 Chenchen Zhao1,2
Chenchen Zhao1,2 Hui Zhao1Shuai Zhang1,3*Junyu Luo1,3Xiangzhen Zhu1Li Wang1Peng Zhao1,2
Hui Zhao1Shuai Zhang1,3*Junyu Luo1,3Xiangzhen Zhu1Li Wang1Peng Zhao1,2 Hongxia Hua2
Hongxia Hua2 Jinjie Cui1,3*
Jinjie Cui1,3*- 1State Key Laboratory of Cotton Biology, Institute of Cotton Research, Chinese Academy of Agricultural Sciences, Anyang, China
- 2Hubei Insect Resources Utilization and Sustainable Pest Management Key Laboratory, College of Plant Science and Technology, Huazhong Agricultural University, Wuhan, China
- 3Zhengzhou Research Base, State Key Laboratory of Cotton Biology, Zhengzhou University, Zhengzhou, China
Chrysoperla sinica (Tjeder) is widely recognized as an important holometabolous natural enemy of various insect pests in different cropping systems and as a non-target surrogate in environmental risk assessment of Bt rice (i.e., genetically modified rice to express a toxin gene from Bacillus thuringiensis). Like other complex organisms, abundant microbes live inside C. sinica; however, to date, microbiome composition and diversity of the whole life cycle in C. sinica has not yet been well characterized. In the current study, we analyze the composition and biodiversity of microbiota across the whole life cycle of C. sinica by using high-throughput Illumina sequencing of the 16S ribosomal RNA gene. Collectively, Proteobacteria and Firmicutes dominated the microenvironment at all stages, but their relative abundances fluctuated by host developmental stage. Interestingly, eggs, neonates, and adults shared similar microbes, including an abundance of Rickettsia and Wolbachia. After larva feeding, Staphylococcus, Enterobacteriaceae, and Serratia were enriched in larvae and pupa, suggesting that food may serve as a major factor contributing to altered microbial community divergence at different developmental stages. Our findings demonstrated that C. sinica harbor a variety of bacteria, and that dynamic changes in community composition and relative abundances of members of its microbiome occur during different life cycle stages. Evaluating the role of these bacterial symbionts in this natural enemy may assist in developing environmental risk assessments and novel biological control strategies.
Introduction
Mutualistic relationships with microbial symbionts have been observed in many insects and are often critical for their survival and development (Kikuchi et al., 2007; Moran et al., 2008; Santos-Garcia et al., 2017). Endosymbionts may profoundly influence insect ecology and evolution and drive the coevolution between plants and herbivores (Duron et al., 2008; Kikuchi et al., 2012; Pietri and Liang, 2018). Furthermore, bacterial symbionts may provide supplemental nutrients, protection from natural enemies, and defense against pathogens and facilitate tolerance of abiotic stress (Rolff and Siva-Jothy, 2003; Scarborough et al., 2005; Douglas, 2009; Gerardo and Parker, 2014; Heyworth and Ferrari, 2015). In addition, symbionts have been shown to play key roles in insect development but the underlying mechanisms are not well understood (Hammer et al., 2017). For example, genetic studies have provided evidence of asymmetric gene transfer from bacteria and fungi to Bombyx mori (silkworm), leading to its increased survival, and reproduction (Sun et al., 2013). Understanding the composition and function of bacterial symbionts and consequential effects on their hosts remain a significant challenge.
The green lacewing (Chrysoperla sinica Tjeder, Chrysopidae: Neuroptera) is widely recognized as a natural enemy of insect pests that are important to various cropping systems (e.g., maize–wheat–maize, cotton–wheat, and wheat–rice). Adult green lacewings are frequently employed for augmentation of biological control (McEwen, 2010; Liu et al., 2011b; Ragsdale et al., 2011). Honey-dew and floral nectar are the preferred food for adult lacewings, while nymphs feed on aphids, planthoppers, coccids, mites, whiteflies, eggs, and early-instar larvae of lepidopterans, as well as many other soft-bodied arthropods (Winterton and Freitas, 2010; Xu et al., 2010). For example, one C. sinica larva can prey on as many as 164 Frankliniella occidentalis nymphs or 238 Bemisia tabaci nymphs per day (Lin et al., 2002; Zhang et al., 2007). Moreover, C. sinica has been selected as a surrogate species in the environmental risk assessment of Bt rice because in vitro rearing and manipulation of C. sinica is not difficult (Wang et al., 2012; Li et al., 2013, 2014; Romeis et al., 2013). Very few studies have explored the gut bacteria of adult C. sinica (Liu et al., 2012). The symbiotic bacteria of C. sinica have rarely been utilized experimentally and their effects on developmental stages are not well understood (Liu et al., 2012; Zhao et al., 2017).
Chrysoperla sinica undergoes differentiated complete metamorphosis where larvae and adults have distinct form and function. To investigate how the microbial community associated with C. sinica varies with life stages, we used high-throughput sequencing of the 16S ribosomal RNA (16S rRNA) gene to conduct a systematic evaluation of the microbial communities associated with C. sinica. The results yielded insights into the influence of microbial symbionts on this important natural enemy, and helped to clearly define the symbiotic changes during the development of this insect.
Materials and Methods
Insect Rearing and Maintenance
Chrysoperla sinica adults were originally collected from cotton plants cultivated at the Institute of Cotton Research, Chinese Academy of Agricultural Sciences (36°5′34.8″N, 114°31′47.19″E). They were then maintained in an environmental chamber and supplied with 10% honey solution, whereas larvae were maintained on a diet of the pea aphid Acyrthosiphon pisum. Rearing conditions were 25 ± 1°C, 65 ± 5% relative humidity, and a 14:10 h light:dark cycle (the suitable environment for feeding) (Li Z. et al., 2015). Generation 6 (G6) C. sinica eggs were used as the starting point for all experiments.
Sampling, DNA Extraction, and 16S rRNA Amplification Sequencing
Eggs, neonates, 1st instar larvae, 2nd instar larvae, 3rd instar larvae, pupae, and adults of C. sinica were randomly drawn from colonies. These seven treatments (stages) were applied with five replicates (each replicate had either 20 insects or 45 eggs). DNA was extracted from each group (whole body—each sample was washed for 5 min in 70% ethanol and rinsed three times with sterile water) using the TIAGEN DNeasy kit (TIANGEN Biotech, Beijing) following the manufacturer’s protocol. We supplemented tissue and cell lysis solutions with lysozyme of 50 mg/ml for efficient extraction. Insect homogenates were incubated at 37°C for 30 min. The quantity and quality of the DNA were measured with a NanoDrop 2000C spectrophotometer (Thermo Fisher Scientific, United States). The V3–V4 hypervariable region of the 16S rRNA gene of bacteria was amplified with the following PCR-cycling conditions: 3 min of denaturation at 95°C, 35 cycles of melting at 95°C for 30 s, annealing at 50°C for 30 s, extension at 72°C for 45 s, and a final extension at 72°C for 10 min, and with the universal primers (338F: 5′-ACTCCTACGGGAGGCAGCAG-3′, 806R: 5′-GGACTACHVGGGTWTCTAAT-3′) that were designed for detecting a broad range of microorganisms (Xu et al., 2016). PCR (GeneAmp 9700, ABI, United States) reactions were performed in triplicate 20-μl mixture containing 4 μl of 5 × FastPfu Buffer, 2 μl of 2.5 mM dNTPs, 0.8 μl of each primer (5 μM), 0.4 μl of FastPfu Polymerase, and 10 ng of template DNA. PCR reactions were subjected to 2% agarose gel electrophoresis. Amplicons of the expected size were excised, purified with the AxyPrep DNA Gel Extraction Kit (Axygen Biosciences, Union City, CA, United States), and quantified with QuantiFluorTM-ST (Promega, United States) according to the manufacturer’s protocol. Then, purified amplicons were pooled at equimolar concentrations (>2 nM) (TruSeqTM DNA Sample Prep Kit was used for library construction) and paired-end sequenced (2 × 300 bp, with an insert size of 468 bp) on an Illumina MiSeq platform (Illumina, San Diego, CA, United States) at Shanghai Majorbio Bio-pharm Technology Co., Ltd. The sequences obtained in this study were deposited in the GenBank short-read archive (SRA), accession number PRJNA5312721.
High-Throughput Analysis
Raw fastq files were quality-filtered via Trimmomatic and merged by FLASH2 with the following criteria: (i) The reads were truncated at any site that received an average quality score <20 over 50 bp (determined by sliding window). (ii) Sequences whose overlaps were longer than 10 bp were merged (allowing two nucleotide mismatching). (iii) Sequences of each sample were separated according to barcodes (exactly matching) and primers (allowing two nucleotide mismatching), and reads containing ambiguous bases were removed. High-quality reads were analyzed using both QIIME (version 1.6.0) (Caporaso et al., 2010) and mothur (version 1.30.1) (Schloss et al., 2009). Reads were assigned to their designated sample and then length-based filtering (<200 bp was excluded from the analysis) and read-quality filtering were performed (using the denoise_wrapper.py script in QIIME) (Chen et al., 2016). Operational taxonomic units (OTUs) were clustered at 97% similarity (Edgar, 2013). The taxonomy of each 16S rRNA gene sequence was analyzed by Performances of Ribosomal Database Project (RDP) Classifier algorithm (vision 2.23) against the SILVA 16S rRNA database (Release1284) (Quast et al., 2013) using a confidence threshold of 70%. Based on the resulting OTU table, we tested for the presence of enterotypes (Li J. et al., 2015) (which were introduced to reflect proportional categories or patterns within the microbiota in different individuals). Samples were clustered using the Jensen–Shannon distance and partitioning around medoid (PAM) clustering. The optimal number of clusters was estimated using the Calinski-Harabasz (CH) index (Arumugam et al., 2011; Koren et al., 2013), and principal coordinates analysis (PCoA, K ≥ 2) was carried out for visualization (Wu et al., 2011).
To avoid heterogeneity among samples due to repeated numbers of samples, and for richness and diversity analyses, rarefied OTUs were sampled. Chao1 and Ace estimators and Shannon and Simpson indices were also calculated for diversity. For normally distributed data, one-way ANOVAs followed by LSD test were performed to compare between treatments. Non-normally distributed samples were subjected to the Kruskal–Wallis test to compare between treatments. Normality was tested using the Shapiro-Wilk test. P values ≤ 0.05 were considered statistically significant. These analyses were performed with the SAS 9.4 software package. For group comparisons, the abovementioned rarefied OTU tables were used to construct Bray–Curtis matrices. Principal coordinate analysis was used for visualization using community membership and relatedness of community members (Lozupone et al., 2011). UniFrac was used for microbial community comparison (Hamady et al., 2010; Lozupone et al., 2011). Using ggplot2 package in R, taxonomic heat maps were constructed using a complete linkage clustering method and Bray–Curtis distance metric (Ginestet, 2011). Detailed methods are in the Supplementary Materials.
Results
Overview of C. sinica Microbiotas
Like all holometabolous insects, C. sinica undergoes complete metamorphosis (Figure 1A). To assess associated microbial communities, DNA from whole insects at different developmental stages (egg; neonate; 1st, 2nd, and 3rd instars; pupa; and adult) was extracted and Illumina sequence of bacterial 16S rRNA was analyzed. Community diversity and composition were analyzed using deep sequencing. We generated 1,729,032 raw reads, 1,340,914 high-quality reads, 590,093,503 base sequence, and an average of 440.07 length (Supplementary Table S1). The sequences were clustered into 262 OTUs that shared ≥97% sequence identity (Supplementary Table S1). A total of 26 OTUs were shared among all seven groups (samples shared 3 OTUs) (Supplementary Table S2).
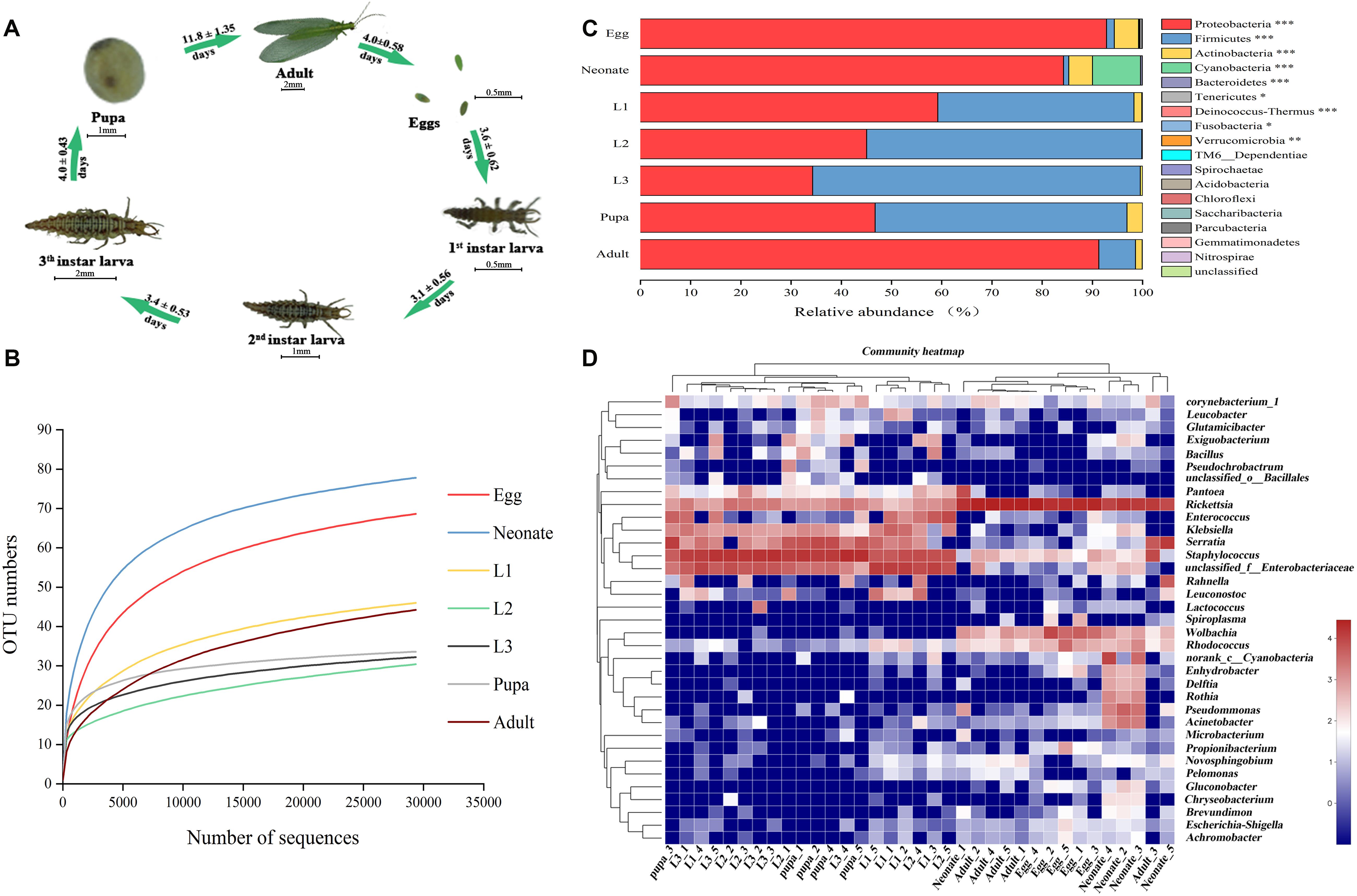
Figure 1. Bacterial community dynamics among different developmental stages in Chrysoperla sinica. (A) Overview of development stages of the C. sinica. (B) Rarefaction curves constructed from randomly subsampled data sets with the same number of 16S sequences. The near-saturated rarefaction curve indicates that the vastness of microbial diversity was captured from each sample. (C) Relative abundance of bacteria communities at the phylum level in different groups (Non-parametric Kruskal–Wallis test ∗0.01 < P ≤ 0.05, ∗∗0.001 < P ≤ 0.01, and ∗∗∗P ≤ 0.001). (D) Heat map of major taxa over the life cycle at the genus level. Cluster analysis using the Bray–Curtis distance and the complete-linkage method. Each column represents a single replicate of each of the seven treatments. Columns were clustered according to the similarity of bacterial abundance profiles. Each row represents an OTU assigned to the genus level. Color gradient represents the proportion of species. Plotting scale, from red to blue, indicates the decrease in richness of bacterial communities. Five replicates are labeled 1–5.
Taxonomic Classification of Microbiota Among Different Development Stages in C. sinica
To assess whether sampling was adequate, we used a rarefaction curve, which indicated that we captured the majority of microbial diversity in all samples (Figure 1B). Taxonomic analysis of all samples showed that Proteobacteria was the most prevalent phylum. In all samples, the top three phyla with highest relative abundances were Proteobacteria, Firmicutes, and Actinobacteria (Figure 1C and Supplementary Table S3). Regardless of age or host species, bacterial ratios varied considerably among the groups. The prevalence of Proteobacteria was significantly higher in eggs (92.69%), neonates (82.40%), and adults (92.12%), while it decreased sharply in the 1st instar larvae (59.26%). The most prevalent phylum in the larvae was Firmicutes, which reached a maximum abundance in the 3rd instar larvae (64.91%). The prevalence of Firmicutes decreased in pupae corresponding with an increase in Proteobacteria (45.63%). After eclosion, relative abundance of Firmicutes decreased (6.50%) in adults while the prevalence of proteobacteria increased. Proteobacteria, Firmicutes, and Actinobacteria showed significantly different relative abundances across a complete life cycle (P < 0.001, Supplementary Table S3).
A heat map was generated to visualize the distribution of multiple OTUs in different groups (Figure 1D). The assigned heat map corresponding to the genus level (35 most abundant) presented a comprehensive overview of the bacterial community composition. Some bacterial taxa were consistent colonizers of C. sinica, including Rickettsia and Staphylococcus. Eggs shared a similar composition of bacterial types compared with neonates and adults, whereas the abundance in larvae appeared to be similar to pupa. Rickettsia was the most abundant bacterial genus in eggs (76.16%), neonates (46.97%), and adults (85.57%). The bacterial community in neonate adults had a greater diversity relative to that of the 1st instar, and this may be the result of feeding. Interestingly, most bacteria were also found to be associated with egg masses, indicating that certain bacteria might be transmitted to the filial generation. In contrast, Staphylococcus, Enterococcus, and Klebsiella had the highest richness of all bacterial taxa in larvae and pupae. Serratia also comprised a major microbial component of pupae (29.73%), but comprised a small portion of other developmental stages (4.04–9.53%), and a very low abundance in eggs (0.05%). After adult eclosion, a significant rise in the abundance of Rickettsia and a decrease of Staphylococcus was observed. Eggs harbored a great abundance of Wolbachia (15.38%), with lower frequencies found in neonates (2.67%) and adults (1.80%). Pseudomonas had a high abundance in neonates and was rarely detected in other developmental stages. Furthermore, some groups (e.g., Leuconstoc) were only present in different stages of larval development. Therefore, it can be concluded that bacterial community composition largely shifted between 1st instars feeding and adult eclosion.
Comparative Assessment of the Microbiota Among Different Developmental Stages
We analyzed α-diversity using several methods, i.e., OTU species counts, ACE index for microbial richness, Chao1 index, the Shannon index, Simpson index, and the Good’s coverage for community coverage (Supplementary Table S1). Good’s coverage is an estimation of the percentage of the total species represented in a sample, and we found an average coverage of 99.98%, which suggested that most of the bacterial phylotypes in the insects were represented in this study. However, the C. sinica-associated bacterial community (designated as microbiota) was observed to be variable across all life stages of the host.
Diversity and richness of bacterial species were found to be significantly different among developmental stages of C. sinica. Egg and neonate had more bacterial species, with 72 unique OTUs and an average of 80 OTUs per sample, while significantly lower species richness was observed in larvae and pupa. Taxon number was lowest in the 2nd instar larvae (32 unique OTUs per sample on average). After metamorphosis, bacterial species richness increased slightly in adults (and 1st instar). Eggs and adults harbored lower diversity (Figures 2A,B), while other developmental stages had a higher diversity of bacterial species. Similarly, Simpson diversity, Chao1, and Ace richness estimators, all exhibited similar patterns (Supplementary Figure S1). Neonates harbored higher species richness and diversity than the other stages: neonates > eggs > 1st instar larvae > adults > pupa > 3rd instar larvae > 2nd instar larvae. The community diversity exhibited the following pattern: 1st instar larvae > pupa > 2nd instar larvae > 3rd instar larvae > neonates > eggs > adults.
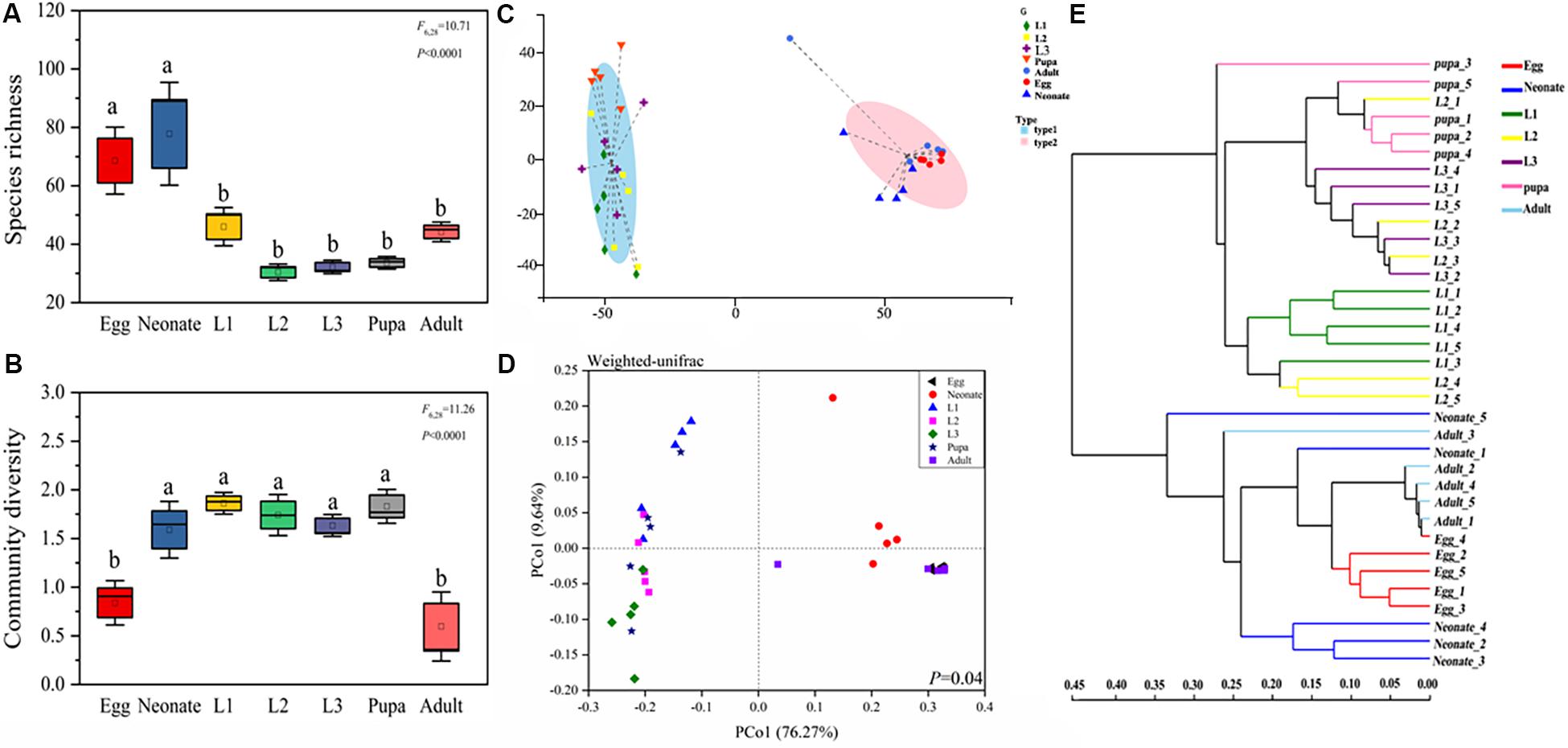
Figure 2. Bacterial community dynamics among different developmental stages in C. sinica. Boxplot of species richness (number of OTUs) (A) and community diversity measured by the Shannon index (B). Different lowercase labels above each group indicates significant differences (one-way ANOVA, LSD post hoc test, P < 0.05) of group mean value. (C) Enterotype analysis of OTU level according to β-diversity metrics of UniFrac. (D) Principal coordinates analysis (PCoA) plot visualizing the data based on β-diversity metrics of UniFrac (NMDS analysis P ≤ 0.05). (E) Unweighted pair-group method with arithmetic means (UPGMA) analysis of microbial community structure based on 16S rRNA gene amplicon sequencing data.
The enterotype analysis suggested that the bacterial colonies in C. sinica were divided into two types (Figure 2C). Larvae and pupa were grouped with one enterotype, whereas eggs, neonates, and adults were grouped with a different enterotype. Similar results were also found in our principal coordinate analysis (Figure 2D), which showed relatively tight clustering of individuals within the same developmental stage, although differences between growth stages were also observed. The results of the unweighted pair-group method using arithmetic averages (UPGMA) analysis indicated two clusters. Cluster I contained four developmental stages (1st instar larva, 2nd instar larva, 3rd instar larva, and pupa), while Cluster II contained three stages (egg, neonate, and adult), which were associated with diet as opposed to developmental stage (larvae fed by pea aphid; adults fed on honey solution; other stages fed on nothing). This suggests that the main factor determining microbial community divergence in different stages is aphids. The PCoA, enterotype, and cluster analysis results indicated that egg and neonate were more similar to adult, while larva were more similar to pupa. These results are consistent with the abovementioned alpha analysis. The 2nd instar acted as a transitional period connecting 1st instar and 3rd instar. Changes in the community composition were correlated with feeding habit as opposed to molting, suggesting that feeding habit drives the microbial community composition.
Dendrograms were generated to illustrate the composition and relatedness of bacterial communities from C. sinica based on OTU information (Figure 2E). As shown by the cluster analysis, there were two branches, the first branch consisted of eggs, neonates, and adults, while the second branch consisted of larvae and pupae. In general, the distances of samples in the same branch were within 0.35 of each other (a value of 0 means they have the same composition and a value of 1 means they do not share any species), which indicates that larvae and pupae, eggs, neonates, and adults shared a high similarity in their bacterial communities.
Discussion
Although microbial diversity in insects has been widely studied, most studies have been taxon specific and focused on the insect gut (Roh et al., 2008; Hulcr et al., 2012; Salem et al., 2013). In the current study, we present a comprehensive examination of the diverse populations of bacteria found in the lacewing C. sinica during its life cycle by using next-generation sequencing. We found that Proteobacteria and Firmicutes were dominant across the entire life cycle, which was similar to observations in other insects (Colman et al., 2012; Yun et al., 2014), such as B. mori (Chen et al., 2018), B. tabaci (Gottlieb et al., 2006), Drosophila melanogaster (Chandler et al., 2011), and Apis mellifera (Engel et al., 2012). Similar results were also found in other species, including Mytilus coruscus (Li et al., 2018), Lytechinus variegatus (Hakim et al., 2015), and humans (Schnorr et al., 2014). Despite significant diversity in the initial developmental stages (egg and neonate mass), significant reductions in bacterial diversity was observed during later developmental stages of C. sinica from eggs to adults (Figure 1C). Overall, the microbiota of C. sinica exhibited low diversity compared to the microbiota of other insects, such as Odontotaenius disjunctus (Ceja-Navarro et al., 2014) and Lymantria dispar (Mason and Raffa, 2014). By consuming large quantities of prey, lacewing larvae may ingest large amounts of potentially harmful microbes. It would thus make sense for such a voracious host to efficiently control its microbiota and quickly clear invading microbes (Chen et al., 2016). Such a phenomenon could explain the relatively simple microbiota observed in C. sinica.
In different developmental stages, the relative abundance and diversity indices varied across stages. In the early stages of development, species richness of bacteria was greater than in the later stages. In contrast, community diversity was greater in the later than in the earlier stages. Because C. sinica is holometabolous, the transition from egg to adult comprises complex changes in structural variation (Figure 1A). The difference in the structure and diversity of bacteria may be due to different physiological states and metabolism of C. sinica at different ages. Microbial symbionts that affect developmental, physiological, and ecological interactions are present in many animals (Hammer et al., 2017).
A shift in the dominant (most abundant) taxa across developmental stages was identified. From Rickettsia and Wolbachia (Alpha-Proteobacteria) in eggs, neonates, and adults to Staphylococcus (Firmicutes), and Enterobacteriaceae and Serratia (Gamma-Proteobacteria) in larvae and pupae. All these dominant taxa have been frequently detected in other insects (Hosokawa et al., 2010; Brumin et al., 2011; Haloi et al., 2016). Thermotolerance had been reported to be associated with Rickettsia, which has vertical transmission in invertebrates (Brumin et al., 2011). Rickettsia can also manipulate host reproduction in Adalia decempunctata (von der Schulenburg et al., 2001), Brachys tessellates (Lawson et al., 2001), Coccotrypes dactyliperda (Zchori-Fein et al., 2006), and Neochrysocharis formosa (Hagimori et al., 2006). Wolbachia are common transovarially transmitted symbionts of arthropods and nematodes (Baldo et al., 2006). In arthropods, Wolbachia have been shown to influence numerous aspects of host biology, including reproductive manipulation (e.g., male killing, feminization, sperm–egg incompatibility and parthenogenesis; Dobson et al., 2002; Bourtzis, 2008; Werren et al., 2008). Wolbachia can also provide benefits (Weeks et al., 2007) and can develop mutualistic relationships with their hosts (Hosokawa et al., 2010). Relative abundance of Wolbachia in eggs adults and neonates was much higher than other stages in current study. Similarly, Wolbachia abundance in adult Aedes albopictus is significantly higher than in larvae or pupae (Wang et al., 2018). Moreover, Wolbachia was shown to be the most dominant genus in pupa and adult stages of Hoplothrips carpathicus, while in larvae, it occurred at a lower frequency (Kaczmarczyk et al., 2018). The mechanism of observed disproportion is unclear. Food, development stage larvae and pupae immunity, incomplete maternal transmission, or selection pressure of Wolbachia could account for such a differential rate of infection on different stages in C. sinica. In the bed bug, Cimex lectularius, Wolbachia densities vary with life stage (high in adults; Fisher et al., 2018). Similarly, Wolbachia densities are altered by diet in Drosophila (Christensen et al., 2019). Infection by Wolbachia may provide a selective advantage for the female flies and little or no advantage for the males (Wolbachia can infect all females, but not all males in Orseolia oryzae) (Behura et al., 2001). Another study found that Wolbachia infection alters the relative abundance of resident bacteria in adult Aedes aegypti, but not larvae (Audsley et al., 2018), which could explain why Wolbachia-infected treatments share lower community diversity (Figure 1D). Pea aphid has been documented to perform as both host and vector for the phytopathogenic bacterium Pseudomonas (Stavrinides et al., 2009). In our study, Pseudomonas had high abundance in neonates, but was rarely detected in other developmental stages, suggesting that the neonate is unable to degrade prey-associated bacteria immediately.
The bacterial composition of adult C. sinica in our study differs from that reported by Liu et al. (2012) for field-collected adult C. sinica based on culture-dependent methods. Ten bacterial taxa, including Morganella, Bacillus pumilus, and Terrabacter, were isolated by Liu et al. (2012), few of which overlapped with the bacterial taxa detected in the present study. However, many of the microbial associates are not readily culturable. It is also well known that bacterial diversities vary from field-collected to lab-reared (Rani et al., 2009) as well as across different geographical regions (Zouache et al., 2011). In previous studies, the bacterial composition and diversity were influenced by the in vitro environment (Zouache et al., 2011), as well as medium selection and an artificial diet (Priya et al., 2012). In addition, microbial diversity could be affected by different physiological and pathological states of the host (Backhed et al., 2004; Ley et al., 2006). C. sinica is also affected by the ecological niche of its host, as well as the environment and its own physiological status (Xu et al., 2002, 2010; Khuhro et al., 2014). Age, mating status, temperature, and humidity affect flight performance and other behaviors (e.g., adult diapause, survival rate, and fecundity) of C. sinica (Xu et al., 2004, 2010; Liu et al., 2011a, b; Chen et al., 2017). Previous observations suggest that prey could alter pre-imaginal development, survival, adult longevity, and fecundity of C. sinica (Khuhro et al., 2012). Moreover, microbial diversity varies greatly among insects that have fed on different food sources, which has been shown for Helicoverpa armigera (Priya et al., 2012), Lymantria dispar (Broderick et al., 2004), and Anopheles stephensi (Rani et al., 2009). In particular, we were interested in whether or not food exerted an effect on the microbial diversity in C. sinica.
In this study, principal coordinate analysis revealed significant changes in the bacterial community compositions under different developmental stages, demonstrating that the variation between aphid feeding stage (initial stage of larvae feeding) and non-feeding stage (after adult eclosion to neonate) was greater than within the same stage. The 2nd instar stage acted as a transitional period that connected the 1st instar and 3rd instar. Although gut morphology was altered sharply by metamorphosis, during different stages of the life cycle (Moll et al., 2001), feeding, and food source may have caused the variation in host-specific microbiota in C. sinica.
The bacterial community exhibited by C. sinica probably results from flexible and facultative associations with free-living and horizontally transmitted bacteria, akin to the phenomenon reported in Drosophila (Chandler et al., 2011). In our study, we used the pea aphid, which is a highly palatable host that fosters lacewing ingestion and development. Previous studies have shown a community composition shift on less preferred species (Xiang et al., 2010).
Transgenic Bt cotton had little effect on the biological characteristics of C. sinica that fed on A. gossypii (Gao et al., 2013). In addition, the insecticidal protein Cry1Ab and Cry2Aa in rice pollen had no significant adverse effects on adult C. sinica (Bai et al., 2005; Wang et al., 2012). Yet, feeding on Spodoptera exigua that were reared on Bt cotton exerted some influence on the life history traits of C. sinica (Shi et al., 2011). Effects of Bt toxin on bacterial diversity of A. mellifera has been a theoretical concern for biosafety assessments of transgenic maize and cotton (Dirk et al., 2010; Weiyu et al., 2010; Geng et al., 2013; Jia et al., 2016). Similar investigations have been undertaken in Nilaparvata lugens (Gao et al., 2008), Holotrichia oblita (Wang et al., 2017), and Chilo suppressalis (Li et al., 2016). Despite being important as a surrogate species in environmental risk assessment, the bacterial diversity of C. sinica had not been well characterized. In the current study, we have provided a better understanding of the symbiont diversity and community composition patterns associated with different life cycle stage of C. sinica. Such knowledge may help advance the manipulation and exploitation of the microbiota, which could result in important practical applications for the development of strategies for the management of insect-related problems (Crotti et al., 2012). For instance, the effects of Bt protein on the bacterial communities of the coccinelid beetle Propylea japonica have been estimated, providing a new aspect of non-target risk assessment of genetically modified crops (Zhang et al., 2019). The present study will help lay the foundation for management of C. sinica-related problems in the future (e.g., the effects of genetically modified organisms on C. sinica bacterial communities).
Data Availability Statement
Publicly available datasets were analyzed in this study. This data can be found here: https://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA531272.
Author Contributions
JC and SZ conceived and designed the work, and edited the manuscript. CZ and HZ performed the experiments and wrote the manuscript. JL, XZ, and LW helped with the theoretical analysis. PZ and HH helped to revise the manuscript.
Funding
This work was supported by grants from the National Special Transgenic Project of China (No. 2016ZX08011-002).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02454/full#supplementary-material
FIGURE S1 | Bacterial community dynamics among C. sinica in different developmental stages. A Boxplot of species richness (Chao1 and Ace richness estimators) and community diversity (Simpson diversity index). Different lowercase above each host species indicate significant differences (one-way ANOVA, LSD post-hoc test, P < 0.05) in the mean values.
TABLE S1 | Summary of high-throughput sequencing read analysis, microbial community diversity richness (OTUs, 97%), sample coverage (Good’s coverage), diversity index (Shannon, ACE, Shannon, Simpson), and estimated OTU richness (Chao1) for community diversity analyses of 35 samples from C. sinica of different development stages.
TABLE S2 | Details of bacterial OTUs shared in C. sinica among different developmental stages.
TABLE S3 | Relative abundance of bacteria communities at the phylum level in different group.
TABLE S4 | Supplementary methods, bioinformatics analysis.
Footnotes
- ^ https://www.ncbi.nlm.nih.gov/sra/?term=PRJNA531272
- ^ http://ccb.jhu.edu/software/FLASH/
- ^ http://sourceforge.net/projects/rdp-classifier/
- ^ http://www.arb-silva.de
References
Arumugam, M., Raes, J., Pelletier, E., Le Paslier, D., Yamada, T., Mende, D. R., et al. (2011). Enterotypes of the human gut microbiome. Nature 473, 174–180. doi: 10.1038/nature09944
Audsley, M. D., Seleznev, A., Joubert, D. A., Woolfit, M., O’Neill, S. L., and McGraw, E. A. (2018). Wolbachia infection alters the relative abundance of resident bacteria in adult Aedes aegypti mosquitoes, but not larvae. Mol. Ecol. 27, 297–309. doi: 10.1111/mec.14436
Backhed, F., Ding, H., Wang, T., Hooper, L. V., Koh, G. Y., Nagy, A., et al. (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc. Natl. Acad. Sci. U.S.A. 101, 15718–15723. doi: 10.1073/pnas.0407076101
Bai, Y., Jiang, M., and Cheng, J. (2005). Effect of transgenic cry1Ab rice pollen on the oviposition and adult longevity of Chrysoperla sinica Tjeder. Acta Phytophylacica Sin. 32, 225–230. doi: 10.13802/j.cnki.zwbhxb.2005.03.001
Baldo, L., Bordenstein, S., Wernegreen, J. J., and Werren, J. H. (2006). Widespread recombination throughout Wolbachia genomes. Mol. Biol. Evol. 23, 437–449. doi: 10.1093/molbev/msj049
Behura, S. K., Sahu, S. C., Mohan, M., and Nair, S. (2001). Wolbachia in the Asian rice gall midge, Orseolia oryzae (Wood-Mason): correlation between host mitotypes and infection status. Insect. Mol. Biol. 10, 163–171. doi: 10.1046/j.1365-2583.2001.00251.x
Bourtzis, K. (2008). Wolbachia-based technologies for insect pest population control. Adv. Exp. Med. Biol. 627, 104–113. doi: 10.1007/978-0-387-78225-6_9
Broderick, N. A., Raffa, K. F., Goodman, R. M., and Handelsman, J. (2004). Census of the bacterial community of the gypsy moth larval midgut by using culturing and culture-independent methods. Appl. Environ. Microbiol. 70, 293–300. doi: 10.1128/aem.70.1.293-300.2004
Brumin, M., Kontsedalov, S., and Ghanim, M. (2011). Rickettsia influences thermotolerance in the whitefly Bemisia tabaci B biotype. Insect. Sci. 18, 57–66. doi: 10.1111/j.1744-7917.2010.01396.x
Caporaso, J. G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F. D., Costello, E. K., et al. (2010). QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 7, 335–336. doi: 10.1038/nmeth.f.303
Ceja-Navarro, J. A., Nguyen, N. H., Karaoz, U., Gross, S. R., Herman, D. J., Andersen, G. L., et al. (2014). Compartmentalized microbial composition, oxygen gradients and nitrogen fixation in the gut of Odontotaenius disjunctus. ISME J. 8, 6–18. doi: 10.1038/ismej.2013.134
Chandler, J. A., Lang, J. M., Bhatnagar, S., Eisen, J. A., and Kopp, A. (2011). Bacterial communities of diverse Drosophila species: ecological context of a host-microbe model system. PLoS Genet. 7:e1002272. doi: 10.1371/journal.pgen.1002272
Chen, B., Du, K., Sun, C., Vimalanathan, A., Liang, X., Li, Y., et al. (2018). Gut bacterial and fungal communities of the domesticated silkworm (Bombyx mori) and wild mulberry-feeding relatives. ISME J. 12, 2252–2262. doi: 10.1038/s41396-018-0174-171
Chen, B., Teh, B. S., Sun, C., Hu, S., Lu, X., Boland, W., et al. (2016). Biodiversity and activity of the gut microbiota across the life history of the insect herbivore Spodoptera littoralis. Sci. Rep. 6:29505. doi: 10.1038/srep29505
Chen, Z., Liu, L., Liu, S., Guo, Q., and Xu, Y.-Y. (2017). Effects of maternal overwintering experience on offspring development and reproduction in Chrysoperla sinica (Neuroptera: Chrysopidae). Acta Entomol. Sin. 60, 553–561. doi: 10.16380/j.kcxb.2017.05.007
Christensen, S., Camacho, M., Sharmin, Z., Momtaz, A. J. M. Z., Perez, L., Navarro, G., et al. (2019). Quantitative methods for assessing local and bodywide contributions to Wolbachia titer in maternal germline cells of Drosophila. BMC Microbiol. 19:206. doi: 10.1186/s12866-019-1579-3
Colman, D. R., Toolson, E. C., and Takacs-Vesbach, C. D. (2012). Do diet and taxonomy influence insect gut bacterial communities? Mol. Ecol. 21, 5124–5137. doi: 10.1111/j.1365-294X.2012.05752.x
Crotti, E., Balloi, A., Hamdi, C., Sansonno, L., Marzorati, M., Gonella, E., et al. (2012). Microbial symbionts: a resource for the management of insect-related problems. Microb. Biotechnol. 5, 307–317. doi: 10.1111/j.1751-7915.2011.00312.x
Dirk, B., David, J., Romeis, J., Franz, B., and Franco, W. (2010). Bacterial community structures in honeybee intestines and their response to two insecticidal proteins. FEMS Microbiol. Ecol. 59, 600–610. doi: 10.1111/j.1574-6941.2006.00249.x
Dobson, S. L., Fox, C. W., and Jiggins, F. M. (2002). The effect of Wolbachia-induced cytoplasmic incompatibility on host population size in natural and manipulated systems. Process. Biol. Sci. 269, 437–445. doi: 10.1098/rspb.2001.1876
Douglas, A. E. (2009). The microbial dimension in insect nutritional ecology. Funct. Ecol. 23, 38–47. doi: 10.1111/j.1365-2435.2008.01442.x
Duron, O., Bouchon, D., Boutin, S., Bellamy, L., Zhou, L., Engelstädter, J., et al. (2008). The diversity of reproductive parasites among arthropods: Wolbachiado not walk alone. BMC Biol. 6:27. doi: 10.1186/1741-7007-6-27
Edgar, R. C. (2013). UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 10, 996–998. doi: 10.1038/nmeth.2604
Engel, P., Martinson, V. G., and Moran, N. A. (2012). Functional diversity within the simple gut microbiota of the honey bee. Proc. Natl. Acad. Sci. U.S.A. 109, 11002–11007. doi: 10.1073/pnas.1202970109
Fisher, M. L., Watson, D. W., Osborne, J. A., Mochizuki, H., Breen, M., and Schal, C. (2018). Growth kinetics of endosymbiont Wolbachia in the common bed bug. Cimex lectularius. Sci. Rep. 8:11444. doi: 10.1038/s41598-018-29682-29682
Gao, F., Liu, X.-H., and Ouyang, F. (2013). Development and reproduction of Chrysopa sinica (Neuroptera: Chrysopidae) reared on Aphis gossypii (Homoptera: Aphidinae) fed transgenic cotton. Appl. Econ. 50, 897–902. doi: 10.7679/j.issn.2095-1353.2013.125
Gao, X., Tian, J., Chen, Y., Hu, C., Peng, Y., and Ye, G. (2008). Impact evaluation of transgenic cry1Ab japonica rice on the diversity of intestinal microbial community in the brown plant thopper Nilaparvata lugens using biolog-ecomethod. Acta Phytophylacica Sinica 35, 327–331. doi: 10.13802/j.cnki.zwbhxb.2008.04.005
Geng, L.-L., Cui, H.-J., Shu, C.-L., Dai, P.-L., Lang, Z.-H., Zhou, T., et al. (2013). The influence of Bt-transgenic maize pollen on the bacterial diversity;in the midgut of Apis mellifera ligustica. Apidologie 44, 198–208. doi: 10.1007/s13592-012-0171-8
Gerardo, N. M., and Parker, B. J. (2014). Mechanisms of symbiont-conferred protection against natural enemies: an ecological and evolutionary framework. Curr. Opin. Insect Sci. 4, 8–14. doi: 10.1016/j.cois.2014.08.002
Ginestet, C. (2011). ggplot2: elegant graphics for data analysis by H. Wickham. J. R. Stat. Soc. 174, 245–246. doi: 10.1111/j.1467-985x.2010.00676_9.x
Gottlieb, Y., Ghanim, M., Chiel, E., Gerling, D., Portnoy, V., Steinberg, S., et al. (2006). Identification and localization of a Rickettsia sp. in Bemisia tabaci (Homoptera: Aleyrodidae). Appl. Environ. Microbiol. 72, 3646–3652. doi: 10.1128/aem.72.5.3646-3652.2006
Hagimori, T., Abe, Y., Date, S., and Miura, K. (2006). The first finding of a Rickettsia bacterium associated with parthenogenesis induction among insects. Curr. Microbiol. 52, 97–101. doi: 10.1007/s00284-005-0092-90
Hakim, J. A., Koo, H., Dennis, L. N., Kumar, R., Ptacek, T., Morrow, C. D., et al. (2015). An abundance of Epsilonproteobacteria revealed in the gut microbiome of the laboratory cultured sea urchin. Lytechinus variegatus. Front. Microbiol. 6:1047. doi: 10.3389/fmicb.2015.01047
Haloi, K., Kalita, M. K., Nath, R., and Devi, D. (2016). Characterization and pathogenicity assessment of gut-associated microbes of muga silkworm Antheraea assamensis Helfer (Lepidoptera: Saturniidae). J. Invertebr. Pathol. 138, 73–85. doi: 10.1016/j.jip.2016.06.006
Hamady, M., Lozupone, C., and Knight, R. (2010). Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 4, 17–27. doi: 10.1038/ismej.2009.97
Hammer, T. J., Janzen, D. H., Hallwachs, W., Jaffe, S. P., and Fierer, N. (2017). Caterpillars lack a resident gut microbiome. Proc. Natl. Acad. Sci. U.S.A. 114, 9641–9646. doi: 10.1073/pnas.1707186114
Heyworth, E. R., and Ferrari, J. (2015). A facultative endosymbiont in aphids can provide diverse ecological benefits. J. Evol. Biol. 28, 1753–1760. doi: 10.1111/jeb.12705
Hosokawa, T., Koga, R., Kikuchi, Y., Meng, X.-Y., and Fukatsu, T. (2010). Wolbachia as a bacteriocyte-associated nutritional mutualist. Proc. Natl. Acad. Sci. U.S.A. 107, 769–774. doi: 10.1073/pnas.0911476107
Hulcr, J., Rountree, N. R., Diamond, S. E., Stelinski, L. L., Fierer, N., and Dunn, R. R. (2012). Mycangia of ambrosia beetles host communities of bacteria. Microb. Ecol. 64, 784–793. doi: 10.1007/s00248-012-0055-55
Jia, H. R., Geng, L. L., Li, Y. H., Wang, Q., Diao, Q. Y., Zhou, T., et al. (2016). The effects of Bt Cry1Ie toxin on bacterial diversity in the midgut of Apis mellifera ligustica (Hymenoptera: Apidae). Sci. Rep. 6:24664. doi: 10.1038/srep24664
Kaczmarczyk, A., Kucharczyk, H., Kucharczyk, M., Kapusta, P., Sell, J., and Zielińska, S. (2018). First insight into microbiome profile of fungivorous thrips Hoplothrips carpathicus (Insecta: Thysanoptera) at different developmental stages: molecular evidence of Wolbachia endosymbiosis. Sci. Rep. 8:14376. doi: 10.1038/s41598-018-32747-x
Khuhro, N. H., Biondi, A., Desneux, N., Zhang, L., Ying, Z., and Chen, H. (2014). Trade-off between flight activity and life-history components in Chrysoperla sinica. Biocontrol 59, 219–227. doi: 10.1007/s10526-014-9560-9564
Khuhro, H. N., Chen, H., Zhang, Y., Zhang, L., and Wang, M. (2012). Effect of different prey species on the life history parameters of Chrysoperla sinica (Neuroptera: Chrysopidae). EJE 109, 175–180. doi: 10.14411/eje.2012.023
Kikuchi, Y., Hayatsu, M., Hosokawa, T., Nagayama, A., Tago, K., and Fukatsu, T. (2012). Symbiont-mediated insecticide resistance. Proc. Natl. Acad. Sci. U.S.A. 109, 8618–8622. doi: 10.1073/pnas.1200231109
Kikuchi, Y., Hosokawa, T., and Fukatsu, T. (2007). Insect-microbe mutualism without vertical transmission: a stinkbug acquires a beneficial gut symbiont from the environment every generation. Appl. Environ. Microbiol. 73, 4308–4316. doi: 10.1128/aem.00067-67
Koren, O., Knights, D., Gonzalez, A., Waldron, L., Segata, N., Knight, R., et al. (2013). A guide to enterotypes across the human body: meta-analysis of microbial community structures in human microbiome datasets. PLoS Comput. Biol. 9:e1002863. doi: 10.1371/journal.pcbi.1002863
Lawson, E. T., Mousseau, T. A., Klaper, R., Hunter, M. D., and Werren, J. H. (2001). Rickettsia associated with male-killing in a buprestid beetle. Heredity 86(Pt 4), 497–505. doi: 10.1046/j.1365-2540.2001.00848.x
Ley, R. E., Turnbaugh, P. J., Klein, S., and Gordon, J. I. (2006). Microbial ecology: human gut microbes associated with obesity. Nature 444, 1022–1023. doi: 10.1038/4441022a
Li, J., Powell, J. E., Guo, J., Evans, J. D., Wu, J., Williams, P., et al. (2015). Two gut community enterotypes recur in diverse bumblebee species. Curr. Biol. 25, R652–R653. doi: 10.1016/j.cub.2015.06.031
Li, Z., Zhang, S., Luo, J., Wang, S., Wang, C., Lv, L., et al. (2015). Identification and expression pattern of candidate olfactory genes in Chrysoperla sinica by antennal transcriptome analysis. Comp. Biochem. Physiol. Part D Genom. Proteom. 15, 28–38. doi: 10.1016/j.cbd.2015.05.002
Li, Y., Chen, X., Hu, L., Romeis, J., and Peng, Y. (2014). Bt rice producing Cry1C protein does not have direct detrimental effects on the green lacewing Chrysoperla sinica (Tjeder). Environ. Toxicol. Chem. 33, 1391–1397. doi: 10.1002/etc.2567
Li, Y., Wang, Y., Romeis, J., Liu, Q., Lin, K., Chen, X., et al. (2013). Bt rice expressing Cry2Aa does not cause direct detrimental effects on larvae of Chrysoperla sinica. Ecotoxicology 22, 1413–1421. doi: 10.1007/s10646-013-1127-1120
Li, Y.-F., Yang, N., Liang, X., Yoshida, A., Osatomi, K., Power, D., et al. (2018). Elevated seawater temperatures decrease microbial diversity in the gut of Mytilus coruscus. Front. Physiol. 9:839. doi: 10.3389/fphys.2018.00839
Li, Z., Han, L., Liu, Y., and Hou, M. (2016). Change in the diversity of bacterial community in larval midguts of the striped stem borer, Chilo suppressalis (Lepidoptera: Crambidae), after treatment with Bacillus thuringiensis insecticidal proteins. Acta Entomologica Sinica 59, 292–300.
Lin, K., Wu, K., Liu, S., Zhang, Y., and Guo, Y. (2002). Functional responses of Chrysopa sinica, Propylaea japonica and Leis axyridis to Bemisia tabaci. Chin. Bull. Entomol. 43, 339–343. doi: 10.3969/j.issn.0452-8255.2006.03.015
Liu, N., Yu-Sheng, L., Wei-Dong, F., and Guo-Liang, Z. (2012). Diversity of intestinal aerobic bacteria in overwintering adults of Chrysoperla sinica. Chin. J. Appl. Entomol. 49, 1287–1292.
Liu, Z., McNeil, J. N., and Wu, K. (2011a). Flight mill performance of the lacewing Chrysoperla sinica (Neuroptera: Chrysopidae) as a function of age, temperature, and relative humidity. J. Econ. Entomol. 104, 94–100. doi: 10.1603/ec10331
Liu, Z., Wyckhuys, K. A. G., and Wu, K. (2011b). Migratory adaptations in Chrysoperla sinica (Neuroptera: Chrysopidae). Environ. Entomol. 40, 449–454. doi: 10.1603/EN10202
Lozupone, C., Lladser, M. E., Knights, D., Stombaugh, J., and Knight, R. (2011). UniFrac: an effective distance metric for microbial community comparison. ISME J. 5, 169–172. doi: 10.1038/ismej.2010.133
Mason, C., and Raffa, K. (2014). Acquisition and structuring of midgut bacterial communities in gypsy moth (Lepidoptera: Erebidae) larvae. Environ. Entomol. 43, 595–604. doi: 10.1603/EN14031
McEwen, P. K. (2010). Lacewings in the crop environment. Zool. J. Linnean Soc. 151, 661–661. doi: 10.1111/j.1096-3642.2007.00317.x
Moll, R. M., Romoser, W. S., Modrzakowski, M. C., Moncayo, A. C., and Lerdthusnee, K. (2001). Meconial peritrophic membranes and the fate of midgut bacteria during mosquito (Diptera: Culicidae) metamorphosis. J. Med. Entomol. 38, 29–32. doi: 10.1603/0022-2585-38.1.29
Moran, N. A., McCutcheon, J. P., and Nakabachi, A. (2008). Genomics and evolution of heritable bacterial symbionts. Annu. Rev. Genet. 42, 165–190. doi: 10.1146/annurev.genet.41.110306.130119
Pietri, J. E., and Liang, D. (2018). The links between insect symbionts and insecticide resistance: causal relationships and physiological tradeoffs. Ann. Entomol. Soc. Am. 111, 92–97. doi: 10.1093/aesa/say009
Priya, N. G., Ojha, A., Kajla, M. K., Raj, A., and Rajagopal, R. (2012). Host plant induced variation in gut bacteria of Helicoverpa armigera. PLoS One 7:e30768. doi: 10.1371/journal.pone.0030768
Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2013). The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res. 41, D590–D596. doi: 10.1093/nar/gks1219
Ragsdale, D. W., Landis, D. A., Brodeur, J., Heimpel, G. E., and Desneux, N. (2011). Ecology and management of the soybean aphid in North America. Annu. Rev. Entomol. 56, 375–399. doi: 10.1146/annurev-ento-120709-144755
Rani, A., Sharma, A., Rajagopal, R., Adak, T., and Bhatnagar, R. K. (2009). Bacterial diversity analysis of larvae and adult midgut microflora using culture-dependent and culture-independent methods in lab-reared and field-collected Anopheles stephensi-an Asian malarial vector. BMC Microbiol. 9:96. doi: 10.1186/1471-2180-9-96
Roh, S. W., Nam, Y.-D., Chang, H.-W., Kim, K.-H., Kim, M.-S., Ryu, J.-H., et al. (2008). Phylogenetic characterization of two novel commensal bacteria involved with innate immune homeostasis in Drosophila melanogaster. Appl. Environ. Microbiol. 74, 6171–6177. doi: 10.1128/AEM.00301-308
Rolff, J., and Siva-Jothy, M. T. (2003). Invertebrate ecological immunology. Science 301, 472–475. doi: 10.1126/science.1080623
Romeis, J., Raybould, A., Bigler, F., Candolfi, M. P., Hellmich, R. L., Huesing, J. E., et al. (2013). Deriving criteria to select arthropod species for laboratory tests to assess the ecological risks from cultivating arthropod-resistant genetically engineered crops. Chemosphere 90, 901–909. doi: 10.1016/j.chemosphere.2012.09.035
Salem, H., Kreutzer, E., Sudakaran, S., and Kaltenpoth, M. (2013). Actinobacteria as essential symbionts in firebugs and cotton stainers (Hemiptera. Pyrrhocoridae). Environ. Microbiol. 15, 1956–1968. doi: 10.1111/1462-2920.12001
Santos-Garcia, D., Silva, F. J., Morin, S., Dettner, K., and Kuechler, S. M. (2017). The all-rounder sodalis: a new bacteriome-associated endosymbiont of the lygaeoid bug Henestaris halophilus (Heteroptera: Henestarinae) and a critical examination of its evolution. Genome Biol. Evol. 9, 2893–2910. doi: 10.1093/gbe/evx202
Scarborough, C. L., Ferrari, J., and Godfray, H. C. (2005). Aphid protected from pathogen by endosymbiont. Science 310:1781. doi: 10.1126/science.1120180
Schloss, P. D., Westcott, S. L., Ryabin, T., Hall, J. R., Hartmann, M., Hollister, E. B., et al. (2009). Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol. 75, 7537–7541. doi: 10.1128/aem.01541-1549
Schnorr, S. L., Candela, M., Rampelli, S., Centanni, M., Consolandi, C., Basaglia, G., et al. (2014). Gut microbiome of the Hadza hunter-gatherers. Nat. Commun. 5, 3654–3654. doi: 10.1038/ncomms4654
Shi, M., Xiuqing, X., Jiang, Q., and Yang, Y. (2011). Impact on the life parameters and functional response of Chrysoperla sinica preying on Spodoptera exigua reared on transgenic Bt cotton. J. Environ. Entomol. 33, 482–487. doi: 10.3969/j.issn.1674-0858.2011.04.011
Stavrinides, J., McCloskey, J. K., and Ochman, H. (2009). Pea aphid as both host and vector for the phytopathogenic bacterium Pseudomonas syringae. Appl. Environ. Microbiol. 75, 2230–2235. doi: 10.1128/aem.02860-2868
Sun, B. F., Xiao, J. H., He, S. M., Liu, L., Murphy, R. W., and Huang, D. W. (2013). Multiple ancient horizontal gene transfers and duplications in lepidopteran species. Insect Mol. Biol. 22, 72–87. doi: 10.1111/imb.12004
von der Schulenburg, J. H., Habig, M., Sloggett, J. J., Webberley, K. M., Bertrand, D., Hurst, G. D., et al. (2001). Incidence of male-killing Rickettsia spp. (alpha-proteobacteria) in the ten-spot ladybird beetle Adalia decempunctata L. (Coleoptera: Coccinellidae). Appl. Environ. Microbiol. 67, 270–277. doi: 10.1128/aem.67.1.270-277.2001
Wang, W., Zhao, D., Guo, W., Li, X., Zhang, Y., and Yan, X. (2017). Effects of Bt Cry8Ea protein on the midgut microbial flora of Holotrichia oblita larvae. Biotechnol. Bull. 33, 126–131. doi: 10.13560/j.cnki.biotech.bull.1985.2017-0236
Wang, X., Liu, T., Wu, Y., Zhong, D., Zhou, G., Su, X., et al. (2018). Bacterial microbiota assemblage in Aedes albopictus mosquitoes and its impacts on larval development. Mol. Ecol.gy 27, 2972–2985. doi: 10.1111/mec.14732
Wang, Y., Li, Y., Romeis, J., Chen, X., Zhang, J., Chen, H., et al. (2012). Consumption of Bt rice pollen expressing Cry2Aa does not cause adverse effects on adult Chrysoperla sinica Tjeder (Neuroptera: Chrysopidae). Biol. Control 61, 246–251. doi: 10.1016/j.biocontrol.2012.02.011
Weeks, A. R., Turelli, M., Harcombe, W. R., Reynolds, K. T., and Hoffmann, A. A. (2007). From parasite to mutualist: rapid evolution of Wolbachia in natural populations of Drosophila. PLoS Biol. 5:e114. doi: 10.1371/journal.pbio.0050114
Weiyu, J., Pingli, D., Yongjun, Z., Ting, Z., Yi, L., Changlong, S., et al. (2010). Effect of transgenic cotton with cry1Ac gene on intestinal bacterial community of Apis mellifera ligustica. Chin. J. Appl. Environ. Biol. 16, 211–215. doi: 10.3724/SP.J.1145.2010.00211
Werren, J. H., Baldo, L., and Clark, M. E. (2008). Wolbachia: master manipulators of invertebrate biology. Nat. Rev. Microbiol. 6, 741–751. doi: 10.1038/nrmicro1969
Winterton, S., and Freitas, S. D. (2010). Molecular phylogeny of the green lacewings (Neuroptera: Chrysopidae). Aus. J. Entomol. 45, 235–243. doi: 10.1111/j.1440-6055.2006.00537.x
Wu, G. D., Chen, J., Hoffmann, C., Bittinger, K., Chen, Y. Y., Keilbaugh, S. A., et al. (2011). Linking long-term dietary patterns with gut microbial enterotypes. Science 334, 105–108. doi: 10.1126/science.1208344
Xiang, Y., Wang, X., Feng, W., Zhou, W., Xie, H., and Wan, Y. (2010). Comparative analysis of the composition of dominant intestinal microflora in silkworm reared with different forages. Acta Ecologica Sinica 30, 3875–3882. doi: 10.7666/d.y1671573
Xu, N., Tan, G., Wang, H., and Gai, X. (2016). Effect of biochar additions to soil on nitrogen leaching, microbial biomass and bacterial community structure. Eur. J. Soil Biol. 74, 1–8. doi: 10.1016/j.ejsobi.2016.02.004
Xu, Y., Cui, H. U., Yuan, M. J., Fu, X. H., and Gang, W. H. (2002). relationship between adult diapause development and over-wintering coloration changes in Chrysoperla sinica (Neuroptera). Acta Ecologica Sinica 22, 1275–1280. doi: 10.1007/s11769-002-0041-9
Xu, Y., JiYuan, M., Cui, H., and Wang, H. (2004). Effects of temperature and relative humidity on survival of the overwintering green lacewing,Chrysoperla sinica (Tjeder) (Neuroptera: Chrysopidae). Acta Ecologica Sinica 2004, 2569–2572. doi: 10.3321/j.issn:1000-0933.2004.11.031
Xu, Y., Mu, J., Hu, C., and Wang, H. (2010). Photoperiodic control of adult diapause in Chrysoperla sinica (tjeder) (Neuroptera: Chrysopidae) -i. Critical photoperiod and sensitive stages of adult diapause induction. Insect Sci. 11, 191–198. doi: 10.1111/j.1744-7917.2004.tb00239.x
Yun, J. H., Roh, S. W., Whon, T. W., Jung, M. J., Kim, M. S., Park, D. S., et al. (2014). Insect gut bacterial diversity determined by environmental habitat, diet, developmental stage, and phylogeny of host. Appl. Environ. Microbiol. 80, 5254–5264. doi: 10.1128/aem.01226-1214
Zchori-Fein, E., Borad, C., and Harari, A. (2006). Oogenesis in the date stone beetle. Coccotrypes dactyliperda, depends on symbiotic bacteria. Physiol. Entomol. 31, 164–169. doi: 10.1111/j.1365-3032.2006.00504.x
Zhang, A., Li, L., Yu, Y., and Men, X. (2007). Function response and searching rate of Chrysopa sinica larvae on Frankliniella occidentalis nymphs. Acta Phytophylacica Sinica 34, 247–251. doi: 10.3321/j.issn:0577-7518.2007.03.005
Zhang, S., Luo, J., Jiang, W., Wu, L., Zhang, L., Ji, J., et al. (2019). Response of the bacterial community of Propylea japonica (Thunberg) to Cry2Ab protein. Environ. Pollut. 254(Pt B), 113063. doi: 10.1016/j.envpol.2019.113063
Zhao, H., Zhang, S., Luo, J., Zhang, L., Wang, A., and Cui, J. (2017). Analysis of endophytic symbiotic bacterial composition in Chrysoperla sinica (Tjeder) adults with 16S rDNA clone library. Chin. J. Biol. Control. 33, 849–856. doi: 10.16409/j.cnki.2095-039x.2017.06.019
Zouache, K., Raharimalala, F. N., Raquin, V., Tran-Van, V., Raveloson, L. H., Ravelonandro, P., et al. (2011). Bacterial diversity of field-caught mosquitoes, Aedes albopictus and Aedes aegypti, from different geographic regions of Madagascar. FEMS Microbiol. Ecol. 75, 377–389. doi: 10.1111/j.1574-6941.2010.01012.x
Keywords: Chrysoperla sinica, development stage, microbiotas, high-throughput sequencing, bacterial diversity
Citation: Zhao C, Zhao H, Zhang S, Luo J, Zhu X, Wang L, Zhao P, Hua H and Cui J (2019) The Developmental Stage Symbionts of the Pea Aphid-Feeding Chrysoperla sinica (Tjeder). Front. Microbiol. 10:2454. doi: 10.3389/fmicb.2019.02454
Received: 30 March 2019; Accepted: 14 October 2019;
Published: 01 November 2019.
Edited by:
Mariana Mateos, Texas A&M University, United StatesReviewed by:
Arturo Vera-Ponce de León, The Ohio State University, United StatesLuis Eduardo Servín, National Autonomous University of Mexico, Mexico
Copyright © 2019 Zhao, Zhao, Zhang, Luo, Zhu, Wang, Zhao, Hua and Cui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shuai Zhang, WmhhbmdzaHVhaTAxQGNhYXMuY24=; Jinjie Cui, YXljdWlqaW5qaWVAMTYzLmNvbQ==