 Gi Yoon Shin1,2
Gi Yoon Shin1,2 Jeffrey K. Schachterle3
Jeffrey K. Schachterle3 Divine Y. Shyntum2
Divine Y. Shyntum2 Lucy N. Moleleki2
Lucy N. Moleleki2 Teresa A. Coutinho1,2*
Teresa A. Coutinho1,2* George W. Sundin3
George W. Sundin3- 1Centre for Microbial Ecology and Genomics, Forestry and Agricultural Biotechnology Institute, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria, South Africa
- 2Forestry and Agricultural Biotechnology Institute, Department of Biochemistry, Genetics and Microbiology, University of Pretoria, Pretoria, South Africa
- 3Department of Plant, Soil and Microbial Sciences, College of Agriculture & Natural Resources, Michigan State University, East Lansing, MI, United States
To successfully infect plant hosts, the collective regulation of virulence factors in a bacterial pathogen is crucial. Hfq is an RNA chaperone protein that facilitates the small RNA (sRNA) regulation of global gene expression at the post-transcriptional level. In this study, the functional role of Hfq in a broad host range phytopathogen Pantoea ananatis was determined. Inactivation of the hfq gene in P. ananatis LMG 2665T resulted in the loss of pathogenicity and motility. In addition, there was a significant reduction of quorum sensing signal molecule acyl-homoserine lactone (AHL) production and biofilm formation. Differential sRNA expression analysis between the hfq mutant and wild-type strains of P. ananatis revealed 276 sRNAs affected in their abundance by the loss of hfq at low (OD600 = 0.2) and high cell (OD600 = 0.6) densities. Further analysis identified 25 Hfq-dependent sRNAs, all showing a predicted Rho-independent terminator of transcription and mapping within intergenic regions of the P. ananatis genome. These included known sRNAs such as ArcZ, FnrS, GlmZ, RprA, RyeB, RyhB, RyhB2, Spot42, and SsrA, and 16 novel P. ananatis sRNAs. The current study demonstrated that Hfq is an important component of the collective regulation of virulence factors and sets a foundation for understanding Hfq-sRNA mediated regulation in the phytopathogen P. ananatis.
Introduction
Pantoea ananatis, formerly described as the pineapple pathogen Erwinia ananas (Serrano, 1928), is a Gram-negative bacterium belonging to the family Enterobacteriaceae. To date, the occurrence of P. ananatis has been reported from various ecological niches spanning both the aquatic and terrestrial environments, including fresh (Morohoshi et al., 2007) and marine water (Jatt et al., 2014) as well as the rhizosphere of crop plants (Oliveira et al., 2008; Marquez-Santacruz et al., 2010). The bacterium exhibits ecologically diverse roles in association with its environment. For example, P. ananatis can be found as an epiphyte of crop and weed plants (Gitaitis et al., 2002) or as an endophyte in maize kernels (Rijavec et al., 2007) and rice seeds (Okunishi et al., 2005). Moreover, the ability of P. ananatis to solubilize phosphate, and produce indole-acetic acid and siderophores, makes the bacterium an ideal plant growth-promoting agent in the production of pepper (Kang et al., 2007), soybean (Kuklinsky-Sobral et al., 2004), and sugarcane (da Silva et al., 2015).
Pantoea ananatis is better known as a phytopathogen affecting the yield of many economically important plant species that causes blight and dieback of Eucalyptus (Coutinho et al., 2002), maize leaf spot disease and brown stalk rot (Goszczynska et al., 2006; Pérez-y-Terrón et al., 2009; Alippi and López, 2010; Krawczyk et al., 2010), leaf blight and bulb rot of onion (Gitaitis and Gay, 1997; Schwartz and Otto, 2000; Goszczynska et al., 2007), palea browning and stem necrosis of rice (Azegami et al., 1983; Cother et al., 2004; Cortesi and Pizzatti, 2007), and fruit rot of netted melon (Kido et al., 2008). P. ananatis has also been considered an emerging plant pathogen due to increasing reports of disease outbreaks in the previously undescribed host and geographical regions (Coutinho and Venter, 2009). This emergence is likely to have resulted from the persistent nature of P. ananatis in diverse environments through its association with a wide range of non-host plant and even insect vectors (Gitaitis et al., 2003; Dutta et al., 2014).
The virulence factors that have been identified as necessary for pathogenesis of P. ananatis in onion are motility for attachment (Weller-Stuart et al., 2016) and quorum sensing (QS) for production of biofilm and exopolysaccharide (EPS) (Morohoshi et al., 2007). In addition, genomic regions named “HiVir” (Asselin et al., 2018) and “Onion Virulence Region” (Stice et al., 2018), encoding enzymes catalyzing phosphonate biosynthetic pathway and cell wall degradation, respectively, have been characterized in the onion pathogenic strains of P. ananatis. For successful infection by P. ananatis, a rapid and collective expression of these virulence genes in response to the surrounding environment is critical as it results in the modulation of cellular pathways that predispose the pathogen for infection, pathogenesis, and survival in the host.
Hfq is an RNA-binding protein that constitutes a key component of post-transcriptional gene regulation exhibited by small non-coding regulatory RNAs (sRNAs) (Vogel and Luisi, 2011). Hfq is a ring-like homohexameric protein that was initially identified as a host factor needed for the replication of RNA bacteriophage Qβ (Franze de Fernandez et al., 1968). It is now known that the chaperone Hfq is essential for the structural stabilization of the class of trans-acting sRNAs whose regulatory mechanisms are dependent on Hfq (Updegrove et al., 2016). The chaperone facilitates imperfect base-pairing between the sRNA and its cognate messenger RNA (mRNA), forming an Hfq–sRNA–mRNA complex that determines the fate of target mRNA translation (Gottesman and Storz, 2011; Storz et al., 2011). Suppression of the protein synthesis is achieved by the formation of a sRNA–mRNA duplex at the 5′-untranslated region (UTR) of the transcript by occlusion of ribosome binding and/or by recruiting ribonucleases for mRNA degradation (De Lay et al., 2013). Conversely, translation of the mRNA is enhanced by Hfq–sRNA complexes that alter the 5′-secondary inhibitory structure of an mRNA, making it more accessible for initiation of translation.
Hfq-dependent sRNAs are typically 50–300 nucleotides in length and are trans-encoded from their cognate mRNAs. They are mostly found in, but not limited to, the intergenic regions of bacterial chromosomes (Argaman et al., 2001; Chao et al., 2012; Guo et al., 2014), and are characterized by often possessing a Rho-independent terminator at the 3′-end, resulting in a poly-uridine tail of sRNA that are recognized by Hfq (Otaka et al., 2011). The cellular functions modulated by Hfq–sRNAs are diverse, ranging from cell membrane integrity, acquisition, and metabolism of nutrients, motility, secretion systems, stress response, and virulence (Chao and Vogel, 2010). Their role in virulence regulation has been extensively studied in bacterial pathogens of animals. For example, in Salmonella typhimurium, motility and expression of the T3SS encoded within Salmonella pathogenicity island SPI-1 and SPI-2 are dependent on Hfq and contribute significantly to the adhesion and invasion of Salmonella into the host cells (Sittka et al., 2007, 2008) whereas in Vibrio cholera, Hfq and its trans acting sRNAs Qrr 1–4 regulate cholera toxin (CT) biosynthesis (Bardill and Hammer, 2012) and QS as an ultrasensitive switch to transition V. cholerae from low to high cell density mode for colonization and disease development (Lenz et al., 2004).
Despite the growing evidence of Hfq and Hfq-dependent sRNAs as a global post-transcriptional gene regulatory complex, the functionality of Hfq and its trans acting sRNAs in plant pathogenic bacteria has only been investigated in a few bacterial species to date, namely in Agrobacterium tumefaciens (Wilms et al., 2012a, b), Burkholderia glumae (Kim et al., 2018), Dickeya dadantii (Yuan et al., 2019), Erwinia amylovora (Zeng et al., 2013; Zeng and Sundin, 2014), Pectobacterium carotovorum (Wang et al., 2018), and Xanthomonas spp. (Schmidtke et al., 2013). Consequently, the functional role of Hfq and the diversity of Hfq-dependent sRNAs in phytopathogens remain largely elusive. We hypothesized that Hfq and Hfq-dependent sRNAs would play a critical role in P. ananatis pathogenesis, through direct regulation of specific virulence traits and through regulation of QS system. In this study, we functionally characterized the role of Hfq as a regulator in the production of acyl-homoserine lactones (AHLs), biofilm development, motility, and virulence, and identified the Hfq-dependent sRNAs that are potentially implicated in the regulation of the virulence traits of the ubiquitous plant pathogen P. ananatis.
Materials and Methods
Bacterial Strains and Growth Conditions
The bacterial strains and plasmids used in this study are listed in Table 1. P. ananatis LMG2665T and Escherichia coli DH5α strains were cultured in Luria-Bertani (LB) broth [1% (w/v) NaCl, 1% (w/v) tryptone, and 0.5% (w/v) yeast extract; pH 7.2] or on LB agar plates [LB broth amended with 1.5% (w/v) agar; pH 7.2] at 28 and 37°C, respectively. The growth medium was supplemented with either ampicillin (100 μg/ml), chloramphenicol (50 μg/ml), gentamicin (20 μg/ml), or kanamycin (50 μg/ml) for plasmid DNA selection and maintenance.
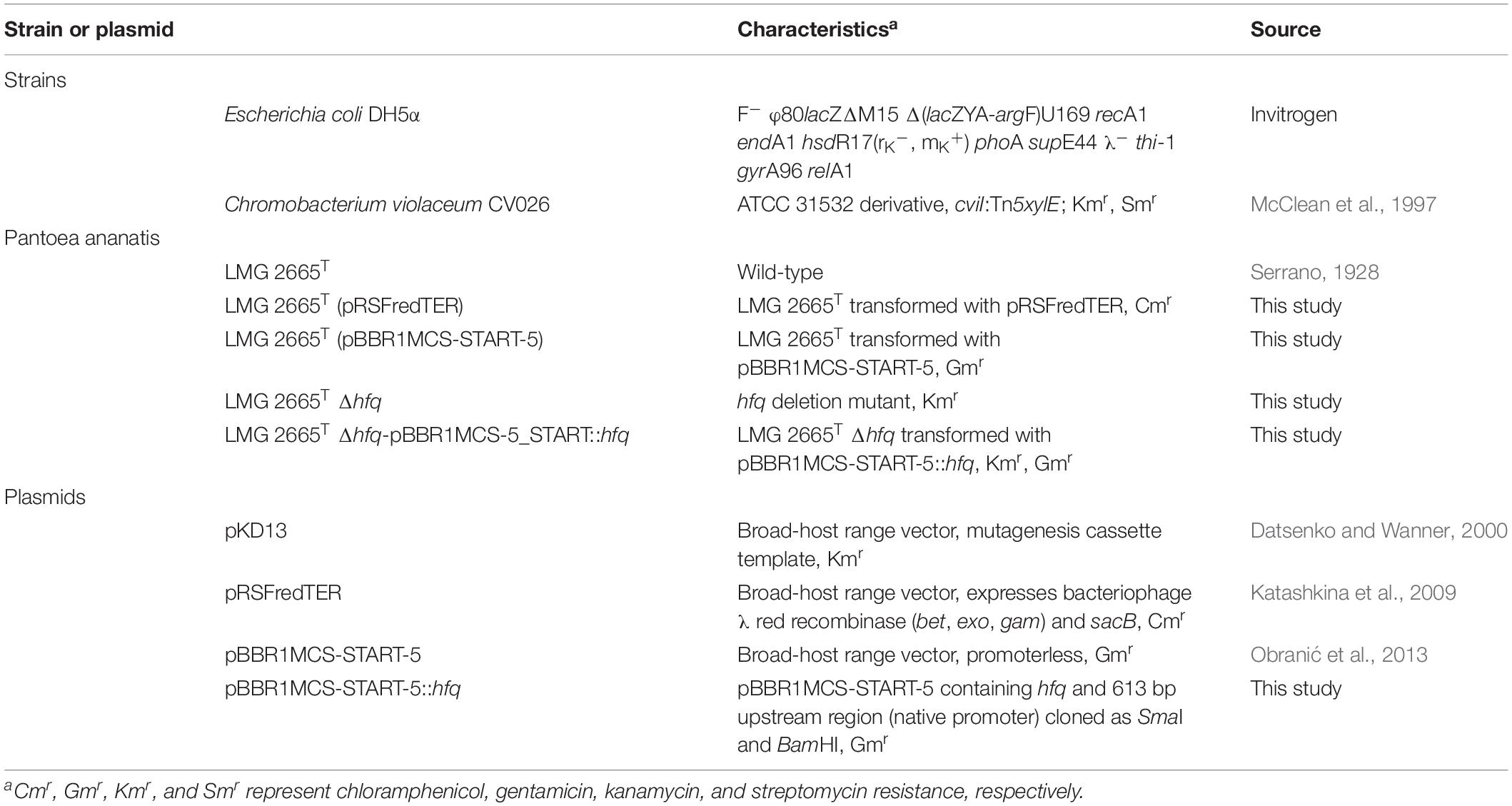
Table 1. A list of strains or plasmids used in this study.
Generation of a P. ananatis hfq Mutant and Complemented Strains
A mutant strain with chromosomal deletion of a single copy gene hfq (locus tag: PANA_RS17940) was constructed as previously described (Katashkina et al., 2009; Shyntum et al., 2015). The modification was made in the preparation of the knockout cassette which was amplified from the pKD13 plasmid using the Kan-F and Kan-R primers (Table 2) consisting of 50 bp homologous sequences of hfq flanking regions and 20 bp of kanamycin resistance gene priming sequences. Insertion of the kanamycin resistance gene was verified by Southern blotting, PCR amplification, and sequencing of the hfq region.
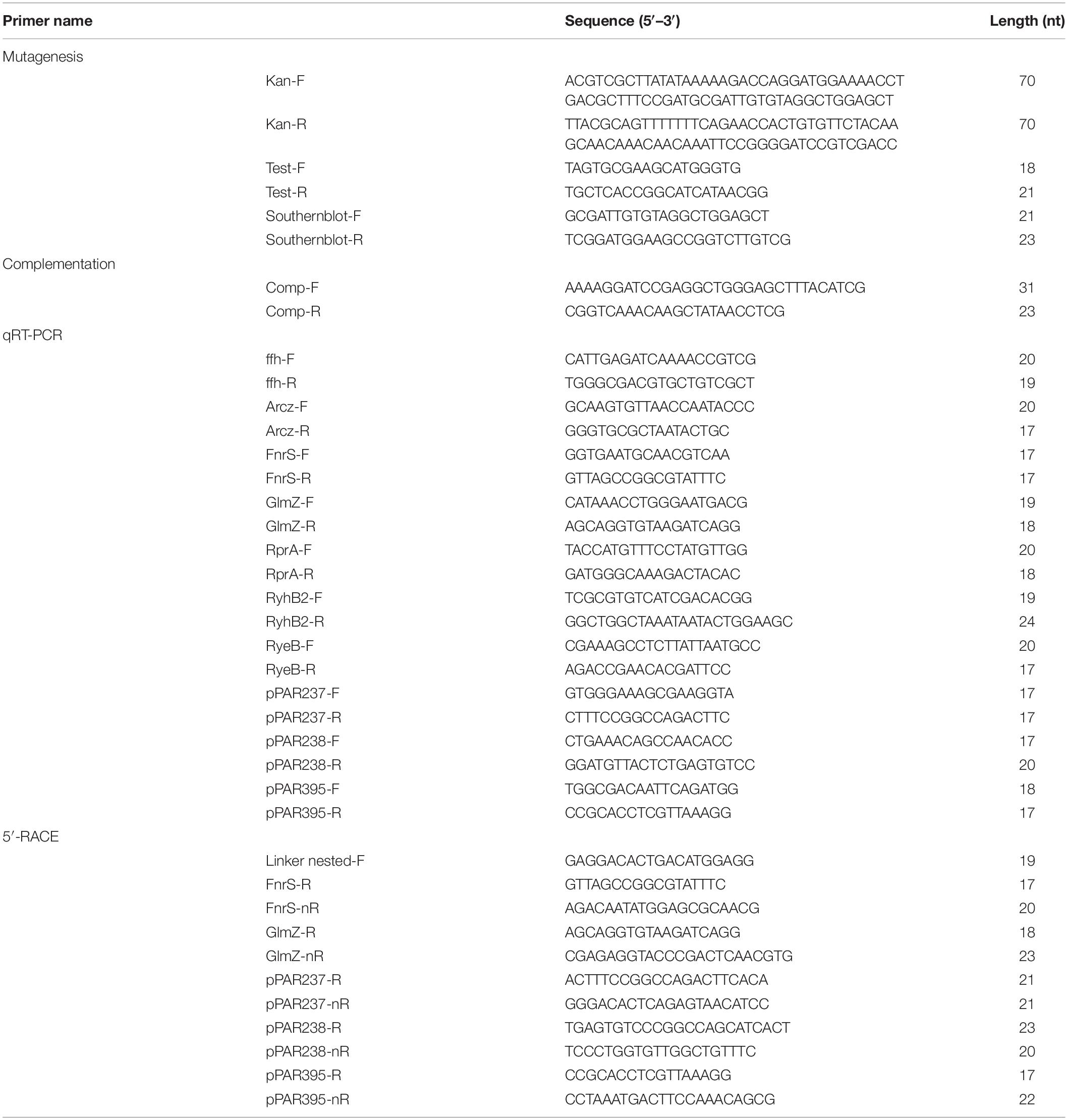
Table 2. A list of primers used in this study.
The promoter sequence of hfq determined in E. coli K12 MG1655 by Kim et al. (2012) was searched against the upstream sequence of hfq start codon in P. ananatis LMG2665T. An amplicon (1038 bp) containing the hfq gene (315 bp), its native promoter (58 bp), and flanking sequences (662 bp) was cloned into a pBBR1MCS-5_START vector (Obranić et al., 2013) restricted with SmaI and BamHI enzymes. Electrocompetent hfq deletion mutant P. ananatis was transformed with hfq complementing plasmid, pBBR1MCS::hfq and the resulting transformants were selected on the gentamicin amended LB agar. The integrity of the hfq complementation was determined by plasmid extraction, PCR, and sequencing using Test-F and Test-R primers (Table 2).
In vitro and in planta Growth Assay
The growth of wild-type P. ananatis with an empty pBBR1MCS-5_START vector (WT), hfq deletion mutant with an empty pBBR1MCS-5_START vector (Δhfq), and hfq complementing pBBR1MCS-5_START::hfq (pBBR1MCS::hfq) strains of P. ananatis was monitored both in vitro and in planta conditions.
Pantoea ananatis strains grown overnight in LB broth were normalized to an OD600nm reading of 0.5. For in vitro growth assay, the normalized cultures were diluted 100-fold in fresh LB medium and incubated with shaking at 200 rpm. The absorbency of each culture was periodically measured. There were three replicates for each culture and the experiment was repeated twice.
The previously described red onion scale assay (Stice et al., 2018) was adapted for quantifying in planta growth of P. ananatis between the WT, Δhfq, and hfq complementing strains. In summary, sliced red onion (Allium cepa L) scales of approximately 9 cm2 in area were surface sterilized in 3% bleach solution for 1 min and were rinsed twice in distilled water. Each scale was inoculated with 1 μl of bacterial cells (1 × 107 CFU/ml) suspended in 1× PBS [0.8% (w/v) NaCl, 0.02% (w/v) KCl, 0.144% (w/v) NaHPO4, 0.024% KH2PO4; pH 7.4] using a sterile pipette tip. Inoculated scales were placed on moistened paper towels in a surface sterilized container and were incubated at room temperature for 5 days. To quantify growth, three onion scales per strain were harvested at 24 h intervals. Each scale was macerated in 1 ml of 1× PBS and the extract was serially diluted and cells were enumerated on LB supplemented with gentamicin. Experiments were repeated in triplicate, and the results were presented as CFU/g of onion tissue. Sterile water was used as a negative control.
Virulence Assay
Virulence assay was performed as previously described for in planta growth assay. The vertical diameter of the water-soaked lesion on onion scales inoculated with WT, Δhfq, and hfq complementing P. ananatis strains was measured at 3 days post inoculation (dpi). The virulence assay was repeated twice, and there were three technical replicates for each P. ananatis strains.
Motility Assay
Overnight cultures of P. ananatis strains (WT, Δhfq, and pBBR1MCS::hfq) were normalized to OD600nm = 0.5 and 1 μl of each culture was inoculated in the center of the soft agar [0.5% (w/v) NaCl, 1% (w/v) tyrptone, and 0.3% (w/v) agar; pH 7.2]. The inoculated plates were incubated at 28°C, and swimming motility was determined after 24 h. Negative control plates were inoculated with sterile water. The swimming motility experiment was repeated three times with three biological replicates in each experiment.
Bioassay Detection of Acyl-Homoserine Lactones
Formation of AHL by WT, Δhfq, and hfq complementing strains of P. ananatis was determined using experimental procedures adapted from McClean et al. (1997). An aliquot (0.5 ml) of AHL reporter strain Chromobacterium violaceum (C. violaceum 026) grown in LB overnight was spread plated on LB agar plates and air-dried. Thereafter, three wells were (three replicates) created on each plate by puncturing the agar with a sterile cork-borer and inoculated with 100 μl of cell-free filtrate of P. ananatis WT, Δhfq, and hfq complementing strains overnight cultures. The inoculated plates were incubated at 28°C for 48 h. The formation of violacein (purple halo) by CV026, around the inoculated wells were indicative of AHL production. The assay was repeated twice and carried out in three technical replicates.
Biofilm Quantification
The biofilm of WT, Δhfq, and hfq complementing strains of P. ananatis was quantified as previously described by Santander and Biosca (2017) with slight modifications. An aliquot of 160 μl broth culture diluted to an OD600nm of 0.5 in half-strength LB [0.5% (w/v) NaCl, 0.5% (w/v) tryptone, and 0.25% (w/v) yeast extract; pH 7.2] was made into each well of a polystyrene 96-well microplate (NuncTM MicroWellTM, Thermo Scientific, Waltham, MA, United States) and incubated for 24 h under static conditions. Eight replicates per P. ananatis strain were included in each experiment with sterile half-strength LB broth serving as a negative control. Thereafter, the inoculated 96-well plates were inverted to remove the excess LB broth, air-dried, and incubated at 60°C for 40 min to heat-fix the biofilms. The biofilms were stained with 1% crystal violet (220 μl) for 15 min before being rinsed with distilled water. After rinsing and invert-air-drying the microplate, 220 μl of ethanol:acetone in 8:2 ratio was added to the wells to solubilize the crystal violet dye for 20 min at room temperature. The solubilized biofilm was measured at OD600 using Safire Microplate Reader (Tecan, Research Triangle Park, NC, United States), and this assay was repeated three times.
RNA Extraction and Transcriptomic Analysis
Total RNA of P. ananatis WT and Δhfq strains grown in LB broth was extracted at OD600nm readings of 0.2 (T1 = low cell density) and 0.6 (T2 = high cell density) using the miRNeasy Mini kit (Qiagen, Hilden, Germany). Genomic DNA was removed by including an on-column DNase digestion step during the RNA extraction. The purity (A260/A280) of extracted RNA was measured by Nanodrop2000 (Thermo Scientific, Sugarland, TX, United States) and RNA integrity was determined by Agilent2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, United States). Illumina Truseq Small RNA Library (Illumina, San Diego, CA, United States) preparation was performed on the RNA samples, and deep sequencing of the library was conducted on Illumina HiSeq2500 platform (single-end, 1 × 50 bp) by Macrogen (South Korea).
Bioinformatic Analysis and sRNA Identification
Raw sequencing reads (BioProject accession number: PRJNA550544) were stringently trimmed and filtered using Trimmomatic (Bolger et al., 2014) to remove adapter sequences and low quality reads. Following adapter trimming and filtering, quality was verified using FastQC (Andrews, 2010) and reads were mapped to the P. ananatis LMG20103 genome (De Maayer et al., 2010) using Bowtie2 (Langmead and Salzberg, 2012), as the genome of LMG 20103 was the only P. ananatis genome with a complete annotation at the time of analysis. For sRNA identification, a custom python script (Supplementary Data Sheet S1, see the section “genic_filter.py” in the Supplementary Material) was compiled to remove reads that mapped to coding sequences, ribosomal RNA, and transfer RNA, or within 120 bases upstream or downstream of these features from the resulting sequence alignment map (SAM) files. The purpose of the 120 base buffer was to reduce the number of sRNAs identified that originated from extended 5′- or 3′-UTR regions. All wild-type sequencing replicates from the same sampling time point were merged into a single gene-filtered SAM file for sRNA identification.
To identify putative sRNAs from gene-filtered SAM files, a custom python script (Supplementary Data Sheet S1, see the section “peak_ID.py” in the Supplementary Material) was used to calculate per base depth relative to the genome-wide per-base sequencing depth by replicate, which was also normalized to library size. A threshold of 10-fold increased abundance above background with a minimum length of 10 nucleotides was chosen for sRNA identification. Using the script, putative sRNAs at the low cell density and high cell density sampling time points were identified and the lists of sRNAs were merged using a custom python script (Supplementary Data Sheet S1, see the section “mergeList.py” in the Supplementary Material), combining any overlapping identified sRNAs into a single sRNA to generate a single list of putative P. ananatis sRNAs (pPARs sRNA).
Computational Prediction of Rho-Independent Terminators
Following established criteria (Zeng and Sundin, 2014), Rho-independent terminators were searched in the P. ananatis LMG20103 genome using a custom python script (Supplementary Data Sheet S1, see the section “RI_term.py” in the Supplementary Material). Briefly, the search was conducted in an effort to detect poly-T regions with at least six continuous Ts and for those that had at least four GC base pairs in the last six bases before the poly-T stretch. Of these, those that had at least 50% GC content in the last 25 bases before the poly-T were considered to be putative Rho-independent terminators.
Differential sRNA Expression in P. ananatis LMG 2665 WT vs. δhfq
Using the genomic coordinates from the BLAST + search of the pPAR sRNAs against the P. ananatis LMG2665 genome (Adam et al., 2014), a gene format file (.gff) for all the pPAR sRNAs was generated. The sRNA sequencing reads that had been trimmed and filtered were mapped to the LMG2665 genome using Bowtie2 (Langmead and Salzberg, 2012). The mapped reads were sorted using SAMtools (Li et al., 2009) and the number of reads mapping to pPAR sRNAs in the LMG2665 genome was counted using HTSeq (Anders et al., 2015). Read counts tables were analyzed for statistically significant differential expression of pPAR sRNAs between WT- and δhfq-mutant samples at corresponding time points using the DESeq R package which utilizes a negative binomial distribution model (Anders and Huber, 2010; R Core Team, 2013). Resulting genes with a false-discovery rate of 0.05 were considered differentially expressed.
sRNA Conservation Analysis
The bacterial genomes were downloaded from NCBI and searched using BLAST+ (Camacho et al., 2009) with all pPAR sRNA sequences as queries. Because BLAST uses local alignment, the global percent identity to pPAR sRNAs was calculated by multiplying the percent identity by the length of the BLAST alignment and dividing by the length of the pPAR sRNA. Heatmaps showing percent identity of sRNA by genome were generated using ClustVis (Metsalu and Vilo, 2015).
qRT-PCR Validation of sRNA Expression
To validate the expression of putative sRNAs identified, a quantitative RT-PCR was conducted on a subset of sRNAs. The 2 μg of total RNA extracted from the two time points (low and high cell density) was converted to cDNA using random primers using the High-Capacity cDNA Synthesis Kit (Applied Biosystems, Carlsbad, CA, United States). Subsequently, PowerUPTM SYBRTM Green Master Mix (Applied Biosystems, Carlsbad, CA, United States) was used to quantify expression levels of the selected sRNAs real time in QuantStudio 12K Flex Real-Time PCR System (Applied Biosystems, Carlsbad, CA, United States). The list of primers used for qRT-PCR is found in Table 2. The relative expression of sRNA was calculated using 2–Δ Δ CT method (Livak and Schmittgen, 2001) with the gene ffh encoding a signal recognition particle protein, serving as an endogenous mRNA control (Takle et al., 2007; Sibanda et al., 2018).
5′-Rapid Amplification cDNA Ends Analysis
The 5′-Rapid Amplification cDNA Ends (RACE) analysis was conducted on the selected putative sRNAs to capture their transcription start sites (TSS). Total RNA (up to 15 μg) of P. ananatis strains grown to high density (OD600 = 0.6) was extracted as above mentioned (see the section “RNA Extraction and Transcriptomic Analysis”). The resulting RNA was ligated to 300 pmol of RNA linker: GACGAGCACGAGGACACUGACAUGGAGGAGGGAGUAG AAA in the presence of RNA 5′-pyrophosphohydrolase (RppH) (New England BioLabs, Ipswich, MA, United States) and T4 RNA ligase (New England BioLabs, Ipswich, MA, United States) at 37°C for 4 h. The linker-ligated RNA was purified using Trizol-chloroform (2:1) extraction method, as described by Rio et al. (2010). The resulting RNA was ethanol precipitated and suspended in 10 μl of RNase-free water. The cDNA of linker-ligated RNA was synthesized as previously described (see the section “qRT-PCR Validation of sRNA Expression”) and Gene Specific PCR (GSP) was performed using nested linker and sRNA-specific PCR primers (Table 2). The GSP using genomic DNA was used as a control and resulting bands from 5′-RACE were gel-purified and cloned into pJET1.2 blunt (Thermo Fischer Scientific Baltics UAB, Vilnius, Lithuania) prior to sequencing.
Secondary Structure and mRNA Target Prediction
The secondary structures of sRNAs, of which their TSS have been determined by 5′-RACE analysis, were predicted in silico using RNAfold web server1 (Hofacker, 2003). The putative target mRNAs of novel sRNAs pPAR237, pPAR238, and pPAR395 and their putative interacting domains were computationally predicted using CopraRNA and IntaRNA2 (Wright et al., 2014). The above information is presented in Supplementary Figure S6 and Supplementary Table S4, respectively.
Image and Statistical Analysis
Images resulting from motility, AHL detection, and virulence assays were analyzed in ImageJ (Schneider et al., 2012) for measurement of halos and lesion diameter. Statistical analyses are performed with R 3.2.6 (R Core Team, 2013) and significance of the data (P < 0.05) were determined by analysis of variance (ANOVA) and Tukey’s honestly significantly difference (HSD) tests. Except where otherwise mentioned, all data shown in this study represent mean values and error bars represent standard error (SE) of the samples.
Results
hfq Mutation Negatively Affects Growth
To investigate the functional role of Hfq in the pathogenesis of P. ananatis, an hfq deletion mutant (Δhfq) was constructed by replacing the hfq gene with a kanamycin resistance marker (the section “Materials and Methods”). Southern blotting (Supplementary Figure S1) and PCR amplification of the hfq region (Supplementary Figure S2) verified a single insertion of the antibiotic marker in the hfq mutant strain. For the construction of the hfq-complementing plasmid, hfq promoter sequence of E. coli K12 was used to search for hfq promoter in P. ananatis and a highly conserved hfq promoter sequence (93% nucleotide identity) of P. ananatis compared to that of E. coli K12 was found overlapping in the coding region of the adjacent gene miaA.
In vitro growth analyses of P. ananatis WT, Δhfq, and hfq complementing strains cultured in LB medium showed that the hfq mutation affected the growth of P. ananatis. The hfq mutant exhibited a slower growth rate relative to the WT and hfq-complementing strains, but similar cell density was reached at stationary phase as the both strains (Supplementary Figure S3). Similarly, in planta growth curves at 12 h showed that WT, Δhfq, and hfq complementing strains of P. ananatis exhibited comparable cell densities to one another (Supplementary Figure S4B) which were at sufficient levels for the onset of symptoms by 3 dpi (Supplementary Figure S4A).
Loss of Hfq Attenuates Virulence in P. ananatis
Virulence assay on red onion scales demonstrated clearing of the red pigment and formation of a water-soaked lesion in the onion scales inoculated with WT P. ananatis while no disease symptoms were observed on the scales infected with the P. ananatis Δhfq mutant (Figure 1). The impaired virulence of the P. ananatis Δhfq mutant was restored to the wild-type levels by trans expression of hfq gene on the plasmid pBBR1MCS-5_START::hfq. The finding that the P. ananatis Δhfq-mutant strain was able to attain in planta population densities equivalent to WT (Supplementary Figure S4) suggests that the lack of disease symptoms is not associated with a growth defect, and that hfq is required for virulence of this strain when inoculated into red onion.
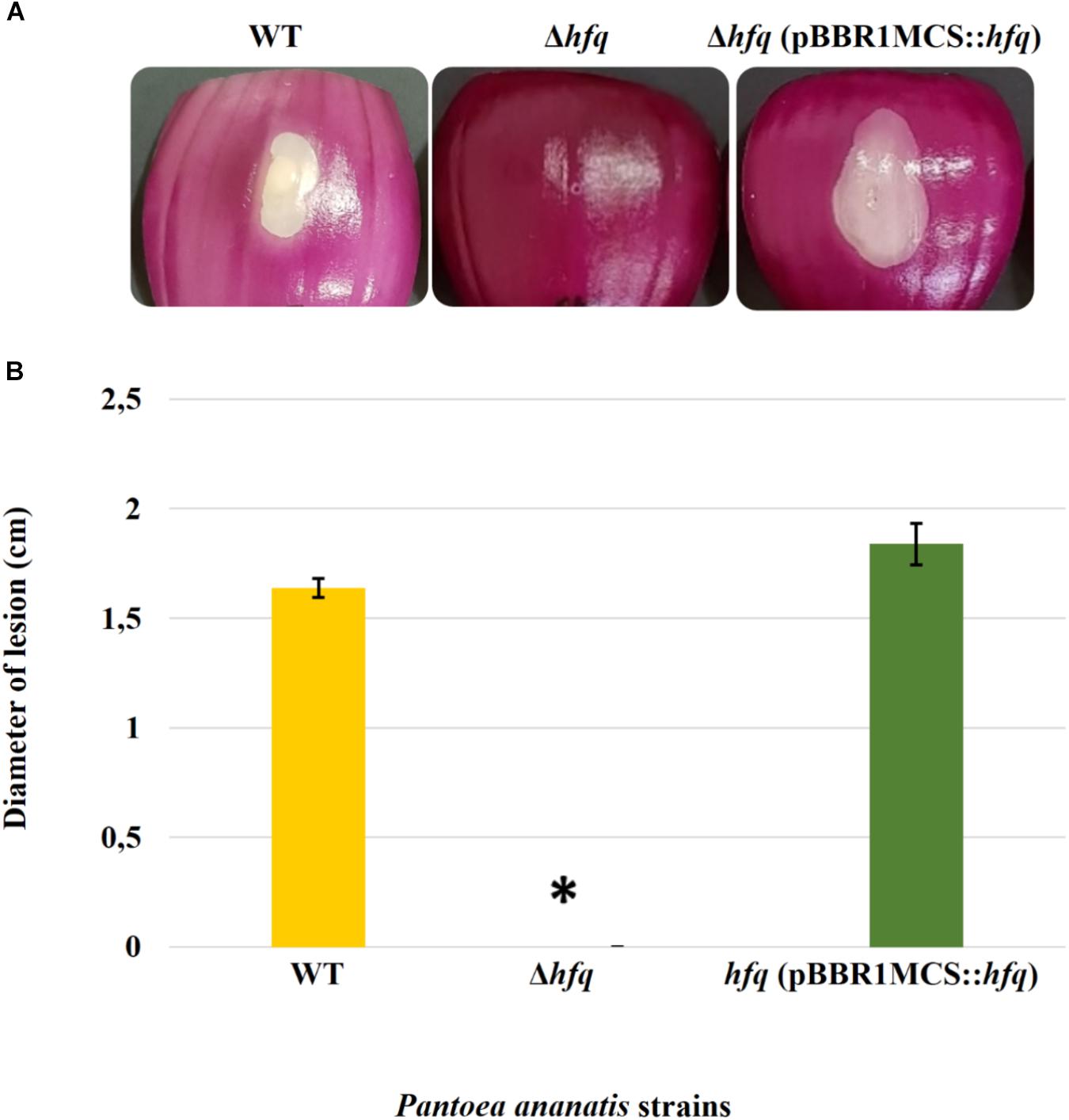
Figure 1. Effect of deletion of hfq on the virulence of P. ananatis. (A) Water-soaked lesion caused by the wild-type (WT), hfq mutant (Δhfq), and hfq complementing [Δhfq (pBBR1MCS::hfq)] strains of P. ananatis LMG 2665T in red onion scales at 3 days post inoculation (dpi). (B) The diameter of the lesion caused by the WT, Δhfq, and Δhfq (pBBR1MCS::hfq) strains was measured at 3 dpi from three replicates for each strain. Mean lesion length from two independent assays is plotted. An asterisk denotes significance differences (P < 0.05) in the lesion size caused by Δhfq relative to WT P. ananatis.
Hfq Regulates Motility, AHL Production, and Biofilm Formation
To determine whether the P. ananatis Hfq regulates virulence traits, swimming motility, production of AHL molecules, and biofilm were quantified in WT, Δhfq, and hfq-complementing strains of P. ananatis. The results show that P. ananatis hfq mutant was impaired in swimming motility relative to the wild-type strain, as determined by the size of the halo that formed on the soft agar (Figure 2). In addition, AHL production, as determined by the production of the purple pigment violacein by the C. violaceum 026 biosensor demonstrated a statistically significant reduction in the size of the purple halo formed by the hfq-mutant strain relative to the wild type, indicating a significant reduction in AHL production by the mutant strain (Figure 3). Furthermore, a threefold reduction (P < 0.05) in the biofilms formed by the hfq-mutant strain relative to the WT P. ananatis was also observed (Figure 4). These findings are consistent with previous studies that showed that AHL molecules are needed as a signal for QS to regulate biofilm formation in P. ananatis (Morohoshi et al., 2007; Sibanda et al., 2016). The phenotypic defects resulting from loss of hfq, which were restored to wild-type levels by trans-complementation of hfq, suggest that Hfq regulates the production of multiple virulence traits in P. ananatis.
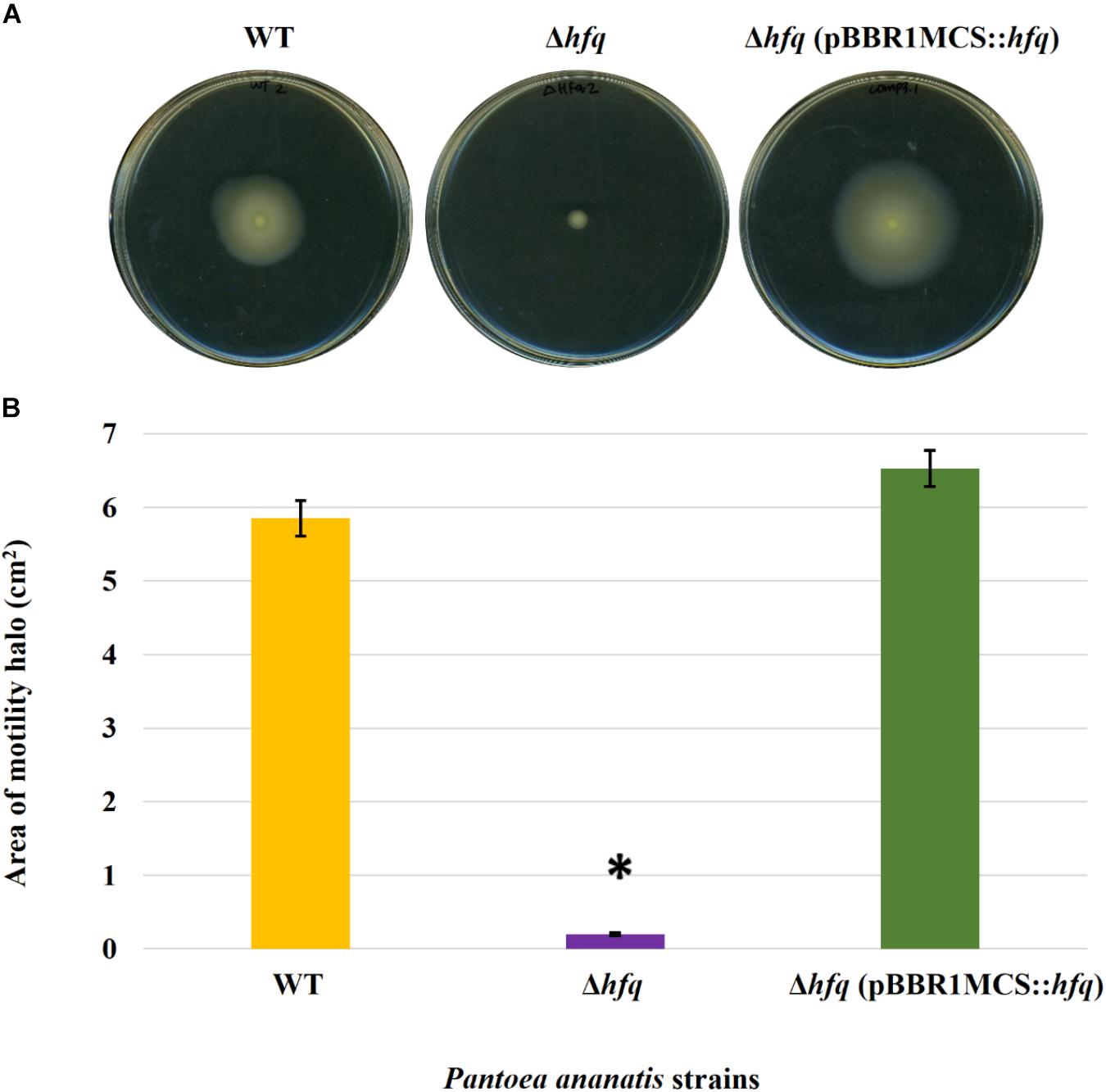
Figure 2. Effect of deletion of hfq on the motility of P. ananatis. (A) Motility of wild-type (WT), hfq mutant (Δhfq), and hfq complementing (Δhfq pBBR1MCS::hfq) strains of P. ananatis LMG 2665T after 24 h on 0.3% swimming agar plates. (B) Motility area of WT, Δhfq, and Δhfq (pBBR1MCS::hfq) strains was measured at 24 hpi. An asterisk denotes significant differences (P < 0.05) in the motility of Δhfq relative to WT P. ananatis.
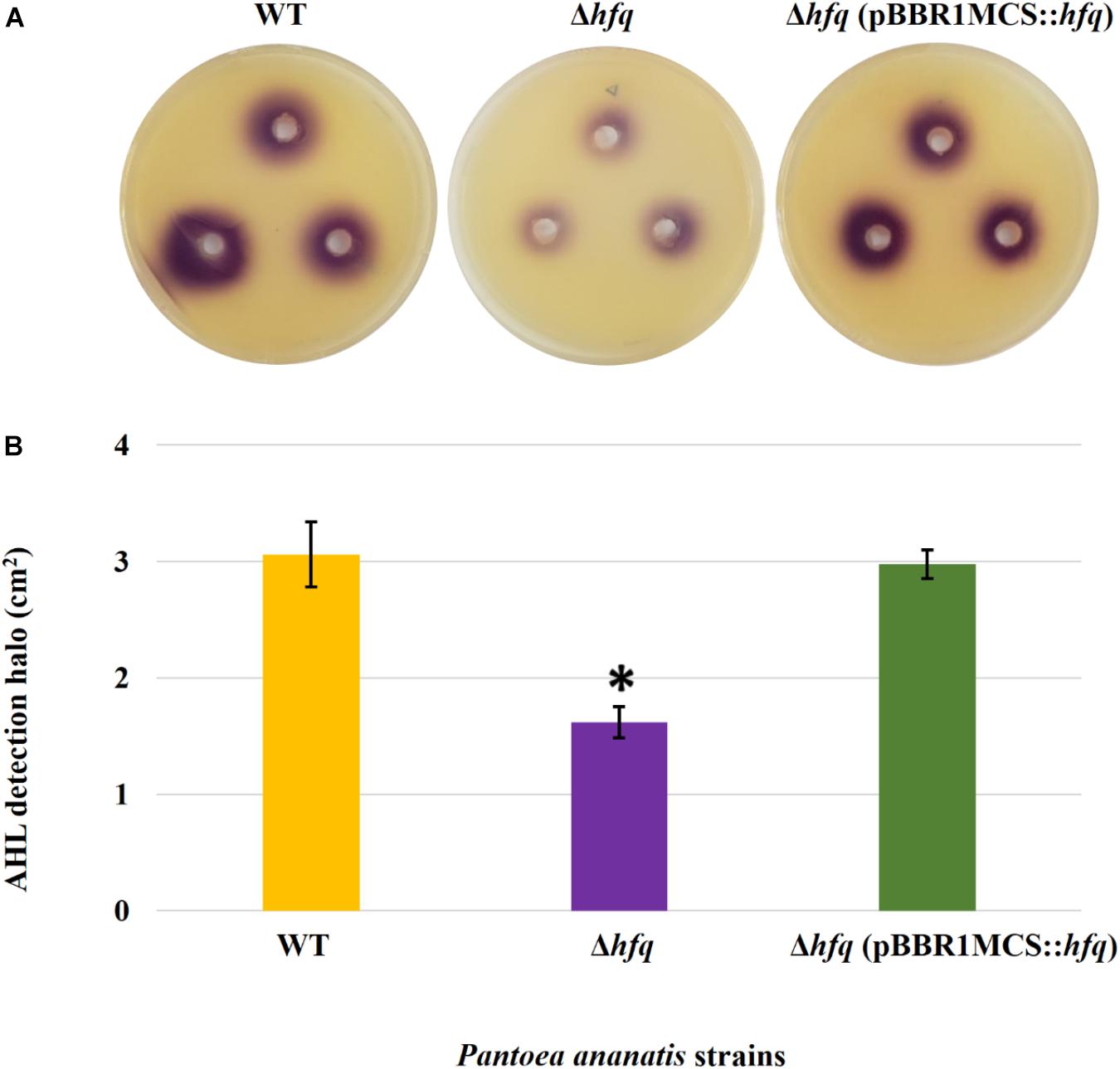
Figure 3. AHL detection bioassay. (A) Detection of AHLs produced by the wild-type (WT), hfq mutant (Δhfq), and hfq complementing [Δhfq (pBBR1MCS::hfq)] strains of P. ananatis LMG 2665T by biosensor Chromobacter violaceum CV026 in the form of a purple (violacein) halo after 48 h. (B) Mean areas of the violacein halo (excluding the area of the well) from two independent experiments were measured for the WT, Δhfq, and Δhfq (pBBR1MCS::hfq) strains. Each plate contained three replicates. An asterisk denotes significant differences (P < 0.05) in the size of violacein halo of Δhfq relative to WT P. ananatis.
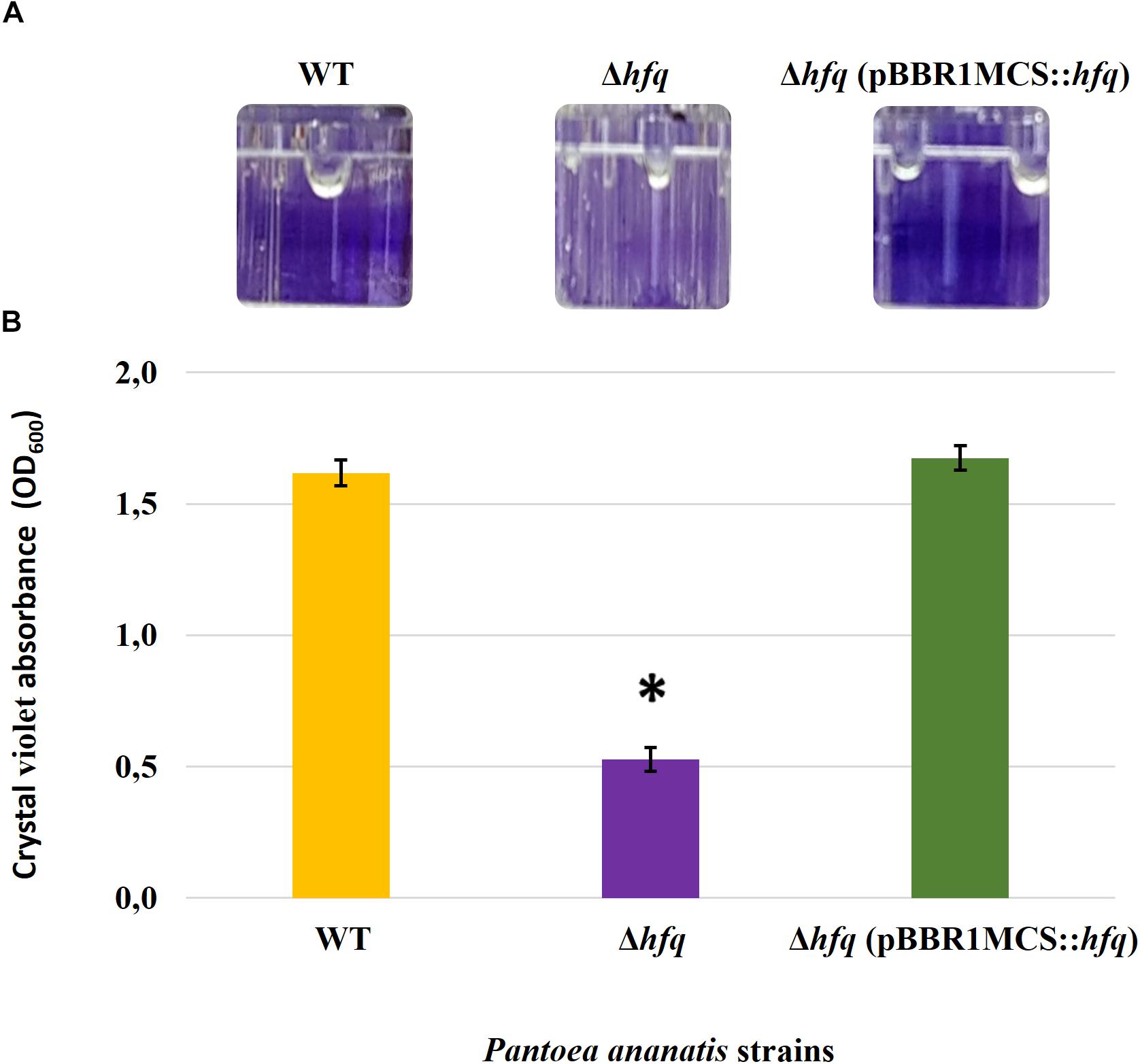
Figure 4. Effect of deletion of hfq on the biofilm-forming ability of P. ananatis. (A) Biofilms formed by the wild-type (WT), hfq mutant (Δhfq), and hfq complementing [Δhfq (pBBR1MCS::hfq)] strains of P. ananatis LMG 2665T in 96-well microtiter plate after 24 h incubation under static conditions. (B) Quantification of the biofilms formed by the (WT), Δhfq, and Δhfq (pBBR1MCS::hfq) strains after 24 h using crystal violet (1%) staining method. The absorbance of solubilized biofilms stained with crystal violet was measured at an optical density wavelength of 600 nm. An asterisk denotes significance differences (P < 0.05) in the amount of biofilm formed by Δhfq relative to WT P. ananatis.
Identification of Putative sRNAs
Due to impaired motility, AHL production, biofilm formation, and virulence caused by the loss of Hfq in P. ananatis LMG 2665, a sRNA sequencing analysis was conducted to identify the regulatory sRNAs that are dependent on Hfq for stability and function. Deep sequencing of the sRNA transcriptomes of WT and Δhfq-mutant P. ananatis strains at low (OD600 = 0.2) and high cell density (OD600 = 0.6) time points resulted in a total of 172.03 million reads. Following trimming of adapters and filtering for high-quality reads (Phred score =30), 66.74 million reads were retained, of which 83.2% mapped uniquely to the P. ananatis LMG20103 genome. Following removal of the reads that mapped to protein coding genes, rRNAs, or tRNAs, 9.72 million reads remained for the sRNA identification and analysis. The distribution of reads across the WT and Δhfq-mutant P. ananatis strains, each with three technical replicates at low and high cell density time points, are included in Supplementary Table S1.
For identification of sRNAs in the transcriptome dataset, the WT sequencing data were utilized and calculated for the per-base depth across the genome relative to the genome-wide average per-base depth. To select a threshold that would allow for sensitive detection of sRNAs while also filtering out noise in the sequencing data, the number of putative sRNAs identified across a broad range of signal-to-noise thresholds was calculated. A strong linear relationship (R2 = 0.9981) between Log10 (Threshold) and Log10 (# sRNAs identified) was found (Supplementary Figure S5), and a signal-to-noise threshold ratio of 10 was selected for calling of putative sRNAs from the sequencing data. Using this threshold, a total of 615 pPARs sRNAs was identified. Of these, 425 pPARs were identified in both time points, 90 were identified only in the low cell density time point, and 100 only in the high cell density time point in P. ananatis LMG2665.
Characterization of pPAR sRNAs
The 615 identified pPARs were further classified as intergenic, antisense, or overlapping. The classification resulted in 249 intergenic pPAR sRNAs, 302 antisense pPAR sRNAs, and 64 overlapping pPAR sRNAs (Figure 5A). The mean length of pPAR sRNAs was 66.4 bases with a median of 42 bases (Figure 5B) and mean GC content of pPAR SRNAs was 52.3% with a median of 52.2% (Figure 5C). Both of these are quite close to the genome average of 53.7% GC bases (De Maayer et al., 2010). Of note, seven pPAR sRNAs had GC content below 30%, and 14 pPAR sRNAs had GC content above 70%, suggesting the potential horizontal acquisition of the genomic regions containing these sRNAs.
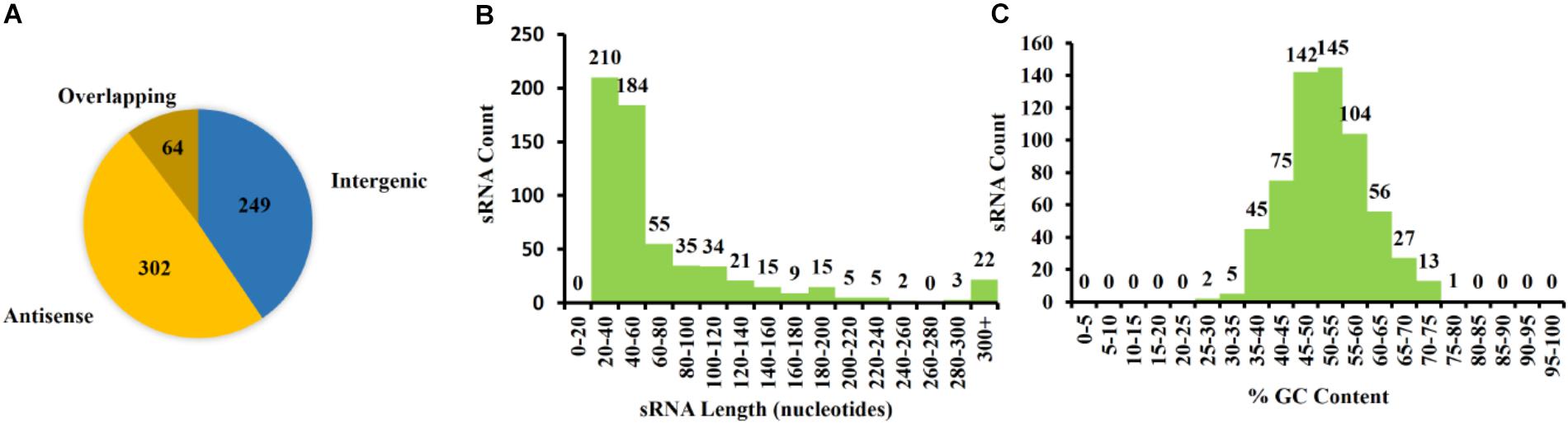
Figure 5. Characterization of Pantoea ananatis sRNAs (pPAR sRNA). (A) A total of 615 putative P. ananatis LMG 2665T sRNAs were classified into 302 antisense, 64 overlappings, and 249 intergenic pPAR sRNAs. (B) The mean length of pPAR sRNAs was 66.4 bases with a median of 42 bases. (C) The mean GC content of pPAR SRNAs was 52.3% with a median of 52.2%.
In addition, we performed a genome-wide computational search for putative Rho-independent terminators that are associated with the transcription termination of Hfq-dependent sRNAs (Otaka et al., 2011). The results revealed that there were 5,002 poly-T stretches with at least 6 continuous Ts and 2,437 of these had four or more GC base pairs in the last 6 bases before the poly-T. A total of 1,842 of poly-T stretches had approximately 50% GC content in the final 25 bases before the poly-T, meeting the established criteria of Rho-independent terminators (Zeng and Sundin, 2014). Based on these criteria, only 569 were associated with protein-coding genes and 69 were associated with the identified pPAR sRNAs. The key features of select pPAR sRNAs are presented in Table 3 and for full data, including genomic coordinates and sRNA sequences, refer to Supplementary Table S2.
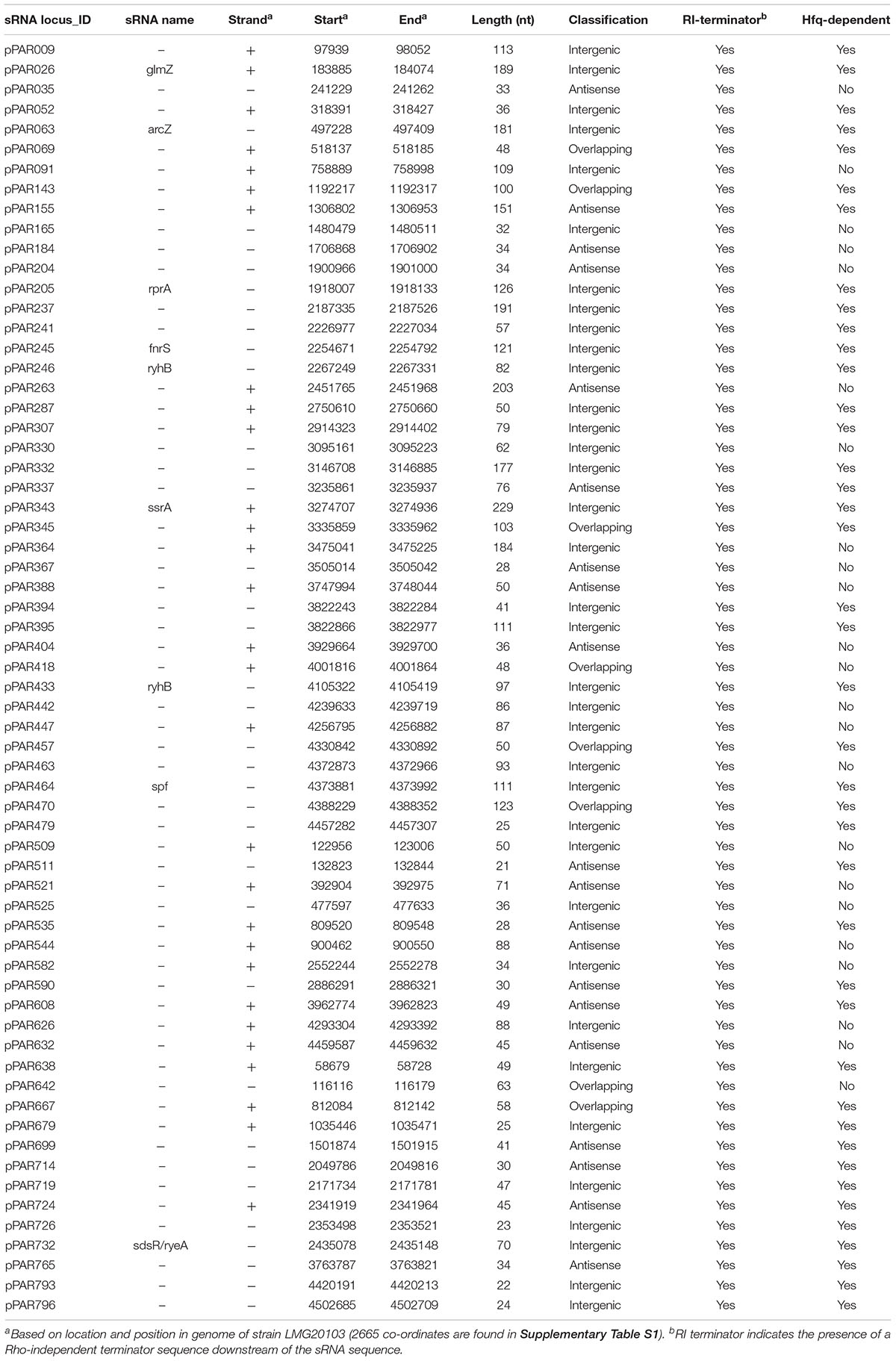
Table 3. A list of selected Pantoea ananatis LMG2665T pPAR sRNAs.
Identification of Hfq-Dependent sRNAs
Because trans encoded sRNAs have been shown to depend on RNA chaperone proteins such as Hfq for stability and activity (Vogel and Luisi, 2011), an hfq mutant was included in the sRNA sequencing experiment in order to determine pPAR sRNAs that are dependent on or influenced by the loss of hfq. The analysis of hfq to WT samples from both low cell density and high cell density samples identified a total of 276 pPAR sRNAs affected in abundance by Hfq. Sixty-four pPAR sRNAs were affected in abundance by loss of hfq in both cell density samples, 58 pPAR sRNAs in low cell density samples, and 154 pPAR sRNAs only in high cell density samples (Figure 6). Of all the Hfq-dependent pPAR sRNAs, 145 had decreased abundance and 131 had increased abundance in the hfq mutant relative to wild type. Overall, results indicate that Hfq affects the abundances of numerous pPAR sRNAs either positively and/or negatively. Supplementary Table S3 lists all pPAR sRNAs affected in abundance by loss of hfq as well as corresponding fold changes in both low and high cell densities.
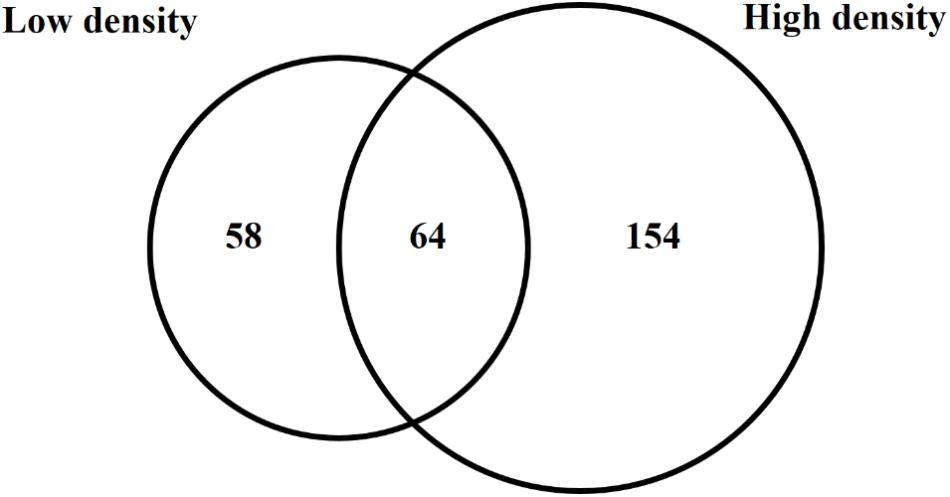
Figure 6. Hfq regulates several Pantoea ananatis sRNAs (pPAR sRNA). Venn-diagram of differentially expressed pPAR sRNAs between by the wild-type (WT) and hfq mutant (Δhfq) P. ananatis LMG 2665T grown in vitro at low (OD600nm = 0.2) or high cell density (OD600nm = 0.6).
Of the pPAR sRNAs affected by the loss of hfq, 41 have predicted Rho-independent terminators. Of these, 25 are intergenic and 16 are antisense, consistent with the classical model that Hfq-dependent sRNAs are frequently intergenic (Vogel and Luisi, 2011). Among the sRNAs detected in intergenic regions and Hfq-dependent with Rho-independent terminator, 9 known sRNAs and 16 novel sRNAs were identified. The known sRNAs included ArcZ, FnrS, GlmZ, RprA, RyeB/SdsR, RyhB, RyhB2, Spot42, and SsrA. The depth plots for a number of selected known and novel pPAR sRNAs of interest were generated, showing per-base sequencing depth across the length of the sRNA (Figure 7). Several pPAR sRNAs have certain regions
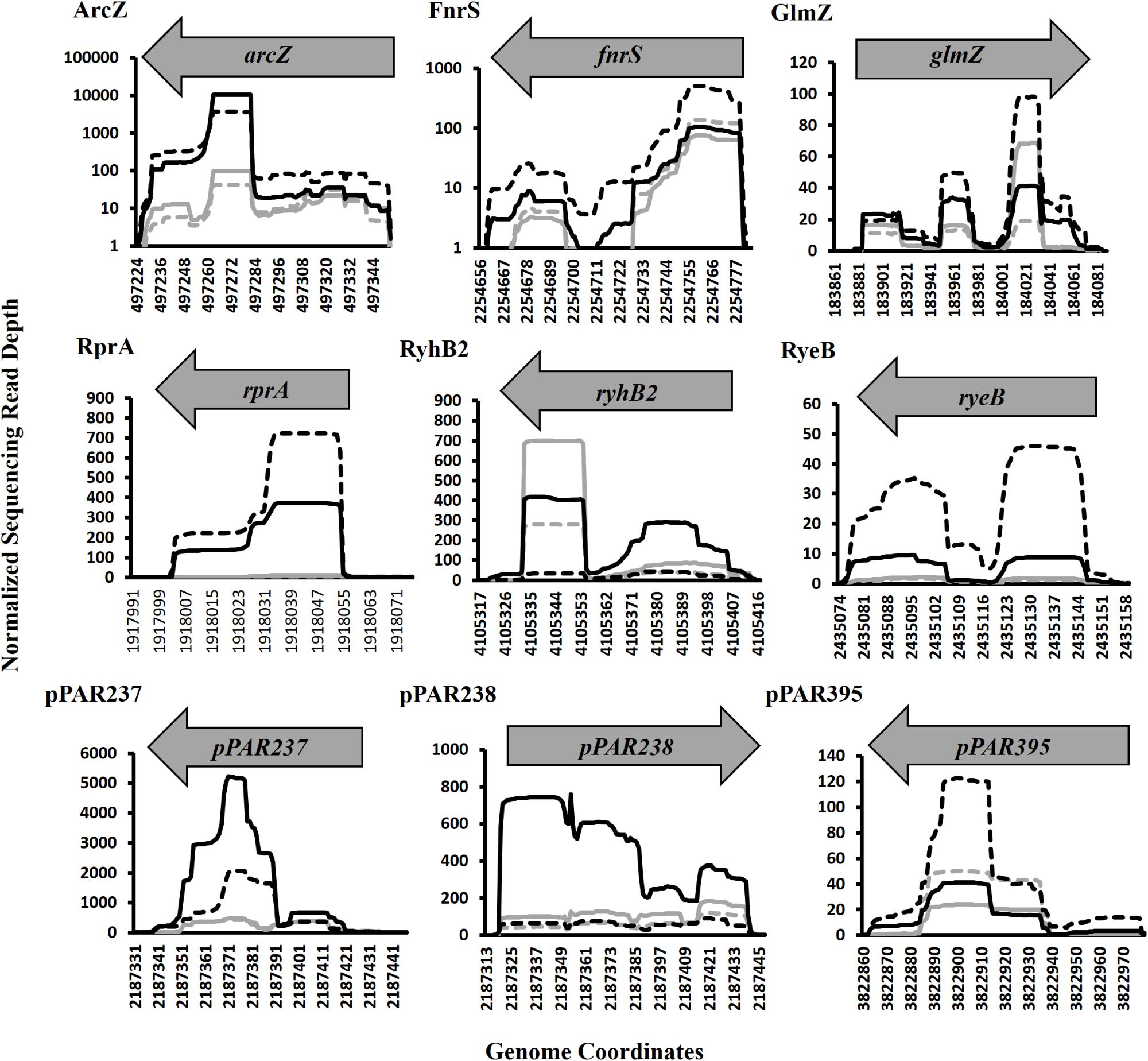
Figure 7. Sequencing read depth plots for selected Pantoea ananatis sRNAs (pPAR sRNA). Per-base sequencing read depth across the length of sRNAs, normalized to the genome-wide average per-base read depth was plotted for selected P. ananatis LMG 2665T sRNAs. Solid black lines represent sRNA sequencing depth in wild-type (WT) P. ananatis at low cell density (OD600nm = 0.2) and dashed black lines represent sRNA sequencing depth in WT at high cell density (OD600nm = 0.6). Solid gray lines represent sRNA sequencing depth in hfq mutant P. ananatis (Δhfq) at low cell density (OD600nm = 0.2) and dashed gray lines represent sRNA sequencing depth in Δhfq at high cell density (OD600nm = 0.6).
with far greater sequencing depth than the rest of the sRNA which suggests that mechanisms such as post-transcriptional processing (Davis and Waldor, 2007; Chao et al., 2017) might be active in P. ananatis, playing a role in sRNA maturation and/or processing.
Conservation of Identified sRNAs
To visualize the degree of conservation of the identified pPAR sRNAs, a genome-wide analysis was performed to identify sequences similar to those of the pPAR sRNAs for several bacterial species both within and outside the genus Pantoea were conducted. Nearly all of the pPAR sRNAs are highly conserved within the P. ananatis strains, and a large portion of sRNAs was also well conserved within the genus Pantoea (Figure 8A). As some P. ananatis genomes are in draft form, it is possible that some sRNAs have not yet been assembled, accounting for low levels of conservation within the species. However, it will be interesting to determine experimentally if some sRNAs are specific within P. ananatis or within the genus Pantoea as far fewer pPAR sRNAs are conserved across different enterobacterial species (Figure 8B).
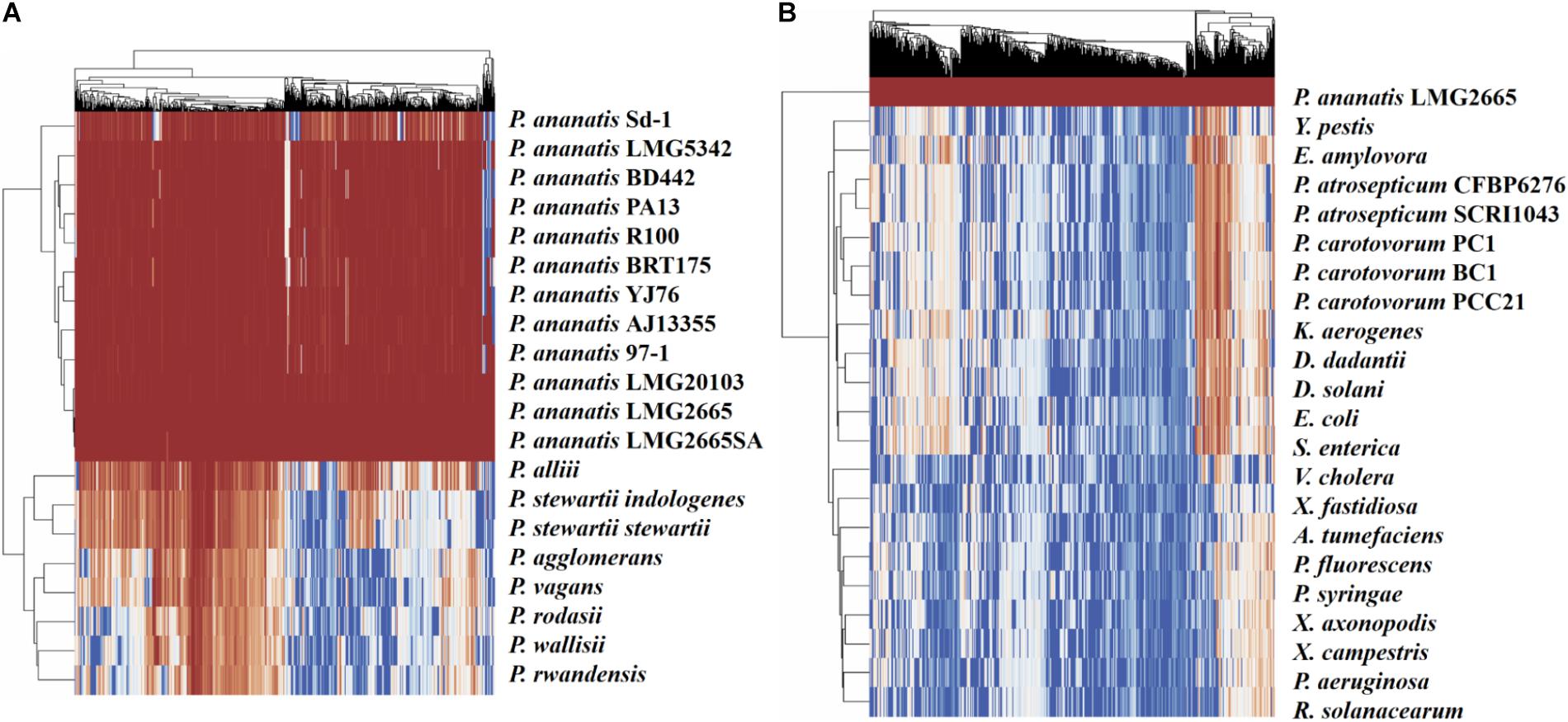
Figure 8. Conservation of Pantoea ananatis LMG 2665T sRNAs (pPAR sRNAs) in genomes of (A) Pantoea species and (B) bacterial species outside of the genus Pantoea. BLAST+ was used to query each genome with each pPAR sRNA. Heatmap scale from 0 to 100 represents percent identity between the best BLAST hit and the P. ananatis pPAR sRNA sequence from strain LMG 2665. Hierarchical clustering was applied to rows and columns by Euclidean distance with no scaling, and heatmaps (x-axis = pPAR sRNAs, y-axis = sRNAs hit in other P. ananatis strain or bacterial species) were generated using ClustVis.
Experimental Validation and Characterization of Individual sRNAs
Expression of the arcZ, fnrS, glmZ, rprA, ryeB, ryhB2, pPAR237, pPAR238, and pPAR395 sRNA genes was quantified in the P. ananatis hfq-mutant strain relative to WT using qRT-PCR (Figure 9). The resulting expression profile of aforementioned sRNA transcript levels (except glmZ and ryhB2) was decreased in the absence of hfq, which was in agreement with the depth plots analysis (Figure 7). In WT P. ananatis, glmZ expression is likely repressed at low cell density (OD600 = 0.2) and increased at high cell density (OD600 = 0.6) in a Hfq-dependent manner as the opposite expression levels were observed in hfq-mutant P. ananatis where abundances of glmZ transcripts were detected at low cell density but not at high cell density condition. Similarly, Hfq may negatively affect ryhB2 expression, as the abundances of ryhB2 transcript in WT at low and high cell density conditions were both low relative to the hfq-mutant ryhB2 levels. The TSS of FnrS, GlmZ, pPAR237, pPAR238, and pPAR395 was determined by 5′-RACE analysis. Their predicted structures, sequence, and targets are reported in Supplementary Figure S6 and Supplementary Table S4.
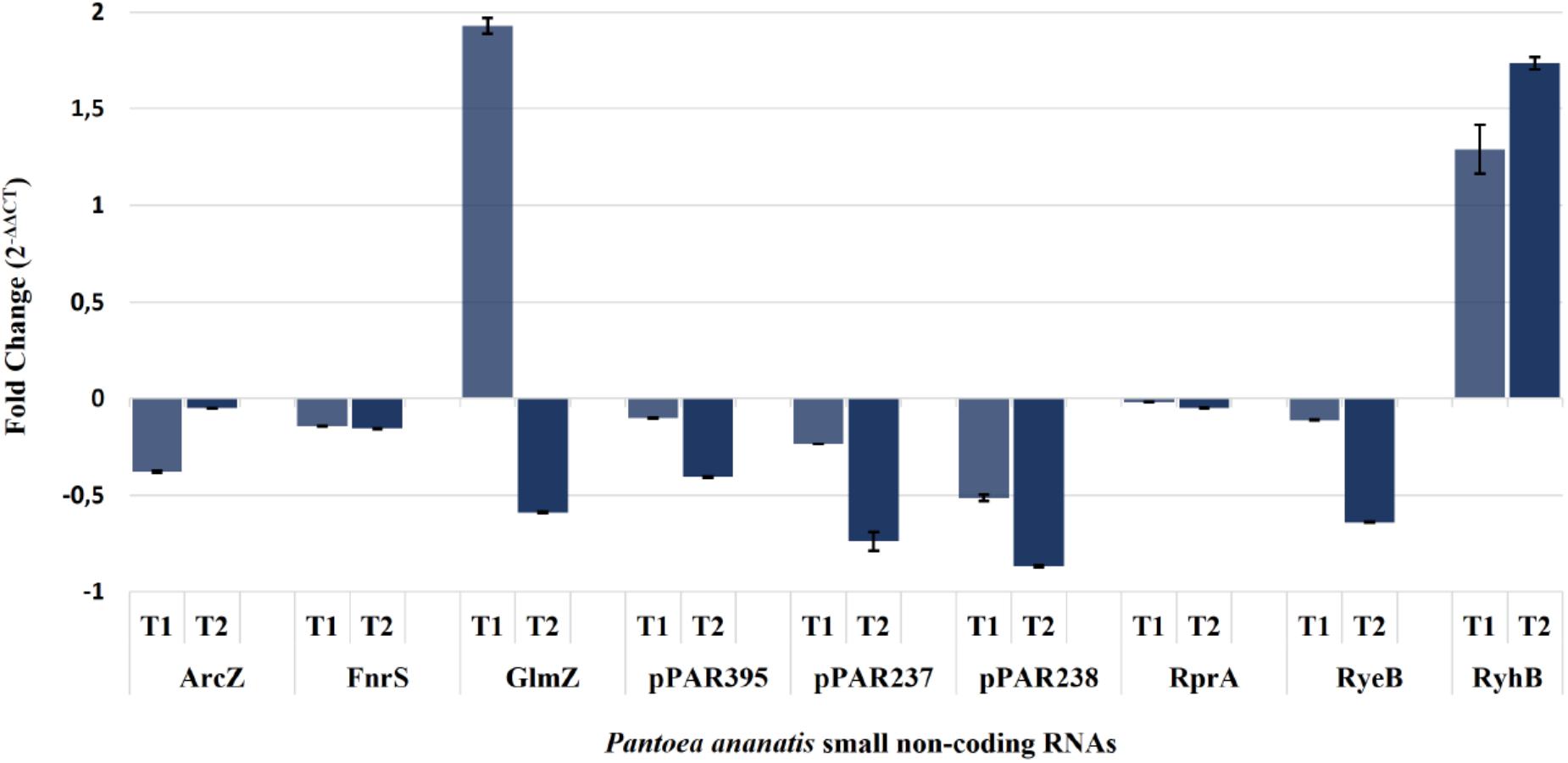
Figure 9. Quantification of selected Pantoea ananatis sRNAs (pPAR sRNAs). Transcript levels of a selected set of sRNAs in the hfq mutant of P. ananatis LMG 2665T (Δhfq) relative to the wild-type (WT) P. ananatis LMG 2665T at low (T1; OD600nm = 0.2) and high cell density (T2; OD600nm = 0.6) were quantified using qRT-PCR. The fold change of sRNA expression in Δhfq calibrated by WT expression is shown.
Discussion
In the present study, we investigated the functional role of Hfq in the pathogenesis of the Gram-negative phytopathogen P. ananatis, and demonstrated that Hfq is important for motility, AHL and biofilm formation, and virulence of the pathogen. We also identified several putative sRNAs, which include known and novel sRNAs that are Hfq-dependent for their abundances in P. ananatis. The pleiotropic phenotypes caused by hfq mutation is due to global post-transcriptional gene regulation operated by Hfq and Hfq-dependent sRNAs that modulate stress response and virulence of numerous bacterial pathogens (Chao and Vogel, 2010). The ability of P. ananatis to survive in diverse ecological niches and to successfully infect susceptible plant hosts requires a timely and collective regulation of cellular functions in response to environmental conditions.
Inactivation of hfq in bacteria generally results in pleiotropic effects, of which growth retardation is common. Decreased growth rate has been reported in hfq-attenuated bacteria such as Acinetobacter baumannii (Kuo et al., 2017), Haemophilus influenzae (Hempel et al., 2013), Yersinia enterocolitica (Kakoschke et al., 2016), and the plant pathogens A. tumefaciens (Wilms et al., 2012a) and P. carotovorum (Wang et al., 2018). This phenotype was consistent with the P. ananatis hfq deletion mutant growing in vitro; however, this alteration did not prevent P. ananatis from entering logarithmic growth phase and eventually reaching the wild-type cell density at a stationary phase which was also observed in planta (Supplementary Figure S4). Unlike the E. amylovora hfq mutant (Zeng et al., 2013), which exhibited reduced growth in an immature pear fruit infection model, the P. ananatis hfq mutant strain was able to reach a population density comparable to that of the wild-type strain when inoculated into onion, indicating that the abolishment of virulence in the hfq-mutant P. ananatis was not due to a growth defect.
The loss of hfq gives rise to impairment of important virulence determinants such as motility in bacterial pathogens. Impaired motility affects the overall fitness of a bacterium as a pathogen as it disables attachment and dispersal of the pathogen in the host. This, in turn, results in the diminished invasion, colonization, and hence virulence (Sittka et al., 2007; Kulesus et al., 2008). In enterobacterial pathogens, Hfq and Hfq-dependent sRNAs control flagellar-based motility. For example, in E. coli, multiple sRNAs including ArcZ, OmrAB, OxyS, and RyeB/SdsR have been shown to modulate the expression and/or translation of flhDC, the master regulator of flagellar biosynthesis (De Lay and Gottesman, 2012). In the phytopathogen, E. amylovora, the sRNAs ArcZ, OmrAB, and RmaA have been found to regulate flhDC at both transcriptional and post-transcriptional levels (Schachterle and Sundin, 2019; Schachterle et al., 2019). In this way, the integration of different environmental cues is achieved through several sRNAs, allowing fine-tuning of flagellar expression and production.
The lack of swimming motility displayed by the P. ananatis hfq mutant clearly demonstrates the role of Hfq in regulating flagellar motility. In a previous study by Weller-Stuart et al. (2016), a P. ananatis flgK mutant deficient in flagellar assembly enzyme, FlgK, was abolished in swimming motility and pathogenicity in onion seedlings. Together with the current study, these findings suggest that flagellar motility is required for the virulence of P. ananatis, and this trait is regulated by functional Hfq. Similarly, in P. carotovorum (Wang et al., 2018) and Serratia sp. ATCC 39006 (Wilf et al., 2013; Hampton et al., 2016), attenuation of flagellar motility was observed in hfq-deletion mutant strains, as an expression of the flhDC genes was dependent on Hfq. Given that sRNAs namely, ArcZ, OmrAB, and RyeB/SdsR, were also identified in our sRNA sequencing data (Supplementary Table S2), and that their wild-type transcript levels are dependent on the functional copy of hfq (Figure 9), we hypothesize that Hfq, in conjunction with the identified sRNAs, may regulate the flagellar motility of P. ananatis in a similar manner as the other enterobacterial species.
In addition to impaired motility, disruption of hfq in Gram-negative pathogens often results in reduced biofilm formation (Kulesus et al., 2008; Monteiro et al., 2012; Zeng et al., 2013; Wang et al., 2018). One possible explanation for this phenotype is an effect on QS-mediated regulation of motility and biofilm formation. As biofilm formation is a developmental and co-operative process, the process necessitates cell to cell communication that enables perception of the signals generated from the community. The signal or information is packaged in the form of autoinducer molecules, acylated homoserine lactones (AHLs), or may be communicated to the QS circuit by secondary signaling molecule such as cyclic dimeric guanosine monophosphate (c-di-GMP) (Castiblanco and Sundin, 2016). Through protein phospho-relay, signals resulting from high cell density reach Hfq-dependent sRNAs which initiate the expression of genes required for the extracellular matrix synthesis and maturation of biofilm (Lenz et al., 2004; Kay et al., 2006; Tu and Bassler, 2007).
Consistent with the findings that in some bacteria, Hfq positively regulates biofilm production, a significantly reduced amount of biofilm was formed by the P. ananatis strain lacking an hfq gene. Interestingly, a decreased level of extracellular AHLs diffused in the supernatant of an hfq mutant overnight culture was detected by the CV026 biosensor, suggesting a potential role of Hfq in the positive regulation of AHL synthesis. According to Pomini et al. (2006), the main P. ananatis-derived AHL molecule is N-hexaonoyl-L-homoserine lactone (C6-HSL) and is synthesized by the LuxI homolog EanI, while LuxR homolog EanR is a transcriptional regulator that down-regulates the expression of eanIR operon in the absence of AHL (Morohoshi et al., 2007). Functional characterization further demonstrated that eanI or AHL synthase was required for the formation of biofilm, EPS, and pathogenicity of P. ananatis (Morohoshi et al., 2007; Sibanda et al., 2016). Thus, decreased production of AHL in the hfq-mutant strain of P. ananatis would mean down-regulation of eanI, virulence-related genes, as well as the genes within the QS regulon (Sibanda et al., 2018).
In the present study, two putative novel Hfq-dependent sRNAs were identified in the vicinity of the eanIR genes namely, pPAR237 and pPAR238 (Supplementary Figure S7A). These sRNAs are partially antisense to each other and are abundantly present in the low cell density condition but are drastically reduced in abundance at high cell density (Figure 9). In contrast to the wild-type expression levels, expression of pPAR237 and pPAR238 was almost non-existent in the two cell density conditions in the P. ananatis hfq-mutant strain (Figure 9). The decreased transcript levels of pPAR237 and pPAR238 in hfq-mutant relative to WT P. ananatis were validated experimentally by qRT-PCR, reinforcing the idea that the expression of these sRNAs is dependent on cell density and Hfq. Potential pairing sites of pPAR237 and pPAR238 to eanI and eanR were predicted in silico using IntaRNA (Supplementary Figures S7B–D). Further experimental confirmation of their interaction will indicate the role of pPAR237 and pPAR238 in QS through modulation of AHL synthesis in P. ananatis. Moreover, it will be also interesting to determine whether there are other upstream and downstream transcriptional or translational regulators of putative sRNAs pPAR237 and pPAR238. This is the case in V. cholera and Pseudomonas aeruginosa whose Hfq-dependent sRNA Qrr 1,2,3, and 4 and RsmY are transcriptionally activated by LuxO and GacA, respectively, and are used to repress transcription of hapR or sequester translational regulator RsmA (Lenz et al., 2004; Kay et al., 2006; Tu and Bassler, 2007; Brencic et al., 2009).
To date, factors that contribute to the pathogenicity of P. ananatis have been characterized, resulting in an expansion in our understanding of virulence mechanisms of this pathogen. A collective regulation of all virulence traits seems likely for the success and persistence of P. ananatis in hostile environments, and this can be achieved through Hfq and its global regulatory networks constituted by Hfq-dependent sRNAs. Overall, this study provided valuable insights into the essential role of Hfq in regulating different virulence traits of P. ananatis. A total of 276 sRNAs were identified that are affected in abundance by Hfq at low and high cell density conditions. These sRNAs include those that are well characterized as well as novel putative sRNAs that may possess novel function involved in the QS of P. ananatis.
Data Availability
The datasets generated for this study can be found in the NCBI BioProject PRJNA550544.
Author Contributions
GS, JS, DS, LM, TC, and GWS conceived and designed the present study. GS conducted the mutagenesis, phenotypic assays, and sRNA validation experiments. JS compiled the custom python script and performed the bioinformatics analyses of sRNA sequencing data. GS and JS wrote the manuscript in consultation with DS, LM, TC, and GWS. All authors contributed to and approved the final version of the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This research, and a research visit by GS to the Michigan State University (MSU), was supported by the National Research Foundation (NRF) of South Africa, and the University of Pretoria. Research at MSU was also supported by the MSU AgBioResearch. Additionally, we thank Professor Gordana Maravić-Vlahoviček at the University of Zagreb for providing broad-host range vectors.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02075/full#supplementary-material
FIGURE S1 | Southern blot validation of hfq knock-out mutation in Pantoea ananatis. Genomic DNA of the wild-type (WT) and hfq mutant (Δhfq) strains of P. ananatis LMG 2665T digested with EcoRI and HindIII restriction enzymes was hybridized to a DIG-labeled probe (a partial amplicon of kanamycin resistance gene). Positive detection of the antibiotic marker was observed in the Δhfq strains of P. ananatis LMG 2665T (lanes 2–8). WT of P. ananatis LMG 2665T DNA was used as a negative control (lane 1) whereas unlabeled probe was used as a positive control (lane 9).
FIGURE S2 | Colony PCR verification of hfq knock-out mutation in Pantoea ananatis. A colony PCR confirmation of insertion of kanamycin resistance gene in the hfq gene region using Test primers (Table 2) hfq mutant (Δhfq) strains of P. ananatis LMG 2665T. L represents a molecular ladder and the sizes of its prominent bands 1, 3, and 6 kilo basepairs (kb) are indicated below. A wild-type (WT) colony of P. ananatis LMG 2665T was used as a negative control (lane 1; 500 bp). Insertion of kanamycin resistance marker is shown in colony PCRs of hfq mutant (Δhfq) strains of P. ananatis LMG 2665T (lanes 2, 3, and 4; 1.5 kb).
FIGURE S3 | In vitro growth assay. Growths of wild-type (WT), hfq mutant (Δhfq), and hfq complementing (Δhfq pBBR1MCS::hfq) strains of Pantoea ananatis LMG 2665T in LB broth at 28°C. The growth was monitored for 20 h at optical density 600 nm (OD600) and the mean OD600 readings of the three replicates for each P. ananatis LMG 2665T strains were plotted. Solid line (yellow) represents WT, dashed line (purple) Δhfq, and dotted line (green) Δhfq pBBR1MCS::hfq. Asterisks denote significance differences (P < 0.05) in the absorbance of Δhfq relative to WT P. ananatis LMG 2665T.
FIGURE S4 | In planta growth assay. (A) Disease progression in onion scales inoculated with wild-type (WT), hfq mutant (Δhfq), and hfq complementing [Δhfq (pBBR1MCS::hfq)] strains of P. ananatis LMG 2665T, and incubated for 5 days post inoculation (dpi). (B) In planta populations of WT, Δhfq, and Δhfq (pBBR1MCS::hfq) strains of P. ananatis LMG 2665T in onion scales measured for 5 dpi. The mean CFUs of three replicates for each strain from two independent experiments were plotted. Solid line (yellow) represents WT, dashed line (purple) Δhfq, and dotted line (green) Δhfq (pBBR1MCS::hfq).
FIGURE S5 | Logarithmic plot of the number of putative small RNAs (sRNAs) identified in Pantoea ananatis LMG 2665T (pPAR sRNA) as a function of the threshold selected for calling sRNAs. This was generated by calling putative sRNAs across a range of thresholds using the custom script (see Supplementary Data Sheet S1 in the section “peak_ID.py”).
FIGURE S6 | In silico prediction of selected Pantoea ananatis sRNAs (pPAR sRNA) secondary structure. Secondary structures of P. ananatis LMG 2665T sRNAs (A) FnrS, (B) GlmZ, (C) pPAR 237, (D) pPAR 238, and (E) pPAR 395 were predicted based on a minimum free energy model provided by RNAfold (http://rna.tbi.univie.ac.at).
FIGURE S7 | Putative interaction of pPAR237 and pPAR238 to eanIR in Pantoea ananatis LMG 2665T. (A) Location of pPAR237 and pPAR238. In silico predicted interaction of pPAR237 (red) to eanIR (black): (B) eanI upstream sequence (energy: −8.62323 kcal/mol; hybridization energy: −23.5). (C) eanR coding sequence (energy: −13.63700 kcal/mol, hybridization energy: −39.4) and (D) in silico predicted interaction of pPAR238 (red) to eanI (black) upstream sequence (energy: −7.83954 kcal/mol, hybridization energy: −12.0).
TABLE S1 | Summary of sRNA sequencing reads obtained and filtered for use in sRNA identification.
TABLE S2 | A list of sRNAs identified, their genomic coordinates, sequences, and selected characteristics.
TABLE S3 | A list of sRNAs that has significant abundance difference between WT and hfq mutant strains of Pantoea ananatis.
TABLE S4 | A list of predicted targets of selected sRNAs.
DATA SHEET S1 | A custom phython script compiled for bioinformatic analyses of sRNA sequencing data.
Footnotes
References
Adam, Z., Tambong, J. T., Lewis, C. T., Lévesque, C. A., Chen, W., Bromfield, E. S., et al. (2014). Draft genome sequence of Pantoea ananatis strain LMG 2665T, a bacterial pathogen of pineapple fruitlets. Genome Announc. 2:e489-14. doi: 10.1128/genomeA.00489-14
Alippi, A. M., and López, A. C. (2010). First report of leaf spot disease of maize caused by Pantoea ananatis in Argentina. Plant Dis. 94:487. doi: 10.1094/PDIS-94-4-0487A
Anders, S., and Huber, W. (2010). Differential expression analysis for sequence count data. Genome Biol. 11:R106. doi: 10.1186/gb-2010-11-10-r106
Anders, S., Pyl, P. T., and Huber, W. (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. doi: 10.1093/bioinformatics/btu638
Andrews, S. (2010). FastQC: A Quality Control Tool for High Throughput Sequence Data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (accessed May 21, 2018).
Argaman, L., Hershberg, R., Vogel, J., Bejerano, G., Wagner, E. G., Margalit, H., et al. (2001). Novel small RNA-encoding genes in the intergenic regions of Escherichia coli. Curr. Biol. 11, 941–950. doi: 10.1016/S0960-9822(01)00270-6
Asselin, J. E., Bonasera, J. M., and Beer, S. V. (2018). Center rot of onion (Allium cepa) caused by Pantoea ananatis requires pepM, a predicted phosphonate-related gene. Mol. Plant Microbe Interact. 31, 1291–1300. doi: 10.1094/MPMI-04-18-0077-R
Azegami, K., Ozaki, K., Matsuda, A., and Ohata, K. (1983). Bacterial palea browning, a new disease of rice caused by Erwinia herbicola (in Japanese with English summary). Bull. Natl. Inst. Agric. Sci. Ser. C 37, 1–12.
Bardill, J. P., and Hammer, B. K. (2012). Non-coding sRNAs regulate virulence in the bacterial pathogen Vibrio cholerae. RNA Biol. 9, 392–401. doi: 10.4161/rna.19975
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brencic, A., McFarland, K. A., McManus, H. R., Castang, S., Mogno, I., Dove, S. L., et al. (2009). The GacS/GacA signal transduction system of Pseudomonas aeruginosa acts exclusively through its control over the transcription of the RsmY and RsmZ regulatory small RNAs. Mol. Microbiol. 73, 434–445. doi: 10.1111/j.1365-2958.2009.06782.x
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Castiblanco, L. F., and Sundin, G. W. (2016). New insights on molecular regulation of biofilm formation in plant-associated bacteria. J. Integr. Plant Biol. 58, 362–372. doi: 10.1111/jipb.12428
Chao, Y., Li, L., Girodat, D., Förstner, K. U., Said, N., Corcoran, C., et al. (2017). In vivo cleavage map illuminates the central role of RNase E in coding and non-coding RNA pathways. Mol. Cell 65, 39–51. doi: 10.1016/j.molcel.2016.11.002
Chao, Y., Papenfort, K., Reinhardt, R., Scharma, C. M., and Vogel, J. (2012). An atlas of Hfq-bound transcripts reveals 3’ UTRs as a genomic reservoir of regulatory small RNAs. EMBO J. 31, 4005–4019. doi: 10.1038/emboj.2012.229
Chao, Y., and Vogel, J. (2010). The role of Hfq in bacterial pathogens. Curr. Opin. Microbiol. 13, 24–33. doi: 10.1016/j.mib.2010.01.001
Cortesi, P., and Pizzatti, C. (2007). Palea browning, a new disease of rice in Italy caused by Pantoea ananatis. J. Plant Pathol. 89:S76.
Cother, E. J., Reinke, R., McKenzie, C., Lanoiselet, V. M., and Noble, D. H. (2004). An unusual stem necrosis of rice caused by Pantoea ananas and the first record of this pathogen on rice in Australia. Aust. Plant Pathol. 33, 495–503. doi: 10.1071/AP04053
Coutinho, T. A., Preisig, O., Mergaert, J., Cnockaert, M. C., Riedel, K.-H., Swings, J., et al. (2002). Bacterial blight and die-back of Eucalyptus species, hybrids and clones in South Africa. Plant Dis. 86, 20–25. doi: 10.1094/PDIS.2002.86.1.20
Coutinho, T. A., and Venter, S. N. (2009). Pantoea ananatis: an unconventional plant pathogen. Mol. Plant Pathol. 10, 325–335. doi: 10.1111/j.1364-3703.2009.00542.x
da Silva, J. F., Barbosa, R. R., de Souza, A. N., da Motta, O. V., Teixeira, G. N., Carvalho, V. S., et al. (2015). Isolation of Pantoea ananatis from sugarcane and characterization of its potential for plant growth promotion. Genet. Mol. Res. 14, 15301–15311. doi: 10.4238/2015.November.30.6
Datsenko, K. A., and Wanner, B. L. (2000). One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U.S.A 97, 6640–6645. doi: 10.1073/pnas.120163297
Davis, B. M., and Waldor, M. K. (2007). RNase E-dependent processing stabilizes MicX, a Vibrio cholerae sRNA. Mol. Microbiol. 65, 373–385. doi: 10.1111/j.1365-2958.2007.05796.x
De Lay, N., and Gottesman, S. (2012). A complex network of small non-coding RNAs regulate motility in Escherichia coli. Mol. Microbiol. 86, 524–538. doi: 10.1111/j.1365-2958.2012.08209.x
De Lay, N., Schu, D. J., and Gottesman, S. (2013). Bacterial small RNA-based negative regulation: Hfq and its accomplices. J. Biol. Chem. 288, 7996–8003. doi: 10.1074/jbc.R112.441386
De Maayer, P., Chan, W. Y., Venter, S. N., Toth, I. K., Birch, P. R., Joubert, F., et al. (2010). Genome sequence of Pantoea ananatis LMG20103, the causative agent of Eucalyptus blight and dieback. J. Bacteriol. 192, 2936–2937. doi: 10.1128/JB.00060-10
Dutta, B., Barman, A. K., Srinivasan, R., Avci, U., Ullman, D. E., Langston, D. B., et al. (2014). Transmission of Pantoea ananatis and P. agglomerans, causal agents of center rot of onion (Allium cepa), by onion thrips (Thrips tabaci) through feces. Phytopathology 104, 812–819. doi: 10.1094/PHYTO-07-13-0199R
Franze de Fernandez, M. T., Eoyang, L., and August, J. T. (1968). Factor fraction required for the synthesis of bacteriophage Qβ-RNA. Nature 219, 588–590. doi: 10.1038/219588a0
Gitaitis, R., Walcott, R., Culpepper, S., Sanders, H., Zolobowska, L., and Langston, D. (2002). Recovery of Pantoea annatis, causal agent of center rot of onion, from weeds and crops in Georgia, USA. Crop Prot. 21, 983–989. doi: 10.1016/S0261-2194(02)00078-9
Gitaitis, R. D., and Gay, J. D. (1997). First report of leaf blight, seed stalk rot, and bulb decay of onion by Pantoea ananas in Georgia. Plant Dis. 81:1096. doi: 10.1094/PDIS.1997.81.9.1096C
Gitaitis, R. D., Walcott, R. R., Wells, M. L., Diaz Perez, J. C., and Sanders, F. H. (2003). Transmission of Pantoea ananatis, causal agent of center rot of onion, by tobacco thrips, Frankliniella fusca. Plant Dis. 87, 675–678. doi: 10.1094/PDIS.2003.87.6.675
Goszczynska, T., Moloto, V. M., Venter, S. N., and Coutinho, T. A. (2006). Isolation and identification of Pantoea ananatis from onion seed in South Africa. Seed. Sci. Technol. 34, 655–668. doi: 10.15258/sst.2006.34.3.12
Goszczynska, T., Venter, S. N., and Coutinho, T. A. (2007). Isolation and identification of the causal agent of brown stalk rot, a new disease of corn in South Africa. Plant Dis. 91, 711–718. doi: 10.1094/PDIS-91-6-0711
Gottesman, S., and Storz, G. (2011). Bacterial small RNA regulators: versatile roles and rapidly evolving variations. RNA 21, 511–512. doi: 10.1261/rna.050047.115
Guo, M. S., Updegrove, T. B., Gogol, E. B., Shabalina, S. A., Gross, C. A., and Storz, G. (2014). MicL, a new σE –dependent sRNA, combats envelope stress by repressing synthesis of Lpp, the major outer membrane lipoprotein. Genes Dev. 28, 1620–1634. doi: 10.1101/gad.243485.114
Hampton, H. G., McNeil, M. B., Paterson, T. J., Ney, B., Williamson, N. R., Easingwood, R. A., et al. (2016). CRISPR-Cas gene-editing reveals RsmA and RsmC act through FlhDC to repress the SdhE flavinylation factor and control motility and prodigiosin production in Serratia. Microbiology 162, 1047–1058. doi: 10.1099/mic.0.000283
Hempel, R. J., Morton, D. J., Seale, T. W., Whitby, P. W., and Stull, T. L. (2013). The role of the RNA chaperone Hfq in Haemophilus influenzae pathogenesis. BMC Microbiol. 13:134. doi: 10.1186/1471-2180-13-134
Hofacker, I. L. (2003). Vienna RNA secondary structure server. Nucleic Acids. Res. 31, 3429–3431. doi: 10.1093/nar/gkg599
Jatt, A. B., Tang, K., Liu, J., Zhang, Z., and Zhang, X.-H. (2014). Quorum sensing in marine snow and its possible influence on production of extracellular hydrolytic enzymes in marine snow bacterium Pantoea ananatis B9. FEMS Microbiol. Ecol. 91, 1–13. doi: 10.1093/femsec/fiu030
Kakoschke, T. K., Kakoschke, S. C., Zeuzem, C., Bouabe, H., Adler, K., Heesemann, K., et al. (2016). The RNA chaperone Hfq is essential for virulence and modulates the expression of four adhesins in Yersinia enterocolitica. Sci. Rep. 6:29275. doi: 10.1038/srep29275
Kang, S. H., Cho, H.-S., Cheong, H., Ryu, C.-M., Kim, J. F., and Park, S.-H. (2007). Two bacterial endophytes eliciting both plant growth promotion and plant defense on pepper (Capsicum annuum L.). J. Microbiol. Biotechnol. 17, 96–103.
Katashkina, J. I., Hara, Y., Golubeva, L. I., Andreeva, I. G., Kuvaeva, T. M., and Mashko, S. V. (2009). Use of the λ Red-recombineering method for genetic engineering of Pantoea ananatis. BMC Mol. Biol. 10:34. doi: 10.1186/1471-2199-10-34
Kay, E., Humair, B., Denervaud, B., Riedel, K., Spahr, S., Eberl, L., et al. (2006). Two GacA-dependent small RNAs modulate the quorum-sensing response in Pseudomonas aeruginosa. J. Bacteriol. 188, 6026–6033. doi: 10.1128/JB.00409-06
Kido, K., Adachi, R., Hasegawa, M., Yano, K., Hikichi, Y., Takeuchi, S., et al. (2008). Internal fruit rot of netted melon caused by Pantoea ananatis (=Erwinia ananas) in Japan. J. Gen. Plant Pathol. 74, 302–312. doi: 10.1007/s10327-008-0107-3
Kim, D., Hong, J. S., Qui, Y., Nagarajan, H., Seo, J.-H., Cho, B.-K., et al. (2012). Comparative analysis of regulatory elements between Escherichia coli and Klebsiella pneumoniae by genome-wide transcription start site profiling. PLoS Genet. 8:e1002867. doi: 10.1371/journal.pgen.1002867
Kim, J., Mannaa, M., Kim, N., Lee, C., Kim, J., Park, J., et al. (2018). The roles of two hfq genes in the virulence and stress resistance of Burkholderia glumae. Plant Pathol. J. 34, 412–425. doi: 10.5423/PPJ.OA.06.2018.0097
Krawczyk, K., Kamasa, J., Zwolinska, A., and Pospieszny, H. (2010). First report of Pantoea ananatis associated with white leaf spot disease of maize in Poland. J. Plant Pathol. 92, 807–811.
Kuklinsky-Sobral, J., Araújo, W. L., Mendes, R., Geraldi, I. O., Pizzirani-Klelner, A. A., and Azevedo, J. L. (2004). Isolation and characterization of soybean-associated bacteria and their potential for plant growth promotion. Environ. Microbiol. 6, 1244–1251. doi: 10.1111/j.1462-2920.2004.00658.x
Kulesus, R. R., Diaz-Perez, K., Slechta, E. S., Eto, D. S., and Mulvey, M. A. (2008). Impact of the RNA chaperone Hfq on the fitness and virulence potential of uropathogenic Escherichia coli. Infect. Immun. 76, 3019–3026. doi: 10.1128/IAI.00022-08
Kuo, H.-Y., Chao, H.-H., Liao, P.-C., Hsu, L., Chang, K.-C., Tung, C.-H., et al. (2017). Functional characterization of Acinetobacter baumannii lacking the RNA chaperone Hfq. Front. Microbiol. 8:2068. doi: 10.3389/fmicb.2017.02068
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9:357. doi: 10.1038/nmeth.1923
Lenz, D. H., Mok, K. C., Lilley, B. N., Kulkarni, R. V., Wingreen, N. S., and Bassler, B. L. (2004). The small RNA chaperone Hfq and multiple small RNAs control quorum sensing in Vibrio harveyi and Vibrio cholerae. Cell 118, 69–82. doi: 10.1016/j.cell.2004.06.009
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Marquez-Santacruz, H. A., Hernandez-Leon, R., Orozco-Mosqueda, M. C., Velazquez-Sepulvedal, I., and Santoyo, G. (2010). Diversity of bacterial endophytes in roots of Mexican husk tomato plants (Physalis ixocarpa) and their detection in the rhizosphere. Genet. Mol. Res. 9, 2372–2390. doi: 10.4238/vol9-4gmr921
McClean, K. H., Winson, M. K., Fish, L., Taylor, A., Chhabra, S. R., Camara, M., et al. (1997). Quorum sensing and Chromobacterium violaceum: exploitation of violacein production and inhibition for the detection of N-acylhomoserine lactones. Microbiology 143, 3703–3711. doi: 10.1099/00221287-143-12-3703
Metsalu, T., and Vilo, J. (2015). ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 43, W566–W570. doi: 10.1093/nar/gkv468
Monteiro, C., Papenfort, K., Hentrich, K., Ahmad, I., Le Guyon, S., Reimann, R., et al. (2012). Hfq and Hfq-dependent small RNAs are major contributors to multicellular development in Salmonella enterica Serovar typhimurium. RNA Biol. 9, 489–502. doi: 10.4161/rna.19682
Morohoshi, T., Nakamura, Y., Yamazaki, G., Ishida, A., Kato, N., and Ikeda, T. (2007). The plant pathogen Pantoea ananatis produces N-acylhomoserine lactone and causes center rot disease of onion by quorum sensing. J. Bacteriol. 189, 8333–8338. doi: 10.1128/JB.01054-07
Obranić, S., Babić, F., and Maravić-Vlahoviček, G. (2013). Improvement of pBBR1MCS plasmids, a very useful series of broad-host-range cloning vectors. Plasmid 70, 263–267. doi: 10.1016/j.plasmid.2013.04.001
Okunishi, S., Sako, K., Mano, H., Imamura, A., and Morisaki, H. (2005). Bacterial flora of endophytes in the maturing seed of cultivated rice (Oryza sativa). Microbes Environ. 20, 168–177. doi: 10.1264/jsme2.20.168
Oliveira, C. A., Alves, V. M. C., Marriel, I. E., Gomes, E. A., Scotii, M. R., Carneiro, N. P., et al. (2008). Phosphate solubilizing microorganisms isolated from rhizosphere of maize cultivated in an oxisol of the Brazilian Cerrado Biome. Soil Biol. Biochem. 41, 1782–1787. doi: 10.1016/j.soilbio.2008.01.012
Otaka, H., Ishikawa, H., Morita, T., and Aiba, H. (2011). PolyU tail of rho-independent terminator of bacterial small RNAs is essential for Hfq action. Proc. Natl. Acad. Sci. U.S.A. 108, 13059–13064. doi: 10.1073/pnas.1107050108
Pérez-y-Terrón, R., Villegas, M. C., Cuellar, A., Muñoz-Rojas, J., Castañeda-Lucio, M., Hernández-Lucas, I., et al. (2009). Detection of Pantoea ananatis, causal agent of leaf spot disease of maize, in Mexico. Aust. Plant Dis. Notes 4, 96–99.
Pomini, A. M., Araújo, W. L., and Marsaioli, A. J. (2006). Structural elucidation and biological activity of acyl-homoserine lactones from the phytopathogen Pantoea ananatis Serrano, 1928. J. Chem. Ecol. 32, 1769–1778. doi: 10.1007/s10886-006-9108-x
R Core Team (2013). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing.
Rijavec, T., Lapanje, A., Dermastia, M., and Rupnik, M. (2007). Isolation of bacterial endophytes from germinated maize kernels. Can. J. Microbiol. 53, 802–808. doi: 10.1139/W07-048
Rio, D. C., Ares, M. Jr., Hannon, G. J., and Nilsen, T. W. (2010). Purification of RNA using TRIzol (TRI reagent). Cold Spring Harb. Protoc. 6:pdb.prot5439. doi: 10.1101/pdb.prot5439
Santander, R. D., and Biosca, E. G. (2017). Erwinia amylovora psychrotrophic adaptations: evidence of pathogenic potential and survival at temperate and low environmental temperatures. PeerJ 5:e3931. doi: 10.7717/peerj.3931
Schachterle, J. K., and Sundin, G. W. (2019). The leucine-responsive regulatory protein participates in virulence regulation downstream of small RNA ArcZ in Erwinia amylovora. mBio 10:e0757-19. doi: 10.1128/mBio.00757-19
Schachterle, J. K., Zeng, Q., and Sundin, G. W. (2019). Three Hfq-dependent small RNAs regulate flagellar motility in the fire blight pathogen Erwinia amylovora. Mol. Microbiol. 111, 1476–1492. doi: 10.1111/mmi.14232
Schmidtke, C., Abendroth, U., Brock, J., Serrania, J., Becker, A., and Bonas, U. (2013). Small RNA sX13: a multifaceted regulator of virulence in the plant pathogen Xanthomonas. PLoS Pathog. 9:e1003626. doi: 10.1371/journal.ppat.1003626
Schneider, C. A., Rasband, W. S., and Eliceiri, K. W. (2012). NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675. doi: 10.1038/nmeth.2089
Schwartz, H. F., and Otto, K. (2000). First report of a leaf blight and bulb decay of onion by Pantoea ananatis in Colorado. Plant Dis. 84:808. doi: 10.1094/PDIS.2000.84.7.808A
Serrano, F. B. (1928). Bacterial fruitlet brown-rot of pineapple in the Philippines. Philipp. J. Sci. 36, 271–324.
Shyntum, D. Y., Theron, J., Venter, S. N., Moleleki, L. N., Toth, I. K., and Coutinho, T. A. (2015). Pantoea ananatis utilizes a type VI secretion system for pathogenesis and bacterial competition. Mol. Plant Microbe Interact. 28, 420–431. doi: 10.1094/MPMI-07-14-0219-R
Sibanda, S., Kwenda, S., Tanui, C. K., Shyntum, D. Y., Coutinho, T. A., and Moleleki, L. N. (2018). Transcriptome profiling reveals the EanI/R quorum sensing regulon in Pantoea ananatis LMG 2665T. Genes 9, 1–17. doi: 10.3390/genes9030148
Sibanda, S., Theron, J., Shyntum, D. Y., Moleleki, L. N., and Coutinho, T. A. (2016). Characterization of two LuxI/R homologs in Pantoea ananatis LMG 2665T. Can. J. Microbiol. 62, 893–903. doi: 10.1139/cjm-2016-0143
Sittka, A., Lucchini, S., Papenfort, K., Sharma, C. M., Rolle, K., Binnewies, T. T., et al. (2008). Deep sequencing analysis of small noncoding RNA and mRNA targets of the global post-transcriptional regulator, Hfq. PLoS Genet. 4:e1000163. doi: 10.1371/journal.pgen.1000163
Sittka, A., Pfeiffer, V., Tedin, K., and Vogel, J. (2007). The RNA chaperone Hfq is essential for the virulence of Salmonella typhimurium. Mol. Microbiol. 63, 193–217. doi: 10.1111/j.1365-2958.2006.05489.x
Stice, S. P., Stumpf, D. S., Gitaitis, R. D., Kvitko, B. H., and Dutta, B. (2018). Pantoea ananatis genetic diversity analysis reveals limited genomic diversity as well as accessory genes correlated with onion pathogenicity. Front. Microbiol. 9:184. doi: 10.3389/fmicb.2018.00184
Storz, G., Vogel, J., and Wassarman, K. M. (2011). Regulation by small RNAs in bacteria. Expanding frontiers. Mol. Cell 43, 880–891. doi: 10.1016/j.molcel.2011.08.022
Takle, G. W., Toth, I. K., and Brurberg, M. B. (2007). Evaluation of reference genes for real-time RT-PCR expression studies in the plant pathogen Pectobacterium atrosepticum. BMC Plant Biol. 7:50. doi: 10.1186/1471-2229-7-50
Tu, K. C., and Bassler, B. L. (2007). Multiple small RNAs act additively to integrate sensory information and control quorum sensing in Vibrio harveyi. Genes Dev. 21, 221–233. doi: 10.1101/gad.1502407
Updegrove, T. B., Zhang, A., and Storz, G. (2016). Hfq: the flexible RNA matchmaker. Curr. Opin. Microbiol. 30, 133–138. doi: 10.1016/j.mib.2016.02.003
Vogel, J., and Luisi, B. F. (2011). Hfq and its constellation of RNA. Nat. Rev. Microbiol. 9, 578–589. doi: 10.1038/nrmicro2615
Wang, C., Pu, T., Lou, W., Wang, Y., Gao, Z., Hu, B., et al. (2018). Hfq, a RNA chaperone, contributes to virulence by regulating plant cell wall–degrading enzyme production, type VI secretion system expression, bacterial competition, and suppressing host defense response in Pectobacterium carotovorum. Mol. Plant Microbe Interact. 31, 1166–1178. doi: 10.1094/MPMI-12-17-0303-R
Weller-Stuart, T., Toth, I., De Maayer, P., and Coutinho, T. A. (2016). Swimming and twitching motility are essential for attachment and virulence of Pantoea ananatis in onion seedlings. Mol. Plant Pathol. 18, 734–745. doi: 10.1111/mpp.12432
Wilf, N. M., Reid, A. J., Ramsay, J. P., Williamson, N. R., Croucher, N. J., Gatto, L., et al. (2013). RNA-seq reveals the RNA binding proteins, Hfq and RsmA, play various roles in virulence, antibiotic production and genomic flux in Serratia sp. ATCC 39006. BMC Genomics 14:822. doi: 10.1186/1471-2164-14-822
Wilms, I., Möller, P., Stock, A., Gurski, R., Lai, E., and Naberhaus, F. (2012a). Hfq influences multiple transport systems and virulence in the plant pathogen Agrobacterium tumefaciens. J. Bacteriol. 194, 5209–5217. doi: 10.1128/JB.00510-12
Wilms, I., Overlöper, A., Nowrousian, M., Sharma, C. M., and Narberhaus, F. (2012b). Deep sequencing uncovers numerous small RNAs on all four replicons of the plant pathogen Agrobacterium tumefaciens. RNA Biol. 9, 446–457. doi: 10.4161/rna.17212
Wright, P. R., Georg, J., Mann, M., Sorescu, D. A., Richter, A. S., Lott, S., et al. (2014). CopraRNA and IntaRNA: predicting small RNA targets, networks and interaction domains. Nucleic Acids Res. 42, 119–123. doi: 10.1093/nar/gku359
Yuan, X., Zeng, Q., Khokhani, D., Tian, F., Severin, G. B., Waters, C. M., et al. (2019). A feed-forward signalling circuit controls bacterial virulence through linking cyclic-di-GMP and two mechanistically distinct sRNAs; ArcZ and RsmB. Environ. Microbiol. 21, 2755–2771. doi: 10.1111/1462-2920.14603
Zeng, Q., McNally, R. R., and Sundin, G. W. (2013). Global small RNA chaperone Hfq and regulatory small RNAs are important virulence regulators in Erwinia amylovora. J. Bacteriol. 195, 1706–1717. doi: 10.1128/JB.02056-12
Keywords: Pantoea ananatis, plant pathogen, Hfq, sRNA, regulation, virulence
Citation: Shin GY, Schachterle JK, Shyntum DY, Moleleki LN, Coutinho TA and Sundin GW (2019) Functional Characterization of a Global Virulence Regulator Hfq and Identification of Hfq-Dependent sRNAs in the Plant Pathogen Pantoea ananatis. Front. Microbiol. 10:2075. doi: 10.3389/fmicb.2019.02075
Received: 28 June 2019; Accepted: 22 August 2019;
Published: 11 September 2019.
Edited by:
Giorgio Gambino, Italian National Research Council, ItalyReviewed by:
Teppei Morita, Suzuka University of Medical Science, JapanVincenzo Scarlato, University of Bologna, Italy
Copyright © 2019 Shin, Schachterle, Shyntum, Moleleki, Coutinho and Sundin. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Teresa A. Coutinho, VGVyZXNhLkNvdXRpbmhvQHVwLmFjLnph