 Ann M. Arfken
Ann M. Arfken Juli Foster Frey
Juli Foster Frey Timothy G. Ramsay
Timothy G. Ramsay- Animal Biosciences and Biotechnology Laboratory, United States Department of Agriculture–Agricultural Research Service, Beltsville, MD, United States
Interactions between the bacteria and fungi in the gut microbiome can result in altered nutrition, pathogenicity of infection, and host development, making them a crucial component in host health. Associations between the mycobiome and bacteriome in the piglet gut, in the context of weaning, remain unknown. Weaning is a time of significant stress, dietary changes, microbial alterations, and a predisposition to infection. The loss of animal health and growth makes potential microbial interventions of interest to the swine industry. Recent studies have demonstrated the diversity and development of the microbiome in the gastrointestinal (GI) tract of piglets during weaning, resulting from the dietary and physiological changes. Despite these advances, the role of the mycobiota in piglet health and its contribution to overall microbiome development remains mostly unknown. In this study we investigated the bacteriome and the mycobiome after weaning in the GI tract organs and feces from 35-day old piglets. Following weaning, the α-diversity and amplicon sequence variants (ASVs) counts of the bacteriome increased, proximally to distally, from the stomach to the feces along the GI tract, while the mycobiome α-diversity and ASV counts were highest in the porcine stomach. β-diversity analyses show distinct clusters based on organ type in the bacteriome and mycobiome, but dispersion remained relatively constant in the mycobiome between organ/fecal sites. Bacteroidetes, Firmicutes, and Epsilonbacteraeota were the most abundant bacterial phyla present in the GI tract and feces based on mean taxonomic composition with high variation of composition found in the stomach. In the mycobiome, the dominant phyla were Ascomycota and Basidiomycota, and the stomach mycobiome did not demonstrate the same high level of variation observed in the bacteriome. Potential interactions between genera were found in the lower piglet GI bacteriome and mycobiome with positive correlations found between the fungus, Kazachstania, and several bacterial species, including Lactobacillus. Aspergillus demonstrated negative correlations with the short chain fatty acid-producing bacteria Butyricoccus, Subdoligranulum, and Fusicatenibacter. This study demonstrates the distinct colonization dynamics between fungi and bacteria in the GI tract and feces of piglets directly following weaning and the potential interactions of these microbes in the porcine gut ecosystem.
Introduction
The microbiome plays a critical role in animal health through its ability to alter nutrition, physiology, immune system development and function, and through bacterial–fungal–host interactions. Fungi in the GI tract of piglets are ubiquitous members of the rare biosphere (Huffnagle and Noverr, 2013; Summers et al., 2019) and disruption of the mycobiome may result in disease, as it does in other species (Ott et al., 2008; Iliev et al., 2012; Mason et al., 2012a; Erb Downward et al., 2013; Li et al., 2014; Li Z. et al., 2019). Additionally, fungi affect gut community structure and function through genetic exchange, interactions with bacterial species, biofilm formation, secondary metabolite secretion, and potential antibiotic creation (Frey-Klett et al., 2011; Suhr and Hallen-Adams, 2015).
Recent studies have demonstrated that commensal fungi can alter host immunity during normal health, as well as modify the severity of some diseases (Mukherjee et al., 2015; Kureljusic et al., 2016; Weissenbacher-Lang et al., 2016; Iliev and Leonardi, 2017; Limon et al., 2017; Richard and Sokol, 2019). Fungi can alter host immune responses through direct and indirect actions in the GI tract via pattern recognition receptors (PRRs), the production of metabolites such as prostaglandin E2 (PGE2), and multiple virulence factors that assist in host tissue invasion and nutrient acquisition. PGE2 is an immunomodulator typically produced by immune cells that can also be secreted by some fungi, such as Candida, leading to extensive immune changes in humans (Kim et al., 2014). Further, studies suggest that commensal fungi may promote immune tolerance to commensal bacteria (Li X.V. et al., 2019). In the context of pigs, studies have documented the effect of mycotoxins, fungal secondary metabolites known for contaminating agricultural feed, on the immune response. Different mycotoxins have the ability to up- or down-regulate the immune response in pigs, and immunosuppressive mycotoxins may increase piglet susceptibility to infectious diseases (Pierron et al., 2016). Due to the known sensitivity of piglets to these fungal metabolites, future studies are vital to understand the role of commensal fungi in porcine health.
The weaning transition is a stressful time in a pig’s life and associated changes in the piglet gut microbiome can result in poor health and reduced growth performance, making it of critical interest to the swine industry (Campbell et al., 2013; Guevarra et al., 2018, 2019). Post-weaning diarrhea and susceptibility to opportunistic pathogens are common consequences of changes to the piglet gastrointestinal (GI) microbiome. Recently, studies have begun to elucidate the normal members of the microbiota in piglets, but details remain unknown as to interactions among members alter immune responses and promote growth performance. This information is necessary to identify potential alternative growth promotants as the use of antibiotics for growth promotion is banned in the United States. While studies have begun to show the importance of weaning and diet changes in the development of the GI microbiome (Bian et al., 2016; Han et al., 2017), the mycobiota remains a poorly understood, yet integral part of the gut ecosystem.
Members of the microbiota interact with each other within the host environment through a variety of means, including physical or chemical interactions, competition for resources or space, production of biofilms, or modulation of the surrounding environment (Krüger et al., 2019). For example, studies have demonstrated the ability of bacterial metabolites to directly inhibit Candida growth and colonization in the gut (Nguyen et al., 2011; Bulgasem et al., 2016) as well as the production of mycotoxins by bacteria residing within the fungal cytosol (Partida-Martinez and Hertweck, 2005). Bacteria can also indirectly inhibit fungal growth by activating different components of the immune system. One such example is the ability of lactobacilli to promote host resistance to gut colonization with Candida spp. through the activation of AhR, a transcription factor that stimulates the release of IL-22 (Kiss et al., 2011; Zelante et al., 2013; Lamas et al., 2016). In humans, Candida albicans can prevent the gut colonization of other fungal and bacterial pathogens (Tso et al., 2018) and Aspergillus fumigatus can inhibit Pseudomonas aeruginosa and alter the pro-inflammatory immune response in co-cultures (Reece et al., 2018). The microbial interplay in the piglet gut may significantly alter the growth and health of pigs long-term due to the numerous potential interactions between fungi and bacteria. Recent studies have demonstrated a link between certain fungal species and weight gain in other mammalian species (Mar Rodriguez et al., 2015), and while currently unknown, potential dietary intervention strategies for piglet weight gain is of great interest to industry (Sam et al., 2017). Previous work from our laboratory has shown that the dominant, post-weaning fungal species is Kazachstania slooffiae, but its role in animal health and development remains to be elucidated (Summers et al., 2019). We hypothesize that the bacteriome and mycobiome will significantly differ between organ sites. The current study investigated the microbiome and mycobiome in piglets 2 weeks post-weaning to evaluate the diversity, populations, and potential interactions between the bacterial and fungal members of the piglet GI tract and feces.
Materials and Methods
Animal Procedures
A 23 Large White × Landrace piglets from 3 litters (L.119 = 8 piglets, L.120 = 8 piglets, and L.126 = 7 piglets) were assessed from birth through day 35 of age and were weaned at day 21. Piglets were not provided with creep feed or milk replacer at any point throughout the experiment. The diet was formulated to meet the National Research Council estimate of nutrient requirements (Supplementary Table S1). From days 21–28, piglets received Nursery Diet 1 followed by Nursery Diet 2 from days 29–35. Piglets were evaluated daily for health and were given free access to feed and water; all piglets used in this study were observed to be healthy. No antibiotics, antifungals, or supplementary additives were administered to the piglets at any time during the experiment. On day 35 of age, piglets were humanely euthanized, and the GI tract was removed from the abdominal cavity and immediately dissected. Sections from the stomach, proximal duodenum, jejunum, distal ileum, cecum, distal colon, and feces were collected under sterile conditions and luminal contents removed with a PBS wash. Mucosal-associated microbial populations were investigated due to their proximity with the host and the potential to alter host tissue responses. Organ sections and feces were placed in sterile cryovials, flash frozen in liquid nitrogen, and stored at −80°C until further processing. Care and treatment of all pigs were approved by the USDA–ARS Institutional Animal Care and Use Committee of the Beltsville Agricultural Research Center.
DNA Extraction and Sequencing
DNA was isolated from 0.25 g feces or organ sections using the MagAttract Power Microbiome Kit (Qiagen, Hilden, Germany) by the Microbial Systems Molecular Biology Laboratory at the University of Michigan. Cells were lysed to isolate DNA using mechanical bead beating for 20 total minutes with 20 frequency/second and extracted using magnetic bead technology according to the Qiagen protocol. The V4 region of the 16S rRNA-encoding gene was amplified from extracted DNA using the barcoded dual-index primers developed previously (Kozich et al., 2013). The ITS region was sequenced utilizing primers ITS3 (5′ GCATCGATGAAGAACGCAGC-3′) AQ3 and ITS4 (5′-TCCTCCGCTTATTGATATGC-3′) with the Illumina adaptor sequence added to the 5′ end (5′-TCGTCGGCAGCGTCAGATGTG TATAAGAGACAG—ITS3-3′) and (5′GCTTCGTGGGCTCGGAGATGTGTATAAG AGACAG—ITS4-3′). Both 16S and ITS regions were sequenced with the Illumina MiSeq Sequencing platform.
Bacteriome (16S) and Mycobiome (ITS) Sequence Processing
Bacteria (16S)
Quality filtering, pairing, denoising, amplicon sequence variants (ASVs) determination, and chimera removal was conducted with the DADA2 plugin (Callahan et al., 2016) in QIIME2 v. 2019.4 (Caporaso et al., 2010). For quality trimming, paired-end sequences were truncated to 240 and 160 bp for forward and reverse reads, respectively, with an average median quality score of 34.8. Taxonomic classification of the ASVs was performed using the pretrained 16S 515F/806R from the Silva 132 database (Yilmaz et al., 2014). ASVs identified as Archaea, chloroplast, mitochondria, or unassigned were removed from further analysis.
Fungi (ITS)
Forward and reverse primers were removed from paired-end reads with cutadapt v 1.18 (Martin, 2011). QIIME2 plugin DADA2 was used to perform similar quality filtering and ASV identification described above for bacterial sequences. Because of the variable nature of fungal ITS sequencing length, however, no quality trimming was conducted on fungal sequences. Average median quality score was 35.9 and 32.3 for forward and reverse reads, respectively. Taxonomic classification was trained and conducted on fungal sequences using the UNITE (Koljalg et al., 2013) developer’s full-length ITS reference sequences in QIIME2. Fungal ASVs without a phylum or higher classification or those identified as unassigned were removed. Additional classification using BLAST1 was performed on removed sequences to confirm non-fungal origin.
Separate rarefaction curves for bacterial and fungal samples were produced using the vegan package (Oksanen et al., 2019) in R v 3.5.12 and visualized in GraphPad Prism v 7 (La Jolla, CA, United States) to determine minimum sequencing depth. A cutoff of 5,000 sequences was determined for bacterial and fungal samples. Samples <5000 sequences were removed (bacteria, n = 30; fungus, n = 11). 130 bacterial and 149 fungal samples were selected for downstream analysis.
Characterization of the Bacteriome and Mycobiome
Calculations of α-diversity were performed on rarefied (n = 5,000 sequences) bacterial and fungal samples using the phyloseq package (McMurdie and Holmes, 2013). Shannon diversity indices and observed ASVs were normalized using box cox and square root transformations, respectively. Satisfaction of normality was tested using the Shapiro–Wilk test. Differences between bacterial and fungal Shannon diversity and observed ASVs were determined using a linear mixed model with organ as the fixed effect and pig as the random effect using the lmer4 and lmerTest. Non-metric multidimensional scaling (NMDS) was conducted using the vegan package on log-transformed bacterial and fungal sequences using Bray–Curtis dissimilarity distances. To reduce potential ASV artifacts, ASVs with <1 sequence in ≤5.0% of samples were removed prior to analysis. NMDs plots were visualized using the ggplot2 package (Wickham, 2016). Pairwise comparisons of mean Bray–Curtis distances to group centroids was calculated using the permutational analysis of multivariate dispersion (PERMDISP) function in vegan and plotted in R. Due to similarities between organ bacteriomes and mycobiomes, samples were recategorized by GI region: duodenum, jejunum, and ileum samples were recategorized as “Upper GI,” cecum and colon were recategorized as “Lower GI,” and stomach and feces remained categorized as “Stomach” and “Feces,” respectively. For visualization purposes, relative abundances of taxa are presented as mean% value by litter for each GI tract region and feces.
Correlation and Network Analyses of the Lower GI
For correlation analysis, samples were rarefied to their corresponding bacterial or fungal sample pair to account for sequencing depth differences between pairs while retaining similar community composition structure. Bacterial (n = 46) and fungal sample pairs (n = 46) were combined and ASVs were merged at the genus level. Genera found <30% of samples were removed to prevent degradation of correlation detection, which increases with increased numbers of 0 counts (Weiss et al., 2017). Correlations between fungus and bacteria were detected using the sparse correlations for composition (SparCC) python module (Friedman and Alm, 2012). Correlation values were visualized using the corrplot package in R. P-values were corrected for multiple comparisons using FDR. A corresponding network analysis of SparCC correlation coefficients was created using the igraph (Csardi and Nepusz, 2005) and the Sparse and Compositionally Robust Inference of Microbial Ecological Networks (Kurtz et al., 2015) R-packages. Only correlations with an absolute value ≥0.4 were plotted. Unless otherwise stated, all statistical tests were performed in R, p-values of <0.05 were considered significant, and errors are given as ±SE. All figures were created with GraphPad Prism 7, unless otherwise indicated.
Results
Composition and Diversity of the Bacteriome and Mycobiome in the Piglet GI Tract
To analyze the microbiota communities in the piglet GI tract, the V4 and ITS2 regions of the bacterial 16S rRNA and fungal ITS genes, respectively, were amplified and sequenced from feces and six organ sections (stomach, duodenum, jejunum, ileum, cecum, and colon) collected from 23 piglets, aged 35 days. A total of 8,363,058 bacterial and 6,670,225 fungal high quality sequences were obtained following the QIIME processing and filtering pipeline. Rarefaction curves showed that a minimum sampling depth of 5,000 sequences was sufficient to capture both bacterial and fungal diversity in organs and feces (Supplementary Figures S2A,B). After removal of samples with <5000 sequences, the number of bacterial and fungal samples was reduced to 130 and 149, respectively (Supplementary Tables S2A,B). A mean sequencing depth of 24,909 ± 1,448 and a total of 2383 ASVs were detected in bacterial samples, and a mean sequencing depth of 35,187 ± 1,585 and a total of 592 ASVs were detected in fungal samples.
Indices for Shannon and observed ASVs were calculated to measure the α-diversity in the bacteriome and mycobiome (Figures 1A,B and Supplementary Tables S3A,B). In the bacteriome, the overall trend showed an increase in diversity and observed ASVs from the stomach to the feces along the GI tract. The mycobiome showed a different trend, with the stomach showing higher diversity and observed ASVs, followed by a decrease in diversity and observed ASVs in the duodenum, jejunum, and ileum and an increase in diversity and observed ASVs in the colon. Compared to the mycobiome, diversity and observed ASVs were significantly higher in the bacteriome (p < 0.001, Supplementary Tables S2A,B).
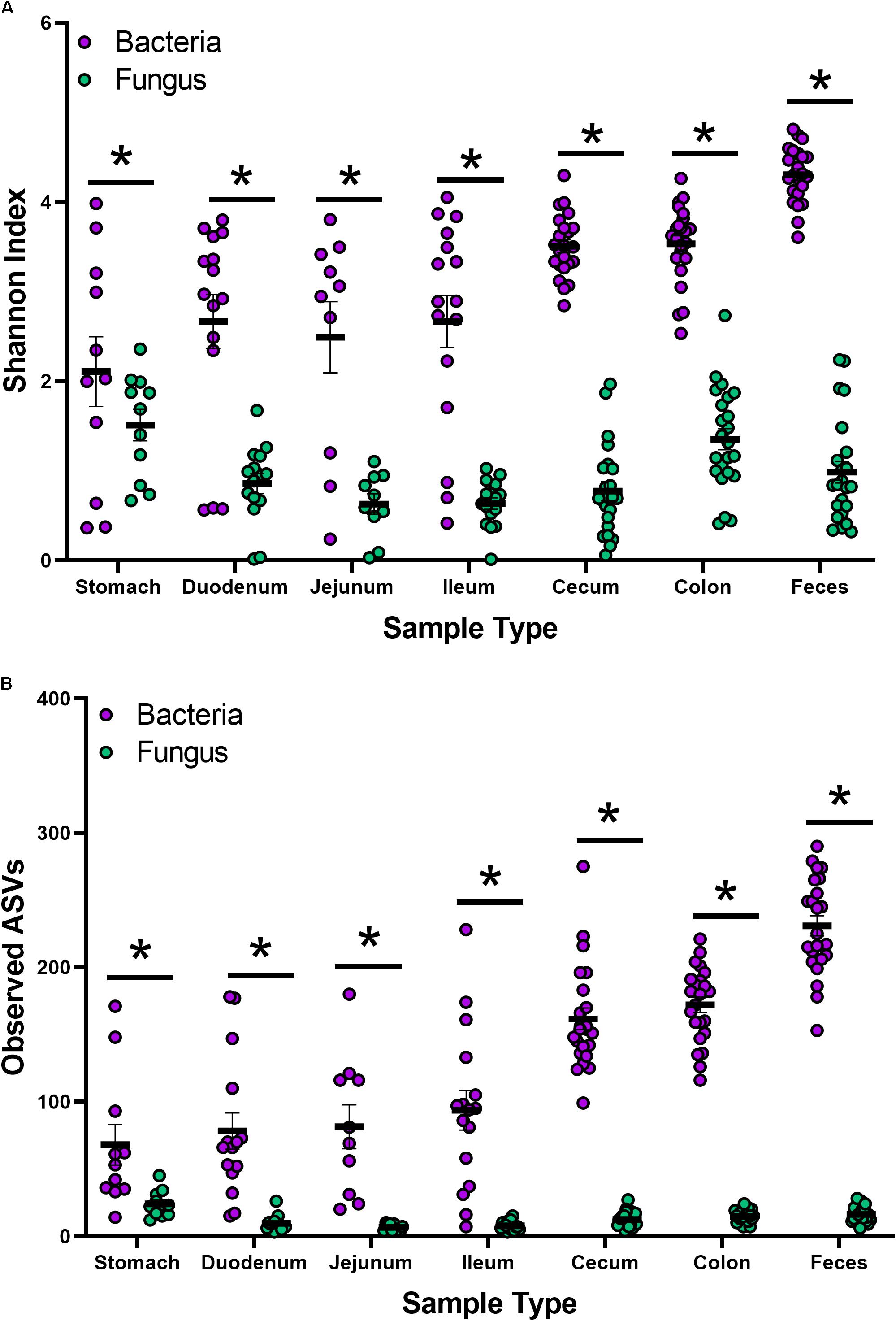
Figure 1. Alpha-diversity of the bacteriome and mycobiome in piglet GI organs. (A) Shannon diversity index values and (B) observed ASV counts for bacterial 16S rRNA and fungal ITS gene sequencing data by sample type. Linear mixed-models were performed to determine differences between bacterial and fungal indices by organ. Only samples with both bacterial 16S rRNA and fungal ITS gene sequencing data were plotted and analyzed. Significance indicated by ∗p < 0.001.
Non-metric multidimensional scaling plot were used to visualize β-diversity between the different regions and organs of the piglet GI (Figures 2A,B). In both the bacteriome and mycobiome, stomach and feces showed distinct clusters from the other organs. The duodenum, jejunum, and ileum organs in the upper GI, and the cecum and colon in the lower GI had a high degree of overlap among their centroids within their respective GI tract region indicating similarities between the microbiota communities. Mean distances between group centroids (dispersion) for each organ were calculated using PERMDISP on Bray–Curtis dissimilarities (Supplementary Tables S4A,B). In the bacteriome, there was a significant decrease in dispersion from the stomach and upper GI tract to the lower GI tract and to the feces, signifying a larger amount of individual variation in the upper GI tract vs. the lower GI tract and feces (p < 0.05, Figure 3A and Supplementary Table S2A). This trend in dispersion directly contrasted with α-diversity, which showed an increase in observed ASVs and Shannon diversity from the stomach to the lower GI as shown previously (Figure 1A). The mycobiome showed no significant trends in dispersion and remained relatively similarly dispersed throughout the piglet GI tract and feces (p ≥ 0.05, Figure 3B, and Supplementary Table S2B).
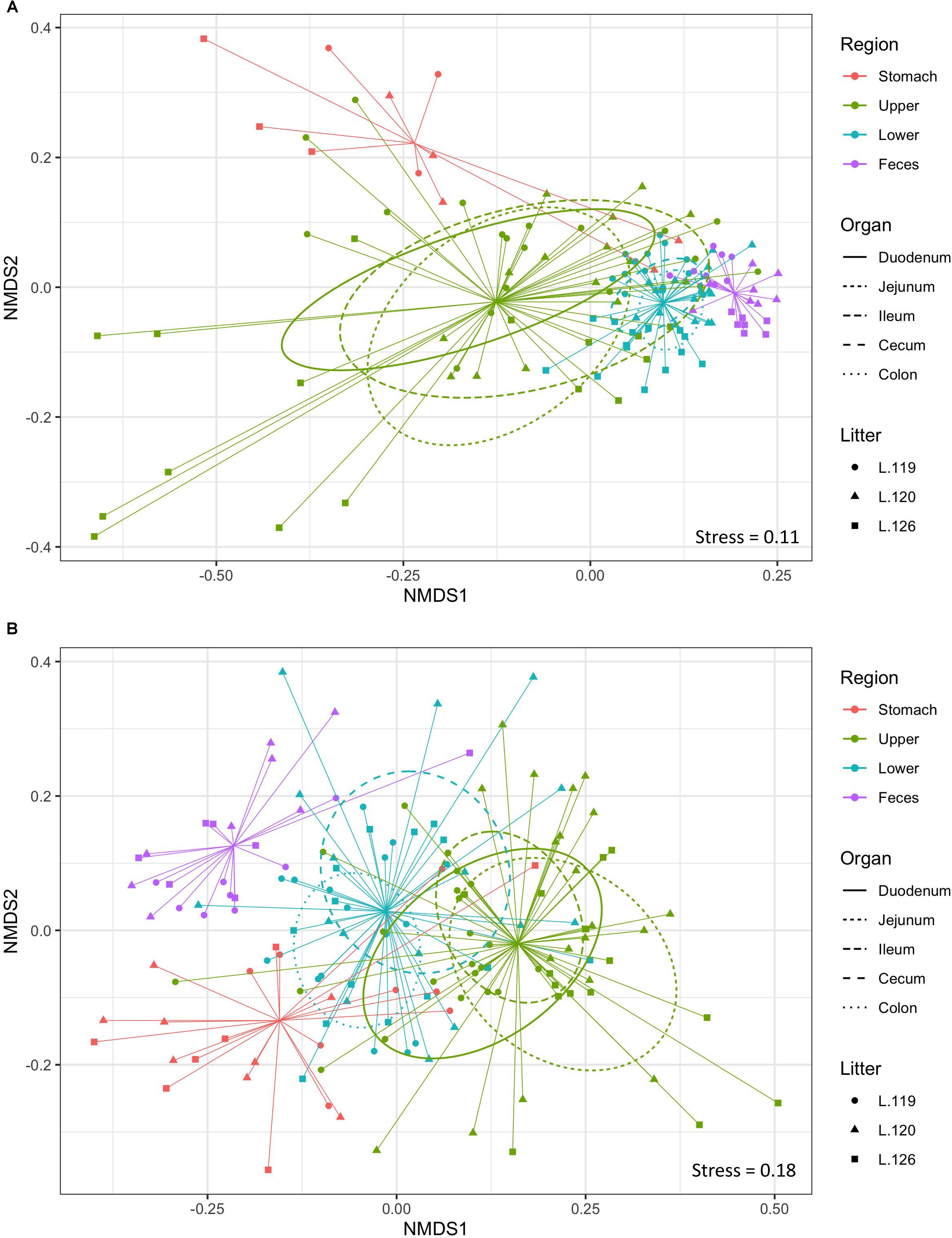
Figure 2. Beta-diversity of gastrointestinal tract organs and litters. Non-metric multidimensional scaling (NMDS) plot of β-diversity based on Bray–Curtis dissimilarities in the (A) bacteriome and (B) mycobiome of the piglet GI tract. Ellipses indicate 1 standard deviation from organ centroid and spiders are drawn to GI tract region centroid. Colors indicate GI tract region, symbols indicate litter, and ellipses line types indicate specific organs of the upper and lower GI.
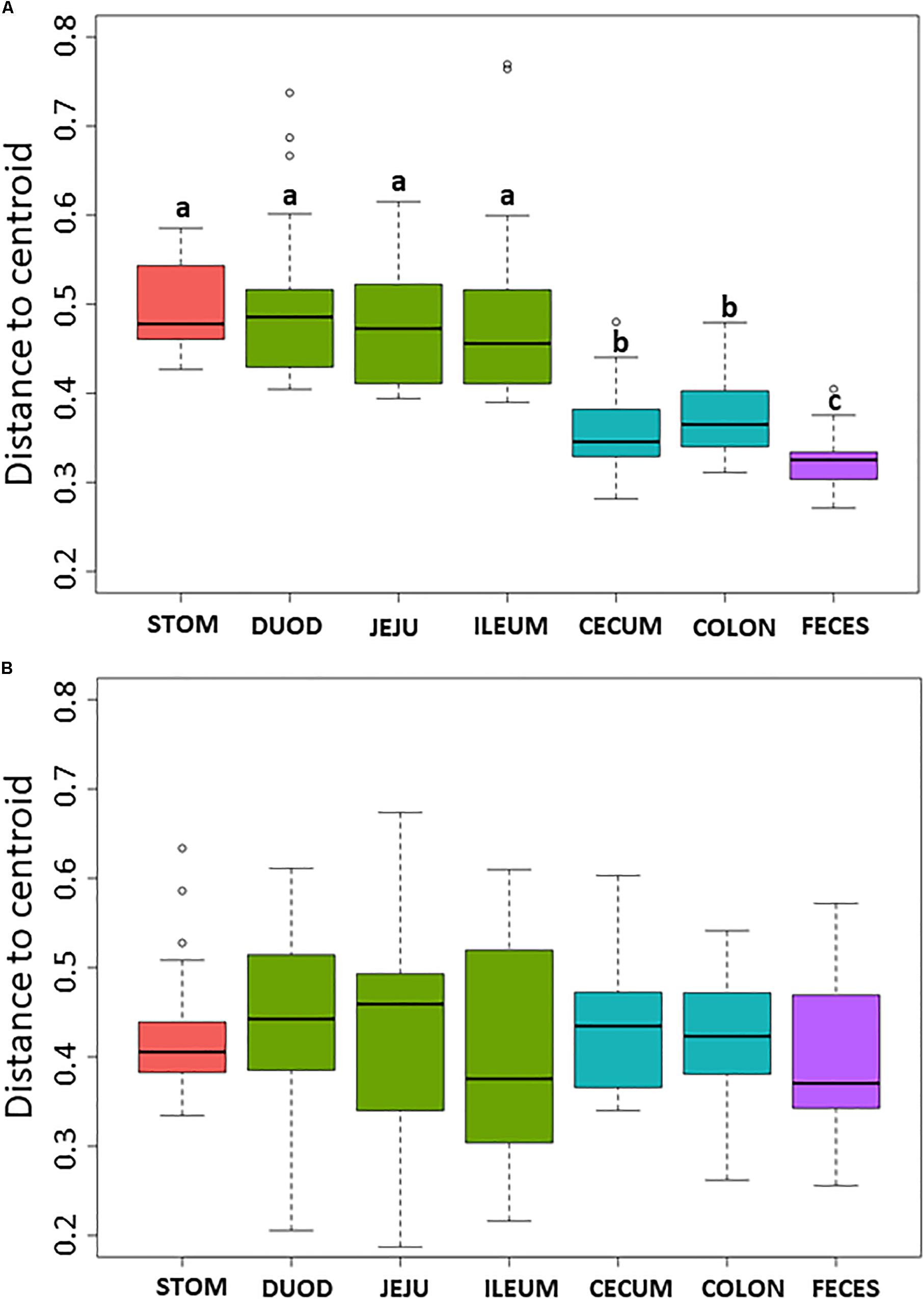
Figure 3. Box plot of pairwise distances between piglet organ centroids. Plots represent the median and interquartile range in the (A) bacteriome and (B) mycobiome. Colors indicate piglet GI tract region: red = stomach, green = upper GI, blue = lower GI, purple = feces. Differences between organ centroids were analyzed using permutational analysis of multivariate dispersion on Bray–Curtis dissimilarities with significance indicated by letters (p < 0.05).
Mean taxonomic composition by litter of bacterial and fungal families present in the piglet GI tract were compared across GI tract and feces (Figures 4A,B). In the bacteriome, the most abundant phyla (Supplementary Figure S2A) present in the GI tract and feces were Bacteroidetes (40.8 ± 1.9%), Firmicutes (37.2 ± 1.9%), and Epsilonbacteraeota (19.5 ± 2.5%) comprising >97% of the piglet bacteriome. Genera Prevotella 9, Prevotella 1, Prevotellaceae NK3B31 group, and Alloprevotella from family Prevotellaceae (38.2 ± 2.0%), Helicobacter from family Helicobacteraceae (17.4 ± 2.4%), Lactobacillus from family Lactobacillaceae (10.3 ± 1.4%), Blautia from family Lachnospiraceae (7.2 ± 0.4%), and Veillonella from family Veillonellaceae (5.2 ± 0.6%) were among the most abundant and prevalent genera and families in the bacteriome (Supplementary Figure S2C). In general, bacterial families Helicobacteraceae and Lactobacillaceae decreased from the stomach and upper GI to the lower GI and feces, while families Prevotellaceae, Lachnospiraceae, and Ruminoccocaceae increased along the GI tract and feces. Relative abundances of Helicobacteraceae in the feces were <1.0%. Of the different GI tract regions, the stomach showed high variation in taxonomic composition among litters, while the lower GI and feces showed relatively consistent taxa across litters. In the mycobiome, Ascomycota (90.7 ± 1.3%) and Basidiomycota (9.0 ± 1.2%) made up the dominant phyla (Supplementary Figure S2B). Genera Kazachstania (sp. sloofiae) from family Saccharomycetaceae (49.6 ± 2.8%), Hyphopichia from family Debaryomycetaceae (23.2 ± 2.3%), and Wallemia from family Wallemiaceae (6.3 ± 1.0%) were the dominant genera and families across all GI tract regions and feces (Figure 4B and Supplementary Figure S2D). Symbiotaphrina from family Symbiotaphrinaceae was dominant in the piglet GI tract organs (9.8 ± 2.0%) but was only found in 3 piglet feces samples at <0.1% abundance. In contrast to the stomach and feces bacteriome, the stomach mycobiome had relatively consistent taxa among the litters, while the feces mycobiome demonstrated a high degree of variation.
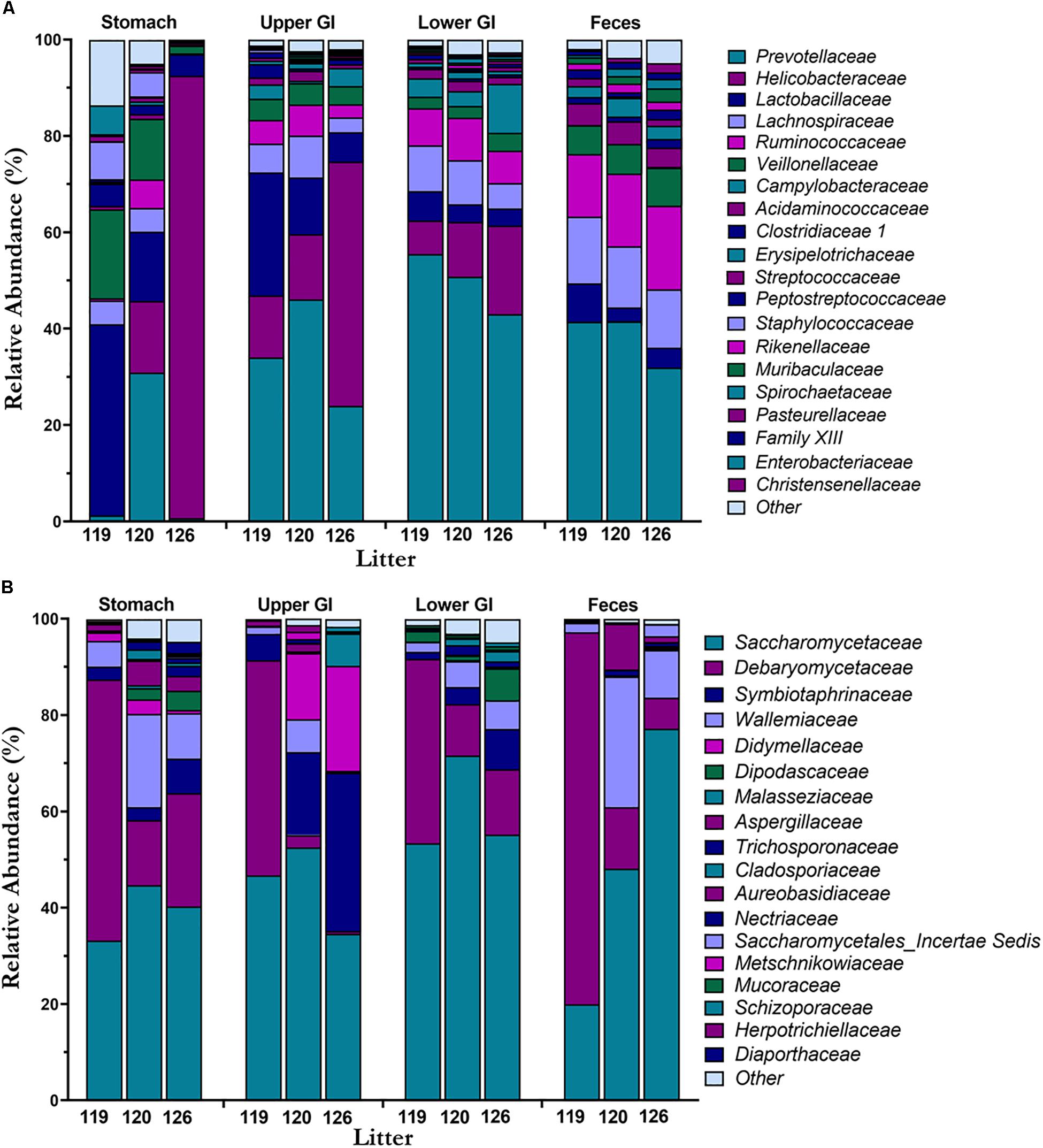
Figure 4. Taxonomic composition of the bacteriome and mycobiome in the piglet GI tract. Mean percent relative abundances by litter at the family level are shown for the most abundant members of the (A) microbiome and (B) mycobiome for each GI tract region.
Interactions Between the Bacteriome and Mycobiome in the Piglet Lower GI
Potential interactions between genera found in the lower piglet GI bacteriome and mycobiome were determined with SparCC correlations and a corresponding network analysis (Figures 5A,B and Supplementary Tables S5A,B). Fungi genus Kazachstania showed significant positive correlations with bacteria genera Alloprevotella, Lactobacillus, Prevotella 9, and Subdoligranulum. Fungi genera Aspergillus, Cladosporium, Hyphopichia, and Wallemia showed mostly negative correlations with other bacteria genera. Aspergillus, in particular, showed predominantly negative correlations with short chain fatty acid-producing bacteria such as Butyricicoccus, Subdoligranulum and Fusicatenibacter. Fungi genera Diopadascus, Symbiotaphrina, and Trichosporon did not show correlations with other fungi or bacteria. Additionally, there were no strong correlations among any of the other fungi genera identified within the piglet gut mycobiome.
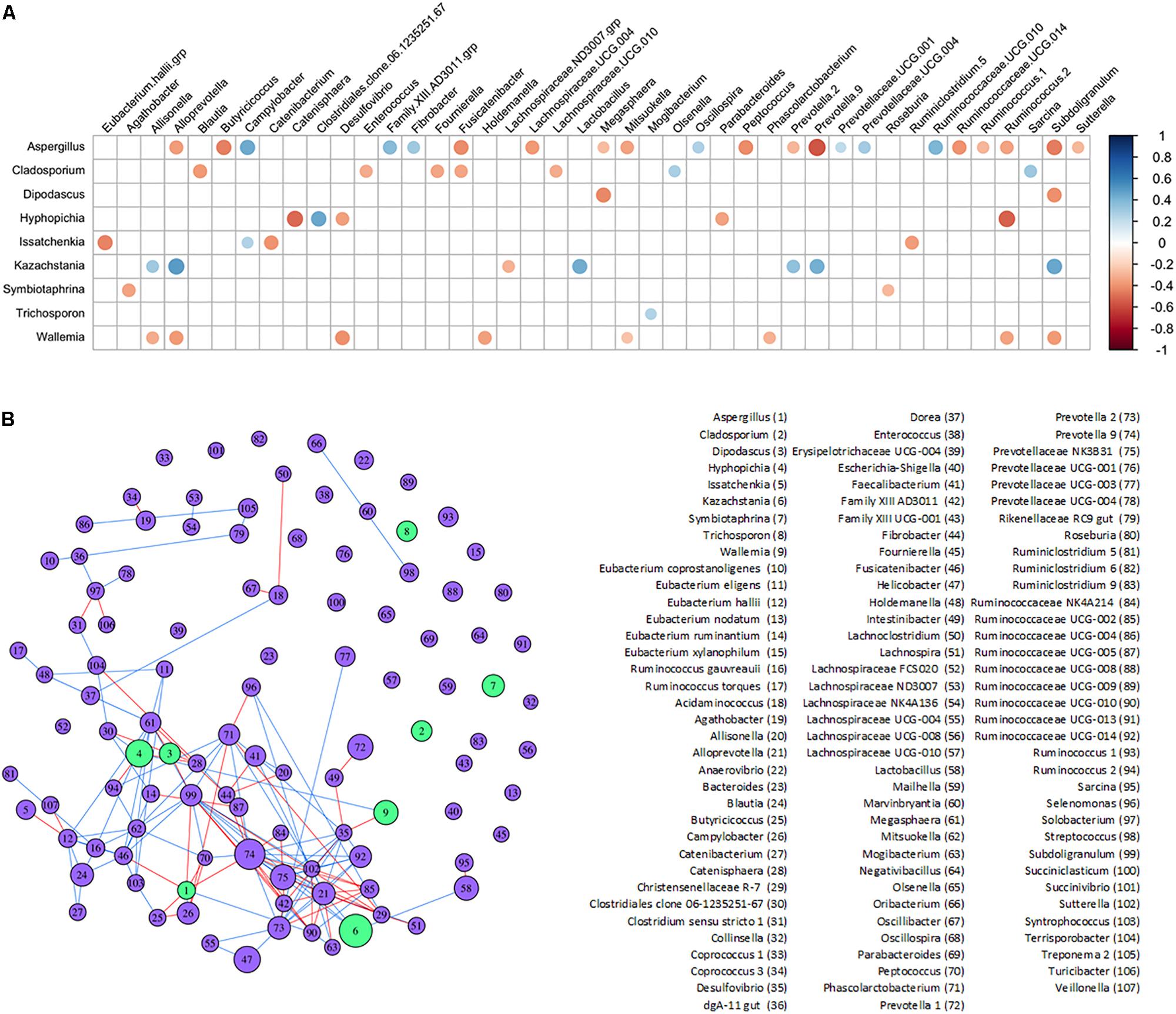
Figure 5. Inferred interactions between the bacteriome and mycobiome in the piglet lower GI tract. (A) SparCC correlation plot showing significant individual correlations between bacterial and fungal genera in the lower GI organs of the post-weaning piglet. Red circles indicate negative correlations and blue circles indicate positive correlations (p < 0.05 after FDR adjustments). The size of circles represents correlation strength while non-significant correlations are not shown. (B) SparCC correlation network between bacterial and fungal genera with plotted correlations with an absolute value ≥0.4. The edge color indicates sign of correlation: negative (red), positive (blue); node color indicates kingdom: bacteria (purple), fungus (green). The size of node is proportional to the mean centered-log ratio abundance for each genus.
Discussion
Fungi, in addition to bacteria, are important members and contributors of the microbiome, and recognition of their role is an essential step forward in elucidating the dynamics of the GI environment. Of the limited gut studies examining bacteria and fungi together, none to our knowledge have explored the interaction of the bacteriome–mycobiome of the piglet GI tract. Our study characterized and compared the mucosal-associated bacteriome and mycobiome of 23 healthy piglets, aged 35 days, from 3 litters along different organs of the GI tract and feces. The lower GI tract, which included the cecum and colon, was further evaluated to assess differences in predicted interactions between the bacteriome and mycobiome and determine associations between bacterial and fungal genera. This research is a critical first step in revealing the complex interactions, including health and growth, promoted by the fungi in the piglet gut during weaning.
While studies investigating the microbiota have become common, extensive studies of the mycobiota have been limited due to a lack of technologies, databases, and consensus in techniques (Li X.V. et al., 2019; Richard and Sokol, 2019). Recently, Li X.V. et al. (2019) reviewed studies characterizing the mycobiome of different human sites and disease states. Despite significant recent advances, methodologies continue to lack consensus for DNA isolation, primer design, sequencing, databases, and analysis of fungal species. Previous work in our laboratory aimed to determine which techniques and fungal primers were effective in studying the piglet mycobiome (Summers et al., 2019). Lower estimated diversity in the mycobiome compared to the bacteriome has been observed in human stool (Nash et al., 2017), piglet feces (Summers et al., 2019), and settling dust in pig farms (White et al., 2019). While there is no established consensus on what constitutes a healthy gut mycobiome, it is widely accepted that fungi are less abundant and demonstrate less diversity than bacteria in the human gut, comprising roughly 0.1% of the microbiome based on shotgun metagenome sequencing (Qin et al., 2010; Huffnagle and Noverr, 2013). In this study, compared to the bacteriome, estimated overall diversity and observed ASVs for all 6 GI organs (stomach, duodenum, jejunum, ileum, cecum, colon) and feces were significantly lower in the mycobiome (Figures 1A,B and Supplementary Figures S1A,B).
Within the bacteriome, there was an increasing trend in α-diversity along the GI tract from the stomach to the colon, with highest α-diversity found in the feces, coupled with a decreasing trend in dispersion (β-diversity) among the organs (Figure 1A and Supplementary Tables S3, S4). These trends are consistent with those seen by Crespo-Piazuelo et al. (2018), which characterized the bacteriomes of 120 day old pigs along the GI tract gradient from the duodenum to the distal colon. In general, the stomach and upper GI tract (duodenum, jejunum, and ileum) host fewer microorganisms than the lower GI tract (colon and cecum) due to shorter retention times for adherence to tissue or mucus (Donaldson et al., 2016) lower pH, and higher concentrations of bile acids (Mackie et al., 1999; Walter and Ley, 2011). The harsher environment of the stomach and upper GI may subsequently select for a smaller number of colonizing bacterial species resulting in reduced diversity. The stomach and organs associated with the upper GI bacteriome also demonstrated higher levels of dispersion than the lower GI and feces, indicating a greater level of individual variation among piglets. Unlike the lower GI, the stomach and upper GI are exposed to new and exogenous bacteria ingested with food particles (Donaldson et al., 2016). The stomach, in particular, serves to block ingested microbes from passing to the intestine (Martinsen et al., 2005). Despite identical piglet diets, individual variation was seen in the stomach and upper GI, potentially due to the amount and timing of the piglet’s meal. Other potential factors, including host immunity or fungal interactions, may influence bacterial variation in the upper GI of the piglet as the small intestine plays a critical role in the development of mucosal and systemic tolerance toward microbes (Villmones et al., 2018).
Much less is known about diversity trends in the gut mycobiome. Unlike the bacteriome, the mycobiome did not follow the same general linear increase in α-diversity along the GI tract (Figure 1B). Instead, the stomach mycobiome had the highest mean diversity, followed by the colon. The average gastric pH of 6-month old pigs fed ad libitum is 4.4, although this level can vary among individuals or timing of meals (Merchant et al., 2011). Compared to many bacteria, fungi are more acid tolerant. Many fungi have adaptive strategies to respond to low pH environments, and some fungi like Aspergillus sp. actively lower the surrounding pH of their environment (Vylkova, 2017). This suggests that the high diversity of the stomach mycobiome may be due to the greater survivability of fungi in highly acidic environments, as well as potentially less competition from bacteria for resources compared to the rest of the GI tract. Individual variation in the mycobiome remained relatively similar along the GI tract based on dispersion estimates (Figure 2B). Unlike the bacteriome, however, there was no reduction in dispersion in the lower GI or feces, and individual variation remained comparable to the upper GI bacteriome. Some studies have suggested that most fungi found in the GI tract are transient via environmental or dietary sources, and are unable to colonize or inhabit the gut long-term (Suhr and Hallen-Adams, 2015; Raimondi et al., 2019). The suspected temporary nature of some fungi in the piglet GI tract, as well as the genetic and immunity factors that affect the bacteriome, may all play a role in the relatively high level of individual variation in the mycobiome. Despite high individual variation within each organ mycobiome, distinct clusters were found for each GI tract region depicted in the NMDS (Figure 2B), indicating that fungal distribution along the GI tract is not random and may indicate different GI environmental niche effects on the mycobiome.
In both the bacteriome and mycobiome, there were dominant taxa throughout most of the piglet GI tract and feces (Figures 3A,B). In the bacteriome, Bacteroidetes, Firmicutes, and Epsilonbactereota were the dominant phyla, consistent with previous studies investigating the pig GI tract (Zhao et al., 2015; Kelly et al., 2017; Crespo-Piazuelo et al., 2018; Zhang et al., 2018). In previous studies, Proteobacteria was considered a dominant phylum, but recently Epsilonbactereota was reclassified as a separate phylum from Proteobacteria (Waite et al., 2017) and Epsilonbactereota was a more dominant phylum in our dataset. In general, there were increases in Prevotellaceae, Lachnospiraceae, and Ruminoccocaceae from the stomach to the lower GI tract, corresponding with a decrease in Lactobacillaceae and Helicobacteraceae. Many of these taxa shifts may be attributed to changing environmental conditions that occur along the GI tract. Measured dissolved oxygen levels undergo a dramatic reduction from the duodenum in the upper GI to the cecum of the lower GI (Hillman et al., 1993), as well as a reduction in pH, an increase in resistant starches (Flint et al., 2012), and slower peristalsis times (Walter and Ley, 2011). Members of Helicobacteraceae and Lactobacillaceae are tolerant of bile acids and oxygen (De Boever and Verstraete, 1999; Okoli et al., 2007; Yasuda et al., 2015), and are able to adhere firmly to the surface of the small intestine (Donaldson et al., 2016) making them suitable for colonizing the upper GI tract. In comparison, Prevotellaceae, Lachnospiraceae, and Ruminoccoccaceae are oxygen-sensitive and are likely more competitive in the lower GI due to their ability to degrade complex carbohydrates (Arumugam et al., 2011; Flint et al., 2012; Liu et al., 2012).
The dominant phyla in the piglet GI tract and feces mycobiome were Ascomycota and Basidiomycota, which are similar to those found in human mycobiome gut studies (Hoffmann et al., 2013; Nash et al., 2017; Raimondi et al., 2019). Unlike human studies, however, commonly found yeasts from genera Candida and Saccharomyces (Hallen-Adams and Suhr, 2017; Sam et al., 2017), were either absent or found at <1% relative abundance in our study samples. Instead, the dominant yeast throughout the piglet GI tract and feces was identified as Kazachstania (sp. sloofiae). K. sloofiae has been previously identified from different parts of the healthy pig GI tract (Uden and Carmo-Sousa, 1962; Urubschurov and Janczyk, 2011) and feces (Summers et al., 2019), and has been shown to establish quickly in the gut of piglets based on fecal analysis (Urubschurov et al., 2015; Urubschurov et al., 2017). Urubschurov et al. (2017) determined K. sloofiae may be responsible for maintaining piglet health by producing peptides, vitamin C, and formic acid in the piglet GI tract. Other dominant fungi genera found throughout the piglet GI tract and feces included Hyphopichia (sp. burtonii) and Wallemia. In contrast to K. sloofiae, both of these fungi are likely non-colonizing, transient fungi of the piglet GI tract. Hyphopichia burtonii, a commonly isolated yeast from corn, wheat, and rice (Kurtzman, 2011), has an estimated maximum growth temperature of 37°C (Burgain et al., 2015), and is unlikely to thrive at the internal temperature of a pigs at around 38.7–40°C. Wallemia, commonly isolated from food sources as well as agricultural dust, is also unlikely to reside in the piglet GI environment due to its extremophilic and xerophilic nature (Zajc and Gunde-Cimerman, 2018).
Complex interactions between bacteria and fungi also occur within the gut. Several significant correlations were found between bacterial and fungal genera in the lower piglet GI tract, suggesting potential bacteriome–mycobiome relationships (Figures 5A,B). The dominant fungal genus in the GI tract of humans is Candida and several species have been found to interact directly with bacterial species like Lactobacillus (Mason et al., 2012b; Allonsius et al., 2017; Hallen-Adams and Suhr, 2017; Rossoni et al., 2018). Kazachstania was strongly and positively correlated with Lactobacillus in the lower GI of our piglets and as Kazachstania is genetically similar to Candida (Kurtzman et al., 2005), it may be a potential porcine analog to Candida in the guts of humans. Future studies will be needed to clarify its role in pig gut health and homeostasis, as well as its potential to act as an opportunistic pathogen. A positive correlation between Kazachstania and Lactobacillus was also found in piglet feces in Urubschurov et al. (2011) using culture methods and PCR-DGGE techniques. In co-cultures of Lactobacillus and some yeasts, Lactobacillus released organic acids that lower the surrounding pH and promoted yeast growth; these yeasts are then stimulated by Lactobacillus to release essential nutrients and vitamins utilized by Lactobacillus (Stadie et al., 2013). A similar mutualistic relationship may exist between the dominant yeast Kazachstania in the piglet GI tract and Lactobacillus.
A strong positive correlation was also observed between Kazachstania and Prevotella 2 and Prevotella 9 genera. It has been hypothesized from observed positive associations between Candida yeasts and Prevotella, that Candida and Prevotella are involved in a mutualistic relationship regarding the degradation and fermentation of complex carbohydrates in the human gut (Hoffmann et al., 2013). In the piglet gut, Kazachstania may fulfill the role of Candida in the potential Candida-Prevotella link to starch metabolism. The corresponding network plot of the bacteriome–mycobiome community correlations showed no interactions between Cladosporium, Symbiotaphrina, and Trichosporon and other microbiota of the bacteriome–mycobiome. Unlike the Kazachstania-bacterial potential relationship, these fungi may be truly transient in the lower piglet GI and pass through without interacting with the gut microbial community. Interestingly, Hyphopichia and Wallemia showed mostly negative correlations with several gut bacteria. While these fungi are also thought to be non-colonizing microbiota of the gut as mentioned previously, they may still have an impact on the gut bacteriome during passage or may be capable of establishing themselves in the lower gut environment. One interesting finding in the lower gut of piglets was the negative association between short chain fatty acid-producing bacteria and Aspergillus. In humans, Aspergillus can exacerbate allergic responses in farm workers and is a well-documented pathogen. Aspergillosis is less common in pigs but has been documented as a rare cause of porcine abortions (Eustis et al., 1981; Todd et al., 1985; Sabino et al., 2012; Li Z. et al., 2019). Aspergillus has been documented to directly interact with bacterial species such as Stenotrophomonas and Pseudomonas and may play a critical role in disease severity (Schroeckh et al., 2009; Melloul et al., 2018; Briard et al., 2019). The negative association of Aspergillus with SCFA-producing bacteria, typically associated with beneficial gut health (Baxter et al., 2019; Peirce and Alvina, 2019) could be explained by the change in Aspergillus spp. behavior following sodium butyrate exposure (Philip et al., 1963).
In this study, we provided a comprehensive overview of the bacteria and fungi present along the piglet GI tract and feces in healthy piglets post-weaning, as well as new insight into potential interactions between the microbiome and mycobiome. The taxonomy and diversity of the mycobiome, in addition to the microbiome, demonstrated distinct differences in diversity between the bacterial and fungal members of the gut. In addition, fungal commensals, such as Candida spp., from the human gut were lacking in the pig. Potential interactions in the porcine gut show that bacteria may be acting in a beneficial way with the fungus, Kazachstania, and through negative interactions with Aspergillus. Further exploration of these significant correlations in the piglet gut will provide a greater understanding of the relationships that exist between the bacteriome and mycobiome that may potentially alter piglet growth and health.
Data Availability Statement
The datasets generated for this study can be found using the Accession number PRJNA558038: https://www.ncbi.nlm.nih.gov/bioproject/PRJNA558038.
Ethics Statement
The animal study was reviewed and approved by the USDA–ARS Institutional Animal Care and Use Committee of the Beltsville Agricultural Research Center.
Author Contributions
AA and KS contributed to the conception and design of the study, and wrote the first draft of the manuscript. AA performed the statistical analysis. All authors performed the animal research and handling, contributed to manuscript revision, read, and approved the submitted version.
Funding
This research did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank Jenile Tapscott, James Woods, and Russ Lange for assistance in maintaining and handling the experimental animals. We would like to thank Dr. Craig Storuzuk and Emily Beall for veterinary care of the experimental animals. We also thank Dr. Jonathan Shao and Matt Kramer for their discussions regarding statistics. This research was supported by work performed by the University of Michigan Microbial Systems Molecular Biology Laboratory.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02286/full#supplementary-material
FIGURE S1 | Rarefaction curves of mean observed ASVs in piglet organs and feces in (A) bacteria and (B) fungi. Error bars represent ±SE.
FIGURE S2 | Taxonomic composition of the bacteriome and mycobiome in the piglet GI tract. Mean percent relative abundances by litter are shown for the top taxa at the (A) bacterial phylum, (B) fungal phylum, (C) bacterial genus, and (D) fungal genus level for each GI tract organ.
TABLE S1 | Composition of diet for weaned piglets.
TABLE S2 | Distribution of bacterial and fungal samples by sample type and litter.
TABLE S3 | Summary results of (A) Shannon diversity and (B) observed ASV differences between bacteria and fungi in the piglet GI tract.
TABLE S4 | Summary results of dispersion comparisons in the (A) bacteriome and (B) mycobiome along the piglet GI tract.
TABLE S5 | Correlation (A) matrix and (B) FDR adjusted p-values between bacteria and fungi in the piglet lower GI tract.
Footnotes
References
Allonsius, C. N., van den Broek, M. F. L., De Boeck, I., Kiekens, S., Oerlemans, E. F. M., Kiekens, F., et al. (2017). Interplay between Lactobacillus rhamnosus gg and candida and the involvement of exopolysaccharides. Microb. Biotechnol. 10, 1753–1763. doi: 10.1111/1751-7915.12799
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Baxter, N. T., Schmidt, A. W., Venkataraman, A., Kim, K. S., Waldron, C., and Schmidt, T. M. (2019). Dynamics of human gut microbiota and short-chain fatty acids in response to dietary interventions with three fermentable fibers. mBio 10:e02566-18. doi: 10.1128/mBio.02566-18
Bian, G., Ma, S., Zhu, Z., Su, Y., Zoetendal, E. G., Mackie, R., et al. (2016). Age, introduction of solid feed and weaning are more important determinants of gut bacterial succession in piglets than breed and nursing mother as revealed by a reciprocal cross-fostering model. Environ. Microbiol. 18, 1566–1577. doi: 10.1111/1462-2920.13272
Briard, B., Mislin, G. L. A., Latge, J. P., and Beauvais, A. (2019). Interactions between aspergillus fumigatus and pulmonary bacteria: current state of the field, new data, and future perspective. J. Fungi 5:48. doi: 10.3390/jof5020048
Bulgasem, B. Y., Lani, M. N., Hassan, Z., Wan Yusoff, W. M., and Fnaish, S. G. (2016). Antifungal activity of lactic acid bacteria strains isolated from natural honey against pathogenic candida species. Mycobiology 44, 302–309. doi: 10.5941/MYCO.2016.44.4.302
Burgain, A., Bensoussan, M., and Dantigny, P. (2015). Validation of a predictive model for the growth of chalk yeasts on bread. Int. J. Food Microbiol. 204, 47–54. doi: 10.1016/j.ijfoodmicro.2015.03.026
Callahan, B. J., McMurdie, P. J., Rosen, M. J., Han, A. W., Johnson, A. J., and Holmes, S. P. (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. doi: 10.1038/nmeth.3869
Campbell, J. M., Crenshaw, J. D., and Polo, J. (2013). The biological stress of early weaned piglets. J. Anim. Sci. Biotechnol. 4:19. doi: 10.1186/2049-1891-4-19
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336.
Crespo-Piazuelo, D., Estelle, J., Revilla, M., Criado-Mesas, L., Ramayo-Caldas, Y., Ovilo, C., et al. (2018). Characterization of bacterial microbiota compositions along the intestinal tract in pigs and their interactions and functions. Sci. Rep. 8:12727. doi: 10.1038/s41598-018-30932-6
Csardi, G., and Nepusz, T. (2005). The igraph software package for complex network research. InterJ. Complex Syst. 1695, 1–9.
De Boever, P., and Verstraete, W. (1999). Bile salt deconjugation by Lactobacillus plantarum 80 and its implication for bacterial toxicity. J. Appl. Microbiol. 87, 345–352.
Donaldson, G. P., Lee, S. M., and Mazmanian, S. K. (2016). Gut biogeography of the bacterial microbiota. Nat. Rev. Microbiol. 14, 20–32. doi: 10.1038/nrmicro3552
Erb Downward, J. R., Falkowski, N. R., Mason, K. L., Muraglia, R., and Huffnagle, G. B. (2013). Modulation of post-antibiotic bacterial community reassembly and host response by Candida albicans. Sci. Rep. 3:2191. doi: 10.1038/srep02191
Eustis, S. L., Kirkbride, C. A., Gates, C., and Haley, L. D. (1981). Porcine abortions associated with fungi, actinomycetes, and Rhodococcus sp. Vet. Pathol. 18, 608–613. doi: 10.1177/030098588101800505
Flint, H. J., Scott, K. P., Duncan, S. H., Louis, P., and Forano, E. (2012). Microbial degradation of complex carbohydrates in the gut. Gut Microb. 3, 289–306.
Frey-Klett, P., Burlinson, P., Deveau, A., Barret, M., Tarkka, M., and Sarniguet, A. (2011). Bacterial-fungal interactions: hyphens between agricultural, clinical, environmental, and food microbiologists. Microbiol. Mol. Biol. Rev. 75, 583–609. doi: 10.1128/MMBR.00020-11
Friedman, J., and Alm, E. J. (2012). Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 8:e1002687. doi: 10.1371/journal.pcbi.1002687
Guevarra, R. B., Hong, S. H., Cho, J. H., Kim, B. R., Shin, J., Lee, J. H., et al. (2018). The dynamics of the piglet gut microbiome during the weaning transition in association with health and nutrition. J. Anim. Sci. Biotechnol. 9:54. doi: 10.1186/s40104-018-0269-6
Guevarra, R. B., Lee, J. H., Lee, S. H., Seok, M. J., Kim, D. W., Kang, B. N., et al. (2019). Piglet gut microbial shifts early in life: causes and effects. J. Anim. Sci. Biotechnol. 10:1. doi: 10.1186/s40104-018-0308-3
Hallen-Adams, H. E., and Suhr, M. J. (2017). Fungi in the healthy human gastrointestinal tract. Virulence 8, 352–358. doi: 10.1080/21505594.2016.1247140
Han, G. G., Lee, J. Y., Jin, G. D., Park, J., Choi, Y. H., Chae, B. J., et al. (2017). Evaluating the association between body weight and the intestinal microbiota of weaned piglets via 16S rRNA sequencing. Appl. Microbiol. Biotechnol. 101, 5903–5911.
Hillman, K., Whyte, A. L., and Stewart, C. S. (1993). Dissolved oxygen in the porcine gastrointesitnal tract. Lett. Appl. Microbiol. 16, 299–302.
Hoffmann, C., Dollive, S., Grunberg, S., Chen, J., Li, H., Wu, G. D., et al. (2013). Archaea and fungi of the human gut microbiome: correlations with diet and bacterial residents. PLoS One 8:e66019. doi: 10.1371/journal.pone.0066019
Huffnagle, G. B., and Noverr, M. C. (2013). The emerging world of the fungal microbiome. Trends Microbiol. 21, 334–341. doi: 10.1016/j.tim.2013.04.002
Iliev, I. D., Funari, V. A., Taylor, K. D., Nguyen, Q., Reyes, C. N., Strom, S. P., et al. (2012). Interactions between commensal fungi and the C-type lectin receptor Dectin-1 influence colitis. Science 336, 1314–1317. doi: 10.1126/science.1221789
Iliev, I. D., and Leonardi, I. (2017). Fungal dysbiosis: immunity and interactions at mucosal barriers. Nat. Rev. Immunol. 17, 635–646.
Kelly, J., Daly, K., Moran, A. W., Ryan, S., Bravo, D., and Shirazi-Beechey, S. P. (2017). Composition and diversity of mucosa-associated microbiota along the entire length of the pig gastrointestinal tract; dietary influences. Environ. Microbiol. 19, 1425–1438. doi: 10.1111/1462-2920.13619
Kim, Y. G., Udayanga, K. G., Totsuka, N., Weinberg, J. B., Nunez, G., and Shibuya, A. (2014). Gut dysbiosis promotes M2 macrophage polarization and allergic airway inflammation via fungi-induced PGE(2). Cell Host Microb. 15, 95–102.
Kiss, E. A., Vonarbourg, C., Kopfmann, S., Hobeika, E., Finke, D., Esser, C., et al. (2011). Natural aryl hydrocarbon receptor ligands control organogenesis of intestinal lymphoid follicles. Science 334, 1561–1565. doi: 10.1126/science.1214914
Koljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Kozich, J. J., Westcott, S. L., Baxter, N. T., Highlander, S. K., and Schloss, P. D. (2013). Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol. 79, 5112–5120. doi: 10.1128/AEM.01043-13
Kureljusic, B., Weissenbacher-Lang, C., Nedorost, N., Stixenberger, D., and Weissenbock, H. (2016). Association between Pneumocystis spp. and co-infections with Bordetella bronchiseptica, Mycoplasma hyopneumoniae and Pasteurella multocida in Austrian pigs with pneumonia. Vet J 207, 177–179. doi: 10.1016/j.tvjl.2015.11.003
Krüger, W., Vielreicher, S., Kapitan, M., Jacobsen, I. D., and Niemiec, M. J. (2019). Fungal-bacterial interactions in health and disease. Pathogens 8:70. doi: 10.3390/pathogens8020070
Kurtz, Z. D., Muller, C. L., Miraldi, E. R., Littman, D. R., Blaser, M. J., and Bonneau, R. A. (2015). Sparse and compositionally robust inference of microbial ecological networks. PLoS Comput. Biol. 11:e1004226. doi: 10.1371/journal.pcbi.1004226
Kurtzman, C. P. (2011). Phylogeny of the ascomycetous yeasts and the renaming of Pichia anomala to Wickerhamomyces anomalus. Antonie Van Leeuwenhoek 99, 13–23. doi: 10.1007/s10482-010-9505-6
Kurtzman, C. P., Robnett, C. J., Ward, J. M., Brayton, C., Gorelick, P., and Walsh, T. J. (2005). Multigene phylogenetic analysis of pathogenic candida species in the Kazachstania (Arxiozyma) telluris complex and description of their ascosporic states as Kazachstania bovina sp. nov., K. heterogenica sp. nov., K. pintolopesii sp. nov., and K. slooffiae sp. nov. J. Clin. Microbiol. 43, 101–111.
Lamas, B., Richard, M. L., Leducq, V., Pham, H. P., Michel, M. L., Da Costa, G., et al. (2016). CARD9 impacts colitis by altering gut microbiota metabolism of tryptophan into aryl hydrocarbon receptor ligands. Nat. Med. 22, 598–605. doi: 10.1038/nm.4102
Li, Q., Wang, C., Tang, C., He, Q., Li, N., and Li, J. (2014). Dysbiosis of gut fungal microbiota is associated with mucosal inflammation in Crohn’s disease. J. Clin. Gastroenterol. 48, 513–523. doi: 10.1097/MCG.0000000000000035
Li, X. V., Leonardi, I., and Iliev, I. D. (2019). Gut mycobiota in immunity and inflammatory disease. Immunity 50, 1365–1379.
Li, Z., Lu, G., and Meng, G. (2019). Pathogenic fungal infection in the lung. Front. Immunol. 10:1524. doi: 10.3389/fimmu.2019.01524
Limon, J. J., Skalski, J. H., and Underhill, D. M. (2017). Commensal fungi in health and disease. Cell Host Microb. 22, 156–165. doi: 10.1016/j.chom.2017.07.002
Liu, H., Ivarsson, E., Dicksved, J., Lundh, T., and Lindberg, J. E. (2012). Inclusion of chicory (Cichorium intybus L.) in pigs’ diets affects the intestinal microenvironment and the gut microbiota. Appl. Environ. Microbiol. 78, 4102–4109. doi: 10.1128/AEM.07702-11
Mackie, R. I., Sghir, A., and Gaskins, H. R. (1999). Developmental microbial ecology of the neonatal gastrointestinal tract. Am. J. Clin. Nutr. 69, 1035S–1045S. doi: 10.1093/ajcn/69.5.1035s
Mar Rodriguez, M., Perez, D., Javier Chaves, F., Esteve, E., Marin-Garcia, P., Xifra, G., et al. (2015). Obesity changes the human gut mycobiome. Sci. Rep. 5:14600. doi: 10.1038/srep14600
Martin, M. (2011). Cutadapt removes adaptor sequences from high-throughput sequencing reads. EMBnet J. 17, 10–12.
Martinsen, T. C., Bergh, K., and Waldum, H. L. (2005). Gastric juice: a barrier against infectious diseases. Basic Clin. Pharmacol. Toxicol. 96, 94–102.
Mason, K. L., Erb Downward, J. R., Falkowski, N. R., Young, V. B., Kao, J. Y., and Huffnagle, G. B. (2012a). Interplay between the gastric bacterial microbiota and Candida albicans during postantibiotic recolonization and gastritis. Infect. Immun. 80, 150–158. doi: 10.1128/IAI.05162-11
Mason, K. L., Erb Downward, J. R., Mason, K. D., Falkowski, N. R., Eaton, K. A., Kao, J. Y., et al. (2012b). Candida albicans and bacterial microbiota interactions in the cecum during recolonization following broad-spectrum antibiotic therapy. Infect. Immun. 80, 3371–3380. doi: 10.1128/IAI.00449-12
McMurdie, P. J., and Holmes, S. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Melloul, E., Roisin, L., Durieux, M. F., Woerther, P. L., Jenot, D., Risco, V., et al. (2018). Interactions of Aspergillus fumigatus and Stenotrophomonas maltophilia in an in vitro Mixed Biofilm Model: Does the strain matter? Front. Microbiol. 9:2850. doi: 10.3389/fmicb.2018.02850
Merchant, H. A., McConnell, E. L., Liu, F., Ramaswamy, C., Kulkarni, R. P., Basit, A. W., et al. (2011). Assessment of gastrointestinal pH, fluid and lymphoid tissue in the guinea pig, rabbit and pig, and implications for their use in drug development. Eur. J. Pharm. Sci. 42, 3–10. doi: 10.1016/j.ejps.2010.09.019
Mukherjee, P. K., Sendid, B., Hoarau, G., Colombel, J. F., Poulain, D., and Ghannoum, M. A. (2015). Mycobiota in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 12, 77–87. doi: 10.1038/nrgastro.2014.188
Nash, A. K., Auchtung, T. A., Wong, M. C., Smith, D. P., Gesell, J. R., Ross, M. C., et al. (2017). The gut mycobiome of the human microbiome project healthy cohort. Microbiome 5:153. doi: 10.1186/s40168-017-0373-4
Nguyen, L. N., Lopes, L. C., Cordero, R. J., and Nosanchuk, J. D. (2011). Sodium butyrate inhibits pathogenic yeast growth and enhances the functions of macrophages. J. Antimicrob. Chemother. 66, 2573–2580. doi: 10.1093/jac/dkr358
Okoli, A. S., Wadstrom, T., and Mendz, G. L. (2007). Bioinformatic study of bile responses in Campylobacterales. FEMS Immunol. Med. Microbiol 49, 101–123.
Oksanen, J., Guillaume Blanchet, F., Friendly, M., Kindt, R., Legendre, P., McGlinn, D., et al. (2019). vegan: Community Ecology Package. Available at: https://cran.r-project.org (accessed July 05, 2019).
Ott, S. J., Kuhbacher, T., Musfeldt, M., Rosenstiel, P., Hellmig, S., Rehman, A., et al. (2008). Fungi and inflammatory bowel diseases: alterations of composition and diversity. Scand. J. Gastroenterol. 43, 831–841.
Partida-Martinez, L. P., and Hertweck, C. (2005). Pathogenic fungus harbours endosymbiotic bacteria for toxin production. Nature 437, 884–888.
Peirce, J. M., and Alvina, K. (2019). The role of inflammation and the gut microbiome in depression and anxiety. J. Neurosci. Res. 97, 1223–1241. doi: 10.1002/jnr.24476
Philip, E. T., Hall, A. N., and Walker, T. K. (1963). Some effects of sodium n-butyrate on the behavior of aspergillus niger growing in a glucose medium. Arch. Biochem. Biophys. 102, 238–241.
Pierron, A., Alassane-Kpembi, I., and Oswald, I. P. (2016). Impact of two mycotoxins deoxynivalenol and fumonisin on pig intestinal health. Porcine Health Manag. 2:21. doi: 10.1186/s40813-016-0041-2
Qin, J., Li, R., Raes, J., Arumugam, M., Burgdorf, K. S., Manichanh, C., et al. (2010). A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464, 59–65. doi: 10.1038/nature08821
Raimondi, S., Amaretti, A., Gozzoli, C., Simone, M., Righini, L., Candeliere, F., et al. (2019). Longitudinal survey of fungi in the human gut: its profiling, phenotyping, and colonization. Front. Microbiol. 10:1575. doi: 10.3389/fmicb.2019.01575
Reece, E., Doyle, S., Greally, P., Renwick, J., and McClean, S. (2018). Aspergillus fumigatus Inhibits Pseudomonas aeruginosa in Co-culture: implications of a mutually antagonistic relationship on virulence and inflammation in the CF airway. Front. Microbiol. 9:1205. doi: 10.3389/fmicb.2018.01205
Richard, M. L., and Sokol, H. (2019). The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat. Rev. Gastroenterol. Hepatol. 16, 331–345. doi: 10.1038/s41575-019-0121-2
Rossoni, R. D., de Barros, P. P., de Alvarenga, J. A., Ribeiro, F. C., Velloso, M. D. S., Fuchs, B. B., et al. (2018). Antifungal activity of clinical Lactobacillus strains against Candida albicans biofilms: identification of potential probiotic candidates to prevent oral candidiasis. Biofouling 34, 212–225. doi: 10.1080/08927014.2018.1425402
Sabino, R., Faisca, V. M., Carolino, E., Verissimo, C., and Viegas, C. (2012). Occupational exposure to Aspergillus by swine and poultry farm workers in Portugal. J. Toxicol. Environ. Health A 75, 1381–1391. doi: 10.1080/15287394.2012.721170
Sam, Q. H., Chang, M. W., and Chai, L. Y. (2017). The fungal mycobiome and its interaction with gut bacteria in the host. Int. J. Mol. Sci. 18:E330. doi: 10.3390/ijms18020330
Schroeckh, V., Scherlach, K., Nutzmann, H. W., Shelest, E., Schmidt-Heck, W., Schuemann, J., et al. (2009). Intimate bacterial-fungal interaction triggers biosynthesis of archetypal polyketides in Aspergillus nidulans. Proc. Natl. Acad. Sci. U.S.A. 106, 14558–14563. doi: 10.1073/pnas.0901870106
Stadie, J., Gulitz, A., Ehrmann, M. A., and Vogel, R. F. (2013). Metabolic activity and symbiotic interactions of lactic acid bacteria and yeasts isolated from water kefir. Food Microbiol. 35, 92–98. doi: 10.1016/j.fm.2013.03.009
Suhr, M. J., and Hallen-Adams, H. E. (2015). The human gut mycobiome: pitfalls and potentials–a mycologist’s perspective. Mycologia 107, 1057–1073.
Summers, K. L., Frey, J. F., Ramsay, T. G., and Arfken, A. M. (2019). The piglet mycobiome during the weaning transition: a pilot study1. J. Anim. Sci. 97, 2889–2900. doi: 10.1093/jas/skz182
Tso, G. H. W., Reales-Calderon, A. J., Tan, A. S. M., Sem, X., Le, G. T. T., Tan, T. G., et al. (2018). Experimental evolution of a fungal pathogen into a gut symbiont. Science 362, 589–595. doi: 10.1126/science.aat0537
Uden, N. V., and Carmo-Sousa, L. D. (1962). On the intestinal yeast flora of free living hippopotami (Hipopotamus amphibius), wart hogs (Phacochoerus aethiopicus) and bush pigs (Potamochoerus choeropotamus). Antonie Van Leeuwenhoek 28, 73–77.
Urubschurov, V., Busing, K., Freyer, G., Herlemann, D. P., Souffrant, W. B., and Zeyner, A. (2017). New insights into the role of the porcine intestinal yeast, Kazachstania slooffiae, in intestinal environment of weaned piglets. FEMS Microbiol. Ecol, 93:fiw245. doi: 10.1093/femsec/fiw245
Urubschurov, V., Busing, K., Janczyk, P., Souffrant, W. B., and Zeyner, A. (2015). Development and evaluation of qPCR assay for quantitation of Kazachstania slooffiae and total yeasts occurring in the porcine gut. Curr. Microbiol. 71, 373–381. doi: 10.1007/s00284-015-0862-2
Urubschurov, V., and Janczyk, C. (2011). “Biodiversity of yeasts in the gastrointestinal ecosystem with emphasis on its importance for the host,” in The Dynamical Processes of Biodiversity - Case Studies of Evolution and Spatial Distribution, eds O. Grillo, and G. Venora (Rijeka: INTECH), 277–302. doi: 10.5772/24108
Urubschurov, V., Janczyk, P., Souffrant, W. B., Freyer, G., and Zeyner, A. (2011). Establishment of intestinal microbiota with focus on yeasts of unweaned and weaned piglets kept under different farm conditions. FEMS Microbiol. Ecol. 77, 493–502. doi: 10.1111/j.1574-6941.2011.01129.x
Villmones, H. C., Haug, E. S., Ulvestad, E., Grude, N., Stenstad, T., Halland, A., et al. (2018). Species level description of the human ileal bacterial microbiota. Sci. Rep. 8:4736. doi: 10.1038/s41598-018-23198-5
Vylkova, S. (2017). Environmental pH modulation by pathogenic fungi as a strategy to conquer the host. PLoS Pathog. 13:e1006149. doi: 10.1371/journal.ppat.1006149
Waite, D. W., Vanwonterghem, I., Rinke, C., Parks, D. H., Zhang, Y., Takai, K., et al. (2017). Comparative genomic analysis of the class Epsilonproteobacteria and proposed reclassification to Epsilonbacteraeota (phyl. nov.). Front. Microbiol. 8:682. doi: 10.3389/fmicb.2017.00682
Walter, J., and Ley, R. (2011). The human gut microbiome: ecology and recent evolutionary changes. Annu. Rev. Microbiol. 65, 411–429.
Weiss, S., Xu, Z. Z., Peddada, S., Amir, A., Bittinger, K., Gonzalez, A., et al. (2017). Normalization and microbial differential abundance strategies depend upon data characteristics. Microbiome 5:27. doi: 10.1186/s40168-017-0237-y
Weissenbacher-Lang, C., Kureljusic, B., Nedorost, N., Matula, B., Schiessl, W., Stixenberger, D., et al. (2016). Retrospective analysis of bacterial and viral co-infections in Pneumocystis spp. positive lung samples of austrian pigs with pneumonia. PLoS One 11:e0158479. doi: 10.1371/journal.pone.0158479
White, J. K., Nielsen, J. L., and Madsen, A. M. (2019). Microbial species and biodiversity in settling dust within and between pig farms. Environ. Res. 171, 558–567. doi: 10.1016/j.envres.2019.01.008
Wickham, H. (2016). ggplot2: Elegant Graphics for Data Analysis. New York, NY: Springer International Publishing.
Yasuda, K., Oh, K., Ren, B., Tickle, T. L., Franzosa, E. A., Wachtman, L. M., et al. (2015). Biogeography of the intestinal mucosal and lumenal microbiome in the rhesus macaque. Cell Host Microb. 17, 385–391. doi: 10.1016/j.chom.2015.01.015
Yilmaz, P., Parfrey, L. W., Yarza, P., Gerken, J., Pruesse, E., Quast, C., et al. (2014). The SILVA and all-species living tree project (LTP) taxonomic frameworks. Nucleic Acids Res. 42, D643–D648. doi: 10.1093/nar/gkt1209
Zajc, J., and Gunde-Cimerman, N. (2018). The genus wallemia-from contamination of food to health threat. Microorganisms 6:E46. doi: 10.3390/microorganisms6020046
Zelante, T., Iannitti, R. G., Cunha, C., De Luca, A., Giovannini, G., Pieraccini, G., et al. (2013). Tryptophan catabolites from microbiota engage aryl hydrocarbon receptor and balance mucosal reactivity via interleukin-22. Immunity 39, 372–385. doi: 10.1016/j.immuni.2013.08.003
Zhang, X., Li, Z., Yang, H., Liu, D., Cai, G., Li, G., et al. (2018). Novel transgenic pigs with enhanced growth and reduced environmental impact. eLife 7:e34286. doi: 10.7554/eLife.34286
Keywords: mycobiome, bacteriome, microbiome, piglet, weaning, swine
Citation: Arfken AM, Frey JF, Ramsay TG and Summers KL (2019) Yeasts of Burden: Exploring the Mycobiome–Bacteriome of the Piglet GI Tract. Front. Microbiol. 10:2286. doi: 10.3389/fmicb.2019.02286
Received: 05 August 2019; Accepted: 19 September 2019;
Published: 08 October 2019.
Edited by:
Jae-Hyuk Yu, University of Wisconsin–Madison, United StatesReviewed by:
Sharon Marie Donovan, University of Illinois at Urbana–Champaign, United StatesMetzler-Zebeli Barbara, University of Veterinary Medicine Vienna, Austria
Copyright © 2019 Arfken, Frey, Ramsay and Summers. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Katie Lynn Summers, a2F0aWUuc3VtbWVyc0B1c2RhLmdvdg==