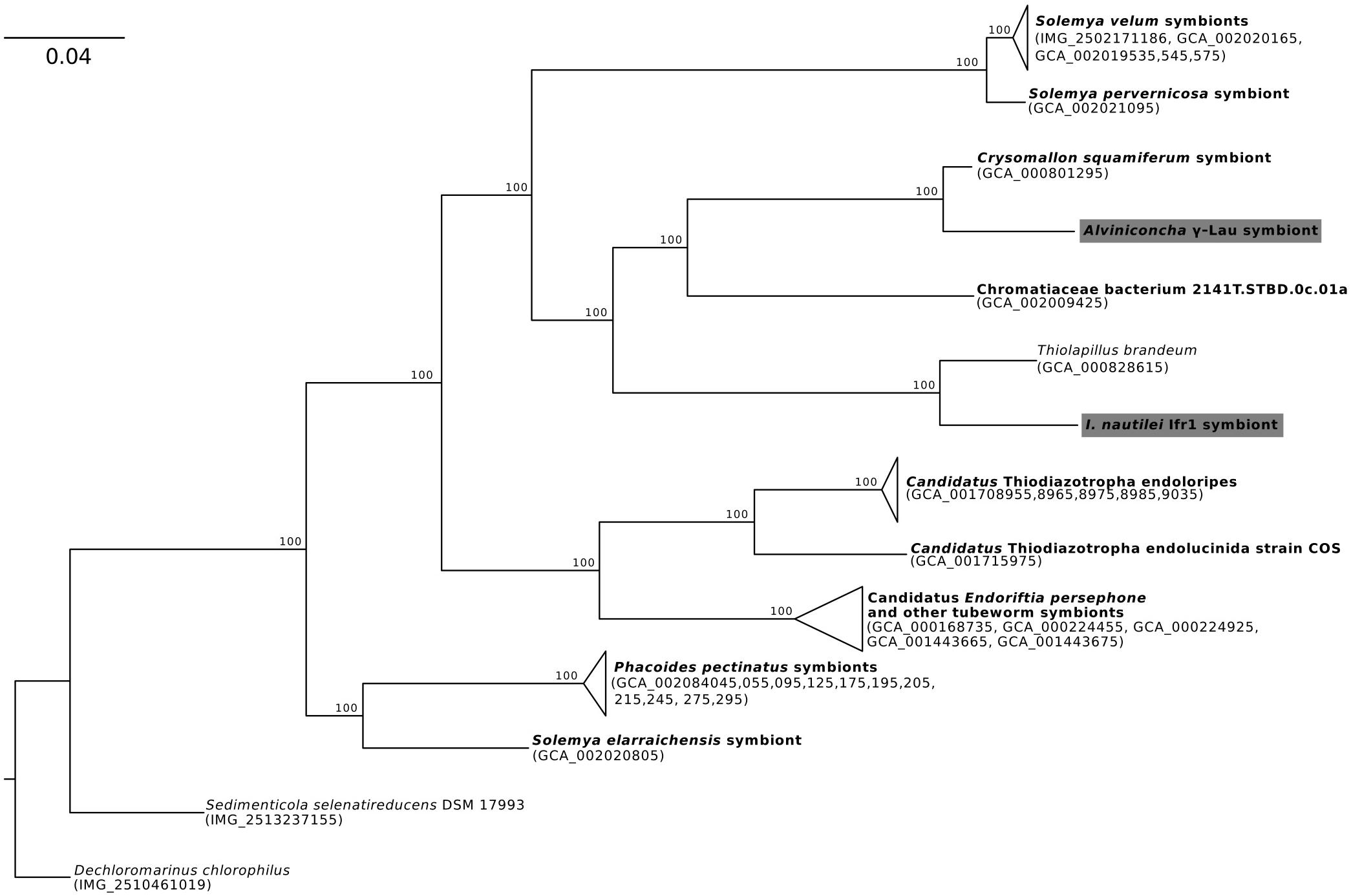
Figure 1. Maximum-likelihood phylogenomic analysis of a 6,989 position concatenated protein alignment of 43 marker genes from the Alviniconcha γ-Lau symbiont, I. nautilei Ifr1 symbiont, and relatives using the LG + I + G + F model of protein evolution. Symbiont genomes from this study are highlighted in gray, and genomes derived from host-associated symbionts are shown in bold. Percent bootstrap support for each clade shown. The scale bar represents the mean number of nucleotide substitutions per site.
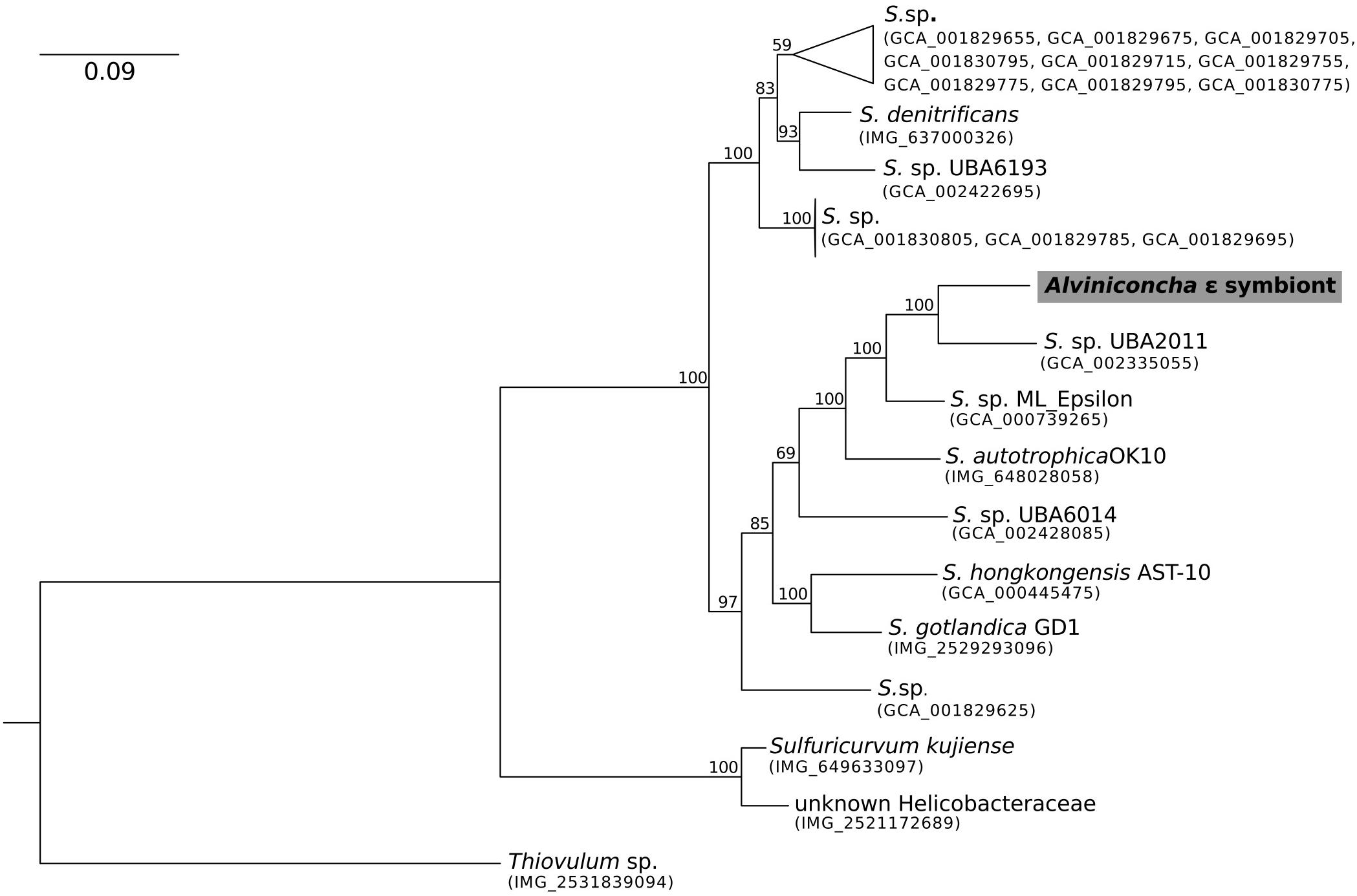
Figure 2. Maximum-likelihood phylogenomic analysis of a 6,989 position concatenated protein alignment of 43 marker genes from the Alviniconcha ε symbiont and its Sulfurimonas relatives using the LG + G + F model of protein evolution. The symbiont genome from this study is highlighted in gray. Percent bootstrap support for each clade shown. The scale bar represents the mean number of nucleotide substitutions per site.
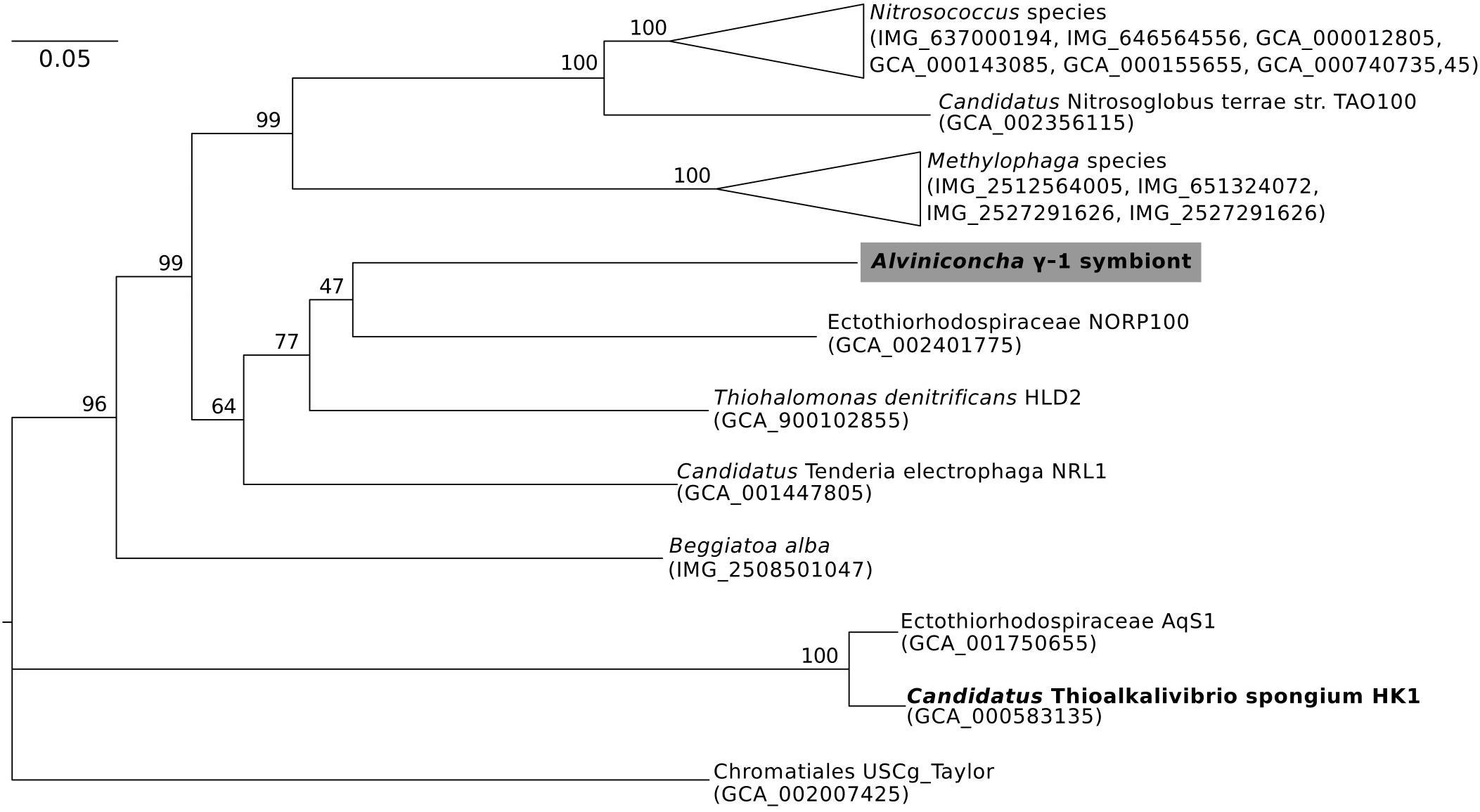
Figure 3. Maximum-likelihood phylogenomic analysis of a 6,989 position concatenated protein alignment of 43 marker genes from Alviniconcha γ-1 and relatives using the LG + G + F model of protein evolution. The symbiont genome from this study is highlighted in gray, and genomes derived from host-associated symbionts are shown in bold. Percent bootstrap support for each clade shown. The scale bar represents the mean number of nucleotide substitutions per site.
In contrast, the closest relatives of the other two Alviniconcha symbiont genomes were from environmental samples, not known to be associated with a host, and were much more divergent from the symbionts (Figures 2, 3). The A. ε symbiont clustered within a clade of bacteria in the genus Sulfurimonas and was most closely related to a Sulfurimonas metagenome-assembled genome (MAG) from a hydrothermal vent plume in the Caribbean (NCBI Assembly: GCA_002335055) (Figure 2). This Sulfurimonas MAG does not contain a 16S rRNA gene, but showed 36% two-way AAI (1139 proteins) with the A. ε symbiont genome. The A. γ-1 symbiont clustered within a larger clade that included both free-living Methylophaga and Nitrosococcus species, but its closest relative was a MAG from an oceanic subsurface aquifer (NCBI Assembly: GCA_002401775) (Figure 3). This MAG does not contain a 16S rRNA gene, but showed 50% two-way AAI (1552 proteins) with the A. γ-1 symbiont.
Overall Gene Content
Between 2072 and 4214 gene coding sequences were found in each symbiont genome assembly (Supplementary Tables S1, S2). Each assembly contained a single-copy 23S rRNA gene in addition to the 16S rRNA gene (reported above). However, the 5S rRNA gene was present in the Ifr1 and A. γ-Lau symbiont assemblies only. Of the protein-coding sequences, approximately half in each genome were annotated as hypothetical proteins (Supplementary Table S1). The overall composition of functional genes (evaluated at the level of SEED category) was relatively similar among genomes – in all genomes, the most abundant gene categories were “Protein Metabolism” and “Amino Acids and Derivatives” (Figure 4). The proportion of genes annotated to the “DNA Metabolism” category was high in all three γ-proteobacterial symbiont genome assemblies, but lower in the A. ε genome assembly (Figure 4). The four symbiont genomes shared 586 core orthologs (Supplementary Tables S2, S3), which represent 36–50% of the genes in each genome. Of the 59% of the core orthologs that could be placed into SEED categories, the most abundant were placed in “Protein metabolism,” “Amino acids and derivatives,” and “Cofactors, Vitamins, Prosthetic Groups, Pigments;” altogether, these three categories represented about a third of the core orthologs (Supplementary Table S3). Each genome also encoded between 129 and 345 genes with unique annotations not found in any of the other three genomes, representing 10–30% of the total annotations in each genome (Supplementary Table S2). In all four genomes, most of the unique genes could not be put into SEED categories. Of the unique genes that could be classified by SEED, most of those from the γ-proteobacterial symbiont genomes were in either the “DNA metabolism” or “Membrane transport” categories (Figure 5), whereas those from the A. ε genome were primarily in the “Motility and chemotaxis” and “Amino acids and derivatives” categories (Figure 5).

Figure 4. Heatmap showing the number of genes annotated to each SEED category within each symbiont genome. Hierarchical clustering based on Euclidean distance with average linkage.
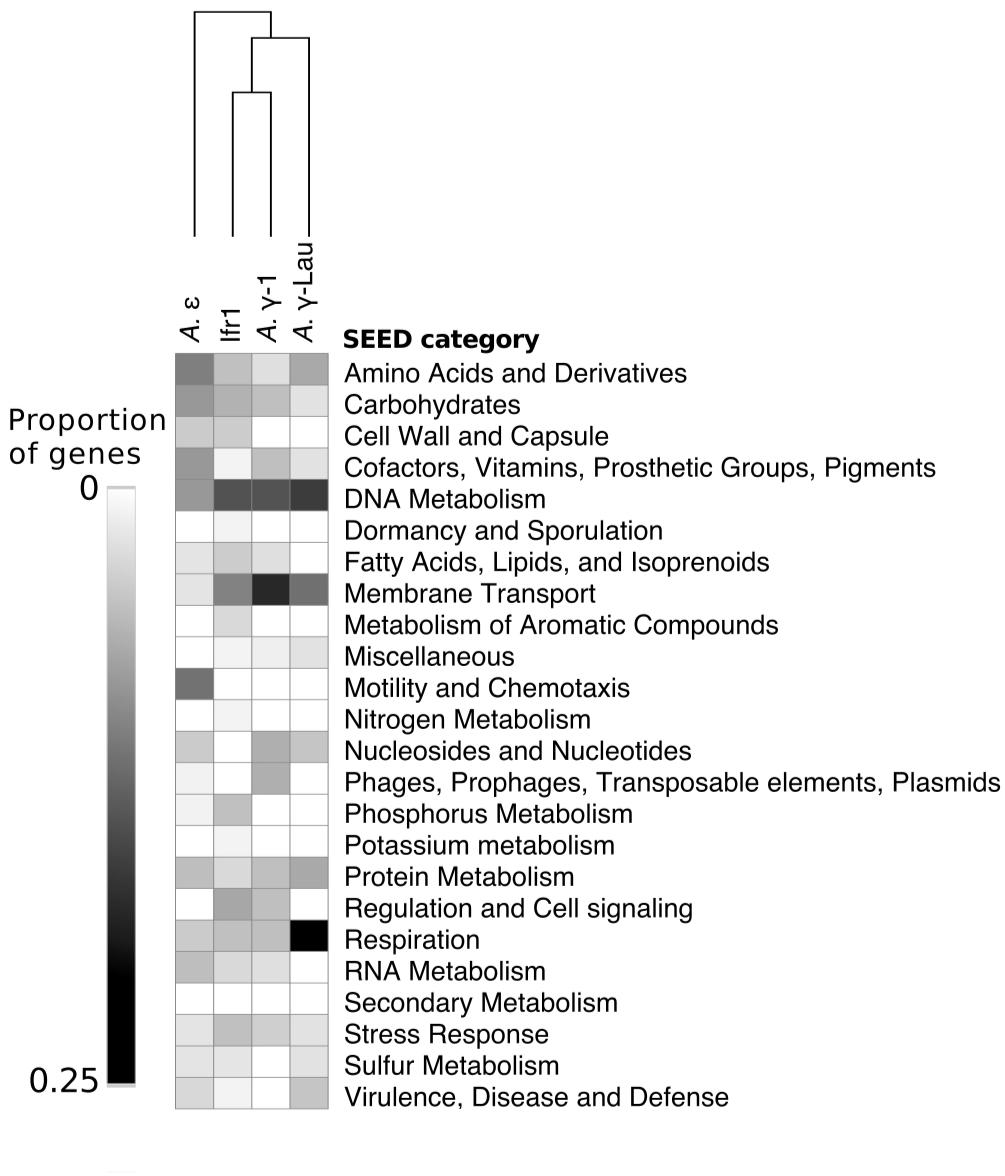
Figure 5. Heatmap showing the proportion of unique genes (i.e., those not present in any other genome) annotated to each SEED category from each symbiont genome. Hierarchical clustering based on Euclidean distance with average linkage.
Sulfur Oxidation
All of the snail symbiont genomes revealed pathways for the oxidation of both hydrogen sulfide and thiosulfate. As suggested in a previous analysis of symbiont gene expression (Sanders et al., 2013; Seston et al., 2016), all four symbiont genomes encoded the thiosulfate-oxidizing Sox multienzyme complex (soxABXYZ), with only the Alviniconcha ε symbiont encoding the genes soxCD. The lack of soxCD in the γ-proteobacterial symbionts is typical of this taxonomic class (Gregersen et al., 2011), and usually results in only partial oxidation of sulfur to elemental sulfur in the periplasm. The γ-proteobacterial symbiont genomes encoded genes for the complete oxidation of this elemental sulfur through the reverse dissimilatory sulfate reduction pathway (rdsrABMKOP), APS-reductase (aprAB), and sulfate adenylyltransferase (sat).
In addition, all four symbiont genomes contained genes for the oxidation of hydrogen sulfide to elemental sulfur in the periplasm. All of the symbiont genomes contained a type VI sulfide:quinone oxidoreductase (sqrF), which has been shown to be important for growth at high sulfide concentrations (>4 mM) in a photosynthetic sulfur-oxidizer (Gregersen et al., 2011). Each genome also contained an additional type of sulfide:quinone oxidoreductase: the γ-proteobacterial symbionts each additionally encoded a type I (sqrA), while the A. ε symbiont encoded a type IV (sqrD). Finally, the Ifr1 symbiont and the A. γ-1 symbiont genome each additionally had a gene for a flavocytochrome c:sulfide dehydrogenase (fcc) which is also thought to oxidize sulfide to elemental sulfur or polysulfides in the periplasm (Chen et al., 1994). These particular sets of sqr and fcc genes are common in the symbionts’ respective taxonomic groups (Gregersen et al., 2011; Han and Perner, 2015), and may be useful for dealing with different concentrations of available sulfide (Han and Perner, 2016).
All four snail symbiont genomes encoded at least one gene for a sulfate permease or transporter, which could be used for the excretion of sulfate (the end-product of sulfur oxidation) or the uptake of thiosulfate (Aguilar-Barajas et al., 2011). Hydrogen sulfide can freely diffuse across membranes (Mathai et al., 2009), while hydrosulfide ions (HS-) must move through a membrane channel (Czyzewski and Wang, 2012). No genes for a hydrosulfide ion channel, which is related to channels for nitrate and formate (Czyzewski and Wang, 2012), were detected in any of the snail symbiont genomes.
Hydrogen Oxidation
Each of the symbiont genomes encoded genes for two types of hydrogenases, a respiratory version that enables the use of hydrogen as an electron donor, and regulatory version that allows for sensing and response to hydrogen concentrations. The A. ε symbiont genome contained a group 1b quinone-reactive NiFe hydrogenase (hydABC), which is a prototypical hydrogenase that is used for anaerobic respiration, for example with nitrate (Søndergaard et al., 2016). Each γ-proteobacterial symbiont genome contained a group 1d oxygen-tolerant NiFe hydrogenase (hyaABC) that typically pairs hydrogen oxidation with the respiration of oxygen or with oxygen-tolerant anaerobic respiration (Søndergaard et al., 2016). In addition to its respiratory hydrogenase, the A. ε symbiont genome contained genes for a NiFe hydrogenase (hyaAB) that is classified within the group 2d Aquificae-type hydrogenases, which currently have unknown function but are thought to either be regulatory or provide reducing power for carbon fixation (Søndergaard et al., 2016). The γ-proteobacterial symbiont genomes also each included a group 2b NiFe sensory hydrogenase (hoxBC/hupUV) that can sense hydrogen and regulate the expression of other hydrogenases (Peters et al., 2015). Based on previously observed variation in symbiont hydrogenase gene expression (Sanders et al., 2013) and the predominance of Alviniconcha species hosting the A. ε symbiont at vent fields with higher hydrogen concentrations (Beinart et al., 2012), it has been hypothesized that Alviniconcha symbionts have differing capacities for the use of hydrogen as an energy source. Notably, the gene content observed here suggests that all of these snail symbionts have the ability to oxidize hydrogen, though it remains to be tested whether hydrogen can provide energy for autotrophy and, if so, how hydrogen use varies depending on environmental conditions.
Respiration
All of the snail symbiont genomes encoded the genes for aerobic respiration. All snail symbiont genomes contained genes for NADH-ubiquinone oxidoreductase (subunits ABCDEFGHIJKLMN, though subunit F is missing from the A. ε symbiont genome). The Ifr1, A. ε, and A. γ-1 symbiont genomes each had some of the genes for a succinate dehydrogenase. All of the symbiont genomes had all necessary genes for cytochrome bc1-type ubiquinol oxidoreductase. The Ifr1 and the A. γ-proteobacterial symbionts all had five genes for different types of cytochrome c, while the A. ε symbiont had only two cytochrome c genes. All had genes for the cytochrome cbb3-type oxidase (ccoNOPQ, though ccoQ was not found in the A. γ-1 symbiont genome). The Ifr1 symbiont genome also encoded subunits I and II for a cytochrome bd-type ubiquinol oxidase, which has a high affinity for oxygen (D’Mello et al., 1996). The presence of a high affinity cytochrome in the Ifr1 symbiont is unexpected, given that I. nautilei typically occupies habitats with relatively high oxygen concentrations (Podowski et al., 2009, 2010). The presence of this gene may indicate that the symbionts of I. nautilei experience relatively low intracellular oxygen concentrations, despite the environmental conditions.
The four symbiont genomes all also encoded genes for anaerobic respiration. For the symbionts of oxygen-respiring animals, the ability to utilize an electron acceptor other than oxygen may be useful as a way to avoid competing with host respiration. Additionally, hydrothermal vent habitats have the potential to be low in oxygen, which could necessitate the use of anaerobic respiration by the symbionts, either during association with a host or a free-living, environmental stage. All symbiont genomes encoded genes for a periplasmic nitrate reductase (napABGH), as well as for the complete denitrification pathway to nitrogen gas: nitric-oxide forming nitrite reductase (nirKN), nitric-oxide reductase (norBC), and nitrous-oxide reductase (nosZ). This suggests that all four symbionts can respire nitrate. In addition, the γ-proteobacterial symbiont genomes all contained genes for an anaerobic dimethyl sulfoxide (DMSO) reductase (dmsABC), which would allow them to respire dimethyl sulfoxide. Dimethyl sulfide (DMS) and other organic sulfur compounds are present around hydrothermal vents (Rogers and Schulte, 2012), and it is possible that DMSO is present in habitats occupied by Alviniconcha and I. nautilei, and could be used for anaerobic respiration by the symbionts.
Carbon Fixation and Heterotrophy
All of the symbiont genomes encoded genes for the fixation of inorganic carbon. The γ-proteobacterial symbiont genomes each contained the genes for the Calvin-Benson-Bassham (CBB) cycle, including genes for a Form II RubisCO (cbbM). The A. ε symbiont genome had the genes for key enzymes involved in the reductive tricarboxylic acid (rTCA) cycle: ATP-citrate lyase (aclAB), 2-oxoglutarate:ferredoxin oxidoreductase (oorABCD), and fumarate reductase/succinate dehydrogenase (SDHAB), as well as all other genes necessary for rTCA except for the gene encoding phosphoenolpyruvate (PEP) synthetase (ppsA). The pathways found in each symbiont genome are typical of their taxonomic groups (i.e., Campylobacteria and γ-proteobacteria) (Hügler and Sievert, 2011). Further, the stable isotopic fingerprints of these pathways are reflected in the carbon stable isotopic composition of Alviniconcha species and I. nautilei, with A. boucheti (hosting the A. ε symbiont) having a much less negative ∂13C than the γ-proteobacteria hosting Alviniconcha species or I. nautilei (Beinart et al., 2012, 2015). Though the CBB cycle is more commonly found in aerobic organisms, while the rTCA cycle is more typical in microaerophilic or anaerobic organisms (Hügler and Sievert, 2011), oxygen concentrations around Alviniconcha species are not significantly different (Beinart et al., 2012) and Alviniconcha species are found at lower oxygen concentrations then I. nautilei, regardless of which symbiont they host (Podowski et al., 2009, 2010). Thus, an interaction between carbon fixation pathways and oxygen habitat is not likely to be important to habitat partitioning in these species.
In addition to autotrophy, all of the snail symbiont genomes indicated the capacity for heterotrophy. All of the symbiont genomes contained the genes for tripartite ATP-independent periplasmic (TRAP) dicarboxylate transporters, which would allow for the acquisition of four-carbon compounds like succinate and fumarate (Mulligan et al., 2011). In addition, both the Ifr1 and the A. γ-1 symbiont genomes contained the gene for an acetate permease (actP), which can transport both acetate and glycolate (Gimenez et al., 2003). The Ifr1 symbiont genome also contained two genes for cytoplasmic proteins involved in the sugar-transporting phosphotransferase system (PTS) (ptsHI). However, the lack of any genes for PTS permeases indicates that the PTS system may function in regulation in these bacteria, not in the import of carbon compounds (Barabote and Saier, 2005).
Nitrogen Assimilation
The four symbiont genomes encoded genes for the assimilation of both nitrate and ammonium into biomass. All of the symbiont genomes encoded genes for an ammonium transporter and a nitrate/nitrate transporter, which suggests these bacteria could take up ammonium or nitrate from the environment (as well as take up any ammonium that is produced by the host). All of the symbiont genomes encoded genes for the reduction of nitrate in the periplasm (napABGH), and for the transport of the resulting nitrite into the cytoplasm (nitrate/nitrate transporter). Nap nitrate reductases are functionally diverse and have been implicated in both nitrate respiration and nitrate scavenging (Sparacino-Watkins et al., 2014). The symbiont genomes all contained cytoplasmic nitrite reductases that produce ammonia from nitrite for assimilation, though the genes were different: the γ-proteobacterial symbionts had the genes for a nitrite reductase (nit-6) that utilizes NAD(P)H as the reductant, while the A. ε symbiont contained a reductase that utilizes ferredoxin (nirA). All the symbionts had genes encoding a glutamine synthetase type I (glnA) for the conversion of ammonia to glutamine, and an NADPH-dependent glutamate synthase (gltAB) to convert glutamine to glutamate (though the gltB gene was not recovered in the Ifr1 symbiont genome). In addition, the Ifr1 symbiont genome contained a gene for a ferredoxin-dependent glutamate synthase (glsF).
Amino Acid Synthesis and Transport
Though Alviniconcha and I. nautilei have gastrointestinal tracts, these systems are noticeably reduced and thought to be mostly non-functional (Beck, 1991; Waren and Bouchet, 1993). Thus, it is likely that all essential amino acids (i.e., amino acids not able to be synthesized by animals) must be provided by their symbionts, either through digestion of symbiont cells or via the excretion of amino acids by symbionts. All of the symbiont genomes encoded the complete or almost complete (i.e., missing 1 or 2 genes but encoding the gene for the final step) pathways to synthesize 15 amino acids, of which seven are considered essential to animals (Table 3). Additionally, all of the Alviniconcha symbionts contained the complete pathways to synthesize the two other essential amino acids, leucine and isoleucine (Table 3). Though the Ifr1 genome was incomplete for the synthesis of these two essential amino acids, it contained all genes in these pathways except for the final step (a branched-chain amino acid aminotransferase), which could indicate that the lack of this gene is due to the incompleteness of the Ifr1 genome assembly.
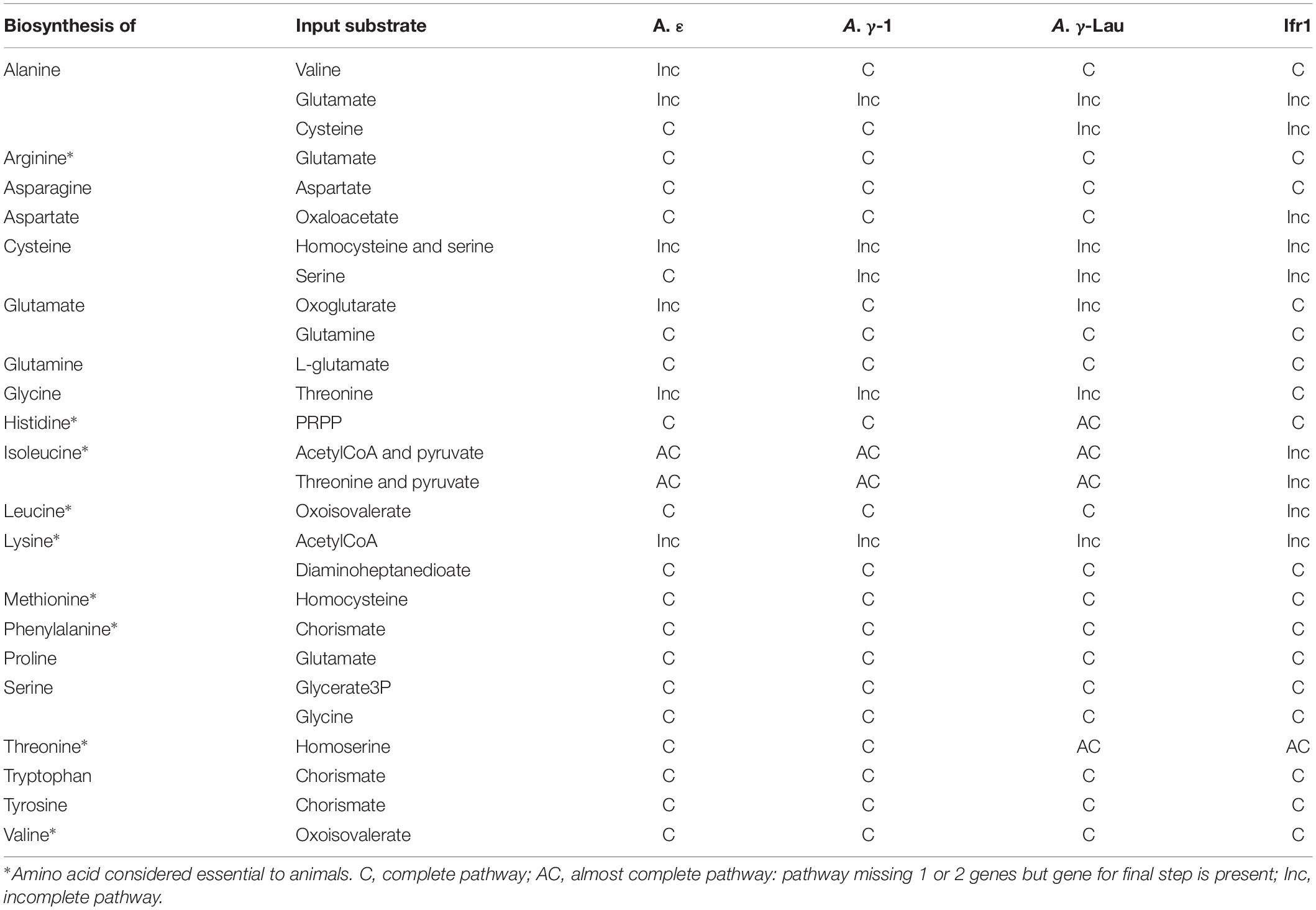
Table 3. Amino acid biosynthesis pathways in the four symbiont genomes.
The A. γ-Lau and Ifr1 symbiont genomes both indicated the capacity for the uptake or excretion of amino acids. Though all of the symbiont genomes contained genes involved in dipeptide transport (dppAC was present in all Alviniconcha symbiont genomes and dppA only in the Ifr1 symbiont genome), they are missing the genes necessary for the complete dipeptide transport system (i.e., dppBDFP). The function of this partial dipeptide transport genes is therefore unclear. However, the A. γ-Lau symbiont genome contained genes for a branched-chain amino acid transporter, an L-amino acid ABC transporter (aapJMQ), and all genes for a cluster 5 ABC transporter that could transport peptides. The Ifr1 symbiont uniquely encoded an uncharacterized amino acid permease from the GabP family, as well as all the genes for an oligopeptide transport system (oppABCDF), which would allow it to take up peptides to use as amino acid sources. Since not all twenty amino acids were able to be synthesized by the symbionts, it is possible that they use these transporters to take up peptides or amino acids from their hosts or the environment. It is also possible that these transporters are being used by the symbionts for the export of peptides or amino acids to their hosts. However, the presence of symbiont-derived fatty acids in host tissue has been used as evidence to hypothesize that the γ-proteobacterial symbionts of Alviniconcha digest their symbionts, rather than rely on the translocation of substrates across the membrane to the host (Suzuki et al., 2005b).
Motility and Adhesion
Though non-motile while intracellular, the symbionts of Alviniconcha spp. and I. nautilei are thought to be horizontally transmitted and, thus, have a free-living stage where motility could be useful for moving to optimal environmental habitats or for finding a host. Moreover, in pathogenic and symbiotic bacteria, motility machinery is often used for adhesion to or interaction with their hosts (reviewed in Chaban et al., 2015). The A. ε symbiont genome was the only symbiont genome that encoded genes for flagella, including genes for the flagellar hook capping protein (flgD), filament (fliC), hook-filament junction (flgLK), hook (flgE), rod (flgBCGF, fliE), ring (flgHI, fliFGMN), and motor (motAB). This genome also contained the paralyzed flagellar protein (pflaA) that is unique to Campylobacterota flagella (Gupta, 2006). The presence of the genes for flagella implies that the A. ε symbiont can be motile during its free-living state outside of the host or possibly during host association. However, no genes for the chemotactic signaling system that is present in motile pathogenic Campylobacterota were found in the A.ε symbiont genome. Flagella and chemotaxis are important in both the colonization and adhesion of pathogenic Campylobacterota (Lertsethtakarn et al., 2011), though flagellar motility is primarily important for colonization of hosts (Gu, 2017). Interestingly, flagellar genes are expressed in host-associated A. ε symbionts (Sanders et al., 2013), suggesting that the symbiont is using the flagella as a secretion system or for penetration into host tissue; such functions have been observed in intracellular pathogens (Chaban et al., 2015).
Both the A. γ-Lau and Ifr1 symbiont genomes encoded genes for type IV pili, which are involved in bacterial motility, adhesion, DNA uptake, and protein secretion (Grohmann et al., 2018). The A. γ-Lau symbiont genome encoded genes for type IV pili biogenesis (pilMNOPY1Z), secretin (pilQ), cassette (pilS), integral membrane protein (pilC), ATPases (pilBTU), and twitching motility proteins (pilGHJT). The Ifr1 symbiont shared all of these genes except pilP, pilS, and pilY1. Interestingly, the gene encoding pilin (pilA), the major structural component of the pilus, was missing from both genomes. Loss of this gene in pathogens impacts virulence (e.g., Essex-lopresti et al., 2005; Forslund et al., 2010), so it is not clear what the lack of this gene indicates for the functioning of the encoded type IV pili in these two symbionts.
Secretion
The A. γ-1 and Ifr1 symbiont encoded genes for secretion systems (T4SSs), which are used to export DNA or proteins into cells and are common in intracellular bacteria (Alvarez-Martinez and Christie, 2009). The A. γ-1 symbiont genome encoded all necessary genes for a type I secretion system (hlyBD, tolc), which is used in the export of a variety of molecules including digestive enzymes, heme-binding proteins, and pore-forming exotoxins (Green and Mecsas, 2016). The A. γ-1 symbiont genome also contained genes for a T4SS IncF plasmid conjugative transfer pilus (traABCDEFGHIKLNUVW). However, it was missing genes involved in DNA processing (traMY) and surface exclusion (traST), which suggests that this pilus may not be used for conjugation but instead for protein secretion (Firth et al., 1996). The Ifr1 symbiont had five of the twelve genes that would be necessary for a VirB/VirD4 T4SS (virB4, virB8, virB10-11, and virD4), a secretion system that is used for the transport of DNA or effector proteins in pathogens (Alvarez-Martinez and Christie, 2009).
Summary and Conclusion
Here, we compared the gene content of distantly related bacteria that are symbiotic with sympatric and closely related host species. The presented genomes presented herein are between 87-96% complete, making it likely that most genes and pathways were recovered in the current assemblies. Nonetheless, the absence of a gene or pathway from these assemblies must be interpreted with caution.
Comparison of the genomes of the symbionts of Alviniconcha species and I. nautilei demonstrated that they were highly similar in their gene content encoding for chemoautotrophic functions. All of the symbiont genomes contained genes for carbon fixation, oxidation of both sulfur and hydrogen, and the respiration of oxygen and nitrate. In most cases, the exact complement of genes found in these pathways was congruent with the metabolic pathways typically employed by other bacteria in their respective taxonomic groups. In addition, all four symbionts encoded genes for the uptake of organic carbon, indicating a capacity for heterotrophy, as well as the assimilation of nitrate and ammonia. The symbiont genomes did differ in gene content that may be related to interactions with their hosts, such as genes related to motility, adhesion, amino acid uptake or excretion, and secretion.
Altogether, our results suggest that, at the broadest level, differences in the presence or absence of the capacity for chemoautotrophic functions (e.g., genes for sulfur and/or hydrogen oxidation, as well as for aerobic and anaerobic respiration) do not of themselves explain the distribution of Alviniconcha and I. nautilei and their symbionts. This is important to consider when using genomic or metagenomic content as a proxy for function; the presence of nearly identical genes among different symbionts does not in and of itself imply comparable metabolic activity. It is likely that the particular homologs or pathways employed by these symbionts vary in their regulation and/or biochemical efficacies under different geochemical regimes, which could easily affect the distribution of their hosts into distinct habitats. For example, though all symbiont genomes contained sulfide:quinone reductase (SQR) genes, differences in the functioning of the types of SQR encoded in the A. ε symbiont compared to the γ-proteobacterial symbiont genomes could influence the optimal hydrogen sulfide concentration for the species hosting this symbiont. Though comparisons of the conditions around the different Alviniconcha species (Beinart et al., 2012), and physiological experiments testing differences between Alviniconcha and I. nautilei (Henry et al., 2008) do not suggest that there are interspecific differences in temperature or oxygen concentration tolerances, we cannot exclude the possibility that differences in host physiology or ecology are driving habitat partitioning in these species. Further work that assesses gene and protein expression, as well as directly compares the metabolism of these symbioses under common conditions, will provide insight into whether physiological differences among the host species and/or their symbionts is impacting their distribution.
Data Availability
Raw sequence data can be found under the NCBI BioProject ID PRJNA523619, and NCBI BioSample IDs SAMN10985003 (A. boucheti), SAMN10985004 (A. kojimai), SAMN10985005 (A. strummeri), and SAMN10985044 (I. nautilei). GenBank SRA accession numbers for the sequence reads are SRR8632448 (A. boucheti), SRR8655138 (A. kojimai), SRR8799392 (A. strummeri), and SRR8799391 (I. nautilei). The assembled and annotated symbiont genomes are publicly available on the RAST server (http://rast.theseed.org/) using the guest login with IDs 6666666.293770 (A. γ-Lau), 6666666.293769 (A. γ-1), 6666666.293768 (A. ε), 6666666.293767 (Ifr1), and 6666666.296237 (Ifr2).
Author Contributions
RB and PG designed the study. RB performed the laboratory work, assembly and binning of the Ifremeria nautilei symbiont genomes, and all other sequence analysis. CL and KK developed the new algorithm for separating host and symbiont sequence reads, and assembled the Alviniconcha symbiont genomes. FS sequenced Ifremeria nautilei symbiont genome. All authors contributed to the writing of the manuscript.
Funding
This material is based upon work supported by the National Science Foundation (DGE-1144152 and OCE-1819530 to RB, ABI-1564559 to FS, OCE-1416673 to KK, and IOS-1257755 and OCE-0732369 to PG) and the Teasley Endowment to the Georgia Institute of Technology (to FS).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to the crews of the R/V Thomas G. Thompson and the ROV JASON II for their support in the collection of the specimens used here.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01818/full#supplementary-material
Footnotes
References
Aguilar-Barajas, E., Díaz-Pérez, C., Ramírez-Díaz, M. I., Riveros-Rosas, H., and Cervantes, C. (2011). Bacterial transport of sulfate, molybdate, and related oxyanions. Biometals 24, 687–707. doi: 10.1007/s10534-011-9421-x
Alvarez-Martinez, C. E., and Christie, P. J. (2009). Biological diversity of prokaryotic type IV secretion systems. Microbiol. Mol. Biol. Rev. 73, 775–808. doi: 10.1128/MMBR.00023-9
Barabote, R. D., and Saier, M. H. (2005). Comparative genomic analyses of the bacterial phosphotransferase system. Microbiol. Mol. Biol. Rev. 69, 608–634. doi: 10.1007/978-3-642-41947-8_3
Beck, B. L. A. (1991). Olgaconcha tufari n. gen. et n. sp. - a new mesogastropod (Gastropoda: Prosobranchia) from hydrothermal vents in the manus backarc basin (Bismarck Sea, Papua New Guinea). Ann. Naturhist. Mus. Wien. 92, 277–287.
Beinart, R. A., Gartman, A., Sanders, J. G., Luther, G. W., and Girguis, P. R. (2015). The uptake and excretion of partially oxidized sulfur expands the repertoire of energy resources metabolized by hydrothermal vent symbioses. Proc. R. Soc. B Biol. Sci. 282:20142811. doi: 10.1098/rspb.2014.2811
Beinart, R. A., Sanders, J. G., Faure, B., Sylva, S. P., Lee, R. W., Becker, E. L., et al. (2012). Evidence for the role of endosymbionts in regional-scale habitat partitioning by hydrothermal vent symbioses. Proc. Natl. Acad. Sci. U.S.A. 109, 241–250. doi: 10.1073/pnas.1202690109
Brettin, T., Davis, J. J., Disz, T., Edwards, R. A., Gerdes, S., Olsen, G. J., et al. (2015). RASTtk: a modular and extensible implementation of the RAST algorithm for annotating batches of genomes. Sci. Rep. 5:8365. doi: 10.1038/srep08365
Chaban, B., Hughes, H. V., and Beeby, M. (2015). The flagellum in bacterial pathogens: for motility and a whole lot more. Semin. Cell Dev. Biol. 46, 91–103. doi: 10.1016/j.semcdb.2015.10.032
Chen, Z. W., Koh, M., Van Driessche, G., Van Beeumen, J. J., Bartsch, R. G., Meyer, T. E., et al. (1994). The structure of flavocytochrome c sulfide dehydrogenase from a purple phototrophic bacterium. Science 266, 430–432. doi: 10.1126/science.7939681
Childress, J. J., and Girguis, P. R. (2011). The metabolic demands of endosymbiotic chemoautotrophic metabolism on host physiological capacities. J. Exp. Biol. 214, 312–325. doi: 10.1242/jeb.049023
Czyzewski, B. K., and Wang, D. N. (2012). Identification and characterization of a bacterial hydrosulphide ion channel. Nature 483, 494–497. doi: 10.1038/nature10881
Desbruyères, D., Alayse-Danet, A. M., Ohta, S. and The scientific parties of Biolau and Starmer cruises. (1994). Deep-sea hydrothermal communities in Southwestern Pacific back-arc basins (the North Fiji and Lau Basins): composition, microdistribution and food web. Mar. Geol. 116, 227–242. doi: 10.1016/0025-3227(94)90178-3
D’Mello, R., Hill, S., and Poole, R. K. (1996). The cytochrome bd quinol oxidase in Escherichia coli has an extremely high oxygen affinity and two oxygen-binding haems: implications for regulation of activity in vivo by oxygen inhibition. Microbiology 142, 755–763. doi: 10.1099/00221287-142-4-755
Dubilier, N., Bergin, C., and Lott, C. (2008). Symbiotic diversity in marine animals: the art of harnessing chemosynthesis. Nat. Rev. Microbiol. 6, 725–740. doi: 10.1038/nrmicro1992
Essex-lopresti, A. E., Boddey, J. A., Thomas, R., Smith, M. P., Hartley, M. G., Atkins, T., et al. (2005). A type IV pilin, pila, contributes to adherence of Burkholderia pseudomallei and virulence in vivo. Infect. Immun. 73, 1260–1264. doi: 10.1128/IAI.73.2.1260
Firth, N., Ippen-ihler, K., and Skurray, R. A. (1996). “Structure and function of the F factor and mechanism of conjugation,” in Escherichia coli and Salmonella: Cellular and Molecular Biology, ed. F. C. Neidhardt (Washington, DC: ASM Press), 2377–2401.
Forslund, A., Salomonsson, E. N., Golovliov, I., Kuoppa, K., Michell, S., Titball, R., et al. (2010). The type IV pilin, PilA, is required for full virulence of Francisella tularensis subspecies tularensis. BMC Microbiol. 10:227. doi: 10.1186/1471-2180-10-227
Gimenez, R., Nuñez, M. F., Badia, J., Aguilar, J., and Baldoma, L. (2003). The gene yjcG, cotranscribed with the gene acs, encodes an acetate permease in Escherichia coli. J. Bacteriol. 185, 6448–6455. doi: 10.1128/JB.185.21.6448-6455.2003
Green, E. R., and Mecsas, J. (2016). Bacterial secretion systems – an overview. Microbiol. Spectr. 4, 215–239. doi: 10.1128/microbiolspec.VMBF-0012-2015.Bacterial
Gregersen, L. H., Bryant, D. A., and Frigaard, N.-U. (2011). Mechanisms and evolution of oxidative sulfur metabolism in green sulfur bacteria. Front. Microbiol. 2:116. doi: 10.3389/fmicb.2011.00116
Grohmann, E., Christie, P. J., Waksman, G., and Backert, S. (2018). Micro review type IV secretion in gram-negative and gram-positive bacteria. Mol. Microbiol. 107, 455–471. doi: 10.1111/mmi.13896
Gu, H. (2017). Role of flagella in the pathogenesis of Helicobacter pylori. Curr. Microbiol. 74, 863–869. doi: 10.1007/s00284-017-1256-4
Gupta, R. S. (2006). Molecular signatures (unique proteins and conserved indels) that are specific for the epsilon proteobacteria (Campylobacterales). BMC Genomics 7:167. doi: 10.1186/1471-2164-7-167
Han, Y., and Perner, M. (2015). The globally widespread genus Sulfurimonas: versatile energy metabolisms and adaptations to redox clines. Front. Microbiol. 6:989. doi: 10.3389/fmicb.2015.00989
Han, Y., and Perner, M. (2016). Sulfide consumption in Sulfurimonas denitrificans and heterologous expression of its three SQR homologs. J. Bacteriol. 198, 1260–1267. doi: 10.1128/JB.01021-15
Henry, M. S., Childress, J. J., and Figueroa, D. (2008). Metabolic rates and thermal tolerances of chemoautotrophic symbioses from Lau Basin hydrothermal vents and their implications for species distributions. Deep. Res. Part I Oceanogr. Res. Pap. 55, 679–695. doi: 10.1016/j.dsr.2008.02.001
Hügler, M., and Sievert, S. M. (2011). Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Ann. Rev. Mar. Sci. 3, 261–289. doi: 10.1146/annurev-marine-120709-142712
Johnson, S. B., Warén, A., Tunnicliffe, V., Dover, C., Van Wheat, G., Schultz, T. F., et al. (2015). Molecular taxonomy and naming of five cryptic species of Alviniconcha snails (Gastropoda: Abyssochrysoidea) from hydrothermal vents. Syst. Biodivers. 13, 278–295. doi: 10.1080/14772000.2014.970673
Keane, T. M., Creevey, C. J., Pentony, M. M., Naughton, T. J. M., and Mclnerney, J. O. (2006). Assessment of methods for amino acid matrix selection and their use on empirical data shows that ad hoc assumptions for choice of matrix are not justified. BMC Evol. Biol. 6:29. doi: 10.1186/1471-2148-6-29
Kleiner, M., Petersen, J. M., and Dubilier, N. (2012). Convergent and divergent evolution of metabolism in sulfur-oxidizing symbionts and the role of horizontal gene transfer. Curr. Opin. Microbiol. 15, 621–631. doi: 10.1016/j.mib.2012.09.003
Konstantinidis, K. T., Rosselló-móra, R., and Amann, R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. doi: 10.1038/ismej.2017.113
Lertsethtakarn, P., Ottemann, K. M., and Hendrixson, D. R. (2011). Motility and Chemotaxis in Campylobacter and Helicobacter. Annu. Rev. Microbiol. 65, 389–410. doi: 10.1146/annurev-micro-090110-102908
Luo, C., Walk, S. T., Gordon, D. M., Feldgarden, M., Tiedje, J. M., and Konstantinidis, K. T. (2011). Genome sequencing of environmental Escherichia coli expands understanding of the ecology and speciation of the model bacterial species. Proc. Natl. Acad. Sci. U.S.A. 108, 7200–7205. doi: 10.1073/pnas.1015622108
Marçais, G., and Kingsford, C. (2011). A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770. doi: 10.1093/bioinformatics/btr011
Mathai, J. C., Missner, A., Kugler, P., Saparov, S. M., Zeidel, M. L., Lee, J. K., et al. (2009). No facilitator required for membrane transport of hydrogen sulfide. Proc. Natl. Acad. Sci. U.S.A. 106, 16633–16638. doi: 10.1073/pnas.0902952106
Matsen, F. A., Kodner, R. B., and Armbrust, E. V. (2010). pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinform. 11:538. doi: 10.1186/1471-2105-11-538
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES Science Gateway for inference of large phylogenetic trees,” in Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, doi: 10.1109/GCE.2010.5676129
Mitarai, S., Watanabe, H., Nakajima, Y., Shchepetkin, A. F., and McWilliams, J. C. (2016). Quantifying dispersal from hydrothermal vent fields in the western Pacific Ocean. Proc. Natl. Acad. Sci. U.S.A. 113, 2976–2981. doi: 10.1073/pnas.1518395113
Mulligan, C., Fischer, M., and Thomas, G. H. (2011). Tripartite ATP-independent periplasmic (TRAP) transporters in bacteria and archaea. FEMS Microbiol. Rev. 35, 68–86. doi: 10.1111/j.1574-6976.2010.00236.x
Nakagawa, S., Shimamura, S., Takaki, Y., Suzuki, Y., Murakami, S., Watanabe, T., et al. (2014). Allying with armored snails: the complete genome of gammaproteobacterial endosymbiont. ISME J. 8, 40–51. doi: 10.1038/ismej.2013.131
Nunoura, T., Takaki, Y., Kazama, H., Kakuta, J., Shimamura, S., Makita, H., et al. (2014). Physiological and genomic features of a novel sulfur-oxidizing gammaproteobacterium belonging to a previously uncultivated symbiotic lineage isolated from a hydrothermal vent. PLoS One 9:e104959. doi: 10.1371/journal.pone.0104959
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., and Tyson, G. W. (2015). CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Peng, Y., Leung, H. C. M., Yiu, S. M., and Chin, F. Y. L. (2012). IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28, 1420–1428. doi: 10.1093/bioinformatics/bts174
Peters, J. W., Schut, G. J., Boyd, E. S., Mulder, D. W., Shepard, E. M., Broderick, J. B., et al. (2015). [FeFe]- and [NiFe]-hydrogenase diversity, mechanism, and maturation. Biochim. Biophys. Acta 1853, 1350–1369. doi: 10.1016/j.bbamcr.2014.11.021
Podowski, E. L., Ma, S., Luther, G. W., Wardrop, D., and Fisher, C. R. (2010). Biotic and abiotic factors affecting distributions of megafauna in diffuse flow on andesite and basalt along the Eastern Lau spreading center. Tonga. Mar. Ecol. Prog. Ser. 418, 25–45. doi: 10.3354/meps08797
Podowski, E. L., Moore, T. S., Zelnio, K. A., Luther, G. W., and Fisher, C. R. (2009). Distribution of diffuse flow megafauna in two sites on the Eastern Lau spreadingcenter, Tonga. Deep Sea Res. Part Oceanogr. Res. Pap. 56, 2041–2056. doi: 10.1016/j.dsr.2009.07.002
Rodriguez-R, L. M., Harvey, W. T., Konstantinidis, K. T., Cole, J. R., Gunturu, S., Tiedje, J. M., et al. (2018). The microbial genomes atlas (MiGA) webserver: taxonomic and gene diversity analysis of archaea and bacteria at the whole genome level. Nucleic Acids Res. 46, W282–W288. doi: 10.1093/nar/gky467
Rodriguez-R, L. M., and Konstantinidis, K. T. (2014). Bypassing cultivation to identify bacterial species. Microbe 9, 111–118. doi: 10.1128/microbe.9.111.1
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ Prepr. 4:e1900v1. doi: 10.7287/peerj.preprints.1900v1
Rogers, K. L., and Schulte, M. D. (2012). Organic sulfur metabolisms in hydrothermal environments. Geobiology 10, 320–332. doi: 10.1111/j.1472-4669.2012.00324.x
Sanders, J. G., Beinart, R. A., Stewart, F. J., Delong, E. F., and Girguis, P. R. (2013). Metatranscriptomics reveal differences in in situ energy and nitrogen metabolism among hydrothermal vent snail symbionts. ISME J. 7, 1556–1567. doi: 10.1038/ismej.2013.45
Seston, S. L., Beinart, R. A., Sarode, N., Shockey, A. C., Ranjan, P., Ganesh, S., et al. (2016). Metatranscriptional response of chemoautotrophic Ifremeria nautilei endosymbionts to differing sulfur regimes. Front. Microbiol. 7:1074. doi: 10.3389/fmicb.2016.01074
Søndergaard, D., Pedersen, C. N. S., and Greening, C. (2016). HydDB: a web tool for hydrogenase classification and analysis. Sci. Rep. 6:34212. doi: 10.1038/srep34212
Sparacino-Watkins, C., Stolz, J. F., and Basu, P. (2014). Nitrate and periplasmic nitrate reductases. Chem. Soc. Rev. 43, 676–706. doi: 10.1039/c3cs60249d
Speer, K., and Thurnherr, A. M. (2012). The lau basin float experiment (LAUB-FLEX). Oceanography 25, 284–285. doi: 10.5670/oceanog.2012.27
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Suzuki, Y., Kojima, S., Sasaki, T., Suzuki, M., Utsumi, T., Watanabe, H., et al. (2006a). Host-symbiont relationships in hydrothermal vent gastropods of the genus Alviniconcha from the Southwest Pacific. Appl. Environ. Microbiol. 72, 1388–1393. doi: 10.1128/AEM.72.2.1388-1393.2006
Suzuki, Y., Kojima, S., Watanabe, H., Suzuki, M., Tsuchida, S., Nunoura, T., et al. (2006b). Single host and symbiont lineages of hydrothermal-vent gastropods Ifremeria nautilei (Provannidae): biogeography and evolution. Mar. Ecol. Prog. Ser. 315, 167–175. doi: 10.3354/meps315167
Suzuki, Y., Sasaki, T., Suzuki, M., Nogi, Y., Miwa, T., Takai, K., et al. (2005a). Novel chemoautotrophic endosymbiosis between a member of the Epsilonproteobacteria and the hydrothermal-vent gastropod Alviniconcha aff. hessleri (Gastropoda: Provannidae) from the Indian Ocean. Appl. Environ. Microbiol. 71, 5440–5450. doi: 10.1128/AEM.71.9.5440
Suzuki, Y., Sasaki, T., Suzuki, M., Tsuchida, S., Nealson, K. H., and Horikoshi, K. (2005b). Molecular phylogenetic and isotopic evidence of two lineages of chemoautotrophic endosymbionts distinct at the subdivision level harbored in one host-animal type: the genus Alviniconcha (Gastropoda: Provannidae). FEMS Microbiol. Lett. 249, 105–112. doi: 10.1016/j.femsle.2005.06.023
Urakawa, H., Dubilier, N., Fujiwara, Y., Cunningham, D. E., Kojima, S., and Stahl, D. A. (2005). Hydrothermal vent gastropods from the same family (Provannidae) harbour epsilon- and gamma-proteobacterial endosymbionts. Environ. Microbiol. 7, 750–754. doi: 10.1111/j.1462-2920.2005.00753.x
Waite, T. J., Moore, T. S., Childress, J. J., Hsu-Kim, H., Mullaugh, K. M., Nuzzio, D. B., et al. (2008). Variation in sulfur speciation with shellfish presence at a Lau Basin diffuse flow vent site. J. Shellfish Res. 27, 163–168. doi: 10.2983/0730-8000(2008)27
Waren, A., and Bouchet, P. (1993). New records, species, genera, and a new family of gastropods from hydrothermal vents and hydrocarbon seeps. Zool. Scr. 22, 1–90. doi: 10.1111/j.1463-6409.1993.tb00342.x
Wu, Y., Tang, Y., Tringe, S. G., Simmons, B. A., and Singer, S. W. (2014). MaxBin: an automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2:26. doi: 10.1186/2049-2618-2-26
Keywords: symbiosis, chemosynthesis, genomics, Campylobacteria, Gammaproteobacteria, gastropod, Alviniconcha, Ifremeria
Citation: Beinart RA, Luo C, Konstantinidis KT, Stewart FJ and Girguis PR (2019) The Bacterial Symbionts of Closely Related Hydrothermal Vent Snails With Distinct Geochemical Habitats Show Broad Similarity in Chemoautotrophic Gene Content. Front. Microbiol. 10:1818. doi: 10.3389/fmicb.2019.01818
Received: 08 April 2019; Accepted: 23 July 2019;
Published: 14 August 2019.
Edited by:
Sébastien Duperron, Muséum National d’Histoire Naturelle, FranceReviewed by:
Horst Felbeck, University of California, San Diego, United StatesSylvie Gaudron, Sorbonne Universités, France
Copyright © 2019 Beinart, Luo, Konstantinidis, Stewart and Girguis. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Roxanne A. Beinart, cmJlaW5hcnRAdXJpLmVkdQ==
†Chengwei Luo, DeepBiome Therapeutics, Inc., Cambridge, MA, United States