 Veronika Koziaeva
Veronika Koziaeva Marina Dziuba
Marina Dziuba Pedro Leão
Pedro Leão Maria Uzun
Maria Uzun Maria Krutkina
Maria Krutkina Denis Grouzdev
Denis Grouzdev- 1Research Center of Biotechnology of the Russian Academy of Sciences, Institute of Bioengineering, Moscow, Russia
- 2Department of Microbiology, University of Bayreuth, Bayreuth, Germany
- 3Instituto de Microbiologia Paulo de Góes, Universidade Federal Do Rio de Janeiro, Rio de Janeiro, Brazil
- 4Faculty of Biology, Lomonosov Moscow State University, Moscow, Russia
Magnetotactic bacteria are widely represented microorganisms that have the ability to synthesize magnetosomes. The magnetotactic cocci of the order Magnetococcales are the most frequently identified, but their classification remains unclear due to the low number of cultivated representatives. This paper reports the analysis of an uncultivated magnetotactic coccus UR-1 collected from the Uda River (in eastern Siberia). Genome analyses of this bacterium and comparison to the available Magnetococcales genomes identified a novel species called “Ca. Magnetaquicoccus inordinatus,” and a delineated candidate family “Ca. Magnetaquicoccaceae” within the order Magnetococcales is proposed. We used average amino acid identity values <55–56% and <64–65% as thresholds for the separation of families and genera, respectively, within the order Magnetococcales. Analyses of the genome sequence of UR-1 revealed a potential ability for a chemolithoautotrophic lifestyle, with the oxidation of a reduced sulfur compound and carbon assimilation by rTCA. A nearly complete magnetosome genome island, containing a set of mam and mms genes, was also identified. Further comparative analyses of the magnetosome genes showed vertical inheritance as well as horizontal gene transfer as the evolutionary drivers of magnetosome biomineralization genes in strains of the order Magnetococcales.
Introduction
Magnetotactic cocci are one type of magnetotactic bacteria (MTB). Since the first description of MTB (Blakemore, 1975), many studies have shown that magnetotactic cocci are the most frequent morphotype among other MTB, both in freshwater (Moench and Konetzka, 1978; Spring et al., 1995; Flies et al., 2005a,b; Lin and Pan, 2009, 2010) and marine habitats (Spring et al., 1998; Abreu et al., 2012; Zhang et al., 2012; Du et al., 2017). However, only five representatives—Magnetococcus marinus MC-1T (Frankel et al., 1997), Magnetofaba australis IT-1 (Morillo et al., 2014), “Ca. Magnetococcus massalia” MO-1 (Lefèvre et al., 2009) and strains PR-3 and SS-1 (Lefèvre et al., 2014) have been isolated in axenic cultures from hypersaline lagoon and marine habitats. Due to the difficulty of isolating MTB in an axenic culture, and especially the freshwater cocci forms, these studies have mostly been carried out using molecular methods for the analysis of environmental samples. However, magnetotactic cocci have a high swimming ability, so they can be easily isolated from non-magnetotactic bacteria using magnetic separation (Lin et al., 2008).
Early studies of the phylogeny of magnetotactic cocci described them as a separate clade within the class Alphaproteobacteria (Amann et al., 2006), but genome sequencing and analysis of three cultivated cocci led to a proposal to classify them as a separate class (“Candidatus Etaproteobacteria”) within the Proteobacteria phylum due to the mosaic origin of their genomes (Ji et al., 2017). Recently, 12 metagenome assembled genomes (MAGs) have been obtained for freshwater strains. The analysis confirmed that the order Magnetococcales belongs to the novel “Ca. Etaproteobacteria” class, rather than to the class Alphaproteobacteria (Lin et al., 2018).
MTB of the order Magnetococcales generally have an ovoid shape and usually synthesize magnetosomes with elongated prismatic and elongated octahedral crystal morphology (Pósfai et al., 2013); however, strain SHHC-1 demonstrates a large variety of magnetosome shapes within the same cell (Zhang et al., 2017). Rod-shaped bacteria potentially belonging to the order Magnetococcales have also been described (Zhang et al., 2013). The magnetosome arrangement within the cells varies from strain to strain: they are organized in one, two, or more chains and can be assembled as a dispersed cluster (Pan et al., 2008; Lefèvre et al., 2009; Lin and Pan, 2009; Abreu et al., 2012; Zhang et al., 2012, 2017; Morillo et al., 2014; Du et al., 2017; Kozyaeva et al., 2017). Many magnetotactic cocci have sulfur globules, indicating their ability to use reduced sulfur compounds, and phosphorus-rich inclusions that sometimes occupy most of the cell volume (Cox et al., 2002; Dziuba et al., 2013; Zhang et al., 2017). Cultivated magnetotactic cocci are microaerophiles and use thiosulfate for autotrophic growth, while using acetate and, in the case of IT-1, succinate for heterotrophic growth (Williams et al., 2006; Lefèvre et al., 2009; Morillo et al., 2014). The ability to accumulate a large amount of phosphorus indicates the important role of magnetotactic cocci in the phosphorus cycle. In particular, they can function as a “bacterial shuttle,” moving phosphorus from the water surface to deeper anaerobic layers (Rivas-Lamelo et al., 2017; Schulz-Vogt et al., 2019).
Magnetotactic cocci are diverse in their phylogeny as well as in their morphology. Attempts have been made to associate the 16S rRNA sequence with the ultrastructure of cells and crystallographic properties of their magnetosomes using fluorescent in situ hybridization (FISH) and other methods of FISH coupled with transmission and scanning electron microscopy (FISH-TEM and FISH-SEM). These approaches have been successfully used to identify cultured and uncultured MTB (Spring et al., 1998; Woehl et al., 2014; Li J. et al., 2017; Zhang et al., 2017; Li et al., 2019), and marine cocci have had their phylogenetic classification associated with their cell morphology and detailed characterization of magnetosome crystals (Abreu et al., 2012; Zhang et al., 2012, 2017). However, no clear results have yet been achieved for freshwater magnetotactic cocci (Spring et al., 1995; Lin and Pan, 2009). The number of genomic sequences associated with magnetotactic cocci has significantly increased, but information remains scarce regarding the cell and magnetosome structure of these MTB (Lin et al., 2018).
This paper reports the culture-independent characterization of an uncultivated magnetotactic coccus isolated from sediments of the Uda River (Eastern Siberia, Russia). FISH-TEM analysis associated its 16S rRNA gene sequence and the phenotype while reconstruction and analysis of the genome of this novel magnetotactic bacterium provided the first link between the genomic data of an uncultivated freshwater magnetotactic coccus with a specific phenotype. In particular, this genome analysis enabled the study of magnetosome biosynthesis genes in this organism in connection with the magnetosome organization within the cells, as revealed by TEM, thereby providing novel data to supplement previously published research on this group of MTB.
A comparison of the genome of the novel magnetotactic coccus with genomes of similar MTB available in public databases revealed several phylogenetic subgroups within the class Magnetococcales at the family level. Using the guidelines for the taxonomic description of uncultivated microbes provided by Konstantinidis et al. (2017), a candidate species for the novel magnetotactic coccus “Ca. Magnetaquicoccus inordinatus” UR-1 is described, and the genus “Ca. Magnetaquicoccus” and the family “Ca. Magnetaquicoccaceae” are proposed.
Materials and Methods
Sample Collection and DNA Extraction
MTB-containing sediment was collected in August 2012 from the Uda River in the city of Ulan-Ude, Eastern Siberia (51.8229 °N, 107.6199 °E). Physicochemical parameters of the water in the sampling site were provided by the Hydrochemical Institute (Rostov-on-Don, Russia). Surface sediments were collected near the shore from a water depth of ~0.5 meters and transferred to a 3-liter glass jar at a sediment: water ratio of ~1:3. The jars were stored in the laboratory at room temperature (~25°C) in dim light for 3 months. The enriched fraction of MTB cells was obtained by placing the south pole of the magnet on the outside of the jar, at the sediment-water interface. After 1 h, the spot of magnetotactic bacteria was collected using a Pasteur pipette. Magnetotactic properties were evaluated by observing the changes in bacterial motion in response to rotation of a magnet located on the stage of an Eclipse E200 light microscope (Nikon, Japan). A fraction of the collected cells (about 10 μl) were used for TEM; some cells were fixed in 3% paraformaldehyde for 1.5 h for coordinated FISH-TEM, and a third fraction was purified from non-magnetotactic microorganisms using the “race track” technique (Wolfe et al., 1987). The purified cells were used to isolate genomic DNA using a modified Birnboim-Doly alkaline method with a Wizard technique (Promega, USA) (Boulygina et al., 2002). Purified DNA was stored at −20°C.
Clonal Library Construction and Phylogenetic Analysis of 16S rRNA Gene Sequences
The 16S rRNA gene sequences were amplified using universal primers 27F (5'-AGAGTTTGATCCTGGCTCAG-3') and 1492R (5'-AAGGAGGTGATCCAGCCGCA-3') (Lane, 1991). The obtained PCR products were purified with a Wizard PCR Prep kit (Promega, USA). Purified PCR products were ligated into a pGEM-T Easy System vector (Promega, USA) and cloned into Escherichia coli strain DH10B cells. Sequencing was performed on an ABI3730 DNA Analyzer sequencer (Applied Biosystems, USA) using the BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems, USA) and the universal primers M13F, M13R, 530F, and 519R (Sambrook et al., 1989). The presence of chimeric sequences was verified using the Bellerophon online service (Huber et al., 2004). The obtained sequences were grouped in operational taxonomic units (OTUs) using identity threshold 99%. Obtained OTUs were aligned with 16S rRNA gene sequences of Magnetococcales strains using MAFFT (Katoh and Standley, 2013), and the maximum-likelihood tree was inferred using the GTR+F+I+G4 model recommended using ModelFinder (Wong et al., 2017) in IQ-Tree (Nguyen et al., 2015). Branch supports were obtained with 10,000 ultrafast bootstraps (Hoang et al., 2017). The 16S rRNA gene sequences of the OTUs were deposited in the GenBank database under accession numbers MK813936 and MK813937.
Phylogenetic and Morphological Correlation of UR-1 Cells
To determine which cell the 16S rRNA sequence named UR-1 came from, a FISH-TEM analysis was performed on the same sample. Approximately 10 μl of fixed magnetically concentrated cells were added to a center-marked copper grid previously covered with Formvar film. After air drying for 5 min, the residual sample not attached to the grid was removed with filter paper, and a thin layer of sputtered carbon was placed on top of the sample (Balzers CED-030, Liechtenstein). The grids were store at room temperature in a vacuum chamber for 2 weeks before being used for FISH. The hybridization reaction was performed using the conditions and buffers described by Pernthaler et al. (2001) using a formamide concentration of 30% in the hybridization buffer, and a probe final concentration of 0.2 μg/ml (Pernthaler et al., 2001). After washing with washing buffer, the sample was stained with 4,6-diamidino-2-phenylindole (DAPI) at a final concentration of 0.1 μg/ml for 5 min and carefully washed with deionized water. For UR-1 cells identification, a specific probe, Uda54-3 (5' Cy3-CAAGAGCAATTCCAGGGTTAAGCCCTGGGCTT-3'), was designed and used as template to retrieve the 16S rRNA sequence MK813936 from the sequence. The positive control for the hybridization reaction was a mixture containing the bacterial universal probes EUB388I, EUB388II, and EUB388III (Daims et al., 1999) labeled with Alexa 488. The negative control was the same hybridization reaction using the Uda54-3 probe and EUB probes in a sample containing only Escherichia coli cells (Supplementary Figure S1). After performing the FISH reaction, the grid was placed between a glass slide and a coverslip and images were obtained using an AxioImager microscope (Zeiss, Germany) equipped with an AxioCam Mrc (Zeiss, Germany). The same grids used to perform FISH were placed on a Morgagni transmission electron microscope (FEI, USA) operated at 80 kV and images were obtained using a MegaView G2 CCD camera (Olympus, Japan) from the same area where the FISH had already been observed.
Transmission Electron Microscopy
For conventional TEM, magnetically enriched cells were added to a Formvar-coated copper grid and imaged on a JEM-100CX (JEOL, Japan) transmission electron microscope operated at 80 kV. the TEM images were processed using ImageJ software to determine cell and magnetosome lengths, widths, and shape factors (width/length). Grids for high-resolution (HR) TEM were prepared as described for conventional TEM, and HR images were acquired using a Tecnai G2 F20 FEG transmission electron microscope (FEI, USA) operated at 200 kV and equipped with a 4k × 4k Gatan UltraScan 1,000 CCD camera. The HR images were analyzed using the Digital Micrograph software (Gatan, USA).
Genome Sequencing, Assembly, and Annotation
DNA libraries were constructed with the NEBNext DNA library prep reagent set for Illumina, following the kit protocol. Sequencing was undertaken using the Illumina HiSeq 1500 platform with pair-end 150-bp reads. Raw reads were quality checked with FastQC v. 0.11.7 (http://www.bioinformatics.babraham.ac.uk/projects/fastqc/), and low-quality reads were trimmed using Trimmomatic v. 0.36 (Bolger et al., 2014). The quality-filtered reads were assembled de novo with metaSPAdes v. 3.12.0 using the default settings (Nurk et al., 2017). Genome statistics were evaluated using an automatic assembly quality evaluation tool (QUAST) (Gurevich et al., 2013). The assembled metagenome of the Uda River was binned using three different tools (MaxBin 2.0 Wu et al., 2015, MyCC Lin and Liao, 2016, and Busy Bee Web Laczny et al., 2017) prior to dereplication and refinement with the DAS Tool (Sieber et al., 2018). The DAS Tool performs a consensus binning and produces the final bin set. Completeness and contamination rates were assessed using CheckM v. 1.0.12 (Parks et al., 2015) with the “lineage wf” command and default settings. Annotation of the UR-1 genome was carried out using the NCBI Prokaryotic Genome Annotation Pipeline (Tatusova et al., 2016). This genome project has been deposited in the DDBJ/ENA/GenBank under the accession number RXIU00000000.
Phylogenetic Analysis and Genome Index Calculation
Phylogenomic analyses of Magnetococcales genomes were conducted with GTDB-Tk v.0.1.3 using the de novo workflow with a set of 120 single-copy marker proteins and the genome taxonomy database (GTDB) (Parks et al., 2018). Concatenated alignments were used to construct a maximum likelihood tree inferred in IQ-Tree using the LG+F+I+G4 model recommended by ModelFinder (Wong et al., 2017), and branch supports were estimated using UFBoot2 (Hoang et al., 2017). The tree was rooted using two Magnetospirillum species, Zetaproteobacteria bacterium PCbin4 and Lambdaproteobacteria bacterium PCRbin3 as outgroups.
The MamA, -B, -E, -H, -I, -K, L, -M, -O, -P, -S, and -T amino-acid sequences were independently aligned using MAFFT (Katoh and Standley, 2013), cleaned with Gblocks v. 0.91b (Castresana, 2000) with an option to allow gap positions in the final blocks, and concatenated. These alignments were used to construct a maximum likelihood tree inferred in IQ-Tree using the LG+F+I+G4 model.
The average nucleotide identity (ANI) and average amino acid identity (AAI) were calculated using the ANI/AAI-Matrix online service (Rodriguez-R and Konstantinidis, 2016). Digital DNA-DNA hybridization (dDDH) values were determined using the Genome-to-Genome Distance Calculator (GGDC) 2.1 online software (Meier-Kolthoff et al., 2013). The pairwise percentage of conserved proteins (POCP) was calculated using the script runPOCP.sh (Pantiukh and Grouzdev, 2017; Grouzdev et al., 2018), based on the previously described approach (Qin et al., 2014).
Genes transferred horizontally were identified using the recentHGT program (Li et al., 2018). This program allows the finding of recently occurred events of horizontal gene transfer between closely related species.
Analyses of Magnetosome Genes and Reconstruction of Metabolic Pathways of Magnetococcales Bacterium UR-1
Computational prediction of CDS and other genomic features, together with functional annotation, was performed using the NCBI automated prokaryotic genome annotation pipeline (https://www.ncbi.nlm.nih.gov/genome/annotation_prok Tatusova et al., 2016). Homologous sequences of magnetosome proteins were identified within the protein database using BLAST searches. The conserved domain structure of putative magnetosome proteins was analyzed with the Batch Web CD-Search Tool with default parameters (Marchler-Bauer et al., 2017). A manual curation of the predicted gene annotations was performed for the metabolic features selected for the current analysis. This was supported by functional analysis with InterProScan and gapped BLAST (Altschul et al., 1997; Quevillon et al., 2005). The metabolic reconstruction was aided by the KEGG Mapper, which included the pathway, BRITE, and MODULE reconstruction tools (Kanehisa et al., 2019).
Results
Enrichment of MTB Cells and Diversity Analysis by 16S rRNA Gene
According to the ecological monitoring data, the waters of the Uda River near the sampling site had a pH of 8.04 and a temperature of 11°C. The water body contained low concentrations of phosphorus, as well as ammonium, nitrite, and nitrate ions. The data on physicochemical composition of water samples are summarized in Supplementary Table S1.
Following the magnetic separation, a visible pellet of magnetotactic bacteria had accumulated. Light microscopy examination revealed only motile cocci present in the cell fraction. Microscopy studies using TEM showed that the magnetically concentrated cells contained the dominant group of magnetotactic cocci with characteristically clustered magnetosomes. The clonal library of the Uda microcosm consisted of 71 16S rRNA gene sequences. On the phylogenetic tree, all the obtained sequences formed two OTUs within the order Magnetococcales. One OTU, designated as Magnetococcales bacterium UR-1 (hereinafter “UR-1”), was dominant (87% of the library) (Figure 1). The level of similarity was 89.6% for the closest validly described organism Mc. marinus MC-1T of the Magnetococcaceae family, order Magnetococcales. This result suggested that the UR-1 might belong to a new family in the order Magnetococcales (Chun et al., 2018).
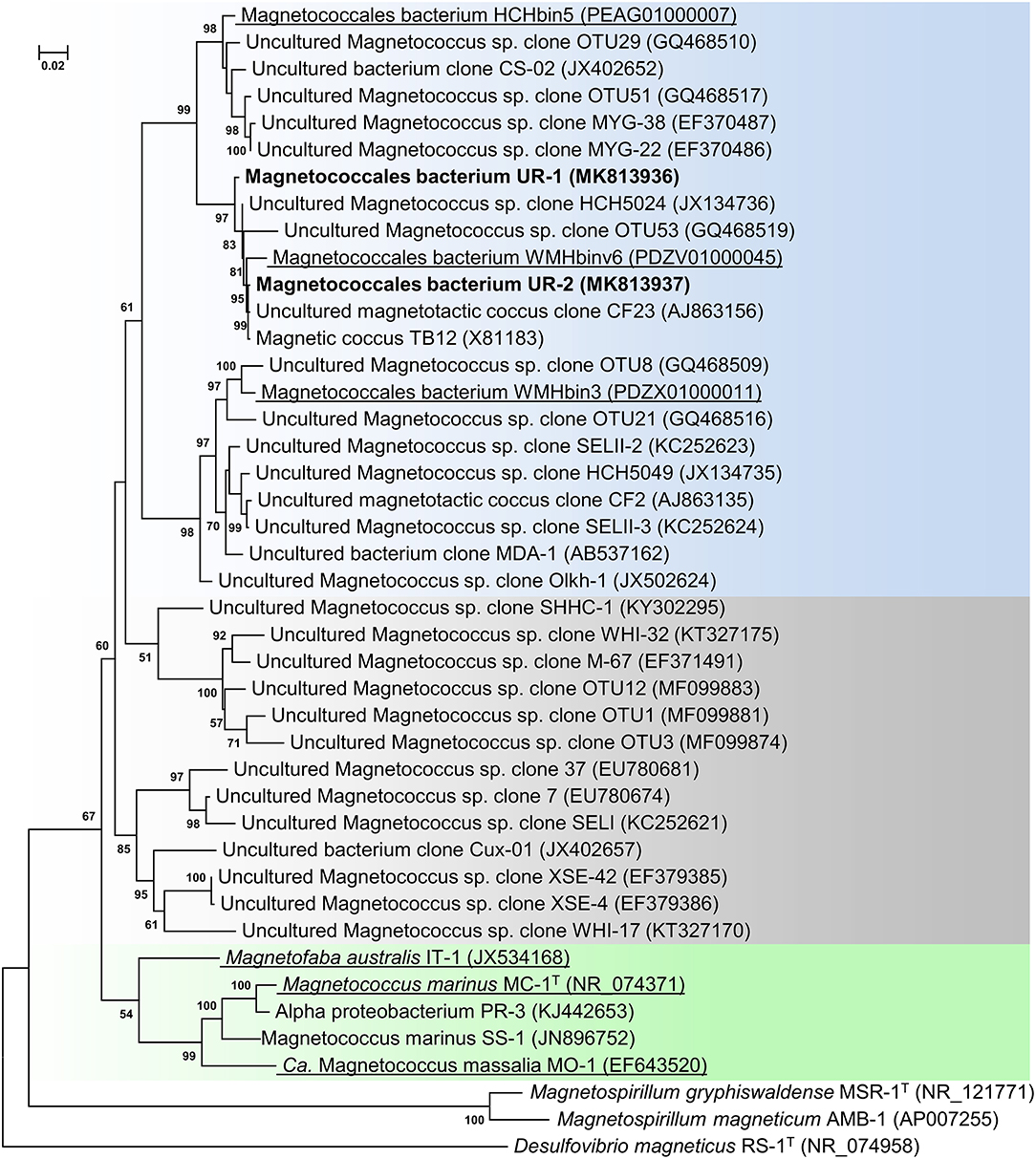
Figure 1. Maximum-likelihood phylogenetic tree based on 16S rRNA gene sequences (1,277 nucleotide sites) reconstructed with evolutionary model GTR+F+I+G4 showing the position of OTUs UR-1 and UR-2 in relation to members of the order Magnetococcales. Bootstrap values (>50%) are listed as percentages at the branching points. The scale bar represents nucleotide substitutions per site. The tree was rooted using Desulfovibrio magneticus RS-1T as outgroup. Bacteria with genomic sequences are underlined.
The phylogenetic tree was constructed using the available 16S rRNA sequences from the MAGs of the freshwater cocci HCHbin5, WMHbinv6, and WMHbin3 (Lin et al., 2018). The level of similarity of UR-1 with WMHbinv6, HCHbin5, and WMHbin3 was 98.2, 96.2, and 93.1%, respectively. The proposed standards for the description of uncultured bacteria are that a level of 16S rRNA sequence similarity of 95–98.6% suggests that analyzed strains belong to the same genus, while 92–95% suggests they belong to the same family (Konstantinidis et al., 2017). Thus, UR-1, HCHbin5, WMHbinv6, and WMHbin3 potentially belong to the same family, within which UR-1, WMHbinv6, and HCHbin5 formed the same genus and WMHbin3 putatively represented another one.
On the phylogenetic tree, the sequence of OTU UR-1 formed a cluster with the sequences HCH5024, CF23, and TB12, which were previously obtained from environmental samples from Germany and China (Spring et al., 1995; Flies et al., 2005b; Wang et al., 2013). The similarity between them was 99%, which indicated their possible affiliation with different strains of the same species or with different very closely related species. The 16S rRNA obtained from the rivers and lakes of Germany, China, and Russia also clustered with the sequences of HCHbin5 and WMHbin3, indicating that the representatives of the putative family, which included OTU UR-1, are widely distributed in freshwater habitats.
Morphology of UR-1 Cells and Magnetosomes
Using FISH, we identified a coccoid cell as the individual associated with the 16S rRNA sequence MK813936 retrieved from strain UR-1 sequencing (Figure 2). The specific probe for the strain UR-1 (Uda54-3) hybridized with a round cell (Figures 2A,B), which was also recognized by EUB probes (Figure 2C) and stained by DAPI (Figure 2D). Probe Uda54-3 did not hybridize with other cell types in the sample (Supplementary Figure S1).
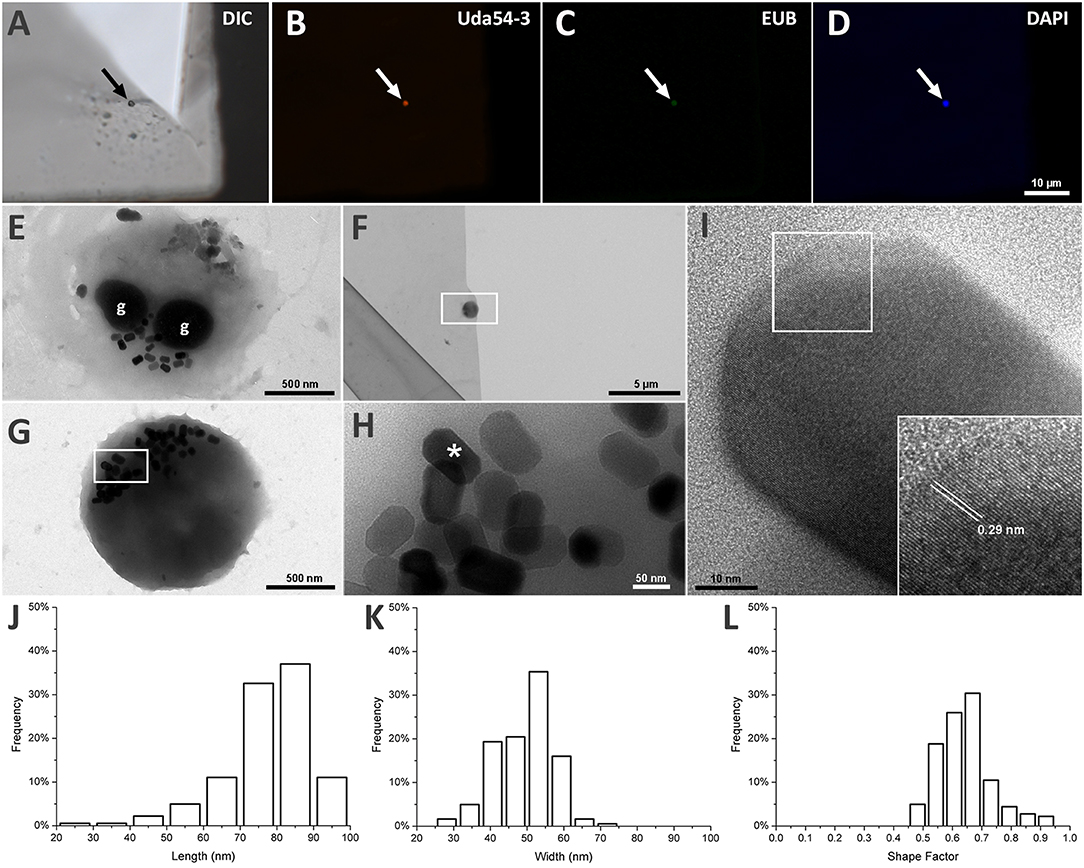
Figure 2. Morphological analysis of strain UR-1 cells and it magnetosomes. (A) DIC microscopy of magnetic enriched MTB sample on top of a TEM grid presenting a round cell on the center (arrow). (B) Fluorescence microscopy image of the same field capture on A showing the round cell hybridize with probe UR54-3 (arrow). (C) Fluorescence microscopy image showing the same cell recognized by probe UR54-3 hybridized with EUB probes (arrow). (D) Fluorescence microscopy of the same field showing the round cell stained by DAPI (arrow). (E) TEM image of UR-1 cell presenting a coccoid morphology and two electron dense inclusions (g) resembling P-rich granules. (F) TEM image of the same coccus present on imaged A by DIC. (G) Higher magnification of the area mark by the rectangle on figure (F). The round cell presented magnetosomes not organized in chains and clustered in one side of the cell. (H) Higher magnification of the area mark by the rectangle on figure (G). Magnetosomes inside the round cell have an elongated shape, with flat corners. (I) HRTEM image of the magnetosome mark with an asterisk on image (H). The insertion shows an interplanar distance of 0.29 nm between the atom columns present on the magnetosome crystal surface, indicating that the crystal is made of magnetite. (J) Length frequency distribution of magnetosomes found on UR-1 cells. (K) Width frequency distribution of magnetosomes found on UR-1 cells. (L) Shape factor frequency distribution of magnetosomes found on UR-1 strain.
The TEM images of the same cell recognized by probe Uda54-3 on Figure 2B are presented in Figures 2F,G and reveal the presence of elongated magnetosomes (Figure 2H); these are not organized in chains but are clustered on one side of the cells (Figures 2E,G). The HRTEM of the magnetosomes showed an interplanar distance between the atom columns of 0.29 nm (Figure 2I), a value associated with the oxide of iron magnetite (Martínez-Mera et al., 2007; Zhuang et al., 2015). The magnetosomes of strain UR-1 presented a mean length of 77.4 nm (SD = 11.8 nm, n = 181), with more than 90% of the magnetosome length between 65 and 95 nm (Figure 2J), a mean width of 46.2 nm (SD = 7.9 nm), with more than 90% of magnetosomes width between 40.6 and 59.3 nm (Figure 2K), and a mean shape factor of 0.64 (SD = 0.09) with more than 90% of the shape factor value between 0.48 and 0.73 (Figure 2L). Electron-dense inclusions were consistently observed in the UR-1 cell cytoplasm (Figure 2E). These structures have been observed in most magnetotactic cocci characterized so far (Dziuba et al., 2013; Abreu et al., 2014) and are correlated with polyphosphate (poly-P) inclusions (Lins and Farina, 1999; Keim et al., 2005).
MAG Statistics
The assembly and binning of data resulted in the MAG of magnetotactic coccus UR-1. It consisted of 546 contigs, with a total length of about 4143644 bp. The GC composition was 52.2 mol%. In accordance with the standards imposed on metagenomic assemblies, the genome of UR-1 had a high quality (completeness > 90%, contamination < 5%) (Supplementary Table S2). In the assembled genome, the 16S rRNA sequence was identified in the contig with a length of 25,034 bp and was identical to the sequence of the dominant OTU UR-1. The specific probe Uda54-3 used for FISH-TEM morphology identification of OTU UR-1 also matched the 16S rRNA of the genome, thereby confirming the link between the genome of the coccus UR-1 and the identified cell phenotype.
Delineation of the “Ca. Magnetaquicoccaceae” Family Within the Class Magnetococcales
Separation of taxa requires the establishment of permissible criteria. Phylogeny based on the genomic sequence is considered to be the main tool (Lang et al., 2013; Chun et al., 2018). The current requirement is that branches must (i) be monophyletic (Rossello-Mora and Amann, 2001) and (ii) result in minimum changes in the current taxonomy (Orata et al., 2018). On the phylogenomic tree, the Magnetococcales genomes formed five clades that were supported by high bootstrap values (100%) (Figure 3A). UR-1 formed a clade together with WMHbinv6, YD0425bin7, HCHbin5, and WMHbin3, which correlated with the 16S rRNA phylogenetic tree. Cultured marine strains MO-1, IT-1, and MC-1T of the Magnetococcaceae family formed a separate branch, consistent with the topology on the 16S rRNA tree. The third group consisted of five genomes: DC0425bin3, WMHbin1, DCbin4, HAa3bin1, HA3dbin1, and DCbin2. The fourth branch was formed solely by ER1bin7, and the fifth group included the genomes DC0425bin3 and HA3dbin3. The five identified clades may represent five families within the order Magnetococcales. These groups were preliminarily designated as “Magnetococcaceae,” “UR-1,” “WMHbin1,” “ER1bin7,” and “DC0425bin3.” These lineages were also confirmed by the analysis available in Genome Taxonomy Database (GTDB), except for “ER1bin7” (Parks et al., 2018). We checked whether the identified clades could represent candidate families by conducting additional studies on the available Magnetococcales genomes. The taxonomic ranks at the family and genus levels were separated using numerical indices based on the amino acid sequences of the genome, AAI and POCP. The AAI analysis showed identity values ranging from 50.1 to 99.0% between all analyzed genomes (Figure 4). The AAI value between the representatives of the identified phylogenetic branches was 50.1 to 55.8%. As the previous studies have shown, AAI has no clear boundaries for taxon separation (Konstantinidis and Tiedje, 2005; Luo et al., 2014). According to Konstantinidis et al. (2017), representatives of the same family may have AAI values of 45–65%, and representatives of the same genus may have values of 65–95%. The use of the lower boundary of 45 and 50% would lead to the unification of all Magnetococcales genomes into one family, but the use of values of 55–56% for the separation of families perfectly correlated with the branching of the phylogenomic tree and confirmed the possibility of separating five families within the order Magnetococcales. If the lower value of 65% is applied for the separation of genera, the cultivated representatives of the genus Magnetococcus, MC-1T and MO-1, would belong to different genera. However, this contradicts the current taxonomy. Thus, setting the limit of differentiation of close genera at 64–65% confirmed the established separation of the genera Magnetofaba and Magnetococcus. When we applied the same criterion to the “UR-1” group, two genera could be identified: the first included UR-1, YD0425bin7, WMHbinv6, and HCHbin5, and the second genus was formed by strain WMHbin3. Similarly, within the clade “WMHbin1,” all representatives formed a single genus, and in the clade DC0425bin3, two genera were distinguished. The proposed AAI ranges can be applied to the constantly increasing number of available genomes of the class “Ca. Etaproteobacteria,” which would enable consistent and reliable classification at the genus and family levels of the sequences derived from axenic cultures or metagenome analyses in the future.
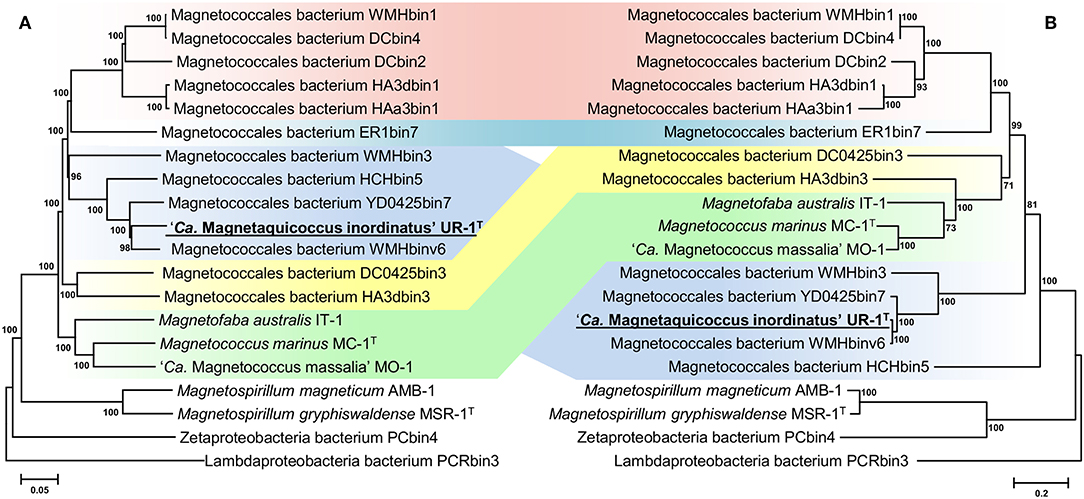
Figure 3. Maximum-likelihood phylogenetic trees inferred from a comparison of (A) concatenated Magnetococcales 120 single copy marker proteins showing the position of the UR-1 and (B) concatenated magnetosome associated protein (MamABEHIKLMOPST) sequences. Both trees reconstructed with evolutionary model LG+F+I+G4. The scale bar represents amino acid substitutions per site.
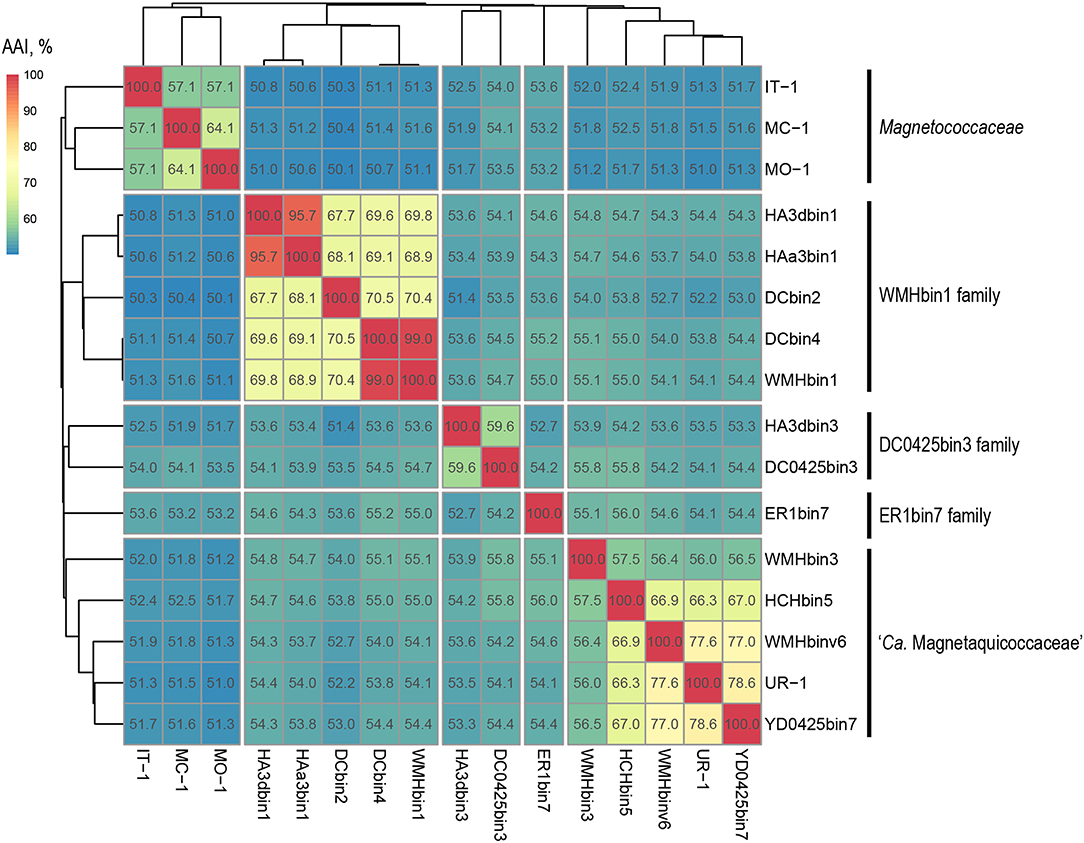
Figure 4. Whole-genome relationships within the order Magnetococcales. The heat map shows pairwise comparisons of AAI between genomes of the Magnetococcales strains. Thermal color scale located at the upper left corner. Boxed regions indicate inferred family clusters based on AAI comparisons. Black lines located at the right indicate a proposed family name.
POCP is another tool for differentiating between genera, and a fixed genus boundary of 50% of conserved proteins has been proposed (Qin et al., 2014). The POCP analysis showed that the values between all cocci ranged from 32.9 to 90.3% (Supplementary Table S3). In general, the POCP results supported the AAI results; however, several cocci had low values of genome completeness, which strongly influences the final result of the POCP calculation and can thus lead to unreliable results. Interestingly, the POCP values between representatives of the two genera Magnetofaba and Magnetococcus were 53–57%, which is higher than the proposed genera separation threshold at 50% (Qin et al., 2014). Thus, a 50% POCP threshold is not applicable for separating genera within the order Magnetococcales. A similar situation was observed for the differentiation within other taxonomic groups, such as the families Methylococcaceae (Orata et al., 2018) and Neisseriaceae (Li Y. et al., 2017).
The strains were distinguished at the species level using the nucleotide sequence-based indexes ANI and dDDH. The ANI analysis showed identity values ranging from 70 to 99.4% between all Magnetococcales genomes (Supplementary Table S4). The ANI values for strain UR-1 were below the standard species separation threshold (<95–96%) with all analyzed sequences, indicating that it belongs to a novel species (Goris et al., 2007). The dDDH analysis showed values ranging from 15.1 to 93.3% (Supplementary Table S5). The dDDH values for strain UR-1 were also below the standard species separation threshold (<70%) with all analyzed sequences, thereby confirming the results of the ANI calculation (Auch et al., 2010). According to the results of ANI and dDDH, within the clade “WMHbin1,” the strains DCbin4 and WMHbin1 belonged to the same species, whereas the values between HA3bin1 and HAa3bin1 (95.3%) are at the boundary for delineation of two species (95–96%); hence, they could not be separated with confidence.
Based on the results obtained by analyzing genome sequences, we propose a delineation of five candidate families within the order Magnetococcales and differentiate strain UR-1 as a novel candidate species within one of these families, for which we propose the name “Ca. Magnetaquicoccaceae.” For strain UR-1, we propose the name “Ca. Magnetaquicoccus inordinatus.”
Genome Analysis of the “Ca. Magnetaquicoccus inordinatus” UR-1
Magnetosome Genes
The assembly of nearly the complete genome of UR-1 allowed identification of parts of the magnetosome genomic island (MAI). Most of the magnetosome synthesis genes were found in the contig RXIU01000008 (44,671 bp). This contig contained 42 genes, 17 of which had a high level of similarity with the mam and mms genes from the representatives of Magnetococcaceae and magnetotactic Alphaproteobacteria (Supplementary Table S6). In addition, the second mamK gene was found in the contig RXIU01000185 (7,100 bp). The mamAB cluster contained the mamK, mamF-like, mamL, mamM, mamN, mamO, mamP, mamA, mamQ, mamB, mamS, and mamT genes (Figure 5). A separate mamHIE cluster was also found, and the mmsF-like and mamD-like genes were located upstream of that. Interestingly, MmsF-like and MamD-like had a high level of similarity with the homologous proteins found in “Ca. Terasakiella magnetica” PR-1 (Monteil et al., 2018) and Magnetospirillum caucaseum SO-1T (Dziuba et al., 2016) (55 and 50%, respectively). Between the mamAB and mamHIE clusters, we identified an ORF, the putative product of which has no predicted function and homology with any of the proteins in the GenBank database. At the end of the contig in which the MAI was located, an incomplete gene was found that has a high similarity to feoA1. The location of this part of the gene was similar to that of the freshwater coccus WMHbinv6, which has feoAB1 genes 7.5 kb upstream of MAI. An incomplete feoB1 gene was also found in UR-1 (contig RXIU01000148), next to which there was a gene with a high similarity to mamB.
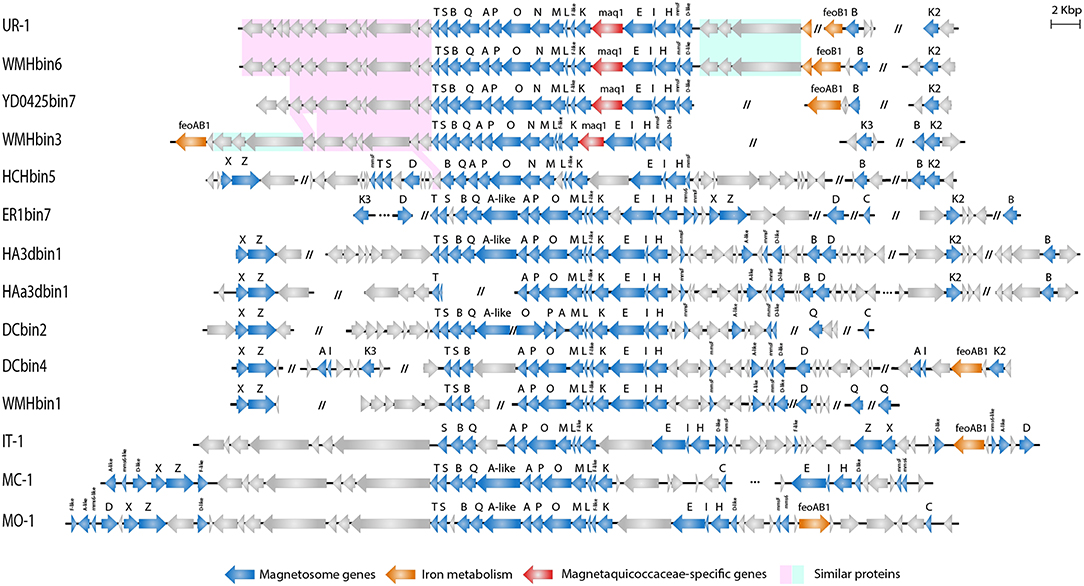
Figure 5. Organization of MAI region in genomes of Magnetococcales strains.
A subsequent phylogenetic analysis showed that the tree based on the concatenated amino acid sequences of the Mam protein was non-collinear with the tree developed on the basis of core genome proteins. On the phylogenetic tree of the magnetosome proteins, UR-1 clustered together with WMHbinv6, UD0425bin7, and WMHbin3 (Figure 3B); however, the clade was external to the branches formed by other strains. In addition, the concatenate of HCHbin5 formed a separate branch and did not cluster with representatives of the same genera. The inconsistency between positions of branches on the obtained trees suggested the horizontal transfer (HGT) of magnetosome genes in “Ca. Magnetaquicoccaceae” and within the family, in HCHbin5. We also provided a comparison of phylogenetic trees based on concatenated protein sequences from mamHIE and mamAB clusters (Supplementary Figure S2). Both trees were mostly congruent, with the exception of HCHbin5 and DC0425bin3 positions. However, the tree of MamHIE proteins had low branch support; therefore, solving the question of how magnetosome clusters evolved in these two strains will require further genome sequences of “Ca. Etaproteobacteria” are needed. In spite of this, the result of the tree topology comparisons suggested that the evolutionary history of both clusters was largely the same in the “Ca. Etaproteobacteria” strains.
In addition to the general non-collinearity of the trees, an interesting result was observed. The phylogenomic tree shows (Figure 3A) that the amino acid substitution rate between strains UR-1 and WMHbinv6 is much higher than that on the phylogenetic tree of the magnetosome proteins (Figure 3B). This discrepancy in branch lengths may be due to recent horizontal gene transfer (Syvanen, 1994; Koonin et al., 2001). The putative horizontal gene transfer was explored using the recentHGT tool, which detects HGT events at the genome level between closely related species (Li et al., 2018). The strain YD0425bin7, closely related to UR-1 and WMHbinv6, was also included in the analysis. The recentHGT strategy allowed calculation of WMHbinv6 and YD0425bin7 sequence similarity values for all homologous genes from strains UR-1. The analysis of the obtained data revealed that all values between strains YD0425bin7 and WMHbinv6, as well as strains UR-1 and YD0425bin7, complied with the Weibull distribution (Figures 6B,C, respectively). Thus, no signs of recent HGT events were detected between these two pairs of strains. However, in case of UR-1 and WMHbinv6, the similarity values of the magnetosome-associated genes did not fit the Weibull distribution (Figure 6A, Supplementary Table S7). The similarity values of the detected genes were 95–100%, which is much higher than the similarity between the housekeeping genes. Thus, the obtained results indicated that MAI genes in UR-1 and WMHbinv6 had undergone a recent HGT event.
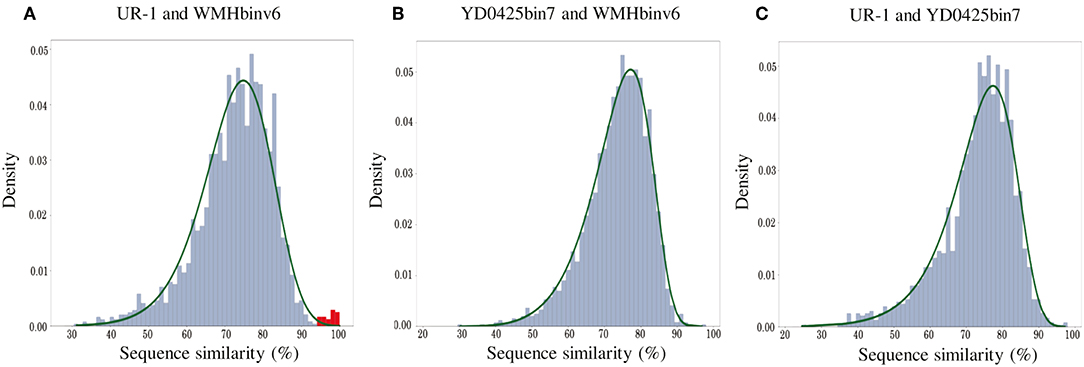
Figure 6. Histograms of sequence similarity values between (A) UR-1 and WMHbinv6, (B) YD0425bin7 and WMHbinv6, (C) UR-1 and YD0425bin7. The x-axis and y-axis represent the values of sequence similarity and density, respectively. The green line shows the boundaries of the Weibull distribution. The distribution of sequence similarity of horizontally transferred genes is highlighted in red.
Comparison of the magnetosome island organization of UR-1 with the other strains demonstrated that representatives of the proposed family “Ca. Magnetaquicoccaceae,” namely UR-1, WMHbinv6, YD0425bin7, WMHbin3, and HCHbin5, had a similar set of magnetosome biomineralization genes. Thus, the mamN gene was included in the mamAB cluster in all of them, unlike in the other Magnetococcales strains. When searching for the closest homologs for the MamN protein, we identified a high level of similarity with Magnetovibrio blakemorei (43.89%) (Trubitsyn et al., 2016) (Supplementary Table S6). The mamCXZ gene cluster, which is present in the genomes of the Magnetococcaceae strains and the other three proposed families, was not found in the UR-1 genome or in the closest related strains WMHbinv6, YD0425bin7, and WMHbin3. The mamA-like gene, which is located inside the mamAB cluster, and the mms6 genes were also absent in genomes of UR-1 and related strains. An additional difference was the presence of a gene located between mamK and mamE. The hypothetical protein encoded by that gene had no homology with any of the proteins in the GenBank database and is probably specific to members of the family “Ca. Magnetaquicoccaceae.” That gene was designated maq1 (“Magnetaquicoccaceae” specific).
Three other genes encoding proteins with unknown functions were found between the mamD-like and feoA1 genes in the UR-1 genome. As predicted by COG and pfam, protein WP_130470120 has an EH signature domain, WP_130470121 was classified as flagellar motor protein MotB, and WP_130470122 contains apolipoprotein A1/A4/E domains. These proteins did not have homology with any of the known proteins of MTB but were also present in WMHbinv6 and WMHbin3. The closest homologs were identified in non-magnetotactic Gamma- and Deltaproteobacteria. The organization and arrangement of these three genes was identical in UR-1 and WMHbinv6; this cluster was 9 kb downstream of the mamT gene in WMHbin3, and the feoAB1 genes were located next to them. Another similarity in MAI organization between UR-1 and WMHbinv6 was observed for 11 genes located downstream of mamT. Eight and seven of them, in the same order, were also found in the strains closest to the UR-1, namely YD0425bin7 and WMHbin3, respectively. Their products are associated with hypothetical and chemotaxis proteins. At the same time, HCHbin5, which belongs to the same candidate genus as UR-1, differed significantly in the MAI structure from the other members of the same group. Another set of the genes flanked the main cluster of MAI, and additional hypothetical proteins were present in the HCHbin5 mamAB cluster. Furthermore, in contrast to the other strains from the proposed family “Ca. Magnetaquicoccaceae,” the mamXZ genes were found in the HCHbin5 genome. In addition, the gene between mamK and mamE in the MAI of HCHbin5 had a low similarity (5% coverage) with the maq1 gene of “Ca. Magnetaquicoccaceae.”
In conclusion, a similar set and organization of magnetosome genes in strains UR-1, WMHbinv6, YD0425bin7, and WMHbin3 may indicate that the strains synthesize magnetosomes with a similar arrangement, whereas the organization of magnetosome genes in HCHbin5 significantly differed from those of the other members of the family “Ca. Magnetaquicoccaceae.” This suggests that magnetosomes might be arranged differently in this strain.
Metabolic Reconstruction
Recovering of the nearly complete genome of UR-1 allowed prediction of the metabolic traits of this novel magnetotactic strain. Comparison of the UR-1genome with the MAGs of other freshwater Magnetococcales allowed the determination of group-specific metabolic features that support the proposed division of the studied strains into distinct phylogenetic groups. Here, we focus on selected metabolic pathways, including carbon fixation, and nitrogen, sulfur, and phosphorus metabolism. The description of the cellular transport, oxidative stress defense strategies, chemotaxis, and motility of UR-1 is provided in Supplementary File 1. The metabolic features of the novel strain are summarized in Figure 7.
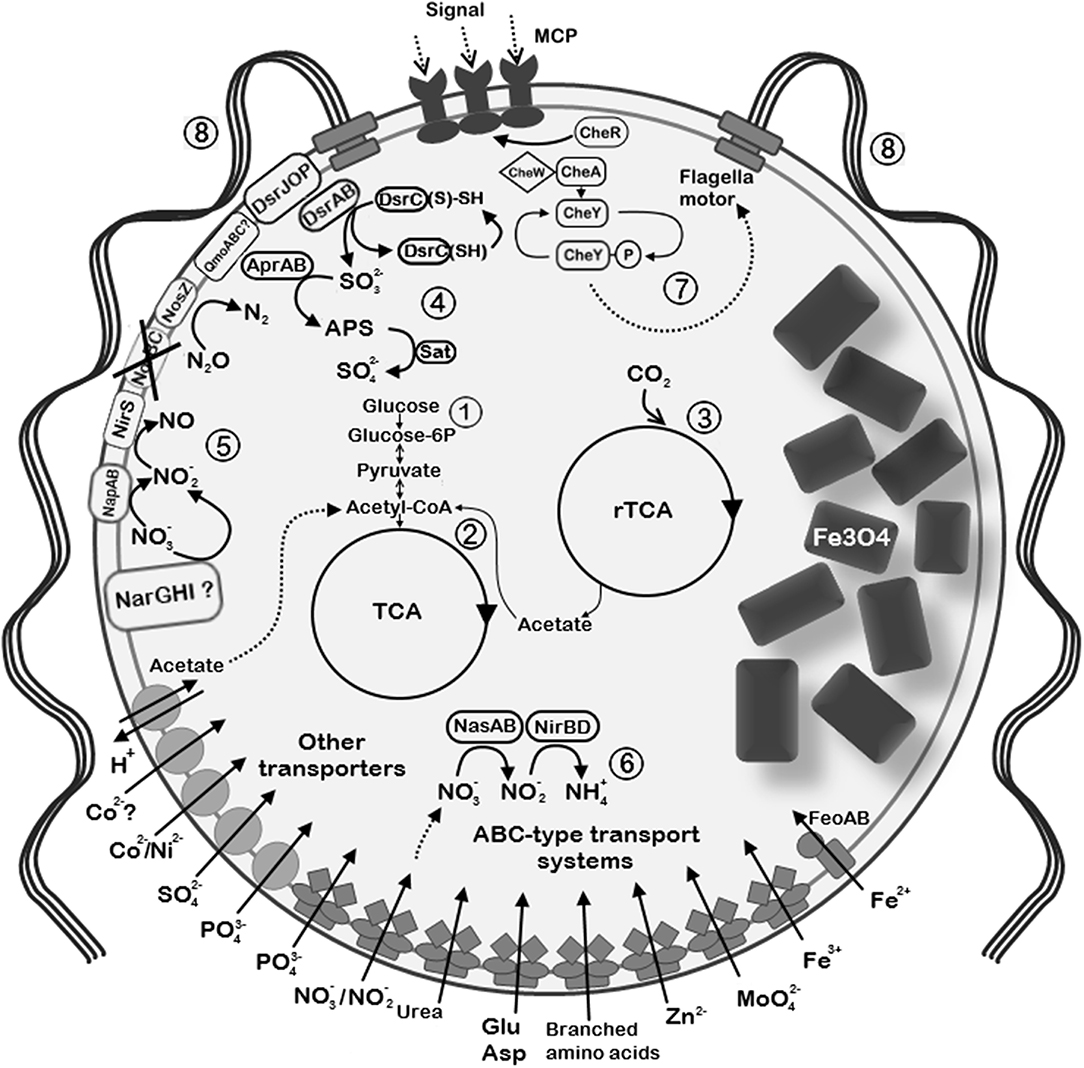
Figure 7. Reconstruction of the selected metabolic and structural features of “Ca. Magnetaquicoccus inordinatus” based on genome analysis and TEM micrographs. The following metabolic pathways are emphasized: (1) glycolysis and glyconeogenesis; (2) tricarboxylic acid cycle (TCA); (3) autotrophic CO2 fixation through reverse TCA (rTCA); (4) sulfide oxidation trough reverse Dsr (dissimilatory sulfite reductase) pathway. Sulfite which is formed via Dsr operation is further oxidized to sulfate through the intermediate 5'-adenylyl sulfate (APS) by APS reductase (AprAB) and sulfate adenylyltransferase (Sat). (5) Dissimilatory nitrate reduction is possible through the periplasmic nitrate reductase (NapAB) and putative membrane-bound nitrate reductase complex (NarGHI). Nitrite reduction is further possible by nitrite reductase NirS. Since norBC genes were not found (crossed out), might be not capable of nitric oxide reduction, whereas nitrous oxide can be reduced to nitrogen by nitrous oxide reductase (NosZ). (6) Assimilatory nitrate and nitrite reduction in the cytoplasm through nitrate reductase (NasA) and nitrite reductase NirBD, respectively. (7) Chemotaxis complex is represented by methyl-accepting chemotaxis proteins (MCP) and Che proteins. (8) The species presumably synthesizes multiple flagella due to the presence of high number of flagellin genes. Since two bunches of flagella are common among magnetotactic cocci, similar organization is assumed in “Ca. Magnetaquicoccus inordinatus”.
Carbon Fixation
Strong experimental evidence supports the utilization of a reverse tricarboxylic acid cycle (rTCA) for CO2 fixation by MC-1T and IT-1 (Williams et al., 2006; Araujo et al., 2016). The genome of MC-1T contains at least three enzymes key to the rTCA: 2-oxoglutarate:acceptor oxidoreductase, pyruvate:acceptor oxidoreductase, and fumarate reductase. However, although citrate cleavage to acetyl-CoA and oxaloacetate is detected in this organism, none of the genes for citryl-CoA synthetase, citryl-CoA lyase, or the bona fide ATP:citrate lyase (ACL) were found in its genome. Nevertheless, two ORFs, which encode subunits of a postulated type II ACL (Mmc1_3638, Mmc1_3639), are present in MC-1T (Schübbe et al., 2009; Hügler and Sievert, 2011). The same is true for the genomes of other cultivated marine magnetotactic cocci “Ca. Mc. massalia” MO-1 and Mf. australis IT-1, suggesting that this group of bacteria employs another type of enzymes for catalyzing citrate cleavage. Our analysis showed that the presence of the genes for the putative type II ACL and the other key enzymes appears to be shared by UR-1 and many other freshwater magnetotactic strains (Supplementary Table S8), implying that rTCA may be a common trait for this group. However, in five MAGs (WMHbin1, DCbin2, DCbin4, HA3dbin1, and HAa3bin1), none of the subunits of the putative ACL were found, while the other key proteins of the pathway are present. This might reflect an incomplete state of the genomes. Interestingly, these strains form a separate group at the family level (Figure 3), which may hint that some other mechanism of carbon fixation, if any occurs, might be intrinsic to this group.
Nitrogen Metabolism
The genome of UR-1 contains the entire set of nif genes for di-nitrogen fixation, a feature that is frequently encountered among MTB, with only a few known exceptions, i.e., Magnetospira sp. QH-2 and Gammaproteobacterial magnetotactic strain SS-5 (Lefèvre et al., 2012b; Ji et al., 2014). The nif cluster in UR-1 comprises 18 genes, including nifVXNEBQHDKTY with the nitrogen fixation transcriptional regulator nifA localized separately, approximately 7 kb downstream of nifY. The nifU gene, which is proposed to have a redox function for assembly of the Fe-S cluster in nitrogenase complex, was also found and is located at a different chromosomal locus (Agar et al., 2000). Unlike the case for MC-1T and MO-1, the nif operon in UR-1 is lacking the gene nifZ, and no homologs could be found outside the nif gene cluster. NifZ is known to play an accessory, but not essential, role in the maturation of MoFe nitrogenase P-clusters (Jimenez-Vicente et al., 2019). Interestingly, nifZ could not be found in the nif operons of several other uncultivated freshwater Magnetococcales strains, which otherwise have essential nif genes: YD0425bin7, WMHbin3, WMHbin1, DC0425bin3, DCbin4, and HA3dbin1. We cannot rule out that nifZ does not constitute a part of the nif operon in these strains; it could be missing due to sequence incompleteness. Alternatively, other mechanism of maturation of the nitrogenase complex could have evolved in these groups of bacteria.
UR-1 is potentially capable of assimilatory and dissimilatory nitrate reduction. The nap operon, which encodes periplasmic nitrate reductase (NAP) together with the genes necessary for its maturation, comprises 6 genes, i.e., napDAGHBC. Interestingly, the nap operon in UR-1 is lacking the gene for the non-heme iron-sulfur protein NapF, which is the first gene in this operon in the majority of denitrifying bacteria, including MC-1T and MO-1. However, in E. coli, NapF is not essential for either catalytic activity or for maturation of the NAP complex, implying that UR-1 can still possess a fully active NAP (Potter and Cole, 1999). In addition to NAP, an operon encoding alpha chain (molybdopterin-containing catalytic subunit), beta chain (iron-sulfur center-containing electron transfer unit) and gamma chain (heme b) of a complex protein belonging to membrane-bound nitrate reductase NarGHI-like anaerobic respiratory enzymes is present in the genome of UR-1 and the closely related freshwater strains WMHbinv6 and YD0425bin7. The gene for a protein with high similarity to TorD-like chaperones, which mediate maturation of the complexes of the molybdopterin respiratory enzymes, was also determined in UR-1 in close proximity to the putative nar operon, suggesting a probably complete assembly and hence functionality of the NarGHI-like enzyme. However, the relatively low similarity of these proteins from UR-1 to the well-studied NAR from other bacteria (e.g., 26.1% for catalytic subunit NarG compared to that of E. coli) does not allow us to predict the type of reduced substrate with confidence, as several other anaerobic respiration oxido-reductases have very similar sequences (e.g., dimethyl sulfoxide, selenate, and chlorate reductase) (Leimkühler and Iobbi-Nivol, 2015). This may suggest an extended ability of UR-1 and related strains to utilize alternative terminal electron acceptors.
The presence of genes for NirBD and NirS in the genome of UR-1 suggests its ability to further reduce nitrite produced by nitrate reductases to ammonia and nitric oxide, respectively. Interestingly, two genes for cd1 cytochrome NO-forming nitrite reductase, NirS, were found in the genome of UR-1. One of the nirS genes (contig RXIU01000046) is followed by nirCFGLHJ, a set of genes encoding the important factors for the biogenesis of the d1 cofactor of NirS. As the gene cluster is located at the end of the contig having nirJ lopped approximately in the middle, determining a precise organization of the nir operon is impossible in UR-1. The gene order in the nir operon may vary among the denitrifying bacteria, with the usually conserved nirFDLGH set and nirN localized as the last genes in the operon (Zajicek et al., 2009). In UR-1, a gene fragment belonging to the putative nirN is localized at the edge of contig RXIU01000360, suggesting that contigs RXIU01000046 and RXIU01000360 may be linked together. Thus, UR-1 possesses almost the entire set of genes needed for the synthesis and functional assembly of nitrite reductase NirS, lacking only the nirE gene. The nirE gene is almost always found adjacent to nirS, and only a few exceptions are known (e.g., Aromatoleum aromaticum) where it is located in a separate operon (Zajicek et al., 2009). Hence, if nirE is localized outside the nir operon in UR-1, it quite possibly was not covered by sequencing.
The second nirS gene is located in contig RXIU01000354, adjacent to a gene for a NirC-like cytochrome. Due to the incomplete state of the genome, we cannot rule out that UR-1 has a second set of the genes for NirS biosynthesis as well that could not be identified in the genome at present. Redundancy in nirS genes may indicate the importance of nitrite respiration in the energy conservation of UR-1.
Curiously, the structural genes for nitric oxide reductase, norBC, were not found in the genome of UR-1 and the related strains WMHbinv6 and HCHbin5 (YD0425bin7 differed, as it had the full set of nor genes, Supplementary Table S8). At the same time, norQ and norD, which are usually parts of the norBCQD operon, can be still found in the genome of UR-1, closely adjacent one to another. The absence of norBC might be explained by the incomplete state of the UR-1, WMHbinv6, and HCHbin5 genomes. Although truncated versions of denitrification are frequent in bacteria from various environments, the lack of nitric oxide reduction is relatively rare due to the danger of accumulation of highly toxic NO (Lycus et al., 2017). Finally, UR-1 appears to be capable of converting N2O to N2 in the last step of denitrification, owing to the presence of the nosZDFYL operon.
In contrast to the marine cocci (e.g., MC-1T, MO-1, and IT-1), UR-1 has the potential ability for assimilatory nitrate reduction as it has the gene set for NADH-dependent assimilatory nitrate and nitrite reductases in its genome. The structural gene for assimilatory nitrate reductase, NasA, was found adjacent to the genes for nitrite reductases nirD and nirB in an operon-like structure, and was followed by genes for the ABC nitrate transporter nrtABC (see section “Transport” in Supplementary File 1), two-component transcriptional regulator nasTS, an HPP-family protein of unknown function, and a fragment of the MFS transporter gene. Interestingly, the presence of nitrate assimilation genes is not common among the available genomes of uncultivated freshwater Magnetococcales strains, being restricted to UR-1 and the closely related WMHbinv6.
Sulfur Metabolism
The genes for the key enzyme of sulfate assimilation, sulfate adenylyltransferase cycN, and cycD, as well as 3'-phosphoadenosine 5'-phosphosulfate synthase PAPSS, were not found in the genome of UR-1 and the other freshwater Magnetococcales strains. This consistence suggests that these genes are more likely to be missing from the analyzed genomes, rather than not being covered by sequencing. The predicted inability to assimilate sulfate by these organisms is consistent with the absence of specialized transport systems for sulfate, which usually occur in bacteria capable of sulfate assimilation (Hryniewicz and Kredich, 1991). Considering that MTB inhabit anoxic and oxic-anoxic transition zones in the aquatic sediments, which are typically rich in reduced sulfur compounds, these organisms should be able to dispense with assimilatory sulfate reduction.
Our analysis predicts that reduced sulfur plays an important role in the metabolism of UR-1, presumably serving as substrate for dissimilatory sulfur oxidation (and possibly reduction as well). MC-1T is well-established as capable of oxidizing thiosulfate or sulfide when it grows chemolithoautotrophically. MC-1T contains a truncated minimalistic soxXYZAB operon and additional putative sox genes distributed in the chromosome in three distinct regions (Schübbe et al., 2009; Bazylinski et al., 2013). The lack of the sulfur dehydrogenase genes soxCD is consistent with accumulation of sulfur deposits in MC-1T (Dahl, 2015). In contrast to marine magnetic cocci, most of the essential sox genes were not found in the genome of UR-1 and other available genomes of “Ca. Etaproteobacteria,” with the exception of HA3dbin3. At the same time, genes for the periplasmic protein SoxYZ were found in almost all the genomes. The fact that soxXYZAB genes usually constitute a single operon and the consistent absence of all sox genes except soxYZ from the analyzed genomes suggest that most of the freshwater magnetotactic cocci more likely lack the SOX pathway, rather than indicating that the sox genes were not covered by sequencing.
The question arises whether the soxYZ genes in these bacteria are non-functional remnants of the complete sox operon or whether they play a role in a different process. A recent study by Dahl et al. demonstrated that SoxYZ alone is involved in sulfite oxidation in Allochromatium vinosum, while the other Sox proteins did not appear to play any role in this process (Dahl et al., 2013). Therefore, in freshwater Magnetococcales, SoxYZ may contribute to the oxidation of sulfite produced by the reverse-acting DsrAB, in parallel with the adenylylsulfate reductase in the 5'-adenylyl sulfate (APS) sulfite oxidation pathway (see below).
UR-1 and the other analyzed strains may be potentially capable of sulfide oxidation through a flavocytochrome c sulfide dehydrogenase (FccAB). That protein consists of a large flavoprotein (FccB) and a smaller cytochrome c (FccA) subunit, which are related to SoxF and SoxE, respectively (Sander and Dahl, 2009). Sulfide oxidation catalyzed by FccAB is expected to form sulfur globules, but none were observed in the available TEM micrographs of UR-1. Sulfur from deposits can be oxidized to sulfite through a reverse-acting dissimilatory sulfite reductase DsrAB, which has an indispensable role in the oxidation of sulfur globules in A. vinosum (Sander and Dahl, 2009). Interestingly, UR-1 contains two copies of the dsrABL genes and a single copy of each dsrC and dsrFEHJOP gene cluster. The presence of dsrEFH and dsrL indicates the reverse (i.e., sulfur oxidizing) type of Dsr pathway in UR-1 and related bacteria (Ghosh and Dam, 2009). This is also confirmed by the phylogenetic analysis, which showed clustering of the dsrAB genes from UR-1 within the sulfur-oxidizing group (Supplementary Figure S3). They formed a monophyletic branch with sulfur-oxidizing Chlorobi and Mc. Marinus, which was consistent with a previous study (Müller et al., 2015). The second copy of DsrAB of the Magnetococcales members generally belongs in the sulfur-oxidizing group, but it formed a separate monophyletic branch. Due to their distant position and the absence of a cultured strain with a known type of sulfur metabolism, the type of the second DsrAB in Magnetococcales requires further confirmation.
In contrast to the marine MTB (e.g., MC-1T and MO-1), UR-1 and other related strains appear to have a potential to further oxidize sulfite produced in dissimilatory sulfur oxidation by indirect AMP-dependent oxidation via the intermediate adenylsulfate (APS). In the reverse direction, this pathway can be also used during dissimilatory sulfate reduction (Matias et al., 2005). Therefore, we cannot rule out that the strains are also capable of respiratory sulfate reduction employing the same enzymes. The APS reductase AprAB catalyzes the formation of APS from sulfite and AMP. In a second step, the AMP moiety is transferred to pyrophosphate by ATP sulfurylase (ATP:sulfate adenylyltransferase) or to phosphate by adenylylsulfate:phosphate adenylyltransferase (Sander and Dahl, 2009). Similar to the genes in some other sulfur-oxidizing bacteria, the genes for ATP sulfurylase (sat) and APS reductase (aprAB) are encoded in the same operon in UR-1 (RXIU01000214). However, the adenylylsulfate:phosphate adenylyltransferase was not found in the genome of UR-1.
In addition to the genes discussed in this section, other gene clusters potentially having accessory functions in sulfur metabolism were found in UR-1. Their descriptions and speculations as to their potential functions are presented in the Supplementary File 1.
Phosphorus Metabolism
Phosphate (Pi) is an essential nutrient that is often scarce in the natural environment. Bacteria take up Pi by low-affinity inorganic phosphate transporters (e.g., PitAB in E. coli), as well as by the high-affinity ABC-type phosphate-specific transporter Pst. Many bacteria are capable of employing the ABC transport system PhnCDE for phosphonates, the compounds containing carbon-phosphorus (C-P) bonds (Villarreal-Chiu et al., 2012). The genomes of the marine magnetotactic cocci include both low-affinity and high-affinity transporters for Pi, as well as the high-affinity PhnCDE for phosphonates, which reflects adaptation of these organisms to the phosphorus limitation common in some marine habitats (Tyrrell, 1999; Schübbe et al., 2009). Multiple (up to 5) low-affinity Pit-like phosphate transporters were found in the genome of UR-1 and the closely related genomes WMHbinv6, YD0425bin7, and HCHbin5. Genes for these transporters were also revealed in WMHbin3 and ER1bin7, but not in the other available MAGs (Supplementary Table S8). The genome of UR-1 contains all the essential genes for the Pst Pi–specific transporter (pstD, A, B, S), as well as for the phosphorus uptake specific transcriptional regulator PhoU. Interestingly, the presence of several copies of these gene clusters appears to be common among freshwater Magnetococcales strains (Supplementary Table S8). In contrast to marine magnetotactic cocci, no phosphonate uptake systems were found in the analyzed freshwater strains. In UR-1 and several other analyzed genomes, genes similar to those for the putative phosphonate-binding periplasmic protein PhnD were found, but the other components of the transporting system were absent, suggesting that these are non-functional remnants or have a different function.
Accumulation of polyphosphate (poly-P) granules is common among MTB, such as MC-1T and Magnetospirillum spp. The TEM micrographs of UR-1 also revealed prominent dark inclusions, reminding poly-P of other bacteria (Figure 2E) (Lins and Farina, 1999; Keim et al., 2005). Poly-P kinase (PPK) is a principle enzyme that catalyzes the transfer of the terminal phosphate of ATP (PPK1) or of GTP (PPK2) to an active site of the protein, the initial step in the progressive synthesis of a long poly-P chain. The reaction is reversible, and in case of PPK2, it is used to synthesize GTP from poly-P (Rao et al., 2009). Many bacteria possess both types of PPK, whereas only one type is found in others. The exclusive presence of genes for PPK2, but not for PPK1, appears to be an intrinsic feature of both marine and freshwater Magnetococcales strains. Interestingly, usually several genes for PPK2 were present. In UR-1, two genes for PPK2 (WP_130472334.1 and WP_130472795.1) shared 49.4% identity with each other. The function of the synthesis and utilization of poly-P may be divided between different PPK2 enzymes in UR-1. Bacteria capable of synthesizing poly-P evolved several mechanisms for efficient poly-P utilization. The enzyme catalyzing the incorporation of Pi into AMP to restore triphosphate, poly-P:AMP phosphotransferase PAP, appears to be absent from the genomes of most Magnetococcales, except for Mf. australis IT-1. At the same time, the marine magnetic cocci, as well as many genomes of freshwater Magnetococcales strains, possess a gene for exopolyphophatase (PPX), which product hydrolyses and progressively releases the terminal phosphates from linear poly-P. Moreover, a high transcription level of the ppx gene was demonstrated in a group of marine magnetotactic cocci that accumulate conspicuous poly-P inclusions and seem to serve as phosphate shuttles within the marine suboxic zone (Schulz-Vogt et al., 2019). Intriguingly, no known genes for PPX were found in UR-1 and the related genomes, WMHbinv6, YD0425bin7, and HCHbin5. Although we cannot rule out that the corresponding genes had not been covered by sequencing, this consistent absence of them in all genomes of the strains closely related to UR-1 suggests that the ability to use poly-P in these organisms might be limited to the predicted GTP synthesis by a PPK2-like enzyme.
Discussion
This paper is the first to attempt to link the cell structure and magnetosome characteristics of an uncultivated freshwater magnetotactic coccus with a high quality MAG using FISH-TEM.
As demonstrated by TEM analysis, strain UR-1 is remarkable for its synthesis of clustered magnetosomes instead of the ordinary magnetosome chains observed in cultivated MTB and most of the uncultured MTB. The magnetosome formation and arrangement is strictly controlled genetically in the model magnetotactic organisms of the genus Magnetospirillum; therefore, we can assume that the peculiar organization of magnetosomes in UR-1 is also determined by the magnetosome gene clusters revealed in this study. The chain formation in Magnetospirillum is mediated by the mamK, mamJ, and mamY genes (Scheffel et al., 2006; Abreu et al., 2014; Toro-Nahuelpan et al., 2019). Among these, mamJ and mamY were not found in the available genomes of Ca. Etaproteobacteria, which indicates that other mechanisms of chain assembly and positioning have evolved in this group of MTB. By contrast, MamK that encodes an actin-like protein important for the chain organization is universally present in MTB, at least in those known to produce one or multiple chains (Komeili et al., 2006; Katzmann et al., 2010; Abreu et al., 2014). Therefore, the mamK gene was expected to be absent from the genome of UR-1, as no clear chains could be identified within the cells. However, the gene was found within magnetosome gene clusters of UR-1 and was even present as two copies.
The “unchained” magnetosomes in some magnetotactic cocci have often been observed to have a location proximal to the flagellar end of the cell rather than being distributed evenly in the cell body (Spring et al., 1995; Lin and Pan, 2009; Abreu et al., 2012; Zhang et al., 2012). One possibility is that the coordinated action of the two MamK proteins provides the specific organization and positioning of magnetosome clusters within the cell; however, which protein links the MamK filament and the magnetosome remains unclear. The MAI of UR-1 contains several genes encoding proteins with unknown functions, which may be candidates for this role. One of these genes, maq1, is located between mamE and mamK in “Ca. Magnetaquicoccaceae,” giving it a similar position to that of mamJ in Magnetospirillum. Our analysis shows that, in addition to maq1, the magnetosome gene clusters of “Ca. Magnetaquicoccaceae” contain a set of 11 genes that could be involved in magnetosome formation and magnetotaxis process. Since little could be predicted from the sequences of these genes, it is very tempting to speculate that other determinants of the clustered magnetosome organization might be found among them. Further research, preferably based on axenic cultures, is necessary to understand the genetic mechanisms of the biomineralization and magnetosome arrangement in magnetotactic cocci, including those forming clustered magnetosomes.
Although the position of UR-1 and WMHbinv6 on the trees based on the concatenated Mam proteins was congruent with the phylogenomic tree, an unusually high similarity rate of the magnetosome genes in comparison to the rest of the core genome was revealed. Since high conservation of magnetosome genes has never been observed, and it would contradict the bulk of data on the evolution of magnetosome genes, this finding suggests that magnetosome gene clusters could be transferred horizontally or from another closely related species.
Another interesting result of the magnetosome protein analyses is a non-congruent position of the branch consisting of “Ca. Magnetaquicoccaceae” magnetosome proteins when compared with the core protein tree. The phylogenetic analysis indicates that the branch consisting of “Ca. Magnetaquicoccaceae” magnetosome proteins was external to all other concatenates; also, the HCHbin5 concatenate did not cluster with those of closely related strains.
These results support the idea of an HGT of magnetosome genes within the order Magnetococcales. As suggested previously, vertical inheritance, followed by multiple independent losses of a magnetosome island, are likely to be the main driving forces for the evolution of magnetosome biomineralization genes. For some MTB (for example, for the genus Magnetospirillum), cases of possible independent HGT events have been demonstrated (Rioux et al., 2010; Komeili, 2012; Lefèvre et al., 2012a; Ji et al., 2017; Lin et al., 2018; Monteil et al., 2018). For example, Mc. marinus MC-1T has a stable MAI (Schübbe et al., 2009), and biomineralization genes of both cultured Magnetococcales strains MC-1T and IT-1 appeared as a result of the vertical inheritance (Morillo et al., 2014). Our results suggest that MAI in Magnetococcales strains could spread due to HGT, which occurred between closely related, as well as more distantly related groups within the order. We found a putative phage-related gene located directly downstream of mamT in the genome of UR-1 that had low identity with the phage capsid protein found in the genome of Pseudoalteromonas neustonica (WP_130470101; 33% identity; 23% coverage). Moreover, genes encoding putative phage-related proteins were also identified directly upstream of feoB1: WP_130471887, WP_130471886, and WP_130471885 contained phage tail, phage cell wall peptidase, and phage_BR0599 domains, respectively. Thus, the influence of bacteriophages could be a possible mechanism for horizontal transfer of magnetosome genes, at least in UR-1.
According to our analysis, HCHbin5 and UR-1 belong to the same genus, but due to putative HGT they had a noticeable difference in the structure of their MAI and had a low level of similarity of magnetosome genes. This may suggest that the magnetosomes of HCHbin5 are also arranged differently. Some studies have shown that magnetotactic cocci with different chain organizations were hybridized with the same specific FISH probe for the 16S rRNA gene (Spring et al., 1995; Cox et al., 2002; Lin and Pan, 2009). This led to the assumption that no strict correlation exists between the magnetosome organization within the cells and the phylogenetic position of magnetotactic cocci. Therefore, the appearance of magnetosomes is not a suitable phenotypic criterion for delimitation of taxonomic groups of MTB (Lin and Pan, 2009). Our analysis supports this assumption, due to the relative frequency of HGT revealed within the order Magnetococcales.
Several decades of studies on the diversity of MTB have led to the accumulation of a large amount of data on the 16S rRNA of “Ca. Etaproteobacteria.” The phylogenetic analysis of the 16S rRNA sequences indicated their significant diversity. The absence of cultivated freshwater representatives hampered classification (new sequences could be classified only at the order level); furthermore, the metabolic features of the representatives of the group remained elusive. Recently, several MAGs of freshwater Magnetococcales members have been reconstructed (Lin et al., 2018), and this has provided new opportunities for comparative genomic analyses and for establishing phylogenetic relationships within the order Magnetococcales.
Based on the results of our genomic analysis, we have proposed AAI thresholds for the separation of taxons of representatives of the class “Ca. Etaproteobacteria” at family and genus levels. Using the AAI values boundaries of 55–56% for the separation of families and 64–65% for the differentiation of close genera perfectly correlated with the branching of the phylogenomic tree. In accordance with these boundaries, five families within the order Magnetococcales were delineated. One of defined families has been named “Ca. Magnetaquicoccaceae” and includes the coccus UR-1 isolated from the Uda River. The comparative genomic analysis conducted in the current research sheds light on the potential metabolic abilities of the strain UR-1 as a representative of the candidate family “Ca. Magnetaquicoccaceae.” In addition, some metabolic diversity has been identified among the representatives of the proposed family (see the description below).
Overall, our analysis shows the fundamental differences in the genetic determinants for sulfur oxidation in UR-1 and the related MTB (with the exception of HA3dbin3), in comparison to the marine magnetic cocci MC-1T, MO-1, and IT-1. The marine forms appear to employ the reverse operating DsrAB in combination with the Sox pathway, whereas we predict that the freshwater forms use the reverse DsrAB, in combination with the APS reductase-mediated oxidation of sulfite.
Several key enzymes for poly-P synthesis were found in various marine and freshwater Magnetococcales, suggesting their important role in the phosphorus cycle (Lins and Farina, 1999; Schulz-Vogt et al., 2019). However, intriguingly, no genes for PPK1, the primary poly-P synthesis enzyme, were revealed in the available genomes. Although PPK2 is considered to have an activity that favors poly-P degradation, in the absence of PPK1, PPK2 can operate as synthesizing enzyme, albeit with less efficiency (Rao et al., 2009). The presence of multiple copies of ppk2 gene in the sequenced genomes of Magnetococcales suggests a plausible scenario in which a novel poly-P synthesis-favoring type of PPK2 could evolve in this group of bacteria. To check this hypothesis, experiments with different PPK2 in the available cultivable representatives (MC-1, MO-1, or IT-1) should be conducted in the future.
In conclusion, the predicted metabolic features—the potential ability for chemolithoautotrophic growth with reduced sulfur compounds, nitrate respiration, the ability to assimilate nitrate and the expected relatively high tolerance to oxygen—may serve as useful guidelines for future attempts to isolate an axenic culture of UR-1 and related strains. These strains will be at the frontier of future research on this group of MTB.
Description of Novel Candidate Family, Genus and Species, and Taxonomic Proposals
Genomic analysis of the UR-1 genome classified it as a novel candidate genus within a novel candidate family of the order Magnetococcales.
We propose the following Latin names for the novel candidate taxa:
Candidatus Magnetaquicoccaceae
Magnetaquicoccaceae (Mag.net.a.qui.coc.ca.ce'ae. N.L. masc. n. Magnetaquicoccus a Candidatus generic name; -aceae ending to denote a family; N.L. fem. pl. n. Magnetaquicoccaceae the (Candidatus) Magnetaquicoccus family).
Ca. “Magnetaquicoccaceae” is characterized by the potential ability for chemolithoautotrophic growth with the oxidation of reduced sulfur compounds through a reverse Dsr pathway (most enzymes from the Sox system are absent) and carbon assimilation by rTCA with the type II ATP:citrate lyase. The genes for nitrogen fixation are not universally found in the members of the family and might be limited to “Ca. Magnetaquicoccus inordinatus,” YD0425bin7 and WMHbin3. The genes for dissimilatory nitrate/nitrite/nitric and nitrous oxide reduction enzymes were found, but not for the entire pathway, indicating that the pathway might be truncated at different steps, depending on the species. The potential ability for assimilation of nitrate by NasA, which is, in general, absent from other Magnetococcales, was predicted for two members of the family: “Ca. Magnetaquicoccus inordinatus” and WMHbinv6.
Candidatus Magnetaquicoccus
Magnetaquicoccus (Mag.net.a.qui.coc'cus. Gr. n. magnes, -etos a magnet; N.L. pref. magneto- pertaining to a magnet; L. fem. n. aqua water; N.L. masc. n. coccus (from Gr. masc. n. kokkos), coccus; N.L. masc. n. Magnetaquicoccus, magnetic coccus from water).
Candidatus Magnetaquicoccus inordinatus
Magnetaquicoccus inordinatus (in.or.di.na' tus. N.L. masc. adj. inordinatus not arranged).
UR-1 cell has a coccoid morphology and represents magnetite magnetosomes not organized in chains and clustered in one side of the cell. Magnetosomes presents mean length of 77.4 nm (SD = 11.8 nm) and mean width of 46.2 nm (SD = 7.9 nm).
Data Availability Statement
The datasets generated for this study can be found in the Genome project has been deposited in DDBJ/ENA/GenBank under the accession number RXIU00000000.
Author Contributions
VK and DG conceived and designed experiments. VK collected the data. MK performed bioinformatics data processing. DG and MU carried out the phylogenetic and comparative genomic analyses. PL performed FISH-TEM analysis. VK, MD, and DG analyzed the data. VK and MD drafted the original manuscript. All authors read and approved the final manuscript.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This study was performed using the scientific equipment of the Core Research Facility Bioengineering (Research Center of Biotechnology RAS). We thank Professor Aharon Oren for his help with Latin names. We thank Jefferson Cypriano, CENABIO, and Unimicro for helping with access to the TEM use in this work.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02290/full#supplementary-material
Supplementary File 1. Genomic reconstruction of cellular transport, oxidative stress defense strategies, chemotaxis and motility of UR-1.
Supplementary Figure S1. Control of FISH reaction using Uda 54-3 probe. (A) DIC image from E. coli culture. (B) E. coli cells stained with DAPI. (C) E. coli recognized by EUB probes. (D) E. coli cells not recognized by Uda 54-3 probe, indicating the specificity of this probe in the hybridization conditions described in this paper.
Supplementary Figure S2. Maximum-likelihood phylogenetic trees inferred from a comparison of concatenated magnetosome associated proteins MamABKLMOPST (2077 amino-acid sites) and MamEHI (885 amino-acid sites). Both trees reconstructed with evolutionary model LG+F+I+G4. The scale bar represents amino acid substitutions per site. Trees were rooted using two Magnetospirillum species, Zetaproteobacteria bacterium PCbin4 and Lambdaproteobacteria bacterium PCRbin3 as outgroup.
Supplementary Figure S3. Maximum-likelihood phylogenetic tree derived from the comparison of DsrAB sequences (1183 sequences) of sulfur-oxidizing and sulfate-reducing bacteria. Phylogenetic analysis was performed with a LG+F+I+G4 model based on 727 amino acid positions. The scale bar represents amino acid substitutions per site.
Supplementary Table S1. Physicochemical parameters of water at the sampling site.
Supplementary Table S2. Genome characteristics of “Ca. Magnetaquicoccus inordinatus” and other Magnetococcales strains used for comparison and phylogenetic reconstructions.
Supplementary Table S3. POCP values between strains of the order Magnetococcales. Black boarders separate the representatives belonging to different putative families; Red boarders separate putative genera.
Supplementary Table S4. ANI values between strains of the order Magnetococcales. Black boarders separate the representatives belonging to different putative families as was predicted by AAI analyses; Red boxes indicate same species; Orange boxes indicate putative same species.
Supplementary Table S5. dDDH values between strains of the order Magnetococcales. Black boarders separate the representatives belonging to different putative families as was predicted by AAI analyses; Red boxes indicate same species; Orange boxes indicate putative same species.
Supplementary Table S6. Results of analyses of putative magnetosome proteins of “Ca. Magnetaquicoccus inordinatus”.
Supplementary Table S7. Putatively horizontally transferred genes between “Ca. Magnetaquicoccus inordinatus UR-1” and Magnetococcales bacterium WMHbinv6. The percentage of genes similarity between UR-1 and WMHbinv6 is higher than between UR-1 and YD0425bin7 and between YD0425bin7 and WMHbinv6. This indicates the presence of horizontal genes transfer between UR-1 and WMHbinv6 and its absence between UR-1 and YD0425bin7 and between YD0425bin7 and WMHbinv6.
Supplementary Table S8. Distribution of the key genes for the major metabolic processes in the “Ca. Etaproteobacteria” members. “Y” (yes) indicates presence of a gene, “N” (no) indicates absence. Bold boarders separate the representatives belonging to different putative families according to the phylogenetic tree in Figure 3.
References
Abreu, F., Araujo, A. C. V., Leão, P., Silva, K. T., Carvalho, F. M., Cunha, O. L., et al. (2012). Culture-independent characterization of novel psychrophilic magnetotactic cocci from Antarctic marine sediments. Arthritis Rheum. 18, 4426–4441. doi: 10.1111/1462-2920.13388
Abreu, N., Mannoubi, S., Ozyamak, E., Pignol, D., Ginet, N., and Komeili, A. (2014). Interplay between two bacterial actin homologs, MamK and MamK-Like, is required for the alignment of magnetosome organelles in Magnetospirillum magneticum AMB-1. J. Bacteriol. 196, 3111–3121. doi: 10.1128/JB.01674-14
Agar, J. N., Yuvaniyama, P., Jack, R. F., Cash, V. L., Smith, A. D., Dean, D. R., et al. (2000). Modular organization and identification of a mononuclear iron-binding site within the NifU protein. J. Biol. Inorg. Chem. 5, 167–177. doi: 10.1007/s007750050361
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Amann, R., Peplies, J., and Schüler, D. (2006). “Diversity and taxonomy of magnetotactic bacteria,” in Magnetoreception and Magnetosomes in Bacteria, ed D. Schüler (Berlin; Heidelberg: Springer, 25–36.
Araujo, A. C. V., Morillo, V., Cypriano, J., Teixeira, L. C. R. S., Leão, P., Lyra, S., et al. (2016). Combined genomic and structural analyses of a cultured magnetotactic bacterium reveals its niche adaptation to a dynamic environment. BMC Genomics 17:726. doi: 10.1186/s12864-016-3243-8
Auch, A. F., von Jan, M., Klenk, H. P., and Göker, M. (2010). Digital DNA-DNA hybridization for microbial species delineation by means of genome-to-genome sequence comparison. Stand. Genomic Sci. 2, 117–134. doi: 10.4056/sigs.531120
Bazylinski, D. A., Williams, T. J., Lefevre, C. T., Berg, R. J., Zhang, C. L., Bowser, S. S., et al. (2013). Magnetococcus marinus gen. nov., sp. nov., a marine, magnetotactic bacterium that represents a novel lineage (Magnetococcaceae fam. nov., Magnetococcales ord. nov.) at the base of the Alphaproteobacteria. Int. J. Syst. Evol. Microbiol. 63, 801–808. doi: 10.1099/ijs.0.038927-0
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Boulygina, E. S., Kuznetsov, B. B., Marusina, A. I., Tourova, T. P., Kravchenko, I. K., Bykova, S. A., et al. (2002). A Study of nucleotide sequences of nifH genes of some methanotrophic bacteria. Microbiology 71, 425–432. doi: 10.1023/A:1019893526803
Castresana, J. (2000). Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol. Biol. Evol. 17, 540–552. doi: 10.1093/oxfordjournals.molbev.a026334
Chun, J., Oren, A., Ventosa, A., Christensen, H., Arahal, D. R., Costa, M. S., et al. (2018). Proposed minimal standards for the use of genome data for the taxonomy of prokaryotes. Int. J. Syst. Evol. Microbiol. 68, 461–466. doi: 10.1099/ijsem.0.002516
Cox, B. L., Popa, R., Bazylinski, D. A., Lanoil, B., Douglas, S., Belz, A., et al. (2002). Organization and elemental analysis of P-, S-, and Fe-rich inclusions in a population of freshwater magnetococci. Geomicrobiol. J. 19, 387–406. doi: 10.1080/01490450290098504
Dahl, C. (2015). Cytoplasmic sulfur trafficking in sulfur-oxidizing prokaryotes. IUBMB Life 67, 268–274. doi: 10.1002/iub.1371
Dahl, C., Franz, B., Hensen, D., Kesselheim, A., and Zigann, R. (2013). Sulfite oxidation in the purple sulfur bacterium Allochromatium vinosum: identification of SoeABC as a major player and relevance of SoxYZ in the process. Microbiology 159, 2626–2638. doi: 10.1099/mic.0.071019-0
Daims, H., Brühl, A., Amann, R., Schleifer, K.-H., and Wagner, M. (1999). The domain-specific probe EUB338 is insufficient for the detection of all Bacteria: development and evaluation of a more comprehensive probe set. Syst. Appl. Microbiol. 22, 434–444. doi: 10.1016/S0723-2020(99)80053-8
Du, H., Zhang, R., Zhang, W., Xu, C., Chen, Y., Pan, H., et al. (2017). Characterization of uncultivated magnetotactic bacteria from the sediments of Yuehu Lake, China. Acta Oceanol. Sin. 36, 94–104. doi: 10.1007/s13131-017-0980-8
Dziuba, M., Koziaeva, V., Grouzdev, D., Burganskaya, E., Baslerov, R., Kolganova, T., et al. (2016). Magnetospirillum caucaseum sp. nov., Magnetospirillum marisnigri sp. nov. and Magnetospirillum moscoviense sp. nov., freshwater magnetotactic bacteria isolated from three distinct geographical locations in European Russia. Int. J. Syst. Evol. Microbiol. 66, 2069–2077. doi: 10.1099/ijsem.0.000994
Dziuba, M. V., Kolganova, T. V., Gorlenko, V. M., and Kuznetsov, B. B. (2013). Species diversity of magnetotactic bacteria from the Ol'khovka River, Russia. Microbiology 82, 335–340. doi: 10.1134/S0026261713030028
Flies, C. B., Jonkers, H. M., De Beer, D., Bosselmann, K., Böttcher, M. E., and Schüler, D. (2005a). Diversity and vertical distribution of magnetotactic bacteria along chemical gradients in freshwater microcosms. FEMS Microbiol. Ecol. 52, 185–195. doi: 10.1016/j.femsec.2004.11.006
Flies, C. B., Peplies, J., and Schüler, D. (2005b). Combined approach for characterization of uncultivated magnetotactic bacteria from various aquatic environments. Appl. Environ. Microbiol. 71, 2723–2731. doi: 10.1128/AEM.71.5.2723-2731.2005
Frankel, R. B., Bazylinski, D. A., Johnson, M. S., and Taylor, B. L. (1997). Magneto-aerotaxis in marine coccoid bacteria. Biophys. J. 73, 994–1000. doi: 10.1016/S0006-3495(97)78132-3
Ghosh, W., and Dam, B. (2009). Biochemistry and molecular biology of lithotrophic sulfur oxidation by taxonomically and ecologically diverse bacteria and archaea. FEMS Microbiol. Rev. 33, 999–1043. doi: 10.1111/j.1574-6976.2009.00187.x
Goris, J., Konstantinidis, K. T., Klappenbach, J. A., Coenye, T., Vandamme, P., and Tiedje, J. M. (2007). DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int. J. Syst. Evol. Microbiol. 57, 81–91. doi: 10.1099/ijs.0.64483-0
Grouzdev, D. S., Rysina, M. S., Bryantseva, I. A., Gorlenko, V. M., and Gaisin, V. A. (2018). Draft genome sequences of “Candidatus Chloroploca asiatica” and “Candidatus Viridilinea mediisalina”, candidate representatives of the Chloroflexales order: phylogenetic and taxonomic implications. Stand. Genomic Sci. 13, 1–9. doi: 10.1186/s40793-018-0329-8
Gurevich, A., Saveliev, V., Vyahhi, N., and Tesler, G. (2013). QUAST: quality assessment tool for genome assemblies. Bioinformatics 29, 1072–1075. doi: 10.1093/bioinformatics/btt086
Hoang, D. T., Chernomor, O., von Haeseler, A., Minh, B. Q., and Vinh, L. S. (2017). UFBoot2: improving the ultrafast bootstrap approximation. Mol. Biol. Evol. 35, 518–522. doi: 10.1093/molbev/msx281
Hryniewicz, M. M., and Kredich, N. M. (1991). The cysP promoter of Salmonella typhimurium: characterization of two binding sites for CysB protein, studies of in vivo transcription initiation, and demonstration of the anti-inducer effects of thiosulfate. J. Bacteriol. 173, 5876–5886. doi: 10.1128/jb.173.18.5876-5886.1991
Huber, T., Faulkner, G., and Hugenholtz, P. (2004). Bellerophon: a program to detect chimeric sequences in multiple sequence alignments. Bioinformatics 20, 2317–2319. doi: 10.1093/bioinformatics/bth226
Hügler, M., and Sievert, S. M. (2011). Beyond the Calvin cycle: autotrophic carbon fixation in the ocean. Ann. Rev. Mar. Sci. 3, 261–289. doi: 10.1146/annurev-marine-120709-142712
Ji, B., Zhang, S., Zhang, W., Rouy, Z., Alberto, F., Santini, C.-L., et al. (2017). The chimeric nature of the genomes of marine magnetotactic coccoid-ovoid bacteria defines a novel group of Proteobacteria. Environ. Microbiol. 19, 1103–1119. doi: 10.1111/1462-2920.13637
Ji, B., Zhang, S. D., Arnoux, P., Rouy, Z., Alberto, F., Philippe, N., et al. (2014). Comparative genomic analysis provides insights into the evolution and niche adaptation of marine Magnetospira sp. QH-2 strain. Environ. Microbiol. 16, 525–544. doi: 10.1111/1462-2920.12180
Jimenez-Vicente, E., Yang, Z.-Y., Martin Del Campo, J. S., Cash, V. L., Seefeldt, L. C., and Dean, D. R. (2019). The NifZ accessory protein has an equivalent function in maturation of both nitrogenase MoFe protein P-clusters. J. Biol. Chem. 294, 6204–6213. doi: 10.1074/jbc.RA119.007905
Kanehisa, M., Sato, Y., Furumichi, M., Morishima, K., and Tanabe, M. (2019). New approach for understanding genome variations in KEGG. Nucleic Acids Res. 47, D590–D595. doi: 10.1093/nar/gky962
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Katzmann, E., Scheffel, A., Gruska, M., Plitzko, J. M., and Schüler, D. (2010). Loss of the actin-like protein MamK has pleiotropic effects on magnetosome formation and chain assembly in Magnetospirillum gryphiswaldense. Mol. Microbiol. 77, 208–224. doi: 10.1111/j.1365-2958.2010.07202.x
Keim, C. N., Solórzano, G., Farina, M., and Lins, U. (2005). Intracellular inclusions of uncultured magnetotactic bacteria. Int. Microbiol. 8, 111–117.
Komeili, A. (2012). Molecular mechanisms of compartmentalization and biomineralization in magnetotactic bacteria. FEMS Microbiol. Rev. 36, 232–255. doi: 10.1111/j.1574-6976.2011.00315.x
Komeili, A., Li, Z., Newman, D. K., and Jensen, G. J. (2006). Magnetosomes are cell membrane imaginations organized by the actin-like protein MamK. Science 311, 242–245. doi: 10.1126/science.1123231
Konstantinidis, K. T., Rosselló-Móra, R., and Amann, R. (2017). Uncultivated microbes in need of their own taxonomy. ISME J. 11, 2399–2406. doi: 10.1038/ismej.2017.113
Konstantinidis, K. T., and Tiedje, J. M. (2005). Towards a genome-based taxonomy for prokaryotes. J. Bacteriol. 187, 6258–6264. doi: 10.1128/JB.187.18.6258-6264.2005
Koonin, E. V., Makarova, K. S., and Aravind, L. (2001). Horizontal Gene transfer in prokaryotes: quantification and classification. Annu. Rev. Microbiol. 55, 709–742. doi: 10.1146/annurev.micro.55.1.709
Kozyaeva, V. V., Grouzdev, D. S., Dziuba, M. V., Kolganova, T. V., and Kuznetsov, B. B. (2017). Diversity of magnetotactic bacteria of the Moskva River. Microbiology 86, 106–112. doi: 10.1134/S0026261717010088
Laczny, C. C., Kiefer, C., Galata, V., Fehlmann, T., Backes, C., and Keller, A. (2017). BusyBee Web : metagenomic data analysis by bootstrapped supervised binning and annotation. Nucleic Acids Res. 45, W171–W179. doi: 10.1093/nar/gkx348
Lane, D. J. (1991). “16S/23S rRNA sequencing,” in Nucleic Acid Techniques in Bacterial Systematics, eds E. Stackebrandt and M. Goodfellow (Chichester: John Wiley & Sons, 115–148.
Lang, J. M., Darling, A. E., and Eisen, J. A. (2013). Phylogeny of bacterial and archaeal genomes using conserved genes : supertrees and supermatrices. PLoS ONE 8:e62510. doi: 10.1371/journal.pone.0062510
Lefèvre, C. T., Bennet, M., Landau, L., Vach, P., Pignol, D., Bazylinski, D. A., et al. (2014). Diversity of magneto-aerotactic behaviors and oxygen sensing mechanisms in cultured magnetotactic bacteria. Biophys. J. 107, 527–538. doi: 10.1016/j.bpj.2014.05.043
Lefèvre, C. T., Bernadac, A., Yu-Zhang, K., Pradel, N., and Wu, L. F. (2009). Isolation and characterization of a magnetotactic bacterial culture from the Mediterranean Sea. Environ. Microbiol. 11, 1646–1657. doi: 10.1111/j.1462-2920.2009.01887.x
Lefèvre, C. T., Schmidt, M. L., Viloria, N., Trubitsyn, D., Schüler, D., and Bazylinski, D. A. (2012a). Insight into the evolution of magnetotaxis in Magnetospirillum spp., based on mam gene phylogeny. Appl. Environ. Microbiol. 78, 7238–7248. doi: 10.1128/AEM.01951-12
Lefèvre, C. T., Viloria, N., Schmidt, M. L., Pósfai, M., Frankel, R. B., and Bazylinski, D. A. (2012b). Novel magnetite-producing magnetotactic bacteria belonging to the Gammaproteobacteria. ISME J. 6, 440–450. doi: 10.1038/ismej.2011.97
Leimkühler, S., and Iobbi-Nivol, C. (2015). Bacterial molybdoenzymes: old enzymes for new purposes. FEMS Microbiol. Rev. 40, 1–18. doi: 10.1093/femsre/fuv043
Li, J., Zhang, H., Liu, P., Menguy, N., Roberts, A. P., Chen, H., et al. (2019). Phylogenetic and structural identification of a novel magnetotactic deltaproteobacteria strain, WYHR-1, from a freshwater lake. Appl. Environ. Microbiol. 85, e00731–e00719. doi: 10.1128/AEM.00731-19
Li, J., Zhang, H., Menguy, N., Benzerara, K., Wang, F., Lin, X., et al. (2017). Single-cell resolution study of uncultured magnetotactic bacteria via fluorescence-coupled electron microscopy. Appl. Environ. Microbiol. 83:e00409-17. doi: 10.1128/AEM.00409-17
Li, X., Tong, W., Wang, L., Rahman, S. U., Wei, G., and Tao, S. (2018). A novel strategy for detecting recent horizontal gene transfer and its application to rhizobium strains. Front. Microbiol. 9:973. doi: 10.3389/fmicb.2018.00973
Li, Y., Xue, H., Sang, S. Q., Lin, C. L., and Wang, X. Z. (2017). Phylogenetic analysis of family Neisseriaceae based on genome sequences and description of Populibacter corticis gen. Nov., sp. Nov., a member of the family Neisseriaceae, isolated from symptomatic bark of Populus × Euramericana canker. PLoS ONE 12:e0174506. doi: 10.1371/journal.pone.0174506
Lin, H.-H., and Liao, Y.-C. (2016). Accurate binning of metagenomic contigs via automated clustering sequences using information of genomic signatures and marker genes. Sci. Rep. 6:24175. doi: 10.1038/srep24175
Lin, W., and Pan, Y. (2009). Uncultivated magnetotactic cocci from Yuandadu Park in Beijing, China. Appl. Environ. Microbiol. 75, 4046–4052. doi: 10.1128/AEM.00247-09
Lin, W., and Pan, Y. (2010). Temporal variation of magnetotactic bacterial communities in two freshwater sediment microcosms. FEMS Microbiol. Lett. 302, 85–92. doi: 10.1111/j.1574-6968.2009.01838.x
Lin, W., Tian, L., Li, J., and Pan, Y. (2008). Does capillary racetrack-based enrichment reflect the diversity of uncultivated magnetotactic cocci in environmental samples? FEMS Microbiol. Lett. 279, 202–206. doi: 10.1111/j.1574-6968.2007.01029.x
Lin, W., Zhang, W., Zhao, X., Roberts, A. P., Paterson, G. A., Bazylinski, D. A., et al. (2018). Genomic expansion of magnetotactic bacteria reveals an early common origin of magnetotaxis with lineage-specific evolution. ISME J. 12, 1508–1519. doi: 10.1038/s41396-018-0098-9
Lins, U., and Farina, M. (1999). Phosphorus-rich granules in uncultured magnetotactic bacteria. FEMS Microbiol. Lett. 172, 23–28. doi: 10.1111/j.1574-6968.1999.tb13444.x
Luo, C., Rodriguez-R, L. M., and Konstantinidis, K. T. (2014). MyTaxa: an advanced taxonomic classifier for genomic and metagenomic sequences. Nucleic Acids Res. 42, 1–12. doi: 10.1093/nar/gku169
Lycus, P., Bøthun, K. L., Bergaust, L., Shapleigh, J. P., Bakken, L. R., and Frostegård, Å. (2017). Phenotypic and genotypic richness of denitrifiers revealed by a novel isolation strategy. ISME J. 11, 2219–2232. doi: 10.1038/ismej.2017.82
Marchler-Bauer, A., Bo, Y., Han, L., He, J., Lanczycki, C. J., Lu, S., et al. (2017). CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203. doi: 10.1093/nar/gkw1129
Martínez-Mera, I., Espinosa-Pesqueira, M. E., Pérez-Hernández, R., and Arenas-Alatorre, J. (2007). Synthesis of magnetite (Fe3O4) nanoparticles without surfactants at room temperature. Mater. Lett. 61, 4447–4451. doi: 10.1016/j.matlet.2007.02.018
Matias, P. M., Pereira, I. A. C., Soares, C. M., and Carrondo, M. A. (2005). Sulphate respiration from hydrogen in Desulfovibrio bacteria: a structural biology overview. Prog. Biophys. Mol. Biol. 89, 292–329. doi: 10.1016/j.pbiomolbio.2004.11.003
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Moench, T. T., and Konetzka, W. A. (1978). A novel method for the isolation and study of a magnetotactic bacterium. Arch. Microbiol. 119, 203–212. doi: 10.1007/BF00964274
Monteil, C. L., Perrière, G., Menguy, N., Ginet, N., Alonso, B., Waisbord, N., et al. (2018). Genomic study of a novel magnetotactic Alphaproteobacteria uncovers the multiple ancestry of magnetotaxis. Environ. Microbiol. 20, 4415–4430. doi: 10.1111/1462-2920.14364
Morillo, V., Abreu, F., Araujo, A. C., De Almeida, L. G. P., Enrich-Prast, A., Farina, M., et al. (2014). Isolation, cultivation and genomic analysis of magnetosome biomineralization genes of a new genus of South-seeking magnetotactic cocci within the Alphaproteobacteria. Front. Microbiol. 5:72. doi: 10.3389/fmicb.2014.00072
Müller, A. L., Kjeldsen, K. U., Rattei, T., Pester, M., and Loy, A. (2015). Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J. 9, 1152–1165. doi: 10.1038/ismej.2014.208
Nguyen, L. T., Schmidt, H. A., Von Haeseler, A., and Minh, B. Q. (2015). IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32, 268–274. doi: 10.1093/molbev/msu300
Nurk, S., Meleshko, D., Korobeynikov, A., and Pevzner, P. A. (2017). metaSPAdes : a new versatile metagenomic assembler. Genome Res. 27, 824–834. doi: 10.1101/gr.213959.116
Orata, F. D., Meier-Kolthoff, J. P., Sauvageau, D., and Stein, L. Y. (2018). Phylogenomic analysis of the Gammaproteobacterial Methanotrophs (Order Methylococcales) calls for the reclassification of members at the genus and species levels. Front. Microbiol. 9:3162. doi: 10.3389/fmicb.2018.03162
Pan, H., Zhu, K., Song, T., Yu-zhang, K., Lefèvre, C., Xing, S., et al. (2008). Characterization of a homogeneous taxonomic group of marine magnetotactic cocci within a low tide zone in the China Sea. Environ. Microbiol. 10, 1158–1164. doi: 10.1111/j.1462-2920.2007.01532.x
Pantiukh, K., and Grouzdev, D. (2017). POCP-Matrix Calculation for a Number Of Genomes. FigShare. Available online at: https://figshare.com/articles/POCP-matrix_calculation_for_a_number_of_genomes/5602957
Parks, D. H., Chuvochina, M., Waite, D. W., Rinke, C., Skarshewski, A., Chaumeil, P.-A., et al. (2018). A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat. Biotechnol. 36, 996–1004. doi: 10.1038/nbt.4229
Parks, D. H., Imelfort, M., Skennerton, C. T., Hugenholtz, P., Tyson, G. W., Centre, A., et al. (2015). CheckM : assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 25, 1043–1055. doi: 10.1101/gr.186072.114
Pernthaler, J., Glöckner, F.-O., Schönhuber, W., and Amann, R. (2001). Fluorescence in situ hybridization (FISH) with rRNA-targeted oligonucleotide probes. Methods Microbiol. 30, 207–226. doi: 10.1016/S0580-9517(01)30046-6
Pósfai, M., Lefèvre, C. T., Trubitsyn, D., Bazylinski, D. A., and Frankel, R. B. (2013). Phylogenetic significance of composition and crystal morphology of magnetosome minerals. Front. Microbiol. 4:344. doi: 10.3389/fmicb.2013.00344
Potter, L. C., and Cole, J. A. (1999). Essential roles for the products of the napABCD genes, but not napFGH, in periplasmic nitrate reduction by Escherichia coli K-12. Biochem. J. 344, 69–76. doi: 10.1042/0264-6021:3440069
Qin, Q. L., Xie, B.-B., Zhang, X. Y., Chen, X. L., Zhou, B. C., Zhou, J., et al. (2014). A proposed genus boundary for the prokaryotes based on genomic insights. J. Bacteriol. 196, 2210–2215. doi: 10.1128/JB.01688-14
Quevillon, E., Silventoinen, V., Pillai, S., Harte, N., Mulder, N., Apweiler, R., et al. (2005). InterProScan: protein domains identifier. Nucleic Acids Res. 33, W116–W120. doi: 10.1093/nar/gki442
Rao, N. N., Gómez-García, M. R., and Kornberg, A. (2009). Inorganic polyphosphate: essential for growth and survival. Annu. Rev. Biochem. 78, 605–647. doi: 10.1146/annurev.biochem.77.083007.093039
Rioux, J. B., Philippe, N., Pereira, S., Pignol, D., Wu, L. F., and Ginet, N. (2010). A second actin-like mamk protein in magnetospirillum magneticum AMB-1 encoded outside the genomic magnetosome island. PLoS ONE 5:e9151. doi: 10.1371/journal.pone.0009151
Rivas-Lamelo, S., Benzerara, K., Lefèvre, C., Monteil, C., Jézéquel, D., Viollier, E., et al. (2017). Magnetotactic bacteria as a new model for P sequestration in the ferruginous Lake Pavin. Geochem. Perspect. Lett. 5, 35–41. doi: 10.7185/geochemlet.1743
Rodriguez-R, L. M., and Konstantinidis, K. T. (2016). The enveomics collection: a toolbox for specialized analyses of microbial genomes and metagenomes. PeerJ 4:e1900v1. doi: 10.7287/peerj.preprints.1900
Rossello-Mora, R., and Amann, R. (2001). The species concept for prokaryotes. FEMS Microbiol. Rev. 25, 39–67. doi: 10.1111/j.1574-6976.2001.tb00571.x
Sambrook, J., Fritsch, E. F., and Maniatis, T. (1989). Molecular Cloning: A Laboratory Manual, 2nd Edn. New York, NY: Cold Spring Harbor.
Sander, J., and Dahl, C. (2009). “Metabolism of inorganic sulfur compounds in purple bacteria,” in The Purple Phototrophic Bacteria, eds C. N. Hunter, F. Daldal, M. C. Thurnauer, and J. T. Beatty (Dordrecht: Springer), 595–622. doi: 10.1007/978-1-4020-8815-5_30
Scheffel, A., Gruska, M., Faivre, D., Linaroudis, A., Plitzko, J. M., and Schüler, D. (2006). An acidic protein aligns magnetosomes along a filamentous structure in magnetotactic bacteria. Nature 440, 110–114. doi: 10.1038/nature04382
Schübbe, S., Williams, T. J., Xie, G., Kiss, H. E., Brettin, T. S., Martinez, D., et al. (2009). Complete genome sequence of the chemolithoautotrophic marine magnetotactic coccus strain MC-1. Appl. Environ. Microbiol. 75, 4835–4852. doi: 10.1128/AEM.02874-08
Schulz-Vogt, H. N., Pollehne, F., Jürgens, K. W., Arz, H., Beier, S., Bahlo, R., et al. (2019). Effect of large magnetotactic bacteria with polyphosphate inclusions on the phosphate profile of the suboxic zone in the Black Sea. ISME J. 13, 1198–1208. doi: 10.1038/s41396-018-0315-6
Sieber, C. M. K., Probst, A. J., Sharrar, A., Thomas, B. C., Hess, M., Tringe, S. G., et al. (2018). Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat. Microbiol. 3:836. doi: 10.1038/s41564-018-0171-1
Spring, S., Amann, R., Ludwig, W., Schleifer, K.-H., Schüler, D., Poralla, K., et al. (1995). Phylogenetic analysis of uncultured magnetotactic bacteria from the Alpha-subclass of Proteobacteria. Syst. Appl. Microbiol. 17, 501–508. doi: 10.1016/S0723-2020(11)80068-8
Spring, S., Lins, U., Amann, R., Schleifer, K. H., Ferreira, L. C. S., Esquivel, D. M. S., et al. (1998). Phylogenetic affiliation and ultrastructure of uncultured magnetic bacteria with unusually large magnetosomes. Arch. Microbiol. 169, 136–147. doi: 10.1007/s002030050553
Syvanen, M. (1994). Horizontal gene transfer: evidence and possible consequences. Annu. Rev. Genet. 28, 237–261. doi: 10.1146/annurev.ge.28.120194.001321
Tatusova, T., Dicuccio, M., Badretdin, A., Chetvernin, V., Nawrocki, P., Zaslavsky, L., et al. (2016). NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 44, 6614–6624. doi: 10.1093/nar/gkw569
Toro-Nahuelpan, M., Giacomelli, G., Raschdorf, O., Borg, S., Plitzko, J. M., Bramkamp, M., et al. (2019). MamY is a membrane-bound protein that aligns magnetosomes and the motility axis of helical magnetotactic bacteria. Nat. Microbiol. doi: 10.1038/s41564-019-0512-8. [Epub ahead of print].
Trubitsyn, D., Abreu, F., Ward, F. B., Taylor, T., Hattori, M., Kondo, S., et al. (2016). Draft genome sequence of Magnetovibrio blakemorei strain MV-1, a marine vibrioid magnetotactic bacterium. Genome Announc. 4, e01330–e01316. doi: 10.1128/genomeA.01330-16
Tyrrell, T. (1999). The relative influences of nitrogen and phosphorus on oceanic primary production. Nature 400, 525–531. doi: 10.1038/22941
Villarreal-Chiu, J. F., Quinn, J. P., and McGrath, J. W. (2012). The genes and enzymes of phosphonate metabolism by bacteria, and their distribution in the marine environment. Front. Microbiol. 3:19. doi: 10.3389/fmicb.2012.00019
Wang, Y., Lin, W., and Pana, Y. (2013). High diversity of magnetotactic Deltaproteobacteria in a freshwater Niche. Appl. Environ. Microbiol. 79, 2813–2817. doi: 10.1128/AEM.03635-12
Williams, T. J., Zhang, C. L., Scott, J. H., and Bazylinski, D. A. (2006). Evidence for autotrophy via the reverse tricarboxylic acid cycle in the marine magnetotactic coccus strain MC-1. Appl. Environ. Microbiol. 72, 1322–1329. doi: 10.1128/AEM.72.2.1322-1329.2006
Woehl, T. J., Kashyap, S., Firlar, E., Perez-Gonzalez, T., Faivre, D., Trubitsyn, D., et al. (2014). Correlative electron and fluorescence microscopy of magnetotactic bacteria in liquid: toward in vivo imaging. Sci. Rep. 4:6854. doi: 10.1038/srep06854
Wolfe, R. S., Thauer, R. K., and Pfennig, N. (1987). A “capillary racetrack”method for isolation of magnetotactic bacteria. FEMS Microbiol. Lett. 45, 31–35. doi: 10.1111/j.1574-6968.1987.tb02335.x
Wong, T. K. F., Jermiin, L. S., Minh, B. Q., Kalyaanamoorthy, S., and von Haeseler, A. (2017). ModelFinder: fast model selection for accurate phylogenetic estimates. Nat. Methods 14, 587–589. doi: 10.1038/nmeth.4285
Wu, Y., Simmons, B. A., and Singer, S. W. (2015). MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32, 605–607. doi: 10.1093/bioinformatics/btv638
Zajicek, R. S., Bali, S., Arnold, S., Brindley, A. A., Warren, M. J., and Ferguson, S. J. (2009). D1 haem biogenesis - assessing the roles of three nir gene products. FEBS J. 276, 6399–6411. doi: 10.1111/j.1742-4658.2009.07354.x
Zhang, H., Menguy, N., Wang, F., Benzerara, K., Leroy, E., Liu, P., et al. (2017). Magnetotactic coccus strain SHHC-1 affiliated to Alphaproteobacteria forms octahedral magnetite magnetosomes. Front. Microbiol. 8:969. doi: 10.3389/fmicb.2017.00969
Zhang, W. Y., Zhou, K., Pan, H. M., Du, H. J., Chen, Y. R., Zhang, R., et al. (2013). Novel rod-shaped magnetotactic bacteria belonging to the class Alphaproteobacteria. Appl. Environ. Microbiol. 79, 3137–3140. doi: 10.1128/AEM.03869-12
Zhang, W. Y., Zhou, K., Pan, H. M., Yue, H. D., Jiang, M., Xiao, T., et al. (2012). Two genera of Magnetococci with bean-like morphology from intertidal sediments of the Yellow Sea, China. Appl. Environ. Microbiol. 78, 5606–5611. doi: 10.1128/AEM.00081-12
Keywords: magnetotactic bacteria (MTB), magnetosome, Candidatus Etaproteobacteria, Candidatus Magnetaquicoccaceae, magnetotactic cocci, Magnetococcales, magnetosome genomic island, AAI classifications
Citation: Koziaeva V, Dziuba M, Leão P, Uzun M, Krutkina M and Grouzdev D (2019) Genome-Based Metabolic Reconstruction of a Novel Uncultivated Freshwater Magnetotactic coccus “Ca. Magnetaquicoccus inordinatus” UR-1, and Proposal of a Candidate Family “Ca. Magnetaquicoccaceae”. Front. Microbiol. 10:2290. doi: 10.3389/fmicb.2019.02290
Received: 05 June 2019; Accepted: 19 September 2019;
Published: 02 October 2019.
Edited by:
Feng Gao, Tianjin University, ChinaReviewed by:
Christopher T. Lefèvre, UMR7265 Biologie Végétale et Microbiologie Environnementales (BVME), FranceJinhua Li, Institute of Geology and Geophysics (CAS), China
Copyright © 2019 Koziaeva, Dziuba, Leão, Uzun, Krutkina and Grouzdev. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Denis Grouzdev, ZGVuaXNncm91emRldkBnbWFpbC5jb20=