 Yuki Matsushima1
Yuki Matsushima1 Fuminori Mizukoshi2
Fuminori Mizukoshi2 Naomi Sakon3
Naomi Sakon3 Yen Hai Doan4
Yen Hai Doan4 Yo Ueki5Yasutaka Ogawa6
Yo Ueki5Yasutaka Ogawa6 Takumi Motoya7
Takumi Motoya7 Hiroyuki Tsukagoshi8Noriko Nakamura9Naoki Shigemoto10Hideaki Yoshitomi11
Hiroyuki Tsukagoshi8Noriko Nakamura9Naoki Shigemoto10Hideaki Yoshitomi11 Reiko Okamoto-Nakagawa12Rieko Suzuki13Rika Tsutsui14Fumio Terasoma15Tomoko Takahashi16Kenji Sadamasu17
Reiko Okamoto-Nakagawa12Rieko Suzuki13Rika Tsutsui14Fumio Terasoma15Tomoko Takahashi16Kenji Sadamasu17 Hideaki Shimizu1Nobuhiko Okabe1
Hideaki Shimizu1Nobuhiko Okabe1 Koo Nagasawa18
Koo Nagasawa18 Jumpei Aso19,20
Jumpei Aso19,20 Haruyuki Ishii20Makoto Kuroda21
Haruyuki Ishii20Makoto Kuroda21 Akihide Ryo22
Akihide Ryo22 Kazuhiko Katayama23*
Kazuhiko Katayama23* Hirokazu Kimura19,22*
Hirokazu Kimura19,22*- 1Division of Virology, Kawasaki City Institute for Public Health, Kawasaki, Japan
- 2Department of Microbiology, Tochigi Prefectural Institute of Public Health and Environmental Science, Utsunomiya, Japan
- 3Department of Microbiology, Osaka Institute of Public Health, Osaka, Japan
- 4Department of Virology II, National Institute of Infectious Diseases, Musashimurayama, Japan
- 5Department of Microbiology, Miyagi Prefectural Institute of Public Health and Environment, Sendai, Japan
- 6Division of Virology, Saitama Institute of Public Health, Saitama, Japan
- 7Ibaraki Prefectural Institute of Public Health, Mito, Japan
- 8Gunma Prefectural Institute of Public Health and Environmental Sciences, Maebashi, Japan
- 9Aichi Prefectural Institute of Public Health, Kita, Japan
- 10Hiroshima Prefectural Technology Research Institute Public Health and Environment Center, Hiroshima, Japan
- 11Fukuoka Institute of Health and Environmental Sciences, Dazaifu, Japan
- 12Yamaguchi Prefectural Institute of Public Health and Environment, Yamaguchi, Japan
- 13Kanagawa Prefectural Institute of Public Health, Chigasaki, Japan
- 14Aomori Prefecture Public Health and Environment Center, Aomori, Japan
- 15Wakayama Prefectural Research Center of Environment and Public Health, Wakayama, Japan
- 16Iwate Prefectural Research Institute for Environmental Sciences and Public Health, Morioka, Japan
- 17Department of Microbiology, Tokyo Metropolitan Institute of Public Health, Shinjuku, Japan
- 18Eastern Chiba Medical Center, Togane, Japan
- 19Graduate School of Health Sciences, Gunma Paz University, Takasaki, Japan
- 20Department of Respiratory Medicine, Kyorin University School of Medicine, Mitaka, Japan
- 21Pathogen Genomics Center, National Institute of Infectious Diseases, Musashimurayama, Japan
- 22Department of Microbiology, Yokohama City University, Graduate School of Medicine, Yokohama, Japan
- 23Laboratory of Viral Infection I, Kitasato Institute for Life Sciences, Graduate School of Infection Control Sciences, Kitasato University, Minato, Japan
Human norovirus (HuNoV) GII.P17-GII.17 (Kawasaki2014 variant) reportedly emerged in 2014 and caused gastroenteritis outbreaks worldwide. To clarify the evolution of both VP1 and RNA-dependent RNA polymerase (RdRp) regions of GII.P17-GII.17, we analyzed both global and novel Japanese strains detected during 2013–2017. Time-scaled phylogenetic trees revealed that the ancestral GII.17 VP1 region diverged around 1949, while the ancestral GII.P17 RdRp region diverged around 2010. The evolutionary rates of the VP1 and RdRp regions were estimated at ~2.7 × 10−3 and ~2.3 × 10−3 substitutions/site/year, respectively. The phylogenetic distances of the VP1 region exhibited no overlaps between intra-cluster and inter-cluster peaks in the GII.17 strains, whereas those of the RdRp region exhibited a unimodal distribution in the GII.P17 strains. Conformational epitope positions in the VP1 protein of the GII.P17-GII.17 strains were similar, although some substitutions, insertions and deletions had occurred. Strains belonging to the same cluster also harbored substitutions around the binding sites for the histo-blood group antigens of the VP1 protein. Moreover, some amino acid substitutions were estimated to be near the interface between monomers and the active site of the RdRp protein. These results suggest that the GII.P17-GII.17 virus has produced variants with the potential to alter viral antigenicity, host-binding capability, and replication property over the past 10 years.
Introduction
Human norovirus (HuNoV) is a major causative pathogen of acute viral gastroenteritis (de Graaf et al., 2016). Although HuNoV is most prevalent during autumn and winter in the northern hemisphere, the virus can be detected throughout the year (Ahmed et al., 2013). A previous report suggested that approximately 680 million people annually suffer from gastroenteritis due to HuNoV infection worldwide (Kirk et al., 2015). Molecular epidemiological studies have indicated that distinct GII.4 variants have recurrently emerged every 2–3 years in the past decade to cause pandemics of gastroenteritis in all aged individuals (Bull et al., 2010). Furthermore, GII.P17-GII.17 (Kawasaki2014 variant) has suddenly been prevalent since 2014 in various countries including Japan, China, South Korea, Italy, Romania, Argentina, Brazil, and the USA (Chan et al., 2015; Fu et al., 2015; Matsushima et al., 2015; Medici et al., 2015; Dang Thanh et al., 2016; Dinu et al., 2016; Cannon et al., 2017; Degiuseppe et al., 2017; Silva et al., 2017). However, the epidemiology of this virus over a long period of time is unclear at present.
HuNoV belongs to the genus Norovirus of the family Caliciviridae, and it is further genetically classified into three genogroups, GI, GII, and GIV. Genetic analyses have shown that HuNoV GI and GII can be further classified into 9 and 22 genotypes, respectively (Kroneman et al., 2013). Of these, many genotypes have been associated with gastroenteritis outbreaks throughout the world (Hoa Tran et al., 2013). The genomes of noroviruses contain three open reading frames and encodes six non-structural proteins, including the RNA-dependent RNA polymerase and the two capsid proteins VP1 and VP2 (de Graaf et al., 2016).
Molecular evolutionary analysis based on advanced bioinformatics technologies is a powerful tool to better understand not only the phylogeny of pathogens, but also their antigenicity (Bok et al., 2009; Siebenga et al., 2010; Boon et al., 2011; Lu et al., 2016; Parra et al., 2017; Tohma et al., 2017; Nagasawa et al., 2018). Furthermore, next-generation sequencing technologies are also useful for the comprehensive analysis of various virus genomes (Quiñones-Mateu et al., 2014). In this study, we used both these methodologies to conduct a detailed molecular evolutionary analysis of the VP1 and RdRp regions of GII.P17-GII.17 strains detected in various countries.
Materials and Methods
Sample Preparation and Ethics Statement
A total of 76 strains of GII.P17-GII.17 detected in Miyagi (16 strains), Kanagawa (11 samples), Saitama (10 samples), Ibaraki (9 strains), Gunma (7 strains), Aichi (7 strains), Hiroshima (5 strains), Tochigi (4 strains), Fukuoka (3 strains), Yamaguchi (3 strains), and Aomori (1 strain) prefectures from 2013 to 2017 were sequenced in this study. Fecal samples were collected from patients with acute gastroenteritis associated with HuNoV infection under compliance with the Food Sanitation Law and the Law Concerning the Prevention of Infections and Medical Care for Patients of Infections of Japan. Informed consent was obtained from all participants, which was acquired from the subjects or their legally acceptable representatives for sample donation. The personal data of the patients was anonymized. To perform extraneous study (this study) and due to the lack of written informed consent, this study obtained ethical approval from the Research and Ethical Committees for the Use of Human Subjects of the National Institute of Infectious Diseases, Tokyo, Japan (No. 576). All methods were conducted in accordance with the approved guidelines. Information on the samples is given in Table S1. RNA was extracted from 10% suspensions of fecal samples in phosphate buffered saline using a QIAamp Viral RNA Mini kit (Qiagen, Hilden, Germany). The extracted RNA was subjected to sequencing as described below.
Sequencing
Sequencing was performed with Sanger and next-generation sequencers. For Sanger sequencing, a reverse transcription–polymerase chain reaction (RT-PCR) was first performed for 30 min at 45°C and then 2 min at 94°C, followed by a total of 45 cycles of 30 s at 98°C, 30 s at 55°C and 90 s at 68°C, and then a final extension of 7 min at 68°C using specific primers for the VP1 and RdRp regions and a PrimeScript II High Fidelity One Step RT-PCR kit (TaKaRa, Shiga, Japan; Table S2). Cycle sequencing was performed for 1 min at 96°C, followed by a total of 30 cycles of 10 s at 96°C, 10 s at 50°C and 2 min at 60°C using a BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems, Carlsbad, California, USA). The DNA sequences were analyzed using a 3500 Genetic Analyser (Applied Biosystems). Full-length nucleotide sequences of the VP1 and RdRp regions were acquired using the primer walking method. Next-generation sequencing was conducted as described previously (Dennis et al., 2014; Ide et al., 2015). Data analysis was performed using CLC Genomics Workbench v8.0.1 (Qiagen). Contigs were assembled from the obtained sequence reads by de novo assembly. HuNoV genotypes were determined using the Norovirus Genotyping Tool (version 2.0) and the Human Calicivirus Typing Tool1 (Kroneman et al., 2011).
Construction of Datasets for Bioinformatics
All full-length nucleotide sequences of the VP1 and RdRp regions of GII.17, including information on sample collection years and no mixed nucleotides, were obtained from GenBank2 (accessed on 29 August, 2017). For the GII.P17-GII.17 genotype, only sequences with information on sample collection years and months were used in this study. Moreover, nine sequences associated with some recent outbreaks of GII.P17-GII.17 (Sakon et al., 2018) were combined with those of the new Japanese strains above. To construct time-scaled phylogenetic tree, we added representative VP1 sequences of all GII genotypes, including porcine NoV GII (GII.11, GII.18, and GII.19) and other HuNoV GII genotypes (18 strains), as well as an outgroup strain of HuNoV GI genotype (GI.1) to the dataset of the VP1 region (resulting in a total of 365 strains). The representative RdRp sequences of the GII genotypes, including porcine NoV (GII.P11 and GII.P18) and other HuNoV GII genotypes (20 strains), as well as an outgroup strain of HuNoV GI genotype (GI.P1), were appended to produce an RdRp dataset consisting of 156 strains (Table S1). The constructed datasets were aligned with MAFFT software (Katoh and Standley, 2013).
Time-Scaled Phylogenetic Trees
Phylogenetic trees with molecular clocks were generated by the Bayesian Markov Chain Monte Carlo (MCMC) method using the BEAST2 package v2.4.8 (Bouckaert et al., 2014). Best substitution models (GTR+Γ+I for both VP1 and RdRp) were determined for the constructed datasets (365 strains for VP1 and 156 strains for the RdRp) by comparison of the Bayesian Information Criterion (BIC) values using jModelTest2 software (Guindon and Gascuel, 2003; Darriba et al., 2012). Appropriate clock and tree prior models (relaxed clock exponential and coalescent exponential population tree prior for both VP1 and RdRp) were selected by path-sampling/stepping stone-sampling marginal-likelihood estimation using the BEAST2 package (Baele et al., 2012). The MCMC runs were conducted with chain lengths of 930,000,000 steps with sampling every 16,000 steps for VP1, and with chain lengths of 465,000,000 steps with sampling every 8,000 steps for RdRp. The analyzed data was evaluated by the effective sample sizes (ESS) values using Tracer3 software, and values of 200 or more were accepted. Maximum clade credibility trees were generated by discarding the first 10% of trees (burn-in) using TreeAnnotator v2.4.8 in the BEAST2 package. The time-scaled phylogenetic trees were visualized by FigTree4 v1.4.0 software. Branch reliability was supported by highest posterior densities (HPDs) of 95%. Moreover, the evolutionary rates of GII.P17-GII.17 strains were estimated as described above. The analyzed parameters with substitution, clock, and tree prior models are shown in Table 1. In this study, the evolutionary rates of overall clusters in the GII.P17-GII.17 strains were calculated since the rate of a cluster could not be estimated due to the limited number of strains available for analysis.

Table 1. Evolutionary rates of GII.P17-GII.17 strains in the VP1 and RdRp regions.
Bayesian Skyline Plot
Transition of effective population sizes was estimated by a Bayesian skyline plot using the BEAST2 package v2.4.8. Appropriate substitution models were selected based on comparison of the BIC values in the dataset, including 337 strains of the Kawasaki2014 variant in the VP1 region. The clock models were appropriately selected on the basis of path-sampling/stepping stone-sampling marginal-likelihood estimation. The MCMC was run on chain lengths of 480,000,000 steps with sampling every 2,000 steps. After evaluation based on the ESS values, the Bayesian skyline plot was generated by Tracer3 software.
Phylogenetic Distances
Phylogenetic trees were created using the maximum likelihood method in MEGA7 software (Kumar et al., 2016). Best substitution models (GTR+Γ+I for VP1 and K80+Γ for RdRp) were selected using the jModelTest2. Branch reliability was supported by 1,000 replications of bootstrap values. Phylogenetic distances between GII.17 strains were calculated by Patristic software (Fourment and Gibbs, 2006).
Construction of Three-Dimensional Structures, Conformational Epitope Analyses, and Selective Pressure Analyses
Structural models of the VP1 and RdRp proteins of HuNoV [Hu/GII/JP/1976/GII.17/Tokyo/27-3: AB684681 (VP1), Hu/GII/NE/1995/GII.P3/Amsterdam/1: KJ194500 (RdRp), Hu/GII/JP/2014/GII.P17-GII.17/Kawasaki323: AB983218 (VP1 and RdRp), Hu/GII/JP/2015/GII.P17-GII.17/Kawasaki308: LC037415 (VP1 and RdRp)] were generated based on homology modeling using the MODELLER software v9.15 (Webb and Sali, 2014). Three crystal structures for VP1 (PDBID: 1IHM, 5F4M and 5LKC) and one for RdRp (PDBID: 1SH0) were used as the templates based on Basic Local Alignment Search Tool5 (BLAST) analyses of the proteins. Amino acid sequences of the templates and the target strains were aligned using MAFFTash software (Standley et al., 2007; Katoh et al., 2009). The constructed structures were minimized using the GROMOS96, implemented by Swiss PDB Viewer v4.1 and evaluated by Ramachandran plots via the RAMPAGE server, which showed the favored regions of 96.1% ± 0.26 (VP1) and 98.0% ± 0.46 (RdRp), the allowed regions of 3.43% ± 0.32 (VP1) and 1.47% ± 0.32 (RdRp), and the outlier regions of 0.47% ± 0.058 (VP1) and 0.57% ± 0.15 (RdRp) (mean ± SD) of all residues in each structure (Guex and Peitsch, 1997; Lovell et al., 2003). The final models were modified and colored using Chimera v1.12 (Pettersen et al., 2004). Conformational epitopes on the VP1 dimer structures of the GII.17 strains were predicted using DiscoTope 2.0, EPCES, and EPSVR programmes (Liang et al., 2007, 2010; Kringelum et al., 2012). Cut-off values were set at −3.1 for the DiscoTope 2.0 and 81 for the EPCES and EPSVR in order to encompass epitopes on the VP1 of GII.17 identified by a previous in vitro study (Lindesmith et al., 2017). Consensus sites identified by more than one of the three tools and regions with two or more closely apposed residues of the sites on the VP1 dimer structures were determined to be conformational epitopes. Positive selection sites in the VP1 region of the GII.17 strains were predicted using the Fast, Unconstrained Bayesian AppRoximation (FUBAR) and Mixed Effects Model of Evolution (MEME) algorithms on the Datamonkey server (Pond and Frost, 2005; Delport et al., 2010; Murrell et al., 2012, 2013; Weaver et al., 2018). Generally, FUBAR hypothesizes constant selection pressure at each site for the entire phylogeny and utilizes a Bayesian approach to estimate non-synonymous (dN) and synonymous (dS) substitution rates at each site. MEME is used to detect positive selection sites under a proportion of branches. Significance levels were set at posterior probabilities of >0.9 for FUBAR and p < 0.05 for MEME.
Median Joining Network Analyses
Transmission links between GII.P17-GII.17 strains were analyzed using a median joining network with PopART software (Bandelt et al., 1999; Leigh and Bryant, 2015). We constructed a dataset that covered nucleotide sequence lengths from the start position of RdRp to the terminal position of VP1 (resulting in a total of 114 strains). The dataset was then processed to detect recombination between the RdRp and VP1 sequences based on seven primary exploratory recombination signal detection methods (RDP, GENECONV, BootScan, MaxChi, Chimera, SiScan, and 3Seq) using RDP4 software (Martin et al., 2015) and the threshold of the p-value for significance was set at 0.001. Recombination regions were assigned when they were identified by more than four of the seven methods; however, this criterion resulted in the identification of no recombinant sequences in the dataset. The median joining networks were analyzed using an epsilon value of zero.
Results
Time-Scaled Phylogeny of the VP1 and RdRp Regions in the GII.P17-GII.17 Strains
We constructed time-scaled phylogenetic trees using the MCMC method based on the full-length of the VP1 (343 strains) and RdRp (133 strains) regions in the GII.17 strains detected in the various countries (Figures 1, 2). The MCMC tree for the VP1 region estimated that the common ancestor of the GII.17, GII.13, and GII.21 diverged in September, 1931 (95% HPD January, 1911–February, 1950). A common ancestor of the GII.17 strains diverged in November, 1948 (95% HPD September, 1934–November, 1961) and further diverged into seven clusters by around October, 2011. Of these, the GII.P17-GII.17 strains belonged to clusters 1 and 2 (Figure 1). The GII.17 strains in clusters 3, 4, 5, and 6 (no information on the RdRp sequence in cluster 7) were composed of the distinct RdRp genotypes, including GII.P3, GII.P16, GII.P4, and GII.Pe, respectively (Figure 1 and Table S1). Most strains of the GII.P17-GII.17 belonged to the cluster 1 (the Kawasaki308 type). The common ancestor of the cluster 1 diverged in October, 2011 (95% HPD May, 2009–August, 2013), while that of cluster 2 (the Kawasaki323 type) emerged in July, 2010 (95% HPD July, 2007–September, 2012) (Figure 1). Moreover, analyses of phylogenetic distances exhibited no overlap between intra- and inter-cluster peaks at a value of 0.035 (Figure 3A). With respect to the RdRp region (Figure 2), the common ancestor of the GII.P17 and GII.P3 diverged in January, 1988 (95% HPD December, 1980–May, 1993). Subsequently, the common ancestor of the GII.P17 diverged in October, 2009 (95% HPD May, 2006–April, 2012) and then further into two clusters by around August, 2012. The GII.P17-GII.17 strains were contained in clusters 1 and 2 in the RdRp region, which was compatible with the classification in the VP1 region. Most strains of the GII.P17-GII.17 also belonged to cluster 1 (the Kawasaki308 type). The common ancestor of cluster 1 diverged in August, 2012 (95% HPD December, 2010–January, 2014), while that of cluster 2 (the Kawasaki323 type) did so in April, 2011 (95% HPD September, 2009–October, 2012). Furthermore, phylogenetic distances overlapped between the intra- and inter-cluster peaks (Figure 3B). These results suggest that the GII.P17-GII.17 strains emerged and formed two variants in approximately the past 10 years.
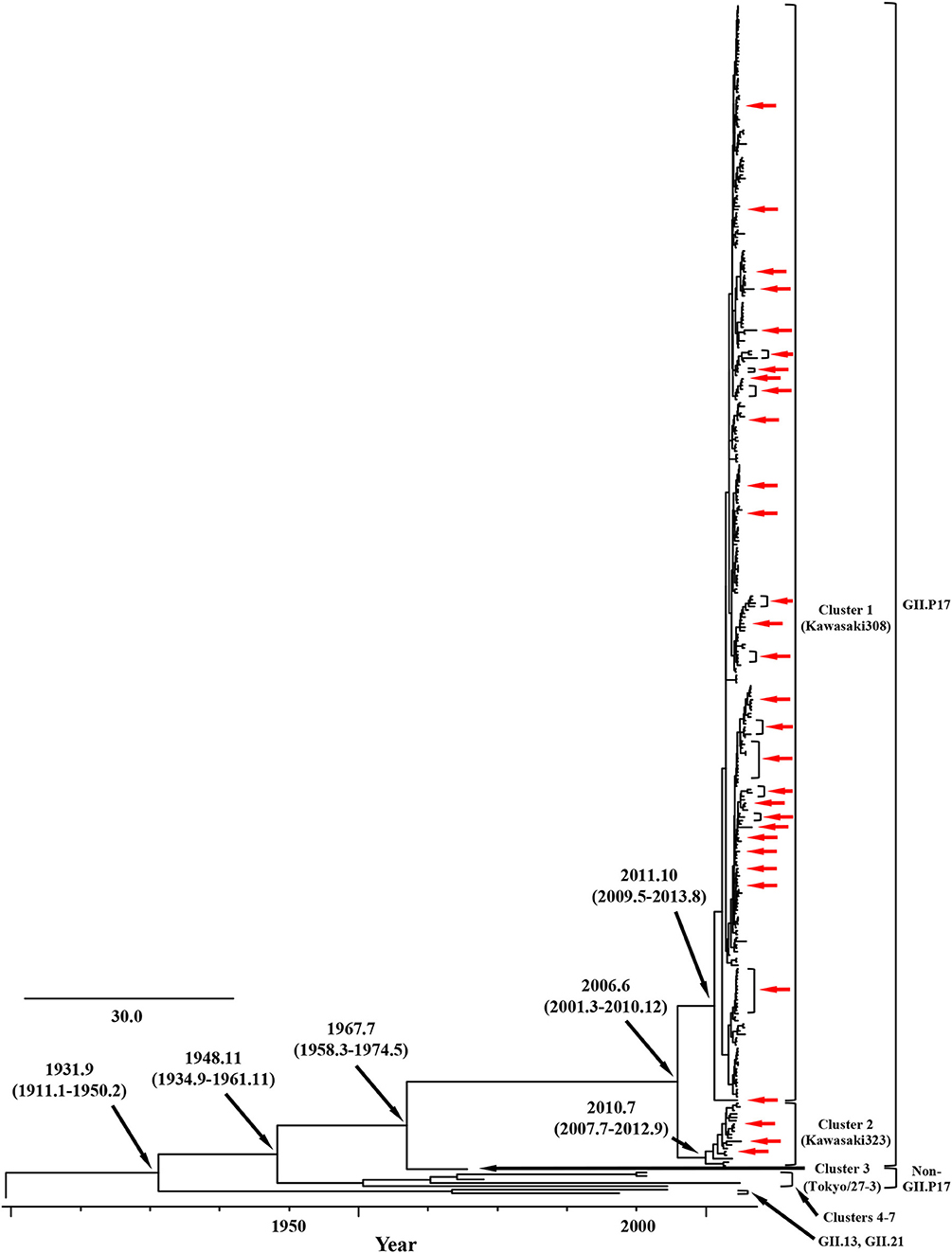
Figure 1. Time-scaled phylogenetic trees of the full-length VP1 region of HuNoV constructed using the Bayesian MCMC inference. The visualization of the tree is enlarged to focus on GII.17, GII.13, and GII.21. The values within parentheses indicate the 95% HPDs for each divergent year, and red arrows indicate the Japanese samples collected in this study.
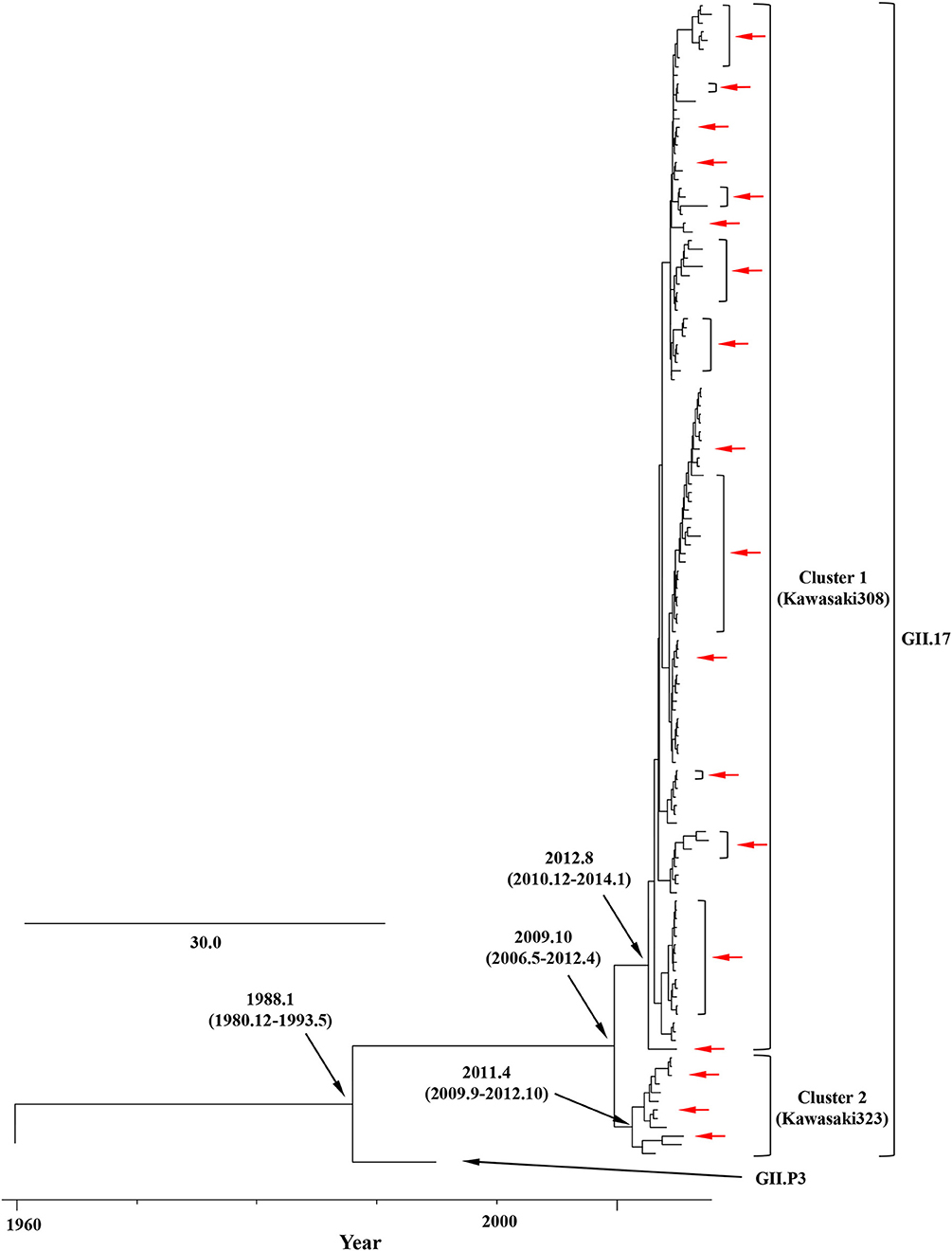
Figure 2. Time-scaled phylogenetic trees of the full-length RdRp region of HuNoV constructed using the Bayesian MCMC inference. The tree visualization is enlarged to focus on GII.P17 and GII.P3. The values within parentheses indicate the 95% HPDs for each divergent year, and red arrows indicate the Japanese samples collected in this study.
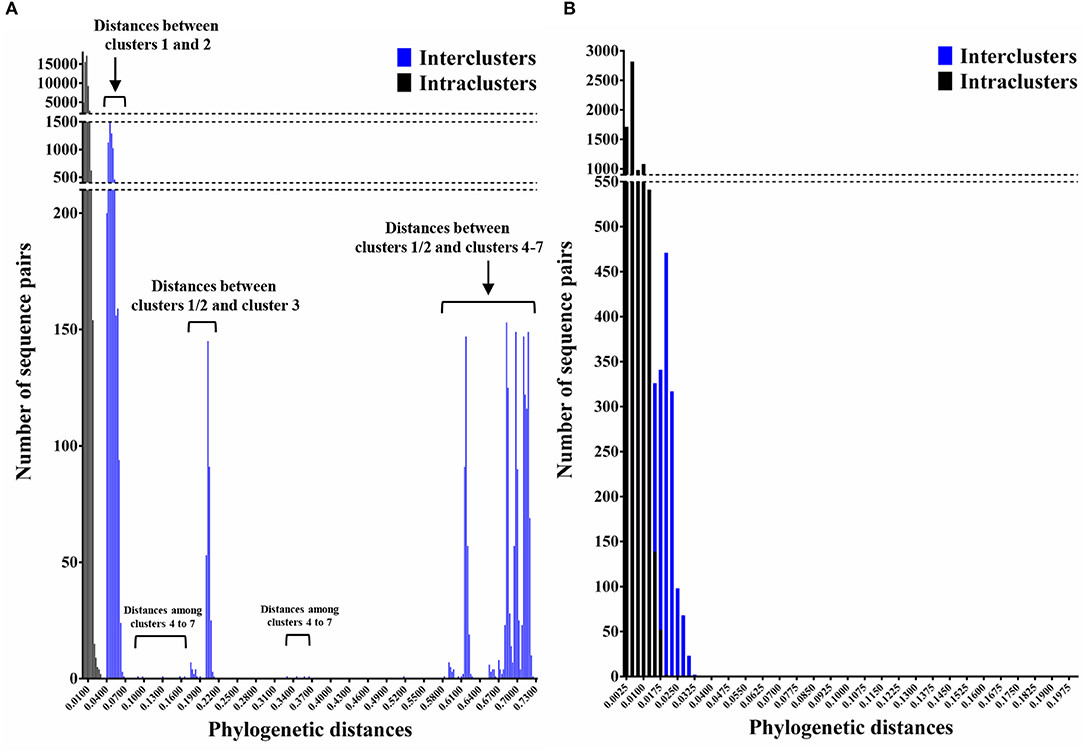
Figure 3. Phylogenetic distances between the nucleotide sequences of GII.17 strains in the full-length VP1 and RdRp regions. (A) Distribution of intra-genotype phylogenetic distances in the VP1 region of the GII.17 strains; (B) Distribution of intra-genotype phylogenetic distances in the RdRp region of the GII.P17 strains. The y-axis shows the number of sequence pairs corresponding to each distance while the x-axis represents phylogenetic distances.
Evolutionary Rates of the VP1 and RdRp Regions in the GII.P17-GII.17 Strains
The evolutionary rates of the VP1 and RdRp regions of the GII.P17-GII.17 strains are presented in Table 1. The rate for the VP1 region was 2.7 × 10−3 substitutions/site/year (95% HPD 2.2–3.2 × 10−3), while that for the RdRp region was 2.3 × 10−3 (95% HPD 1.5–3.2 × 10−3). These results indicate that the VP1 and RdRp regions of the GII.P17-GII.17 strains evolved with similar rates.
Phylodynamics of GII.P17-GII.17 Strains in the VP1 Region
We estimated the transition of population sizes of GII.P17-GII.17 in the VP1 region using a Bayesian skyline plot. The population of this region increased sharply in around 2014 and remained constant from 2016 after an immediate reduction (Figure 4). These results suggest that this was compatible between the fluctuation of the plot and actually epidemiological GII.17 prevalence.
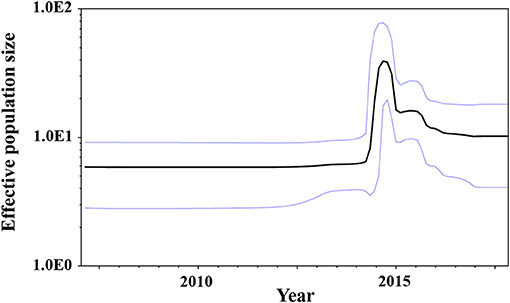
Figure 4. Bayesian skyline plots for the VP1 sequences of GII.P17-GII.17 strains. The y-axis represents the effective population sizes on a logarithmic scale, whereas the x-axis denotes time in years. The solid black line indicates the mean posterior value and the intervals with 95% HPDs are shown by blue lines.
Prediction of Conformational Epitopes and Selective Pressures in the VP1 in GII.P17-GII.17 Strains
Conformational epitopes and selective pressures in the VP1 protein were analyzed in order to assess the possibility of antigenicity changes at pre and post emergence of GII.P17-GII.17 based on the time-scaled phylogeny for the VP1 region in Figure 1. As a result, common epitopes among strains of clusters 1, 2 (GII.P17-GII.17 clusters) and 3 [a closely related cluster (GII.P3-GII.17 strain) to the GII.P17-GII.17 clusters] were found in the shell domain and the protruding 2 (P2) domain. Although the amino acid epitope positions were similar among the strains, many amino acid substitutions were identified (Figure 5 and Table 2). Substitutions in epitopes were also identified in strains belonging to the same cluster. Of these substitution residues, amino acids (aa) 375, aa376, aa394, aa396, and aa442 are located close to the histo-blood group antigen (HBGA) binding sites of VP1, which are utilized for attachment of HuNoV to host cells. Additionally, three positive selection sites were identified in the P2 domain (Asn342Ser, Glu376Asn/Asp, and Arg412Leu/Glu/Pro). Of these sites, aa342 and aa376 were at predicted epitopes (Figure 5). These results indicate that GII.P17-GII.17 could evolve with changes in antigenicity and binding affinity to HBGAs.
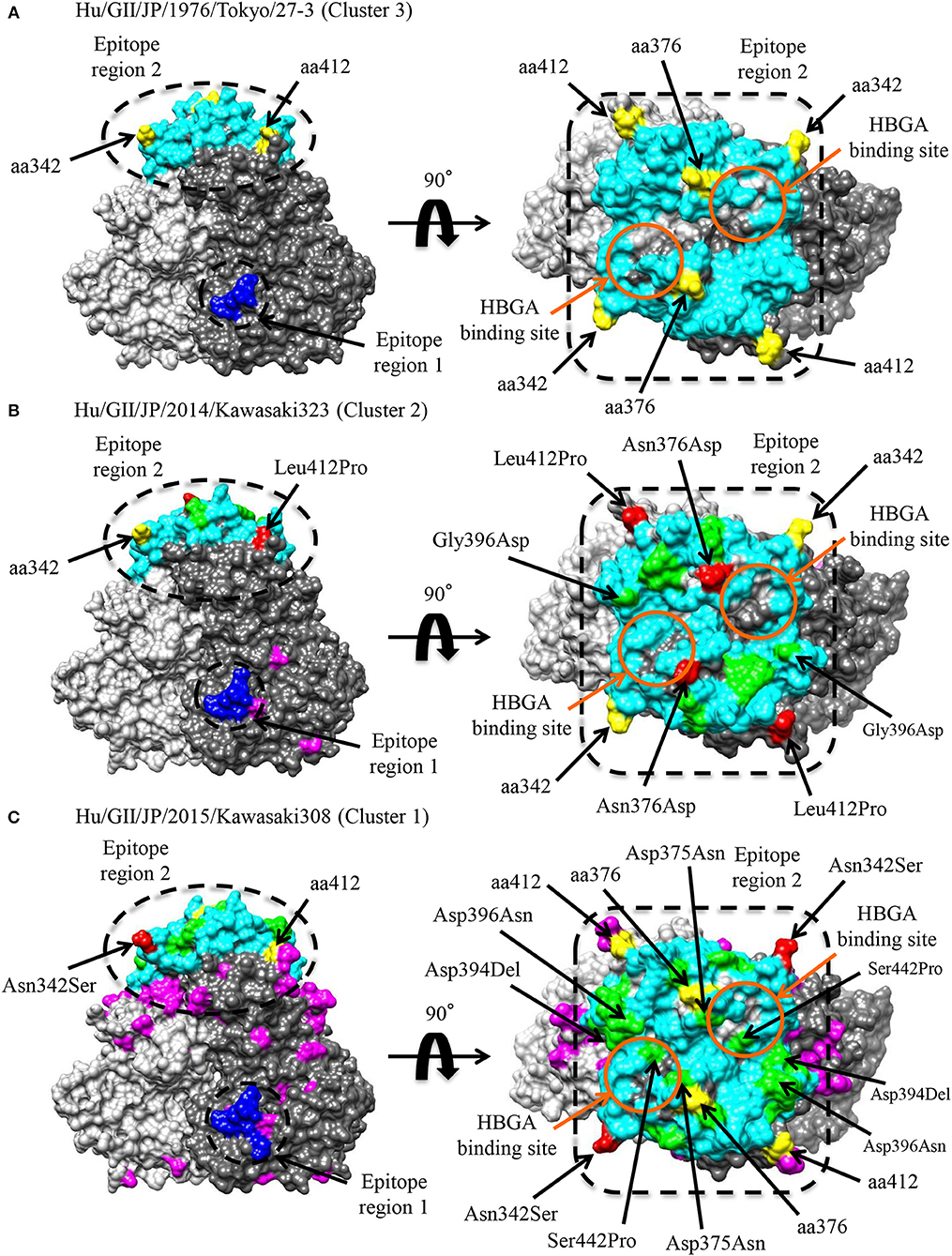
Figure 5. Structural models of the VP1 protein for representative GII.17 strains in the clusters of the Kawasaki2014 variant and its ancestor. Three-dimensional VP1 dimer structures for Tokyo/27-3 [ancestor; cluster 3; (A)], Kawasaki323 [cluster 2; (B)], and Kawasaki308 [cluster 1; (C)] viruses are shown. Each monomeric chain that comprise the dimer structures is colored gray (chain A) and dim gray (chain B). The predicted epitope regions of the strains are circled in black and the amino acids of the epitopes with no substitutions are colored blue for the shell domain and cyan for the P2 domain. Positive selection sites are colored yellow with the positions under the order of alignments. Orange circles represent the HBGA binding sites on the structures. Amino acid substitutions on the epitopes, non-epitopes, and positive selection sites within the clusters are colored green, magenta, and red, respectively.
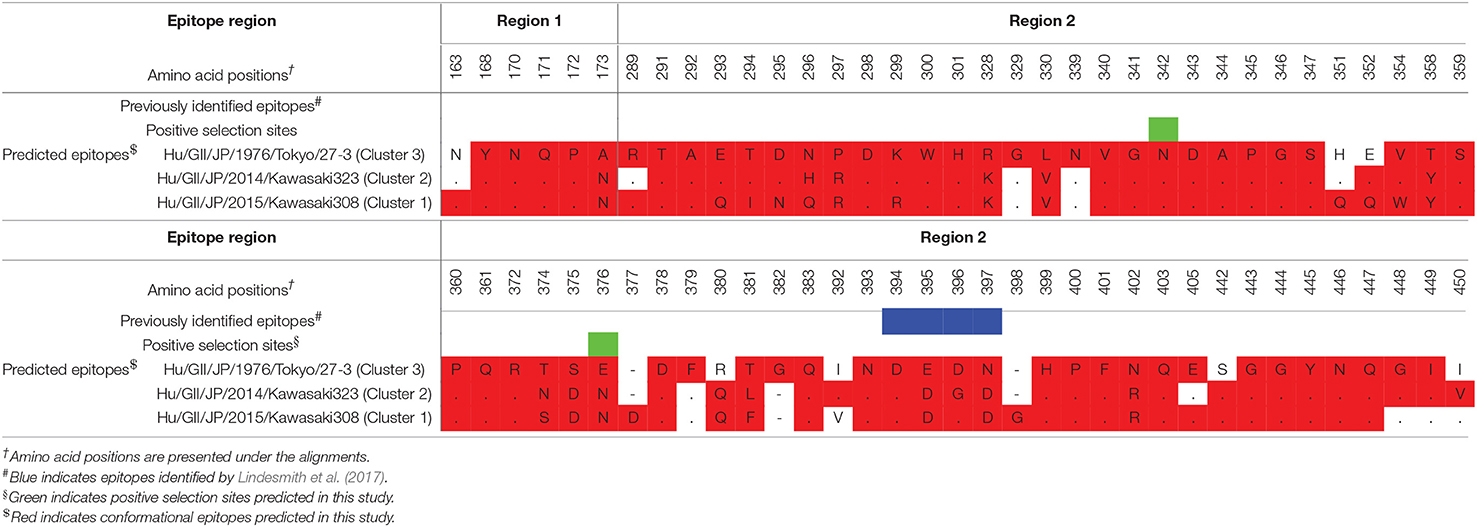
Table 2. Conformational epitopes of GII.17 variants in the VP1 protein.
Mapping of Amino Acid Substitutions of GII.P17-GII.17 to the RdRp Protein
To assess the possibility of changes of RNA replication properties at pre and post emergence of GII.P17-GII.17 based on the time-scaled phylogeny for the RdRp region in Figure 2, we mapped the amino acid substitutions between GII.P17 strains and other RdRp genotypes onto three-dimensional structures of the RdRp protein (Figure 6). A total of 48 amino acid substitutions were identified among the GII.P17 and GII.P4 strains (Accession number; FJ537137). Of these, the substitutions at aa33, aa34, aa49, and aa434 were located close to the interface between monomers, while substitutions at aa206, aa215, aa409, aa438, and aa442 were located close to the active site (Figure 6A). Moreover, a total of 16 amino acid substitutions were estimated among the GII.P17 and GII.P3 (a closely related genotype to GII.P17) strains (Accession number; KJ194500). The substitution at aa49 was located adjacent to the interface between monomers, whereas the substitutions at aa206, aa215, aa407, and aa409 were located proximal to the active site (Figure S1). For intra-GII.P17 genotype, Phe206Leu and Lys405Arg substitutions were found around the active site in the strains of cluster 2 (12 strains) (Figure 6B). Thirty one substitutions were also found in the strains belonging to the cluster 1 (121 strains), and a Ser427Asn substitution was located proximally at the interface between monomers. Ile193Val, Ile215Val, Asp224Glu, Ile346Val, Thr394Ala, Val395Ile, Asn409Ser, Val438Met, Gly445Ser, and Phe503Leu substitutions were located close to the active sites (Figure 6C).
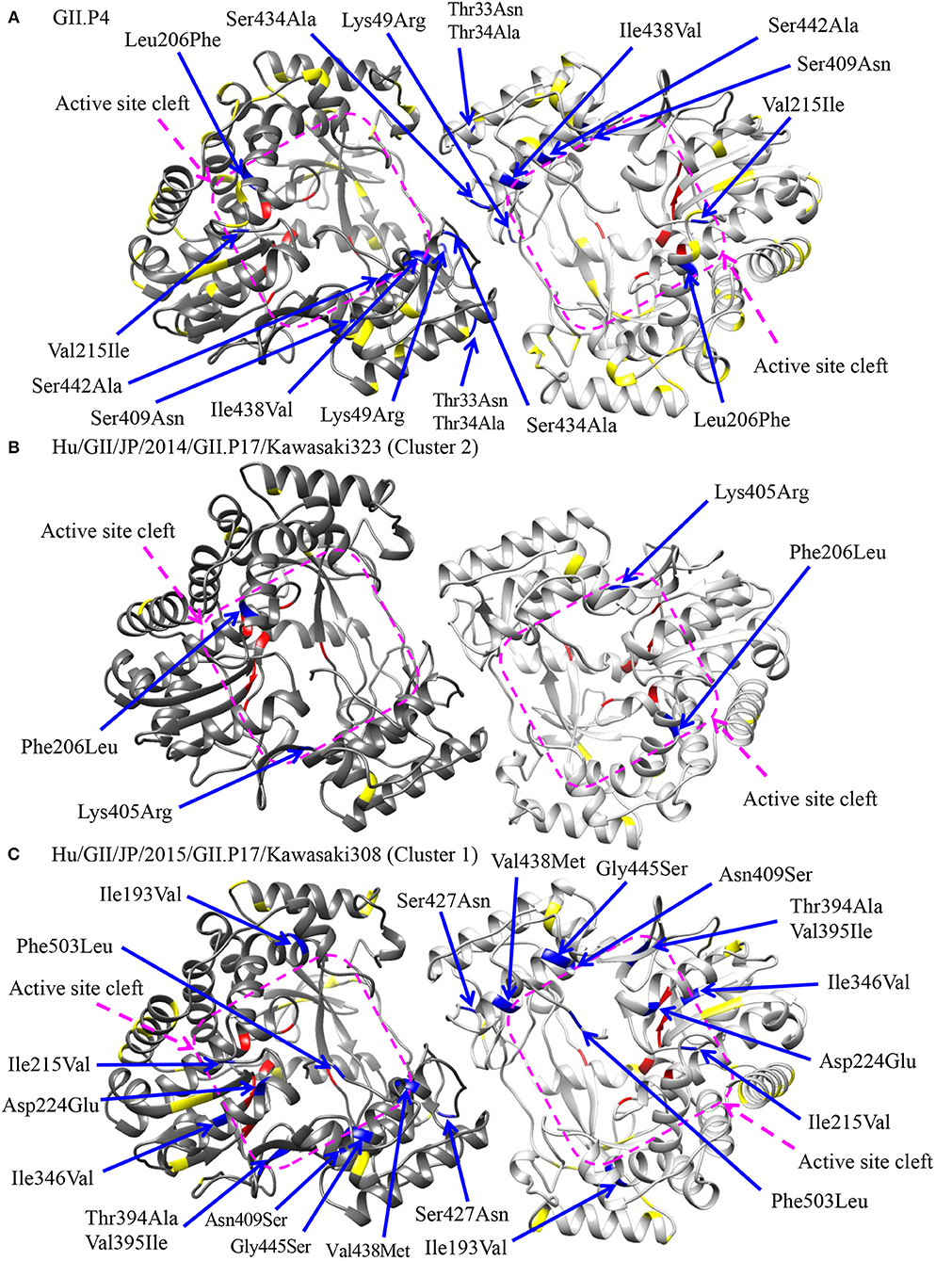
Figure 6. Structural models of the RdRp protein for representative GII.P17 strains. Homodimer structures of the RdRp of GII.P4 [PDBID 1SH0; (A)], Kawasaki323 [cluster 2; (B)], and Kawasaki308 [cluster 1; (C)] are shown. Each monomeric chain that compose the dimer structure are colored gray (chain A) and dim gray (chain B). Amino acid substitutions between GII.P17 and GII.P4 (A) or within the GII.P17 clusters (B,C) that are close to the interface between monomers and to the active site cleft are colored blue with the positions under the order of alignments, whereas the substitutions distant from these regions are shown in yellow. The residues of active sites for RNA replication are colored red. The purple quadrilaterals highlight the region of the active site cleft.
Transmission Network Between the GII.P17-GII.17 Strains
Links in circulation of prevalent GII.P17-GII.17 (cluster 1) in Japanese regions and in other countries were analyzed using a median joining network. The strains analyzed in this study were divided into four clusters. Strains belonging to cluster 1 were mainly found from Japan, while those belonging to clusters 2 and 3 were found in various countries including Japan, China, Hong Kong, Taiwan, and Australia. The strains in cluster 4 were detected only in Miyagi prefecture in Japan. Notably, the network contained the key strains that were linked to the many other strains (Figure 7). These results suggest that the GII.P17-GII.17 strains form a broad network and that some strains are associated with the prevalence of the virus.
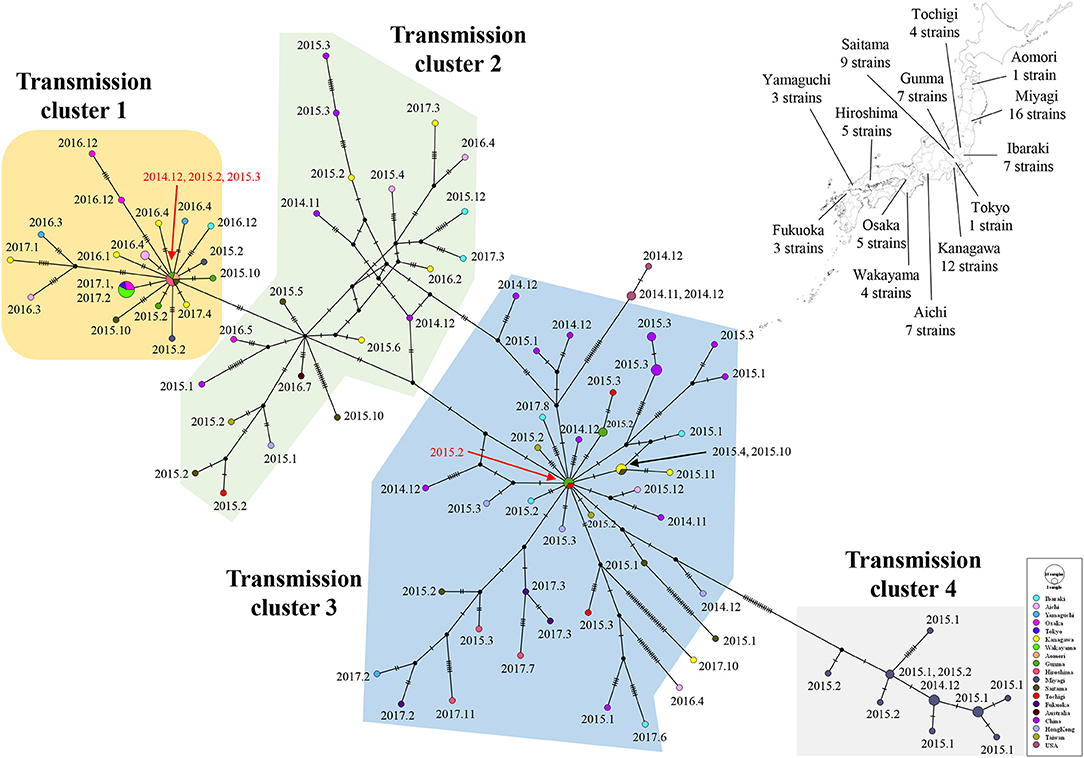
Figure 7. A median joining network for the major epidemic cluster of GII.P17-GII.17 strains based on nucleotide sequences from the RdRp to VP1 (3133 bp). The detected areas for Japanese strains used in the analysis are shown with the number of strains on a Japanese map. The filled circles represent viral haplotypes, including collection years and months for the strains. The sizes of the circles represent the number of strains within the haplotype. Red arrows indicate strains associated with the production of many haplotypes.
Discussion
In this study, we demonstrated the molecular evolution of both the VP1 and RdRp regions of GII.P17-GII.17 since the emergence of the virus. First, a MCMC time-scaled evolutionary phylogenetic tree based on GII.17 VP1 nucleotide sequences showed that all the present strains grouped into a total of seven clusters (Figure 1). Trees also indicated that the recently emerged GII.P17-GII.17 strains uniquely formed 2 clusters (clusters 1 and 2) based on both the VP1 and RdRp regions. The common ancestor of the VP1 region of the GII.P17-GII.17 viruses diverged first, followed by divergence of the RdRp region after a several-year delay (Figures 1, 2). Previous reports have also suggested a gap in the divergence of GII.P16-GII.2 strains (Mizukoshi et al., 2017; Nagasawa et al., 2018). To resolve this issue, we analyzed the phylogenetic distances of the VP1 and RdRp regions and found that the distance of the VP1 region was longer than that of RdRp, indicating that the genetic diversity of VP1 in GII.P17-GII.17 is larger than that of RdRp (Figure 3). However, the evolutionary rates for the overall cluster in GII.P17-GII.17 were similar between the VP1 and RdRp regions (Table 1). Thus, we also calculated the rates of the strains belonging to the cluster 1, which resulted in reduction of the value in the RdRp region, but not in the VP1 region (data not shown). These results suggest that the differences of divergent times and genetic distances between these regions may be associated with those in the changes of the evolutionary rates. Furthermore, we analyzed the time-scaled MCMC phylogenetic trees by adding 343 sequences of GII.17 for the VP1, 133 sequences of GII.P17 for the RdRp and data of not only collection years but also months, which produced confidence intervals (95% HPD values) that were smaller in our analyses than in an earlier report (Sang and Yang, 2018). Thus, more precise collection data, as well as the use of a large number of virus strains, may facilitate the construction of more precise MCMC phylogeny.
The evolutionary rates of the VP1 and RdRp regions in the GII.P17-GII.17 were ~2.7 and ~2.3 × 10−3 substitutions/site/year, respectively (Table 1). The rate for GII.17 is likely similar to that for GII.2 in the VP1 region, but lower than that of GII.4 (Bok et al., 2009; Siebenga et al., 2010; Mizukoshi et al., 2017; Motoya et al., 2017). Furthermore, the rate for GII.P17 is likely analogous to that for GII.P16, but lower than that of GII.P4 (Nagasawa et al., 2018; Ozaki et al., 2018). Interestingly, it has been suggested that GII.P4-GII.4 virus prevalence is associated with the rapid evolution of the VP1 regions with a high ratio of non-synonymous amino acid substitutions to synonymous substitutions (Bull et al., 2010; Parra et al., 2017). Previous reports have also suggested that the rates of non-synonymous amino substitutions in the VP1 protein differ between GII.17 and GII.4, with GII.4 being greater than GII.17 (Mori et al., 2017); therefore, the rates of antigenicity change between GII.17 and GII.4 viruses may be distinct. In contrast, it has been estimated that the number of negative selection sites in GII.P4 RdRp are larger than that in GII.P17 (Ozaki et al., 2018), and thus variations in nucleotide substitution in GII.P4 RdRp may be restricted compared to GII.P17. However, despite such restrictions, we observed a lower evolutionary rate in GII.P17 than GII.P4. This may be due to differences in replication activities and/or in replication errors of the RdRp protein between GII.P17 and GII.P4. Additional in vitro studies may be needed to address these possibilities.
We also assessed the phylodynamics of GII.P17-GII.17 and found that the genome population sizes of GII.P17-GII.17 increased rapidly during 2014–2015 and then decreased rapidly thereafter (Figure 4). These fluctuations may be associated with epidemics of the newly emerged HuNoV and the acquisition of herd immunity. For example, the fluctuations in the phylodynamics of the GII.4 virus could be associated with the emergence of a variant virus with altered antigenicity and the acquisition of herd immunity to the VP1 capsid protein of that virus (Debbink et al., 2013; Lindesmith et al., 2013; Motoya et al., 2017). Thus, it is possible that the phylodynamic fluctuations in GII.P17-GII.17 identified in this study are associated with epidemics in various regions and subsequent acquisition of herd immunity (Chan et al., 2015; Fu et al., 2015; Matsushima et al., 2015; Dang Thanh et al., 2016). These results suggest that continuous phylodynamic analysis may provide early notice of other HuNoV epidemics and repeat prevalence of HuNoV, including GII.17.
We additionally found amino acid substitutions in the conformational epitopes of the VP1 capsid protein of GII.P17-GII.17 (Figure 5 and Table 2). Such substitutions were also identified around the HBGA binding sites. Such evolutionary substitutions may induce both changes in antigenicity and in HBGA binding ability (Tan et al., 2009; Chen et al., 2011; de Rougemont et al., 2011; Lindesmith et al., 2012; Debbink et al., 2013; Jin et al., 2015). Indeed, Lindesmith et al. showed that some amino acid substitutions (corresponding to aa394-aa397 in our alignments) are located adjacent of the HBGA binding sites and result in changes in the antigenicity of the GII.17 capsid protein (Lindesmith et al., 2017). Jin et al. also reported changes to the antigenicities and HBGA binding affinities of GII.17 capsid proteins belonging to different phylogenetic clusters (Jin et al., 2016). Moreover, an amino acid substitution (aa376) located adjacent the HBGA binding sites was identified as a possible positive selection site. Previous reports have suggested that aa375 and aa377 are associated with the HBGA binding ability (He et al., 2017; Koromyslova et al., 2017); however, to the best of our knowledge, there have been no reports regarding possible roles of the aa376 substitution in antigenicity and the HBGA binding capability, which is a subject for possible further study.
We also showed that GII.P17-GII.17 contains amino acid substitutions in residues adjacent to the RdRp active sites and the contact surfaces between RdRp monomers (Figure 6). Ng et al. showed that the amino acids around the active sites regulate viral genome replication (Ng et al., 2008). Moreover, substitutions around the monomer contact surfaces of RdRp affect the stability of the dimer and its RNA binding abilities (Chen et al., 2009). Thus, the similar substitutions found in the present study warrant additional investigation into changes in the properties of the RdRp protein. Previous studies have also revealed key amino acids associated with the efficiency of HuNoV genome replication (Bull et al., 2010; Eden et al., 2011). Of these, the threonine residue at aa33 is phosphorylated by a host factor, Akt, and is involved in producing high replication rates (Eden et al., 2011). This residue is located around the interface between monomers in the RdRp structure. The RdRp protein of GII.P4 contains a threonine residue at the position, while that of GII.P17 contains an asparagine (data not shown). This may suggest that the enzyme activity of RdRp differs between the GII.P4 and GII.P17 genotypes.
We constructed a genome network of the GII.P17-GII.17 strains examined in this study and found four major clusters (Figure 7). Notably, the strains belonging to clusters 1 and 4 were detected exclusively in Japan, while the strains belonging to clusters 2 and 3 were detected from various countries. Moreover, the two haplotype strains found in Japan might give rise to many variant types. However, we could not determine specific amino acid substitutions in the strains belonging to clusters 1 and 3, which contained the haplotype strains (data not shown). Thus, these haplotype strains may exhibit no phenotypes acquiring high infectivity due to the mutations, although the reason for this remains unclear at present. Moreover, the strains belonging to cluster 4 had unique amino acid substitutions in p48, p22, protease, RdRp, and VP2 proteins (data not shown). These viruses were found only in Miyagi prefecture in Japan, during short periods and was never detected in other areas, perhaps because these mutations are deleterious to the propagations of the virus. As a limitation, the results of the present analyses may partially be affected by selection bias introduced in the collection of the strains.
In conclusion, the GII.P17-GII.17 virus has evolved differently to GII.P4-GII.4. However, this virus may have the potential to alter its antigenicity, host-binding capability (i.e., HBGA) and genome replication efficiency. Such changes could recurrently generate variants of GII.17 with the potential to produce pandemics such as those caused by GII.4 variant strains. Thus, additional and continuous evolutionary analyses of this genotype should be needed in the future.
Data Availability Statement
The datasets generated for this study can be found in the GenBank and the accession numbers are as follow; LC369214-LC369222, LC369224, LC369225, LC369227-LC369249, LC369251-LC369258, LC486737-LC486770.
Ethics Statement
The studies involving human participants were reviewed and approved by the Research and Ethical Committees for the Use of Human Subjects of the National Institute of Infectious Diseases, Tokyo, Japan (No. 576). Informed consent was obtained from all participants, which was acquired from the subjects or their legally acceptable representatives for sample donation.
Author Contributions
YM, KK, and HK designed this study. YM conducted all bioinformatic analyses and experiments. YD and MK performed the next-generation sequencing. YM, FM, NSa, YU, YO, TM, HT, NN, NSh, HY, RO-N, RS, RT, FT, TT, KS, HS, KN, JA, and HI collected samples and conducted the Sanger sequencing. YM and HK wrote the manuscript. HS, NO, AR, KK, and HK supervised this study. All authors read and approved the manuscript.
Funding
This work was partly supported by a commissioned project for Research on Emerging and Re-emerging Infectious Diseases from Japan Agency for Medical Research and Development, AMED (Grant numbers; 19fk0108103h0201 and 19fk0108033h0003).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Enago6 for the English language review.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02189/full#supplementary-material
Footnotes
1. ^Available online at: https://norovirus.phiresearchlab.org/
2. ^https://www.ncbi.nlm.nih.gov/genbank/
3. ^Available online at: http://tree.bio.ed.ac.uk/software/tracer/
4. ^Available online at: http://tree.bio.ed.ac.uk/software/figtree/
5. ^Available online at: https://blast.ncbi.nlm.nih.gov/Blast.cgi
6. ^Available online at: https://www.enago.jp/
References
Ahmed, S. M., Lopman, B. A., and Levy, K. (2013). A systematic review and meta-analysis of the global seasonality of norovirus. PLoS ONE 8:e75922. doi: 10.1371/journal.pone.0075922
Baele, G., Lemey, P., Bedford, T., Rambaut, A., Suchard, M. A., and Alekseyenko, A. V. (2012). Improving the accuracy of demographic and molecular clock model comparison while accommodating phylogenetic uncertainty. Mol. Biol. Evol. 29, 2157–2167. doi: 10.1093/molbev/mss084
Bandelt, H., Forster, P., and Röhl, A. (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. doi: 10.1093/oxfordjournals.molbev.a026036
Bok, K., Abente, E. J., Realpe-Quintero, M., Mitra, T., Sosnovtsev, S. V., Kapikian, A. Z., et al. (2009). Evolutionary dynamics of GII.4 noroviruses over a 34-year period. J. Virol. 83, 11890–11901. doi: 10.1128/JVI.00864-09
Boon, D., Mahar, J. E., Abente, E. J., Kirkwood, C. D., Purcell, R. H., Kapikian, A. Z., et al. (2011). Comparative evolution of GII.3 and GII.4 norovirus over a 31-year period. J. Virol. 85, 8656–8666. doi: 10.1128/JVI.00472-11
Bouckaert, R., Heled, J., Kühnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Bull, R. A., Eden, J. S., Rawlinson, W. D., and White, P. A. (2010). Rapid evolution of pandemic noroviruses of the GII.4 lineage. PLoS Pathog. 6:e1000831. doi: 10.1371/journal.ppat.1000831
Cannon, J. L., Barclay, L., Collins, N. R., Wikswo, M. E., Castro, C. J., Magaña, L. C., et al. (2017). Genetic and epidemiologic trends of norovirus outbreaks in the United States from 2013 to 2016 demonstrated emergence of novel GII.4 recombinant viruses. J. Clin. Microbiol. 55, 2208–2221. doi: 10.1128/JCM.00455-17
Chan, M. C., Lee, N., Hung, T. N., Kwok, K., Cheung, K., Tin, E. K., et al. (2015). Rapid emergence and predominance of a broadly recognizing and fast-evolving norovirus GII.17 variant in late 2014. Nat. Commun. 6:10061. doi: 10.1038/ncomms10061
Chen, B., Fang, S., Tam, J. P., and Liu, D. X. (2009). Formation of stable homodimer via the C-terminal alpha-helical domain of coronavirus nonstructural protein 9 is critical for its function in viral replication. Virology 383, 328–337. doi: 10.1016/j.virol.2008.10.032
Chen, Y., Tan, M., Xia, M., Hao, N., Zhang, X. C., Huang, P., et al. (2011). Crystallography of a Lewis-binding norovirus, elucidation of strain-specificity to the polymorphic human histo-blood group antigens. PLoS Pathog. 7:e1002152. doi: 10.1371/journal.ppat.1002152
Dang Thanh, H., Than, V. T., Nguyen, T. H., Lim, I., and Kim, W. (2016). Emergence of norovirus GII.17 variants among children with acute gastroenteritis in South Korea. PLoS ONE 11:e0154284. doi: 10.1371/journal.pone.0154284
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat Methods 9:772. doi: 10.1038/nmeth.2109
de Graaf, M., van Beek, J., and Koopmans, M. P. (2016). Human norovirus transmission and evolution in a changing world. Nat. Rev. Microbiol. 14, 421–433. doi: 10.1038/nrmicro.2016.48
de Rougemont, A., Ruvoen-Clouet, N., Simon, B., Estienney, M., Elie-Caille, C., Aho, S., et al. (2011). Qualitative and quantitative analysis of the binding of GII.4 norovirus variants onto human blood group antigens. J. Virol. 85, 4057–4070. doi: 10.1128/JVI.02077-10
Debbink, K., Lindesmith, L. C., Donaldson, E. F., Costantini, V., Beltramello, M., Corti, D., et al. (2013). Emergence of new pandemic GII.4 Sydney norovirus strain correlates with escape from herd immunity. J. Infect. Dis. 208, 1877–1887. doi: 10.1093/infdis/jit370
Degiuseppe, J. I., Gomes, K. A., Hadad, M. F., Parra, G. I., and Stupka, J. A. (2017). Detection of novel GII.17 norovirus in Argentina, 2015. Infect. Genet. Evol. 47, 121–124. doi: 10.1016/j.meegid.2016.11.026
Delport, W., Poon, A. F., Frost, S. D., and Kosakovsky Pond, S. L. (2010). Datamonkey 2010: a suite of phylogenetic analysis tools for evolutionary biology. Bioinformatics 26, 2455–2457. doi: 10.1093/bioinformatics/btq429
Dennis, F. E., Fujii, Y., Haga, K., Damanka, S., Lartey, B., Agbemabiese, C. A., et al. (2014). Identification of novel Ghanaian G8P[6] human-bovine reassortant rotavirus strain by next generation sequencing. PLoS ONE 9:e100699. doi: 10.1371/journal.pone.0100699
Dinu, S., Nagy, M., Negru, D. G., Popovici, E. D., Zota, L., and Oprişan, G. (2016). Molecular identification of emergent GII.P17-GII.17 norovirus genotype, Romania, 2015. Eur. Surveill. 21:30141. doi: 10.2807/1560-7917.ES.2016.21.7.30141
Eden, J. S., Sharpe, L. J., White, P. A., and Brown, A. J. (2011). Norovirus RNA-dependent RNA polymerase is phosphorylated by an important survival kinase, Akt. J. Virol. 85, 10894–10898. doi: 10.1128/JVI.05562-11
Fourment, M., and Gibbs, M. J. (2006). PATRISTIC: a program for calculating patristic distances and graphically comparing the components of genetic change. BMC Evol. Biol. 6:1. doi: 10.1186/1471-2148-6-1
Fu, J., Ai, J., Jin, M., Jiang, C., Zhang, J., Shi, C., et al. (2015). Emergence of a new GII.17 norovirus variant in patients with acute gastroenteritis in Jiangsu, China, September 2014 to March 2015. Eur. Surveill. 20:21157. doi: 10.2807/1560-7917.ES2015.20.24.21157
Guex, N., and Peitsch, M. C. (1997). SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis 18, 2714–2723. doi: 10.1002/elps.1150181505
Guindon, S., and Gascuel, O. (2003). A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
He, Z., Liu, B., Tao, Y., Li, C., Xia, M., Zhong, W., et al. (2017). Norovirus GII.17 natural infections in rhesus monkeys, China. Emerg. Infect. Dis. 23, 316–319. doi: 10.3201/eid2302.161077
Hoa Tran, T. N., Trainor, E., Nakagomi, T., Cunliffe, N. A., and Nakagomi, O. (2013). Molecular epidemiology of noroviruses associated with acute sporadic gastroenteritis in children: global distribution of genogroups, genotypes and GII.4 variants. J. Clin. Virol. 56, 185–193. doi: 10.1016/j.jcv.2012.11.011
Ide, T., Komoto, S., Higo-Moriguchi, K., Htun, K. W., Myint, Y. Y., Myat, T. W., et al. (2015). Whole genomic analysis of human G12P[6] and G12P[8] rotavirus strains that have emerged in Myanmar. PLoS ONE 10:e0124965. doi: 10.1371/journal.pone.0124965
Jin, M., Tan, M., Xia, M., Wei, C., Huang, P., Wang, L., et al. (2015). Strain-specific interaction of a GII.10 norovirus with HBGAs. Virology 476, 386–394. doi: 10.1016/j.virol.2014.12.039
Jin, M., Zhou, Y. K., Xie, H. P., Fu, J. G., He, Y. Q., Zhang, S., et al. (2016). Characterization of the new GII.17 norovirus variant that emerged recently as the predominant strain in China. J. Gen. Virol. 97, 2620–2632. doi: 10.1099/jgv.0.000582
Katoh, K., Asimenos, G., and Toh, H. (2009). Multiple alignment of DNA sequences with MAFFT. Methods Mol. Biol. 537, 39–64. doi: 10.1007/978-1-59745-251-9_3
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kirk, M. D., Pires, S. M., Black, R. E., Caipo, M., Crump, J. A., Devleesschauwer, B., et al. (2015). World Health Organization estimates of the global and regional disease burden of 22 foodborne bacterial, protozoal, and viral diseases, 2010: a data synthesis. PLoS Med. 12:e1001940. doi: 10.1371/journal.pmed.1001940
Koromyslova, A., Tripathi, S., Morozov, V., Schroten, H., and Hansman, G. S. (2017). Human norovirus inhibition by a human milk oligosaccharide. Virology 508, 81–89. doi: 10.1016/j.virol.2017.04.032
Kringelum, J. V., Lundegaard, C., Lund, O., and Nielsen, M. (2012). Reliable B cell epitope predictions: impacts of method development and improved benchmarking. PLoS Comput. Biol. 8:e1002829. doi: 10.1371/journal.pcbi.1002829
Kroneman, A., Vega, E., Vennema, H., Vinjé, J., White, P. A., Hansman, G., et al. (2013). Proposal for a unified norovirus nomenclature and genotyping. Arch. Virol. 158, 2059–2068. doi: 10.1007/s00705-013-1708-5
Kroneman, A., Vennema, H., Deforche, K., v d Avoort, H., Peñaranda, S., Oberste, M. S., et al. (2011). An automated genotyping tool for enteroviruses and noroviruses. J. Clin. Virol. 51, 121–125. doi: 10.1016/j.jcv.2011.03.006
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Viol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Leigh, J. W., and Bryant, D. (2015). PopART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116. doi: 10.1111/2041-210X.12410
Liang, S., Liu, S., Zhang, C., and Zhou, Y. (2007). A simple reference state makes a significant improvement in near-native selections from structurally refined docking decoys. Proteins 69, 244–253. doi: 10.1002/prot.21498
Liang, S., Zheng, D., Standley, D. M., Yao, B., Zacharias, M., and Zhang, C. (2010). EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results. BMC Bioinformatics 11:381. doi: 10.1186/1471-2105-11-381
Lindesmith, L. C., Beltramello, M., Donaldson, E. F., Corti, D., Swanstrom, J., Debbink, K., et al. (2012). Immunogenetic mechanisms driving norovirus GII.4 antigenic variation. PLoS Pathog. 8:e1002705. doi: 10.1371/journal.ppat.1002705
Lindesmith, L. C., Costantini, V., Swanstrom, J., Debbink, K., Donaldson, E. F., Vinj,é, J., et al. (2013). Emergence of a norovirus GII.4 strain correlates with changes in evolving blockade epitopes. J. Virol. 87, 2803–2813. doi: 10.1128/JVI.03106-12
Lindesmith, L. C., Kocher, J. F., Donaldson, E. F., Debbink, K., Mallory, M. L., Swann, E. W., et al. (2017). Emergence of novel human norovirus GII.17 strains correlates with changes in blockade antibody epitopes. J. Infect. Dis. 216, 1227–1234. doi: 10.1093/infdis/jix385
Lovell, S. C., Davis, I. W., Arendall, W. B. 3rd, de Bakker, P. I., Word, J. M., Prisant, M. G., et al. (2003). Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450. doi: 10.1002/prot.10286
Lu, J., Fang, L., Zheng, H., Lao, J., Yang, F., Sun, L., et al. (2016). The evolution and transmission of epidemic GII.17 noroviruses. J. Infect. Dis. 214, 556–564. doi: 10.1093/infdis/jiw208
Martin, D. P., Murrell, B., Golden, M., Khoosal, A., and Muhire, B. (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1:vev003. doi: 10.1093/ve/vev003
Matsushima, Y., Ishikawa, M., Shimizu, T., Komane, A., Kasuo, S., Shinohara, M., et al. (2015). Genetic analyses of GII.17 norovirus strains in diarrheal disease outbreaks from December 2014 to March 2015 in Japan reveal a novel polymerase sequence and amino acid substitutions in the capsid region. Eur. Surveill. 20:21173. doi: 10.2807/1560-7917.ES2015.20.26.21173
Medici, M. C., Tummolo, F., Calderaro, A., Chironna, M., Giammanco, G. M., De Grazia, S., et al. (2015). Identification of the novel Kawasaki 2014 GII.17 human norovirus strain in Italy, 2015. Eur. Surveill. 20:30010. doi: 10.2807/1560-7917.ES.2015.20.35.30010
Mizukoshi, F., Nagasawa, K., Doan, Y. H., Haga, K., Yoshizumi, S., Ueki, Y., et al. (2017). Molecular evolution of the RNA-dependent RNA polymerase and capsid genes of human norovirus genotype GII.2 in Japan during 2004-2015. Front. Microbiol. 8:705. doi: 10.3389/fmicb.2017.00705
Mori, K., Motomura, K., Somura, Y., Kimoto, K., Akiba, T., and Sadamasu, K. (2017). Comparison of genetic characteristics in the evolution of Norovirus GII.4 and GII.17. J. Med. Virol. 89, 1480–1484. doi: 10.1002/jmv.24791
Motoya, T., Nagasawa, K., Matsushima, Y., Nagata, N., Ryo, A., Sekizuka, T., et al. (2017). Molecular evolution of the VP1 gene in human norovirus GII.4 variants in 1974-2015. Front. Microbiol. 8:2399. doi: 10.3389/fmicb.2017.02399
Murrell, B., Moola, S., Mabona, A., Weighill, T., Sheward, D., Kosakovsky Pond, S. L., et al. (2013). FUBAR: a fast, unconstrained bayesian approximation for inferring selection. Mol. Biol. Evol. 30, 1196–1205. doi: 10.1093/molbev/mst030
Murrell, B., Wertheim, J. O., Moola, S., Weighill, T., Scheffler, K., and Kosakovsky Pond, S. L. (2012). Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 8:e1002764. doi: 10.1371/journal.pgen.1002764
Nagasawa, K., Matsushima, Y., Motoya, T., Mizukoshi, F., Ueki, Y., Sakon, N., et al. (2018). Genetic analysis of human norovirus strains in Japan in 2016-2017. Front. Microbiol. 9:1. doi: 10.3389/fmicb.2018.00001
Ng, K. K., Arnold, J. J., and Cameron, C. E. (2008). Structure-function relationships among RNA-dependent RNA polymerases. Curr. Top. Microbiol. Immunol. 320, 137–156. doi: 10.1007/978-3-540-75157-1_7
Ozaki, K., Matsushima, Y., Nagasawa, K., Motoya, T., Ryo, A., Kuroda, M., et al. (2018). Molecular evolutionary analyses of the RNA-dependent RNA polymerase region in norovirus genogroup II. Front. Microbiol. 9:3070. doi: 10.3389/fmicb.2018.03070
Parra, G. I., Squires, R. B., Karangwa, C. K., Johnson, J. A., Lepore, C. J., Sosnovtsev, S. V., et al. (2017). Static and evolving norovirus genotypes: implications for epidemiology and immunity. PLoS Pathog. 13:e1006136. doi: 10.1371/journal.ppat.1006136
Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., et al. (2004). UCSF Chimera–a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612. doi: 10.1002/jcc.20084
Pond, S. L., and Frost, S. D. (2005). Datamonkey: rapid detection of selective pressure on individual sites of codon alignments. Bioinformatics 21, 2531–2533. doi: 10.1093/bioinformatics/bti320
Quiñones-Mateu, M. E., Avila, S., Reyes-Teran, G., and Martinez, M. A. (2014). Deep sequencing: becoming a critical tool in clinical virology. J. Clin. Virol. 61, 9–19. doi: 10.1016/j.jcv.2014.06.013
Sakon, N., Sadamasu, K., Shinkai, T., Hamajima, Y., Yoshitomi, H., Matsushima, Y., et al. (2018). Foodborne outbreaks caused by human norovirus GII.P17-GII.17-contaminated nori, Japan, 2017. Emerg. Infect. Dis. 24, 920–923. doi: 10.3201/eid2405.171733
Sang, S., and Yang, X. (2018). Evolutionary dynamics of GII.17 norovirus. PeerJ. 6:e4333. doi: 10.7717/peerj.4333
Siebenga, J. J., Lemey, P., Kosakovsky Pond, S. L., Rambaut, A., Vennema, H., and Koopmans, M. (2010). Phylodynamic reconstruction reveals norovirus GII.4 epidemic expansions and their molecular determinants. PLoS Pathog. 6:e1000884. doi: 10.1371/journal.ppat.1000884
Silva, L. D., Bandeira, R. D., Junior, E. C., Lima, I. C., da Penha Júnior, E. T., Teixeira, D. M., et al. (2017). Detection and genetic characterization of the emergent GII.17_2014 norovirus genotype among children with gastroenteritis from Northern Brazil. Infect. Genet. Evol. 48, 1–3. doi: 10.1016/j.meegid.2016.11.027
Standley, D. M., Toh, H., and Nakamura, H. (2007). ASH structure alignment package: sensitivity and selectivity in domain classification. BMC Bioinformatics 8:116. doi: 10.1186/1471-2105-8-116
Tan, M., Xia, M., Chen, Y., Bu, W., Hegde, R. S., Meller, J., et al. (2009). Conservation of carbohydrate binding interfaces: evidence of human HBGA selection in norovirus evolution. PLoS ONE 4:e5058. doi: 10.1371/journal.pone.0005058
Tohma, K., Lepore, C. J., Ford-Siltz, L. A., and Parra, G. I. (2017). Phylogenetic analyses suggest that factors other than the capsid protein play a role in the epidemic potential of GII.2 norovirus. mSphere 2:e00187-17. doi: 10.1128/mSphereDirect.00187-17
Weaver, S., Shank, S. D., Spielman, S. J., Li, M., Muse, S. V., and Kosakovsky Pond, S. L. (2018). Datamonkey 2.0: a modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 35, 773–777. doi: 10.1093/molbev/msx335
Keywords: GII.P17-GII.17, human norovirus, molecular evolution, RdRp, VP1
Citation: Matsushima Y, Mizukoshi F, Sakon N, Doan YH, Ueki Y, Ogawa Y, Motoya T, Tsukagoshi H, Nakamura N, Shigemoto N, Yoshitomi H, Okamoto-Nakagawa R, Suzuki R, Tsutsui R, Terasoma F, Takahashi T, Sadamasu K, Shimizu H, Okabe N, Nagasawa K, Aso J, Ishii H, Kuroda M, Ryo A, Katayama K and Kimura H (2019) Evolutionary Analysis of the VP1 and RNA-Dependent RNA Polymerase Regions of Human Norovirus GII.P17-GII.17 in 2013–2017. Front. Microbiol. 10:2189. doi: 10.3389/fmicb.2019.02189
Received: 12 June 2019; Accepted: 05 September 2019;
Published: 27 September 2019.
Edited by:
Stefan Taube, Universität zu Lübeck, GermanyReviewed by:
Janet Mans, University of Pretoria, South AfricaSandra Niendorf, Robert Koch Institute, Germany
Copyright © 2019 Matsushima, Mizukoshi, Sakon, Doan, Ueki, Ogawa, Motoya, Tsukagoshi, Nakamura, Shigemoto, Yoshitomi, Okamoto-Nakagawa, Suzuki, Tsutsui, Terasoma, Takahashi, Sadamasu, Shimizu, Okabe, Nagasawa, Aso, Ishii, Kuroda, Ryo, Katayama and Kimura. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kazuhiko Katayama, a2F0YXlhbWFAbGlzY2kua2l0YXNhdG8tdS5hYy5qcA==; Hirokazu Kimura, aC1raW11cmFAcGF6LmFjLmpw