 Sarah Temmam1
Sarah Temmam1 Delphine Chrétien1
Delphine Chrétien1 Thomas Bigot1,2
Thomas Bigot1,2 Evelyne Dufour3Stéphane Petres3
Evelyne Dufour3Stéphane Petres3 Marc Desquesnes4,5,6Elodie Devillers7Marine Dumarest1Léna Yousfi7Sathaporn Jittapalapong8Anamika Karnchanabanthoeng8Kittipong Chaisiri9Léa Gagnieur1Jean-François Cosson7
Marc Desquesnes4,5,6Elodie Devillers7Marine Dumarest1Léna Yousfi7Sathaporn Jittapalapong8Anamika Karnchanabanthoeng8Kittipong Chaisiri9Léa Gagnieur1Jean-François Cosson7 Muriel Vayssier-Taussat7
Muriel Vayssier-Taussat7 Serge Morand10,11
Serge Morand10,11 Sara Moutailler7
Sara Moutailler7 Marc Eloit1,12*
Marc Eloit1,12*- 1Institut Pasteur, Biology of Infection Unit, Inserm U1117, Pathogen Discovery Laboratory, Paris, France
- 2Institut Pasteur – Bioinformatics and Biostatistics Hub – Computational Biology Department, Institut Pasteur, USR 3756 CNRS, Paris, France
- 3Institut Pasteur, Production and Purification of Recombinant Proteins Technological Platform – C2RT, Paris, France
- 4Centre de Coopération Internationale en Recherche Agronomique pour le Développement (CIRAD), UMR InterTryp, Bangkok, Thailand
- 5InterTryp, Institut de Recherche pour le Développement (IRD), CIRAD, University of Montpellier, Montpellier, France
- 6Department of Parasitology, Faculty of Veterinary Medicine, Kasetsart University, Bangkok, Thailand
- 7UMR BIPAR, Animal Health Laboratory, ANSES, INRA, Ecole Nationale Vétérinaire d'Alfort, Université Paris-Est, Maisons-Alfort, France
- 8Faculty of Veterinary Medicine, Kasetsart University, Bangkok, Thailand
- 9Faculty of Tropical Medicine, Mahidol University, Bangkok, Thailand
- 10Institut des Sciences de l'Evolution, CNRS, CC065, Université Montpellier, Montpellier, France
- 11CIRAD ASTRE, Faculty of Veterinary Technology, Kasetsart University, Bangkok, Thailand
- 12National Veterinary School of Alfort, Paris-Est University, Maisons-Alfort, France
Emerging zoonoses caused by previously unknown agents are one of the most important challenges for human health because of their inherent inability to be predictable, conversely to emergences caused by previously known agents that could be targeted by routine surveillance programs. Emerging zoonotic infections either originate from increasing contacts between wildlife and human populations, or from the geographical expansion of hematophagous arthropods that act as vectors, this latter being more capable to impact large-scale human populations. While characterizing the viral communities from candidate vectors in high-risk geographical areas is a necessary initial step, the need to identify which viruses are able to spill over and those restricted to their hosts has recently emerged. We hypothesized that currently unknown tick-borne arboviruses could silently circulate in specific biotopes where mammals are highly exposed to tick bites, and implemented a strategy that combined high-throughput sequencing with broad-range serological techniques to both identify novel arboviruses and tick-specific viruses in a ticks/mammals interface in Thailand. The virome of Thai ticks belonging to the Rhipicephalus, Amblyomma, Dermacentor, Hyalomma, and Haemaphysalis genera identified numerous viruses, among which several viruses could be candidates for future emergence as regards to their phylogenetic relatedness with known tick-borne arboviruses. Luciferase immunoprecipitation system targeting external viral proteins of viruses identified among the Orthomyxoviridae, Phenuiviridae, Flaviviridae, Rhabdoviridae, and Chuviridae families was used to screen human and cattle Thai populations highly exposed to tick bites. Although no positive serum was detected for any of the six viruses selected, suggesting that these viruses are not infecting these vertebrates, or at very low prevalence (upper estimate 0.017% and 0.047% in humans and cattle, respectively), the virome of Thai ticks presents an extremely rich viral diversity, among which novel tick-borne arboviruses are probably hidden and could pose a public health concern if they emerge. The strategy developed in this pilot study, starting from the inventory of viral communities of hematophagous arthropods to end by the identification of viruses able (or likely unable) to infect vertebrates, is the first step in the prediction of putative new emergences and could easily be transposed to other reservoirs/vectors/susceptible hosts interfaces.
Introduction
Among the list of human-infecting pathogens, two-thirds are zoonotic agents (Woolhouse et al., 2012). With increasing contacts between humans, wildlife, and their associated arthropods (due to changes in land use, global climate warming, or urbanization), the frequency of human infections with animal viruses is expected to increase (Cutler et al., 2010). Wolfe et al. noted that the emergence of a pathogen of vertebrate into a new susceptible vertebrate population goes through four main steps: (1) spillover of a pathogen from its animal reservoir to sporadic human cases, (2) limited interhuman transmission of a zoonotic pathogen, (3) large-scale spread of a zoonotic pathogen through interhuman transmissions, and (4) adaptation of the pathogen to humans (Wolfe et al., 2007). This is not the case for arboviruses: arbovirus emergence is linked to the spread of infected arthropods (directly or via the geographical expansion of their vertebrate host, as for ticks) into new areas or to the incursion of humans in new biotopes. Most recent epidemics that emerged were linked to arboviruses that were already known but described in limited areas, as the recent example of Zika virus rapid expansion (Liu et al., 2019).
Ticks are major hematophagous arthropod vectors that harbor multiple infectious agents. Some of these agents are known to infect humans, leading to several tick-borne diseases such as Lyme borreliosis (Borrelia burgdorferi sensu lato), tick-borne encephalitis (due to several flaviviruses such as tick-borne encephalitis virus or Powassan virus), and spotted fever (several Rickettsia) (Moyer, 2015; Nelder et al., 2016). However, asymptomatic or pauci-symptomatic infections with non-specific syndromes (for example, asthenia, fever, myalgia, etc.) occurring after tick bites are frequent and have become a serious issue (Moyer, 2015). The same situation is observed for domestic animals: since several tick species, mainly ixodid ticks, are able to feed both on domestic animals and humans (Kiewra and Lonc, 2012), they can transmit zoonotic pathogens common to animals and humans. Moreover, due to global and local environmental changes, the geographical repartition of ticks is increasing, leading to exposure of naive populations to new pathogens (Ogden et al., 2013). The progresses in usage of high-throughput sequencing (HTS) have allowed an increase in the knowledge of infectious agents, and particularly viruses, carried by ticks. Recent studies have indeed reported the identification of new tick-borne viruses, belonging either to known or completely new viral families, for which the potential zoonotic risk for humans or domestic animals is still unknown (Tokarz et al., 2014a, 2018; Xia et al., 2015; Moutailler et al., 2016; Shi et al., 2016; Pettersson et al., 2017). For example, among the Phenuiviridae, known to contain tick-borne pathogens such as the severe fever with thrombocytopenia syndrome phlebovirus (SFTS virus) (Silvas and Aguilar, 2017), recent bisegmented phleboviruses have been described, such as Lihan tick phlebovirus (LTPV) primarily identified in Rhipicephalus microplus ticks from China, Brazil, and Trinidad and Tobago (Li C. X. et al., 2015; Souza et al., 2018; Sameroff et al., 2019) and further detected in Turkish Hyalomma marginatum (Dinçer et al., 2017) and Rhipicephalus sanguineus ticks (Brinkmann et al., 2018). Flaviviridae-related tick-borne viruses (e.g., Bole tick virus 4, BLTV4) were primarily associated with Hyalomma asiaticum and Rh. sanguineus ticks (Shi et al., 2015; Sameroff et al., 2019). This virus presents a genome 1.5 times larger than other Flaviviridae tick-borne viruses and could constitute, with other related flaviviruses that present large genomes, at least a new genus among the Flaviviridae family. In complement to known rhabdoviruses transmitted by ticks (Labuda and Nuttall, 2004) [including several viruses pathogenic for humans (Menghani et al., 2012)], novel single-stranded RNA (ssRNA) negative-strand viruses belonging to the dimarhabdovirus group within the Rhabdoviridae family were also identified in Rhipicephalus [Rh. microplus (Li C. X. et al., 2015), Rh. annulatus (Li C. X. et al., 2015; Brinkmann et al., 2018)] ticks [for example, Wuhan tick virus 1 (WhTV-1)]. In addition to these viral families known to contain tick-borne viruses, new viruses identified by HTS and constituting novel viral families recently recognized by the ICTV were reported. It is the case of the Chuviridae family, a group of viruses belonging to the Jingchuvirales order [class Monjiviricetes, phylum Negarnaviricota, (Siddell et al., 2019)] and constituted of circular or segmented ssRNA negative-strand viruses primarily associated with Dermacentor sp., Haemaphysalis parva (Li C. X. et al., 2015; Brinkmann et al., 2018), and Rh. sanguineus (Sameroff et al., 2019) ticks [e.g., Changping tick virus 2 (CpTV2)] or Rh. microplus ticks from China, Brazil, and Trinidad and Tobago [Wuhan tick virus 2 (WhTV2)] (Li C. X. et al., 2015; Souza et al., 2018; Sameroff et al., 2019).
We hypothesized that currently unknown tick-borne arboviruses could silently circulate in specific biotopes where mammals (including humans) are highly exposed to tick bites and used wide range identification techniques to track them in a tick/mammal interface in Thailand. Despite the fact that the description of the virome of ticks is a prerequisite to the evaluation of the risk of spillover, few studies have tried to go further and characterize, among the viral communities infecting ticks, which viruses would more likely be transmissible to vertebrates. Starting from the inventory of viruses infecting tick vectors, the first step in the understanding of the mechanisms of viral emergence is therefore to identify which viruses can cross the species barrier and infect vertebrates, even without any reported clinical signs. Serological techniques are useful tools for getting insights into arbovirus exposure history of new hosts without the limits of genomic tests, which need to collect biological samples in the course of a viremic phase. However, the recognition of the antigen (Ag) by its specific antibodies (Ab) requires good conservation of epitopes conformation, a problem frequently encountered in solid phase Ab/Ag assays (Burbelo et al., 2010). In 2010, Burbelo et al. have established a sensitive liquid phase format assay for profiling Ab responses to various antigens (Burbelo et al., 2010). Luciferase immunoprecipitation system (LIPS) assays use as antigens viral open reading frames (ORFs) fused to the luciferase (Luc) reporter gene expressed in mammalian cells. Fusion proteins are harvested as cell lysates under native conditions and used as antigens. Mixed with serum samples, the resulting immune complexes are then immunoprecipitated using protein A/G-coated beads and revealed by the substrate of the Luc. The measure of Luc activity is an index of the initial Ab titer. Owing to high affinity to the Fc region of their immunoglobulins to staphylococcal A and streptococcal G proteins, LIPS assay can be performed either on human and several animals sera (such as rabbits, dogs, monkeys, cows, mice, etc.) (Burbelo et al., 2010), resulting in the possibility to use one single tool to monitor the viral cycle both in its reservoir (frequently animal) and in its spillover (human) host.
In Thailand, several infectious agents such as protozoa (i.e., Babesia and Theileria) and bacteria (Anaplasma, Ehrlichia, Rickettsia, and Bartonella, for example) are carried by ticks. Viruses belonging to the Reoviridae, Togaviridae, Flaviviridae, Nairoviridae, and Orthomyxoviridae were also reported (Cornet et al., 2004; Ahantarig et al., 2008). We hypothesized that other tick-borne viruses (new or already known) are silently crossing the barrier and sporadically infect humans and/or domestic animals living in the vicinity of humans (as cattle or dogs for example) in specific ecosystems where populations are in close contact with ticks. The aim of this study is therefore to evaluate the ability of the strategy that consists in coupling HTS with serological screenings to identify which viruses are (and are not) able to cross the species barrier and infect new vertebrate hosts.
Materials and Methods
Sample Collection and Processing
Ticks Samples
A total of 266 ticks (18 males, 197 females, and 51 nymphs) were collected in Ban Huay Muang village (Tha Wang Pha district, Nan province, Thailand) in November 2012. Ticks were morphologically identified at least at the genus level: 40 Rh. sanguineus, 22 Amblyomma sp., 148 Boophilus sp., 16 Dermacentor marginatus, 22 Hyalomma sp., and 18 Haemaphysalis sp. To determine the species of Amblyomma sp., Boophilus sp., Hyalomma sp., and Haemaphysalis sp. ticks, all trimmed reads were mapped onto the Ixodidae Barcode of Life Data Systems (BOLD) database, de novo assembled after extraction of mapped reads, and submitted to the BOLD Identification System. Boophilus sp. ticks were identified as Rh. microplus, and Haemaphysalis sp. were identified as Haemaphysalis hystricis.
All of the genera collected here contain several tick species that can either bite wildlife or domestic animals (including cattle) and even humans (Kiewra and Lonc, 2012). Most of the ticks were engorged (222 out of 266) and were collected as follows: Amblyomma ticks were collected on wild boars and rodents; Rh. microplus ticks were collected on cattle (except for two ticks collected by flagging); D. marginatus ticks were collected on wild boars (except for one tick collected by flagging); Ha. hystricis ticks were collected either on wild boars (N = 11), on dogs (N = 2), and by flagging (N = 3); Hyalomma ticks were either collected on dogs (N = 3) or on wild boars (N = 15); and Rh. sanguineus ticks were either collected on dogs (N = 12) or on the floor of houses were dogs lived (N = 4).
All ticks were washed as previously described (Vayssier-Taussat et al., 2013) to remove external contaminants and homogenized individually in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% of fetal calf serum. After clarification, DNA and RNA extractions were performed from 100 μl of supernatant using the Nucleospin Tissue or the Nucleospin RNA II kits, respectively (Macherey-Nagel, Germany), according to the manufacturer's recommendations. Purified DNA and RNA were eluted into 50 μl of elution buffer and RNAse-free water, respectively. Extracts were kept at −80°C before HTS libraries preparation and further amplifications.
Human Serum Samples
The study procedures concerning human sample collection, laboratory investigation, interviews, and questionnaires were approved by the Ethical Committee of the Faculty of Tropical Medicine, Mahidol University (Bangkok, Thailand), document no 0517.1116/661.
Public health personnel made appointment with the villager participants in Saen Thong subdistrict, Nan province. Voluntary participation in the research project was obtained after explanation of the purposes of the study and by signing the informed consent/assent form. Blood samples were collected once by local nurses from a primary care unit, as previously described (Chaisiri et al., 2018).
Cattle Serum Samples
With the support and under the control of the local veterinary services from the Department of Livestock Development, 70 cattle were blood sampled in the Tha Wang Pha district, Nan province, in November 2012. When animals were infested, ticks were collected. Animals were tested by ELISA against Trypanosoma evansi as previously described (Desquesnes et al., 2009), and seropositive samples were tested by PCR using Trypanosoma brucei primers previously published (Masiga et al., 1992; Pruvot et al., 2010, 2013) to confirm active infection.
High-Throughput Sequencing and Bioinformatics Analyses of Tick Virome
Individual RNA tick extracts were combined to form one pool of total RNA that was used as template for random reverse transcription followed by random amplification using Qiagen QuantiTect Whole Transcriptome kit. Complementary DNA (cDNA) was used for library preparations and sequenced on an Illumina HiSeq2000 sequencer in a single-read 100-bp format outsourced to DNAVision Company.
A total of 222,152,554 of raw reads were processed with an in-house bioinformatics pipeline comprising quality check and trimming [based on AlienTrimmer package (Criscuolo and Brisse, 2014)], reads normalization [using BBnorm program (https://jgi.doe.gov/data-and-tools/bbtools)], de novo assembly (using Megahit tool Li D. et al., 2015), and ORF prediction of contigs and singletons (https://figshare.com/articles/translateReads_py/7588592). A BLAST-based similarity search was performed for all contigs and singletons against the protein Reference Viral database (Bigot et al., 2019) followed by a BlastP-based verification of the accuracy of the viral taxonomic assignation against the whole protein NCBI/nr database. A final BLAST-based verification was performed against NCBI/nt to confirm that no better hit was obtained in non-coding regions of NCBI/nt.
The quantification of abundance of each viral taxon was obtained by summing the length (in amino acids) of all sequences being associated to this taxon instead of summing the raw number of sequences, to take into account the identification of long viral contigs. In addition, to take into account that contigs are the results of the assembly of numerous reads, we weighted the identification of viral hits coming from contigs by multiplying the length (in amino acids) of contigs with their respective k-mer coverage.
Phylogenetic reconstructions were performed on conserved non-structural RNA-dependent RNA polymerase (RdRP) gene. Complete ORFs were aligned along with other representative sequences of viral orders/families using MAFFT (Multiple Alignment using Fast Fourier Transform) aligner under the L-INS-i or G-INS-i parameters (Katoh et al., 2017). The best amino acids substitution models that fitted the data were determined with ATGC Smart Model Selection (Lefort et al., 2017) implemented in http://www.atgc-montpellier.fr/phyml-sms/ using the corrected Akaike information criterion. Phylogenetic trees were constructed using maximum likelihood method implemented through RAxML program under the CIPRES Science Gateway portal (Miller et al., 2010) according to the selected substitution model. Nodal support was evaluated using the “automatic bootstrap replicates” parameter. For Orthomyxoviridae-related phylogenetic analyses, neighbor-joining reconstruction method, p-distance, and 10,000 bootstraps replications were used.
Genome Finishing and Prevalence Study in Ticks
The complete ORFs were obtained by conventional PCR and Sanger sequencing after designing specific primers targeting the identified viruses. Briefly, viral RNA was reverse transcribed using the SuperScript IV reverse transcriptase (Invitrogen, USA), and cDNA was subsequently used to fill the gaps in the genomes using the Phusion High Fidelity DNA polymerase (New England Biolabs, France). Positive PCR products were further purified and sequenced by Sanger sequencing on the Eurofins Segenic Cochin platform. When start and stop codons were lacking, rapid amplification of cDNA ends PCR were performed using the 5′/3′ RACE kit, second generation (Roche Applied Science, Germany).
For prevalence study, individual RNA extracts were reverse transcribed into cDNA using the qScript cDNA Supermix kit according to the manufacturer's instructions (Quanta Biosciences, USA). Briefly, the reaction was performed in a final volume of 5 μl containing 1 μl of qScript cDNA Supermix 5×, 1 μl of RNA, and 3 μl of RNase-free water. Cycling condition was as follows: one cycle at 25°C for 5 min, one cycle at 42°C for 30 min, and one final cycle at 85°C for 5 min. Specific Taqman qPCR systems targeting the polymerase gene were designed for six relevant viruses (the sequences of primers and probes are available upon request). Real-time Taqman qPCR assays were performed in a final volume of 12 μl using the LightCycler® 480 Probe Mastermix (Roche Applied Science) at 1× final concentration, with primers and probes at 200 nM, and 2 μl of cDNA. Negative (water) control was included in each run. Thermal cycling conditions were as follows: 95°C for 5 min, 45 cycles at 95°C for 10 s, then 60°C for 15 s, with a final cooling step at 40°C for 10 s.
To identify possible endogenous viral elements (EVEs) originating from arthropod hosts that could have been sequenced during the process, nested qPCRs were performed on tick-borne total DNA without any RT step targeting the polymerase gene of the six selected as relevant viruses. Positive or suspicious results were further analyzed by Sanger sequencing.
Serological Screening of Human and Cattle Sera
Choice of Target Antigens
To maximize the probability of detecting antibodies specific to the viruses detected in ticks and minimize the risk of cross-reactions, we decided to target viral external proteins, meaning glycoproteins (GP) or nucleoproteins (NP) when GP were not identified. The annotation of the genes and the consecutive identification of surface proteins were first performed by position-specific iterated Blast (Psi-Blast). For viral ORFs for which we could not determine the function of the gene by Psi-Blast, Swiss Model was used to model the structure of the viral protein and to compare it to known structures deposited onto the PDB database (https://swissmodel.expasy.org/interactive) (Arnold et al., 2006; Biasini et al., 2014). Finally, for hypothetical proteins for which neither Psi-Blast nor Swiss Model gave positive results, online prediction of possible N- and O-glycosylation sites was performed both on the NetNGlyc and NetOGlyc webservers (http://www.cbs.dtu.dk/services/NetNGlyc/ or http://www.cbs.dtu.dk/services/NetOGlyc/) (Gupta et al., 2002).
The identified GP or NP were then analyzed with the Transmembrane Prediction tool of CLC Main Workbench 7 (Qiagen Bioinformatics) to identify extracellular regions of the proteins. These regions were used to produce viral antigens. The coordinates of the extracellular regions expressed for relevant viruses are presented in Supplemental Table 1.
Cloning
Complete extracellular regions of viral glycoproteins and/or nucleoproteins were cloned into pFC32K vector (Promega, France), at the N-terminal end of the NanoLuc luciferase gene, using the Gibson Assembly (GA) kit (NEBuilder® HiFi DNA Assembly Master Mix, New England Biolabs), according to the manufacturer's instructions. First, the Kosak sequence (GCCACC, in bold in Supplemental Table 2) and the start codon (ATG, in italic in Supplemental Table 2) were added by PCR in 5′ of the target fragment along with a 28- and a 27-nt vector region overlapping the sequence of the vector and containing the restrictions sites of the multiple cloning sites (underlined in Supplemental Table 2, SgfI and EcoICRI, respectively, for forward and reverse primers). Great attention was paid to maintain the reading frame between the target fragment and the NanoLuc gene. Then, PCR products were gel purified with the Nucleospin Gel and PCR cleanup kit (Macherey-Nagel) and quantified by Qubit with the ds DNA High Sensitivity kit (Invitrogen).
One hundred nanograms of pre-linearized pFC32K vector (digested by SgfI and EcoICRI restriction enzymes, Carboxy Flexi Enzyme Blend, Promega) was used to clone by GA the target fragments at a ratio of 1:3 to 1:10 (depending on the constructs) in XL1 competent cells (Agilent Technologies, USA). Positive clones were screened by PCR with primers designed in the vector and flanking the inserts and verified by Sanger sequencing. PureLink HiPure midiprep kit (Invitrogen) was used to extract plasmids from 100 ml of bacterial overnight cultures grown in LB medium supplemented with 25 μg/ml of kanamycin.
Expression of Fusion Proteins and LIPS Assay
A modified version of Burbelo's protocol was developed for expression of fusion proteins: HEK-293A cells (kindly provided by Bernard Klonjkowski, Veterinary School, Maisons-Alfort, France) were transfected with polyethylenimine (PEI, Polyscience Inc., USA) instead of Cos-1 cells and Fugene-6 transfecting reagent (Burbelo et al., 2007, 2009). Cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 1% sodium pyruvate, 1% penicillin-streptomycin mix, 1% amphotericin B, and 1% non-essential amino acids (Invitrogen) with two passages a week. Cells (4 × 105 per well) were plated on a six-well plate the day before transfection, and 5 μg of plasmid DNA was transfected with 20 μl of 1 mg/ml PEI. Two days post-transfection, fusion proteins were harvested as previously described (Burbelo et al., 2007, 2009). Protease inhibitors (complete protease inhibitor cocktail minitablets, Roche Applied Science) and 10% of glycerol were added to the cell lysates to enhance protein stability. The luciferase activity of the fusion proteins was measured onto a Centro XS3 LB 960 luminometer (Berthold Technologies, France) by adding the NanoLuc substrate (NanoGlo reagent, Promega) to 10-fold dilutions of the lysates. Titers (in light unit LU/ml) were corrected for background by subtracting LU values produced by negative controls constituted of cells transfected with the same amount of PEI without plasmid DNA.
LIPS assay was performed as previously described by Burbelo et al. (2007, 2009) except that human and cattle sera were not diluted. Sera of 30 healthy French donors living in Paris area who did not report travel to Thailand or any tick bite (kindly provided by ICAReB platform of Institut Pasteur, Paris, France) were screened for the presence of antibodies against the targeted viruses as a non-exposed group control. Residual background was measured as the mean of 10 negative controls (without serum), and positivity threshold was defined as the mean of these controls + 5 standard deviations.
Statistical Analyses
Significant differences between exposed and non-exposed groups of human sera tested by LIPS were calculated using the Student t-test (95% confidence interval). The maximal prevalence of viral infection in each exposed group was estimated assuming a sensitivity of 90% and a specificity of 99% of the test and the Blaker confidence interval calculation using the True Prevalence calculator implemented in the EpiTools portal (https://epitools.ausvet.io/trueprevalence).
Accessions Numbers
Complete coding regions of the six viruses characterized were deposited into the GenBank database under the numbers MN095535 to MN095546.
Results
The Viral Transcriptome of Thai Ticks Is Mainly Constituted of Negative-Strand ssRNA Viruses
The transcriptome of Thai ticks was sequenced at a depth of 222 × 106 reads. Most sequences were assigned to the Eukaryota domain while bacteria and viruses represented 0.09 and 0.001% of validated taxonomic assignation of reads, respectively. Of these, more than 99% of viral sequences were composed of RNA viruses, while few other related viral sequences were identified (Table 1). ssRNA negative-strand viruses represented 97.20% of all viruses, followed by ssRNA positive strand viruses (1.94%), unclassified viruses (0.70%), and dsRNA viruses (0.14%). Only few sequences were assigned to DNA viruses (ssDNA: 0.01%), reflecting the specificity of RNA extraction. The identification of ssDNA viruses would more likely correspond to the sequencing of ssDNA viral transcripts, reflecting the replicative form of these viruses.
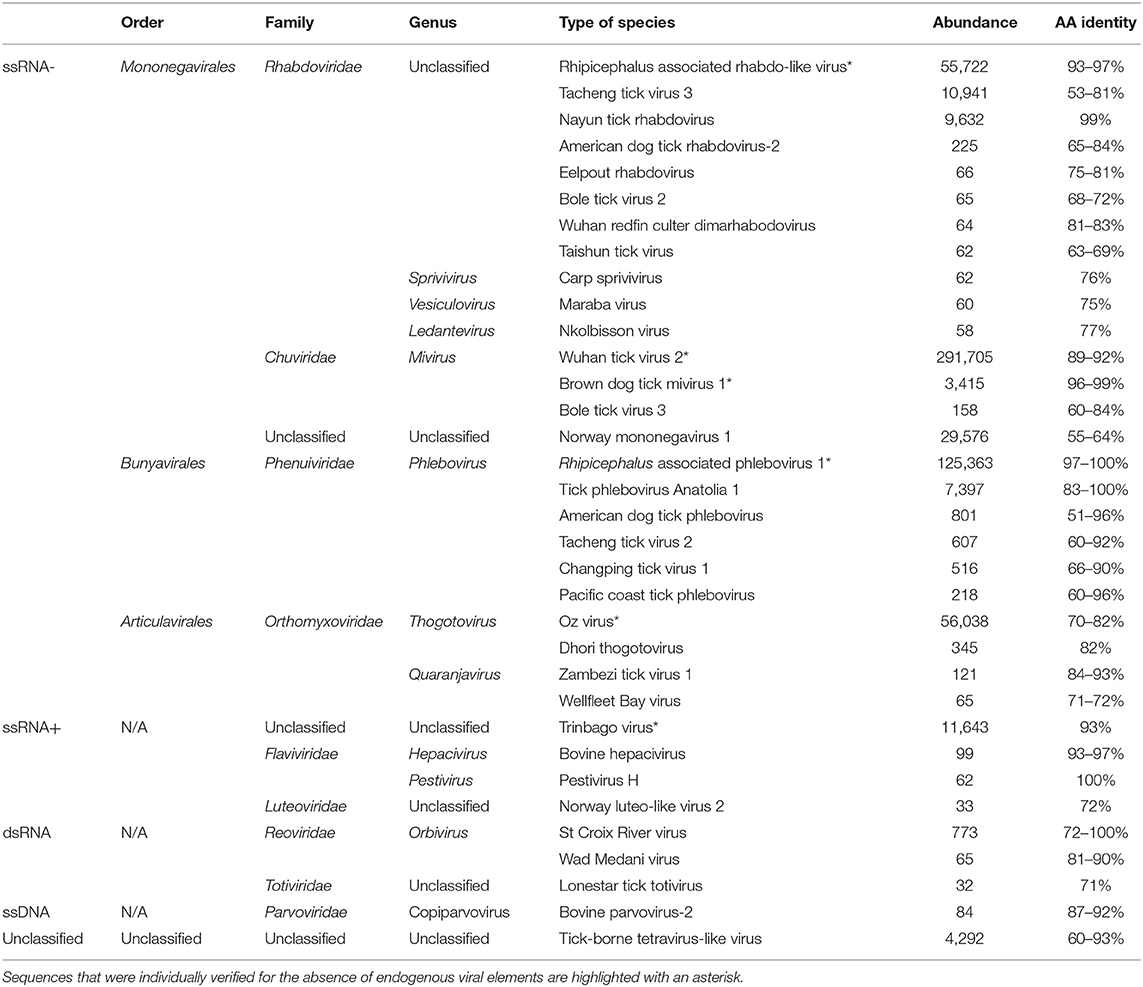
Table 1. Viral sequences identified in ticks from Thailand by high-throughput sequencing.
Among the ssRNA viruses, positive-strand RNA viruses were in the minority (1.96% of ssRNA viruses). More than 98% of sequences were taxonomically related to Trinbago virus, a BLTV4-related virus identified in Trinidad and Tobago Rh. sanguineus ticks (Sameroff et al., 2019). The strain identified in Thai ticks, tentatively named BLTV4-Thailand virus (accession no. MN095535) presented a mean amino-acid identity of 93% with the polyprotein of Trinbago virus (Table 1). Other positive-strand ssRNA viruses were less frequently identified and were assigned to the Hepacivirus and Pestivirus genera (more likely corresponding to cattle-infecting viruses that are detected through the bloodmeal of ticks), or to the Luteoviridae family (Table 1).
The negative-strand ssRNA viruses constituted more than 97% of viral sequences (Table 1, Supplemental Figure 1). Among these, more than 66% of ssRNA viruses were assigned to the nonsegmented Mononegavirales or Jingchuvirales orders. For example, sequences assigned to the Rhabdoviridae family mainly mapped onto a novel strain of Wuhan tick virus 1 (WhTV1), with amino-acid identities ranging from 93 to 97%, depending on the gene (WhTV1-Thailand virus, accession no MN095536); while other unclassified Rhabdoviridae-related sequences were detected (as for example a distant Tacheng tick virus 3 virus or a new strain of Nayun tick rhabdovirus). Few sequences, were assigned to the Sprivivirus, the Vesiculovirus, and the Ledantevirus Rhabdoviridae genera. Viruses belonging to the Chuviridae mapped onto Wuhan tick virus 2 (WhTV2) and Brown dog mivirus 1 (a new strain of Changping tick virus 2 [CpTV2]), with amino-acid identities ranging from 89 to 99% (WhTV2-Thailand virus: MN095546; and CpTV2-Thailand virus: MN095545). Finally, a novel unclassified mononegavirus genome presented distant homologies with Norway mononegavirus 1 (amino-acid identities ranging from 55 to 64%, depending on the gene).
Up to 23% of ssRNA viruses were assigned to the Bunyavirales, and more precisely to the Phenuiviridae family. More than 92% of sequences were related to Rhipicephalus-associated phlebovirus 1, a novel strain of Lihan tick phlebovirus (LTPV) primarily associated with Rh. microplus Chinese ticks. Tentatively named LTPV-Thailand virus, this viral genome presented amino-acid identities ranging from 97 to 100% with its closest viral relative, depending on the gene (accession no MN095537-38). Several sequences close to Tick phlebovirus Anatolia 1 were also detected.
Finally, Orthomyxoviridae-related sequences (more than 9% of total ssRNA viruses), either belonging to the Thogotovirus or the Quaranjavirus genera were detected, but Oz virus-related viral sequences were the most abundant. Tentatively named Thailand tick thogotovirus (accession no MN095539-44), this virus presented amino-acid identities ranging from 70 to 82% depending on the genes (Table 1) with Oz virus, a recently reported tick-borne virus identified in Amblyomma testudinarium ticks from Japan that is phylogenetically close to Bourbon and Dhori thogotoviruses (Ejiri et al., 2018).
For each strain of BLTV4-Thailand virus, WhTV1-Thailand virus, WhTV2-Thailand virus, CpTV2-Thailand virus, LTPV-Thailand virus, and Thailand tick thogotovirus described hereafter, the presence of the virus in the initial RNA pool used to produce HTS library was validated by specific reverse transcription PCR targeting different portions of the genome (data not shown).
Genomic Organization and Phylogenetic Analyses of Rhabdoviridae-, Chuviridae-, Phenuiviridae-, Flaviviridae-, and Orthomyxoviridae-Related Viruses
Rhabdoviridae/WhTV1-Thailand Virus
Rhabdoviridae is a family of RNA viruses infecting a large spectrum of hosts, ranging from plants to invertebrates and vertebrates. Tick-borne rhabdoviruses include viruses belonging to the recognized Ledantevirus and Ephemerovirus, and the putative Sawgravirus genera (Labuda and Nuttall, 2004; Walker et al., 2015; Tokarz et al., 2018). Rhabdoviridae viruses present a general genome organization of linear negative-strand ssRNA genome usually encoding five proteins, in the order N (nucleoprotein)/P (phosphoprotein)/M (matrix)/G (glycoprotein)/L (RNA-dependent RNA polymerase). WhTV1-Thailand virus presents a similar gene organization, although only four ORFs were identified (Figure 1). ORF1 code for a protein of 488 aa corresponds to the putative nucleoprotein of the virus. ORF2 code for a hypothetical protein of 363 aa presenting numerous O-glycosylation sites. Its size (in the range of those observed for other Rhabdoviridae phosphoproteins), the absence of transmembrane domains, and its richness in acidic amino acids (PI = 4.89) suggest that ORF2 could code for the putative P protein of WhTV1-Thailand virus. ORF3 codes for a small protein of 205 aa corresponding to the putative matrix protein of the virus. The last ORF codes for a large protein of 2,183 aa corresponding to the RNA polymerase of the virus. WhTV1-Thailand virus present therefore an N–P–M–L gene organization, suggesting that a major evolution event in its emergence was the loss of G gene. Walker et al. noted that gain and/or loss of genes was common and a major driver of rhabdoviruses evolution (Walker et al., 2015), and several assumptions were made to explain the mechanism of entry in hosts cells of such viruses presenting the loss of G protein, as for example the use of a helper virus (Sameroff et al., 2019). WhTV1-Thailand viral ORFs present genetic identity ranging from 93 to 97% at the protein level, depending on the gene, with its closest viral relative Rhipicephalus associated rhabdo-like virus (Table 2).
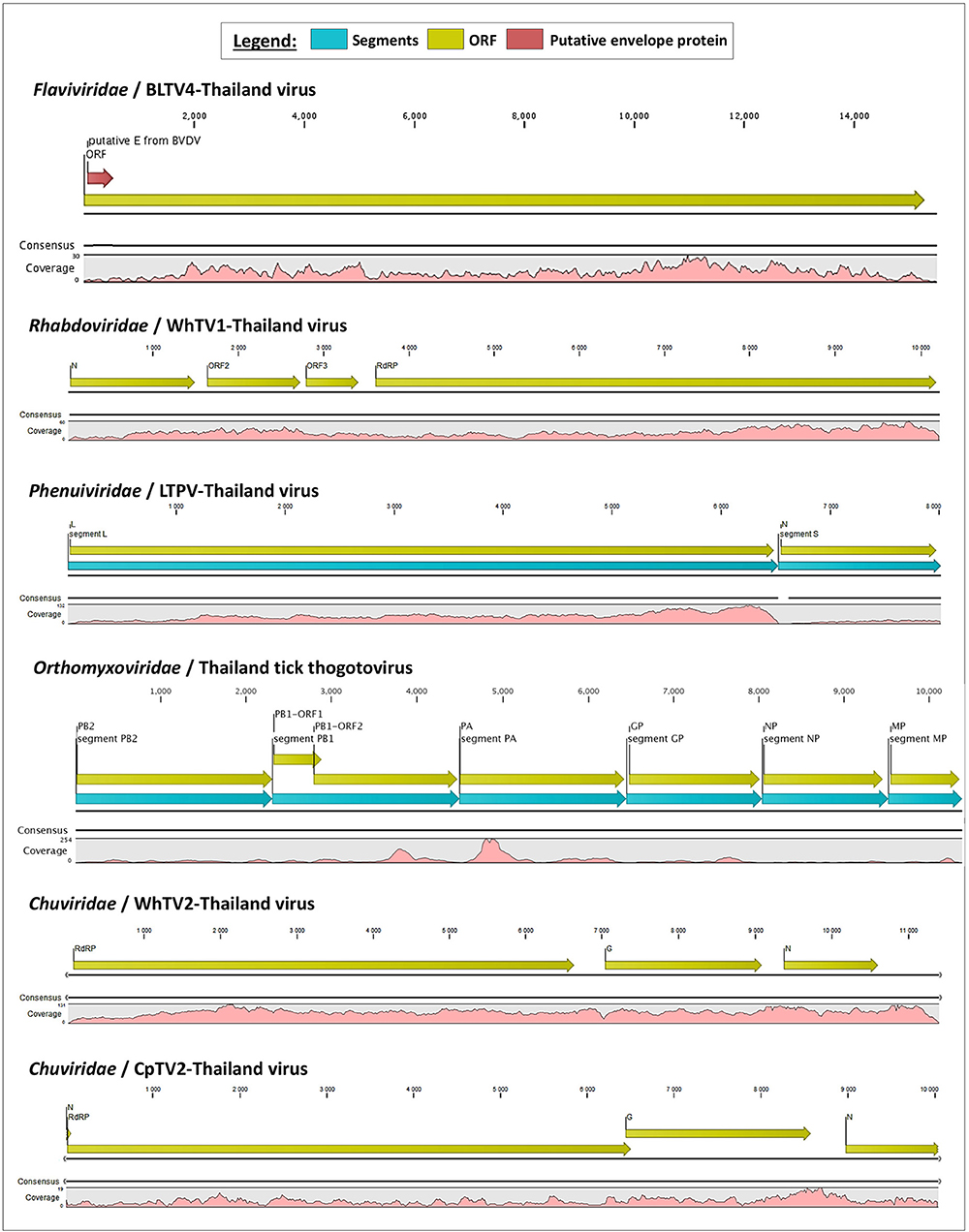
Figure 1. Schematic organization of the six viral genomes identified in Thai ticks. The open reading frames (ORFs) are indicated with yellow arrows, and genome coverage is indicated in pink. Segmented viruses were presented as concatenated sequences for better clarity (blue arrows represent the different segments). The putative envelope glycoprotein (GP) of Thailand tick flavivirus is highlighted by a red arrow in the polyprotein ORF.
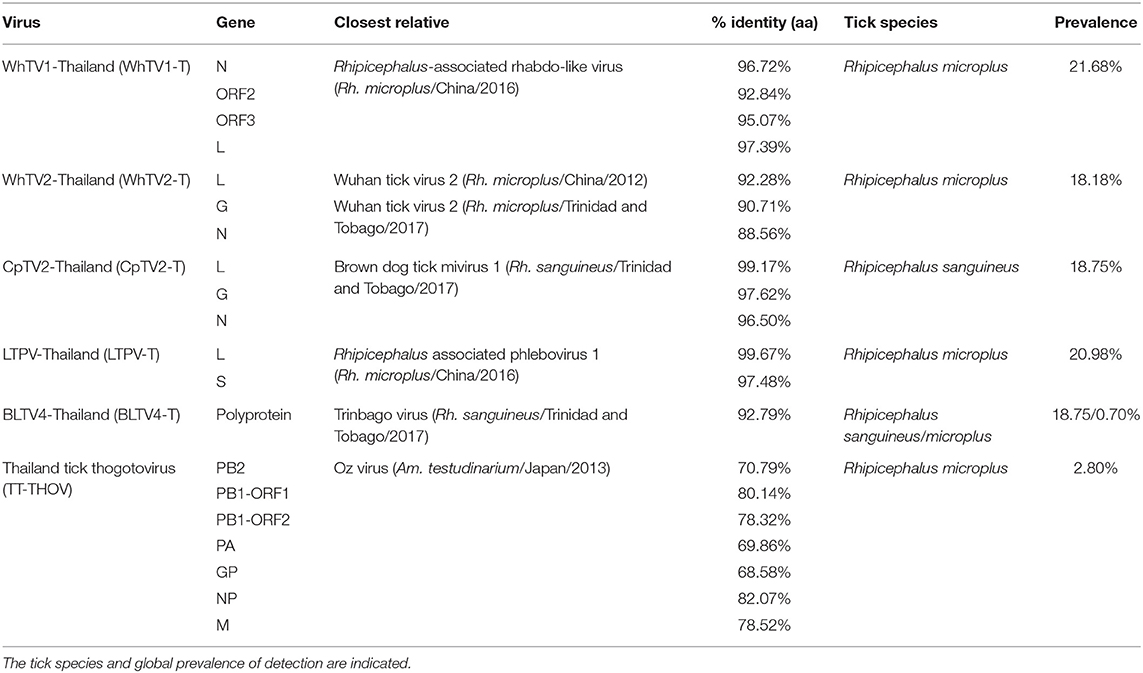
Table 2. Amino-acid identity of Flaviviridae-, Rhabdoviridae-, Chuviridae-, Phenuiviridae-, and Orthomyxoviridae-related viruses with their closest viral reference genome.
Recently, numerous tick-associated rhabdoviruses were reported worldwide, but they are still unassigned to a genus and fall in several distinct clades that possibly constitute new genera in the family (Tokarz et al., 2014b; Li C. X. et al., 2015; Xia et al., 2015; Brinkmann et al., 2018). Phylogenetic analyses performed on the complete amino-acid sequence of the RNA-dependent RNA polymerase revealed that WhTV1-Thailand virus falls in a clade, distant from classical rhabdoviruses, that comprised viruses identified exclusively in ticks from a wide range of geographical origins (China, USA, Turkey and Norway) (Figure 2). One should note that the genome organization of several viruses within this subclade of tick-borne viruses (comprising WhTV1-Thailand virus) present also gene loss (Figure 2, left inset).
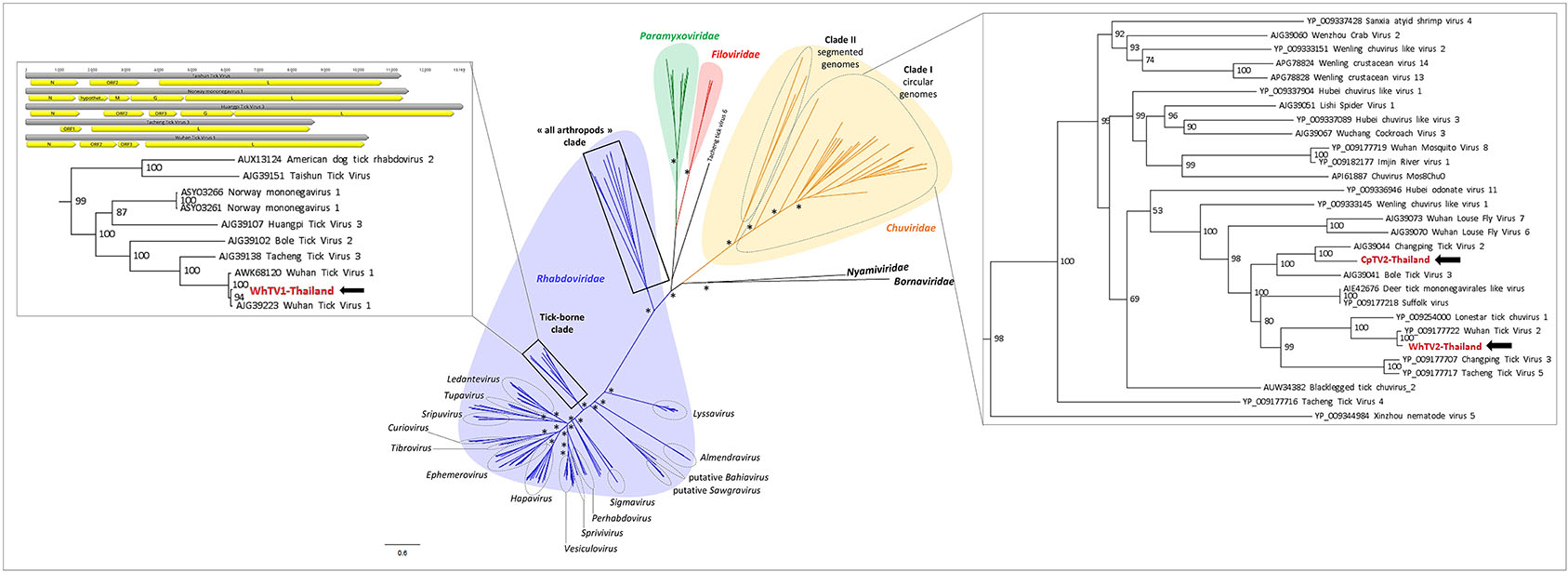
Figure 2. Phylogenetic relationship of Mononegavirales- and Jingchuvirales-related viral genomes identified in Thai ticks with other representative viruses. Nodes with bootstrap values >50 are noted with an asterisk. Phylogenetic reconstruction was performed by maximum likelihood on the complete RNA-dependent RNA polymerase (RdRP) amino-acid gene (model: LG + G + I + F).
Chuviridae/WhTV2- and CpTV2-Thailand Viruses
Chuviridae, a recently recognized viral family among the Jingchuvirales that was primarily assigned to the Mononegavirales order forms a monophyletic group of segmented, non-segmented, and circular viruses (Siddell et al., 2019) at median distance with segmented and unsegmented negative-strand RNA viruses (Li C. X. et al., 2015). They infect a wide variety of invertebrate hosts, including Crustacea, Nematoda, Insecta, Myriapoda, Arachnida, and Ixodida. Among tick-borne chuviruses, both Argasidae (Argas mivirus) and Ixodidae ticks are infected by tick-borne chuviruses (e.g., Bole, Changping, Dermacentor, Lonestar, Suffolk, and Wuhan mivirus). Chuviridae viruses present a large variety of genome organization, from a circular L–G–N order of genes (clade I, Figure 2) to segmented or linear G–N–L order of genes (clade II, Figure 2) (Li C. X. et al., 2015). WhTV2-Thailand virus present a genome of 11.4 kb with three ORFs: the first ORF codes for a large protein of 2,189 aa corresponding to the RNA-dependent RNA polymerase of the virus; ORF2 code for the putative glycoprotein of 683 aa and ORF3 codes for the nucleoprotein of 411 aa (Figure 1). WhTV2-Thailand virus genome was confirmed to belong to the circular L–G–N form of Chuviridae by performing PCR with the forward primer located in 3′ of the genome and the reverse primer in the 5′ part (data not shown). CpTV2-Thailand virus presents similar genome organization as WhTV2-Thailand virus, with a circular ssRNA genome, except that its genome is shorter (10 kb) and that the G ORF overlaps the L ORF on 61 nucleotides (this junction was confirmed by specific amplification followed by Sanger sequencing). The L gene code similarly for the RdRP of 2,164 aa, the G gene codes for the glycoprotein of 710 aa and the N gene codes for the nucleoprotein of 374 aa (Figure 1).
WhTV2-Thailand virus presents amino acid identity ranging from 89 to 92% with the prototype Wuhan tick virus 2 while CpTV2-Thailand virus is close to Brown dog tick mivirus 1, a Changping tick virus 2-related virus (Table 2). Phylogenetic analyses performed on the complete amino-acid sequence of the RdRP confirmed that WhTV2-Thailand and CpTV2-Thailand chuviruses belong to the circular clade I of the Chuviridae (Figure 2). In addition, with a highly supported bootstrap of 100, they both placed in a subclade composed exclusively of viruses identified in ticks (Figure 2, right inset).
Phenuiviridae/LTPV-Thailand Virus
Phenuiviridae is a family of segmented negative-strand ssRNA viruses which comprise Goukovirus and Phasivirus (insect-specific viruses), Tenuivirus (plant arboviruses), and Phlebovirus (animal arboviruses) genera. This latter is the unique known viral genus among the family able to infect vertebrates, including humans and domestic animals. Recognized tick-borne phleboviruses (TBPVs) are currently clustered into three main serogroups: SFTS virus, Bhanja virus, and Uukuniemi virus serogroups (Yu et al., 2011; Matsuno et al., 2013; Palacios et al., 2013). The ecological cycle of TBPVs implies Ixodidae ticks as the main vector, wild small animals (e.g., Soricidae or Erinaceidae) or migratory birds (Li et al., 2016) as putative reservoirs and/or amplifying hosts (also responsible of the dissemination of the virus over long distances), and accidental mammalian hosts. SFTS-like viruses are able to infect humans, while other TBPVs usually infect domestic animals like sheep, goat, and cattle (Hubálek et al., 2014), and, in rare specific cases, humans (Calisher and Goodpasture, 1975). Viruses belonging to this genus are trisegmented, with L segment coding for the viral RdRP, M segment coding for the glycoprotein precursor which will be further matured in two glycoproteins (GP) and a non-structural protein (NS), and the S segment coding for the nucleoprotein (NP) and a NS protein. Recently, bi-segmented phlebovirus-like viruses without any M segment were reported in ticks from various origins (Li C. X. et al., 2015). LTPV-Thailand virus presents the same bisegmented genome architecture, with L and S segments of 6,517 and 1,406 nt, respectively (which is in the range of trisegmented phleboviruses), coding for the expected RdRP and NP genes. As for other bisegmented phleboviruses, LTPV-Thailand virus lacked the M segment. It has been proposed that a high degree of divergence of M segments of these phleboviruses may explain that usual Blast-based methods of taxonomic assignation of reads have missed this segment (Li C. X. et al., 2015). As a consequence, the only identified structural protein of LTPV-Thailand virus was NP, which interestingly present structural homologies with the nucleoprotein of SFTS virus, a tick-borne phlebovirus causing severe symptoms in humans and close to the Uukuniemi virus serocomplex (Yu et al., 2011).
LTPV-Thailand virus presents amino acid identity of 97 and 100% with Rhipicephalus-associated phlebovirus 1, a new strain of Lihan tick phlebovirus identified in Rh. microplus Chinese ticks (Table 2). Phylogenetic analyses performed on the complete RdRP amino acid gene revealed that LTPV-Thailand virus is located in a clade at the root of the Uukuniemi virus group, a tick-borne phlebovirus primarily isolated in cattle-infesting Ixodes ricinus ticks from Finland that could sporadically infect cattle and humans (Saikku, 1973; Traavik and Mehl, 1975) (Figure 3). Two subclades of bisegmented phlebovirus-like viruses are placed at the root of UUKV, one only composed of tick-borne viruses identified in Ixodes (I. ricinus and I. scapularis) ticks from USA and Norway (Pettersson et al., 2017; Tokarz et al., 2018), while the other (containing LTPV-Thailand virus) is formed by viruses detected in Rhipicephalus, Dermacentor, Haemaphysalis, and Hyalomma ticks from China, Brazil, USA and Turkey (Tokarz et al., 2014a; Li C. X. et al., 2015; Dinçer et al., 2017; Brinkmann et al., 2018; Souza et al., 2018), suggesting a possible association between the tick species and the virus and a putative coevolution of bisegmented phleboviruses with their respective tick host (Figure 3, inset).
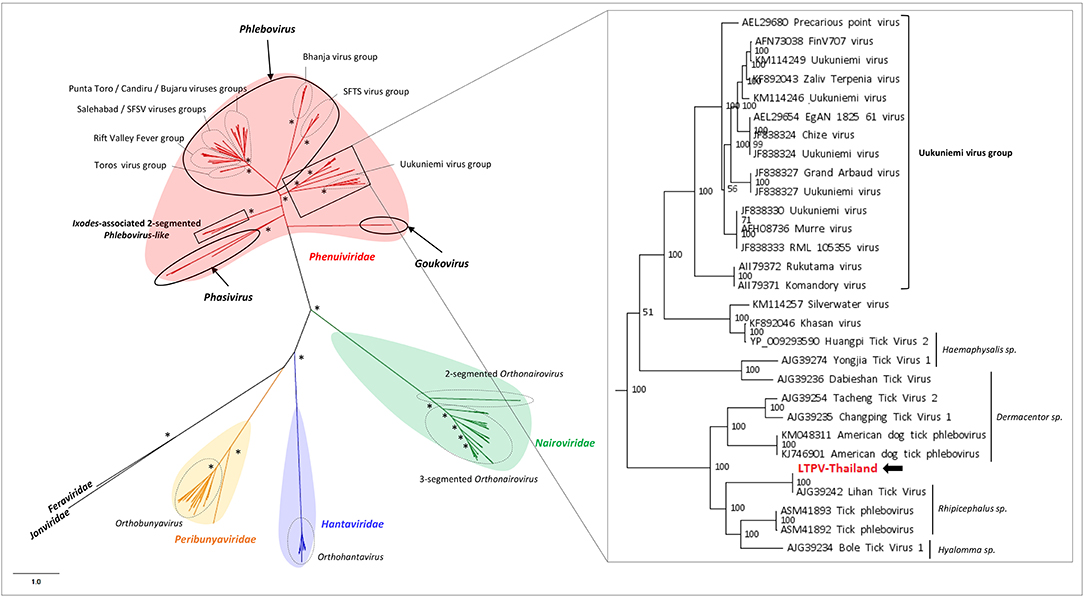
Figure 3. Phylogenetic relationship of Bunyavirales-related viral genomes identified in Thai ticks with other representative viruses. Nodes with bootstrap values >50 are noted with an asterisk. Phylogenetic reconstruction was performed by maximum likelihood on the complete RNA-dependent RNA polymerase (RdRP) amino-acid gene (model: LG + G + I + F).
Flaviviridae/BLTV4-Thailand Virus
Flaviviridae is a family of positive-strand ssRNA viruses composed of four recognized genera: Flavivirus (arboviruses infecting both vertebrates and invertebrates), Pegivirus (mammalian viruses including primates, humans, pigs, and bats), Pestivirus (animal viruses such as bovine diarrhea virus or classical swine fever virus), and Hepacivirus (human Hepatitis C virus). Tick-borne flaviviruses (TBFVs) are divided into mammalian- and seabird-infecting viruses (Brackney and Armstrong, 2016). Mammalian TBFVs are transmitted by Ixodidae hard ticks, while seabird TBFVs are transmitted by Argasidae soft ticks. The flavivirus viral genome of ~10–12 kb codes for a unique polyprotein that will be cleaved during maturation by viral and host proteases into viral structural and non-structural proteins. Recently, Shi et al. reported the identification of divergent flavivirus-like viruses in arthropods with genome size ranging from 16 to 26 kb with similar genome organizations, including Bole tick virus 4 (BLTV4) virus primarily identified in Hy. asiaticum Chinese ticks and later in Rh. sanguineus ticks from Trinidad and Tobago (Trinbago virus) (Shi et al., 2015; Sameroff et al., 2019). We identified in Thai ticks a genome close to Trinbago virus, tentatively named BLTV4-Thailand virus. Its genome of 15.5 kb (shorter than BLTV4 genome of 16.2 kb) codes for a unique polyprotein of 5,089 aa.
Phylogenetic analyses performed on the RdRP region of representative sequences of Flaviviridae placed BLTV4-Thailand virus in a sister clade of the Pestivirus genus that contains viruses only identified in invertebrates, as previously described (Sameroff et al., 2019). Putatively forming the fifth genus of the Flaviviridae family, this group of viruses infecting arthropods placed at the root of the four recognized genera (Figure 4A), supporting the hypothesis of Shi et al. that flaviviruses may originate from arthropods (Shi et al., 2015). Interestingly, with a highly supported node of 96, two subclades compose this putative genus: one restricted to the class of Insecta and the second, including BLTV4-Thailand virus, composed of viruses infecting all types of arthropods, ranging from Chelicerata to Myriapoda and Hexapoda, suggesting a possible more ancestral position of this virus in the evolution of flaviviruses (Figure 4A, inset) and a later divergence of insect-specific Flaviviridae-related viruses.
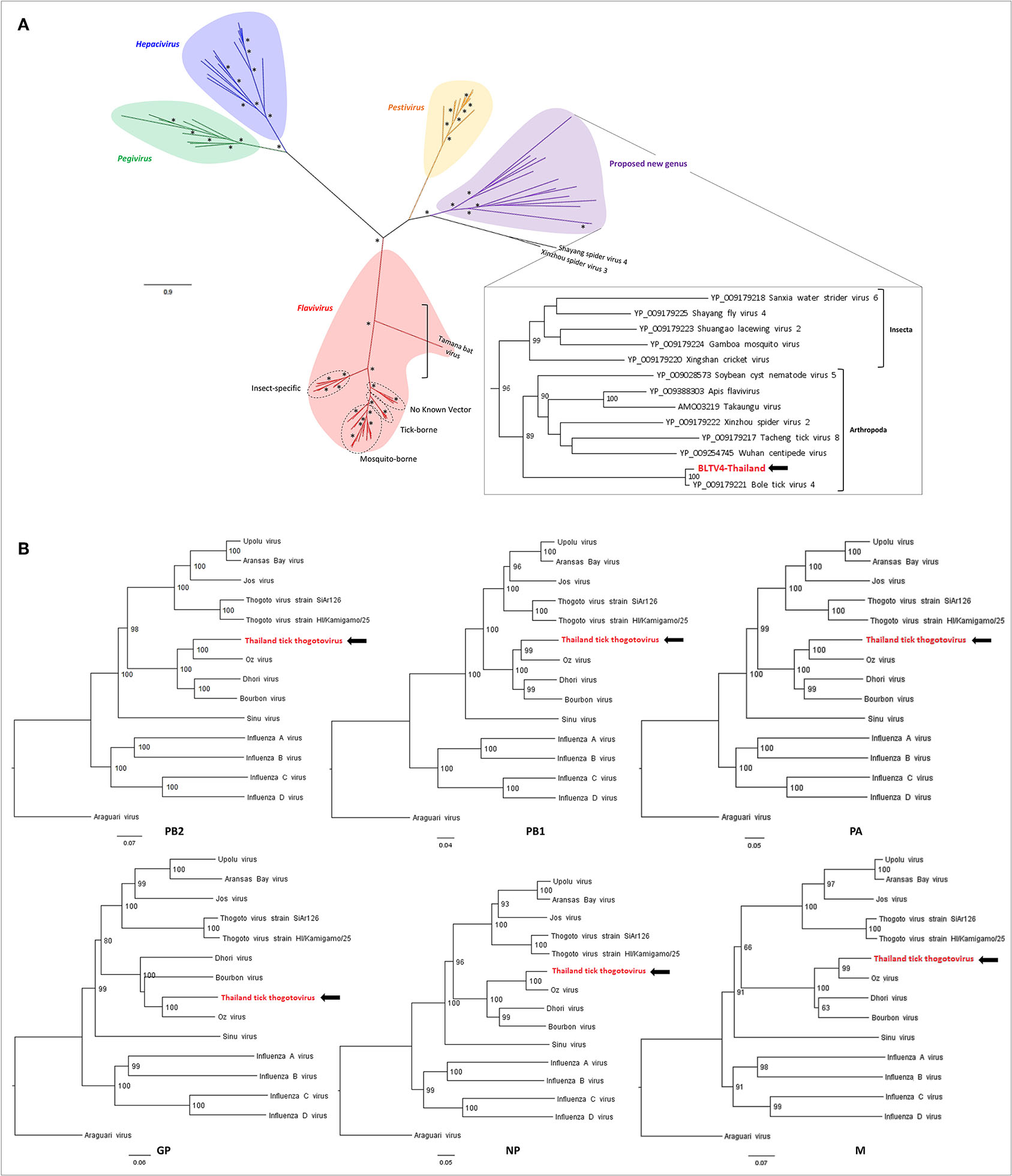
Figure 4. Phylogenetic relationship of viral genomes identified in Thai ticks with other representative viruses. (A) Maximum likelihood (ML) tree of the complete RNA-dependent RNA polymerase (RdRP) amino-acid sequence of representative Flaviviridae viruses (model: LG + G + I + F). Nodes with bootstrap values >50 are noted with an asterisk. (B) Neighbor joining (NJ) trees of the complete PB2–PB1–PA–GP–NP–M amino-acid sequences of representative Orthomyxoviridae viruses (model: p-distance).
Orthomyxoviridae/Thailand Tick Thogotovirus
The Orthomyxoviridae family is composed of seven genera: types A/B/C and D Influenzavirus, Isavirus, Quaranjavirus, and Thogotovirus. Thogotoviruses, arthropod-borne viruses infecting mammals mainly transmitted by ticks, have been identified in Africa, North America, Southern Europe, the Middle East, and in Far East Asia, either in ticks and/or in humans (for Thogoto virus, Dhori virus, and Bourbon virus) (Ejiri et al., 2018). Thogotoviruses are composed of six negative-strand ssRNA segments that code from the polymerase complex (segments PB2, PB1, and PA), the glycoprotein (GP), the nucleoprotein (NP) and the matrix protein (M). Although influenza viruses present a surface GP constituted of hemagglutin and neuraminidase proteins, the GP of thogotoviruses present structural similarities with the gp64 envelope glycoprotein of alphabaculoviruses (Morse et al., 1992; Kadlec et al., 2008). We identified in Thai ticks a viral genome close to Oz virus (OZV), a thogotovirus recently isolated from Am. testudinarium ticks in Japan (Ejiri et al., 2018). Provisionally named Thailand tick thogotovirus, this virus presents the characteristics of a Thogotovirus member, meaning: (i) the six segments genome architecture [segment 1 codes for the putative PB2 protein of 764 aa; segment 2 for two ORFs (of 187 and 559 aa, respectively)] constituting the PB1 protein complex; segment 3 for the PA protein of 637 aa; segment 4 for the GP protein of 510 aa; segment 5 for the NP protein of 465 aa; and segment 6 for the M protein of 252 aa); and (ii) the structural conformation of Thailand tick thogotovirus GP close to the baculovirus fusion protein gp64 (data not shown) (Morse et al., 1992; Kadlec et al., 2008). Thailand tick thogotovirus presents an amino-acid identity with OZV ranging from 69% (GP) to 82% (NP) (Table 2).
Phylogenetic analyses performed on the six segments of Thailand tick thogotovirus and representative Thogotovirus genomes resulted in the same topology, i.e., a clustering of Thailand tick thogotovirus with OZV in a sister clade of the group formed by Bourbon and Dhori viruses, suggesting that no reassortment events occurred during Thailand tick thogotovirus evolution (Figure 4B).
Prevalence of Flaviviridae-, Rhabdoviridae-, Chuviridae-, Phenuiviridae-, and Orthomyxoviridae-Related Viruses
Among the 266 tick extracts used to generate the pool sequenced, 231 were available and screened individually for the presence of Thailand tick thogotovirus and other LTPV-, BLTV4-, WhTV1-, WhTV2-, and CpTV2-Thailand viruses. Results are presented in Table 3A. LTPV-Thailand phlebovirus was only detected in Rh. microplus ticks at a global prevalence of 21%. BLTV4-Thailand virus was mainly detected in Rh. sanguineus ticks (19%) and in one Rh. microplus tick. Interestingly, these two viruses are genetically close but presented only 82% of nucleotide identity in the RdRP gene. Thogotovirus-related virus was identified at low prevalence (<3%) in adult Rh. microplus ticks, while WhTV2-Thailand chuvirus was more prevalent (18%). CpTV2-Thailand virus was only detected in Rh. sanguineus ticks at a global prevalence of nearly 19%. Finally, WhTV1-Thailand virus was only detected in more than 21% of Rh. microplus ticks (Tables 2, 3A).
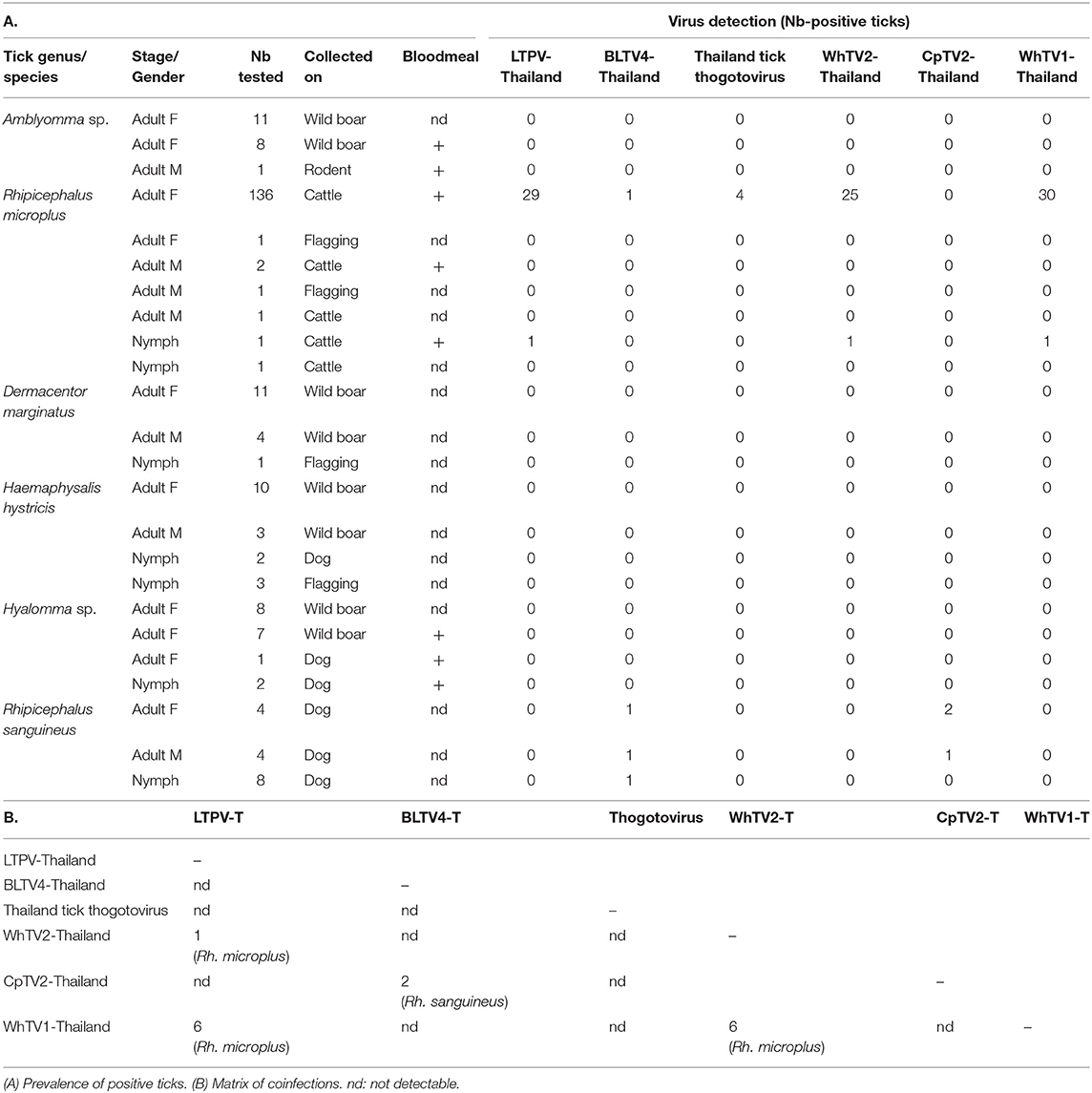
Table 3. Results of prevalence studies of Flaviviridae-, Rhabdoviridae-, Chuviridae-, Phenuiviridae-, and Orthomyxoviridae-related viruses.
Few coinfections of ticks were detected. The presence of two viruses (most frequently WhTV1-Thailand virus associated with LTPV-Thailand virus or WhTV2-Thailand virus) in the same individual tick was identified in 13 Rh. microplus and in 2 Rh. sanguineus ticks (Table 3B). Four coinfections with three different viruses were also detected in Rh. microplus ticks (three coinfections LTPV-/WhTV1-/WhTV2-viruses, and one coinfection LTPV-/thogoto-/WhTV2-viruses).
Ticks harbor numerous EVEs within their genome (Katzourakis and Gifford, 2010; Li C. X. et al., 2015), including several sequences phylogenetically related to viruses identified in the present study. Therefore, ticks positive either for LTPV-, BLTV4-, WhTV1-, WhTV2-, CpTV2-, and thogoto-Thailand viruses were individually screened for the presence of EVE using their corresponding DNA extract. No positive result was obtained, showing that the viruses identified by next generation sequencing are exogenous viruses (data not shown).
LIPS-Based Serological Screening of Human and Cattle Sera
To test if one or more viruses identified among Thai ticks is able to infect selected mammalian species (humans and cattle) highly exposed to tick bites, and therefore could constitute putative novel tick-borne arboviruses, we developed LIPS-based serological screening against Thailand tick thogotovirus and LTPV-, BLTV4-, WhTV1-, WhTV2-, and CpTV2-Thailand viruses. Results are presented in Figure 5.
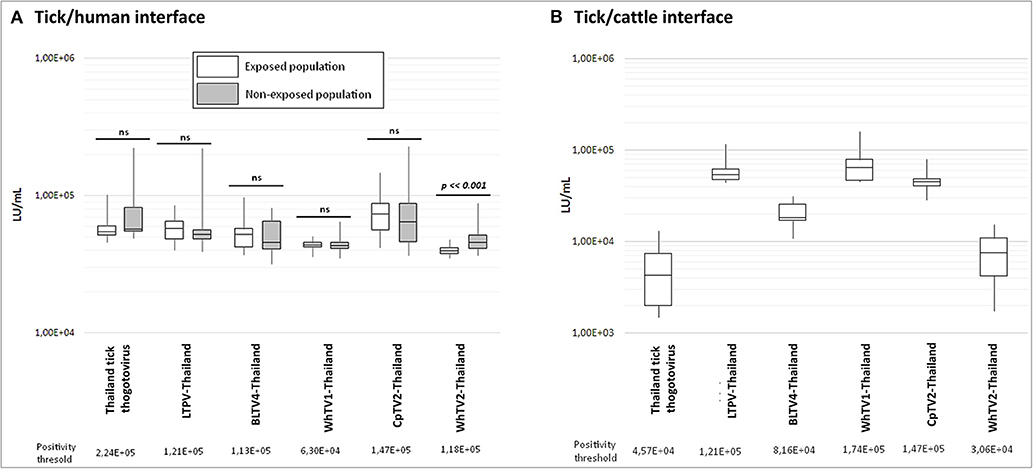
Figure 5. Luciferase activity (in LU/ml) distribution of measures after luciferase immunoprecipitation system (LIPS) performed in (A) tick/human interface and (B) tick/cattle interface. In white: human and cattle populations exposed to tick bites; in gray: non-exposed human populations. Positivity threshold is indicated for each antigen construct. t-test statistical analysis (α = 0.05) was used to compare the mean LU/ml measure of both exposed and non-exposed human groups.
For the tick/human interface, no significant difference was observed between exposed and non-exposed groups (Figure 5A), and no serum exceeded the positivity threshold, except for WhTV2-Thailand chuvirus antigen, which presents a higher luciferase activity in the non-exposed group than in the exposed group (p = 0.0003), while none of the sera in the exposed nor non-exposed groups exceeded the positivity threshold. Similarly, none of the cattle sera presented a luciferase activity higher than the positivity threshold (Figure 5B), showing that no antibodies against one of the six viruses tested were detected in human and cattle sera. The maximal prevalence of sera reacting with at least one of the six viral constructs was estimated at 0.017% and 0.047% (p = 0.05) in humans and cattle, respectively, meaning that these infections of vertebrates, if they occur, are very rare.
Discussion
The increasing accessibility of HTS technology and the resulting description of viral communities in various environmental samples pave the way for a better understanding of viral spillovers. However, identifying a list of viruses at higher risk of emergence requires first the characterization of the ecological cycle of the virus, including its putative reservoir hosts, hematophagous arthropod vectors (in the case of arboviruses), and vertebrate recipient hosts (Temmam et al., 2014). Usual practices combine field surveillance of vectors and wildlife, description of viral communities present in these animals, analyses of bloodmeal origin in arthropods, and phylogenetic analyses of viruses. Recently, novel approaches tried to predict vertebrate and invertebrate hosts using computational modeling (Babayan et al., 2018). In this study, we combined field surveillance of arthropods and virome characterization with comprehensive phylogenetic analyses to select relevant viruses close to tick-borne arboviruses. We further characterized putative spillover events by applying a systematic serological survey in human and cattle populations highly exposed to these ticks. Among the viral communities characterized in Thai ticks, we selected viruses belonging to four viral families known to contain zoonotic viruses (Orthomyxoviridae, Phenuiviridae, Rhabdoviridae, and Flaviviridae families) and a recently recognized viral family for which the zoonotic potential was still unknown (Chuviridae).
The Phenuiviridae, Flaviviridae, Orthomyxoviridae, Rhabdoviridae, and Chuviridae viruses detected in Rh. microplus and Rh. sanguineus Thai ticks were previously reported in other ticks species and genera, highlighting a low degree of host restriction and an increase in the vector host range of these viruses (Table 4). For example, CpTV2-Thailand chuvirus identified in Rh. sanguineus was previously reported in Chinese Dermacentor sp. and Ha. parva ticks (Li C. X. et al., 2015), in Turkish Ha. parva ticks (Brinkmann et al., 2018), and in Rh. sanguineus ticks from Trinidad and Tobago (Sameroff et al., 2019). Similarly, Thailand tick thogotovirus, identified in Rh. microplus ticks, is phylogenetically related to Oz virus isolated in Japan from Am. testudinarium tick (Ejiri et al., 2018). LTPV-Thailand phlebovirus presented also a low degree of host specificity, as regards to its reports in Thai Rh. microplus ticks, in Dermacentor nitens from Colombia, in Rh. microplus from China (Li C. X. et al., 2015), Brazil (Souza et al., 2018), and Trinidad and Tobago (Sameroff et al., 2019), and in Hy. marginatum (Dinçer et al., 2017) and Rh. sanguineus (Brinkmann et al., 2018) from Turkey. Finally, BLTV4-Thailand flavivirus, identified in Thai Rh. microplus and Rh. sanguineus ticks, was either previously reported in Hy. asiaticum (Shi et al., 2015), Hyalomma detritum, and D. marginatus ticks from China, and in Rh. sanguineus ticks from Trinidad and Tobago (Sameroff et al., 2019). This low degree of host restriction [either suggesting multiple hosts switches or reflecting the non-viremic transmission of viruses occurring during cofeeding onto the same host (Labuda and Nuttall, 2004)], added to the phylogenetic placement of these viruses at the root of known tick-borne arboviruses, raised the question of their possible ability to infect vertebrate hosts.
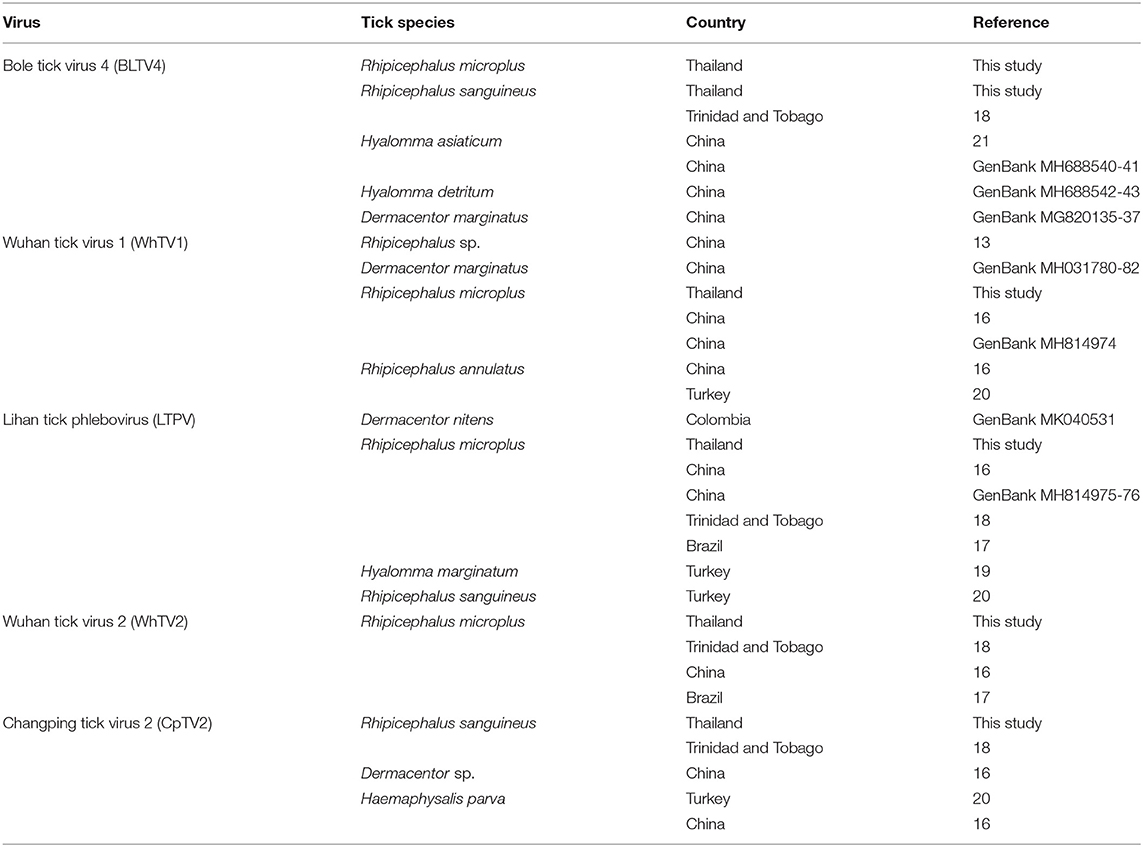
Table 4. Tick spectrum and geographical origin of Flaviviridae-, Rhabdoviridae-, Chuviridae-, and Phenuiviridae-related viruses.
The serological approach employed in the present study to assess the exposure of humans and mammals to viruses carried by ticks is highly dependent of the conservation of epitopes conformation. LIPS test developed by Burbelo et al. allows the recovery of viral antigens under native conditions using expression system in mammalian cells, limiting therefore this issue (Burbelo et al., 2010). Since it was impossible to obtain any positive control, and therefore to assess the analytical sensitivity of LIPS assay for each of the targeted antigens, we have successfully validated the protocol using the same approach targeting Hepatitis E virus (HEV), a prevalent zoonotic virus in France (Capai et al., 2019). Briefly, recombinant viral antigens expressing the external domain of the capsid of HEV were produced and further used for screening of French human sera by LIPS. The assay showed the same sensitivity as the one observed when human sera were tested in parallel by the commercial ELISA (Supplemental Table 3). In addition, LIPS sensitivity has been extensively described in different studies and demonstrated to be at least as sensitive as plaque reduction neutralization test and ELISA (Burbelo et al., 2012; Tin et al., 2017; Crim et al., 2019). We therefore conclude that the different viruses we studied are not (or very rarely) transmitted to humans and cattle (<0.017% and <0.047%, respectively). Therefore, the six selected viruses may possibly either infect other vertebrates exposed to tick bites [and especially the role of rodents and migratory birds has to be evaluated, as regards to the trophic preference of ticks (Mlera and Bloom, 2018; Tomassone et al., 2018)], or be restricted to their tick hosts.
Tick-borne flavivirus, phlebovirus, rhabdovirus, and chuviruses described in this study presented a high global prevalence (ranging from 18 to 22%), which could suggest that they are tick endosymbionts (Bouquet et al., 2017; Tokarz et al., 2018; Sameroff et al., 2019). In addition, the low degree of host restriction of these viruses, previously identified in various tick species and genera from distant geographical origins, supports this hypothesis (Table 4). Our serological results in humans and cattle also are in favor of this hypothesis. Our results show that LTPV-Thailand phlebovirus, although it has phylogenetic relatedness with Uukuniemi virus [able to infect cattle and humans (Saikku, 1973; Traavik and Mehl, 1975)], seems not be able to infect these species (or at very low prevalence). This virus, highly prevalent in Rh. microplus ticks, and other related bisegmented phleboviruses belonging to the same clade, should more likely be considered as tick endosymbionts that represent viral ancestors of the known trisegmented tick-borne phleboviruses, as previously suggested (Li C. X. et al., 2015). Similar conclusions could be drawn for Thai strains of tick-borne flavivirus, rhabdovirus, and chuviruses described here. However, it has been suggested that viruses presented a low degree of host restriction, and a high prevalence in ticks populations could be considered as vertebrate-borne viruses, reflecting the origin of bloodmeal of ticks (Sameroff et al., 2019). Further investigations are therefore needed to conclude on the host spectrum and get primary insights into the ecological cycle of these viruses.
The prevalence of Thailand tick thogotovirus in Rh. microplus ticks was inferior to 3%, suggesting that this virus possibly infects vertebrate hosts, according to Bouquet hypothesis linking prevalence of a given virus in ticks with its putative infectivity in vertebrates (Bouquet et al., 2017). Indeed, thogotoviruses are zoonotic tick-borne viruses, among which Bourbon, Dhori, and Thogoto viruses are able to infect several mammals (cattle, sheep, donkeys, camels, and buffalos) including humans, causing for the latter from febrile illness to encephalitis (McCauley et al., 2012). Apart from ticks, many arthropods were reported as alternative vectors, including mosquitoes [Batken and Sinu viruses (Frese et al., 1997; Contreras-Gutiérrez et al., 2017)] and biting midges (Temmam et al., 2016). Several evidence of a possible role of rodents as amplifying hosts or reservoirs were reported (Ejiri et al., 2018). Our survey shows that human and cattle infections were absent (or at most very rare), although this virus is phylogenetically close to known human pathogens. This finding raised the question of the mechanism of host restriction of tick-borne viruses (and more specifically of Thailand tick thogotovirus and other Oz-related tick-borne viruses) and more generally of the mechanisms that underlined the adaptation of tick-specific viruses to vertebrate-infecting tick-borne arboviruses (Li C. X. et al., 2015; Shi et al., 2015).
Conclusions
Although identifying novel zoonotic viruses is of great importance and a prerequisite to monitor silent viral emergences, it is of equal importance to determine, among the huge number of viruses composing the virome of vectors and reservoirs, which viruses are not able to cross the species because it could lead to a better understanding of the mechanisms underlying viral emergence, and especially to understand why some viruses that are phylogenetically close to zoonotic viruses are not able to spill over. The strategy developed in this pilot study starting from the inventory of viral communities infecting a given vector by HTS to finally identify reactive antibodies possibly present in exposed vertebrate populations by luciferase immunoprecipitation is well-adapted to such considerations and could easily be transposable to other vectors/mammals interfaces.
Data Availability Statement
The datasets generated for this study can be found in the NCBI GenBank database MN095535 to MN095546.
Ethics Statement
The studies involving human participants were reviewed and approved by Ethical Committee of the Faculty of Tropical Medicine, Mahidol University, Thailand. The patients/participants provided their written informed consent to participate in this study. The animal study was reviewed and approved by Department of Livestock Development, Thailand.
Author Contributions
ST, MDu, J-FC, MV-T, SMou, and ME designed and conceived the study. ST, DC, EDu, MDu, EDe, MDe, LY, and LG performed the experiments. ST and TB performed the bioinformatics analyses. MDe, SJ, AK, KC, J-FC, MV-T, and SMor realized fieldwork in Thailand. ST wrote the manuscript. ST, DC, TB, SP, MDu, SMor, SMou, MDe, and ME reviewed the manuscript.
Funding
This work was supported by Laboratoire d'Excellence Integrative Biology of Emerging Infectious Diseases (grant no. ANR-10-LABX-62-IBEID), by the Direction Internationale de l'Institut Pasteur, and by the ANR projects BiodivHealthSEA and FutureHealthSEA (ANR-17-CE35-0003-02) for fieldwork studies in Thailand.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors want to thank Jacques Bellalou and his team of the Production and Purification of Recombinant Proteins Technological Platform from the Institut Pasteur for their help in expression of recombinant proteins, Yves Jacob and Mélanie Dos Santos for their technical assistance with the luminometer, the ICAReB Platform from the Institut Pasteur for giving access to human control sera, Yves Gaudin for his analysis of Thailand tick rhabdovirus genome structure, Nicole Pavio for providing HEV positive and negative serum controls used to validate LIPS assay, and all the people involved in sample collection in Thailand.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02315/full#supplementary-material
References
Ahantarig, A., Trinachartvanit, W., and Milne, J. R. (2008). Tick-borne pathogens and diseases of animals and humans in Thailand. Southeast Asian J. Trop. Med. Public Health. 39, 1015–1032. Available online at: https://www.tm.mahidol.ac.th/seameo/2008-39-6-full/10-4238.pdf
Arnold, K., Bordoli, L., Kopp, J., and Schwede, T. (2006). The SWISS-MODEL workspace: a web-based environment for protein structure homology modelling. Bioinformatics. 22, 195–201. doi: 10.1093/bioinformatics/bti770
Babayan, S. A., Orton, R. J., and Streicker, D. G. (2018). Predicting reservoir hosts and arthropod vectors from evolutionary signatures in RNA virus genomes. Science. 362, 577–580. doi: 10.1126/science.aap9072
Biasini, M., Bienert, S., Waterhouse, A., Arnold, K., Studer, G., Schmidt, T., et al. (2014). SWISS-MODEL: modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 42, 252–258. doi: 10.1093/nar/gku340
Bigot, T., Temmam, S., Pérot, P., and Eloit, M. (2019). RVDB-prot, a reference viral protein database and its HMM profiles [version 1; peer review: awaiting peer review]. F1000Research. 8:530. doi: 10.12688/f1000research.18776.1
Bouquet, J., Melgar, M., Swei, A., Delwart, E., Lane, R. S., and Chiu, C. Y. (2017). Metagenomic-based surveillance of Pacific coast tick Dermacentor occidentalis identifies two novel Bunyaviruses and an emerging human Ricksettsial pathogen. Sci. Rep. 7:12234. doi: 10.1038/s41598-017-12047-6
Brackney, D. E., and Armstrong, P. M. (2016). Transmission and evolution of tick-borne viruses. Curr. Opin. Virol. 21, 67–74. doi: 10.1016/j.coviro.2016.08.005
Brinkmann, A., Dinçer, E., Polat, C., Hekimoglu, O., Hacioglu, S., Földes, K., et al. (2018). A metagenomic survey identifies Tamdy orthonairovirus as well as divergent phlebo-, rhabdo-, chu- and flavi-like viruses in Anatolia, Turkey. Ticks Tick Borne Dis. 9, 1173–1183. doi: 10.1016/j.ttbdis.2018.04.017
Burbelo, P. D., Ching, K. H., Bush, E. R., Han, B. L., and Iadarola, M. J. (2010). Antibody-profiling technologies for studying humoral responses to infectious agents. Expert Rev. Vaccines. 9, 567–578. doi: 10.1586/erv.10.50
Burbelo, P. D., Ching, K. H., Klimavicz, C. M., and Iadarola, M. J. (2009). Antibody profiling by luciferase immunoprecipitation systems (LIPS). J Vis Exp. 7:1549. doi: 10.3791/1549
Burbelo, P. D., Ching, K. H., Mattson, T. L., Light, J. S., Bishop, L. R., and Kovacs, J. A. (2007). Rapid antibody quantification and generation of whole proteome antibody response profiles using LIPS (luciferase immunoprecipitation systems). Biochem. Biophys. Res. Commun. 352, 889–895. doi: 10.1016/j.bbrc.2006.11.140
Burbelo, P. D., Dubovi, E. J., Simmonds, P., Medina, J. L., Henriquez, J. A., Mishra, N., et al. (2012). Serology-enabled discovery of genetically diverse hepaciviruses in a new host. J. Virol. 86, 6171–6178. doi: 10.1128/JVI.00250-12
Calisher, C. H., and Goodpasture, H. C. (1975). Human infection with Bhanja virus. Am. J. Trop. Med. Hyg. 24, 1040–1042. doi: 10.4269/ajtmh.1975.24.1040
Capai, L., Falchi, A., and Charrel, R. (2019). Meta-analysis of human IgG anti-HEV seroprevalence in industrialized countries and a review of literature. Viruses. 11:E84. doi: 10.3390/v11010084
Chaisiri, K., Jollivet, C., Della Rossa, P., Sanguankiat, S., Wattanakulpanich, D., Lajaunie, C., et al. (2018). Parasitic infections in relation to practices and knowledge in a rural village in Northern Thailand with emphasis on fish-borne trematode infection. Epidemiol. Infect. 15, 1–12. doi: 10.1017/S0950268818002996
Contreras-Gutiérrez, M. A., Nunes, M. R. T., Guzman, H., Uribe, S., Suaza Vasco, J. D., Cardoso, J. F., et al. (2017). Sinu virus, a novel and divergent orthomyxovirus related to members of the genus Thogotovirus isolated from mosquitoes in Colombia. Virology 501, 166–175. doi: 10.1016/j.virol.2016.11.014
Cornet, J. P., Kittayapong, P., and Gonzalez, J. P. (2004). Risk of arbovirus transmission by ticks in Thailand. Med. Trop. 64:43–49. Available online at: https://www.researchgate.net/publication/8483012_Risk_of_arbovirus_transmission_by_ticks_in_Thailand
Crim, R. L., Kumari, S., Jayanti, P., Audet, S., Kulkarni, A., and Beeler, J. (2019). Development of luciferase immunoprecipitation systems (LIPS) assay to detect IgG antibodies against human respiratory syncytial virus G-glycoprotein. Vaccines. 7:16. doi: 10.3390/vaccines7010016
Criscuolo, A., and Brisse, S. (2014). AlienTrimmer removes adapter oligonucleotides with high sensitivity in short-insert paired-end reads. Commentary on Turner (2014) Assessment of insert sizes and adapter content in FASTQ data from NexteraXT libraries. Front. Genet. 5:130. doi: 10.3389/fgene.2014.00130
Cutler, S. J., Fooks, A. R., and van der Poel, W. H. (2010). Public health threat of new, reemerging, and neglected zoonoses in the industrialized world. Emerg. Infect. Dis. 16, 1–7. doi: 10.3201/eid1601.081467
Desquesnes, M., Kamyingkird, K., Pruvot, M., Kengradomkij, C., Bossard, G., Sarataphan, N., et al. (2009). Antibody-ELISA for Trypanosoma evansi: application in a serological survey of dairy cattle, Thailand, and validation of a locally produced antigen. Prev. Vet. Med. 90, 233–241. doi: 10.1016/j.prevetmed.2009.04.011
Dinçer, E., Brinkmann, A., Hekimoglu, O., Hacioglu, S., Földes, K., Karapinar, Z., et al. (2017). Generic amplification and next generation sequencing reveal Crimean-Congo hemorrhagic fever virus AP92-like strain and distinct tick phleboviruses in Anatolia, Turkey. Parasit. Vectors. 10:335. doi: 10.1186/s13071-017-2279-1
Ejiri, H., Lim, C. K., Isawa, H., Fujita, R., Murota, K., Sato, T., et al. (2018). Characterization of a novel thogotovirus isolated from Amblyomma testudinarium ticks in Ehime, Japan: a significant phylogenetic relationship to Bourbon virus. Virus Res. 249, 57–65. doi: 10.1016/j.virusres.2018.03.004
Frese, M., Weeber, M., Weber, F., Speth, V., and Haller, O. (1997). Mx1 sensitivity: Batken virus is an orthomyxovirus closely related to Dhori virus. J. Gen. Virol. 78, 2453–2458. doi: 10.1099/0022-1317-78-10-2453
Gupta, R., Jensen, L. J., and Brunak, S. (2002). Orphan protein function and its relation to glycosylation. Ernst. Schering. Res. Found Workshop. 38, 276–294. doi: 10.1007/978-3-662-04747-7_13
Hubálek, Z., Rudolf, I., and Nowotny, N. (2014). Arboviruses pathogenic for domestic and wild animals. Adv. Virus Res. 89, 201–275. doi: 10.1016/B978-0-12-800172-1.00005-7
Kadlec, J., Loureiro, S., Abrescia, N. G., Stuart, D. I., and Jones, I. M. (2008). The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 15, 1024–1030. doi: 10.1038/nsmb.1484
Katoh, K., Rozewicki, J., and Yamada, K. D. (2017). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Katzourakis, A., and Gifford, R. J. (2010). Endogenous viral elements in animal genomes. PLoS Genet. 6:e1001191. doi: 10.1371/journal.pgen.1001191
Kiewra, D., and Lonc, E. (2012). Epidemiological consequences of host specificity of ticks (Ixodida). Ann. Parasitol. 58, 181–187. Available online at: https://www.annals-parasitology.eu/go.live.php/download_default/D532/2012-58-4_181.pdf
Labuda, M., and Nuttall, P. A. (2004). Tick-borne viruses. Parasitology. 129, S221–45. doi: 10.1017/S0031182004005220
Lefort, V., Longueville, J. E., and Gascuel, O. (2017). SMS: smart model selection in PhyML. Mol. Biol. Evol. 34, 2422–2424. doi: 10.1093/molbev/msx149
Li, C. X., Shi, M., Tian, J. H., Lin, X. D., Kang, Y. J., Chen, L. J., et al. (2015). Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife. 4:05378. doi: 10.7554/eLife.05378
Li, D., Liu, C. M., Luo, R., Sadakane, K., and Lam, T. W. (2015). MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics. 31, 1674–1676. doi: 10.1093/bioinformatics/btv033
Li, Z., Bao, C., Hu, J., Liu, W., Wang, X., Zhang, L., et al. (2016). Ecology of the tick-borne phlebovirus causing severe fever with thrombocytopenia syndrome in an endemic area of China. PLoS Negl. Trop. Dis. 10:e0004574. doi: 10.1371/journal.pntd.0004574
Liu, Z. Y., Shi, W. F., and Qin, C. F. (2019). The evolution of Zika virus from Asia to the Americas. Nat. Rev. Microbiol. 17:131–139. doi: 10.1038/s41579-018-0134-9
Masiga, D. K., Smyth, A. J., Hayes, P., Bromidge, T. J., and Gibson, W. C. (1992). Sensitive detection of trypanosomes in tsetse flies by DNA amplification. Int. J. Parasitol. 22, 909–918. doi: 10.1016/0020-7519(92)90047-O
Matsuno, K., Weisend, C., Travassos da Rosa, A. P., Anzick, S. L., Dahlstrom, E., Porcella, S. F., et al. (2013). Characterization of the Bhanja serogroup viruses (Bunyaviridae): a novel species of the genus Phlebovirus and its relationship with other emerging tick-borne phleboviruses. J. Virol. 87, 3719–3728. doi: 10.1128/JVI.02845-12
McCauley, J. W., Hongo, S., Kaverin, N. V., Kochs, G., Lamb, R. A., Matrosovich, M. N., et al. (2012). Family Orthomyxoviridae, in Virus Taxonomy: Ninth Report of the International Committee on Taxonomy of Viruses, 1st Edn (London: Elsevier), 749–761. doi: 10.1016/B978-0-12-384684-6.00061-6
Menghani, S., Chikhale, R., Raval, A., Wadibhasme, P., and Khedekar, P. (2012). Chandipura Virus: an emerging tropical pathogen. Acta Trop. 124, 1–14. doi: 10.1016/j.actatropica.2012.06.001
Miller, M. A., Pfeiffer, W., and Schwartz, T. (2010). “Creating the CIPRES science gateway for inference of large phylogenetic trees”. in Proceedings of the Gateway Computing Environments Workshop (GCE), 14 Nov. 2010 (New Orleans, LA), 1–8. doi: 10.1109/GCE.2010.5676129
Mlera, L., and Bloom, M. E. (2018). The role of mammalian reservoir hosts in tick-borne flavivirus biology. Front. Cell Infect. Microbiol. 8:298. doi: 10.3389/fcimb.2018.00298
Morse, M. A., Marriott, A. C., and Nuttall, P. A. (1992). The glycoprotein of Thogoto virus (a tick-borne orthomyxo-like virus) is related to the baculovirus glycoprotein GP64. Virology 186, 640–646. doi: 10.1016/0042-6822(92)90030-S
Moutailler, S., Popovici, I., Devillers, E., Vayssier-Taussat, M., and Eloit, M. (2016). Diversity of viruses in Ixodes ricinus, and characterization of a neurotropic strain of Eyach virus. N. Microbes N. Infect. 11, 71–81. doi: 10.1016/j.nmni.2016.02.012
Moyer, M. W. (2015). The growing global battle against blood-sucking ticks. Nature. 524, 406–408. doi: 10.1038/524406a
Nelder, M. P., Russell, C. B., Sheehan, N. J., Sander, B., Moore, S., Li, Y., et al. (2016). Human pathogens associated with the blacklegged tick Ixodes scapularis: a systematic review. Parasit. Vectors. 9:265. doi: 10.1186/s13071-016-1529-y
Ogden, N. H., Mechai, S., and Margos, G. (2013). Changing geographic ranges of ticks and tick-borne pathogens: drivers, mechanisms and consequences for pathogen diversity. Front. Cell Infect. Microbiol. 3:46. doi: 10.3389/fcimb.2013.00046
Palacios, G., Savji, N., Travassos da Rosa, A., Guzman, H., Yu, X., Desai, A., et al. (2013). Characterization of the Uukuniemi virus group (Phlebovirus: Bunyaviridae): evidence for seven distinct species. J. Virol. 87, 3187–3195. doi: 10.1128/JVI.02719-12
Pettersson, J. H., Shi, M., Bohlin, J., Eldholm, V., Brynildsrud, O. B., Paulsen, K. M., et al. (2017). Characterizing the virome of Ixodes ricinus ticks from northern Europe. Sci. Rep. 7:10870. doi: 10.1038/s41598-017-11439-y
Pruvot, M., Kamyingkird, K., Desquesnes, M., Sarataphan, N., and Jittapalapong, S. (2010). A comparison of six primer sets for detection of Trypanosoma evansi by polymerase chain reaction in rodents and Thai livestock. Vet. Parasitol. 171, 185–193. doi: 10.1016/j.vetpar.2010.04.001
Pruvot, M., Kamyingkird, K., Desquesnes, M., Sarataphan, N., and Jittapalapong, S. (2013). The effect of the DNA preparation method on the sensitivity of PCR for the detection of Trypanosoma evansi in rodents and implications for epidemiological surveillance efforts. Vet. Parasitol. 191, 203–208. doi: 10.1016/j.vetpar.2012.09.010
Saikku, P. (1973). Arboviruses in Finland. 3. Uukuniemi virus antibodies in human, cattle, and reindeer sera. Am. J. Trop. Med. Hyg. 22, 400–403. doi: 10.4269/ajtmh.1973.22.400
Sameroff, S., Tokarz, R., Charles, R. A., Jain, K., Oleynik, A., Che, X., et al. (2019). Viral diversity of tick species parasitizing cattle and dogs in Trinidad and Tobago. Sci. Rep. 9:10421. doi: 10.1038/s41598-019-46914-1
Shi, M., Lin, X. D., Tian, J. H., Chen, L. J., Chen, X., Li, C. X., et al. (2016). Redefining the invertebrate RNA virosphere. Nature. 540, 539–543. doi: 10.1038/nature20167
Shi, M., Lin, X. D., Vasilakis, N., Tian, J. H., Li, C. X., Chen, L. J., et al. (2015). Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and Related Viruses. J. Virol. 90, 659–669. doi: 10.1128/JVI.02036-15
Siddell, S. G., Walker, P. J., Lefkowitz, E. J., Mushegian, A. R., Adams, M. J., Dutilh, B. E., et al. (2019). Additional changes to taxonomy ratified in a special vote by the International Committee on Taxonomy of Viruses. Arch. Virol. 164, 943–946. doi: 10.1007/s00705-018-04136-2
Silvas, J. A., and Aguilar, P. V. (2017). The emergence of severe fever with thrombocytopenia syndrome virus. Am. J. Trop. Med. Hyg. 97, 992–996. doi: 10.4269/ajtmh.16-0967
Souza, W. M., Fumagalli, M. J., Torres Carrasco, A. O., Romeiro, M. F., Modha, S., Seki, M. C., et al. (2018). Viral diversity of Rhipicephalus microplus parasitizing cattle in southern Brazil. Sci. Rep. 8:16315. doi: 10.1038/s41598-018-34630-1
Temmam, S., Davoust, B., Berenger, J. M., Raoult, D., and Desnues, C. (2014). Viral metagenomics on animals as a tool for the detection of zoonoses prior to human infection? Int. J. Mol. Sci. 15, 10377–10397. doi: 10.3390/ijms150610377
Temmam, S., Monteil-Bouchard, S., Robert, C., Baudoin, J. P., Sambou, M., Aubadie-Ladrix, M., et al. (2016). Characterization of viral communities of biting midges and identification of novel thogotovirus species and Rhabdovirus genus. Viruses. 8:77. doi: 10.3390/v8030077
Tin, C. M., Yuan, L., Dexter, R. J., Parra, G. I., Bui, T., Green, K. Y., et al. (2017). A Luciferase Immunoprecipitation system (LIPS) assay for profiling human norovirus antibodies. J. Virol. Methods 248, 116–129. doi: 10.1016/j.jviromet.2017.06.017
Tokarz, R., Sameroff, S., Leon, M. S., Jain, K., and Lipkin, W. I. (2014b). Genome characterization of Long Island tick rhabdovirus, a new virus identified in Amblyomma americanum ticks. Virol. J. 11:26. doi: 10.1186/1743-422X-11-26
Tokarz, R., Sameroff, S., Tagliafierro, T., Jain, K., Williams, S. H., Cucura, D. M., et al. (2018). Identification of novel viruses in Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis Ticks. mSphere. 3, e00614–e00617. doi: 10.1128/mSphere.00614-17
Tokarz, R., Williams, S. H., Sameroff, S., Sanchez-Leon, M., Jain, K., and Lipkin, W. I. (2014a). Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate viruses. J. Virol. 88, 11480–11492. doi: 10.1128/JVI.01858-14
Tomassone, L., Berriatua, E., De Sousa, R., Duscher, G. G., Mihalca, A. D., Silaghi, C., et al. (2018). Neglected vector-borne zoonoses in Europe: into the wild. Vet. Parasitol. 251, 17–26. doi: 10.1016/j.vetpar.2017.12.018
Vayssier-Taussat, M., Moutailler, S., Michelet, L., Devillers, E., Bonnet, S., Cheval, J., et al. (2013). Next generation sequencing uncovers unexpected bacterial pathogens in ticks in Western Europe. PLoS ONE. 8:e81439. doi: 10.1371/journal.pone.0081439
Walker, P. J., Firth, C., Widen, S. G., Blasdell, K. R., Guzman, H., Wood, T. G., et al. (2015). Evolution of genome size and complexity in the Rhabdoviridae. PLoS Pathog. 11:e1004664. doi: 10.1371/journal.ppat.1004664
Wolfe, N. D., Dunavan, C. P., and Diamond, J. (2007). Origins of major human infectious diseases. Nature. 447, 279–283. doi: 10.1038/nature05775
Woolhouse, M., Scott, F., Hudson, Z., Howey, R., and Chase-Topping, M. (2012). Human viruses: discovery and emergence. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2864–2871. doi: 10.1098/rstb.2011.0354
Xia, H., Hu, C., Zhang, D., Tang, S., Zhang, Z., Kou, Z., et al. (2015). Metagenomic profile of the viral communities in Rhipicephalus spp. ticks from Yunnan, China. PLoS ONE. 10:e0121609. doi: 10.1371/journal.pone.0121609
Keywords: virome, tick, emergence, spillover, LIPS
Citation: Temmam S, Chrétien D, Bigot T, Dufour E, Petres S, Desquesnes M, Devillers E, Dumarest M, Yousfi L, Jittapalapong S, Karnchanabanthoeng A, Chaisiri K, Gagnieur L, Cosson J-F, Vayssier-Taussat M, Morand S, Moutailler S and Eloit M (2019) Monitoring Silent Spillovers Before Emergence: A Pilot Study at the Tick/Human Interface in Thailand. Front. Microbiol. 10:2315. doi: 10.3389/fmicb.2019.02315
Received: 17 July 2019; Accepted: 23 September 2019;
Published: 17 October 2019.
Edited by:
Erna Geessien Kroon, Federal University of Minas Gerais, BrazilReviewed by:
Rafal Tokarz, Columbia University, United StatesNicholas Johnson, Animal and Plant Health Agency, United Kingdom
Copyright © 2019 Temmam, Chrétien, Bigot, Dufour, Petres, Desquesnes, Devillers, Dumarest, Yousfi, Jittapalapong, Karnchanabanthoeng, Chaisiri, Gagnieur, Cosson, Vayssier-Taussat, Morand, Moutailler and Eloit. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marc Eloit, bWFyYy5lbG9pdEBwYXN0ZXVyLmZy