 Cristina Merla1,2
Cristina Merla1,2 Carla Rodrigues3
Carla Rodrigues3 Virginie Passet3
Virginie Passet3 Marta Corbella1
Marta Corbella1 Harry A. Thorpe4
Harry A. Thorpe4 Teemu V. S. Kallonen5,6
Teemu V. S. Kallonen5,6 Zhiyong Zong7
Zhiyong Zong7 Piero Marone1
Piero Marone1 Claudio Bandi8,9
Claudio Bandi8,9 Davide Sassera10Jukka Corander5,6,11Edward J. Feil4
Davide Sassera10Jukka Corander5,6,11Edward J. Feil4 Sylvain Brisse3*
Sylvain Brisse3*- 1Fondazione IRCCS Policlinico San Matteo, Unità Operativa Complessa Microbiologia e Virologia, Pavia, Italy
- 2Scuola di Specializzazione in Microbiologia e Virologia, Università degli Studi di Pavia, Pavia, Italy
- 3Biodiversity and Epidemiology of Bacterial Pathogens, Institut Pasteur, Paris, France
- 4Department of Biology and Biochemistry, The Milner Centre for Evolution, University of Bath, Bath, United Kingdom
- 5Infection Genomics, Wellcome Sanger Institute, Cambridge, United Kingdom
- 6Department of Biostatistics, University of Oslo, Oslo, Norway
- 7Center of Infectious Diseases, West China Hospital, Sichuan University, Chengdu, China
- 8Department of Biosciences, University of Milan, Milan, Italy
- 9Pediatric Clinical Research Center “Romeo ed Enrica Invernizzi”, University of Milan, Milan, Italy
- 10Dipartimento di Biologia e Biotecnologie “L. Spallanzani”, Università di Pavia, Pavia, Italy
- 11Department of Mathematics and Statistics, Helsinki Institute for Information Technology HIIT, University of Helsinki, Helsinki, Finland
Klebsiella oxytoca causes opportunistic human infections and post-antibiotic haemorrhagic diarrhea. This Enterobacteriaceae species is genetically heterogeneous and is currently subdivided into seven phylogroups (Ko1 to Ko4 and Ko6 to Ko8). Here we investigated the taxonomic status of phylogroups Ko3 and Ko4. Genomic sequence-based phylogenetic analyses demonstrate that Ko3 and Ko4 formed well-defined sequence clusters related to, but distinct from, Klebsiella michiganensis (Ko1), K. oxytoca (Ko2), K. huaxiensis (Ko8), and K. grimontii (Ko6). The average nucleotide identity (ANI) of Ko3 and Ko4 were 90.7% with K. huaxiensis and 95.5% with K. grimontii, respectively. In addition, three strains of K. huaxiensis, a species so far described based on a single strain from a urinary tract infection patient in China, were isolated from cattle and human feces. Biochemical and MALDI-ToF mass spectrometry analysis allowed differentiating Ko3, Ko4, and Ko8 from the other K. oxytoca species. Based on these results, we propose the names Klebsiella spallanzanii for the Ko3 phylogroup, with SPARK_775_C1T (CIP 111695T and DSM 109531T) as type strain, and Klebsiella pasteurii for Ko4, with SPARK_836_C1T (CIP 111696T and DSM 109530T) as type strain. Strains of K. spallanzanii were isolated from human urine, cow feces, and farm surfaces, while strains of K. pasteurii were found in fecal carriage from humans, cows, and turtles.
Introduction
The genus Klebsiella, a member of the Enterobacteriaceae family, includes Gram-negative, non-motile (except K. aerogenes) and non-spore-forming capsulated bacteria. Bacteria belonging to the genus Klebsiella are found in water, soil and plants, and as commensals in the gut of animals including humans (Schmitz et al., 2002; Brisse et al., 2006; Caltagirone et al., 2017). In humans, Klebsiella species are frequently associated with hospital-acquired infections and are increasingly multidrug-resistant (Paczosa and Mecsas, 2018). Klebsiella oxytoca is the second most common Klebsiella species causing disease in humans, after K. pneumoniae (Broberg et al., 2014). K. oxytoca carries a chromosomally encoded β-lactamase gene (blaOXY) that confers resistance to amino- and carboxypenicillins (Fournier and Roy, 1997). This gene was shown to have diversified in parallel to housekeeping genes, and variants were classified into seven groups (blaOXY–1 to blaOXY–7) (Granier et al., 2003a, b; Fevre et al., 2005; Izdebski et al., 2015). K. oxytoca phylogenetic lineages were named Ko1, Ko2, Ko3, Ko4, Ko6, and Ko7 reflecting which blaOXY variant they carry; note that Ko5 was not defined, as isolates carrying blaOXY–5 represent a sublineage of Ko1 (Fevre et al., 2005). Taxonomic work has shown that K. oxytoca (sensu lato, i.e., as commonly identified in clinical microbiology laboratories) is in fact a complex of species, with K. oxytoca (sensu stricto) corresponding to phylogroup Ko2, K. michiganensis to Ko1 (Saha et al., 2013) and K. grimontii to Ko6 (Passet and Brisse, 2018). The closely related K. huaxiensis (Hu et al., 2019) represents yet another phylogroup, which we here denominate as Ko8 and which carries blaOXY–8. Phylogroups Ko3, Ko4, Ko7, and K. huaxiensis were so far described only based on a single strain (Fevre et al., 2005; Hu et al., 2019), which has limited our ability to define their genotypic and phenotypic characteristics. While analyzing a large number of Klebsiella strains from multiple human, animal and environmental sources in and around the Northern Italian town of Pavia, we identified 3 Ko3, 13 Ko4, and 3 K. huaxiensis strains. The aim of this work was to define the taxonomic status of K. oxytoca phylogroups Ko3 and Ko4 and provide identification biomarkers for all members of the K. oxytoca species complex.
Materials and Methods
Bacterial Strains
Novel strains (3 Ko3, 13 Ko4, and 3 Ko8) were isolated through enrichment in Luria-Bertani broth supplemented with 10 μg/mL of amoxicillin, followed by isolation on Simmons citrate agar with 1% inositol (SCAI) medium (Van Kregten et al., 1984) and re-isolation on MacConkey agar. Additional strains, including type and reference strains of each K. oxytoca phylogroup and the type strain of K. pneumoniae (Brisse et al., 2014) were included in the study (Table 1). Strain SG271 (internal strain bank identifier, SB3356) and SG266 (SB3355) were included as reference strains for the phylogroups Ko3 and Ko4, respectively (Fevre et al., 2005).
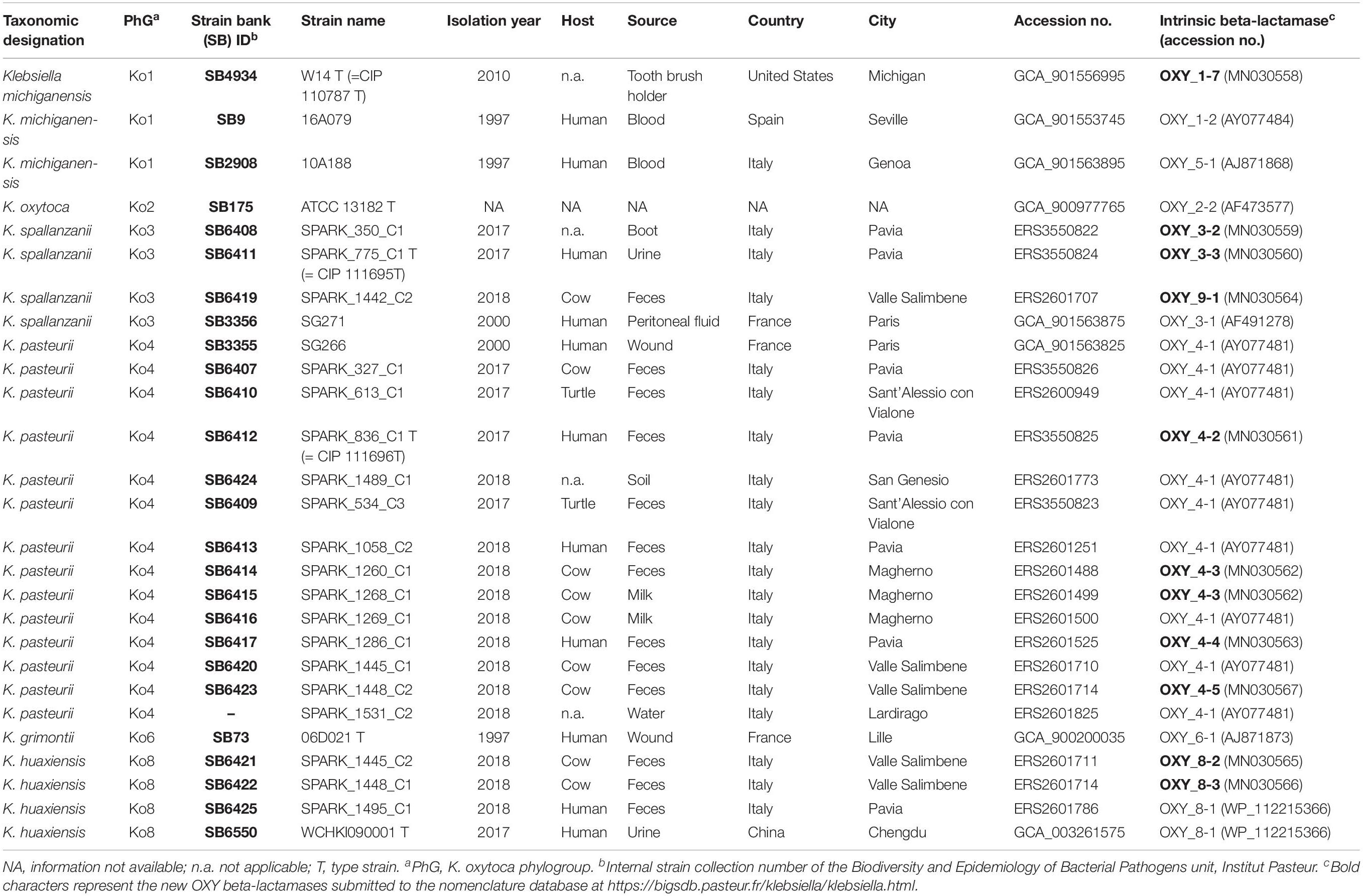
Table 1. Strains included in the study, with provenance, and genomic information.
Genome Sequencing and Analyses
Colonies from the novel strains grown on MacConkey agar were collected and resuspended in distilled water for DNA purification, which was performed using QIAsymphony automated instrument with the kit QIAsymphony DSP Virus/Pathogen following the manufacturer’s recommendation. DNA was stored at −20°C until sequencing on an Illumina HiSeq X Ten platform with a 2 × 150 nt paired-end protocol. Reads were assembled using SPAdes v3.11 and the assemblies were annotated using Prokka v1.12 (Seemann, 2014). JSpeciesWS (Richter et al., 2016) was used to calculate the average nucleotide identity (ANI) using the BLAST algorithm (ANIb), whereas in silico DNA-DNA hybridization (isDDH) was performed through GGDC tool1 (formula 2) (Meier-Kolthoff et al., 2013). Sequences of gyrA and rpoB genes were obtained from genome assemblies using BLASTN, while 16S rRNA gene sequences were obtained using Barrnap2. The chromosomal blaOXY sequences were also extracted, and the new amino-acid sequence variants were submitted to the Institut Pasteur MLST nomenclature database3 for variant number attribution, and to NCBI for accession number attribution. 16S rRNA, gyrA, rpoB, and blaOXY beta-lactamase gene sequences were aligned using Muscle (Edgar, 2004), concatenated (in the case of rpoB and gyrB) and phylogenetic relationships were assessed using MEGA v7.0 (Kumar et al., 2016). Genetic distances were inferred using the neighbor-joining method with the Jukes-Cantor correction (Jukes and Cantor, 1969) in the case of nucleotide sequences or maximum-likelihood with Jones-Taylor-Thornton (JTT) (Jones et al., 1992) model in the case of the beta-lactamase protein sequences. The genome-based phylogenetic analysis was performed on the concatenation of 3,814 core genes defined using Roary v3.12 (Page et al., 2015) with a BLASTP identity cut-off of 80% and presence in more than 90% of the isolates. K. pneumoniae ATCC 13883T (GCA_000742135.1) was used as outgroup. An approximate maximum-likelihood phylogenetic tree was inferred using FastTree v2.1 (Price et al., 2010).
Biochemical and Proteomic Analyses
A representative subset of strains (n = 30, 7 Ko1, 5 Ko2, 4 Ko3, 5 Ko4, 6 Ko6, and 3 Ko8) of phylogroups of the K. oxytoca complex was subjected to API20E (BioMérieux) and to phenotype microarray characterization using plates PM1 and PM2 (Biolog, Hayward, CA, United States) in aerobic conditions as previously described by Blin et al. (2017). The same subset of strains was also used to perform a MALDI-ToF mass spectrometry (MS) analysis following the protocol described by Rodrigues et al. (2018). Briefly, cell extracts were spotted onto an MBT Biotarget 96 target plate, air dried and overlaid with 1 μL of a saturated α-cyano-4-hydroxycinnamic acid (HCCA). Mass spectra were acquired on a Microflex LT mass spectrometer (Bruker Daltonics, Bremen, Germany) using the default parameters, preprocessed (applying smoothing and baseline subtraction) with FlexAnalysis software, and then imported and analyzed in a dedicated BioNumerics v7.6 (Applied-Maths, Belgium) database.
Results
The phylogenomic analysis based on the concatenation of 3,814 core genes (Figure 1) showed six distinct and highly supported branches. The thirteen Ko4 strains were clustered with Ko4 reference strain SG266 (SB3355) and this group was related to, but clearly distinct from, K. grimontii (Ko6). The three Ko3 strains (SPARK_350_C1, SPARK_775_C1 and SPARK_1442_C2) formed a well-defined cluster with Ko3 reference strain SG271 (SB3356, Figure 1), whereas the remaining three strains (SPARK_1445_C2, SPARK_1448_C1, SPARK_1495_C1) clustered with K. huaxiensis, which formed a distinct phylogroup that we here name Ko8. We therefore identified novel strains of these three phylogroups, which were each previously recognized based on a single strain. Furthermore, genome-based phylogeny revealed that Ko4 shares a common ancestor with K. grimontii, K. michiganensis and K. oxytoca, whereas Ko3 and K. huaxiensis share a common ancestor distinct from the Ko1/Ko4/Ko6 one (Figure 1).
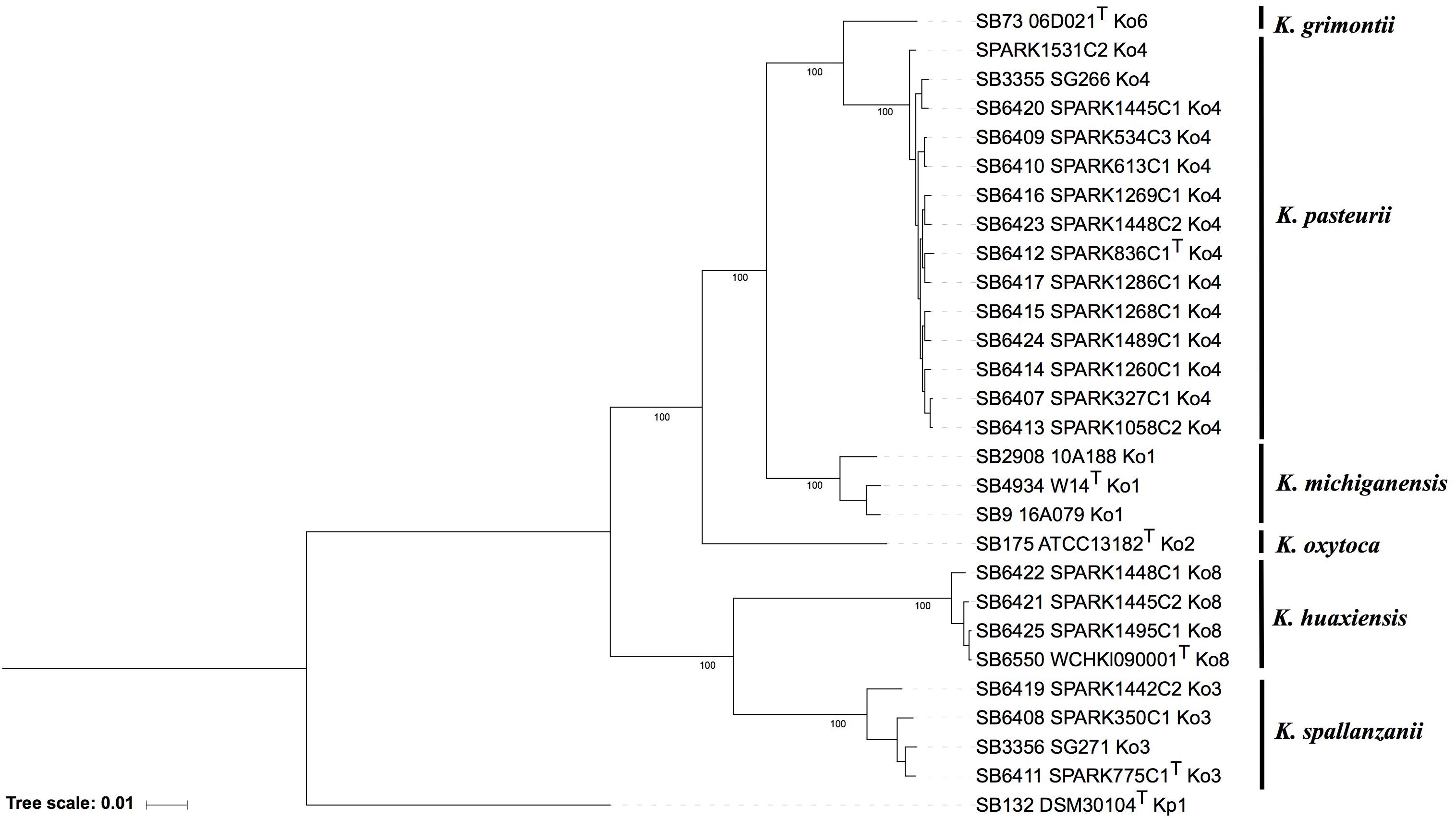
Figure 1. Maximum likelihood phylogenetic tree inferred based on the concatenated nucleotide sequence alignments of 3,814 core genes. The tree was rooted using K. pneumoniae DSM 30104T (=ATCC 13883T). Taxonomic groups are indicated in front of the branches. Branch lengths represent the number of nucleotide substitutions per site (scale, 0.01 substitution per site). Bootstrap values are indicated at major nodes. Strain labels are given as Strain Bank ID (e.g., SB73) followed by original strain name, followed by the phylogroup. A “T” after the strain name indicates that the strain is the type strain of its taxon.
To determine how previously used phylogenetic markers (Brisse and Verhoef, 2001; Granier et al., 2003a, b; Fevre et al., 2005) would group these novel strains, the sequences of internal portions of the housekeeping genes gyrA (383 nt) and rpoB (501 nt), as well as the rrs (1,454 nt) sequence coding for 16S rRNA, were extracted from genomic sequences and compared to previously characterized sequences of reference and type strains from the K. oxytoca complex (Table 1). The clustering of Ko4 strains and Ko3 strains was supported by phylogenetic analysis of combined gyrA and rpoB gene sequences (Figure 2), as well as by single gene phylogenies (Supplementary Figures S1, S2), showing that either gene used alone would allow reliable identification. The phylogeny of the chromosomal OXY beta-lactamase gene (Supplementary Figure S3) was also in concordance with previous phylogenetic analyses. However, phylogroup Ko1 and Ko3 each harbored two different types of blaOXY, coding for OXY-1/OXY-5 and OXY-3/OXY-9, respectively (Supplementary Figure S3). As previously reported (Boye and Hansen, 2003; Naum et al., 2008; Passet and Brisse, 2018), the phylogeny based on the rrs gene was not reliable for species or phylogroup identification (type strain sequences were >97.8% similar), with only a few informative variable sites (Supplementary Figure S4).
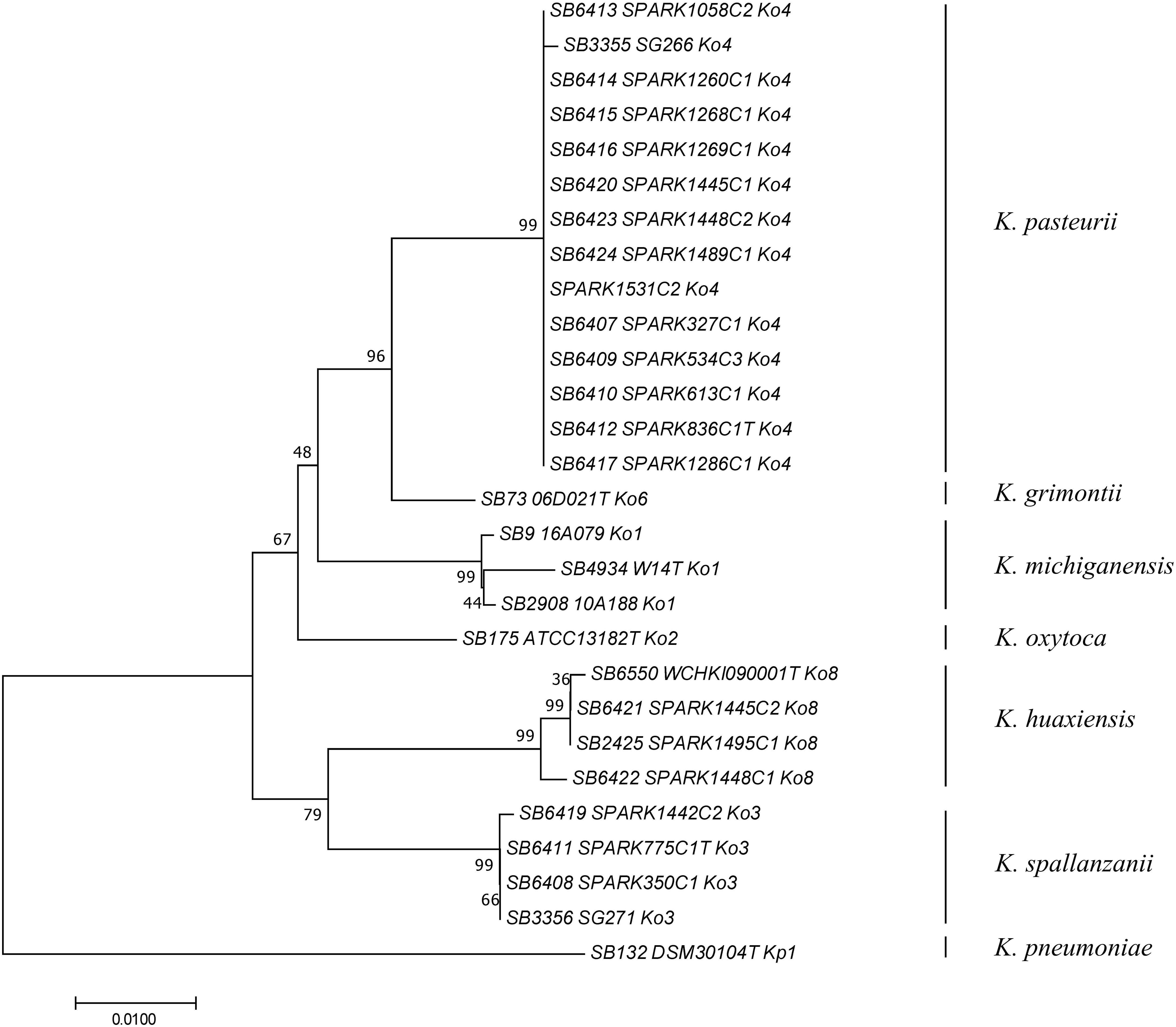
Figure 2. Phylogenetic relationships (neighbor-joining method, Jukes-Cantor correction) based on the concatenated sequences of gyrA and rpoB genes. The tree was rooted using K. pneumoniae DSM 30104T (=ATCC 13883T). Taxonomic groups are indicated in front of the branches. Bootstrap proportions obtained after 1000 replicates are indicated at the nodes. Branch lengths represent the number of nucleotide substitutions per site (scale, 0.01 substitution per site). Strain labels are given as Strain Bank ID (e.g., SB73) followed by original strain name, followed by phylogroup. A “T” after the strain name indicates that the strain is the type strain of its taxon.
Average nucleotide identity was estimated between Ko3 and Ko4, and the type strains of species of the K. oxytoca complex (Table 2). The three Ko3 strains, including SPARK_775_C1T, shared high identity (above 98%) with the Ko3 strain SG271 (SB3356) (data not shown). The ANI values of SPARK_775_C1T (Ko3) strain with K. huaxiensis (WCHKl090001T), K. michiganensis (W14T), K grimontii (06D021T) and K. oxytoca (ATCC 13182T) were 90.7, 88.4, 88.3, and 87.9%, respectively (Table 2). The novel Ko4 strains showed approximately 98% ANI with Ko4 strain SG266 (SB3355). The ANI values of SPARK_836_C1T (Ko4) with K. grimontii, K. michiganensis, K. oxytoca and K. huaxiensis were 95.5, 93.3, 90.6, and 87.1%, respectively (Table 2). Finally, the three Ko8 strains presented ANI values >99% with the type strain of K. huaxiensis (WCHKl090001T), showing that they belong to this recently described species. The isDDH relatedness range between the Ko3 and Ko4 type strains and other species was 36.3–44.1% and 34.3–67.8%, respectively. In conclusion, both ANI and isDDH values were below the thresholds proposed (Rossello-Mora and Amann, 2015) for species distinction (∼95–96% in the case of ANI, ∼70% in the case of isDDH), indicating that Ko3 and Ko4 represent two new species.
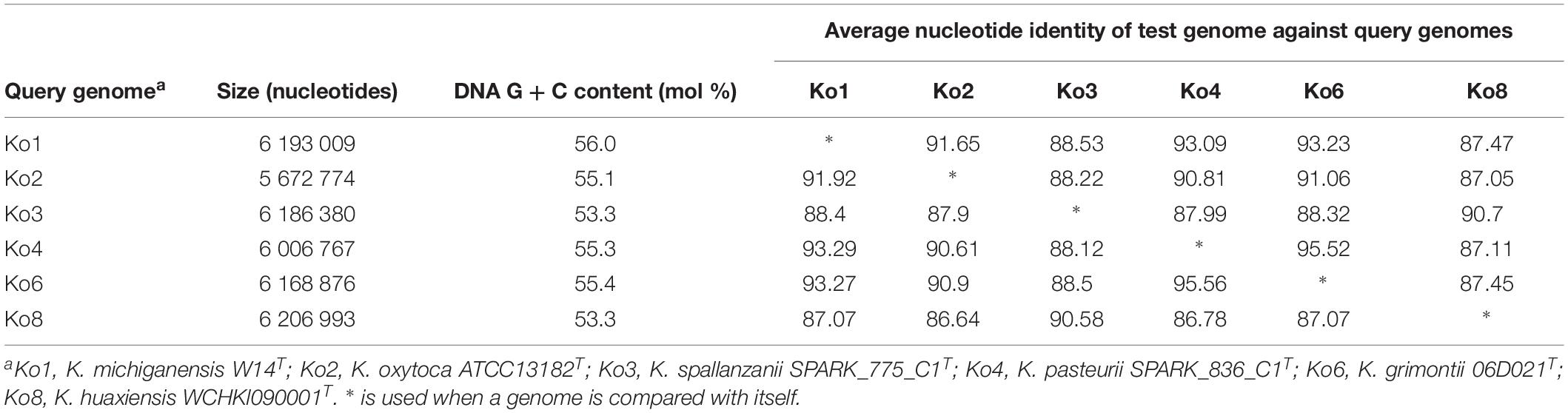
Table 2. Average nucleotide identity (ANI) values obtained among the type strains of members of the Klebsiella oxytoca complex.
The phenotypic characteristics of Ko3 and Ko4 strains were analyzed and compared with those of other Klebsiella isolates. We confirmed that all strains were non-motile by microscopy and that all isolates were positive for indole, lactose, mannitol, malonate, lysine decarboxylase, and the ONPG test, and reduced nitrate to nitrite, whereas they were all negative for ornithine decarboxylase. Ko8 and Ko3 isolates were negative for Voges–Proskauer test and Ko3 isolates were urease positive (similar to Ko2). To define further the biochemical features of the K. oxytoca phylogroups, their carbon source utilization profiles were analyzed. Among 190 substrates, several appeared useful for differentiating the phylogroups among themselves and to differentiate Ko3 and Ko4 strains from other groups (Table 3 and Supplementary Figure S5). The inability to metabolize L-proline and tricarballylic acid differentiated Ko3 strains from other phylogroups except Ko8, which can be differentiated based on its unique ability to utilize 3-O-methyl-glucose. Ko4 had a weak but unique capacity to utilize glyoxylic acid, and differed from Ko6 (K. grimontii) by its inability to metabolize D-melezitose; Ko4 was otherwise similar to Ko6 for many features, consistent with their phylogenetic association.
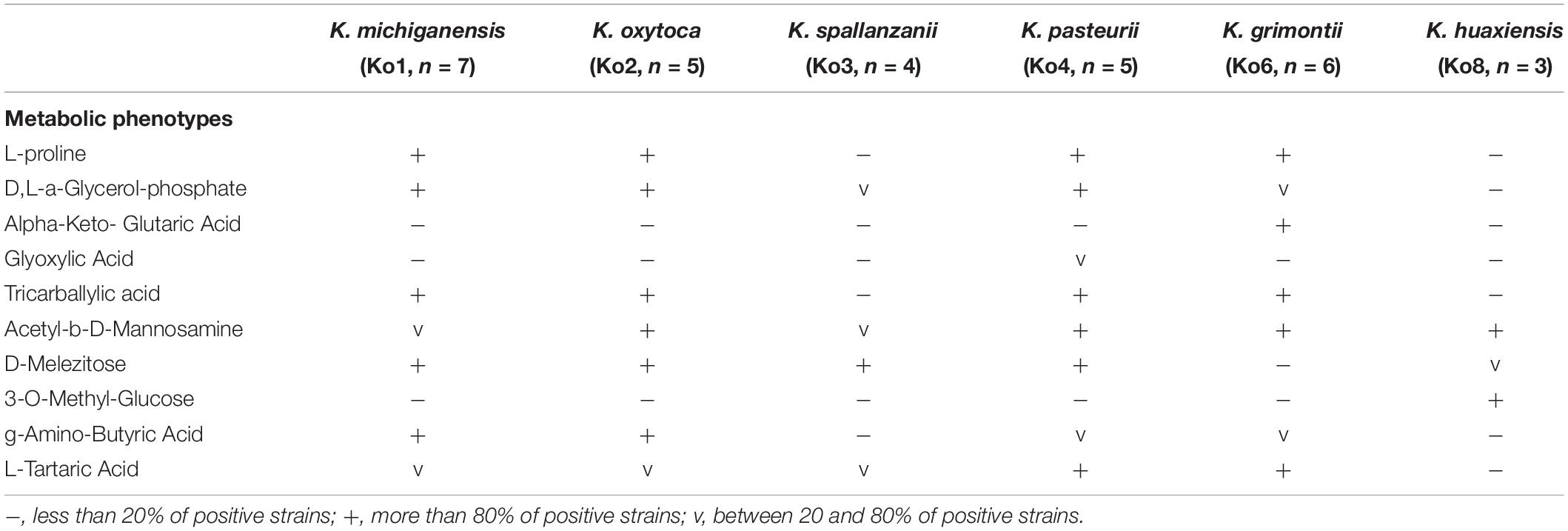
Table 3. Differential biochemical characteristics of the taxa under study.
We also analyzed the MALDI-ToF MS peak patterns of the different members of the K. oxytoca complex. Based on the MALDI Biotyper Compass database version 4.1.80 (Bruker Daltonics, Bremen, Germany), the thirty strains were identified either as K. oxytoca (23 strains, all belonging to Ko1, Ko2, Ko4, and Ko6) or as Raoultella ornithinolytica (7 strains, all strains of Ko3 and Ko8). These misidentifications can be explained by the lack of reference spectra of most phylogroups in the reference database. Supplementary Figure S6 summarizes the peak positions found in each strain. A total of 31 biomarkers (2383–10152 m/z) associated with specific members of the K. oxytoca complex were identified (Supplementary Table S1 and Supplementary Figure S6). Consistent with genetic and biochemical findings, we also observed that Ko4 shared most of its spectral peaks with Ko1 and Ko6, presenting only one specific peak (which was variably present) at 3681 m/z, whereas Ko3 shared six peaks with only Ko8 and presented two unique peaks at 5178 and 6795 m/z. For the remaining phylogroups, specific peaks were observed for Ko2 and Ko8, whereas Ko1 and Ko6 could be identified by specific peak combinations. Based on the current dataset, the specificity and sensitivity of their distribution among phylogroups ranged between 60–100% and 80–100%, respectively (Supplementary Table S1). This finding paves the way to identify isolates of the K. oxytoca complex at the species (or phylogroup) level based on MALDI-ToF MS analysis, pending incorporation of reference spectra of the various taxa into reference spectra databases.
Based on the above genomic, phenotypic and proteomic characteristics, we propose Ko3 and Ko4 to be considered as two novel species, which we propose to name K. spallanzanii and K. pasteurii, respectively.
Description of Klebsiella spallanzanii sp. nov.
Klebsiella spallanzanii (spal. lan.za ‘ni.i N. L. gen. n. referring to Lazzaro Spallanzani, Italian biologist, important contributor to the experimental study of bodily functions and of animal reproduction. He provided what is considered the first disproval of the theory of the spontaneous generation of microbes).
The description is based on 4 strains. Cells are Gram-negative, non-motile, non-spore-forming, straight, rod-shaped and capsulated. Colonies are smooth, circular, white, dome-shaped, and glistening. The general characteristics are as described for the genus Klebsiella. Indole-positive, ONPG-positive, lysine decarboxylase positive and ornithine decarboxylase negative. Differentiated from the other species of the K. oxytoca complex by the urease-positive (similar to Ko2) and Voges–Proskauer test negative (also negative for Ko8). Distinguished from the other members of K. oxytoca complex also by the characteristics listed in Table 3. Distinguishable from K. huaxiensis by the ability to use D-melezitose and the inability to ferment 3-O-methyl-glucose, and from the remaining K. oxytoca members by the inability to use L-proline. K. spallanzanii isolates were recovered from human urine and cow feces.
The type strain is strain SPARK_775_C1T (=SB6411, CIP 111695T, DSM 109531T), isolated in 2017 from the urine of a patient in Pavia, Italy. The INSDC (GenBank/ENA/DDBJ) accession numbers of the gyrA, rpoB and rrs (coding for 16S rRNA) genes are MN076620, MN076626, and MN091365, respectively. The genome sequence accession number is ERS3550824. The DNA G + C content of the type strain is 53.3%.
Description of Klebsiella pasteurii sp. nov.
Klebsiella pasteurii (pas. teu ‘ri.i N. L. gen. n. referring to Louis Pasteur, a French microbiologist, who made seminal contributions to microbiology and infectious diseases, vaccination and pasteurization. He contributed decisively to disprove the theory of the spontaneous generation of microbes).
The description is based on 14 strains. Cells are Gram-negative, non-motile, non-spore-forming, straight, rod-shaped and capsulated. Colonies are smooth, circular, white, dome-shaped, and glistening. The general characteristics are as described for the genus Klebsiella. Indole-positive, urease-negative, ONPG-positive, Voges–Proskauer test positive, lysine decarboxylase positive, and ornithine decarboxylase negative. They can be distinguished from the other members of K. oxytoca complex by the characteristics listed in Table 3. They are distinguishable from K. grimontii by the ability to ferment D-melezitose and inability to ferment alpha-keto-glutaric acid, and from the remaining K. oxytoca groups by the unique weak ability to ferment glyoxylic acid. K. pasteurii isolates were recovered from feces of cows, turtles and humans.
The type strain is strain SPARK_836_C1T (=SB6412, CIP 111696T, and DSM 109530), isolated in 2017 from the feces of a patient in Pavia, Italy. The INSDC (GenBank/ENA/DDBJ) accession numbers of the gyrA, rpoB, and rrs (coding for 16S rRNA) genes are MN076619, MN076625 and MN091366, respectively. The genome sequence accession number is ERS3550825. The DNA G + C content of the type strain is 55.3%.
Data Availability Statement
The nucleotide sequences generated in this study were deposited in European Nucleotide Archive (ENA) and are available through the INSDC databases under accession numbers MN091365 (SB6411T = SPARK775C1T), MN091366 (SB6412T = SPARK836C1T), MN104661 to MN104677 (16S rRNA), MN076606 to MN076643 (gyrA and rpoB), and MN030558 to MN030567 (blaOXY). Complete genomic sequences were submitted to European Nucleotide Archive under the BioProject number PRJEB15325.
Ethics Statement
The approval of Ethical Committee of the San Matteo Hospital in Pavia was granted under number 20170001787 in date 25/05/2017, proceeding number is 2017000759. The internal code of the project is 0890170117. The ethical procedure includes written informed consent from all the patients participating in the study.
Author Contributions
CM, MC, PM, CB, and DS isolated Klebsiella from diverse sources. CM, CR, VP, and MC performed the microbiological characterization of isolates. CM, HT, TK, and DS performed the genomic sequencing. CM, CR, HT, and TK analyzed the sequence data. CR and VP performed the MALDI-TOF analyses. VP and SB performed the phenotypic microarray analyses. CM, CR, and SB wrote the initial version of the manuscript. All authors revised the manuscript. EF, SB, JC, CB, and DS acquired funding for this study.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Funding
This work was funded by the SpARK project “The rates and routes of transmission of multidrug resistant Klebsiella clones and genes into the clinic from environmental sources,” which has received funding under the 2016 JPI-AMR call “Transmission Dynamics” (MRC reference MR/R00241X/1); and by the French Government’s Investissement d’Avenir program Laboratoire d’Excellence “Integrative Biology of Emerging Infectious Diseases” (ANR-10-LABX-62-IBEID). CR was financially supported by the MedVetKlebs project, a component of European Joint Programme One Health EP, which has received funding from the European Union’s Horizon 2020 Research and Innovation Programme under Grant Agreement No. 773830. JC was funded by the ERC grant no. 742158 and the Norwegian Research Council JPIAMR grant no. 144501.
Acknowledgments
We acknowledge Marie-Hélène Nicolas-Chanoine and Alan McNally for providing strains included in this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02360/full#supplementary-material
FIGURE S1 | Phylogenetic relationships (neighbor-joining method, Jukes-Cantor correction) based on the aligned sequences of gyrA gene. The tree was rooted using K. pneumoniae DSM 30104T (=ATCC 13883T). Taxonomic groups are indicated in front of the branches. Bootstrap proportions obtained after 1000 replicates are indicated at the nodes. Branch lengths represent the number of nucleotide substitutions per site (scale, 0.01 substitution per site). Strain labels are given as Strain Bank ID (e.g., SB73) followed by original strain name, followed by phylogroup. A “T” after the strain name indicates that the strain is the type strain of its taxon.
FIGURE S2 | Phylogenetic relationships (neighbor-joining method, Jukes-Cantor correction) based on the aligned sequences of rpoB gene. The tree was rooted using K. pneumoniae DSM 30104T (=ATCC 13883T). Taxonomic groups are indicated in front of the branches. Bootstrap proportions obtained after 1000 replicates are indicated at the nodes. Branch lengths represent the number of nucleotide substitutions per site (scale, 0.05 substitution per site). Strain labels are given as Strain Bank ID (e.g., SB73) followed by original strain name, followed by phylogroup. A “T” after the strain name indicates that the strain is the type strain of its taxon.
FIGURE S3 | Phylogenetic tree based on chromosomal OXY beta-lactamase amino-acid sequences (maximum-likelihood method, Jones-Taylor-Thornton model). Branch lengths represent the number amino-acid substitutions per site (scale, 0.01 substitution per site). Taxonomic groups are indicated in front of the branches. Strain labels are given as Strain Bank ID (e.g., SB73) followed by original strain name, phylogroup, and the OXY beta-lactamase variant. A “T” after the strain name indicates type strains. Node labels indicate bootstrap values based on 1000 replicates.
FIGURE S4 | Phylogenetic relationships (neighbor-joining method, Jukes-Cantor correction) based on the sequence of the rrs gene coding for 16S rRNA, and the respective multiple sequence alignment restricted to the variable positions. The tree was rooted using K. pneumoniae DSM 30104T (=ATCC 13883T). Bootstrap proportions obtained after 1000 replicates are indicated at the nodes. Branch lengths represent the number of nucleotide substitutions per site (scale, 0.001 substitution per site). Strain labels are given as Strain Bank ID (e.g., SB73) followed by original strain name, followed by phylogroup. A “T” after the strain name indicates that the strain is the type strain of its taxon.
FIGURE S5 | Metabolic phenotypes for the most discriminant carbon sources among members of the K. oxytoca complex. Black squares correspond to substrate utilization; white square to absence of utilization; and gray squares to intermediate values.
FIGURE S6 | Peak positions (m/z) observed for each strain. Stars denote the peaks that are useful for discrimination among phylogroups, as detailed in Supplementary Table S1. 1Strains added for MALDI-ToF experiments.
TABLE S1 | MALDI-ToF mass spectrometry peaks, which are useful biomarkers to discriminate phylogroups of the Klebsiella oxytoca species complex. CI, confidence interval. 1Position in the spectra using. 2Double-charged ion.
Abbreviations
ANI, average nucleotide identity; HCCA, a-cyano -4-hydroxycinnamic acid; isDDH, in silico DNA-DNA hybridization; SCAI, simmons citrate agar with inositol; MALDI-ToF MS, matrix-assisted laser desorption/ionization time of flight mass spectrometry.
Footnotes
References
Blin, C., Passet, V., Touchon, M., Rocha, E. P. C., and Brisse, S. (2017). Metabolic diversity of the emerging pathogenic lineages of Klebsiella pneumoniae. Environ. Microbiol. 19, 1881–1898. doi: 10.1111/1462-2920.13689
Boye, K., and Hansen, D. S. (2003). Sequencing of 16S rDNA of Klebsiella: taxonomic relations within the genus and to other Enterobacteriaceae. Int. J. Med. Microbiol. 292, 495–503. doi: 10.1078/1438-4221-00228
Brisse, S., Grimont, F., and Grimont, P. A. D. (2006). “The genus Klebsiella,” in The Prokaryotes-A Handbook on the Biology of Bacteria, eds M. Dworkin, S. Falkow, E. Rosenberg, K.-H. Schleifer, and E. Stackebrandt, (New York, NY: Springer), 159–197.
Brisse, S., Passet, V., and Grimont, P. A. (2014). Description of Klebsiella quasipneumoniae sp. nov., isolated from human infections, with two subspecies, Klebsiella quasipneumoniae subsp. Quasipneumoniae subsp. nov. and Klebsiella quasipneumoniae subsp. similipneumoniae subsp. nov., and demonstration that Klebsiella singaporensis is a junior heterotypic synonym of Klebsiella variicola. Int. J. Syst. Evol. Microbiol. 64, 3146–3152. doi: 10.1099/ijs.0.062737-0
Brisse, S., and Verhoef, J. (2001). Phylogenetic diversity of Klebsiella pneumoniae and Klebsiella oxytoca clinical isolates revealed by randomly amplified polymorphic DNA, gyrA and parC genes sequencing and automated ribotyping. Int. J. Syst. Evol. Microbiol. 51, 915–924. doi: 10.1099/00207713-51-3-915
Broberg, C. A., Palacios, M., and Miller, V. L. (2014). Klebsiella: a long way to go towards understanding this enigmatic jet-setter. F1000 Prime Rep. 6:64. doi: 10.12703/P6-64
Caltagirone, M., Nucleo, E., Spalla, M., Zara, F., Novazzi, F., Marchetti, V. M., et al. (2017). Occurrence of extended spectrum β-Lactamases, KPC-Type, and MCR-1.2-producing enterobacteriaceae from wells, river water, and wastewater treatment plants in Oltrepò Pavese Area, Northern Italy. Front. Microbiol. 8:2232. doi: 10.3389/fmicb.2017.02232
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fevre, C., Jbel, M., Passet, V., Weill, F. X., Grimont, P. A., and Brisse, S. (2005). Six groups of the OXY b-lactamase evolved over millions of years in Klebsiella oxytoca. Antimicrob. Agents Chemother. 49, 3453–3462. doi: 10.1128/AAC.49.8.3453-3462.2005
Fournier, B., and Roy, P. H. (1997). Variability of chromosomally encoded b-lactamases from Klebsiella oxytoca. Antimicrob. Agents Chemother. 41, 1641–1648. doi: 10.1128/aac.41.8.1641
Granier, S. A., Leflon-Guibout, V., Goldstein, F. W., and Nicolas-Chanoine, M. H. (2003a). New Klebsiella oxytoca beta-lactamase genes bla(OXY-3) and bla(OXY-4) and a third genetic group of K oxytoca based on bla (OXY-3). Antimicrob. Agents Chemother. 47, 2922–2928. doi: 10.1128/AAC.47.9.2922-2928.2003
Granier, S. A., Plaisance, L., Leflon-Guibout, V., Lagier, E., Morand, S., Goldstein, F. W., et al. (2003b). Recognition of two genetic groups in the Klebsiella oxytoca taxon on the basis of chromosomal b-lactamase and housekeeping gene sequences as well as ERIC-1 R PCR typing. Int. J. Syst. Evol. Microbiol. 53, 661–668. doi: 10.1099/ijs.0.02408-0
Hu, Y., Wei, L., Feng, Y., Xie, Y., and Zong, Z. (2019). Klebsiella huaxiensis sp. nov., recovered from human urine. Int. J. Syst. Evol. Microbiol. 69, 333–336. doi: 10.1099/ijsem.0.003102
Izdebski, R., Fiett, J., Urbanowicz, P., Baraniak, A., Derde, L. P., Bonten, M. J., et al. (2015). Phylogenetic lineages, clones and β-lactamases in an international collection of Klebsiella oxytoca isolates non susceptible to expanded-spectrum cephalosporins. J. Antimicrob. Chemother. 70, 3230–3237. doi: 10.1093/jac/dkv273
Jones, D. T., Taylor, W. R., and Thornton, J. M. (1992). The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 8, 275–282. doi: 10.1093/bioinformatics/8.3.275
Jukes, T. H., and Cantor, C. R. (1969). “Evolution of protein molecules,” in Mammalian Protein Metabolism, ed. H. N. Munro, (New York, NY: Academic Press), 21–132. doi: 10.1016/b978-1-4832-3211-9.50009-7
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Meier-Kolthoff, J. P., Auch, A. F., Klenk, H. P., and Göker, M. (2013). Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinformatics 14:60. doi: 10.1186/1471-2105-14-60
Naum, M., Brown, E. W., and Mason-Gamer, R. J. (2008). Is 16S rDNA a reliable phylogenetic marker to characterize relationships below the family level in the Enterobacteriaceae? J. Mol. Evol. 66, 630–642. doi: 10.1007/s00239-008-9115-3
Paczosa, M. K., and Mecsas, J. (2018). Klebsiella pneumoniae: going on the offense with a strong defense. Microbiol. Mol. Biol. Rev. 80, 629–661. doi: 10.1128/MMBR.00078-15
Page, A. J., Cummins, C. A., Hunt, M., Wong, V. K., Reuter, S., Holden, M. T. G., et al. (2015). Roary: rapid large-scale prokaryote pan genome analysis. Bioinformatics 31, 3691–3693. doi: 10.1093/bioinformatics/btv421
Passet, V., and Brisse, S. (2018). Description of Klebsiella grimontii sp. nov. Int. J. Syst. Evol. Microbiol. 68, 377–381. doi: 10.1099/ijsem.0.00251
Price, M. N., Dehal, P. S., and Arkin, A. P. (2010). FastTree 2 - approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. doi: 10.1371/journal.pone.0009490
Richter, M., Rosselló-Móra, R., Glöckner, F. O., and Peplies, J. (2016). JSpeciesWS: a web server for prokaryotic species circumscription based on pairwise genome comparison. Bioinformatics 32, 929–931. doi: 10.1093/bioinformatics/btv681
Rodrigues, C., Passet, V., Rakotondrasoa, A., and Brisse, S. (2018). Identification of Klebsiella pneumoniae, Klebsiella quasipneumoniae, Klebsiella variicola and related phylogroups by MALDI-TOF mass spectrometry. Front. Microbiol. 9:3000. doi: 10.3389/fmicb.2018.03000
Rossello-Mora, R., and Amann, R. (2015). Past and future species definitions for bacteria and archaea. Syst. Appl. Microbiol. 38, 209–216. doi: 10.1016/j.syapm.2015.02.001
Saha, R., Farrance, C. E., Verghese, B., Hong, S., and Donofrio, R. S. (2013). Klebsiella michiganensis sp. nov., a new bacterium isolated from a toothbrush holder. Curr. Microbiol. 66, 72–78. doi: 10.1007/s00284-012-0245-x
Schmitz, R. A., Klopprogge, K., and Grabbe, R. (2002). Regulation of nitrogen fixation in Klebsiella pneumoniae and Azotobacter vinelandii: NifL, transducing two environmental signals to the nif transcriptional activator NifA. J. Mol. Microbiol. Biotechnol. 4, 235–242.
Seemann, T. (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. doi: 10.1093/bioinformatics/btu153
Keywords: Klebsiella oxytoca complex, phylogeny, taxonomy, genome sequencing, blaOXY, MALDI-ToF mass spectrometry
Citation: Merla C, Rodrigues C, Passet V, Corbella M, Thorpe HA, Kallonen TVS, Zong Z, Marone P, Bandi C, Sassera D, Corander J, Feil EJ and Brisse S (2019) Description of Klebsiella spallanzanii sp. nov. and of Klebsiella pasteurii sp. nov. Front. Microbiol. 10:2360. doi: 10.3389/fmicb.2019.02360
Received: 05 July 2019; Accepted: 27 September 2019;
Published: 25 October 2019.
Edited by:
Andrés Moya, University of Valencia, SpainReviewed by:
Valeria Souza, National Autonomous University of Mexico, MexicoMaher Gtari, Carthage University, Tunisia
Copyright © 2019 Merla, Rodrigues, Passet, Corbella, Thorpe, Kallonen, Zong, Marone, Bandi, Sassera, Corander, Feil and Brisse. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sylvain Brisse, c3lsdmFpbi5icmlzc2VAcGFzdGV1ci5mcg==