 Marine Feyereisen1*
Marine Feyereisen1* Jennifer Mahony1,2
Jennifer Mahony1,2 Horst Neve3
Horst Neve3 Charles M. A. P. Franz3
Charles M. A. P. Franz3 Jean-Paul Noben4
Jean-Paul Noben4 Tadhg O’Sullivan5Viktor Boer5
Tadhg O’Sullivan5Viktor Boer5 Douwe van Sinderen1,2*
Douwe van Sinderen1,2*- 1School of Microbiology, University College Cork, Cork, Ireland
- 2APC Microbiome Ireland, University College Cork, Cork, Ireland
- 3Department Microbiology and Biotechnology, Federal Research Centre of Nutrition and Food, Max Rubner-Institut, Kiel, Germany
- 4Department Physiology Biochemistry and Immunology, Biomedical Research Institute, Hasselt University, Diepenbeek, Belgium
- 5HEINEKEN Global Innovation and Research, Heineken Supply Chain B.V, Zoeterwoude, Netherlands
Lactobacillus brevis is a lactic acid bacterium that is known as a food and beverage spoilage organism, and more specifically as a beer-spoiler. Phages of L. brevis have been described, but very limited data is available regarding temperate phages of L. brevis. Temperate phages may exert benefits to the host, while they may also be employed to combat beer spoilage. The current study reports on the incidence of prophage sequences present in nineteen distinct L. brevis genomes. Prophage induction was evaluated using mitomycin C exposure followed by genome targeted-PCR, electron microscopy and structural proteome analysis. The morphological and genome sequence analyses revealed significant diversity among L. brevis prophages, which appear to be dominated by members of the Myoviridae phage family. Based on this analysis, we propose a classification of L. brevis phages into five groups.
Introduction
The most prevalent spoilage bacteria associated with beer fermentations are members of the lactic acid bacteria (LAB), which account for approximately 70% of all microbial spoilage incidents (Vaughan et al., 2005; Bokulich and Bamforth, 2013; Garofalo et al., 2015). Lactobacillus brevis strains are frequently reported to be the cause of such spoilage events as they have developed mechanisms to survive and grow in beer (Suzuki et al., 2006). Strains of this species can be found on raw materials used in breweries and represent a major microbial contaminant during the production and storage of beer. Bacterial strains have acquired features throughout evolution allowing them to become more robust including resistance to virulent bacteriophages (Forde and Fitzgerald, 1999; Brüssow, 2001; Mahony et al., 2016b). Moreover, the chromosomes of the majority of LAB are known to harbor one or more prophage regions and their presence may benefit the host by providing resistance attributes to its environment (Canchaya et al., 2003). Upon induction, these phages enter the lytic cycle leading to bacterial cell death and formation of intact phage particles (Davidson et al., 1990), and therefore it is relevant to examine their presence and functionality (Kelleher et al., 2018). Prophages of LAB have been widely studied and particularly so in Lactococcus lactis (Kelleher et al., 2018) and Streptococcus thermophilus (Husson-Kao et al., 2000). Temperate phages of the genus Lactobacillus have been sequenced and are primarily classified as members of the Siphoviridae family (Villion and Moineau, 2009), being characterized by a non-contractile tail (Forsman, 1993). Successful induction of such prophages has been reported using UV, thermal exposure or treatment with DNA damaging/antimicrobial compounds such as mitomycin C and bacteriocins (Chopin et al., 1989, 2001; Meijer et al., 1998; Madera et al., 2009).
While the presence of prophages in LAB genomes is widely described, only a small number of studies have investigated their integrity and inducibility within these strains (Mittler, 1996; Lunde et al., 2003). In the case of beer spoilage by L. brevis, prophage induction represents a beneficial attribute that could be harnessed as a natural alternative to chemical compounds in the eradication of spoilage bacteria. Indeed, induction of prophages is expected to cause bacterial cell lysis thus avoiding the development of spoilage organisms in the brewing industry. Although prophages represent a reservoir for adaptation, prophages of L. brevis are currently poorly described. To date, the genome of a single temperate phage of L. brevis, namely LBR48, has been sequenced and characterized. The prophage was induced from L. brevis strain C30 using mitomycin C. LBR48 is 48 Kb long containing 90 putative open reading frames (ORFs) and was classified as a member of the Myoviridae family. Interestingly, the LBR48 genome does not show protein sequence similarity with any other Lactobacillus phages (Jang et al., 2011).
Advances in genome sequencing technologies have considerably increased the number of available bacterial genome sequences, improving also the detection capability of prophage-encoded regions within these genomes. In the current study, nineteen publically available L. brevis genome sequences were used to identify prophage-encoding regions. Five of these strains were available for prophage induction trials, allowing the assessment of the ability of these temperate phages to form intact phage particles and cause host cell lysis. The diversity of L. brevis temperate phages was studied to establish relatedness between L. brevis phages (temperate and virulent), resulting in a proposed classification scheme of L. brevis phages based on morphology and genome sequence data.
Materials and Methods
Bacterial Strains and Growth Conditions
Lactobacillus brevis strains used in this study are detailed in Table 1. Bacteria were cultured in MRS broth (Oxoid Ltd., Hampshire, United Kingdom) at 30°C.
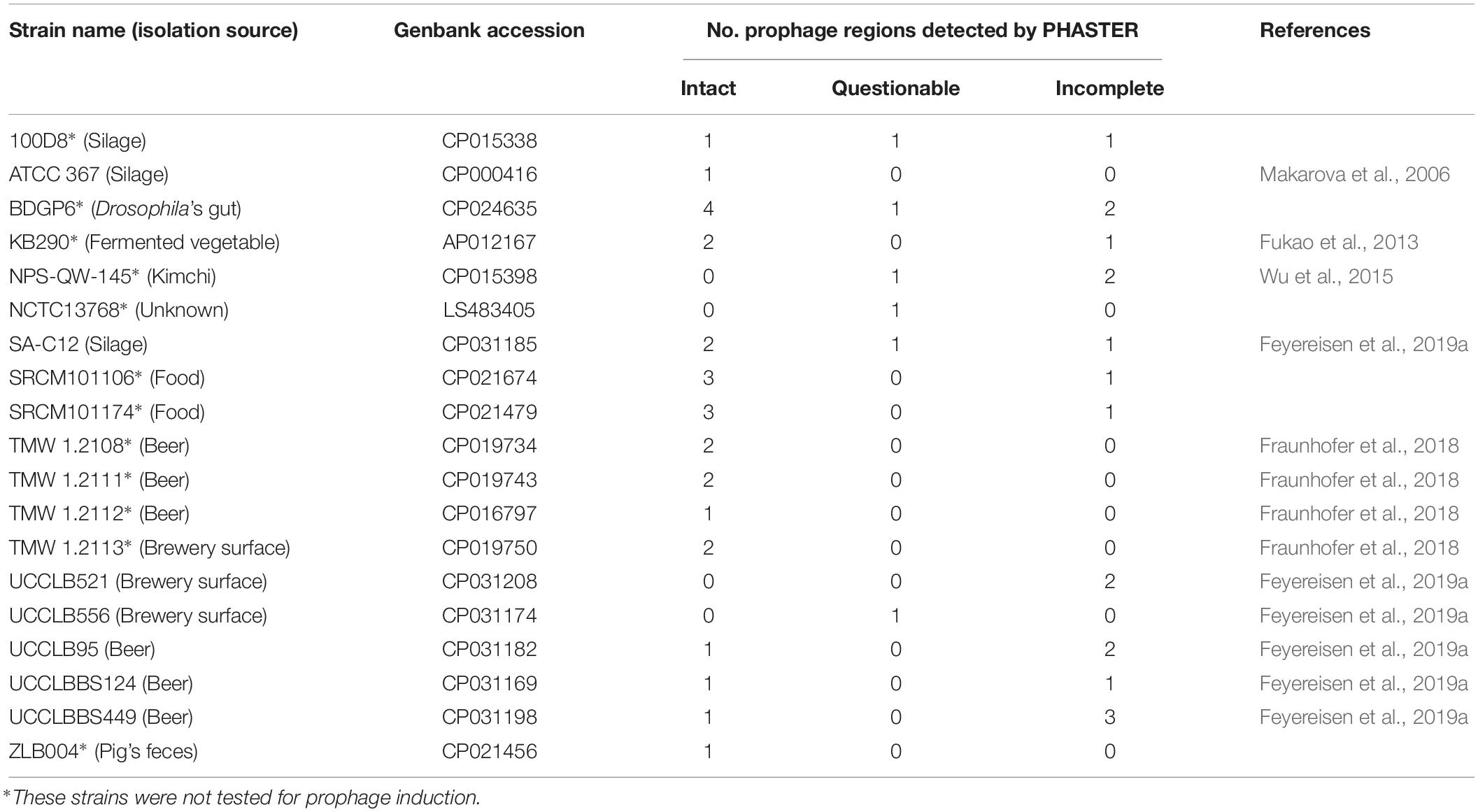
Table 1. Lactobacillus brevis strains used in this study and prophage regions predicted by PHASTER.
Prophage Identification and Genome Annotation
PHAge search tool enhanced release (PHASTER) (Zhou et al., 2011; Arndt et al., 2016) was used to screen for prophage-specifying DNA regions within the genome of available L. brevis strains. Intact prophages were manually annotated to confirm the presence of all expected genes required to produce a fully functional phage particle including genes encoding proteins associated with replication functions (e.g., replisome and DNA-binding proteins) packaging (small and large terminases), morphogenesis (e.g., capsid and tail) and lysis (holin and lysin). Genes required for lysogeny maintenance (e.g., integrase and repressor) were also investigated (Kelleher et al., 2018). Integration sites of prophages (attL and attR) were recorded and are presented in Supplementary Table S1. Prophage genome sequences were retrieved and annotated as previously described (Kelleher et al., 2018). Briefly, ORF prediction was performed using the Prodigal prediction software (Hyatt et al., 2010) and confirmed using BLASTX alignments (Altschul et al., 1990). The automatic annotations were refined using Artemis v16.0.0 to allow visual inspection of ORF predictions (Rutherford et al., 2000). Moreover, BLASTP (Altschul et al., 1997) and HHPred (Söding et al., 2005) analyses were performed to assign functional annotations to the predicted ORFs. Transfer RNA (tRNA) genes were predicted using tRNA-scan-SE v2.0 (Schattner et al., 2005) and added manually using Artemis.
Phylogenetic Analysis
A proteomic tree was constructed using a concatenated amino acid sequence of all encoded proteins for each of the L. brevis phages sequenced to date. The concatenated amino acid sequence begins with the ORF encoding the small terminase subunit (TerS) (Casey et al., 2015). The concatenated sequences were aligned using ClustalW (Thompson et al., 2002). The phylogenetic tree was constructed using the neighbor-joining method and bootstrapped employing 1,000 replicates. The final tree was visualized using MEGA7 (Kumar et al., 2008).
Genome Characterization and Organization
Following phylogenetic analysis, genomes of representative temperate phages were selected for further analysis where overall genome content and organization were studied. The genome content and architecture were analyzed based on the observation of the modular organization of the genomes into the following modules: packaging, morphogenesis, lysis, lysogeny, and replication. Protein sequences of representative phages were compared using all-against-all, bi-directional BLAST alignments (Altschul et al., 1990). An alignment cut-off E-value of 0.0001, and a similarity cut-off level of at least 30% amino acid identity across 80% of the sequence length was applied. This analysis allowed the amino acid similarity assignment between temperate phage genomes and the study of the overall genome similarity/diversity among L. brevis phages.
Prophage Induction Trials
To assess the functionality of the identified prophage-encoding regions, prophage induction trials were performed using the DNA crosslinking agent mitomycin C (MitC). For this assay, five L. brevis strains were available for testing: ATCC367, SA-C12, UCCLB95, UCCLBBS124, and UCCLBBS449. 10 mL MRS broth was inoculated with 2% of a fresh overnight culture of the relevant bacterial strain. Cultures were incubated at 30°C until an OD600nm of 0.1, 0.2, or 0.3 was reached at which point 0.1, 0.2, or 0.3 μg/mL MitC (final concentration) was added. A high concentration of 2 μg/mL MitC (final concentration) was applied when cells reached an OD600nm of 0.2. This was performed to ascertain if cell lysis occurred due to prophage induction or lethal MitC toxicity. Indeed, MitC levels of between 0.1 and 0.3 μg/mL are relatively low and when induction occurs it would be considered genuine prophage-induction mediated cell lysis. Conversely, higher concentrations of MitC (e.g., 2 μg/mL MitC) are expected to cause growth arrest and cell death due to acute toxicity. Cultures were maintained at room temperature for 30 h during which the OD600nm was recorded at 60 min time intervals for the first 8 h and then at 15, 20, 25, and 30 h.
Using the same protocol as MitC prophage induction, potential induction regimes using other stress-inducing chemicals (2% v/v NaOH, 2% v/v formic acid or 2% v/v acetic acid) or physical treatment (direct UV light exposure (254 nm) of 30 min at a distance of 5 cm on a 1 cm height culture suspension) that resemble circumstances in the brewing industry for cleaning and sanitizing purposes were also assessed (Schmidt, 1997; Loeffler, 2006).
Validation of Prophage Induction by DNA Sequencing and Electron Microscopy
To validate prophage induction from L. brevis strains cited above, the DNA derived from cell-free supernatants was extracted and sequenced. Phage DNA was isolated using a previously described phage DNA extraction protocol (Lavelle et al., 2018). Primers were designed based on prophage sequences in order to confirm the presence of induced prophages in the cell-free supernatants of MitC-treated cultures (Table 2).

Table 2. Primer sequences used to amplify specific regions of induced prophages.
To validate prophage induction using electron microscopy, MitC-treated cultures were harvested by centrifugation at 5000 × g for 10 min, after which the supernatant was filtered twice through a 0.45 μm filter prior to electron microscopy analysis. Transmission electron microscopy of the samples was performed as previously described (Casey et al., 2015). Negative staining was performed using 2% (w/v) uranyl acetate on freshly prepared ultrathin carbon films. Grids were analyzed in a Tecnai 10 transmission electron microscope (FEI Thermo Fisher Scientific, Eindhoven, Netherlands) at an acceleration voltage of 80 kV. Micrographs were taken with a MegaView G2 charge-coupled device camera (Emsis, Muenster, Germany).
Phage Structural Proteome and Mass-Spectrometry
An aliquot (30 μL) of CsCl-purified phage sample was mixed with 10 μL of SDS loading buffer containing 50 mM β-mercaptoethanol. The structural protein profile was generated by standard Tris-glycine sodium dodecyl sulfate (SDS)–12% polyacrylamide gel electrophoresis (PAGE). Gel slices were then excised, trypsinized, and analyzed using electrospray ionization tandem mass spectrometry (ESI-MS/MS), as previously described (Ceyssens et al., 2008; Holtappels et al., 2015).
Genome Accession Numbers
Lactobacillus brevis 100D8: CP015338, L. brevis ATCC 367: CP000416, L. brevis BDGP6: CP024635, L. brevis KB290: AP012167, L. brevis NCTC13768: LS483405, L. brevis NPS-QW-145: CP015398, L. brevis SA-C12: CP031185, L. brevis SRCM101106: CP021674, L. brevis SRCM101174: CP021479, L. brevis TMW 1.2108: CP019734, L. brevis TMW 1.2111: CP019743, L. brevis TMW 1.2112: CP016797, L. brevis TMW 1.2113: CP019750, L. brevis UCCLB521: CP031208, L. brevis UCCLB556: CP031174, L. brevis UCCLB95: CP031182, L. brevis UCCLBBS124: CP031169, L. brevis UCCLBBS449: CP031198, L. brevis ZLB004: CP021456, L. brevis phage 3-521: MK504444, L. brevis phage 521B: MK504443, L. brevis phage 3-SAC12: MK504442, L. brevis phage SAC12B: MK504446, L. brevis phage ATCCB: MK504445, L. brevis phage SA-C12: KU052488, and L. brevis phage LBR48: GU967410.
Results and Discussion
Prophage Identification and Characterization
Lactobacillus brevis contamination of beer is a consistent threat for breweries as its survival and growth in beer cause spoilage thus leading to product withdrawal and economic loss. The food and beverage industries aim to apply more natural, environmentally friendly and safer food preservation methods. Phage bioremediation or sanitation may represent a potential method to prevent bacterial growth and spoilage. LAB strains are known to carry prophage regions which, upon induction, may cause phage particle release and bacterial cell death.
The genomes of nineteen completely sequenced L. brevis strains were screened for the presence of prophage-encoding regions using PHASTER (Table 1). Of the nineteen bacterial strains of L. brevis, twenty-seven intact prophage sequences were predicted ranging from one to four prophage regions per strain. Twenty-three partial (marked as questionable and incomplete according to PHASTER analysis (Zhou et al., 2011; Arndt et al., 2016)) prophage regions were also identified among these strains. Four L. brevis strains do not appear to harbor intact prophage regions in their sequences, yet are predicted to carry remnant prophage sequences. The high number of prophage regions (intact and partial) identified shows that prophages are a very common occurrence in L. brevis genomes. Predicted intact prophage regions were manually examined and extracted for further analysis (general genome features are detailed in Table 3). Among the fifteen L. brevis strains whose genomes contain predicted intact prophage regions, L. brevis BDGP6 presented the highest number with four such prophage regions ranging in size from 42 to 74 Kb (Table 3). Interestingly, L. brevis strains SRCM101106 and SRCM101174 harbor an identical prophage, designated TPSRCM101106-3 and TPSRCM101174-3, respectively (100% nucleotide similarity across the full length of their genomes), this can be explained by the similarity between the two L. brevis host strains which share 99.8% nt sequence identity across 94% of their genomes. Similarly, L. brevis TMW1.2108 harbors two prophage regions that are nearly identical to those present in the genome of TMW1.2111 (TPTMW1-4 and TPTMW1-6, and TPTMW1-5 and TPTMW1-7 bearing 99.99 and 100% nucleotide similarity, respectively) and in L. brevis TMW1.2112 and TMW1.2113 sharing a 99.99% nucleotide identical prophage region (TPTMW1-1 and TPTMW1-2, respectively). These results are perhaps unsurprising as these L. brevis strains are more than 99% identical in their genome sequences but mostly differ in their plasmid content (Feyereisen et al., 2019a). The similarities observed in prophage content for some L. brevis strains might also be due to the common environment from which these strains have been isolated, i.e., food (South Korea) for L. brevis SRCM101106 and SRCM101174 and beer (Germany) for L. brevis TMW1.2108, TMW1.2111, TMW1.2112, and TMW1.2113. Integration sites (attL and attR sites) of prophages were identified and are presented in Supplementary Table S1. Diversity is observed among these integration sites; however, closely related prophages share the same sites, such as TPSRCM101106-3 and TPSRCM101174-3 or TPBDGP6-1 and TPSAC12-2. Several integration site sequences seem to be shared by certain prophages such as 5′-aaatcctgtactctcctt-3′ which was identified in five genomes. The presence of such sites acts as indicators of potential phage integration and movement, which is important in the context of phage and host evolution, fitness and adaptability in its ecological niche.
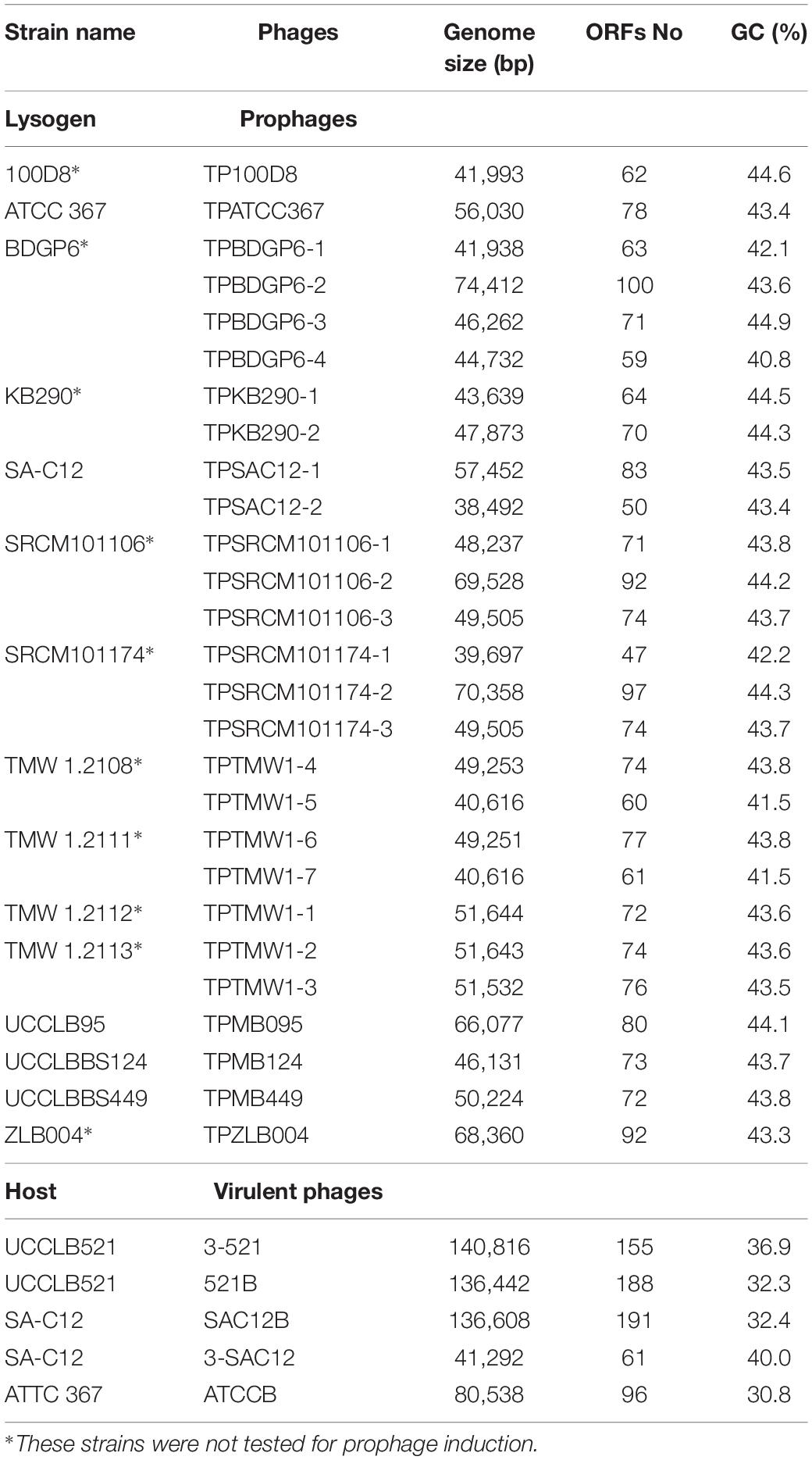
Table 3. General genome features of Lactobacillus brevis intact prophage regions and virulent phages.
Temperate phages can be detrimental for the host if, for example, they switch to a lytic state but they can also be beneficial to the host by carrying genes that will help the strain survive in its environment. These genes may encode phage-resistance systems such as Abi system or Sie proteins as previously identified (Mahony et al., 2008). Abortive infection systems were shown to block phage multiplication leading to the release of few particles and the death of the infected cells allowing the survival of the bacterial overall population (Chopin et al., 2005). Superinfection proteins were identified in Lactococcus lactis strains where they were shown to prevent DNA injection of certain phages without affecting phage adsorption (Mahony et al., 2008). Of the 19 L. brevis strains studied, nine strains were predicted to carry potential prophage-encoded Sie systems. Meanwhile six were predicted to carry potential prophage-encoded Abi system such as observed in the prophages TPMB124 and TPSRCM101174-3 based on BlastN analysis. These two prophage regions harbor the same potential Abi system which show similarity to the AbiL system identified in Lactococcus lactis (Deng et al., 1999). The presence of these potential resistance systems is predicted to confer resistance to the host against phage infection thus increasing the overall host fitness. The absence of such systems in L. brevis UCCLB521 or SA-C12 may explain their higher sensitivity to lytic phage infection. Conversely, the presence of potential phage-resistance systems (i.e., Abi) in the prophage of the beer-spoiling L. brevis strain UCCLBBS124 could explain its resistance against lytic phage infection (Feyereisen et al., 2019b).
Prophage Inductions
Small scale prophage induction trials were performed for L. brevis strain UCCLBBS124. These trials were furthermore applied to ascertain the accuracy of the bioinformatic predictions of likely intact prophages within these genomes. MitC exposure was employed at sub-lethal (0.1–0.3 μg/mL) or lethal concentrations (2 μg/mL) to distinguish between genuine prophage induction-mediated cell lysis and cell death due to acute MitC toxicity. Prophage induction trials with L. brevis UCCLBBS124 generated different induction profiles (Figure 1), where both sub-lethal and lethal doses of MitC caused cell lysis, indicating that prophage induction may have occurred. Using the lowest concentration of MitC required for prophage induction in UCCLBBS124, phage inductions in other L. brevis strains were performed as described: cultures were grown and MitC was added at a sub-lethal concentration of 0.1 μg/mL MitC when the culture reached an OD600nm of 0.1. L. brevis strains ATCC 367, SA-C12, UCCLB95, UCCLBBS124, and UCCLBBS449 exhibited lysis upon addition of 0.1 μg/mL MitC indicating prophage induction, cell lysis and phage particle release.
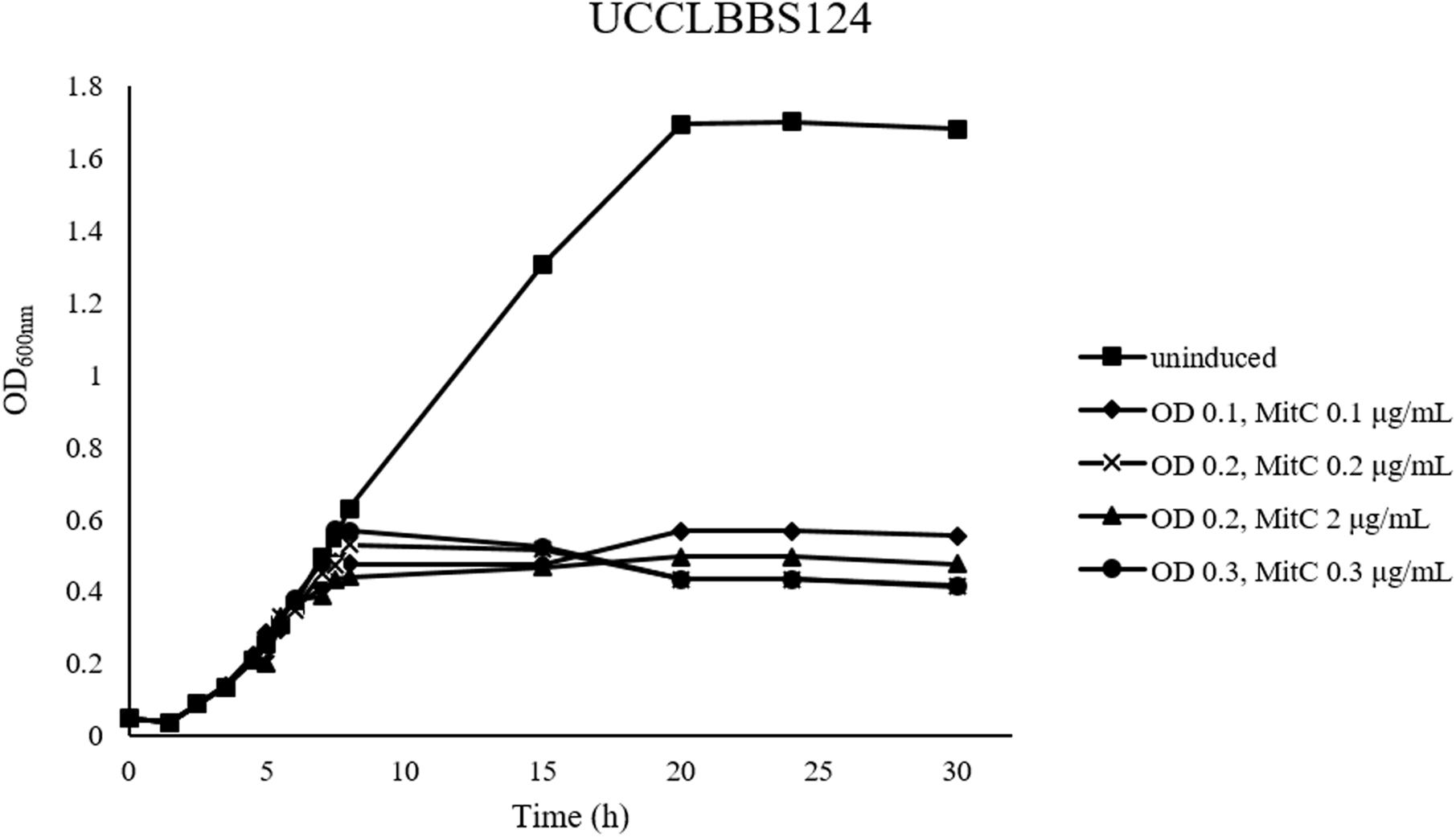
Figure 1. MitC induction profiles of Lactobacillus brevis UCCLBBS124. Different concentrations of the inducing agent MitC: 0.1, 0.2, 2, and 0.3 μg/mL were added to the culture after growth of the bacterial strain at an OD600nm of 0.1, 0.2, and 0.3, respectively. An uninduced culture was included as a control (indicated as “uninduced”).
Prophage inductions using 2% v/v formic acid or UV light exposure for 30 min were successful as indicated by PCR validation after phage DNA isolation. However, no apparent prophage induction was observed when acetic acid (2% v/v) or sodium hydroxide (2% v/v) was used as a potential inducing agent (data not shown). While in some cases formic acid and UV treatment appeared to cause cell death, phage particles were not observed by electron microscopy despite positive PCR assay results. This may be due to the detection limit of the microscopy approach limiting the ability to visualize the particles or to the poor growth characteristics of certain cultures (which appear to lyse) thus representing bacteriostasis rather than lysis.
Validation of Prophage Induction
The five L. brevis strains that were available for testing showed lysis following induction using 0.1 μg/mL MitC. To further validate that the observed lysis corresponds to phage particle release, filtered cell free supernatants of the induced cultures were analyzed by (i) electron microscopy, (ii) DNA extraction to confirm the prophage sequence using a PCR-based technique, and (iii) structural proteome analysis using mass spectrometry. This was also performed to match a specific prophage sequence to virion morphology in cases where more than one intact prophage region was identified in a bacterial strain such as L. brevis SA-C12, which harbors two predicted prophage regions (Table 3).
Prophage induction attempts for five L. brevis strains, i.e., UCCLBBS124, ATCC 367, SA-C12, UCCLBBS449 and UCCLB95, resulted in the identification of intact virions which in one case was shown to bear morphological characteristics of Siphoviridae phages: TPSAC12-2 (induced from SA-C12) which is characterized by a long thin, non-contractile tail (Figure 2 and Table 3), while in three cases Myoviridae phages were obtained: TPMB124 (induced from UCCLBBS124), TPATCC367 (induced from ATCC 367), TPMB449 (induced from UCCLBBS449) characterized by a decorated contractile-tailed phage (Figure 2 and Table 3). No phage particles were visible following induction of strain UCCLB95, which indicates that the prophage may not be inducible (to produce detectable virions) under the tested conditions despite causing cell lysis highlighting the limitations of in silico analysis. Such predictions have previously been shown to require manual evaluation and assessment in lactococcal prophages thus necessitating induction trials employing various chemical agents and/or UV treatment (Kelleher et al., 2018).
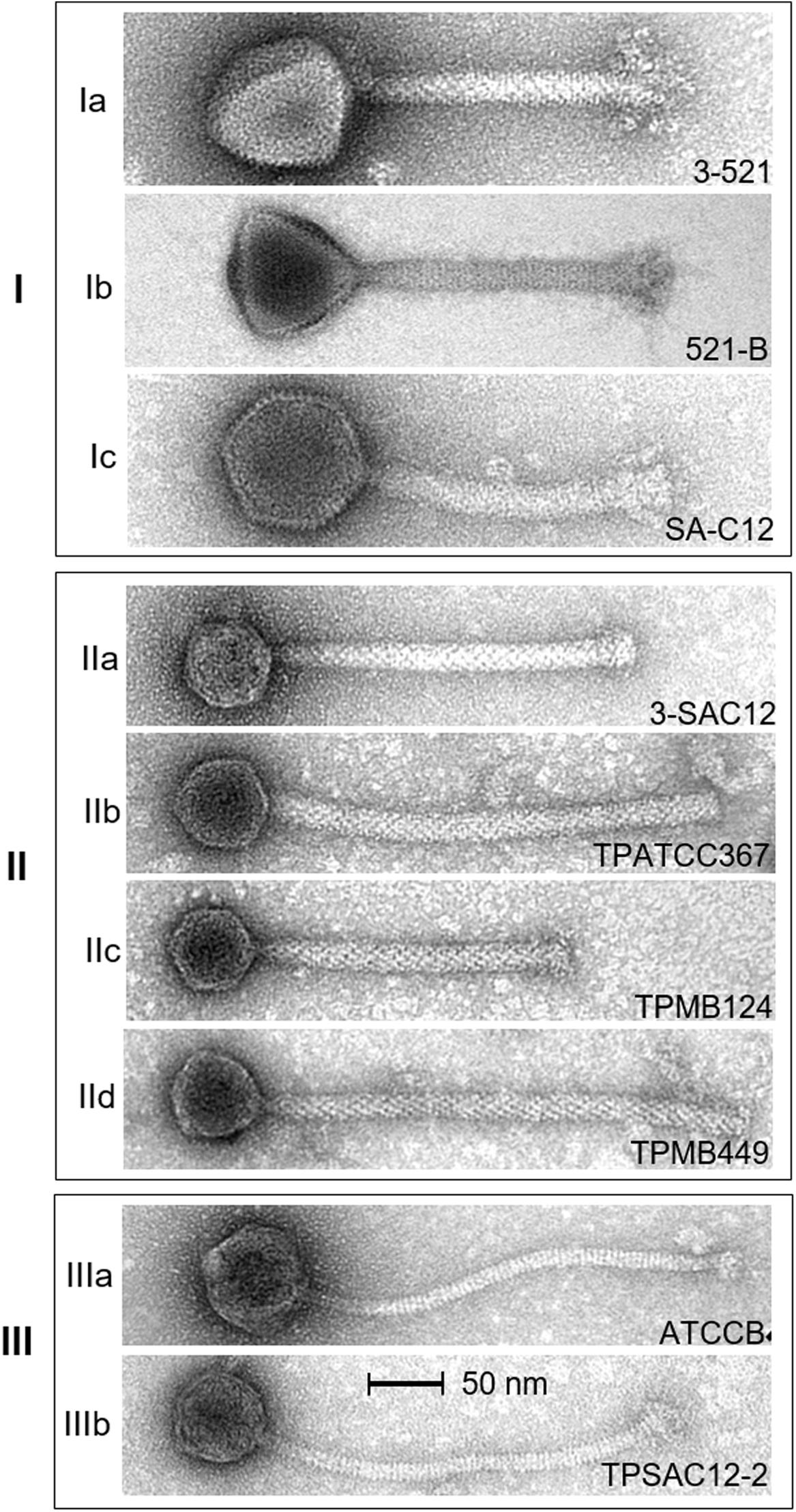
Figure 2. Electron micrographs of L. brevis phages representing morphotypes I to III. Ia, virulent phage 3-521; Ib, virulent phage 521B (Feyereisen et al., 2019b); and Ic, virulent phage SA-C12 (Deasy et al., 2011). IIa, virulent phage 3-SAC12 (Feyereisen et al., 2019b); IIb, temperate phage TPATCC367 induced from the L. brevis strain ATCC 367; IIc, temperate phage TPMB124 induced from the L. brevis strain UCCLBBS124; and IId, temperate phage TPMB449 induced from the L. brevis strain UCCLBBS449. IIIa, virulent phage ATCCB (Feyereisen et al., 2019b) and IIIb, temperate phage TPSAC12-2 induced from the L. brevis strain SA-C12. Temperate phages were induced from L. brevis strains using 0.1 μg/mL MitC.
In parallel, PCRs targeting the gene encoding the Tape Measure Protein (TMP) of the temperate phages (Table 2) as well as phage structural proteome analysis using mass spectrometry (Table 4 and Figure 4) further validated the induction and prophage identification findings. Predicted proteins encoded within the morphogenesis module of the phage genomes of TPSAC12-2, TPMB124, TPATCC367, and TPMB449 were confirmed as structural proteins. Tail proteins, the major capsid protein and the portal protein were identified as structural proteins of the temperate phage TPSAC12-2 confirming the induction of this prophage from strain SA-C12 (no structural proteins matching those encoded by the prophage region TPSAC12-1 were identified, suggesting that this prophage was not induced upon MitC treatment) (Table 4). The minor capsid protein and three hypothetical proteins were confirmed as structural proteins of the prophage region TPMB124 (Table 4). More than ten proteins of TPATCC367 were identified as structural proteins among which the tape measure protein, the head protein and the major capsid protein (Table 4). Structural proteins of the temperate phage TPMB449 were identified by mass spectrometry including the capsid, head, and portal proteins. Some predicted structural proteins were not identified in the experimentally determined proteome, most likely due to their small size or their low relative abundance.
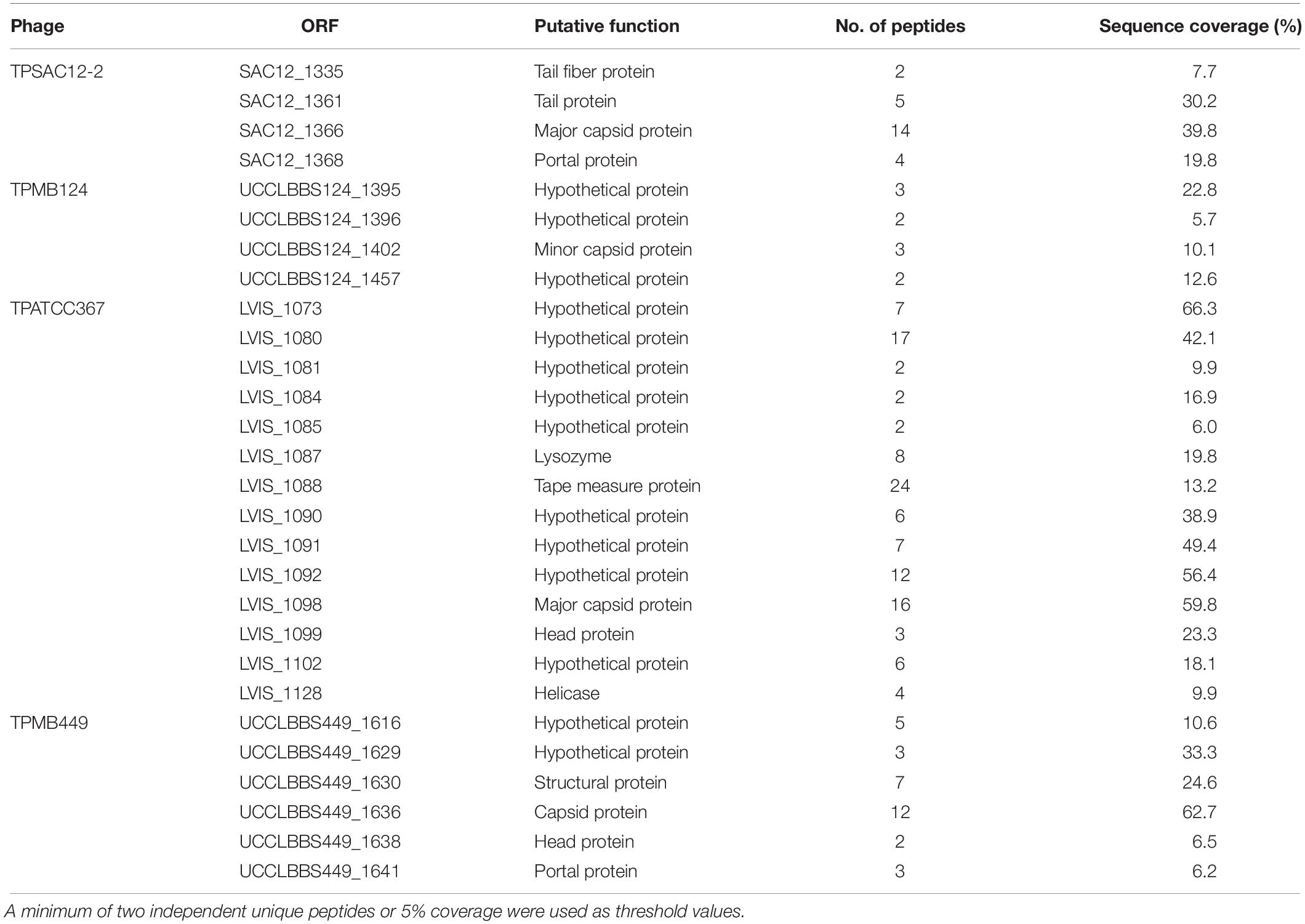
Table 4. Structural proteins extracted from purified phage particles by ESI-MS/MS.
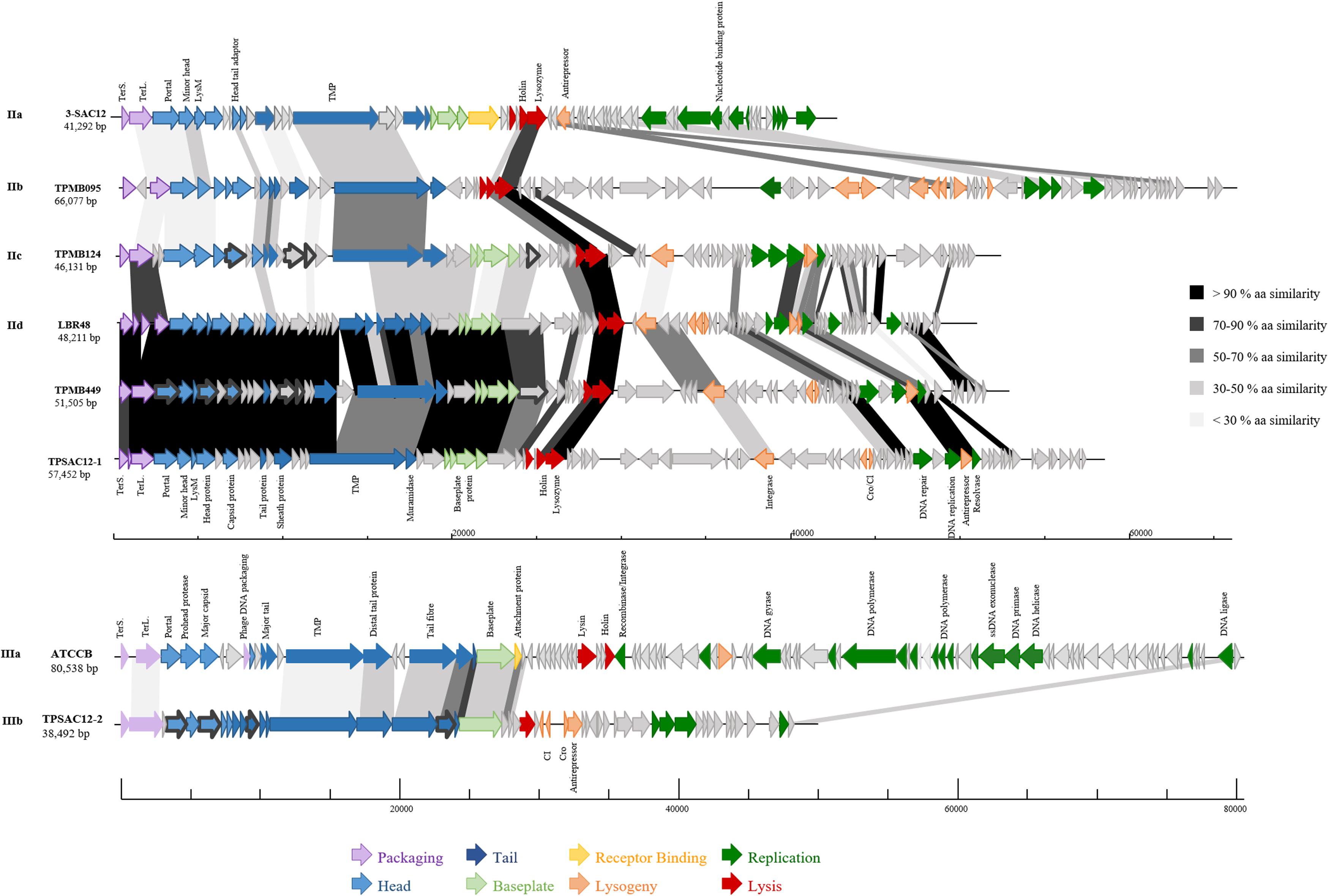
Figure 4. Genomic organization of representatives of L. brevis phages belonging to group II (Myoviridae Morphotype II phages): (IIa) 3-SAC12, (IIb) TPMB095, (IIc) TPMB124, (IId) LBR48, TPMB449 and TPSAC12-1, and group III: (IIIa) ATCCB and (IIIb) TPSAC12-2. The scale at the bottom of genomes is in base pairs. Each arrow represents an ORF, with the color representing the putative function of the encoded protein. Shaded boxes match the percentage of amino acid similarity between ORFs (TerS, small terminase; TerL, large terminase; and TMP, tape measure protein). Confirmed structural protein-encoding genes from mass spectrometry analysis are also highlighted (dark gray bold outline).
Morphology of L. brevis Phages
Electron microscopic analysis of L. brevis phages available to date were gathered, providing insights into the morphological diversity among L. brevis phages. From these analyses, three distinct morphologies were observed (Figure 2). Firstly, Myoviridae phages which exhibit imposing head structures and contractile tails ranging from 166 to 201.9 nm incorporating an organelle at the tail tip called a baseplate were termed Myoviridae Morphotype I phages (Figure 2). They are represented by the virulent phages 3-521, 521B, SAC12B (Feyereisen et al., 2019b), and SA-C12 (Deasy et al., 2011).
Secondly, L. brevis phages such as 3-SAC12, TPATCC367, TPMB124 and TPMB449 also belong to the Myoviridae family, although their common morphology differs from that of the virulent Myoviridae Morphotype I phages mentioned above (Feyereisen et al., 2019b). In this case, their morphology is represented by a small head structure and a decorated tail with a discrete baseplate, termed here as Myoviridae Morphotype II phages (Figure 2). This morphology is similar to the one observed for the prophage LBR48 (Jang et al., 2011) and interestingly, varying tail lengths were observed for phages of this morphotype (Figure 2).
Finally, some L. brevis phages were classified as members of the Siphoviridae family. Phages from this group, ATCCB and TPSAC12-2, are characterized as possessing a long non-contractile tail and an icosahedral head and were termed Morphotype III phages (Figure 2).
L. brevis Phage Phylogeny
In order to gain insight into the phylogeny of L. brevis prophages, a proteomic tree was constructed with all available sequences of L. brevis phages (virulent and temperate). Phylogenetic analysis resulted in the identification of five different groups (Figure 3) highlighting the apparent uniqueness of L. brevis phages. The virulent Myoviridae phages previously studied (Feyereisen et al., 2019b) were gathered in group I. Group II comprises twenty-one (one virulent and twenty temperate phages) L. brevis phages and phages of this group for which the family is known are all part of the Myoviridae family. Meanwhile group III gathers phages of the Siphoviridae family (when phage family is known). Groups IV and V each comprise of a single prophage sequence with distinct genetic composition and as yet unknown morphology (Figure 3). Interestingly, the majority of phages belong to the Myoviridae family, which is unusual among phages of LAB where Siphoviridae phages are more typically reported (Brüssow and Desiere, 2001; Casey et al., 2015; Kelleher et al., 2018).
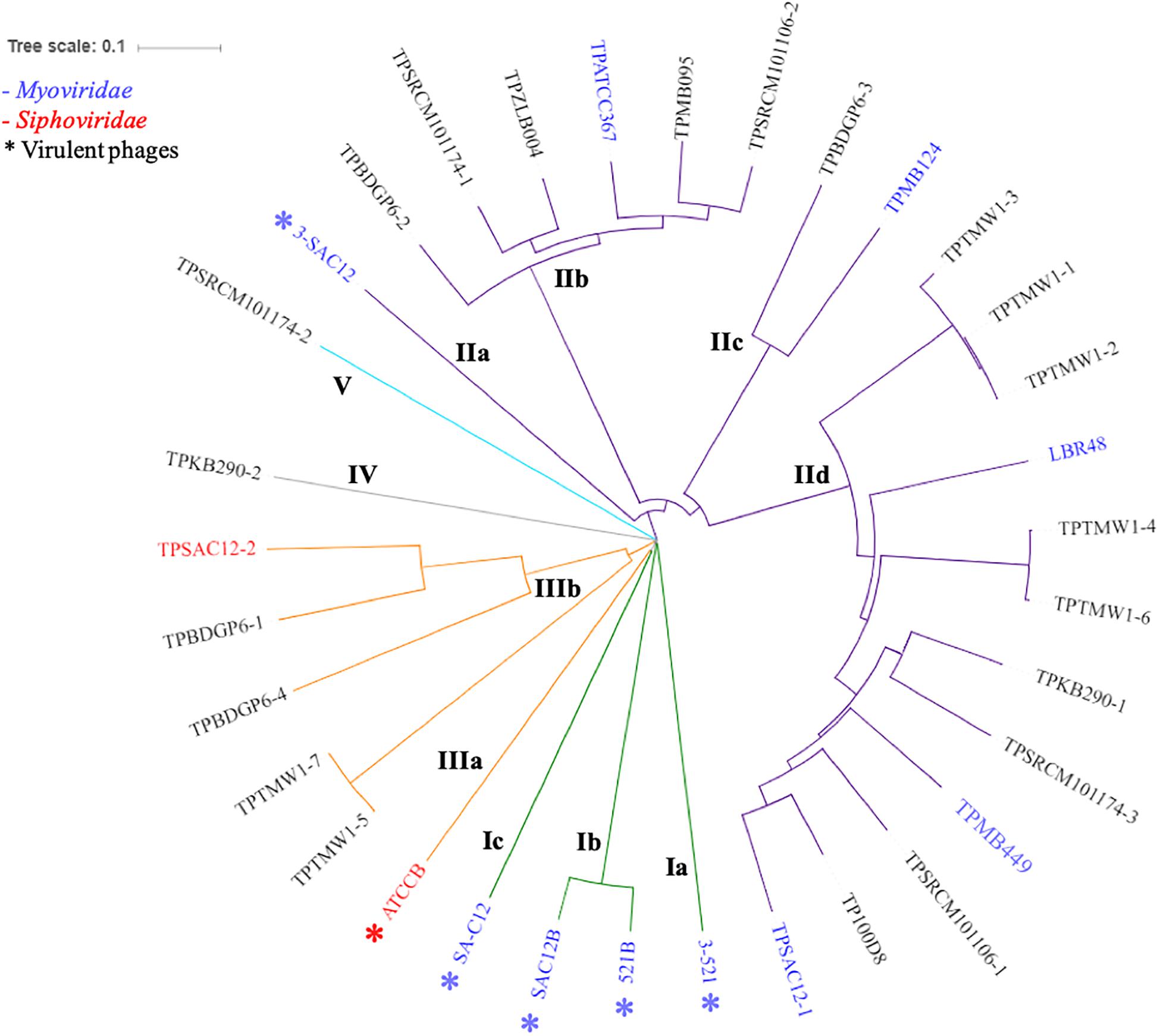
Figure 3. Proteomic tree of L. brevis phages available so far: virulent (annotated with ∗) and temperate phages. The phylogenetic analysis revealed five distinct groups (I to V) highlighted by different colors on the tree (green I, purple II, orange III, gray IV, and light blue V). Myoviridae phages are labeled in blue, while Siphoviridae phages are labeled in red.
Classification of L. brevis Phages
To date, limited studies of L. brevis phages have been undertaken despite the commercial relevance of this bacterial species and its associated bacteriophages (Deasy et al., 2011; Jang et al., 2011). Here, we propose a classification of L. brevis phages similar to what has previously been undertaken for Leuconostoc phages (Kot et al., 2014) based on morphology, phylogeny and genomic diversity. The classification suggested here, divides L. brevis phages into five groups, I to V, linking the phylogeny and morphology analyses as described above.
The group I observed on the proteomic tree (Figure 3), gathers virulent Myoviridae Morphotype I phages (Figure 2). They are further divided into three subgroups based on their genetic diversity level, phage 3-521 (Ia), phages 521B and SAC12B (Ib) sharing 97% nucleotide similarity (88% coverage) and finally SA-C12 (Ic). The previously described (Feyereisen et al., 2019b) phages 3-521, 521B and SAC12B are characterized by a large genome size (>136 Kb), probably in line with their imposing head structure, and a high degree of synteny throughout their genomes. Phage SA-C12 presents a similar morphology, yet harbors a smaller genome (79 Kb) (Deasy et al., 2011). The genome of SA-C12 is quite divergent and seems to be missing a certain number of genes encoding for hypothetical proteins and proteins involved in the replication process compared to other phages of this group.
Group II (Figure 3) is represented by Myoviridae Morphotype II phages (Figure 2), thereby encompassing over half of the L. brevis phages. Genomic and morphological analysis of representative isolates suggest that all members of this group belong to the Myoviridae family. Based on their genetic diversity, group II phages are divided here into four subgroups (Figure 3). The subgroup IIa is comprised of the virulent phage 3-SAC12 (40 Kb) which has previously been described (Feyereisen et al., 2019b). Subgroup IIb is composed of six temperate phages, including TPATCC367 (Figure 2), which share around 90% nucleotide similarity (78% sequence coverage) and harbor a larger genome (average of 62 Kb) compared to other group II members. Subgroup IIc gathers temperate phages TPBDGP6-3 and TPMB124 with an average genome size of 46 Kb and sharing 92% nucleotide similarity (56% sequence coverage). The biggest subgroup, subgroup IId, comprises twelve temperate phages sharing at least 90% nucleotide similarity (55% sequence coverage) and harboring a genome with an average size of 49 Kb.
In depth comparative analysis highlighted the degree of amino acid similarity between these Myoviridae Morphotype II phages. Although divergence occurs within these phages, genomic synteny is observed and the genomes are organized into modules corresponding to DNA packaging, structure, lysis/lysogeny and replication (Figure 4). Different tail lengths were observed for phages belonging to the group II (Figure 2) and, as previously described, the length of the tail seems to be linked to the size of the TMP gene (Mahony et al., 2016a). Indeed, phages belonging to the group IIc and presenting the smallest tail (Figure 2), have on average a TMP-encoding gene of 4.82 Kb, while phages belonging to the group IIb and IId which present the longest tailed-phages (Figure 2) possess a TMP-encoding gene with an average size of 5.67 and 5.50 Kb, respectively (Figure 4). Proteins encoding holin and lysozyme (lysin) are highly conserved across the representative phages (more than 70% amino acid (aa) similarity). The virulent phage 3-SAC12 (IIa) and the temperate phages TPMB095 (IIb) and TPMB124 (IIc) revealed low levels of similarity (between 30 and 50% aa similarity) across their structural modules. Surprisingly, only temperate phage TPMB095 lacks sequence homology with other phages of this group in the region encoding the baseplate structure. The absence of such genes may be partly responsible for the inability of TPMB095 to form functional phage and thus was not inducible. Furthermore, the genome appears quite decayed with several transposase elements interjecting the genome and the overall genome appears to lack architectural conservation compared to many other phages of LAB. L. brevis prophages LBR48, TPMB449, and TPSAC12-1 (IId) share a high level of similarity (more than 70% aa similarity) across the entire packaging, structural and lysis module (Figure 4). They also share between 30 and 50% aa similarity between their predicted integrase proteins.
Group III (Figure 3) is represented by temperate and virulent phages, and gathers members of the Siphoviridae family (e.g., ATCCB and TPSAC12-2) characterized as Morphotype III (Figure 2). Other lysogenic bacteria carrying prophages of this group were not available for prophage induction, therefore their morphotype remains unknown. It is likely that they belong to the Siphoviridae family as they are most closely related to the siphophage TPSAC12-2. The group III is divided into two subgroups separating the virulent phage ATCCB (IIIa) with a genome size of approximately 80 Kb and five temperate phages (IIIb) harboring smaller genomes (size of around 40 Kb) and among which the siphophage TPSAC12-2 can be found (Figure 3). Representatives of group III for which electron microscopy images (where available) were chosen for further comparative analysis (Figure 4). Virulent phage ATCC-B and temperate phage TPSAC12-2 share synteny in terms of genome organization with the DNA packaging module followed by the structural module, the lysis/lysogeny module and the replication module. The two phages share a low level of similarity, yet synteny and aa similarity of around 30% across the DNA packaging and structural modules with proteins encoding terminase, capsid, and tail morphogenesis-associated functions were observed. Temperate phage TPSAC12-2, unlike virulent phage ATCC-B, harbors ORFs encoding predicted lysogeny functions, such as a Cro/Cl repressor and an antirepressor (Figure 4).
Groups IV and V contain single members, i.e., prophages TPKB290-2 and TPSRCM101174-2, respectively, and do not share any significant sequence similarity to the other groups. The morphology of these phages is unknown and based on their genome analysis it is difficult to derive assumptions on the morphology/classification of TPSRCM101174-2. However, based on TPKB290-2 sequence data, a gene encoding a sheath protein is predicted suggesting that this phage is a Myoviridae member.
Induced temperate phages were tested for their potential ability to infect the seven L. brevis strains that were available in our collection (Table 1). However, these phages did not show any activity against the tested strains. Interestingly, the repressors encoded by prophages of L. brevis do not share widespread sequence homology indicating that homo-immunity based on the activity of repressors is not the basis of the observed phage-resistance. It is likely that the observed phage-resistance is due to the use of alternative receptors on the cell surface by different phage groups or through the activity of phage-resistance mechanisms such as abortive infection systems.
In order to evaluate the diversity of L. brevis phages in relation to other Lactobacillus phages, a proteomic tree was created gathering L. brevis phages described in this study as well as previously sequenced Lactobacillus phages (Supplementary Figure S1). The phylogenetic tree clearly shows the distinct grouping of L. brevis phages separately to other Lactobacillus phages. However, some L. brevis phages showed similarity with phages infecting other species such as observed for the L. brevis phage SA-C12 and the Lb. plantarum phage 8014-B2.
Conclusion
The genomes of nineteen bacterial strains of L. brevis analyzed in this study all harbor predicted prophage regions and twenty-seven intact prophage regions were identified. Only four L. brevis strains do not appear to contain intact prophage regions in their genomes. These numbers reveal the high incidence of prophages among L. brevis genomes with an average of 1.4 prophage region per strain. Of the five L. brevis strains available for prophage induction trials, four prophages were successfully induced and morphologically characterized by electron microscopy indicating a significant incidence of inducibility of these temperate phages. Electron microscopy observations, genome sequence analyses, and phylogeny allowed the classification of L. brevis phages into five groups, I to V. The results show substantial diversity among L. brevis phages and interestingly these entities are mostly represented by members of the Myoviridae family, unlike the majority of LAB phages.
The potential of prophages as antimicrobial agents in beer fermentation is a promising alternative to currently employed processes. Prophage induction in beer-spoiling L. brevis strain could be used during the cleaning process (such as coupled with sanitizers and/or UV treatment). However, this approach presents challenges and hurdles (i.e., scale-up for industrial settings and phage-encoded resistance mechanisms) that need to be addressed before their use in industry as bacterial spoilage control. To date, very few L. brevis phages have been characterized and the identification and characterization of additional phages will provide greater insights into L. brevis phage biodiversity and their potential application and role in food spoilage prevention.
Data Availability Statement
All datasets generated for this study are included in the manuscript/Supplementary Files.
Author Contributions
MF performed the experiments, annotation, genomic, and proteomic analysis. HN and CF carried out the electron microscopy analysis. J-PN carried out the mass spectrometry analysis. DS, JM, TO’S, and VB provided the materials and strains. MF, JM, TO’S, and DS were involved in project design and wrote the manuscript. All authors read and approved the final manuscript.
Funding
MF is the recipient of an Irish Research Council Enterprise Partnership Scheme postgraduate scholarship (Ref. No. EPSPG/2015/7). DS is supported by a Principal Investigator award (Ref. No. 450 13/IA/1953) through the Science Foundation Ireland (SFI). JM is in receipt of a Starting Investigator Research Grant (SIRG) (Ref. No. 15/SIRG/3430) funded by the Science Foundation Ireland (SFI).
Conflict of Interest
VB and TO’S are employees of Heineken.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors acknowledge the technical help of Angela Back (Max Rubner-Institut, Kiel) in the electron microscopic analysis and Johan Mathijs for technical assistance in mass spectrometry.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02396/full#supplementary-material
References
Altschul, S., Gish, W., Miller, W., Myers, E., and Lipman, D. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/s0022-2836(05)80360-2
Altschul, S. F., Madden, T. L., Schaffer, A. A., Zhang, J., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Arndt, D., Grant, J. R., Marcu, A., Sajed, T., Pon, A., Liang, Y., et al. (2016). PHASTER: a better, faster version of the PHAST phage search tool. Nucleic Acids Res. 44, W16–W21. doi: 10.1093/nar/gkw387
Bokulich, N. A., and Bamforth, C. W. (2013). The microbiology of malting and brewing. Microbiol. Mol. Biol. Rev. 77, 157–172. doi: 10.1128/MMBR.00060-12
Brüssow, H. (2001). Phages of dairy bacteria. Ann. Rev. Microbiol. 55, 283–303. doi: 10.1146/annurev.micro.55.1.283
Brüssow, H., and Desiere, F. (2001). Comparative phage genomics and the evolution of Siphoviridae: insights from dairy phages. Mol. Microbiol. 39, 213–222.
Canchaya, C., Proux, C., Fournous, G., Bruttin, A., and Brüssow, H. (2003). Prophage genomics. Microbiol. Mol. Biol. Rev. 67, 238–276.
Casey, E., Mahony, J., Neve, H., Noben, J.-P., Dal Bello, F., and van Sinderen, D. (2015). Novel phage group infecting Lactobacillus delbrueckii subsp. lactis, as revealed by genomic and proteomic analysis of bacteriophage Ldl1. Appl. Environ. Microbiol. 81, 1319–1326. doi: 10.1128/aem.03413-14
Ceyssens, P.-J., Mesyanzhinov, V., Sykilinda, N., Briers, Y., Roucourt, B., Lavigne, R., et al. (2008). The genome and structural proteome of YuA, a new Pseudomonas aeruginosa phage resembling M6. J. Bacteriol. 190, 1429–1435. doi: 10.1128/jb.01441-07
Chopin, A., Bolotin, A., Sorokin, A., Ehrlich, S. D., and Chopin, M. C. (2001). Analysis of six prophages in Lactococcus lactis IL1403: different genetic structure of temperate and virulent phage populations. Nucleic Acids Res. 29, 644–651. doi: 10.1093/nar/29.3.644
Chopin, M.-C., Chopin, A., and Bidnenko, E. (2005). Phage abortive infection in lactococci: variations on a theme. Curr. Opin. Microbiol. 8, 473–479. doi: 10.1016/j.mib.2005.06.006
Chopin, M. C., Chopin, A., Rouault, A., and Galleron, N. (1989). Insertion and amplification of foreign genes in the Lactococcus lactis subsp. lactis chromosome. Appl. Environ. Microbiol. 55, 1769–1774.
Davidson, B. E., Powell, I. B., and Hillier, A. J. (1990). Temperate bacteriophages and lysogeny in lactic acid bacteria. FEMS Microbiol. Lett. 87, 79–90. doi: 10.1016/0378-1097(90)90698-p
Deasy, T., Mahony, J., Neve, H., Heller, K. J., and van Sinderen, D. (2011). Isolation of a virulent Lactobacillus brevis phage and its application in the control of beer spoilage. J. Food Prot. 74, 2157–2161. doi: 10.4315/0362-028x.jfp-11-262
Deng, Y.-M., Liu, C. Q., and Dunn, N. (1999). Genetic organization and functional analysis of a novel phage abortive infection system, AbiL, from Lactococcus lactis. J. Biotechnol. 67, 135–149. doi: 10.1016/S0168-1656(98)00175-8
Feyereisen, M., Mahony, J., Kelleher, P., Roberts, R. J., O’Sullivan, T., Geertman, J.-M. A., et al. (2019a). Comparative genome analysis of the Lactobacillus brevis species. BMC Genomics 20:416. doi: 10.1186/s12864-019-5783-1
Feyereisen, M., Mahony, J., Lugli, G. A., Ventura, M., Neve, H., Franz, C. M. A. P., et al. (2019b). Isolation and characterization of Lactobacillus brevis phages. Viruses 11:E393.
Forde, A., and Fitzgerald, G. F. (1999). Bacteriophage defence systems in lactic acid bacteria. Antonie van Leeuwenhoek 76, 89–113. doi: 10.1023/a:1002027321171
Forsman, P. (1993). Characterization of a prolate-headed bacteriophage of Lactobacillus delbrueckii subsp. lactis, and its DNA homology with isometric-headed phages. Arch. Virol. 132, 321–330. doi: 10.1007/bf01309542
Fraunhofer, M. E., Geissler, A. J., Wefers, D., Bunzel, M., Jakob, F., and Vogel, R. F. (2018). Characterization of β-glucan formation by Lactobacillus brevis TMW 1.2112 isolated from slimy spoiled beer. Int. J. Biol. Macromol. 107, 874–881. doi: 10.1016/j.ijbiomac.2017.09.063
Fukao, M., Oshima, K., Morita, H., Toh, H., Suda, W., Kim, S. W., et al. (2013). Genomic analysis by deep sequencing of the probiotic Lactobacillus brevis KB290 harboring nine plasmids reveals genomic stability. PLoS One 8:e60521. doi: 10.1371/journal.pone.0060521
Garofalo, C., Osimani, A., Milanovic, V., Taccari, M., Aquilanti, L., and Clementi, F. (2015). The occurrence of beer spoilage lactic acid bacteria in craft beer production. J. Food Sci. 80, 2845–2852. doi: 10.1111/1750-3841.13112
Holtappels, M., Vrancken, K., Schoofs, H., Deckers, T., Remans, T., Noben, J. P., et al. (2015). A comparative proteome analysis reveals flagellin, chemotaxis regulated proteins and amylovoran to be involved in virulence differences between Erwinia amylovora strains. J. Proteom. 123, 54–69. doi: 10.1016/j.jprot.2015.03.036
Husson-Kao, C., Mengaud, J., Cesselin, B., van Sinderen, D., Benbadis, L., and Chapot-Chartier, M.-P. (2000). The Streptococcus thermophilus autolytic phenotype results from a leaky prophage. Appl. Environ. Microbiol. 66, 558–565. doi: 10.1128/aem.66.2.558-565.2000
Hyatt, D., Chen, G.-L., LoCascio, P. F., Land, M. L., Larimer, F. W., and Hauser, L. J. (2010). Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. doi: 10.1186/1471-2105-11-119
Jang, S. H., Yoon, B. H., and Chang, H. I. (2011). Complete nucleotide sequence of the temperate bacteriophage LBR48, a new member of the family Myoviridae. Arch. Virol. 156, 319–322. doi: 10.1007/s00705-010-0841-7
Kelleher, P., Mahony, J., Schweinlin, K., Neve, H., Franz, C., and van Sinderen, D. (2018). Assessing the functionality and genetic diversity of lactococcal prophages. Int. J. Food Microbiol. 272, 29–40. doi: 10.1016/j.ijfoodmicro.2018.02.024
Kot, W., Neve, H., Heller, K. J., and Vogensen, F. K. (2014). Bacteriophages of Leuconostoc, Oenococcus, and Weissella. Front. Microbiol. 5:186. doi: 10.3389/fmicb.2014.00186
Kumar, S., Nei, M., Dudley, J., and Tamura, K. (2008). MEGA: a biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 9, 299–306. doi: 10.1093/bib/bbn017
Lavelle, K., Murphy, J., Fitzgerald, B., Lugli, G. A., Zomer, A., Neve, H., et al. (2018). A decade of Streptococcus thermophilus phage evolution in an Irish dairy plant. Appl. Environ. Microbiol. 84:e02855-17. doi: 10.1128/aem.02855-17
Loeffler, D. (2006). “Modern brewery sanitation,” in Brewing New Technologies, ed C. W. Bamforth (Sawston: Woodhead Publishing).
Lunde, M., Blatny, J. M., Lillehaug, D., Aastveit, A. H., and Nes, I. F. (2003). Use of real-time quantitative PCR for the analysis of φLC3 prophage stability in lactococci. Appl. Environ. Microbiol. 69, 41–48. doi: 10.1128/aem.69.1.41-48.2003
Madera, C., García, P., Rodríguez, A., Suárez, J. E., and Martínez, B. (2009). Prophage induction in Lactococcus lactis by the bacteriocin Lactococcin 972. Int. J. Food Microbiol. 129, 99–102. doi: 10.1016/j.ijfoodmicro.2008.11.004
Mahony, J., Alqarni, M., Stockdale, S., Spinelli, S., Feyereisen, M., Cambillau, C., et al. (2016a). Functional and structural dissection of the tape measure protein of Lactococcal phage TP901-1. Sci. Rep. 6:36667. doi: 10.1038/srep36667
Mahony, J., McDonnell, B., Casey, E., and van Sinderen, D. (2016b). Phage-host interactions of cheese-making lactic acid bacteria. Ann. Rev. Food Sci. Technol. 7, 267–285. doi: 10.1146/annurev-food-041715-0
Mahony, J., McGrath, S., Fitzgerald, G. F., and van Sinderen, D. (2008). Identification and characterization of lactococcal-prophage-carried superinfection exclusion genes. Appl. Environ. Microbiol. 74, 6206–6215. doi: 10.1128/aem.01053-08
Makarova, K., Slesarev, A., Wolf, Y., Sorokin, A., Mirkin, B., and Koonin, E. (2006). Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. U.S.A. 103, 15611–15616. doi: 10.1073/pnas.0607117103
Meijer, W., Dobbelaar, C., and Hugenholtz, J. (1998). Thermoinducible lysis in Lactococcus lactis subsp. cremoris SK110: implications for cheese ripening. Int. Dairy J. 8, 275–280. doi: 10.1016/S0958-6946(98)00052-1
Mittler, J. E. (1996). Evolution of the genetic switch in temperate bacteriophage. I. basic theory. J. Theor. Biol. 179, 161–172. doi: 10.1006/jtbi.1996.0056
Rutherford, K., Parkhill, J., Crook, J., Horsnell, T., Rice, P., Rajandream, M., et al. (2000). Artemis: sequence visualization and annotation. Bioinformatics 16, 944–945. doi: 10.1093/bioinformatics/16.10.944
Schattner, P., Brooks, A. N., and Lowe, T. M. (2005). The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 33, W686–W689. doi: 10.1093/nar/gki366
Schmidt, R. H. (1997). Basic Elements of Equipment Cleaning and Sanitizing in Food Processing and Handling Operations. Florida: University of Florida.
Söding, J., Biegert, A., and Lupas, A. N. (2005). The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 33, W244–W248. doi: 10.1093/nar/gki408
Suzuki, K., Iijima, K., Sakamoto, K., Sami, M., and Yamashita, H. (2006). A review of hop resistance in beer spoilage lactic acid bacteria. J. Inst. Brew. 112, 173–191. doi: 10.1002/j.2050-0416.2006.tb00247.x
Thompson, J. D., Gibson, T. J., and Higgins, D. G. (2002). Multiple sequence alignment using ClustalW and ClustalX. Curr. Protoc. Bioinform. 2.3.1–2.3.22. doi: 10.1002/0471250953.bi0203s00
Vaughan, A., O’Sullivan, T., and van Sinderen, D. (2005). Enhancing the microbiological stability of malt and beer – a review. J. Inst. Brew. 111, 355–371. doi: 10.1002/j.2050-0416.2005.tb00221.x
Wu, Q., Law, Y. S., and Shah, N. P. (2015). Dairy Streptococcus thermophilus improves cell viability of Lactobacillus brevis NPS-QW-145 and its γ-aminobutyric acid biosynthesis ability in milk. Sci. Rep. 5:12885. doi: 10.1038/srep12885
Keywords: lactic acid bacteria, beer spoilage, bacteriophage, lysogeny, prophage, genomic analysis, induction, Myoviridae
Citation: Feyereisen M, Mahony J, Neve H, Franz CMAP, Noben J-P, O’Sullivan T, Boer V and van Sinderen D (2019) Biodiversity and Classification of Phages Infecting Lactobacillus brevis. Front. Microbiol. 10:2396. doi: 10.3389/fmicb.2019.02396
Received: 29 August 2019; Accepted: 03 October 2019;
Published: 16 October 2019.
Edited by:
Robert Czajkowski, University of Gdańsk, PolandReviewed by:
Stefan Hertwig, Federal Institute for Risk Assessment (BfR), GermanySimon J. Labrie, SyntBioLab Inc., Canada
Copyright © 2019 Feyereisen, Mahony, Neve, Franz, Noben, O’Sullivan, Boer and van Sinderen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Marine Feyereisen, MTE2MjIxMjA5QHVtYWlsLnVjYy5pZQ==; Douwe van Sinderen, ZC52YW5zaW5kZXJlbkB1Y2MuaWU=