 Mehmet Tatli1,2†
Mehmet Tatli1,2† Daniel Amador-Noguez
Daniel Amador-Noguez- 1DOE Great Lakes Bioenergy Research Center, University of Wisconsin-Madison, Madison, WI, United States
- 2Department of Bacteriology, University of Wisconsin-Madison, Madison, WI, United States
- 3Genome Center of Wisconsin, Madison, WI, United States
- 4Department of Biomolecular Chemistry, University of Wisconsin-Madison, Madison, WI, United States
- 5Department of Chemistry, University of Wisconsin-Madison, Madison, WI, United States
- 6Morgridge Institute for Research, Madison, WI, United States
Protein phosphorylation is a post-translational modification with widespread regulatory roles in both eukaryotes and prokaryotes. Using mass spectrometry, we performed a genome wide investigation of protein phosphorylation in the non-model organism and biofuel producer Zymomonas mobilis under anaerobic, aerobic, and N2-fixing conditions. Our phosphoproteome analysis revealed 125 unique phosphorylated proteins, belonging to major pathways such as glycolysis, TCA cycle, electron transport, nitrogen metabolism, and protein synthesis. Quantitative analysis revealed significant and widespread changes in protein phosphorylation across growth conditions. For example, we observed increased phosphorylation of nearly all glycolytic enzymes and a large fraction of ribosomal proteins during aerobic and N2-fixing conditions. We also observed substantial changes in the phosphorylation status of enzymes and regulatory proteins involved in nitrogen fixation and ammonia assimilation during N2-fixing conditions, including nitrogenase, the Rnf electron transport complex, the transcription factor NifA, GS-GOGAT cycle enzymes, and the PII regulatory protein. This suggested that protein phosphorylation may play an important role at regulating all aspects of nitrogen metabolism in Z. mobilis. This study provides new knowledge regarding the specific pathways and cellular processes that may be regulated by protein phosphorylation in this important industrial organism and provides a useful road map for future experiments that investigate the physiological role of specific phosphorylation events in Z. mobilis.
Introduction
Zymomonas mobilis, a facultatively anaerobic alphaproteobacterium, possesses several desirable characteristics for industrial biofuel production. These include fast glucose catabolism, high ethanol yield (up to 96% of consumed glucose), low biomass production, resilience to inhibitors present in lignocellulosic hydrolysates, and tolerance to high ethanol and sugar concentrations (Rogers et al., 1980; Panesar et al., 2006; Pan et al., 2014; Yang et al., 2016a; Martien et al., 2019). In addition, Z. mobilis possesses nitrogenase and can efficiently fix atmospheric nitrogen (N2) without affecting ethanol yield, which adds potential economic and environmental benefits to ethanol production (Kremer et al., 2015). However, regulation of nitrogen fixation and metabolism in Z. mobilis remains largely unexplored (Jack et al., 2001; Huergo et al., 2013).
Unlike most anaerobic biofuel producers, Z. mobilis is highly tolerant to oxygen, although its growth rate and ethanol yield decrease significantly under aerobic conditions (Tanaka et al., 1990; Yang et al., 2009; Martien et al., 2019). Recent studies have indicated that during aerobic growth, oxidative damage to iron-sulfur (FeS) clusters constitutes a major factor influencing Z. mobilis metabolism and that respiratory enzymes and the ability to form multicellular aggregates are important for its survival (Jones-Burrage et al., 2019; Martien et al., 2019). Despite these and other recent advances (Yang et al., 2009; Rutkis et al., 2016; Strazdina et al., 2018), much remains to be learned about the regulation of Z. mobilis physiology during aerobic growth.
There is growing interest in redirecting Z. mobilis highly catabolic metabolism toward the production of advanced biofuels such as higher alcohols (e.g., isobutanol) and isoprenoid fuels derived from the methylerythritol phosphate (MEP) pathway (Yang et al., 2016b; Agrawal et al., 2017; Martien et al., 2019). However, a deeper understanding of the control and regulation of Z. mobilis metabolism will be required to achieve this.
Protein phosphorylation is best known for its widespread regulatory role in eukaryotes. However, a growing body of research now demonstrates that this post-translational modification is highly prevalent in bacteria, with potentially critical regulatory functions (Macek and Mijakovic, 2011; Soufi et al., 2012; Pisithkul et al., 2015; Ravikumar et al., 2015; Soares and Blackburn, 2016). Propelled by recent advances in mass spectrometry techniques, genome-wide phosphoproteome analyses have been reported in several industrially and medically relevant bacteria, including Escherichia coli, Bacillus subtilis, Klebsiella pneumoniae, Streptomyces coelicolor, Rhodopseudomonas palustris (Soung et al., 2009; Parker et al., 2010; Manteca et al., 2011; Lin et al., 2015), and others (Soufi et al., 2008; Aivaliotis et al., 2009; Ravichandran et al., 2009). These studies have identified large numbers of phosphorylation events on Ser, Thr, and Tyr residues, many of them in metabolic enzymes. Although, the regulatory functions of protein phosphorylation in bacteria are still largely undefined, these phosphoproteomics studies have provided the necessary foundation to investigate the physiological function of specific phosphorylation events in these bacterial species (Macek et al., 2007b; Soufi et al., 2008; Aivaliotis et al., 2009; Manteca et al., 2011; Ravikumar et al., 2014).
Here, using mass spectrometry, we report the first genome wide investigation of protein phosphorylation in the biofuel producer Z. mobilis across three different growth conditions: anaerobic growth, aerobic growth, and N2-fixing conditions. We identified 125 unique phosphorylated proteins distributed among primary metabolic pathways and cellular processes such as glycolysis, TCA cycle, protein biosynthesis, electron transport, and nitrogen fixation. Our analysis also revealed widespread changes in protein phosphorylation across these three growth conditions, providing novel insights regarding the specific pathways and cellular processes that may be regulated by protein phosphorylation in this important industrial organism.
Materials and Methods
Strain and Growth Conditions
Zymomonas mobilis ZM4 (ATCC 31821) strain was first grown on a rich medium plate anaerobically at 30°C for 3 days. This was followed by inoculation of rich medium with a single colony from the plate. A 2 mL ZM4 minimal medium was inoculated with liquid rich medium overnight culture and grown for 16 h. This culture was then used to inoculate a 25 mL minimal medium culture to a starting optical density of 0.05 (measured at 600 nm). The cells were grown to O.D600 of 0.5 before being collected by centrifugation for 15 min at 4500 rpm (Allegra X-30R, Beckman Coulter) (Supplementary Figure S1); cell pellets were stored at −80°C. Anaerobic growth experiments were performed in an anaerobic glove bag with an atmosphere of 5% H2 and 5% CO2, and 90% N2; oxygen level was kept < 50 ppm. For aerobic growth experiments, all steps were the same as anaerobic conditions with the exception that cultures were moved to ambient atmosphere at O.D600 of 0.2 and allowed to grow by stirring at 30°C until the cells reached to O.D600 of 0.5 at which point they were collected by spinning down at 4500 rpm for 15 min. For N2-fixing conditions, all steps used were the same as the anaerobic conditions, but 1 g/L NH4SO4 was omitted from the minimal medium. Rich media plates were prepared using 10 g/L yeast extract, 2 g/L KH2PO4, 18 g/L agar, and 20 g/L glucose. Minimal medium contained 1 g/L K2HPO4, 1 g/L KH2PO4, 0.5 g/L NaCl, 1 g/L NH4SO4, 0.2 g/L MgSO4 7H2O, 25 mg/L Na2MoO4 2H2O, 2.5 mg/L FeSO4 7H2O, 20 mg/L CaCl2 2H2O, 1 mg/L Calcium Pantothenate, and 20 g/L glucose.
Lysis and Digestion
Lysis and digestion were performed as previously described (Ghosh et al., 2019). Briefly, the cell pellets were first re-suspended in 1 mL 6 M guanidine hydrochloride (GnHCl) to lyse the cells and precipitate the protein, MeOH was added to 90% final concentration. The samples were then centrifuged at 3,000 g for 15 min. The supernatant was discarded, and pellets were allowed to dry for approximately 5 min. The protein pellets were re-suspended in 1 mL 8 M urea, 100 mM Tris pH 8.0, 10 mM TCEP, and 40 mM chloroacetamide followed by dilution to 2 M urea using 50 mM Tris pH 8. Trypsin was added at approximate 100:1 ratio, and the samples were incubated overnight at ambient temperature. Trypsin was added again at 100:1 ratio, and the samples were incubated at ambient temperature for 1 h. Each sample was acidified with trifluoroacetic acid (TFA) and pelleted. The supernatant was desalted over a PS-DVB cartridge and dried down. The final peptide yield was estimated by absorbance at 205 nm using a nanodrop system (extinction coefficient = 31).
Phosphorylation Enrichment
Phosphopeptide enrichment was performed using a previously described titanium (IV) immobilized metal affinity chromatography (Ti-MAC) method (Hebert et al., 2018). In brief, for each sample, 1 mg of peptides was combined with 100 μL of Ti-IMAC beads (ResynBio) in 6% TFA/80% ACN, washed three times with this buffer, then washed twice with 80% ACN, once with 0.5 M glycolic acid/80% ACN, and lastly washed twice in 80% ACN. Phosphopeptides were eluted with 50% ACN/1% ammonium hydroxide. The eluate was dried and then desalted over a PS-DVB cartridge.
LC-MS/MS
For each phosphorylation analysis, 25% of the sample was loaded onto a 75 μm i.d. 30 cm long capillary with an imbedded electrospray emitter and packed with 1.7 μm C18 BEH stationary phase. Peptides were eluted with a gradient of acetonitrile over 100 min (Mobile phase A: 0.2% formic acid, Mobile phase B: 70% ACN with 0.2% formic acid as previously described (Hebert et al., 2014). Eluting peptides were analyzed with an Orbitrap Fusion Lumos. Survey scans were performed with the Orbitrap at 60,000 resolution. Data dependent top speed (1 s cycle time) MS/MS sampling of peptide precursors, with charge states +2 to +4, was performed with dynamic exclusion set to 30 s. The MS/MS sampling was performed with quadrupole isolation = 1.6 m/z, fragmentation by higher-energy collisional dissociation (HCD) with normalized collision energy (NCE) = 30, maximum injection time = 118 ms, AGC = 2 × 105 and fragment ions were analyzed by the Orbitrap with R (resolution) = 60,000. Each sample was analyzed with and without the advanced precursor determination (APD) toggled (Hebert et al., 2018).
Software and Data Analysis
Raw files were analyzed with MaxQuant software program; default settings were applied except for match between runs and label free quantitation, which were toggled on (Cox and Mann, 2008). The intensity values for each phosphosite were averaged across the replicate analyses for each sample. Phosphosites that were not localized or quantified in all bioreplicates of at least one condition were excluded from the quantitative analysis. A phosphosite was considered quantified if an MS1 signal for the peptide precursor was observed in back to back scans. This data was then processed with Perseus by first performing a log2 transformation, followed by missing value imputation using the default parameters (Tyanova et al., 2016). Student’s T-tests were performed across aerobic vs. anaerobic growth and N2-fixing conditions vs. anaerobic growth). These p-values were converted to q-values using permutation-based FDR correction. For normalization to protein abundance, phosphorylation log2 signals were mean normalized across each sample for each site independently, and the process was repeated for protein log2 label-free quantitation signals. A protein normalized signal was then calculated for each sample by subtracting the mean normalized protein value from the corresponding mean normalized phosphorylation site value, followed by calculating the fold changes and q-values in Perseus software platform.
Results
Analysis of Zymomonas mobilis Phosphoproteome
We performed a genome wide investigation of protein phosphorylation in Z. mobilis across three different growth conditions: anaerobic, aerobic, and N2-fixing conditions. Cells were grown in defined minimal media using glucose as the single carbon source and ammonia as the nitrogen source, except for N2-fixing conditions in which no ammonia was added. For each condition, five biological replicates were generated. Samples for phosphoproteome analyses were taken during mid-exponential growth (i.e., OD600 ∼0.5) (Supplementary Figure S1). Tryptic peptides generated from these samples were enriched for phosphopeptides using a Ti(IV)-IMAC protocol described previously (Riley and Coon, 2016). Enriched phosphopeptides were analyzed by nano LC-MS/MS on an Orbitrap Fusion Lumos instrument and the resulting identifications were quantitatively compared by label free quantitation. In total, we identified 363 phosphorylation sites (Supplementary Table S1), of which 226 were confidently localized (≥75% probability of single residue localization) and 197 were both localized and quantified. To be considered quantified, the phosphopeptide must have a measurable MS1 signal (see section “Materials and Methods”) in all five biological replicates of at least one growth condition. The identified sites were distributed across 125 unique phosphorylated proteins (Supplementary Table S2). In parallel to phosphoproteome analyses, we performed comprehensive quantitative proteomics analyses to evaluate if changes in phosphorylation across growth conditions were stoichiometric (Supplementary Table S3). The number of phosphorylated proteins identified comprised 6.6% of all Z. mobilis proteins (125/1892 proteins). This fraction is similar to the fraction of phosphorylated proteins previously reported in other bacteria such as E. coli (9.1%) B. subtilis (4.2%), and K. pneumoniae (5.4%) (Table 1; Lin et al., 2015). We categorized the phosphoproteins that we identified according to their biological functions and found them to be distributed across essential cellular and metabolic processes such as glycolysis, TCA cycle, amino acid, nucleotide, and protein biosynthesis, nitrogen fixation, and ammonia assimilation (Figure 1). The serine/threonine/tyrosine (STY) distribution of phosphorylation sites across all phosphoproteins was 73% serine, 21% threonine, and 6% tyrosine (Table 1). This was very similar to the STY distributions found in E. coli, B. subtilis, and K. pneumoniae (Ravikumar et al., 2014; Lin et al., 2015; Potel et al., 2018), but somewhat different than R. palustris (STY: 63.5/16/19.5) and S. coelicolor (STY: 46.8/48/5.2) (Manteca et al., 2011; Hu et al., 2012; Table 1). Similarly to reports from other bacteria, we observed an enrichment of N-terminal phosphorylation sites in Z. mobilis: 15% of all phosphoproteins were phosphorylated on Ser2 or Thr2 (Supplementary Table S4). Interestingly, this fraction is higher than E. coli (6.12%), B. subtilis (6.71%), and K. pneumoniae (4.64%). We also observed that nearly all (>95%) proteins phosphorylated on Ser2 and Thr2 had their first methionine removed, as it has been observed in other bacteria (Lin et al., 2015; Supplementary Table S4).
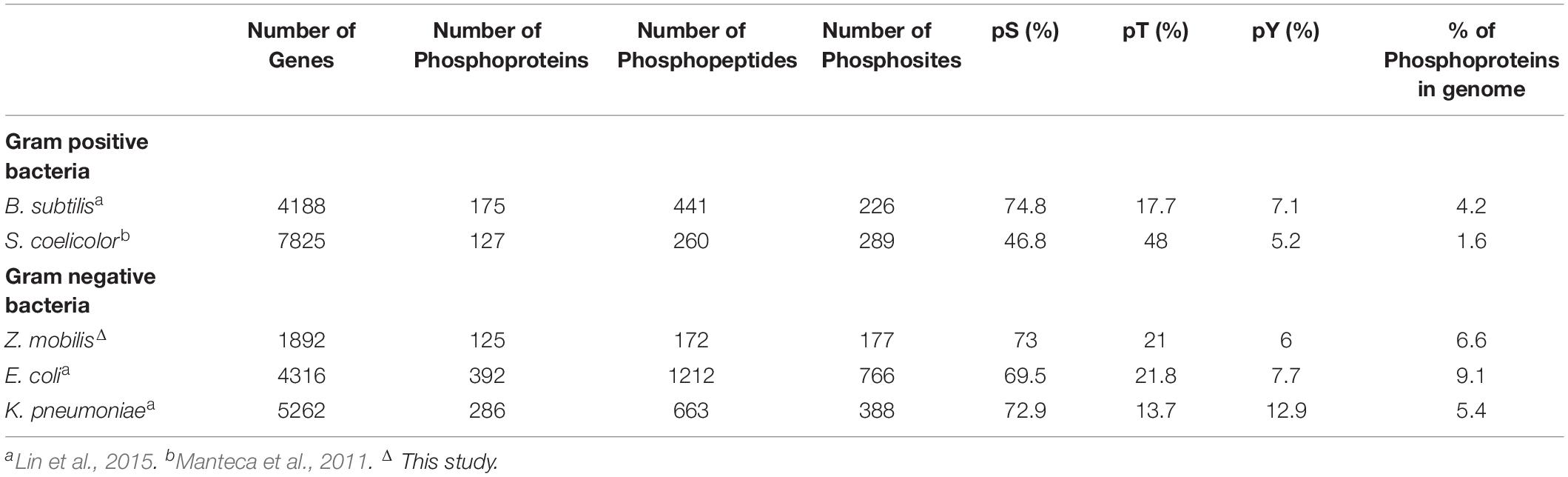
Table 1. STY distribution of phosphorylation sites across all phosphoproteins.
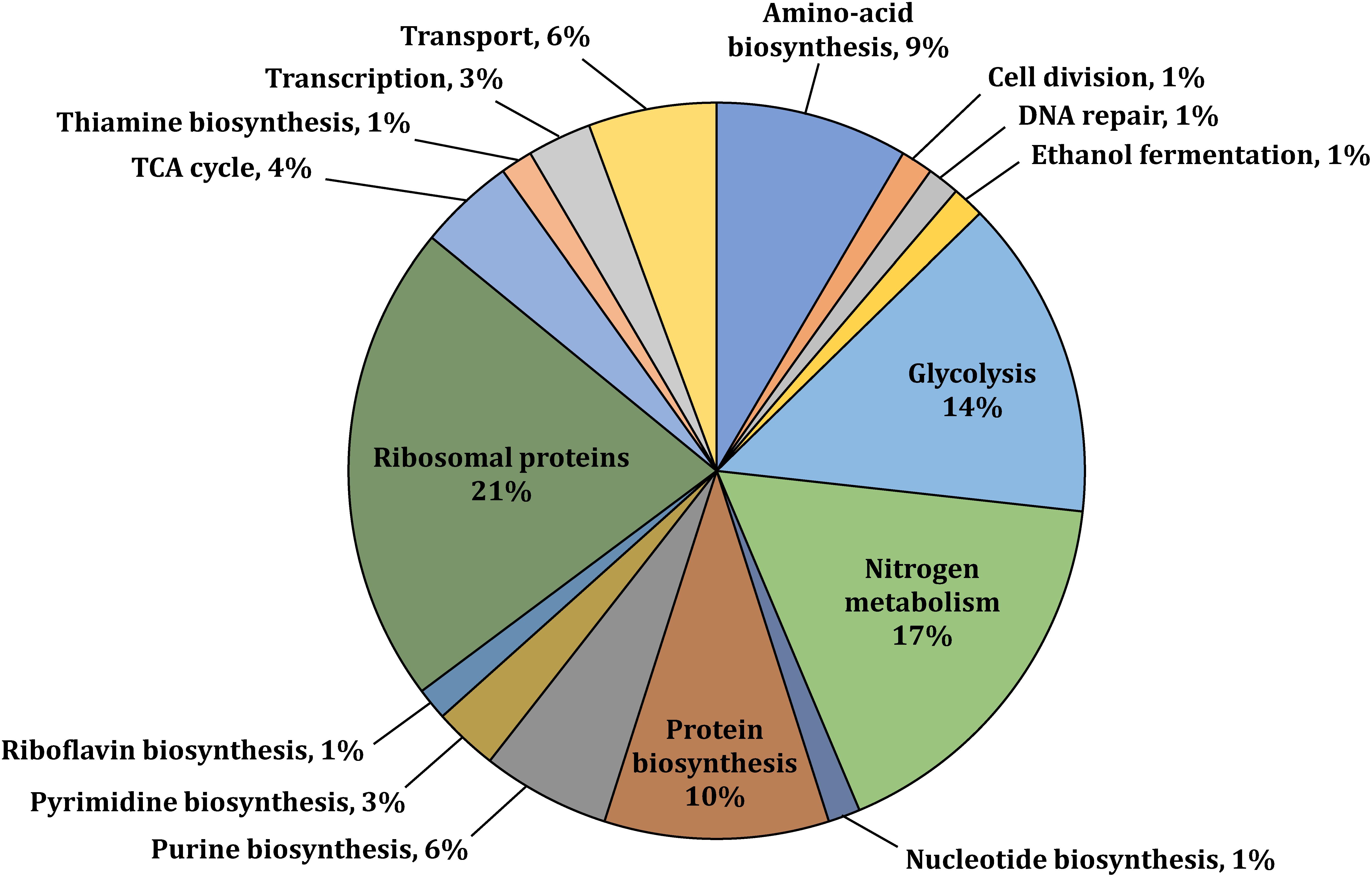
Figure 1. Functional classification of selected phosphoproteins with available gene annotation in UniProt or the KEGG Pathway databases (UniProt Consortium, 2008; Kanehisa et al., 2016). Phosphoproteins were identified in at least one of the three studied growth conditions (i.e., aerobic, anaerobic, or N2-fixing conditions).
Differential Phosphorylation Across Growth Conditions
Our analysis revealed widespread changes in protein phosphorylation across anaerobic, aerobic, and N2-fixing growth conditions that affected several major pathways and cellular processes (Figure 2). Below we summarize a subset of the most significant alterations.
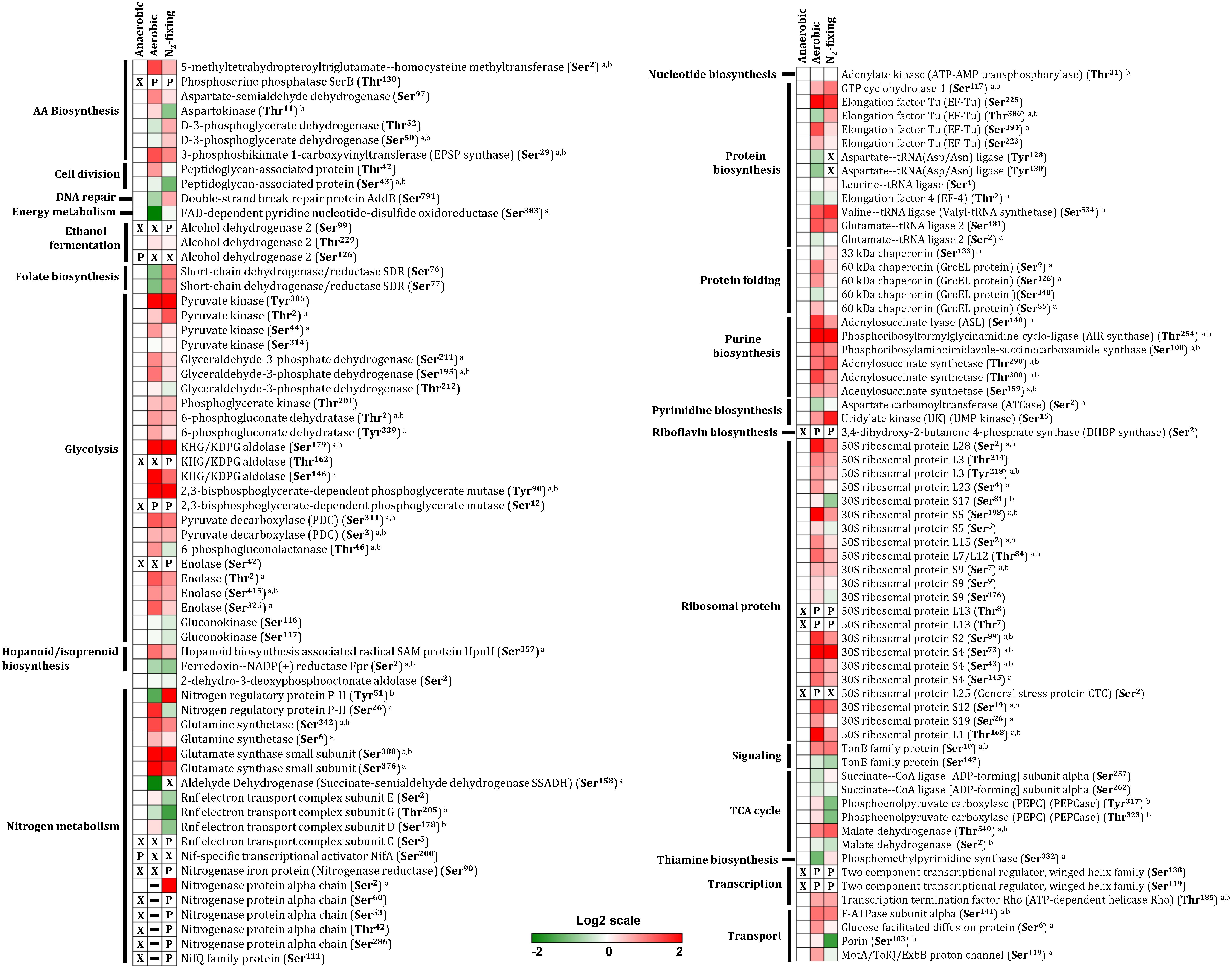
Figure 2. Differential protein phosphorylation across aerobic, anaerobic and N2-fixing growth conditions. The heatmap displays data for 79 selected phosphoproteins. Multiple phosphorylation sites were identified for several proteins. The red (high) and green (low) color scale indicates relative changes (log2 fold-changes vs. anaerobic samples) in protein phosphorylation across growth conditions. Data represents the average of 5 biological replicates. For proteins that were not phosphorylated in the anaerobic control samples and in at least one more growth condition, fold-change comparisons were not made; instead, changes in phosphorylation status are shown as follows: X indicates no phosphorylation was detected, P indicates site is phosphorylated, and a dash (–) indicates that protein level was not quantitated due to its low abundance in the specified growth condition and phosphorylation was also not detected. For fold-change comparisons, the superscripts a and b indicate significant (adjusted q < 0.05, see section “Materials and Methods”) changes in phosphorylation under aerobic and N2-fixing conditions, respectively.
Glycolysis and TCA Cycle
Zymomonas mobilis catabolizes sugars via the Entner–Doudoroff (ED) pathway (Figure 3A) instead of the well-known Embden–Meyerhof–Parnas (EMP) pathway found in model organisms such as E. coli and Saccharomyces cerevisiae (Sprenger, 1996; Inoshima et al., 2012). The ED and EMP pathways share a common set of reactions in lower glycolysis that convert glyceraldehyde-3-phosphate (GAP) into pyruvate but each pathway has unique reactions in its initial steps. The ED pathway starts with phosphorylation of glucose to generate glucose-6-phosphate (G6P), which is then oxidized and dehydrated to 2-keto-3-deoxy-6-phosphogluconate (KDPG). KDPG is then cleaved by KDPG aldolase (KDPGA, ZMO0997) into pyruvate and GAP. Z. mobilis converts >95% of the pyruvate produced from glucose into ethanol via pyruvate decarboxylase (PDC, ZMO1360) and alcohol dehydrogenase (ADH 1, ZMO1236; ADH 2, ZMO1596) (Figure 3A; Panesar et al., 2006).
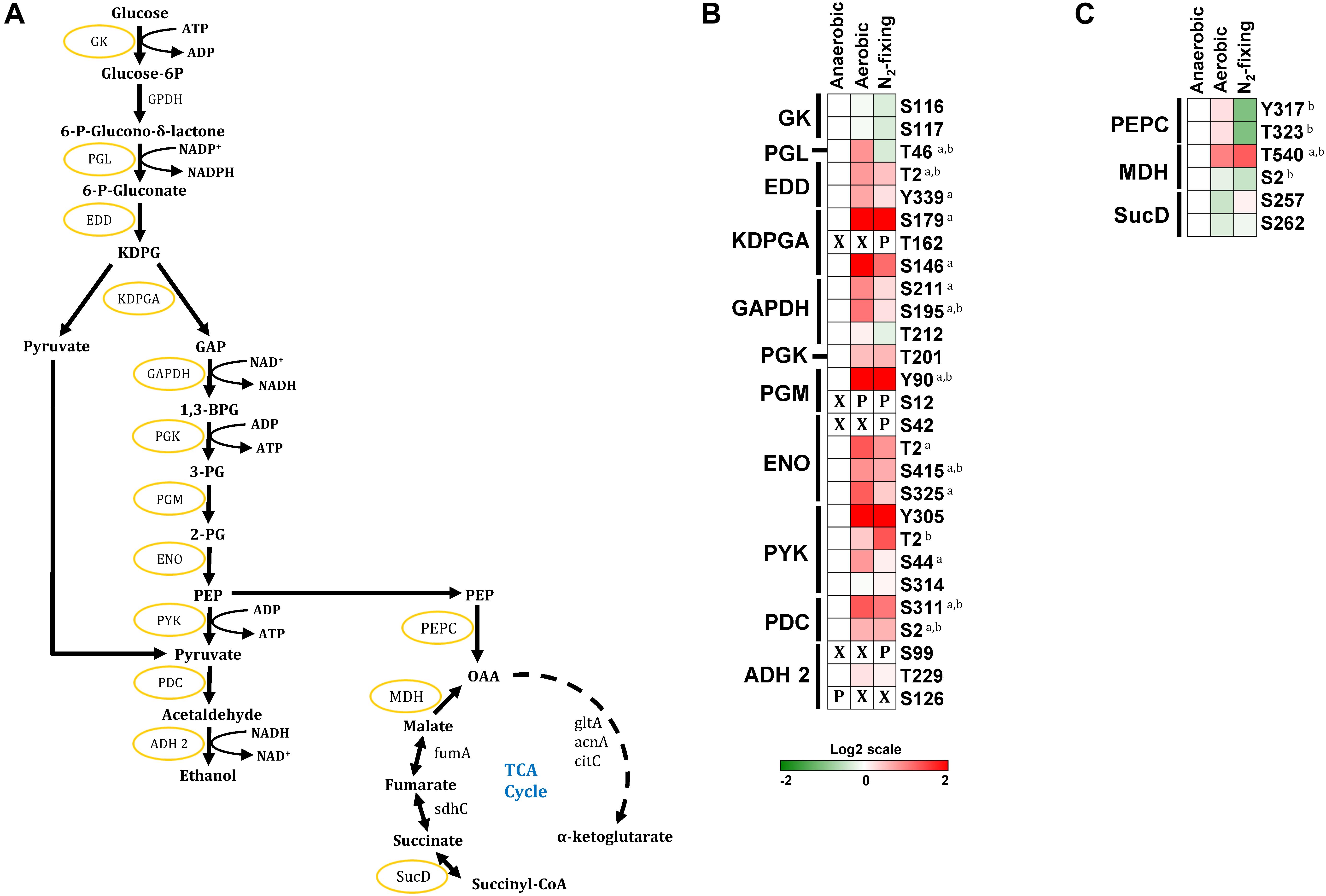
Figure 3. Phosphorylation of central carbon metabolic enzymes. (A) The Entner–Doudoroff glycolytic pathway, TCA cycle, and ethanol fermentation pathway in Z. mobilis. Yellow circles indicate phosphorylated proteins identified in this study. (B,C) Heatmaps showing differential phosphorylation of glycolytic, ethanol fermentation, and TCA cycle enzymes across growth conditions. The red (high) and green (low) color scale indicates relative changes (log2 fold-changes vs. anaerobic samples) in protein phosphorylation across growth conditions. Data represents the average of 5 biological replicates. For proteins that were not phosphorylated in the anaerobic control samples and in at least one more growth condition, fold-change comparisons were not made; instead, changes in phosphorylation status are shown as follows: X indicates no phosphorylation was detected, P indicates site is phosphorylated. For fold-change comparisons, the superscripts a and b indicate significant (adjusted q < 0.05, see section “Materials and Methods”) changes in phosphorylation under aerobic and N2-fixing conditions, respectively. Abbreviations: GK, Glucokinase; GPDH, Glucose-6-phosphate dehydrogenase; PGL, Phosphogluconolactonase; EDD, 6-phosphogluconate dehydratase; KDPGA, 2-keto-3-deoxy-6-phosphogluconate aldolase; GAPDH, Glyceraldehyde-3-phosphate dehydrogenase; PGK, Phosphoglycerate kinase; PGM, phosphoglycerate mutase; ENO, Enolase; PYK, Pyruvate kinase; PDC, Pyruvate decarboxylase; ADH 2, Alcohol dehydrogenase 2; PEPC, Phosphoenolpyruvate carboxylase; MDH, Malate dehydrogenase; fumA, Fumarate hydratase; SucD, Succinate–CoA ligase [ADP-forming] subunit alpha; gltA, Citrate synthase; acnA, Aconitate hydratase; citC, Isocitrate dehydrogenases; sdhC, Succinate dehydrogenase.
We found that with the exception of glucose-6-phosphate dehydrogenase (GPDH, ZMO0367), all other enzymes in the ED glycolytic pathway were phosphorylated under at least one growth condition (Figure 3). We identified multiple phosphorylation sites in most enzymes in glycolysis (Figure 3B). For example, 6-phosphogluconate dehydratase (EDD, ZMO0368) had two phosphorylation sites (Thr2, Tyr339), glyceraldehyde phosphate dehydrogenase (GAPDH, ZMO0177) had three phosphorylation sites (Thr212, Ser211 and Ser195), and pyruvate kinase (PYK, ZMO0152) had four phosphorylation sites (Tyr305, Thr2, Ser44, Ser314). Interestingly, we observed a general trend for increased phosphorylation in nearly all glycolytic enzymes during aerobic and N2-fixing growth (Supplementary Table S4). Some phosphorylation sites were found only in specific growth conditions. For example, KDPGA and Enolase (ENO, ZMO1608) were phosphorylated at Thr162 and Ser42, respectively, only under N2-fixing conditions; while phosphoglycerate mutase (PGM, ZMO1240) was phosphorylated at Ser12 under aerobic and N2-fixing conditions but not under anaerobic growth. The high prevalence of phosphorylation sites in Z. mobilis glycolytic enzymes bears similarity to previous phosphoproteome analyses of other bacteria (i.e., S. coelicolor, Lactococcus lactis, E. coli, B. subtilis, and K. pneumoniae) showing that most of their glycolytic enzymes are phosphorylated (Macek et al., 2007a, b; Soufi et al., 2008; Manteca et al., 2011; Lin et al., 2015).
Within the ethanol fermentation pathway, we found multiple phosphorylation sites in PDC (Ser2 and Ser311) and ADH 2 (Ser99, Thr229, and, Ser126). PDC displayed significantly increased phosphorylation on both Ser2 and Ser311 under aerobic and N2-fixing conditions. In contrast, ADH 2 was specifically phosphorylated at Ser126 only under anaerobic growth and at Ser99 only under N2-fixing conditions (Figure 3B). Previous reports showed that ethanol yield is reduced to <40% under aerobic growth while it stayed largely unaffected under N2-fixing conditions (Kremer et al., 2015; Martien et al., 2019); thus, ADH 2 phosphorylation at Ser99 and Ser126 might play a regulatory role on ethanol biosynthesis in Z. mobilis.
In contrast to glycolytic enzymes, only a few enzymes associated with the TCA cycle were phosphorylated. Phosphoenolpyruvate carboxylase (PEPC, ZMO1496) was phosphorylated at Tyr317 and Thr323; succinate-CoA ligase (SucD, ZMO0567) was phosphorylated at Ser257 and Ser262, and putative malate dehydrogenase (MDH, ZMO1955) was phosphorylated at Ser2 and Thr540 (Figure 3C). Unlike glycolytic enzymes, we did not observe a generalized increase in phosphorylation of TCA cycle-related enzymes under aerobic and N2-fixing conditions (Figure 3C). Only MDH phosphorylation at Thr540 increased significantly under both aerobic and N2-fixing conditions while phosphorylation of PEPC (Tyr317 and Thr323) decreased significantly under N2-fixing conditions (Figure 3C).
Nitrogen Fixation
Biological nitrogen fixation (i.e., reduction of atmospheric nitrogen to ammonia) in bacteria and archaea is carried out by the highly conserved nitrogenase complex. In most nitrogen-fixing bacteria, including Z. mobilis, the nitrogenase complex is encoded by the NifHDK genes. NifHDK expression is controlled by the transcription factor NifA, a master regulator that also controls expression of many other genes involved in nitrogen fixation (Curatti et al., 2005; Heiniger et al., 2012; Tsoy et al., 2016). In Z. mobilis, all NifA-regulated genes are located within a single chromosomal region (ZMO1808-37) that includes the NifA gene, the nitrogenase operon (nifHDKENX-fdxB-nifQ), two operons involved in nitrogenase maturation (nifB-fdxN and iscN-nifUSVW-modD), and the Rnf operon (rnfABCDGEH) encoding the Rnf electron transport complex (Figure 4A; Tsoy et al., 2016).
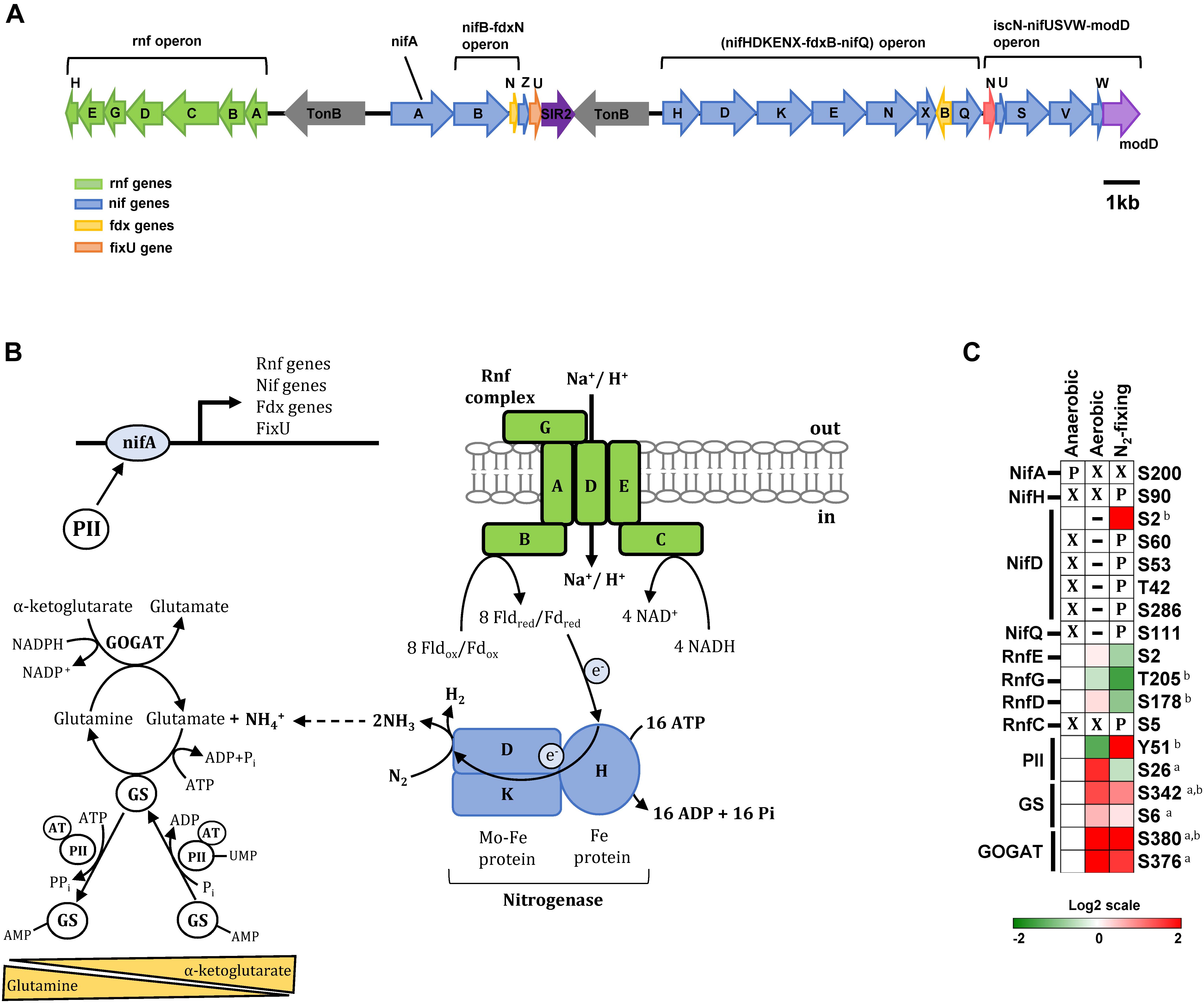
Figure 4. Phosphorylation of enzymes and regulatory proteins involved in nitrogen fixation and ammonia assimilation. (A) Nitrogen fixation genes are located within a single chromosomal region that includes the master regulator NifA, the nitrogenase operon (nifHDKENX-fdxB-nifQ), two operons involved in nitrogenase maturation (nifB-fdxN and iscN-nifUSVW-modD), and the Rnf operon (rnfABCDGEH) encoding the Rnf electron transport complex. (B) Schematic representation of proteins involved in the nitrogen fixation and ammonia assimilation networks in Z. mobilis. Shown are the transmembrane Rnf complex (RnfABCDFE) thought to transfer electrons to nitrogenase via ferredoxin/flavodoxin, the nitrogenase complex (nifHDK), and the GS/GOGAT cycle of ammonia assimilation. Also depicted is the transcription factor NifA acting as a master regulator controlling the expression of nitrogen fixation genes and the putative role of regulatory protein PII at regulating NifA activity. (C) Heatmap showing differential phosphorylation of nitrogen metabolism enzymes and regulatory proteins across growth conditions. The red (high) and green (low) color scale indicates relative changes (log2 fold-changes vs. anaerobic samples) in protein phosphorylation across growth conditions. Data represents the average of 5 biological replicates. For proteins that were not phosphorylated in the anaerobic control samples and in at least one more growth condition, fold-change comparisons were not made; instead, changes in phosphorylation status are shown as follows: X indicates no phosphorylation was detected, P indicates site is phosphorylated, and a dash (–) indicates that protein level was not quantitated due to its low abundance in the specified growth condition and phosphorylation was also not detected. For fold-change comparisons, the superscripts a and b indicate significant (adjusted q < 0.05, see section “Materials and Methods”) changes in phosphorylation under aerobic and N2-fixing conditions, respectively. Abbreviations: GS, Glutamine synthase; GOGAT, Glutamate synthase; NifH, Nitrogenase iron protein; NifD, Nitrogenase molybdenum iron protein; PII, Nitrogen regulatory protein; NifA, Nif specific regulatory protein; NifL, Nitrogen fixation regulatory protein; NifQ, Nitrogen fixation Q protein; RnfEGDC, Electron transport complex subunits.
We observed substantial changes in phosphorylation of the nitrogenase subunits NifH (Fe protein, ZMO1823) and NifD (Fe-Mo protein α-subunit, ZMO1824) during N2-fixing growth. NifH was phosphorylated at Ser90 and NifD was phosphorylated at Ser60, Ser53, Thr42, and Ser286 exclusively under N2-fixing conditions (Figure 4C). NifD phosphorylation at Ser2 was observed in both anaerobic and N2-fixing conditions but displayed a significant increase in phosphorylation under N2-fixing conditions (Figure 4C). NifQ (ZMO1831), potentially involved in the incorporation of molybdenum into nitrogenase, was also phosphorylated (Ser111) only under N2-fixing conditions (Figure 4C).
In contrast to nitrogenase proteins, NifA (ZMO1816) was phosphorylated at Ser200 only during anaerobic growth (Figure 4C). In γ-proteobacteria, NifA activation is regulated via an interacting partner protein, NifL, whereas in α-proteobacteria and some β-proteobacteria that lack the nifL gene, activation of NifA is controlled by other regulatory mechanisms, mainly involving regulatory PII proteins acting in response to the cellular nitrogen and carbon status (Jack et al., 2001; Dixon and Kahn, 2004; Martinez-argudo et al., 2004; Radchenko and Merrick, 2011). Z. mobilis lacks the nifL gene and the mechanism of NifA regulation remains unexplored. It is possible that dephosphorylation of NifA at Ser200 under aerobic and N2-fixing conditions, as well as the phosphorylation changes in the regulatory protein PII described in the next section, may play a role on regulating the activity of this transcription factor in Z. mobilis.
Rnf proteins (RnfABCDGEH) form a transmembrane complex that catalyzes reverse electron flow from NADH to reduce ferredoxin/flavodoxin, which acts as the electron donor to nitrogenase (Curatti et al., 2005; Biegel et al., 2011; Bueno Batista and Dixon, 2019) (Figure 4B). We observed a significant decrease in phosphorylation of RnfD (Ser178) and RnfG (Thr205) proteins in N2-fixing conditions. In contrast, RnfC was phosphorylated (Ser5) exclusively under N2-fixing conditions (Figure 4C). Although the role of Rnf proteins has not been studied in Z. mobilis, changes in their phosphorylation status under N2-fixing conditions may affect their activity in transferring electrons to nitrogenase.
Ammonia Assimilation (GS/GOGAT Cycle) and Regulatory Protein PII
Ammonia (NH4+) assimilation into glutamate takes places via the glutamine synthetase/glutamate synthase cycle (GS/GOGAT, ZMO0493, and ZMO1116). GS catalyzes the ATP-dependent production of glutamine from NH4+ and glutamate. GOGAT then takes this glutamine and α-ketoglutarate to produce two molecules of glutamate (Figure 4B). Glutamate may also be produced from NH4+ and α-ketoglutarate in a single reaction catalyzed by glutamate dehydrogenase (GDH), but this enzyme appears to be missing in Z. mobilis. GS activity is extensively regulated in response to intracellular levels of glutamine, α-ketoglutarate, and other metabolites. Post-translationally, GS is regulated via adenylyation/deadenylylation by the bifunctional adenylyl transferase (AT). AT activity is directly modulated by glutamine, which favors GS adenylyation (i.e., inactivation). AT activity is also regulated by regulatory protein PII: uridylyated PII interacts with AT to promote GS deadenylylation (i.e., activation) while non-uridylyated PII favors adenylyation. Uridylylation of PII by uridylyl transferase (UT, ZMO0766) is in turn favored by low glutamine and high α-ketoglutarate levels (Figure 4B; Bueno Batista and Dixon, 2019).
We found that GS was phosphorylated at Ser6 and Ser342 and GOGAT was phosphorylated at Ser376 and Ser380 (Figure 4C). Under aerobic conditions, phosphorylation of GS and GOGAT increased significantly at all sites. Under N2-fixing conditions, only Ser342 in GS and Ser380 in GOGAT displayed significant increase in phosphorylation, although there was also a clear trend for increased phosphorylation of GOGAT at Ser376 (Figure 4C). Phosphorylation of GS/GOGAT enzymes may constitute a regulatory mechanism controlling activity of this ammonia assimilation cycle under aerobic and N2-fixing conditions.
In addition to GS/GOGAT enzymes, we also found regulatory protein PII (ZMO0492) to be differentially phosphorylated at Tyr51 and Ser26 under different growth conditions (Figure 4C). Tyr51 phosphorylation increased significantly during N2-fixing conditions while phosphorylation on Ser26 was significantly higher in aerobic growth (Figure 4C). Thus, it is possible that these two PII phosphorylation events may have unique, and potentially opposite, regulatory effects in N2-fixing vs. aerobic growth conditions in Z. mobilis.
Ribosomal Proteins and Protein Biosynthesis
Ribosomal proteins, together with rRNA, make up the small and large ribosomal subunits, termed 30S and 50S in bacteria, responsible for cellular protein synthesis. We found that 15 out of 57 ribosomal proteins were phosphorylated in at least one growth condition. Within the large subunit, L1, L3, L13, L15, L23, L25, L28, and L7/L12 stalk complex were phosphorylated; within the small subunit, S2, S4, S5, S9, S12, S17, and S19 were phosphorylated (Figure 5A). Phosphorylation of ribosomal proteins has been observed in several bacteria, including L. lactis, B. subtilis, M. tuberculosis, E. coli, and R. palustris (Macek et al., 2007b; Soufi et al., 2008; Aivaliotis et al., 2009; Ravichandran et al., 2009; Soung et al., 2009; Parker et al., 2010; Manteca et al., 2011; Hu et al., 2012; Lin et al., 2015). All of the ribosomal proteins that we found phosphorylated in Z. mobilis, except for L15, have also been reported to be phosphorylated in E.coli (Macek et al., 2007a, b; Soufi et al., 2008; Prisic et al., 2010; Hu et al., 2012; Lin et al., 2015). Interestingly, we observed a clear trend of increased phosphorylation in ribosomal proteins during aerobic and N2-fixing growth. 12 out of 15 phosphorylated ribosomal proteins displayed increased phosphorylation on single or multiple sites under aerobic and N2-fixing growth conditions (Figure 5B).
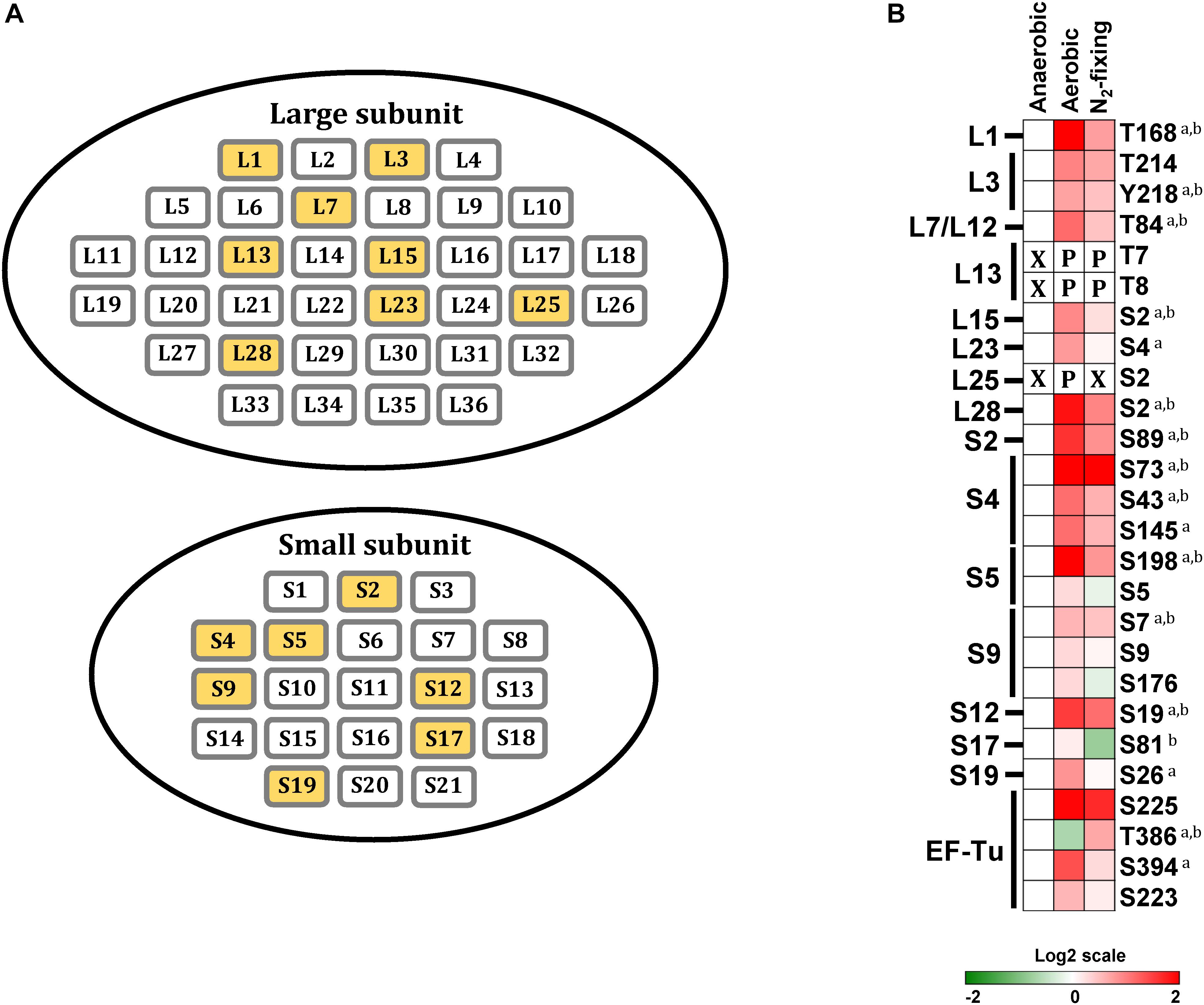
Figure 5. Phosphorylation of ribosomal proteins. (A) Ribosomal proteins of Z. mobilis. Yellow colored boxes indicate the phosphoproteins identified in this study. (B) Heatmap showing differential phosphorylation of ribosomal proteins and Elongation factor-Tu (EF-Tu) across growth conditions. The red (high) and green (low) color scale indicates relative changes (log2 fold-changes vs. anaerobic samples) in protein phosphorylation across growth conditions. Data represents the average of 5 biological replicates. For proteins that were not phosphorylated in the anaerobic control samples and in at least one more growth condition, fold-change comparisons were not made; instead, changes in phosphorylation status are shown as follows: X indicates no phosphorylation was detected, P indicates site is phosphorylated. For fold-change comparisons, the superscripts a and b indicate significant (adjusted q < 0.05, see section “Materials and Methods”) changes in phosphorylation under aerobic and N2-fixing conditions, respectively.
The elongation factor EF-Tu catalyzes binding of aminoacyl-tRNAs (aa-tRNA) to the ribosome (Sajid et al., 2011). EF-Tu forms a ternary complex with GTP via its N-terminal domain (domain I) and aminoacyl-tRNA (aa-tRNA) through its β-barrel domains (domain II and III) (Nissen et al., 1995). EF-Tu phosphorylation at Thr382 in E.coli, Thr63 in B. subtilis, and Thr118 in M. tuberculosis, has been shown to block protein synthesis by interfering with GTP binding (Sajid et al., 2011; Pereira et al., 2015; Talavera et al., 2018). Here we observed that EF-Tu phosphorylation at Thr386, equivalent to Thr382 in E. coli, decreased significantly during aerobic growth but increased under N2-fixing conditions (Figure 5B), suggesting that Z. mobilis might also regulate protein synthesis via EF-Tu phosphorylation. Additionally, we found three other phosphorylation sites (Ser223, Ser225, and Ser394) on EF-Tu that were located on domains II and III. These phosphorylation events could potentially modulate EF-Tu interaction with aa-tRNAs (Nissen et al., 1995). These three sites displayed a trend of increased phosphorylation under both aerobic and N2-fixing conditions (Figure 5B). Interestingly, we also found multiple phosphorylation sites on several aa-tRNA ligases that displayed differential phosphorylation under different growth conditions, but the potential significance of this is unclear (Figure 2).
The L7/L12 stalk complex is known to physically interact with elongation factors EF-Tu and EF-G to promote ternary complex binding to the ribosome (Agrawal et al., 1999, 2002; Ban et al., 2000; Kothe et al., 2004; Savelsbergh et al., 2005). Lys84 of L7/L12 is a highly conserved surface residue that is thought to be critical for its interaction with EF-Tu; its mutation to alanine has been reported to significantly decrease binding efficiency of the ternary complex to the ribosome (Kothe et al., 2004). Interestingly, Lys84 is replaced with threonine in Z. mobilis. We found that phosphorylation of this Thr84 residue increased significantly under aerobic and N2-fixing conditions (Figure 5B), which could potentially affect the interaction of L7/L12 with EF-Tu and thereby ternary complex binding to the ribosome.
L3 and L13 are part of a protein cluster in the large ribosomal subunit that provides important interaction sites for elongation factors (Ban et al., 2000). We found that L13 was phosphorylated (Thr7 and Thr8) exclusively under aerobic and N2-fixing conditions while L3 (Thr214 and Tyr218) phosphorylation increased under these two growth conditions (Figure 5B), which may potentially affect the interaction with elongation factors and influence ribosomal function.
Discussion
This study represents the first genome wide phosphoproteome analysis of Z. mobilis. We identified 125 unique phosphorylated proteins belonging to metabolic pathways and cellular processes such as glycolysis, TCA cycle, protein biosynthesis, electron transport, nitrogen fixation, and ammonia assimilation (Figure 1). Quantitative analysis revealed widespread changes in protein phosphorylation across anaerobic, aerobic, and N2-fixing growth conditions (Figure 2).
Phosphorylation of glycolytic enzymes appears to be prevalent in bacteria, highlighting its potential as a conserved regulatory mechanism of this pathway (Macek et al., 2007a, b; Soufi et al., 2008; Manteca et al., 2011; Lin et al., 2015). For example, the activity of pyruvate phosphate dikinase (PPDK), an enzyme involved in glycolysis/gluconeogenesis and CO2 assimilation, is positively regulated by phosphorylation (Thr487) in the phototrophic bacterium R. palustris (Hu et al., 2012). Also, the activity of the glycolytic enzyme enolase has been shown to decrease by phosphorylation of Ser336, Thr363, and Ser367 in the pathogen Bacillus anthracis (Virmani et al., 2019). In Z. mobilis, we observed a generalized increase, compared to baseline anaerobic growth, in the phosphorylation of most glycolytic enzymes during aerobic and N2-fixing growth conditions (Figure 3). The specific rate of glucose consumption changes substantially in these two growth conditions (Kremer et al., 2015; Strazdina et al., 2018), and it is possible that some of the phosphorylation events that we identified may regulate glycolytic enzyme activity leading to overall changes in flux.
We observed substantial changes in the phosphorylation status of enzymes and regulatory proteins involved in nitrogen fixation and ammonia assimilation, including nitrogenase, the Rnf electron transport complex, the transcription factor NifA, GS-GOGAT cycle enzymes, and the PII regulatory protein (Figure 4). These observations suggest that protein phosphorylation may play a critical role at regulating all aspects of nitrogen metabolism in Z. mobilis. For example, increased phosphorylation of the nitrogenase complex during N2-fixation suggests that these phosphorylation events may promote its activation. There are several known mechanisms that regulate the activity of the nitrogenase complex, including regulation by PII and ADP-ribosylation (Huergo et al., 2012, 2013; Sarkar et al., 2012; Moure et al., 2013). However, to the best of our knowledge, changes in the phosphorylation status of nitrogenase have never been reported in any bacteria. Similarly, we have not come across any reported example of NifA phosphorylation. The activity of the GS-GOGAT cycle is also extensively regulated through various mechanisms, including allosteric regulation and PTM; our results suggests that in Z. mobilis, GS and GOGAT may also be regulated by phosphorylation.
In proteobacteria, PII signal transduction proteins are known to regulate both nitrogen fixation and nitrogen assimilation. PII activity is allosterically regulated by intracellular glutamine, ADP, ATP, and a-ketoglutarate levels as well as by post-translational modification of its flexible T-loop region, which induces conformational changes that modulate its ability to interact with target proteins (Huergo et al., 2013). Specifically, the highly conserved T-loop residue Tyr51 may be modified in response to changes in cellular nitrogen status by uridylation (E. coli and other bacteria), adenylylation (S. coelicolor, Corynebacterium glutamicum) or nitration (anabaena) (Forchhammer and De Marsac, 1994; Kloft and Forchhammer, 2005; Huergo et al., 2013). In addition, phosphorylation of T-loop residue Ser49 under N2–fixing conditions or in response to high AKG concentrations has been observed in cyanobacteria (S. elongatus and Synechocystis sp.) (Forchhammer and De Marsac, 1994, 1995; Kloft and Forchhammer, 2005). In Z. mobilis, we observed differential phosphorylation of PII at Tyr51 across growth conditions (Figure 4C). Phosphorylation of PII at Tyr51 has not been reported in any bacteria, but it is possible that Z. mobilis utilizes this phosphorylation instead of other PTMs to regulate PII activity and nitrogen metabolism. Similarly, although outside of the T-loop, differential PII phosphorylation at Ser26 could also play a role in regulating nitrogen metabolism. Finally, it has been reported that PII can bind to RnfC in the Rnf complex to inactivate its activity (Sarkar et al., 2012) It would be interesting to investigate whether the observed changes in PII or RnfC phosphorylation in Z. mobilis modulate Rnf complex inhibition by PII and disrupt electron flow to the nitrogenase complex.
Paralleling observations in other bacteria (Aivaliotis et al., 2009; Soung et al., 2009; Mikulík et al., 2011; Lin et al., 2015), we found that a significant fraction of ribosomal proteins (24%) are phosphorylated in Z. mobilis (Figure 5). In vitro experiments have shown that phosphorylation of ribosomal proteins can alter ribosomal activity (Traugh and Traut, 1972; Mikulík et al., 2001, 2011). Therefore, it is possible that the generalized increase in phosphorylation of ribosomal proteins during aerobic and N2-fixing growth may play a role at regulating translation in Z. mobilis under these conditions. For example, changes in phosphorylation of L7/L12, L3, and L13 could potentially affect ribosomal activity by modulating interactions with elongation factors EF-Tu and EF-G. Similarly, differential phosphorylation of EF-Tu across growth conditions may also serve as a regulatory mechanism of protein synthesis (Figure 5B).
Our results show that overall protein phosphorylation in Z. mobilis increases under what may be considered “non-optimal” growth conditions, i.e., aerobic and N2-fixing conditions vs. anaerobic growth with abundant ammonia. This suggests that phosphorylation may be particularly important for regulating metabolism during adverse growth conditions.
Conclusion
The genome-wide phosphoproteome analysis of Z. mobilis that we have presented here provides new knowledge regarding the specific proteins, pathways, and cellular processes that may be regulated by phosphorylation in this important industrial organism. The results of this study provide a useful road map and a unique hypothesis-generating resource to establish future genetic and biochemical experiments that investigate the physiological role of specific phosphorylation events in Z. mobilis.
Data Availability
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner repository with the dataset identifier PXD014065 and can be accessed via this link: http://www.ebi.ac.uk/pride/archive/projects/PXD014065.
Author Contributions
MT, AH, JC, and DA-N designed the study. MT prepared the samples for the analysis. AH performed the mass spectrometry experiments. MT, AH, and DA-N analyzed the data. MT and DA-N wrote the manuscript. All authors read and approved the final version of the manuscript.
Funding
This material is based upon the work supported by the Great Lakes Bioenergy Research Center, U.S. Department of Energy, Office of Science, Office of Biological and Environmental Research under Award Number DE-SC0018409.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.01986/full#supplementary-material
References
Agrawal, M., Dunn, K. L., and Rao, C. V. (2017). Engineering of Microorganisms for the Production of Chemicals and Biofuels from Renewable Resources. Berlin: Springer, 1–200.
Agrawal, R. K., Heagle, A. B., Penczek, P., Grassucci, R. A., and Frank, J. (1999). EF-G-dependent GTP hydrolysis induces translocation accompanied by large conformational changes in the 70S ribosome. Nat. Struct. Biol. 6, 643–647. doi: 10.1038/10695
Agrawal, R. K., Penczek, P., Grassucci, R. A., and Frank, J. (2002). Visualization of elongation factor G on the Escherichia coli 70S ribosome: the mechanism of translocation. Proc. Natl. Acad. Sci. U.S.A. 95, 6134–6138. doi: 10.1073/pnas.95.11.6134
Aivaliotis, M., Macek, B., Gnad, F., Reichelt, P., Mann, M., and Oesterhelt, D. (2009). Ser/Thr/Tyr protein phosphorylation in the archaeon Halobacterium salinarum - A representative of the third domain of life. PLoS One 4:e4777. doi: 10.1371/journal.pone.0004777
Ban, N., Nissen, P., Hansen, J., Moore, P. B., and Steitz, T. A. (2000). The complete atomic structure of the large ribosomal subunit at 2.4 Å resolution. Science 289, 905–920. doi: 10.1126/science.2479982
Biegel, E., Schmidt, S., González, J. M., and Müller, V. (2011). Biochemistry, evolution and physiological function of the Rnf complex, a novel ion-motive electron transport complex in prokaryotes. Cell. Mol. Life Sci. 68, 613–634. doi: 10.1007/s00018-010-0555-558
Bueno Batista, M., and Dixon, R. (2019). Manipulating nitrogen regulation in diazotrophic bacteria for agronomic benefit. Biochem. Soc. Trans. 47, 603–614. doi: 10.1042/BST20180342
Cox, J., and Mann, M. (2008). MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372. doi: 10.1038/nbt.1511
Curatti, L., Brown, C. S., Ludden, P. W., and Rubio, L. M. (2005). Genes required for rapid expression of nitrogenase activity in Azotobacter vinelandii. Proc. Natl. Acad. Sci. U.S.A. 102, 6291–6296. doi: 10.1073/pnas.0501216102
Dixon, R., and Kahn, D. (2004). Genetic regulation of biological nitrogen fixation. Nat. Rev. Microbiol. 2, 621–631. doi: 10.1038/nrmicro954
Forchhammer, K., and De Marsac, N. T. (1994). The P(II) protein in the cyanobacterium Synechococcus sp. strain PCC 7942 is modified by serine phosphorylation and signals the cellular N-status. J. Bacteriol. 176, 84–91. doi: 10.1128/jb.176.1.84-91.1994
Forchhammer, K., and De Marsac, N. T. (1995). Functional analysis of the phosphoprotein P(II) (glnB gene product) in the cyanobacterium Synechococcus sp. strain PCC 7942. J. Bacteriol. 177, 2033–2040. doi: 10.1128/jb.177.8.2033-2040.1995
Ghosh, I. N., Martien, J., Hebert, A. S., Zhang, Y., Coon, J. J., Amador-Noguez, D., et al. (2019). OptSSeq explores enzyme expression and function landscapes to maximize isobutanol production rate. Metab. Eng. 52, 324–340. doi: 10.1016/j.ymben.2018.12.008
Hebert, A. S., Richards, A. L., Bailey, D. J., Ulbrich, A., Coughlin, E. E., Westphall, M. S., et al. (2014). The one hour yeast proteome. Mol. Cell. Proteom. 13, 339–347. doi: 10.1074/mcp.m113.034769
Hebert, A. S., Thöing, C., Riley, N. M., Kwiecien, N. W., Shiskova, E., Huguet, R., et al. (2018). Improved precursor characterization for data-dependent mass spectrometry. Anal. Chem. 90, 2333–2340. doi: 10.1021/acs.analchem.7b04808
Heiniger, E. K., Oda, Y., Samanta, S. K., and Harwood, C. S. (2012). How posttranslational modification of nitrogenase is circumvented in Rhodopseudomonas palustris strains that produce hydrogen gas constitutively. Appl. Environ. Microbiol. 78, 1023–1032. doi: 10.1128/AEM.07254-7211
Hu, C. W., Lin, M. H., Huang, H. C., Ku, W. C., Yi, T. H., Tsai, C. F., et al. (2012). Phosphoproteomic analysis of Rhodopseudomonas palustris reveals the role of pyruvate phosphate dikinase phosphorylation in lipid production. J. Proteome Res. 11, 5362–5375. doi: 10.1021/pr300582p
Huergo, L. F., Chandra, G., and Merrick, M. (2013). PII signal transduction proteins: nitrogen regulation and beyond. FEMS Microbiol. Rev. 37, 251–283. doi: 10.1111/j.1574-6976.2012.00351.x
Huergo, L. F., Pedrosa, F. O., Muller-Santos, M., Chubatsu, L. S., Monteiro, R. A., Merrick, M., et al. (2012). P II signal transduction proteins: pivotal players in post-translational control of nitrogenase activity. Microbiology 158, 176–190. doi: 10.1099/mic.0.049783-0
Inoshima, I., Inoshima, N., Wilke, G., Powers, M., Frank, K., Wang, Y., et al. (2012). A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 17, 1310–1314. doi: 10.1038/nm.2451
Jack, R., Arconde, T., and Merrick, M. (2001). P II signal transduction proteins, pivotal players in microbial nitrogen control. Microbiol. Mol. Biol. Rev. 65, 80–105. doi: 10.1128/MMBR.65.1.80
Jones-Burrage, S. E., Kremer, T. A., and McKinlay, J. B. (2019). Cell aggregation and aerobic respiration are important for Zymomonas mobilis ZM4 survival in an aerobic minimal medium. Appl. Environ. Microbiol. 85, 1–12. doi: 10.1128/aem.00193-119
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2016). KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462. doi: 10.1093/nar/gkv1070
Kloft, N., and Forchhammer, K. (2005). Signal transduction protein P II phosphatase PphA is required for light-dependent control of nitrate utilization in in Synechocystis sp. strain PCC 6803. J. Bacteriol. 187, 6683–6690. doi: 10.1128/JB.187.19.6683
Kothe, U., Wieden, H. J., Mohr, D., and Rodnina, M. V. (2004). Interaction of helix d of elongation factor tu with helices 4 and 5 of protein L7/12 on the ribosome. J. Mol. Biol. 336, 1011–1021. doi: 10.1016/j.jmb.2003.12.080
Kremer, T. A., LaSarre, B., Posto, A. L., and McKinlay, J. B. (2015). N 2 gas is an effective fertilizer for bioethanol production by Zymomonas mobilis. Proc. Natl. Acad. Sci. 112, 2222–2226. doi: 10.1073/pnas.1420663112
Lin, M. H., Sugiyama, N., and Ishihama, Y. (2015). Systematic profiling of the bacterial phosphoproteome reveals bacterium-specific features of phosphorylation. Sci. Signal. 8, 1–9. doi: 10.1126/scisignal.aaa3117
Macek, B., Gnad, F., Soufi, B., Kumar, C., Olsen, J. V., Mijakovic, I., et al. (2007a). Phosphoproteome analysis of E. coli Reveals evolutionary conservation of bacterial Ser/Thr/Tyr phosphorylation. Mol. Cell. Proteom. 7, 299–307. doi: 10.1074/mcp.m700311-mcp200
Macek, B., Mijakovic, I., Olsen, J. V., Gnad, F., Kumar, C., Jensen, P. R., et al. (2007b). The serine/threonine/tyrosine phosphoproteome of the model Bacterium Bacillus subtilis. Mol. Cell. Proteom. 6, 697–707. doi: 10.1074/mcp.m600464-mcp200
Macek, B., and Mijakovic, I. (2011). Site-specific analysis of bacterial phosphoproteomes. Proteomics 11, 3002–3011. doi: 10.1002/pmic.201100012
Manteca, A., Ye, J., Sánchez, J., and Jensen, O. N. (2011). Phosphoproteome analysis of streptomyces development reveals extensive protein phosphorylation accompanying bacterial differentiation. J. Proteom. Res. 10, 5481–5492. doi: 10.1021/pr200762y
Martien, J. I., Hebert, A. S., Stevenson, D. M., Regner, M. R., Khana, D. B., Coon, J. J., et al. (2019). Systems level analysis of oxygen exposure in Zymomonas mobilis: implications for isoprenoid production. mSystems 4, 1–23. doi: 10.1128/msystems.00284-218
Martinez-argudo, I., Little, R., Shearer, N., Johnson, P., and Dixon, R. (2004). The NifL-NifA system: a multidomain transcriptional regulatory complex that integrates environmental signals. J. Bacteriol. 186, 1–10. doi: 10.1128/JB.186.3.601
Mikulík, K., Bobek, J., Ziková, A., Smtáková, M., and Bezouková, S. (2011). Phosphorylation of ribosomal proteins influences subunit association and translation of poly (U) in Streptomyces coelicolor. Mol. Biosyst. 7, 817–823. doi: 10.1039/c0mb00174k
Mikulík, K., Suchan, P., and Bobek, J. (2001). Changes in ribosome function induced by protein kinase associated with ribosomes of Streptomyces collinus producing kirromycin. Biochem. Biophys. Res. Commun. 289, 434–443. doi: 10.1006/bbrc.2001.6017
Moure, V. R., Danyal, K., Yang, Z. Y., Wendroth, S., Müller-santos, M., Pedrosa, F. O., et al. (2013). The nitrogenase regulatory enzyme dinitrogenase reductase adpribosyltransferase (DraT) is activated by direct interaction with the signal transduction protein glnb. J. Bacteriol. 195, 279–286. doi: 10.1128/JB.01517-2
Nissen, P., Kjeldgaard, M., Thirup, S., Polekhina, G., Clark, B. F. C., Nyborg, J., et al. (1995). Published by: american association for the advancement of science crystal structure of the ternary complex of Phe-tRNAPhe, EF-Tu and a GTP analog. Science 270, 1464–1472.
Pan, K., Wu, B., Tang, X., He, M., Wang, J., Tan, F., et al. (2014). Zymomonas mobilis: a novel platform for future biorefineries. Biotechnol. Biofuels 7, 101. doi: 10.1186/1754-6834-7-101
Panesar, P. S., Marwaha, S. S., and Kennedy, J. F. (2006). Zymomonas mobilis: an alternative ethanol producer. J. Chem. Technol. Biotechnol. 81, 623–635. doi: 10.1002/jctb.1448
Parker, J. L., Jones, A. M. E., Serazetdinova, L., Saalbach, G., Bibb, M. J., and Naldrett, M. J. (2010). Analysis of the phosphoproteome of the multicellular bacterium Streptomyces coelicolor A3(2) by protein/peptide fractionation, phosphopeptide enrichment and high-accuracy mass spectrometry. Proteomics 10, 2486–2497. doi: 10.1002/pmic.201000090
Pereira, S. F. F., Gonzalez, R. L., and Dworkin, J. (2015). Protein synthesis during cellular quiescence is inhibited by phosphorylation of a translational elongation factor. Proc. Natl. Acad. Sci. U.S.A. 112, E3274–E3281. doi: 10.1073/pnas.1505297112
Pisithkul, T., Patel, N. M., and Amador-Noguez, D. (2015). Post-translational modifications as key regulators of bacterial metabolic fluxes. Curr. Opin. Microbiol. 24, 29–37. doi: 10.1016/j.mib.2014.12.006
Potel, C. M., Lin, M. H., Heck, A. J. R., and Lemeer, S. (2018). Widespread bacterial protein histidine phosphorylation revealed by mass spectrometrybased proteomics. Nat. Methods 15, 187–190. doi: 10.1038/nmeth.4580
Prisic, S., Dankwa, S., Schwartz, D., Chou, M. F., Locasale, J. W., Kang, C.-M., et al. (2010). Extensive phosphorylation with overlapping specificity by Mycobacterium tuberculosis serine/threonine protein kinases. Proc. Natl. Acad. Sci. U.S.A. 107, 7521–7526. doi: 10.1073/pnas.0913482107
Radchenko, M., and Merrick, M. (2011). The role of effector molecules in signal transduction by P II proteins. Biochem. Soc. Trans. 39, 189–194. doi: 10.1042/bst0390189
Ravichandran, A., Sugiyama, N., Tomita, M., Swarup, S., and Ishihama, Y. (2009). Ser/Thr/Tyr phosphoproteome analysis of pathogenic and non-pathogenic Pseudomonas species. Proteomics 9, 2764–2775. doi: 10.1002/pmic.200800655
Ravikumar, V., Jers, C., and Mijakovic, I. (2015). Elucidating host-pathogen interactions based on post-translational modifications using proteomics approaches. Front. Microbiol. 6:1313. doi: 10.3389/fmicb.2015.01312
Ravikumar, V., Shi, L., Krug, K., Derouiche, A., Jers, C., Cousin, C., et al. (2014). Quantitative phosphoproteome analysis of bacillus subtilis reveals novel substrates of the kinase PrkC and phosphatase PrpC. Mol. Cell. Proteomics 13, 1965–1978. doi: 10.1074/mcp.m113.035949
Riley, N. M., and Coon, J. J. (2016). Phosphoproteomics in the age of rapid and deep proteome profiling. Anal. Chem. 88, 74–94. doi: 10.1021/acs.analchem.5b04123
Rogers, P. L., Lee, K. J., Lefebvre, M., and Tribe, D. E. (1980). High productivity ethanol fermentations with Zymomonas Mobilis using continuous cell recycle. Biotechnol. Lett. 492, 487–492.
Rutkis, R., Strazdina, I., Balodite, E., Lasa, Z., Galinina, N., and Kalnenieks, U. (2016). The low energy-coupling respiration in Zymomonas mobilis accelerates flux in the entner-doudoroff pathway. PLoS One 11:e0153866. doi: 10.1371/journal.pone.0153866
Sajid, A., Arora, G., Gupta, M., Singhal, A., Chakraborty, K., Nandicoori, V. K., et al. (2011). Interaction of Mycobacterium tuberculosis elongation factor Tu with GTP is regulated by phosphorylation. J. Bacteriol. 193, 5347–5358. doi: 10.1128/JB.05469-11
Sarkar, A., Köhler, J., Hurek, T., and Reinhold-Hurek, B. (2012). A novel regulatory role of the Rnf complex of Azoarcus sp. strain BH72. Mol. Microbiol. 83, 408–422. doi: 10.1111/j.1365-2958.2011.07940.x
Savelsbergh, A., Mohr, D., Kothe, U., Wintermeyer, W., and Rodnina, M. V. (2005). Control of phosphate release from elongation factor G by ribosomal protein L7/12. EMBO J. 24, 4316–4323. doi: 10.1038/sj.emboj.7600884
Soares, N. C., and Blackburn, J. M. (2016). Mass spectrometry targeted assays as a tool to improve our understanding of post-translational modifications in pathogenic bacteria. Front. Microbiol. 7:1216. doi: 10.3389/fmicb.2016.01216
Soufi, B., Gnad, F., Jensen, P. R., Petranovic, D., Mann, M., Mijakovic, I., et al. (2008). The Ser/Thr/Tyr phosphoproteome of Lactococcus lactis IL1403 reveals multiply phosphorylated proteins. Proteomics 8, 3486–3493. doi: 10.1002/pmic.200800069
Soufi, B., Soares, N. C., Ravikumar, V., and Macek, B. (2012). Proteomics reveals evidence of cross-talk between protein modifications in bacteria: focus on acetylation and phosphorylation. Curr. Opin. Microbiol. 15, 357–363. doi: 10.1016/j.mib.2012.05.003
Soung, G. Y., Miller, J. L., Koc, H., and Koc, E. C. (2009). Comprehensive analysis of phosphorylated proteins of Escherichia coli ribosomes. J. Proteome Res. 8, 3390–3402. doi: 10.1021/pr900042e
Sprenger, G. A. (1996). a Catabolic highway with some scenic routes. FEMS Microbiol. Lett. 145, 301–307.
Strazdina, I., Balodite, E., Lasa, Z., Rutkis, R., Galinina, N., and Kalnenieks, U. (2018). Aerobic catabolism and respiratory lactate bypass in Ndh-negative Zymomonas mobilis. Metab. Eng. Commun. 7, 1–9. doi: 10.1016/j.mec.2018.e00081
Talavera, A., Hendrix, J., Versées, W., Jurenas, D., Van Nerom, K., Vandenberk, N., et al. (2018). Phosphorylation decelerates conformational dynamics in bacterial translation elongation factors. Sci. Adv. 4, eaa9714. doi: 10.1126/sciadv.aap9714
Tanaka, H., Ishikawa, H., Osuga, K., and Takagi, Y. (1990). Fermentative ability of Zymomonas mobilis under various oxygen supply conditions in batch culture. J. Ferment. Bioeng. 69, 234–239. doi: 10.1016/0922-338X(90)90219-M
Traugh, J. A., and Traut, R. R. (1972). Phosphorylation of ribosomal proteins of Escherichia coli by protein kinase from rabbit skeletal muscle. Biochemistry 11, 2503–2509. doi: 10.1021/bi00763a019
Tsoy, O. V., Ravcheev, D. A., Čuklina, J., and Gelfand, M. S. (2016). Nitrogen fixation and molecular oxygen: comparative genomic reconstruction of transcription regulation in alphaproteobacteria. Front. Microbiol. 7:1343. doi: 10.3389/fmicb.2016.01343
Tyanova, S., Temu, T., Sinitcyn, P., Carlson, A., Hein, M. Y., Geiger, T., et al. (2016). The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 13, 731–740. doi: 10.1038/nmeth.3901
UniProt Consortium (2008). The universal protein resource (UniProt) 2009. Nucleic Acids Res. 37, D169–D174. doi: 10.1093/nar/gkn664
Virmani, R., Sajid, A., Singhal, A., Gaur, M., Joshi, J., Bothra, A., et al. (2019). The Ser/Thr protein kinase PrkC imprints phenotypic memory in Bacillus anthracis spores by phosphorylating the glycolytic enzyme enolase. J. Biol. Chem 294, 8930–8941. doi: 10.1074/jbc.ra118.005424
Yang, S., Fei, Q., Zhang, Y., Contreras, L. M., Utturkar, S. M., Brown, S. D., et al. (2016a). Zymomonas mobilis as a model system for production of biofuels and biochemicals. Microb. Biotechnol. 9, 699–717. doi: 10.1111/1751-7915.12408
Yang, S., Mohagheghi, A., Franden, M. A., Chou, Y. C., Chen, X., Dowe, N., et al. (2016b). Metabolic engineering of Zymomonas mobilis for 2,3-butanediol production from lignocellulosic biomass sugars. Biotechnol. Biofuels 9, 1–15. doi: 10.1186/s13068-016-0606-y
Keywords: Z. mobilis, protein phosphorylation, glycolysis, nitrogen fixation, ammonia assimilation, phosphoproteome
Citation: Tatli M, Hebert AS, Coon JJ and Amador-Noguez D (2019) Genome Wide Phosphoproteome Analysis of Zymomonas mobilis Under Anaerobic, Aerobic, and N2-Fixing Conditions. Front. Microbiol. 10:1986. doi: 10.3389/fmicb.2019.01986
Received: 04 June 2019; Accepted: 13 August 2019;
Published: 04 September 2019.
Edited by:
Uldis Kalnenieks, University of Latvia, LatviaReviewed by:
Catherine Duport, University of Avignon, FranceAndris Zeltins, Latvian Biomedical Research and Study Centre (BMC), Latvia
Copyright © 2019 Tatli, Hebert, Coon and Amador-Noguez. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Amador-Noguez, YW1hZG9ybm9ndWV6QHdpc2MuZWR1
†These authors have contributed equally to this work