 Maria Pain
Maria Pain Erik Hjerde
Erik Hjerde Claus Klingenberg
Claus Klingenberg Jorunn Pauline Cavanagh
Jorunn Pauline Cavanagh- 1Pediatric Infections Research Group, Department of Clinical Medicine, UiT The Arctic University of Norway, Tromsø, Norway
- 2Department of Chemistry, Norstruct, UiT The Arcic University of Norway, Tromsø, Norway
- 3Department of Paediatrics, University Hospital of North Norway, Tromsø, Norway
Staphylococcus haemolyticus is a skin commensal gaining increased attention as an emerging pathogen of nosocomial infections. However, knowledge about the transition from a commensal to an invasive lifestyle remains sparse and there is a paucity of studies comparing pathogenicity traits between commensal and clinical isolates. In this study, we used a pan-genomic approach to identify factors important for infection and hospital adaptation by exploring the genomic variability of 123 clinical isolates and 46 commensal S. haemolyticus isolates. Phylogenetic reconstruction grouped the 169 isolates into six clades with a distinct distribution of clinical and commensal isolates in the different clades. Phenotypically, multi-drug antibiotic resistance was detected in 108/123 (88%) of the clinical isolates and 5/46 (11%) of the commensal isolates (p < 0.05). In the clinical isolates, we commonly identified a homolog of the serine-rich repeat glycoproteins sraP. Additionally, three novel capsular polysaccharide operons were detected, with a potential role in S. haemolyticus virulence. Clinical S. haemolyticus isolates showed specific signatures associated with successful hospital adaption. Biofilm forming S. haemolyticus isolates that are resistant to oxacillin (mecA) and aminoglycosides (aacA-aphD) are most likely invasive isolates whereas absence of these traits strongly indicates a commensal isolate. We conclude that our data show a clear segregation of isolates of commensal origin, and specific genetic signatures distinguishing the clinical isolates from the commensal isolates. The widespread use of antimicrobial agents has probably promoted the development of successful hospital adapted clones of S. haemolyticus clones through acquisition of mobile genetic elements or beneficial point mutations and rearrangements in surface associated genes.
Introduction
Staphylococcus haemolyticus is an emerging pathogen of nosocomial infections, and the most frequently isolated coagulase-negative staphylococcal (CoNS) species alongside Staphylococcus epidermidis (Hope et al., 2008; Pereira et al., 2014; Nanoukon et al., 2017; Teeraputon et al., 2017). S. haemolyticus infections particularly affect immunocompromised patients and mainly occur as bloodstream and device-associated infections. Nosocomial S. haemolyticus isolates are ranked as the most antibiotic resistant species among the CoNS, and antibiotic therapy choices are therefore very limited (Hope et al., 2008; Kresken et al., 2011; Barros et al., 2012).
Compared to the more virulent Staphylococcus aureus, S. haemolyticus possesses few typical virulence factors (Takeuchi et al., 2005). Formation of biofilm (Fredheim et al., 2009; Giormezis et al., 2014; Pereira et al., 2014), production of phenol-soluble modulins (Da et al., 2017) and frequent phenotypic rearrangements due to a large number of insertion sequences (IS) (Takeuchi et al., 2005) have been suggested as important S. haemolyticus virulence determinants. However, these traits have not yet been linked explicitly to strains of clinical origin. The oriC environ is a chromosomal region of staphylococci proposed to be important for the evolution and differentiation of each staphylococcal species. There is little homology between the oriC environ of the different staphylococcal species, and the region does not contain genes essential for viability. The oriC environ is significantly larger in S. haemolyticus compared to that of S. aureus and Staphylococcus epidermidis. Moreover, almost half of the candidate coding sequences (CDS) for virulence are located within the oriC environ, encoding e.g., surface adhesins and capsular polysaccharides, factors that can modulate adherence and contribute to phagocytosis resistance (Takeuchi et al., 2005; Flahaut et al., 2008).
Despite the advancing clinical relevance of S. haemolyticus, knowledge about the transition from a commensal to an invasive lifestyle remains sparse. Moreover, there is a paucity of studies comparing pathogenicity traits between commensal and clinical invasive isolates. Predicting invasiveness of staphylococcal strains by use of marker genes is one approach to differentiate isolates with different pathogenicity. For S. epidermidis, the ica operon encoding biofilm formation and the insertion sequence element IS256; associated with aminoglycoside resistance, have been proposed as markers for invasive strains of S. epidermidis (Kozitskaya et al., 2004; Rohde et al., 2004). More recently, Méric et al. (2018) presented how calculation of a genotype risk score can predict pathogenicity in S. epidermidis isolates with 80% accuracy. With the advances in sequencing technologies and more available bacterial genomes new methods for analysis have emerged. The total number of genes in a bacterial population, collectively called the pan-genome, allows identification of genes more present and important in pathogenic strains (Tettelin et al., 2005). Studies of S. haemolyticus to date have focused predominantly on clinical isolates. The aim of this study was to identify factors important for infection and hospital adaptation by exploring the genomic variability of clinical and commensal S. haemolyticus using a pan-genomic approach.
Materials and Methods
Bacterial Isolates, Species Identification, Antibiotic Susceptibility and Biofilm Testing
This study includes 169 S. haemolyticus isolates; 123 clinical isolates and 46 commensal isolates. The clinical isolates (mainly from blood cultures, invasive catheters and wounds), in addition to five of the commensal isolates, were from different hospital units and had different geographical origins. They were collected between 1988 and 2010, and have been described previously (Cavanagh et al., 2014). The remaining 41 commensal isolates were skin samples isolated from healthy volunteers with no antibiotic exposure and no hospitalization or health care affiliation during the three previous months (Cavanagh et al., 2016). This was a separate collection from one geographical location (Tromsø, Norway) and collected between 2013 and 2014 (Cavanagh et al., 2016). An overview of all the isolates used and their characteristics can be found in Supplementary Data S1. Species identification was done by 16s rRNA sequencing and/or matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) using a Microflex LT instrument (Bruker Daltonics, Bremen, Germany), Flex Control software and the MALDI Biotyper 3.1 software (Bruker Daltonics, Bremen, Germany). Antibiotic susceptibility testing was performed as previously described (Cavanagh et al., 2014, 2016), and interpreted according to the 9th version of the EUCAST guidelines1. Isolates resistant to three or more classes of antibiotics were classified as multidrug resistant (MDR). Semi-quantitative determination of biofilm formation was performed with the modified Christensen assay, as described previously (Fredheim et al., 2009). Isolates were considered biofilm positive if they had an optical density (OD) value >0.2 above the negative control.
Whole Genome Sequencing (WGS), Assembly and Annotation
The WGS procedure for all clinical isolates and five of the 46 commensal isolates is described in a previous study (Cavanagh et al., 2014). For the remaining 41 commensal isolates, bacterial DNA was extracted and prepared for WGS using the Wizard Genomic DNA purification kit (Promega, Madison, United States) according to the manufacturer’s instructions. WGS was performed on index-tagged libraries for each S. haemolyticus strain by paired-end sequencing at the Norwegian Sequencing Centre on an Illumina MiSeq (Illumina Inc., San Diego, California, United States). Subsequently, all 169 genomes were (re-assembled using SPAdes version 3.7 software (Bankevich et al., 2012), with some modification to the default parameters. Contigs >500 bp were reordered relative to the only complete fully annotated closed reference genome (JCSC 1435) (Takeuchi et al., 2005) using ABACAS (version 1.3.1). Protein CDSs were predicted using Prokka v1.12 (Seemann, 2014) using default settings. The sequences are deposited in the European Nucleotide Archive2; study accession no ERP000943 and ERP114853.
Phylogeny and Molecular Typing
Subtyping of the 169 isolates was performed using kSNP3 to identify single-nucleotide polymorphisms (SNPs) in the core genomes and to reconstruct a parsimony phylogenomic tree (Gardner et al., 2015). Phylogenetic trees were visualized using the online tool iTol3.
Pan-Genome and Pan-Genome-Wide Association Study (Pan-GWAS) of S. haemolyticus
Pan-genome analyses of all 169 isolates were performed using the Roary software package with default settings (Page et al., 2015). The program generates a file containing all the predicted gene clusters and the sequence identifier of each isolate containing said gene. Based on this file, core, accessory and unique genes were extracted and saved as individual lists. The accessory list was subdivided based on clusters common for both clinical and commensal isolates, in addition to clusters unique to each group. The unique list was further subdivided based on genes identified in clinical and commensal isolates. These lists were uploaded to eggNOG to get cluster of orthologs groups (COG) identifications (Huerta-Cepas et al., 2016).
Files created by Roary, were used as input for Scoary, a microbial pan-GWAS tool that calculates the association between all genes in the accessory genome and traits defined by the users. For our purpose we only used one trait; whether a given isolate were of clinical or commensal origin. Based on this information Scoary reported a list of genes sorted by strength of association per gene (Brynildsrud et al., 2016).
In silico Analysis and Statistics
The resistance gene identifier in the comprehensive antibiotic resistance database (CARD; version 1.1.1; Department of Biochemistry and Biomedical Science; McMaster University, Canada (Jia et al., 2017) was used to predict genes presumed to confer antibiotic resistance, and the results were further compared with the phenotypic susceptibility test results. Potential virulence factors were identified by homology searches against the virulence factor database (VFDB) together with putative virulence factors previously predicted by Takeuchi et al. (2005) and Chen et al. (2016). Identification of IS elements was performed using the ISsaga program (Varani et al., 2011). The presence of IS elements were confirmed by sequence search against the complete S. haemolyticus sequence collection (both on contigs and CDS). Sequence coverage of contigs harboring IS elements was used to quantify the copy numbers: the coverage of contigs with IS elements divided by the overall average coverage of its respective genome. Identification of putative plasmids was performed by screening the genome assemblies for plasmid replicon (rep) genes using the PlasmidFinder database (Carattoli et al., 2014) with coverage settings set to default of 75%. Identification of prophages, potentially important for horizontal gene transfer (HGT), was performed by using PHASTER (Arndt et al., 2016). Plasmid replicon sequences with more than 80% coverage and predicted intact phages identified in more than 10 isolates were further investigated by sequence search in order to confirm the presence of plasmid and phage genes, respectively.
Data were also analyzed using IBM-SPSS statistical software (IBM Corp. Released 2015. IBM SPSS Statistics for Windows, Version 23.0. Armonk, NY: IBM Corp.). Categorical data are displayed as ratios and frequency (%), and analyzed using the Chi square test. Pathogenicity is a complex multifactorial property. We used biological knowledge to identify known pathogenicity-associated traits enriched in clinical isolates. In particular, we focused on antibiotic resistance genes (ARGs) and previously established virulence factors. We developed different scores including the following four traits; aacA-aphD, mecA, folP and phenotypic biofilm production. In order to find a pragmatic score that could differentiate between a clinical and a commensal isolate we calculated the area under receiver operating characteristic (ROC) curves, and its 95% confidence interval.
Results
Genome Composition and Genetic Variability
The average size of the assembled genomes was 2.52 Mb (2.32–2.86 Mb), with an average of 120 (32–415) contigs per genome. Each genome had on average 2,457 (2,239–2,816) predicted protein sequences (CDSs) with an average GC content of 32%.
The pan-genome of the 169 S. haemolyticus isolate dataset comprised 9,092 Cluster of Orthologous groups (COGs). We divided the pan-genome into core genes (genes shared by all strains), accessory genes (genes shared by some but not all strains) and unique genes only present in one genome (Figure 1A). The pan-genome sub-groups were annotated and sorted into COG categories (Figure 1B). Gene accumulation curves showed that the core genome plateaued at 1,522 genes reflecting a stable core and an open pan-genome where the addition of each new genome increases the total gene pool (Figure 1C).
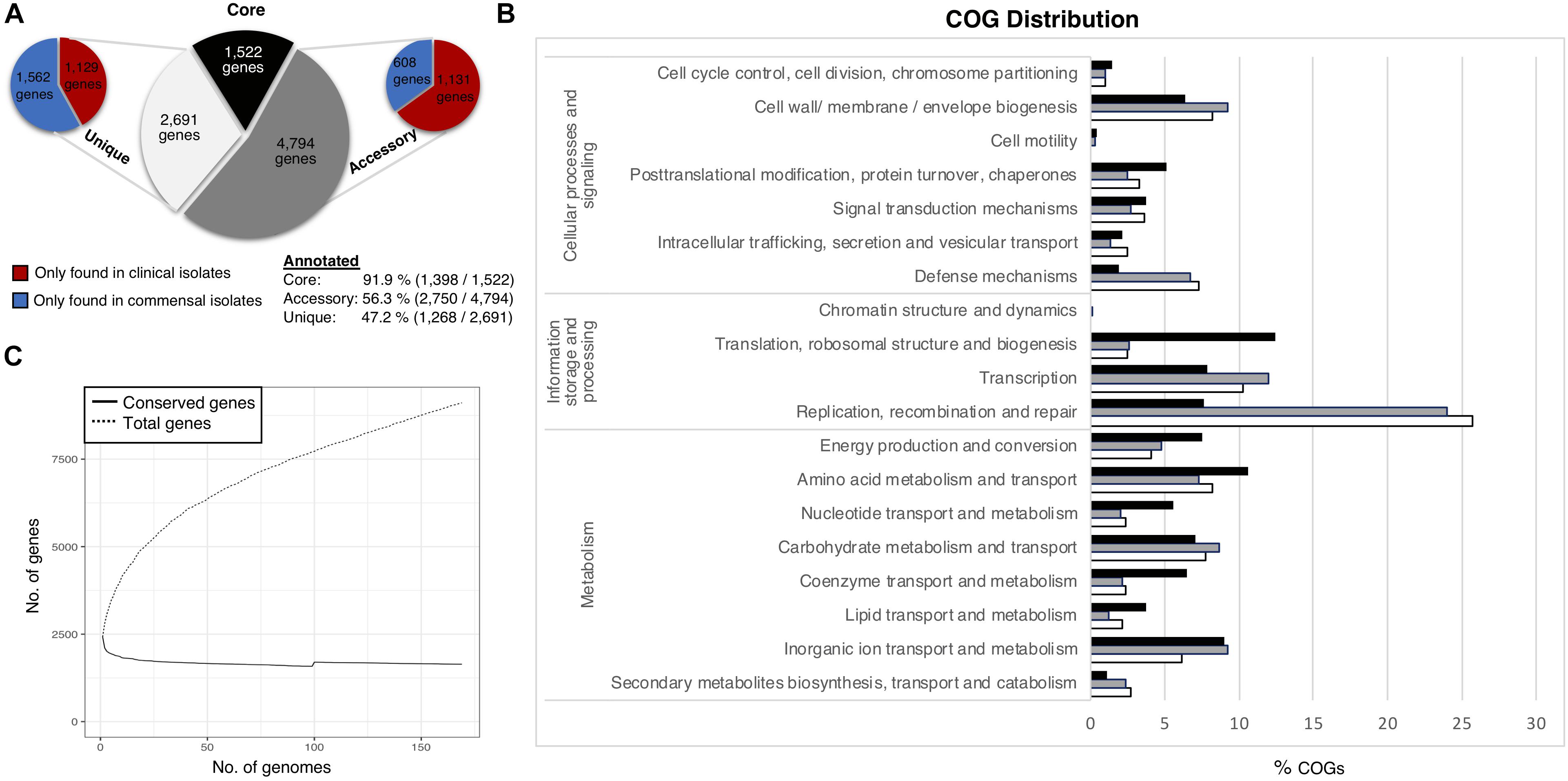
Figure 1. Staphylococcus haemolyticus pangenome statistics. (A) The size and distribution of the pangenome into the subgroups; core (shared by all isolates), accessory (shared by some isolates) and unique (found only in one isolate). (B) Collective distribution of core (black), accessory (gray) and unique (white) genes in COG. (C) Pangenome curve generated by plotting total number of gene families in the pan and core genome.
Thirty-two percent of the annotated genes were categorized as function unknown, and this class was excluded from the graphical representation of the COG categories. The most abundant categories in the core genome were genes involved in housekeeping functions, like transcription and translation, and different metabolism categories. The accessory genome had a larger portion of genes associated with mobile genetic elements (MGE) such as transposons and bacteriophages (transcription, replication, recombination and repair (24%), which was also the most enriched category amongst the unique genes (25.7%). Genes involved in transcription (12%), genes associated with cell wall and membrane biogenesis (9.2%) and defense mechanisms (6.8%) were also considerably more prevalent in the accessory and unique gene pool, compared to the core gene pool. No significant differences of the COG distribution were found between the clinical and commensal group (data not shown).
Phylogeny and Population Structure
The phylogenetic reconstruction grouped the 169 isolates into six clades (A–F). There was a distinct distribution of clinical and commensal isolates into the different clades (Figure 2). The two largest clades, A and C, consisted almost exclusively of clinical isolates (88/90; 98%), while the majority of the commensal isolates (39/46; 85%) were found in clades D and F. The clinical isolates in clade A–C were more closely related compared to the commensal isolates. Twenty-eight of 123 (22.8%) clinical isolates were grouped in the three clades (D–F) predominantly consisting of commensal isolates. A long branch separating the “commensal heavy” clade F (78% commensal isolates) from the rest of the isolates underlines the high variability observed in the commensal isolates, compared to the clustered clinical isolates (Figure 2). We observed that with the exception of clade F, the isolates in all the other clades were closely related, independent of country of origin. The commensal isolates in clade F, mainly originating from the same geographic location in Norway, were in contrast very diverse.
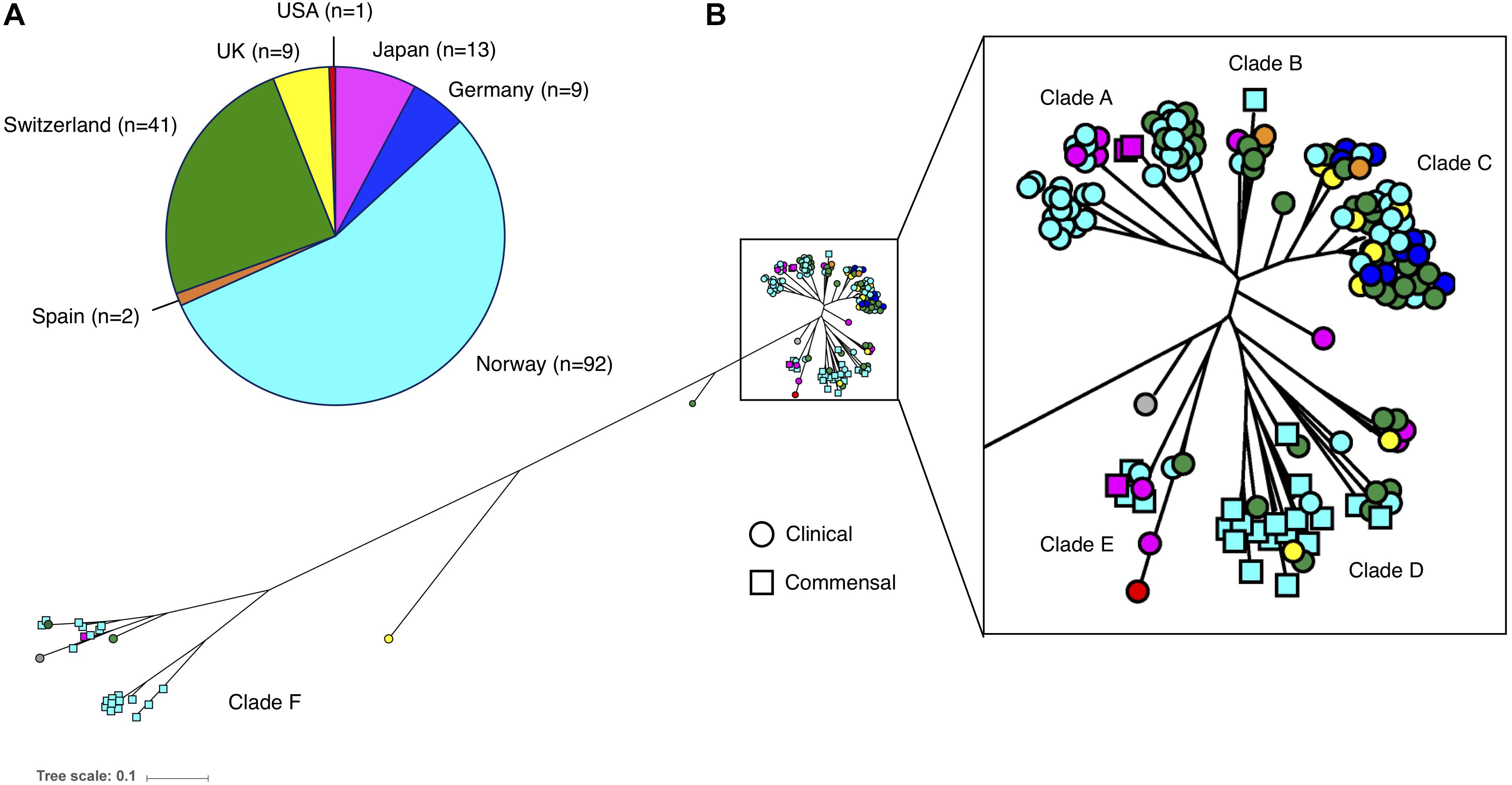
Figure 2. (A) Number and origin of isolates included in this study. (B) Phylogenetic tree of the 169 S. haemolyticus isolates, based on SNPs in the core genome. Each isolate is color coded based on country of origin, as demonstrated in the pie chart. Clinical isolates are displayed as circles and commensal isolates as squares.
Antibiotic Resistance and Mobile Genetic Elements (MGE)
Phenotypically, 113/169 (68%) of the isolates were classified as MDR; 108/123 (88%) of the clinical isolates (Cavanagh et al., 2014) and 5/46 (11%) of the commensal isolates (Cavanagh et al., 2016) (p < 0.05). Using a genotypic approach, we identified the ARGs for most of the observed phenotypes, and overall, phenotypic and genotypic resistance correlated well. However, ARGs toward eight additional antibiotic classes were also detected, but not phenotypically tested (Figure 3).
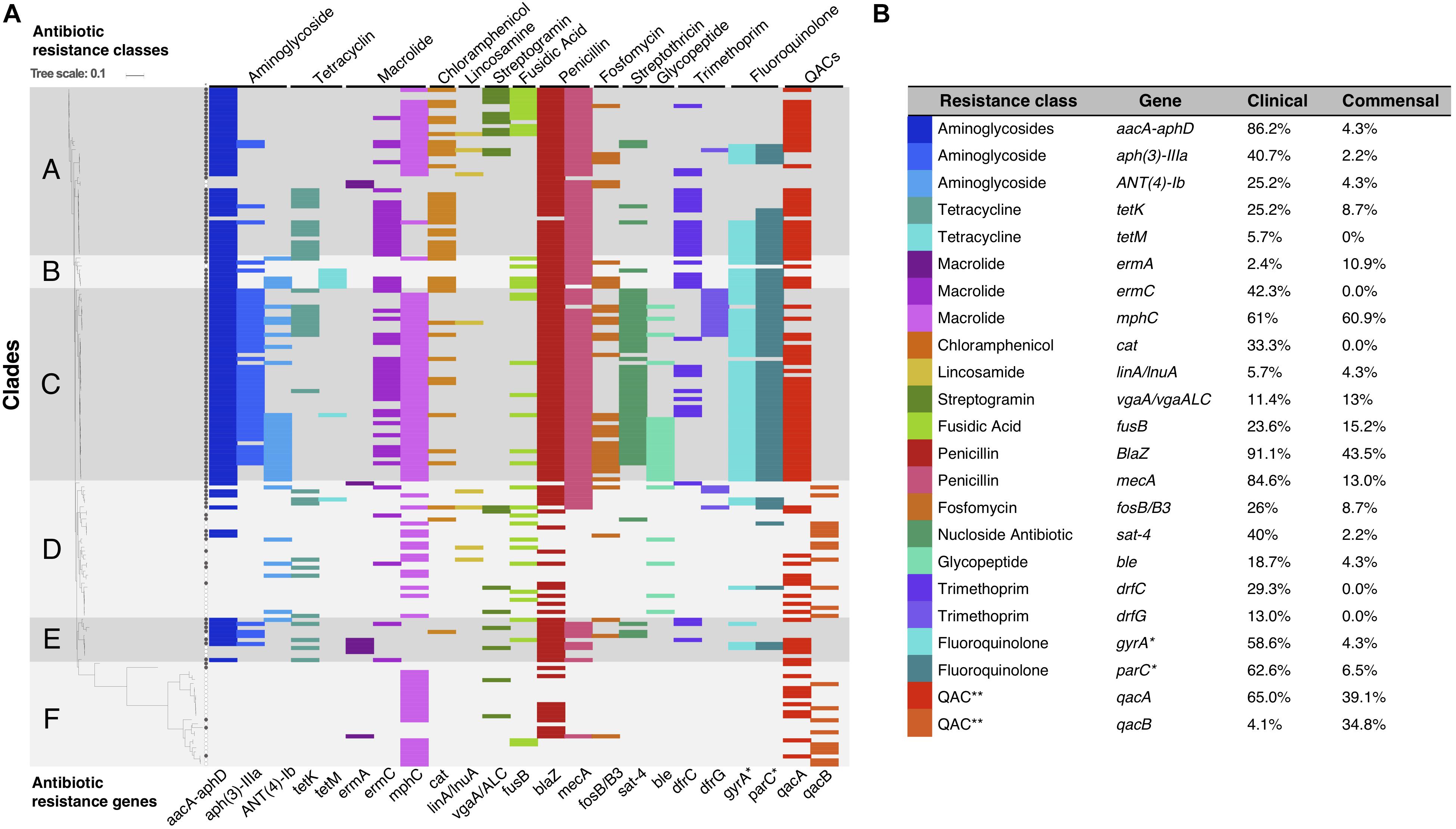
Figure 3. (A) Graphical representation of S. haemolyticus antibiotic resistance genotype plotted onto the phylogenetic SNP-based core tree. The figure shows the presence and absence of ARGs and classes across the different clades. ∗Mutations in gyrA and parC gives resistance to fluoroquinolones. ∗∗ QACs: quaternary ammonium compounds. (B) The percentage of each ARG present in the clinical and commensal subgroups.
ARGs were among the most common genes among all clinical isolates. In particular mecA, aacA/aphD, blaZ were predominantly found in invasive isolates (Table 1). Phenotypic macrolide resistance was common in both the commensal (30/46; 65%) and the clinical (100/123; 81%) isolates. However, the majority of commensal isolates carried the macrolide resistance gene mphC (28/46; 61%) and few carried ermA (5/46; 11%), while the clinical isolates had both mphC (75/123; 61%) and ermC (52/123; 42.3%). The antiseptic resistance genes qacA and qacB were found in both clinical and commensal isolates, however qacB was identified predominantly in the commensal isolates whereas qacA was detected predominantly in clinical isolates (Figure 3).
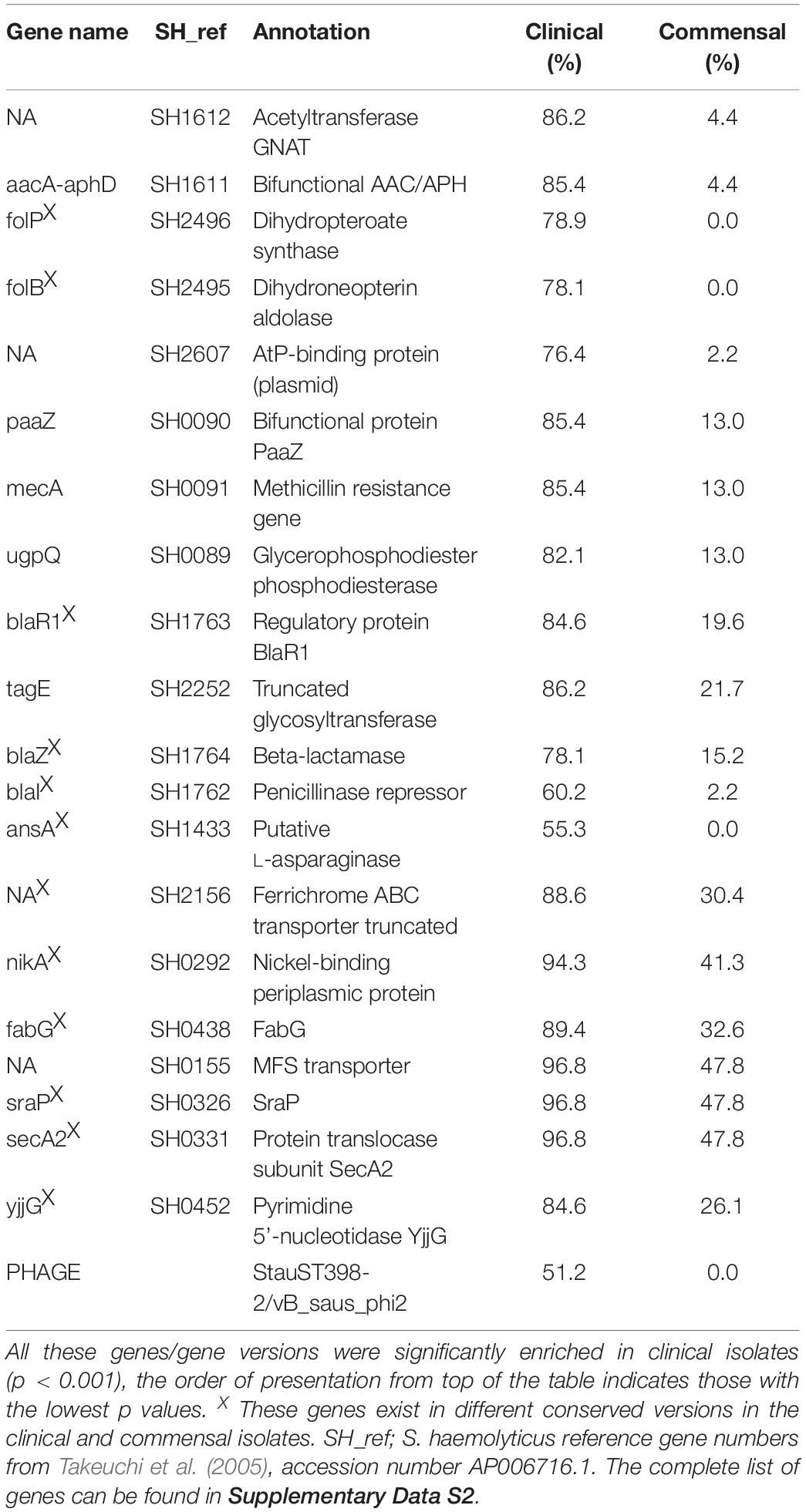
Table 1. Genes and gene versions enriched in 123 clinical isolates, sorted by significance.
For the plasmid analysis, we found a total of 51 replicon sequences, with an average of 5.5 replicon per clinical isolates and 7.6 average replicon in commensal isolates. When a replication sequence was identified in more than 10 isolates, we examined the presence of the entire plasmid. For smaller plasmids presence was easily determined, and numbers in clinical and commensal isolates were calculated and presented in Table 2. Larger plasmid sequences were often split over several contigs, thus not all of the plasmid genes were identified. If the whole plasmid could not be identified, we looked for the presence of its cargo genes instead. The replicon sequence of S. haemolyticus plasmid pSHaeB, carrying ermC, was only found in the clinical isolates (55/123; 45%). The S. aureus plasmid replicon for pUB110, carrying kanamycin and bleomycin resistance genes, was identified in 23/123 (19%) clinical and 2/46 (4%) commensal isolates. Chloramphenicol resistance plasmid pC221 was predicted based on the replicon sequence, and the chloramphenicol gene (cat) was identified in 39 isolates, all of clinical origin. In addition, the plasmids pSHaeA and pSHAaeC, the former carrying the fosfomycin resistance gene, were identified in our collection but in less than 1/3 of all isolates, and with no clear difference between clinical and commensal isolates. Several larger plasmids were predicted based on replicon sequence but none of these were identified in their entirety. We extracted what we considered interesting cargo genes from these plasmids and determined the prevalence of these (Table 3).
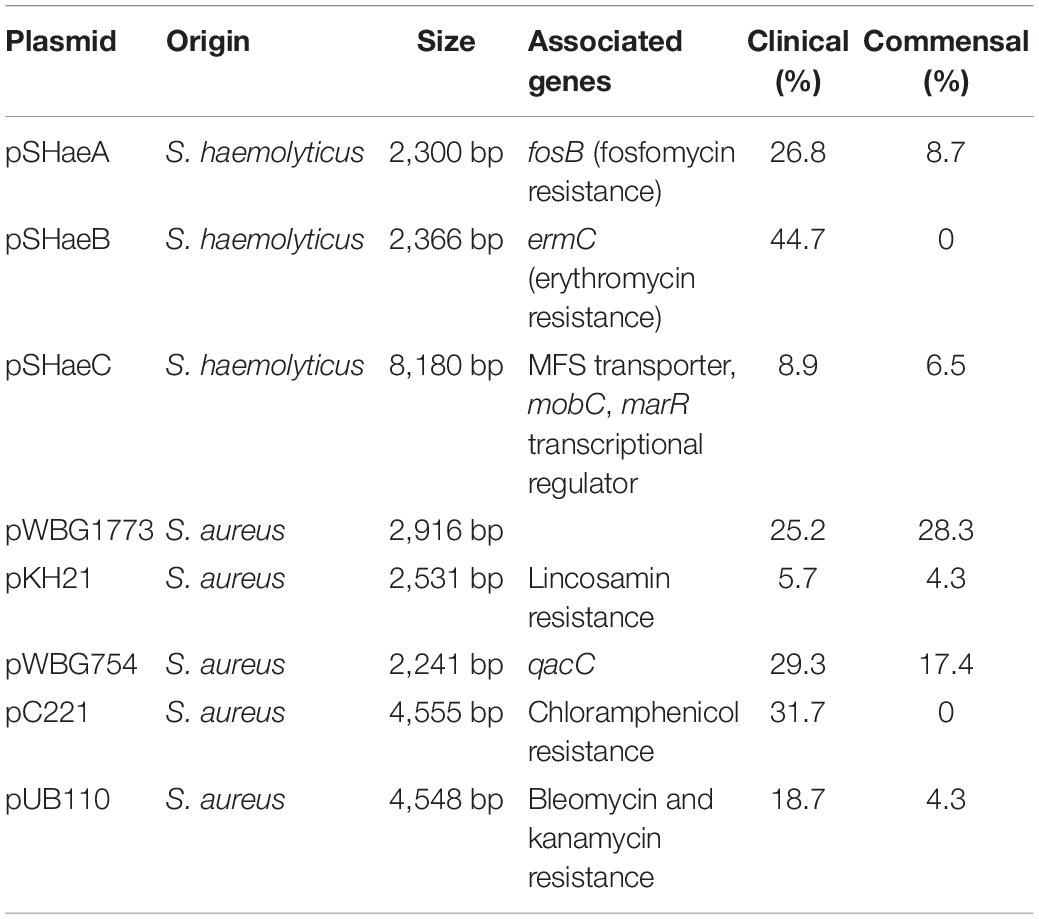
Table 2. Smaller plasmids and their distribution in clinical (n = 123) and commensal (n = 46) isolates.
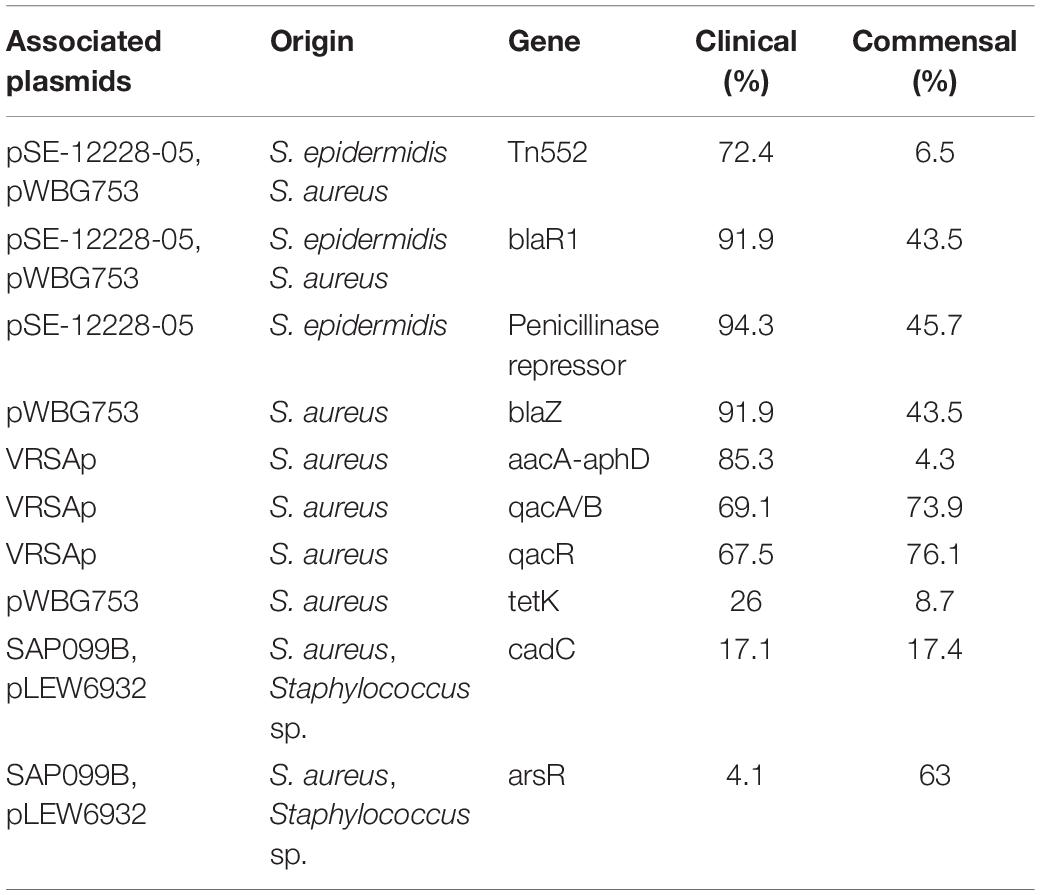
Table 3. Distribution of genes associated with larger plasmids in the isolates.
We predicted 13 different intact prophages in our collection, identified in 68% (114/169) of the isolates (clinical 94/123; 76% and commensal 17/46; 37%). Five of these prophages were found in more than 10 isolates. The most prevalent phage, exclusively found in only clinical isolates (63/123; 51%), was predicted as staphylococcal phage vB_Saus_phi2 and S. aureus phage StauST398-2. Sequence searches of the two phage genes matched the same S. haemolyticus genes. Staphylococcus phage SPbeta-like is a phage of 127,726 bp with 156 CDS, and was identified in 21 clinical isolates and in one commensal isolate. This phage carries the genes aacA-aphA and dfrC, encoding resistance to gentamicin and trimethoprim, in addition to five IS256 and one IS431 element (CDS 146–156). Of all the isolates carrying this phage, we could identify the first 145 genes in the same order as in the reference phage.
We identified a large number of IS elements, ranging from 15 to 88 per each isolate. ISSha1 and IS1272 were found as several copies in all isolates. IS256 was found almost exclusively in clinical isolates (106/123; 86%) and rarely (5/46; 11%) in commensal isolates. The transposon Tn552/IS481 was also identified predominantly in clinical isolates (89/123; 72%) compared to commensal isolates (6/46; 13%).
Biofilm Production and Biofilm Encoding Genes
Phenotypic biofilm production was significantly more prevalent among clinical isolates (83/123; 67%) versus commensal isolates (16/46; 35%), p < 0.001. However, we did not detect ica genes which typically encode biofilm production in other CoNS species.
Putative Surface Proteins
The C-terminal part of a hypothetical serine-rich surface protein (SH0326) named sraP (serine rich adhesin for platelets) was present in 119/123 (97%) of the clinical isolates and in 22/46 (48%) of commensal isolates. The partial sraP gene was located up-streams of secY2, the first gene in the accessory sec system (aSec); a structure dedicated to transport and modification of this surface protein. The N-terminal was located on a different contig which was, in most cases, correctly assembled directly upstream of the aSec contig. The central part of SraP, known to contain long stretches of repeated patterns was lost during assembly. Mapping sequence reads on assemblies of isolates, predicted to contain sraP, against the full length SH0326 showed that these strains did indeed have the full sequence. However, there appeared to be individual differences in the length of these repeat sequences. The complete aSec operon was identified in 119 clinical isolates (97%) and 34 (74%) of commensal isolates by performing sequence read mapping. Isolates without sraP and aSec belonged to the commensal clades D (12 isolates) and F (3 isolates), and one commensal isolate in clade B.
Putative Capsule Polysaccharide (CP) Operons
We identified four different structures of the CP operons among the 169 isolates. Seventeen isolates, all of clinical origin, had the capA-M operon structure similar to that described in S. haemolyticus JCSC 1435 (Takeuchi et al., 2005). Another 52 isolates had three potential novel CP operons (Figure 4). These three novel CP operons were homologs to the capA-G of S. haemolyticus JCSC 1435 (65–100% CDS identity). Each novel CP operon contained a capH-K region unique to its group, followed by the region capMII-P (CDS 51–78% identity) which was common to all the three novel versions. The last gene in the operon, named capM in JCSC1435 was present in all four CP operon versions. capM identified in the three novel versions were named capMII to distinguish it from the already annotated capM found in JCSC 1435. capL-MII shared homology with S. aureus cap5/8. The novel CP version 1 was lacking the capH gene and capI-K did not show considerable homology to any other known CP genes. However, the novel CP version 2 capH-K showed homology to S. aureus cap8 (CDS 55–72% identity) and novel CP version 3 to S. aureus cap5 (CDS 57–68% identity).
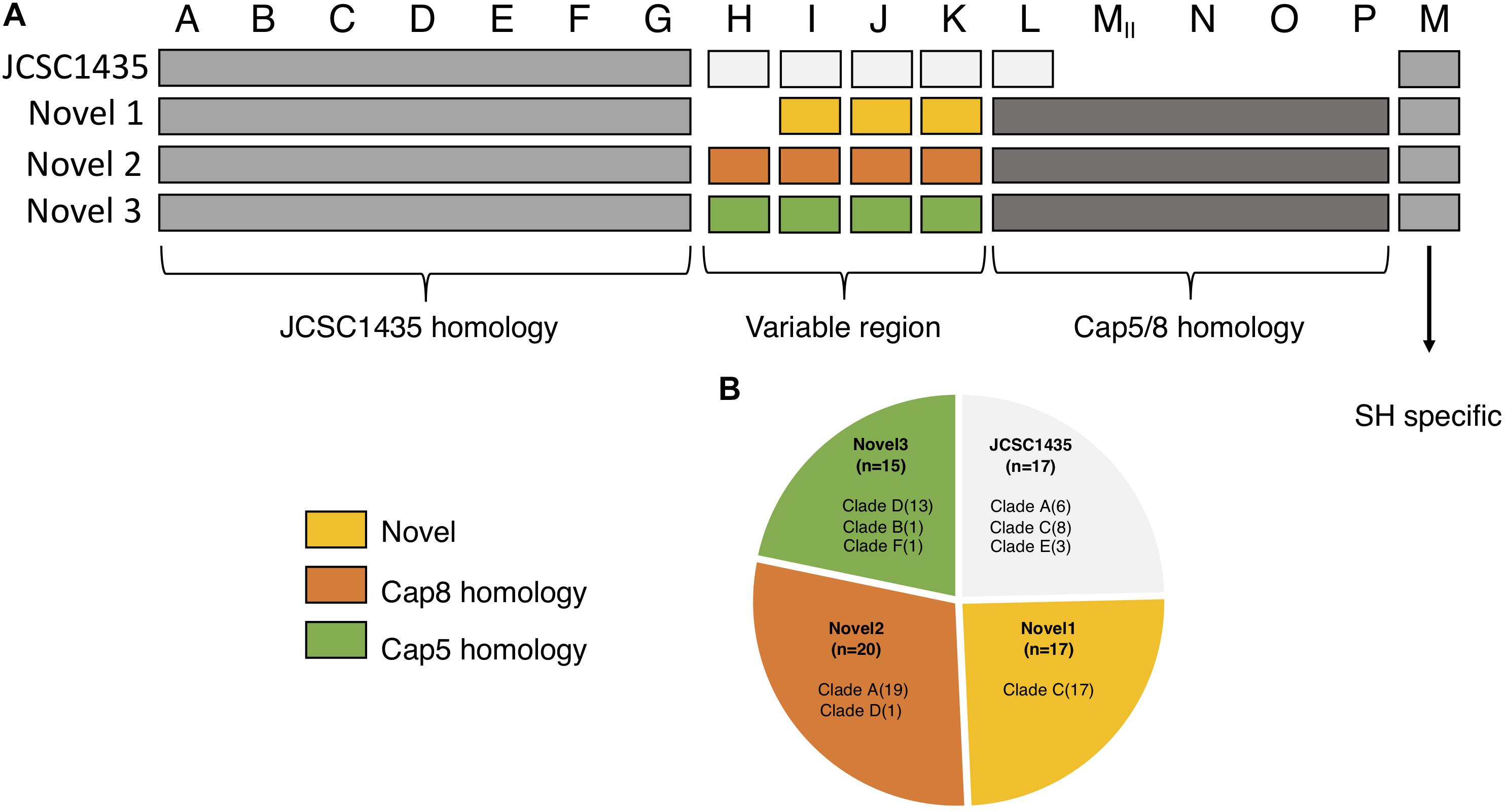
Figure 4. Organization of the different polysaccharide capsules (CP) operons identified in our collection. (A) The three novel varies in their homology to the CP in JCSC1435. capA-G is homologs among all isolates with identified capsule operon. The region capH-K is what separates the four types from one another; novel 1 with no known homology, novel 2 shows homology to S. aureus cap8 and novel 3 with homology to S. aureus cap5. The region capL-P is homologs to S. aureus and is found in all three novel types, a region absent in JSCS1435. (B) Pie chart showing the distribution of all identified CP operons in this study.
Genes and MGE Significantly Different Between Clinical and Commensal Isolates
In order to identify a specific signature in the gene repertoire of the clinical isolates important for hospital adaptation, a pan-GWAS analysis was performed on the accessory genome. 1887 predicted genes were statistically different between the two groups (p < 0.05), and the genes found most enriched for each group are listed in Tables 1, 2. As the population of S. haemolyticus is diverse, the gene sequence of genes with the same function varied significantly between the isolates, resulting in several orthologs clusters for one gene. Two enzymes in the folate pathway, dihydropteroate synthase (folP) and dihydroneopterin aldolase (folB) are a representative example of this. One version was only present in clinical isolates (96/123 isolates, 78%), and not found in any commensal isolates. A different version was found in all 46 commensal isolates, and 27/123 (22%) of clinical isolates. The majority of the clinical isolates of the latter group belonged to the commensal clade D and F (19 isolates), with 8 isolates scattered amongst clade A, B and E. Extracting these CDSs from each isolate and aligning them displays conserved differences between the genes in the two groups.
The most prevalent genes with a known function enriched in the clinical isolates included several ARGs (Table 1). The most prevalent genes with a known function enriched in the commensal isolates included several genes involved in metal control, transposases, and generally different versions of genes found in clinical isolates (Table 4). Using scores including one or more ARGs and phenotypic biofilm formation resulted in ROC curves with area under the curve (AUC) values >0.9, indicating a high discriminatory capacity to differentiate between clinical and commensal isolates (Figure 5).
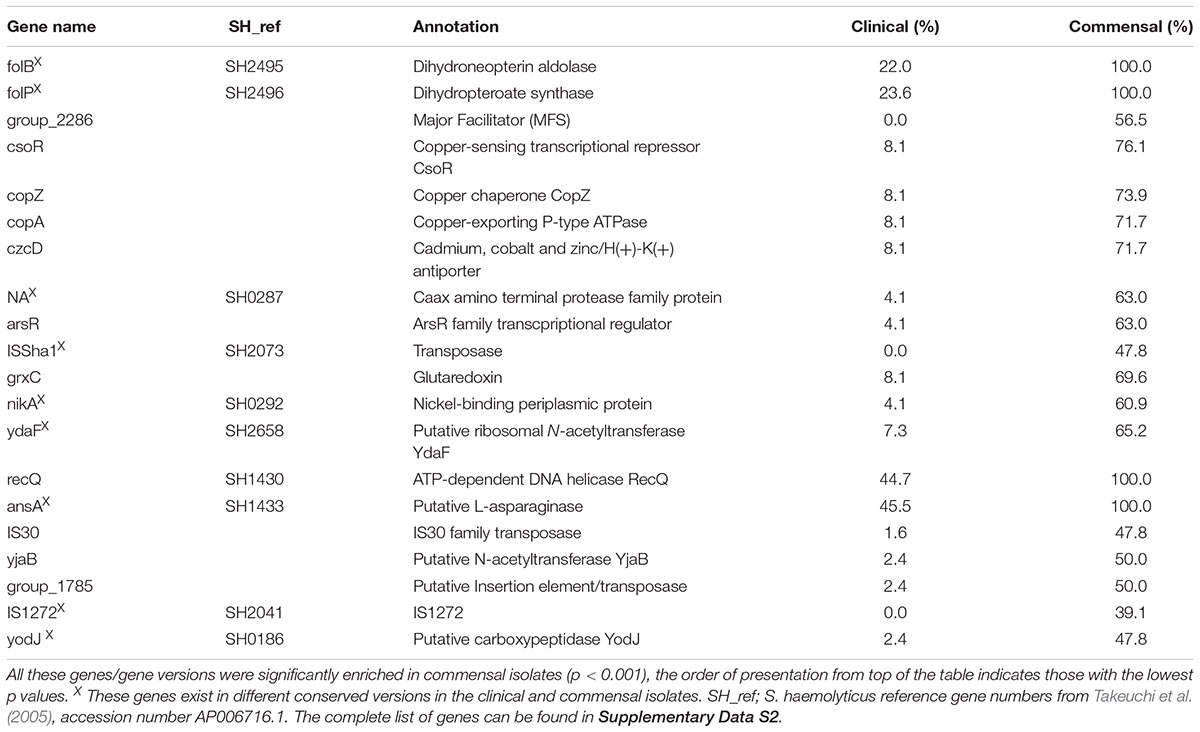
Table 4. Genes and gene versions enriched in 46 commensal isolates, sorted by significance.
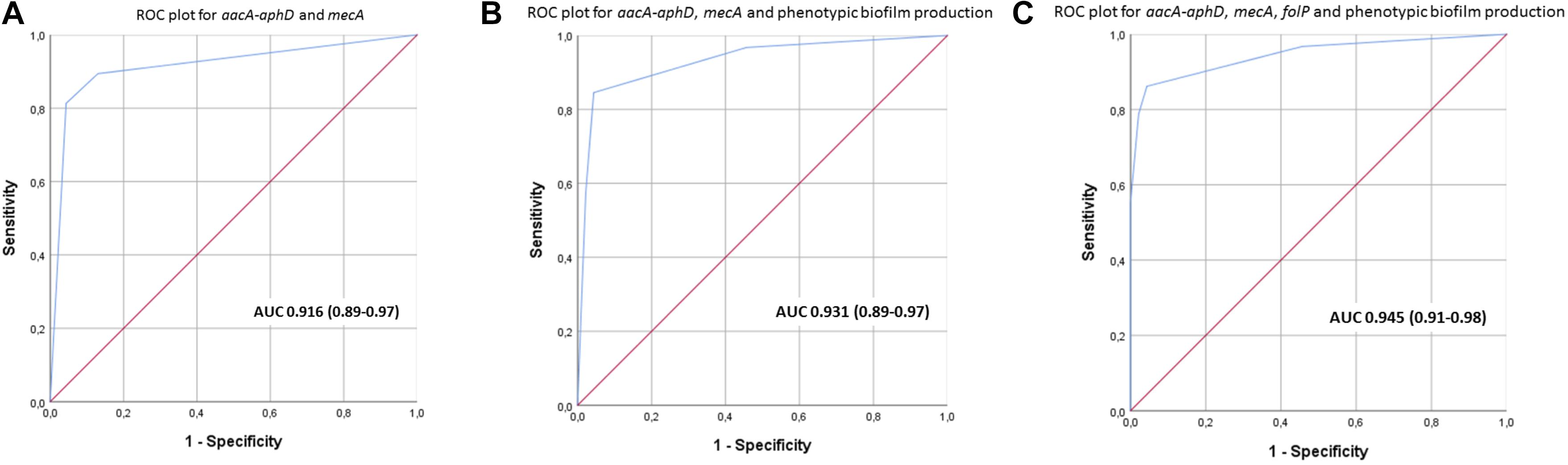
Figure 5. (A–C) Receiver operating characteristics (ROC) curves with area under curve (95% confidence interval) for scores using different combinations of aacA-aphD, mecA, folP and phenotypic biofilm production in order to discriminate between clinical and commensal isolates.
Discussion
There is a significant lack of knowledge about S. haemolyticus pathogenicity. In the present comparative genome study of clinical and commensal isolates we identified several genetic determinants and genotypes associated with the pathogenicity and the success of S. haemolyticus in the hospital environment. Resistance to commonly used antimicrobial agents and disinfectants, in addition to genes encoding proteins involved in adhesion and human immune defense escape predominates in the clinical isolates.
Population Structure – Distinct Signatures of Clinical and Commensal Isolates
The establishment of hospital adapted S. haemolyticus clones was previously reported by our group (Cavanagh et al., 2014). In the current study the majority of the clinical and commensal isolates form distinct phylogenetic groups when phylogeny is reconstructed based on SNPs in the core genome. In clades where clinical isolates are grouped with commensal isolates, they display a signature more similar to the commensals. In S. epidermidis there has been more research on phylogenetic relationship between clinical isolates, but data reported are conflicting. Conlan et al. (2012) showed that commensal and invasive isolates were grouped as distinct groups. In contrast, Méric et al. (2018) recently demonstrated that pathogenic clones of S. epidermidis can arise from various commensal backgrounds. This was shown by the absence of specific disease associated clades in a comparative study of invasive and commensal isolates. Staphylococcus haemolyticus and S. epidermidis are the two most recovered CoNS species from infection. Abundance of S. epidermidis on skin enhances the probability of contamination of indwelling devices. While S. haemolyticus also is a skin commensal, the population structure observed among our isolates suggests that specific persistent hospital adapted clones of S. haemolyticus are the major sources of infections, and that skin contamination is less likely (Tognetti et al., 2012; Kaspar et al., 2016; Byrd et al., 2018).
Surveillance and molecular typing are important in order to monitor the epidemiology of established and developing S. haemolyticus hospital clones. The availability of online resources, such as PubMLST4, Enterobase5, and BacWGSTdb6, for bacterial typing offers rapid classification and source tracing, which is increasingly important in a globalized community (Ruan and Feng, 2016; Alikhan et al., 2018; Jolley et al., 2018; Ruan et al., 2019). Currently, it is possible to investigate S. haemolyticus epidemiology using the traditional MLST database7, but extended MLST schemes are unavailable. As S. haemolyticus is an emerging nosocomial pathogen with extended antibiotic resistance, an online resource offering rapid typing and phylogenetic relatedness linked to antibiotic resistance genes and clinical data would be very useful.
The accessory gene repertoire of the clinical isolates is characterized by high prevalence of ARGs. This was expected as S. haemolyticus infections are commonly caused by MDR isolates (Barros et al., 2012; Chang et al., 2018). Creating a score with ARGs and phenotypic biofilm formation clearly separated the invasive from the commensal isolates. This can be useful in clinical microbiology when conveying results of a positive S. haemolyticus culture back to the treating clinician. Biofilm forming S. haemolyticus isolates that are resistant to oxacillin (mecA) and aminoglycosides (aacA-aphD) are most likely invasive isolates whereas absence of these traits strongly indicates a commensal isolate.
The interpretation of the folB/folP results is complex. We found distinct conserved differences in folB and folP clearly separating clinical from commensal S. haemolyticus isolates. In S. aureus five mutations in folP have been shown to directly contribute to sulfonamide resistance (Griffith et al., 2018). Combination treatment of trimetoprim-sulfamethoxazole has been used in infections caused by methicillin resistant staphylococci, and the rates of trimetoprim-sulfamethoxazole resistance is increasing (Khamash et al., 2018). Neither of the two versions of folP that we identified in our S. haemolyticus collection had the mutations shown to increase resistance to sulfonamides in S. aureus. However, there is substantial sequence variations between the S. haemolyticus and S. aureus folB and folP genes, and other mutations could also lead to decreased susceptibility. The importance of the folP versions in S. haemolyticus needs further evaluation, and extensive testing on sulfonamide resistance is needed.
In the successful epidemic S. aureus EMRSA-15, mecA acquisition, mutations in gyrA and glrA; resulting in fluoroquinolone resistance, and uptake of plasmids carrying ermC, was shown to be strong drivers of evolution (Holden et al., 2013). Uptake of plasmid pSHaeB carrying ermC was only observed for the clinical S. haemolyticus isolates, and has also been shown to be prevalent in several clinically important staphylococcal species (Águila-Arcos et al., 2017). The observed population structure in S. haemolyticus appears more similar to S. aureus than to S. epidermidis. In S. aureus defined pathogenic clones have adapted to the specific challenges of the hospital environment (Rasmussen et al., 2013), and our data support that similar phenomenon occurs in S. haemolyticus.
IS256, found in the majority of clinical S. haemolyticus isolates, has also been reported to be more common in nosocomial isolates of S. epidermidis, and has been associated with gentamicin resistance due to co-localization on the transposon Tn4001, in addition to biofilm formation (Kozitskaya et al., 2004; Conlan et al., 2012; Sharma et al., 2018). IS256 can shape the genome by affecting gene expression. In several successful virulent MRSA STs, such as ST 247, the presence of IS256 has been linked to increased virulence, vancomycin resistance and formation of small-colony variants (Kleinert et al., 2017). The opposite was demonstrated in Brazilian MRSA ST 239 isolates where IS256 was integrated near global regulatory genes (agr and mgr) likely leading to rapid changes of bacterial traits, resulting in reduced virulence (Botelho et al., 2019). IS256 has been suggested as a marker for molecular typing and identification of nosocomial, invasive S. epidermidis isolates (Gu et al., 2005; Yao et al., 2005; Montanaro et al., 2007). There is also some evidence indicating that IS256 is not only enriched within invasive isolates, but also more prevalent in isolates with poor treatment outcome (Post et al., 2017).
The transposon Tn552/IS481, was also predominantly identified in clinical S. haemolyticus isolates, and this MGE often carry antiseptic resistance and beta lactamase genes (Anthonisen et al., 2002). The pandemic S. aureus ST239 has acquired resistance to multiple antibiotics and antiseptics, among them qac A and B (Chang et al., 2017). Qac proteins are efflux pumps that protect bacteria not only from a variety of toxic substances but also from fluorquinolones and beta-lactams (Prag et al., 2014; Wassenaar et al., 2015; Taheri et al., 2016). In our study, qacA was detected more often in clinical isolates, while qacB was almost exclusive to the commensal isolates. qacA has been reported to have a broader spectrum of resistance than qacB, and this might be the reason why we found qacA more often in the clinical samples (in 65% clinical isolates and 39% commensal isolates), obtained from an environment with higher antibiotic pressure (Wassenaar et al., 2015).
The Adhesion Protein SraP Is Highly Prevalent in Clinical Isolate
In the clinical isolates we commonly identified a homolog (SH0326) of the serine-rich repeat glycoproteins SraP of S. aureus and the accessory sec system, dedicated for the export of SraP. SraP belongs to a highly conserved family of serine-rich surface glycoproteins of Gram-positive bacteria. Expression of SraP has been linked to adhesion to different types of cells, including human platelets and is associated with infective endocarditis (Siboo et al., 2005; Yang et al., 2014; Bensing et al., 2016). The higher prevalence of the aSec system and sraP is likely advantageous for the clinical S. haemolyticus isolates as they are mainly isolated from blood. In S. epidermidis, genome signatures linked to pathogenicity identified the aSec gene asp3 as one of four strong virulence predictors. In their full list of pathogenicity-associated signature genes sraP was also identified (Méric et al., 2018).
A Novel Capsular Polysaccharide Operon
The polysaccharide capsule (CP) in S. haemolyticus has been shown to play a role in the protection against uptake and killing by human neutrophils (Flahaut et al., 2008). In S. aureus it was demonstrated to modulate adherence to endothelial surfaces in vitro, and to promote bacterial colonization and persistence on mucosal surfaces in animal models (Riordan and Lee, 2004).
In this study we found four different capsule operons of which only the first type has been described in S. haemolyticus JCSC1435 previously (Takeuchi et al., 2005). Two of the three novel CP (capNOP) structures have homology to the S. aureus cap 5/8 genes, and identification of capO in S. haemolyticus has not previously been reported. The third type has no homology to any previously described cap version. The new CP versions in S. haemolyticus have a variable region, capH-K which is unique to each of the four versions with little or no homology between them. The GC content of this variable region is also significantly lower than the surrounding cap genes, indicating the variable region was acquired by HGT. Both CP genes and SCC are located in the oriC environ, a region of high genomic flexibility, allowing staphylococci to maintain or acquire genes needed for the adaption to on-going environmental changes (Hiramatsu et al., 2014). The new CP structures were mainly found in isolates belonging to two clinical clades. The four different CP types were shown to be clade specific, which has also been shown for S. aureus. In S. aureus 13 putative capsular operons have been reported, but only CP type 5 and 8 have been associated with disease (Mohamed et al., 2019), thus the three novel CP types now identified in S. haemolyticus need to be further investigated for their role in virulence.
Taken together, our findings point toward HGT as a driving force in S. haemolyticus evolution and in response to the selective pressure of broad-spectrum antibiotics used in hospitals.
The S. haemolyticus Pan-Genome Distribution Reflects the High Variation of Commensal Isolates
The pan-genome analysis reflected a relatively stable core genome which is comparable to what is observed in S. epidermidis, S. aureus and Staphylococcus lugdunensis. In S. haemolyticus the core genome is slightly smaller which could be explained by the higher number of unique genes in the commensal isolates (Méric et al., 2015, 2018; Argemi et al., 2018). Similar to S. haemolyticus, S. aureus and S. epidermidis also have an open pan-genome in contrast to S. lugdunensis (Bosi et al., 2016; Argemi et al., 2018). However, the pan-genome accumulation curves for S. epidermidis and S. aureus are not as steep as we see for S. haemolyticus (Conlan et al., 2012; Méric et al., 2015; Sharma et al., 2018). The relatively large pan-genome of S. haemolyticus is, at least in part, due to the variation seen in genes of the same function, resulting in two or more clusters for one gene. The most abundant categories in the core genome were genes involved in housekeeping functions, like transcription and translation, and different metabolism categories, a result similar to reports on S. aureus and S. epidermidis (Bosi et al., 2016; Sharma et al., 2018). The large repertoire of genes in the accessory genome confer advantages in highly variable environmental conditions.
Strengths and Limitation With the Study
This study is the largest comparative study of clinical and commensal S. haemolyticus isolates to date, and the clinical isolates have wide spatial and temporal distribution. We have used state of the art technology to analyze pathogenicity traits and the genetic signatures of clinical and invasive isolates. The study also has limitations. First, the majority of the commensal isolates are from one geographic location, and more recently collected than the invasive isolates. Still, the very diverse commensal population indicates that commensal isolates may lack the pathogenic traits of hospital-adapted clones. Second, the invasive isolates are collected over a wide time-span and some of the isolates originated from infections two decades ago. Still we see a very clear phylogenetic clustering, and invasive isolates from different geographical origin cluster together. We believe this clearly indicates the emergence of disease-causing isolates with a homogenous genetic signature. Third, it would have been of interest to collect skin samples from hospitalized patients without infection, in order to see whether colonization of more pathogenic isolates emerges after hospitalization. We did not have the opportunity to collect such samples in this study, but we will pursue this in the future. Finally, we do not know whether some of the clonal groups are still circulating or might have been replaced by new clones. This phenomenon was observed in S. aureus where the pandemic clone ST239-MRSA-III that circulated for several decades (Monecke et al., 2018) was replaced with clones with increased fitness (Hsu et al., 2015). Still, we believe that our current study has identified important features of hospital adapted S. haemolyticus clones. Future sampling of both commensal and invasive isolates is needed in order to monitor the evolution of S. haemolyticus.
Conclusion
In this study we have gained a deeper understanding of the mechanism of adaption of S. haemolyticus in the hospital environment by phylogenetic and pan-genome analysis. We have found a clear segregation of isolates of commensal origin, and specific genetic signatures distinguishing the clinical isolates from the commensal isolates. It is highly likely that the widespread use of antimicrobial agents has promoted the development of MDR clones of S. haemolyticus persisting in the hospital environment, and that these isolates have responded through acquisition of mobile genetic elements or beneficial point mutations and rearrangements in surface associated genes. Defining pathogen-associated signatures is an important step in infection control. Continuous surveillance and molecular typing are important in order to monitor the spread and evolution of the S. haemolyticus hospital clones in the future.
Data Availability
The datasets generated for this study can be found in the European Nuclotide Archive (www.ebi.ac.uk/ena) study accession no. ERP000943 for the clinical isolates and ERP114853 for the commensal isolates. The DNA accession number for the individual sequences are found in Supplementary Data S2.
Author Contributions
MP organized and performed the bioinformatic analysis, took part in the study design, wrote the first version of the manuscript, and revised the manuscript. EH participated in bioinformatical analyzes and revised the manuscript. JC and CK conceptualized and designed the study and revised the final manuscript. All authors approved the final manuscript as submitted and agreed to be accountable for all aspects of the work.
Funding
MP was funded by UiT The Arctic University of North Norway. The publication charges for this article have been funded by a grant from the publication fund of UiT The Arctic University of Norway.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Elizabeth Aarag Fredheim and Runa Wolden for sampling of volunteers for the collection of commensal S. haemolyticus isolates.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02096/full#supplementary-material
Abbreviations
ARG, antibiotic resistance genes; aSec, accessory secretion system; CARD, comprehensive antibiotic resistance database; CDS, coding sequence; COG, cluster of orthologous groups; CoNS, coagulase negative staphylococci; CP, capsule polysaccharide; ENA, European Nucleotide Archive; IS, insertion sequences; MDR, multi drug resistant; Orf, open reading frame; WGS, whole genome sequence.
Footnotes
- ^ http://www.eucast.org/
- ^ www.ebi.ac.uk/ena
- ^ https://itol.embl.de/
- ^ http://pubmlst.org
- ^ http://enterobase.warwick.ac.uk
- ^ http://bacdb.org/BacWGSTdb
- ^ https://pubmlst.org/shaemolyticus/
References
Águila-Arcos, S., Álvarez-Rodríguez, I., Garaiyurrebaso, O., Garbisu, C., Grohmann, E., and Alkorta, I. (2017). Biofilm-forming clinical Staphylococcus isolates harbor horizontal transfer and antibiotic resistance genes. Front. Microbiol. 8:2018. doi: 10.3389/fmicb.2017.02018
Alikhan, N. F., Zhou, Z., Sergeant, M. J., and Achtman, M. (2018). A genomic overview of the population structure of Salmonella. PLoS Genet. 14:e1007261. doi: 10.1371/journal.pgen.1007261
Anthonisen, I.-L., Sunde, M., Steinum, T. M., Sidhu, M. S., and Sørum, H. (2002). Organization of the antiseptic resistance gene qacA and Tn552-related beta-lactamase genes in multidrug- resistant Staphylococcus haemolyticus strains of animal and human origins. Antimicrob. Agents Chemother. 46, 3606–3612. doi: 10.1128/AAC.46.11.3606
Argemi, X., Matelska, D., Ginalski, K., Riegel, P., Hansmann, Y., Bloom, J., et al. (2018). Comparative genomic analysis of Staphylococcus lugdunensis shows a closed pan-genome and multiple barriers to horizontal gene transfer. BMC Genomics 19:621. doi: 10.1186/s12864-018-4978-4971
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bankevich, A., Nurk, S., Antipov, D., Gurevich, A. A., Dvorkin, M., Kulikov, A. S., et al. (2012). SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. doi: 10.1089/cmb.2012.0021
Barros, E. M., Ceotto, H., Bastos, M. C. F., Dos Santos, K. R. N., and Giambiagi-deMarval, M. (2012). Staphylococcus haemolyticus as an important hospital pathogen and carrier of methicillin resistance genes. J. Clin. Microbiol. 50, 166–168. doi: 10.1128/JCM.05563-5511
Bensing, B. A., Loukachevitch, L. V., Mcculloch, K. M., Yu, H., Vann, K. R., Wawrzak, Z., et al. (2016). Structural basis for sialoglycan binding by the Streptococcus sanguinis SrpA adhesin. J. Biol. Chem. 291, 7230–7240. doi: 10.1074/jbc.M115.701425
Bosi, E., Monk, J. M., Aziz, R. K., Fondi, M., Nizet, V., and Palsson, B. Ø. (2016). Comparative genome-scale modelling of Staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. U.S.A. 113, E3801–E3809. doi: 10.1073/pnas.1523199113
Botelho, A. M. N., Cerqueira e Costa, M. O., Moustafa, A. M., Beltrame, C. O., Ferreira, F. A., Côrtes, M. F., et al. (2019). Local diversification of methicillin-resistant Staphylococcus aureus ST239 in South America after its rapid worldwide dissemination. Front. Microbiol. 10:82. doi: 10.3389/fmicb.2019.00082
Brynildsrud, O., Bohlin, J., Scheffer, L., and Eldholm, V. (2016). Rapid scoring of genes in microbial pan-genome-wide association studies with Scoary. Genome Biol. 17:238. doi: 10.1186/s13059-016-1108-1108
Byrd, A. L., Belkaid, Y., and Segre, J. A. (2018). The human skin microbiome. Nat. Rev. Microbiol. 16, 143–155. doi: 10.1038/nrmicro.2017.157
Carattoli, A., Zankari, E., Garciá-Fernández, A., Larsen, M. V., Lund, O., Villa, L., et al. (2014). In Silico detection and typing of plasmids using plasmidfinder and plasmid multilocus sequence typing. Antimicrob. Agents Chemother. 58, 3895–3903. doi: 10.1128/AAC.02412-14
Cavanagh, J. P., Hjerde, E., Holden, M. T. G., Kahlke, T., Klingenberg, C., Flaegstad, T., et al. (2014). Whole-genome sequencing reveals clonal expansion of multiresistant Staphylococcus haemolyticus in European hospitals. J. Antimicrob. Chemother. 69, 2920–2927. doi: 10.1093/jac/dku271
Cavanagh, J. P., Wolden, R., Heise, P., Esaiassen, E., Klingenberg, C., and Aarag Fredheim, E. G. (2016). Antimicrobial susceptibility and body site distribution of community isolates of coagulase-negative staphylococci. APMIS 124, 973–978. doi: 10.1111/apm.12591
Chang, P. H., Liu, T. P., Huang, P. Y., Lin, S. Y., Lin, J. F., Yeh, C. F., et al. (2018). Clinical features, outcomes, and molecular characteristics of an outbreak of Staphylococcus haemolyticus infection, among a mass-burn casualty patient group, in a tertiary center in northern Taiwan. J. Microbiol. Immunol. Infect 51, 847–855. doi: 10.1016/j.jmii.2018.07.004
Chang, S.-C., Lee, M.-H., Yeh, C.-F., Liu, T.-P., Lin, J.-F., Ho, C.-M., et al. (2017). Characterization of two novel variants of staphylococcal cassette chromosome mec elements in oxacillin-resistant Staphylococcus lugdunensis. J. Antimicrob. Chemother. 72, 3258–3262. doi: 10.1093/jac/dkx291
Chen, L., Zheng, D., Liu, B., Yang, J., and Jin, Q. (2016). VFDB 2016: hierarchical and refined dataset for big data analysis - 10 years on. Nucleic Acids Res. 44, D694–D697. doi: 10.1093/nar/gkv1239
Conlan, S., Mijares, L. A., Becker, J., Blakesley, R. W., Bouffard, G. G., Brooks, S., et al. (2012). Staphylococcus epidermidis pan-genome sequence analysis reveals diversity of skin commensal and hospital infection-associated isolates. Genome Biol. 13:R64. doi: 10.1186/gb-2012-13-7-r64
Da, F., Joo, H.-S., Cheung, G. Y. C., Villaruz, A. E., Rohde, H., Luo, X., et al. (2017). Phenol-soluble modulin toxins of Staphylococcus haemolyticus. Front. Cell. Infect. Microbiol. 7:206. doi: 10.3389/fcimb.2017.00206
Flahaut, S., Vinogradov, E., Kelley, K. A., Brennan, S., Hiramatsu, K., and Lee, J. C. (2008). Structural and biological characterization of a capsular polysaccharide produced by Staphylococcus haemolyticus. J. Bacteriol. 190, 1649–1657. doi: 10.1128/JB.01648-1647
Fredheim, E. G. A., Klingenberg, C., Rohde, H., Frankenberger, S., Gaustad, P., Flaegstad, T., et al. (2009). Biofilm formation by Staphylococcus haemolyticus. J. Clin. Microbiol. 47, 1172–1180. doi: 10.1128/JCM.01891-1898
Gardner, S. N., Slezak, T., and Hall, B. G. (2015). kSNP3.0: SNP detection and phylogenetic analysis of genomes without genome alignment or reference genome. Bioinformatics 31, 2877–2878. doi: 10.1093/bioinformatics/btv271
Giormezis, N., Kolonitsiou, F., Foka, A., Drougka, E., Liakopoulos, A., Makri, A., et al. (2014). Coagulase-negative staphylococcal bloodstream and prosthetic-device-associated infections: the role of biofilm formation and distribution of adhesin and toxin genes. J. Med. Microbiol. 63, 1500–1508. doi: 10.1099/jmm.0.075259-0
Griffith, E. C., Wallace, M. J., Wu, Y., Kumar, G., Gajewski, S., Jackson, P., et al. (2018). The structural and functional basis for recurring sulfa drug resistance mutations in Staphylococcus aureus dihydropteroate synthase. Front. Microbiol. 9:1369. doi: 10.3389/fmicb.2018.01369
Gu, J., Li, H., Li, M., Vuong, C., Otto, M., Wen, Y., et al. (2005). Bacterial insertion sequence IS256 as a potential molecular marker to discriminate invasive strains from commensal strains of Staphylococcus epidermidis. J. Hosp. Infect 61, 342–348. doi: 10.1016/j.jhin.2005.04.017
Hiramatsu, K., Katayama, Y., Matsuo, M., Sasaki, T., Morimoto, Y., Sekiguchi, A., et al. (2014). Multi-drug-resistant Staphylococcus aureus and future chemotherapy. J. Infect. Chemother. 20, 593–601. doi: 10.1016/j.jiac.2014.08.001
Holden, M. T. G., Hsu, L., Kurt, K., Weinert, L. A., Mather, A. E., Harris, S. R., et al. (2013). A genomic portrait of the emergences, evolution, and global spread of methicillin-resistant Staphylococcus aureus pandemic. Genome Res. 23, 653–664. doi: 10.1101/gr.147710.112
Hope, R., Livermore, D. M., Brick, G., Lillie, M., and Reynolds, R. (2008). Non-susceptibility trends among staphylococci from bacteraemias in the UK and Ireland, 2001-06. J. Antimicrob. Chemother. 62, ii65–ii74. doi: 10.1093/jac/dkn353
Hsu, L.-Y., Harris, S. R., Chlebowicz, M. A., Lindsay, J. A., Koh, T.-H., Krishnan, P., et al. (2015). Evolutionary dynamics of methicillin-resistant Staphylococcus aureus within a healthcare system. Genome Biol. 16, 1–13. doi: 10.1186/s13059-015-0643-z
Huerta-Cepas, J., Szklarczyk, D., Forslund, K., Cook, H., Heller, D., Walter, M. C., et al. (2016). EGGNOG 4.5: a hierarchical orthology framework with improved functional annotations for eukaryotic, prokaryotic and viral sequences. Nucleic Acids Res. 44, D286–D293. doi: 10.1093/nar/gkv1248
Jia, B., Raphenya, A. R., Alcock, B., Waglechner, N., Guo, P., Tsang, K. K., et al. (2017). CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 45, D566–D573. doi: 10.1093/nar/gkw1004
Jolley, K. A., Bray, J. E., and Maiden, M. C. J. (2018). Open-access bacterial population genomics: BIGSdb software, the PubMLST.org website and their applications [version 1; referees: 2 approved]. Wellcome Open Res. 3, 1–20. doi: 10.12688/wellcomeopenres.14826.1
Kaspar, U., Kriegeskorte, A., Schubert, T., Peters, G., Rudack, C., Pieper, D. H., et al. (2016). The culturome of the human nose habitats reveals individual bacterial fingerprint patterns. Environ. Microbiol. 18, 2130–2142. doi: 10.1111/1462-2920.12891
Khamash, D. F., Voskertchian, A., Tamma, P. D., Akinboyo, I. C., Carroll, K. C., and Milstone, A. M. (2018). Increasing clindamycin and trimethoprim-sulfamethoxazole resistance in pediatric Staphylococcus aureus infections. J. Pediatric Infect. Dis. Soc. doi: 10.1093/jpids/piy062 [Epub ahead of print].
Kleinert, F., Kallies, R., Hort, M., Zweynert, A., Szekat, C., Nagel, M., et al. (2017). Influence of IS 256 on genome variability and formation of small-colony variants in Staphylococcus aureus. Antimicrob. Agents Chemother. 61:e00144-17. doi: 10.1128/AAC.00144-117
Kozitskaya, S., Cho, S.-H., Dietrich, K., Marre, R., Naber, K., and Ziebuhr, W. (2004). The bacterial insertion sequence element IS256 occurs preferentially in nosocomial Staphylococcus epidermidis isolates: association with biofilm formation and resistance to aminoglycosides. Infect. Immun. 72, 1210–1215. doi: 10.1128/IAI.72.2.1210-1215.2004
Kresken, M., Becker, K., Seifert, H., Leitner, E., Körber-Irrgang, B., Von Eiff, C., et al. (2011). Resistance trends and in vitro activity of tigecycline and 17 other antimicrobial agents against Gram-positive and Gram-negative organisms, including multidrug-resistant pathogens, in Germany. Eur. J. Clin. Microbiol. Infect. Dis. 30, 1095–1103. doi: 10.1007/s10096-011-1197-y
Méric, G., Mageiros, L., Pensar, J., Laabei, M., Yahara, K., Pascoe, B., et al. (2018). Disease-associated genotypes of the commensal skin bacterium Staphylococcus epidermidis. Nat. Commun. 9:5034. doi: 10.1038/s41467-018-07368-7367
Méric, G., Miragaia, M., de Been, M., Yahara, K., Pascoe, B., Mageiros, L., et al. (2015). Ecological overlap and horizontal gene transfer in Staphylococcus aureus and Staphylococcus epidermidis. Genome Biol. Evol. 7, 1313–1328. doi: 10.1093/gbe/evv066
Mohamed, N., Timofeyeva, Y., Jamrozy, D., Rojas, E., Hao, L., Silmon de Monerri, N. C., et al. (2019). Molecular epidemiology and expression of capsular polysaccharides in Staphylococcus aureus clinical isolates in the United States. PLoS One 14:e0208356. doi: 10.1371/journal.pone.0208356
Monecke, S., Slickers, P., Gawlik, D., Müller, E., Reissig, A., Ruppelt-Lorz, A., et al. (2018). Molecular typing of ST239-MRSA-III from diverse geographic locations and the evolution of the SCCmec III element during its intercontinental spread. Front. Microbiol. 9:1436. doi: 10.3389/fmicb.2018.01436
Montanaro, L., Campoccia, D., Pirini, V., Ravaioli, S., Otto, M., and Aricola, C. R. (2007). Antibiotic multiresistance strictly associated with IS256 and ica genes in Staphylococcus epidermidis strains from implant orthopedic infections. J. Biomed. Mater. Res. 83A, 813–818. doi: 10.1002/jbm.a.31399
Nanoukon, C., Argemi, X., Sogbo, F., Orekan, J., Keller, D., Affolabi, D., et al. (2017). Pathogenic features of clinically significant coagulase-negative staphylococci in hospital and community infections in Benin. Int. J. Med. Microbiol. 307, 75–82. doi: 10.1016/j.ijmm.2016.11.001
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Pereira, P. M. A., Binatti, V. B., Sued, B. P. R., Ramos, J. N., Peixoto, R. S., Simões, C., et al. (2014). Staphylococcus haemolyticus disseminated among neonates with bacteremia in a neonatal intensive care unit in Rio de Janeiro. Brazil. Diagn. Microbiol. Infect. Dis. 78, 85–92. doi: 10.1016/j.diagmicrobio.2013.06.026
Post, V., Harris, L. G., Morgenstern, M., Mageiros, L., Hitchings, M. D., Méric, G., et al. (2017). Comparative genomics study of Staphylococcus epidermidis isolates from orthopedic-device-related infections correlated with patient outcome. J. Clin. Microbiol. 55, 3089–3103. doi: 10.1128/JCM.00881-17
Prag, G., Falk-Brynhildsen, K., Jacobsson, S., Hellmark, B., Unemo, M., and Söderquist, B. (2014). Decreased susceptibility to chlorhexidine and prevalence of disinfectant resistance genes among clinical isolates of Staphylococcus epidermidis. APMIS 122, 961–967. doi: 10.1111/apm.12239
Rasmussen, G., Monecke, S., Ehricht, R., and Söderquist, B. (2013). Prevalence of clonal complexes and virulence genes among commensal and invasive Staphylococcus aureus isolates in Sweden. PLoS One 8:e7747. doi: 10.1371/journal.pone.0077477
Riordan, K. O., and Lee, J. C. (2004). Staphylococcus aureus capsular polysaccharides. Clin. Microbiol. Rev. 17, 218–234. doi: 10.1128/CMR.17.1.218
Rohde, H., Kalitzky, M., Kröger, N., Scherpe, S., Horstkotte, M. A., Knobloch, J. K. M., et al. (2004). Detection of virulence-associated genes not useful for discriminating between invasive and commensal Staphylococcus epidermidis strains from a bone marrow transplant unit. J. Clin. Microbiol. 42, 5614–5619. doi: 10.1128/JCM.42.12.5614-5619.2004
Ruan, Z., and Feng, Y. (2016). BacWGSTdb, a database for genotyping and source tracking bacterial pathogens. Nucleic Acids Res. 44, D682–D687. doi: 10.1093/nar/gkv1004
Ruan, Z., Yu, Y., and Feng, Y. (2019). The global dissemination of bacterial infections necessitates the study of reverse genomic epidemiology. Brief. Bioinform. 00, 1–10. doi: 10.1093/bib/bbz010
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Sharma, S., Chaudhry, V., Kumar, S., and Patil, P. B. (2018). Phylogenomic based comparative studies on Indian and American commensal Staphylococcus epidermidis isolates. Front. Microbiol 9:333. doi: 10.3389/fmicb.2018.00333
Siboo, I. R., Chambers, H. F., and Sullam, P. M. (2005). Role of SraP, a serine-rich surface protein of Staphylococcus aureus, in binding to human platelets. Infect. Immun. 73, 2273–2280. doi: 10.1128/IAI.73.4.2273
Taheri, N., Ardebili, A., Amouzandeh-Nobaveh, A., and Ghaznavi-Rad, E. (2016). Frequency of antiseptic resistance among Staphylococcus aureus and coagulase-negative staphylococci isolated from a university hospital in Central Iran. Oman Med. J. 31, 426–432. doi: 10.5001/omj.2016.86
Takeuchi, F., Watanabe, S., Baba, T., Yuzawa, H., Morimoto, Y., Kuroda, M., et al. (2005). Whole-genome sequencing of Staphylococcus haemolyticus uncovers the extreme plasticity of its genome and the evolution of human-colonizing staphylococcal species. J. Bacteriol. 187, 7292–7308. doi: 10.1128/JB.187.21.7292
Teeraputon, S., Santanirand, P., Wongchai, T., Songjang, W., Lapsomthob, N., Jaikrasun, D., et al. (2017). Prevalence of methicillin resistance and macrolide–lincosamide–streptogramin B resistance in Staphylococcus haemolyticus among clinical strains at a tertiary-care hospital in Thailand. New Microbes New Infect. 19, 28–33. doi: 10.1016/j.nmni.2017.05.007
Tettelin, H., Masignani, V., Cieslewicz, M. J., Donati, C., Medini, D., Ward, N. L., et al. (2005). Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial “pan-genome.”. Proc. Natl. Acad. Sci. U.S.A. 102, 13950–13955. doi: 10.1073/pnas.0506758102
Tognetti, L., Martinelli, C., Berti, S., Hercogova, J., Lotti, T., Leoncini, F., et al. (2012). Bacterial skin and soft tissue infections: review of the epidemiology, microbiology, aetiopathogenesis and treatment. J. Eur. Acad. Dermatol. Venereol. 26, 931–941. doi: 10.1111/j.1468-3083.2011.04416.x
Varani, A. M., Siguier, P., Gourbeyre, E., Charneau, V., and Chandler, M. (2011). ISsaga is an ensemble of web-based methods for high throughput identification and semi-automatic annotation of insertion sequences in prokaryotic genomes. Genome Biol. 12:R30. doi: 10.1186/gb-2011-12-3-r30
Wassenaar, T. M., Ussery, D., Nielsen, L. N., and Ingmer, H. (2015). Review and phylogenetic analysis of qac genes that reduce susceptibility to quaternary ammonium compounds in Staphylococcus species. Eur. J. Microbiol. Immunol. 5, 44–61. doi: 10.1556/eujmi-d-14-00038
Yang, Y. H., Jiang, Y. L., Zhang, J., Wang, L., Bai, X. H., Zhang, S. J., et al. (2014). Structural insights into SraP-mediated Staphylococcus aureus adhesion to host cells. PLoS Pathog 10:e1004169. doi: 10.1371/journal.ppat.1004169
Keywords: Staphylococcus haemolyticus, pangenome, multidrug resistance, bacterial genomics, pathogenicity, antibiotic resistance genes
Citation: Pain M, Hjerde E, Klingenberg C and Cavanagh JP (2019) Comparative Genomic Analysis of Staphylococcus haemolyticus Reveals Key to Hospital Adaptation and Pathogenicity. Front. Microbiol. 10:2096. doi: 10.3389/fmicb.2019.02096
Received: 16 April 2019; Accepted: 26 August 2019;
Published: 10 September 2019.
Edited by:
David W. Ussery, University of Arkansas for Medical Sciences, United StatesReviewed by:
Zhi Ruan, Zhejiang University, ChinaRobin Anderson, Agricultural Research Service, United States Department of Agriculture, United States
Copyright © 2019 Pain, Hjerde, Klingenberg and Cavanagh. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jorunn Pauline Cavanagh, UGF1bGluZS5jYXZhbmFnaEB1aXQubm8=