 Juan Chen
Juan Chen Jia-Mei Li
Jia-Mei Li Yan-Jing Tang
Yan-Jing Tang Yong-Mei Xing
Yong-Mei Xing Peng Qiao
Peng Qiao Yang Li
Yang Li Pei-Gui Liu3
Pei-Gui Liu3- 1Key Laboratory of Bioactive Substances and Resources Utilization of Chinese Herbal Medicine, Ministry of Education, Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China
- 2Shandong Institute of Sericulture, Shandong Academy of Agricultural Sciences, Yantai, China
- 3Key Laboratory for Plant Diversity and Biogeography of East Asia, Kunming Institute of Botany, Chinese Academy of Sciences, Kunming, China
It is well known that the microbes associated with truffle fruiting bodies play a very important role during the truffle lifecycle. Tuber indicum, commonly called Chinese black truffle, is a species endemic to Eastern Asia and in the genus of Tuber. Here, we reported the bacterial communities of T. indicum from different geographical regions and described the bacterial diversity from three compartments (soil, ectomycorrhizae and ascocarps) of T. indicum using high-throughput sequencing combined tissue culture. The results revealed that Bradyrhizobium was the dominant genus in fruiting bodies of T. indicum from nine geographical sites in China, and the microbes in T. indicum ascocarps were influenced by geological locations and soil characteristics. More specific bacterial taxa were enriched in the fruiting bodies than in the ectomycorrhizae and soil. In addition, 60 cultural bacteria were isolated from T. indicum fruiting bodies (4 families, 24 genera), and Pseudomonas, Alcaligenes faecalis, Microbacterium, and Arthrobacter were dominant. One of 13 strains that have potential nitrogen-fixation activities was further verified by an acetylene reduction assay (ARA). Together, this research provides new and important data for better understanding of the interaction between truffle and associated microbe and the biology of truffle itself.
Introduction
Tuber is a genus belonging to Tuberaceae (Ascomycota) and usually produces hypogeous edible fruiting bodies, known as “true truffle” (Pegler et al., 1993). Some Tuber species have high economic value for their unique and delightful aromas, such as Italian white truffle (T. magnatum) and Perigord black truffle (T. melanosporum), which costing as much as 600€–6000€ per kilogram and are recognized as the most expensive foodstuffs in the world (Wang and Marcone, 2011; Torregiani et al., 2017). In addition, the summer truffle (T. aestivum) and the Chinese black truffle (T. indicum complex) are also very popular in the edible mushroom market. Tuber spp. can establish a tightly ectomycorrhizal symbiotic associations with Pinaceae (pines), Fagaceae (oak/beech), Myrtaceae (eucalyptus) and Salicaceae (willows/poplar) and benefit the growth of the host plants (Bonito and Smith, 2016); thus, Tuber spp. play an important role in nutrition recycling and utilization of forest ecosystems. Currently, truffles are cultivated in native habitats such as Spain, France, and Italy, as well as in non-native areas such as Australia and New Zealand by the mean of inoculating seedlings with spore suspension (Murat, 2015). Truffle cultivation involves environmental, economic, and social components that benefit tree species and forest ecosystems, providing high market value fruiting bodies as food and medicine and attracting interest from various social, ethnic, economic and age groups (Benucci et al., 2012). Moreover, as a means of land-use stability and reforestation and rural economic development, truffles are cultivated as long-term economic crops in some European countries such as France, Spain, and Italy (Bonet et al., 2006). Unfortunately, the yields harvested are low and highly variable due to climate factors, low genetic diversity, the quality of inoculums on nursery trees and the distribution of truffle mating types in truffle orchards (Linde and Selmes, 2012). In China, the natural production of truffle is gradually decreasing because of climate change (e.g., drought and low precipitation) and destruction of human activity (over-harvesting immature truffle), while artificial cultivation on a large scale is still not successful.
Previous studies on truffle evolution, species and genetic diversity, chemical composition and cultivation have made outstanding progress, but the biology of truffle fruiting bodies is still mysterious. It takes at least 5–7 years for truffle to complete a complex lifecycle from haploid spore germination to haploid free-living mycelium growth in soil, which includes the establishment of a mutualistic symbiotic association with roots of host plants and final formation and development of the fruiting body (Selosse et al., 2017). During the whole process, truffle interacts closely with microbes in the surrounding soil. Fluorescent in situ hybridization (FISH) confirmed the occurrence of dominant bacteria within mature ascocarps of T. melanosporum (Mello et al., 2013; Antony-Babu et al., 2014). Roux et al. (2016) suggested that bacteria of the genus Rhodopseudomonas (Bradyrhizobiaceae) establish obligate symbiosis with mycelia cultures of the black truffles T. melanosporum.
In past decades, microbial communities of several commercial truffle species have has been characterized with culture-dependent and 16S rRNA sequencing methods, including T. borchii (Barbieri et al., 2005), T. aestivum (Gryndler et al., 2013; Splivallo et al., 2019), T. magnatum (Barbieri et al., 2010; Leonardi et al., 2013; Amicucci et al., 2018), and T. melanosporum (Belfiori et al., 2012; Antony-Babu et al., 2013; Mello et al., 2013; Deveau et al., 2016; Roux et al., 2016). These studies demonstrated that truffle fruiting bodies harbor abundant and diverse microbial communities. Studies on the bacterial community of fruiting bodies of truffle indicated that Pseudomonas, Bacteroides and gram-positive bacteria were dominant in culturable bacterial communities (Sbrana et al., 2002; Barbieri et al., 2005), while Proteobacteria, Bacteroidetes, Firmicutes, and Actinobacteria formed the core component of the complex bacterial communities of all truffle species analyzed so far by culture-dependent and independent method techniques (Vahdatzadeh et al., 2015).
It has been reported that the bacterial community composition of truffle is potentially influenced by truffle species (Ye et al., 2018), geography (Benucci and Bonito, 2016), lifecycle stage (mycelium, ectomycorrhiza, and fruiting body) (Antony-Babu et al., 2014), host plant physiology (Li et al., 2018), seasons (Deveau et al., 2016), postharvest storage conditions (Vahdatzadeh et al., 2019), samples number and methodologies (Splivallo et al., 2019). Moreover, different tissues of fruiting bodies (e.g., peridium and gleba) were also enriched in specific bacteria (Antony-Babu et al., 2014). The microbial community associated with the truffle fruiting body has been deemed a complement of the external mycelium, which can promote the establishment of ectomycorrhizal symbiosis, modulate competition with other symbiotic and saprotrophic microbes and mobilize nutrients from minerals (Gryndler et al., 2013). Barbieri et al. (2010) demonstrated that nitrogen fixation occurs inside fruiting bodies of the white truffle T. magnatum, which may be related to presence of fixing bacteria associated with T. magnatum ascomata. Moreover, some microorganisms are believed to affect the production of the unique aroma of truffle (Buzzini et al., 2005). The unique aromas emitted from the fruiting body of truffle are responsible for its high economic value in the world market and determine the quality of the truffle. Early studies showed that yeasts isolated from the ascocarps of black and white truffles can produce volatile organic compounds (VOCs), speculating that these unique aromas might result from the interaction of truffle with their microbiome (Splivallo et al., 2011). Recently, it was demonstrated that bacteria isolated from truffle fruiting bodies were able to produce thiophene volatiles, which are sulfur-containing volatiles and characteristic of T. borchii fruiting bodies, suggesting that thiophene volatiles are exclusively synthesized by bacteria and not by the truffle extract during the sexual stage of truffles (Splivallo et al., 2015). Furthermore, correlations were detected between the changes in the volatile profiles of truffle and specific bacterial classes during the storage period of T. aestivum (Vahdatzadeh et al., 2019). These results highlighted the role of bacterial community as a most important contributor to the truffle aroma.
Tuber indicum, known as Chinese black truffle, is endemic to Eastern Asian and mainly distributes in China. The phylogenetic position of T. indicum belongs to the Melanosporum clade, which comprises species in Europe (T. melanosporum and T. brumale), Asia (T. indicum and T. pseudoexcavatum) and America (T. regimontanum) (Bonito et al., 2013). Among over 60 Tuber species of China, T. indicum is the most important species with the widest distribution and the largest production in China. Since the 1990s, T. indicum has been exported to the European market as the most primary trade wild edible mushroom of China. Due to the very close similarity with T. melanosporum in macro-and micro-morphological characteristics (black ascomata with conspicuous warts and spiny ascospores) and lower cost, T. indicum have attracted an increasing gastronomic and scientific interest in the past 20 years.
Previous studies on T. indicum have mainly focused on taxonomy (Liu and Tao, 1996), phylogeny (Wang et al., 2006; Zhang et al., 2010; Chen et al., 2011; Kinoshita et al., 2018), population genetics (Feng et al., 2016; Qiao et al., 2018) and mycorrhizal synthesis (Kinoshita et al., 2018). Only a few studies have attempted to explore the microbial community of T. indicum in the last 2 years. For example, Deng et al. (2018) investigated the bacterial community of T. indicum collected in Southwestern China using the traditional culture method and high-throughput sequencing and revealed that Flavobacterium, Agromyces, and Ensifer were dominant in fruiting bodies. Li et al. (2018) reported the microbial community structures of the ectomycorrhizosphere of T. indicum and host tree Quercus aliena. However, few studies have been carried out on the bacterial communities of T. indicum from different geographical regions systematically. Thus, the aims of this study are to compare the bacterial community of T. indicum from nine different geographical regions, to describe the bacterial community structure of ectomycorrhizae, fruiting bodies and soil in the same region using the high-throughput sequencing technique, to isolate the cultural bacteria from fruiting bodies and identify potential bacterial partners with biological activity. This study will provide useful information for understanding truffle development and realizing artificial cultivation of Chinese black truffles in the future.
Materials and Methods
Sample Collection
Fruiting bodies of T. indicum were collected from nine different geographical sites from October to November 2017. In addition, ectomycorrhizae and soil, and fruiting bodies, were collected from a Castanea mollissima orchard in Hebei Province. All samples were stored on ice during transport to the laboratory, after gently washed with sterile distilled water and then deposited at −80°C or used directly for subsequent morphological examination under a light microscope. A total of 120 fruiting bodies (FBs) of T. indicum were morphologically examined and classification of the maturation stage depends on the percentage of asci containing mature spores (Zeppa et al., 2002). Of them, fifty-four FBs were selected for bacterial community sequencing. Ectomycorrhizae of T. indicum were also collected under microscope according to the morphological characteristics (Geng et al., 2009). Rhizosphere soil adhering to the ectomycorrhizae was collected for microbial analysis and the bulk soils surrounding the fruiting bodies were collected for soil parameter analysis.
DNA Extraction and Sequencing
For DNA extraction, 0.5 cm3 samples of healthy T. indicum fruiting bodies (no insect larvae galleries or animal injuries, etc.) from each geographical region were collected. Total DNA of ascocarps and ectomycorrhizae were extracted using a CTAB Plant DNA Extraction Kit (Aidlab Biotechnologies Co., Ltd., Beijing, China) according to the extraction protocol. Total DNA of soil adhering to the ascocarps (0.5–1 g) was extracted using the Power Soil DNA Isolation Kit (Mo Bio Laboratories, Carlsbad, CA, United States) according to the manufacturer’s protocol. DNA quality and quantity were assessed by the ratios of 260 nm/280 nm. For the 16S rRNA V3 + V4 region sequencing, samples from each region had 3–9 replicates. Amplification libraries were generated using the universal primers 338F (5′-ACTCCTACGGGAGGCAGCA-3′) and 806R (5′-GGACTACHVGGGTWTCTAAT-3′) combined with adapter sequences and barcode sequences (Mori et al., 2014). High-throughput sequencing analysis of bacterial rRNA genes was performed on the purified, pooled sample using the Illumina HiSeq 2500 platform (2 × 250 paired ends) at Biomarker Technologies Corporation, Beijing, China.
Bacterial Community Analysis
The raw paired-end reads from the original DNA fragments were filtered and merged using FLASH v1.2.11 (Tanja and Salzberg, 2011), and the sequences were quality evaluated with PRINSEQ v0.20.4 (Schmieder and Edwards, 2011). High-quality reads from each sample were clustered into operational taxonomic units (OTUs) based on a 97% sequence similarity according to UCLUST (Version 1.2.22) (Edgar, 2010). Taxonomic assignments were performed against the RDP 16S rDNA sequence reference database with the naïve Bayesian RDP classifier and a minimum confidence level of 0.8 (Wang et al., 2007).
For alpha diversity analysis, we rarefied the OTUs to several metrics, including rare faction curve and Shannon curve (Wang et al., 2012), as well as rank abundance curve (Kõljalg et al., 2013). Shannon index (Rodrigues et al., 2014), Simpson index (Simpson, 1949), ACE index (Hughes et al., 2001) and Chao1 index (Chao et al., 2005) were calculated and plotted in the R statistical environment with the package phyloseq (Mcmurdie and Susan, 2013). For beta diversity analysis, a heatmap of RDA-identified key OTUs, principal component analysis (PCA) and Linear discriminate analysis effect size (LEfSe) analyses were performed for the quantitative analysis of biomarkers among each group. Briefly, for LEfSe analysis with an LDA threshold of >4, the non-parametric factorial Kruskal-Wallis (KW) sum-rank test was used, and the (unpaired) Wilcoxon rank-sum test was used to identify the most differently abundant taxa.
Statistical Analysis
The effects of the geographic sites on the structures of the bacterial communities were determined by ANOVA at a threshold level of P = 0.05 and Fisher’s test. All statistical analyses were performed with SPSS (Version 23.0.0.0). Permutational multivariate analysis of variance (Anderson, 2010) was implemented in the R package vegan (Oksanen et al., 2019), and adonis and beta dispersion functions were used to test the null hypothesis that there are no differences among bacterial communities of Tuber species.
Isolation and Identification of Cultural Bacteria
Ascocarp samples were briefly flamed, and approximately 1 g of gleba tissue was cut from the ascocarps using a sterile scalpel, and the sample was homogenized in 10 mL of filter-sterilized physiologic solution (0.85% NaCl) before shaking the flasks at 160 rpm for 2 h. Serial dilutions were prepared from 10–2 to 10–6, and 100 μL aliquots (10–4 to 10–6) were chosen to spread on Tryptone Soya Agar (TSA), and the plates were inoculated at 28°C to isolate the culturable bacteria. Three replications were carried out for each concentration. One hundred microliters of sterile water was used as a blank control. The plates were incubated for 2–5 days at 28°C. Morphologically different colonies that appeared in the medium were isolated and purified on plates containing TSA medium. The isolated strains were routinely subcultured every 4 weeks and stored in TSA medium amended with 25% glycerol at −80°C (Navarroródenas et al., 2016). The physiological and biochemical characteristics of the bacterial isolates were examined according to the methods described in Bergey’s Manual of Determinative Bacteriology Eighth Edition (Balows, 1975), and the bacterial isolates were also identified based on 16S rDNA sequencing. Bacterial DNA was extracted with a Genomic DNA Extraction Kit (TIANGEN, Beijing, China), and 16S rDNAs were amplified with primers 27F (5′AGAGTTTGATCMTGGCTCAG3′) and 1492R (5′TACGGYTACCTTGTTACGACTT3′) primers (Loaces et al., 2011).
Soil Analysis
The soil was collected with ectomycorrhizal roots from sites where truffle fruiting occurred, including Hebei, Liaoning, Yunnan, and Heilongjiang Provinces. The samples were dried and used to determine the soil properties. Analyses were performed with standard protocols at the Institute of Plant Nutrition and Resource, Beijing Academy of Agriculture and Forestry Sciences and also followed the methods of Benucci et al. (2016). Organic matter (OM), total nitrogen (N), assimilable phosphorus (P), exchangeable potassium (K), magnesium (Mg), calcium (Ca), soil granularity and pH were measured. Significant differences between different sites were tested using one-way ANOVA.
Characterization of Nitrogen Fixation and Phosphorus-Solubilizing Traits
For the cultured bacteria isolated from T. indicum, nitrogen-fixation activity was tested in two ways: whether it can grow on Burk’s N-free medium (Wilson and Knight, 1974; Park et al., 2005), and whether nifH gene can be amplified by PolF/PolR primer set on bacterial DNA (Poly et al., 2001). A single colony of each purified strain was spotted on National Botanical Research Institute’s phosphate growth medium (NBRIP) plates, and observed for halo/zone formation (Ramachandran et al., 2007; Dastager et al., 2011). Furthermore, the nitrogen-fixation activity of those bacteria growing on N-free medium was verified with an ARA in the State Key Laboratory of Agricultural Microbiology of Huazhong Agricultural University according to previously described methods for legume root nodules (Wych and Rains, 1978; Das and De Kumar, 2018) and combined with a modified method by Barbieri et al. (2010). In brief, the cultured bacteria strains were incubated for 2 h at 28°C in 30 ml glass bottles containing 2 ml of C2H2. The amount of ethylene was measured using an East & West Analytical Instrument GC 4000A gas chromatograph.
Results
The Morphological Characteristics and Phylogenetic Relationship Between the Analyzed Samples of T. indicum
Tuber indicum is a typical Chinese black truffle species with great morphological variation and wide distribution. In this study, we collected 120 fruiting bodies of T. indicum from nine sites across Northeastern and Southwestern China, including Zhaotong County, Qujing City, Gongshan County (Yunnan Province), Liangshan County (Sichuan Province), Linzhi County (Tibet), Zunhua City (Hebei Province), Mudanjiang City and Qiqihar City (Heilongjiang Province) and Chaoyang City (Liaoning Province) (Figure 1). The morphological characteristics and phylogenetic relationships of the representative samples of T. indicum analyzed in our study are shown in Figure 2. All samples collected from the nine sites had similar morphological features, namely, black ascomata with conspicuous warts, ellipsoidal ascospores with sparse spine. Based on ITS sequencing and phylogenetic analysis, all samples analyzed belong to the same clade with high bootstrap values (100%), indicating the relatively consistent genetic background of all samples.
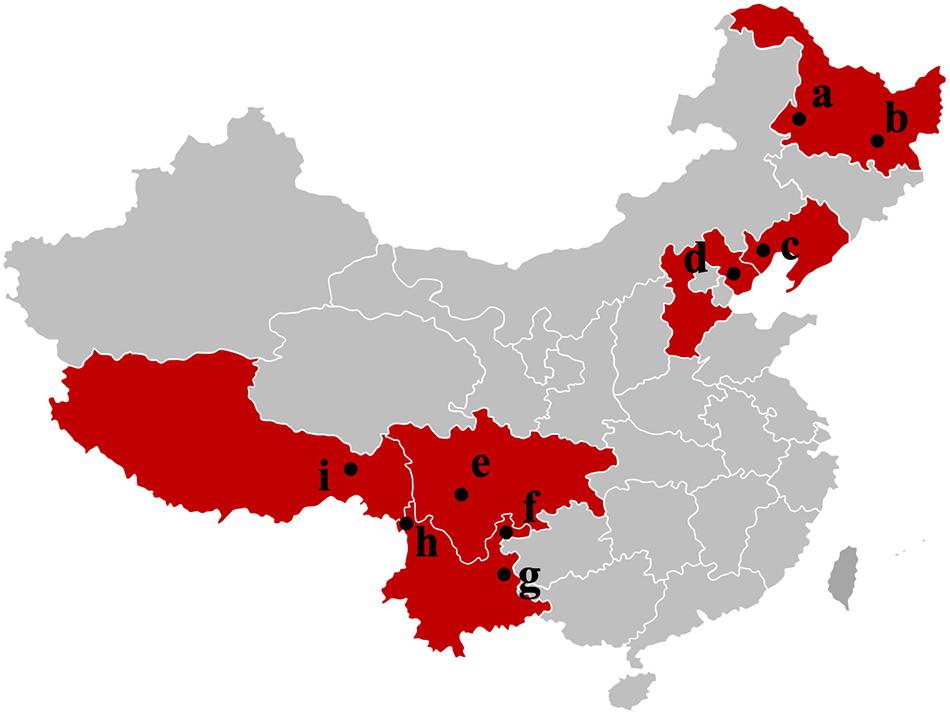
Figure 1. Samples map of T. indicum fruiting bodies collected in this study. a: Qiqihar city, Heilongjiang Province; b: Mudanjiang city, Heilongjiang Province; c: Chaoyang city, Liaoning Province; d: Zunhua city, Hebei Province; e: Liangshan Yi Autonomous Prefecture, Sichuan Province; f: Zhaotong city, Yunnan Province; g: Qujing city, Yunnan Province; h: Gongshan Derung and Nu Autonomous county, Yunnan Province; i: Linzhi city, Tibet.

Figure 2. Morphological characteristics of ascocarps and ascospores of T. indicum (A) and phylogenetic analysis of all samples from different regions of China based on ITS sequence using the Neighbor-joining analysis (B). Morphological characters of ascocarps (black with conspicuous warts) (A1–A7) and ascospores (brown to dark, ornamented with spine) (B1–B7), respectively. Samples collected from Gongshan Derung and Nu Autonomous County, Yunnan Province (A1,B1); Zunhua County, Hebei Province (A2,B2); Liaoning Province (A3,B3); Mudanjiang City, Heilongjiang Province (A4,B4); Qiqihar City, Heilongjiang Province (A5,B5), Qujing County, Yunnan Province (A6,B6); Tibet (A7,B7). (A8,B8) showed the host tree: Quercus mongolica and Castanea mollissima, respectively. (B) The neighbor-joining tree based on ITS sequences of T. indicum from different regions. △HB1-HB3: samples from Hebei Province; ■LN1-LN3: samples from Liaoning Province; ⚫QJ1-QJ3: samples from Qujing county, Yunnan Province; ▲MDJ1-MDJ3: samples from Mudanjiang, Heilongjiang Province; ○GS1-GS3: samples from Gongshan, Yunnan Province; □XZ2-1-XZ2-3: samples from Linzhi city, Tibet; ▼QQ2, QQ4: samples from Qiqihar, Heilongjiang Province; ▽SC1-3: samples from Liangshan, Sichuan Province.
Diversity of T. indicum Bacterial Communities From Different Regions
High throughput sequencing of the V3 + V4 region of 16S rDNA was carried out on 54 fruiting bodies of T. indicum (3–9 replicates each site) with the Illumina HiSeq platform. A total of 3981692 reads were obtained from the representative fruiting bodies of T. indicum from the nine sites. Although it did not capture the entire community, the rare faction curve and Shannon curve indicated that the sequencing depth basically covered the bacterial species of the samples (Supplementary Figure S1). After quality filtering, 3450293 sequences were clustered into 695 OTUs. The results of OTUs taxonomy and relative abundances are listed in Table 1. Comparatively, the bacterial communities of T. indicum ascocarps from different regions showed that the number of OTUs was highest in the Hebei region (310 ± 11) and the lowest in the Liaoning region (274 ± 18), but no significant differences were observed between the samples from different geographic locations. The Shannon index of the bacterial community was highest in samples from Yunnan Province (3.5039 ± 0.0418) and significantly different from other regions. All of the OTUs were distributed across 20 phyla, 153 families, and 307 genera. Among the 307 genera, 180 genera were shared between samples from different regions, no specific genus was distributed in the Sichuan, Tibet and Liaoning regions, and only 2 genera were specific in samples from Yunnan and Hebei Province, and 11 genera exhibited geographical enrichment in Heilongjiang samples, including Barnesiella, Allobaculum, Nitrosomonas, Plasticicumulans, Rheinheimera, and other unclassified bacteria (Figure 3A).
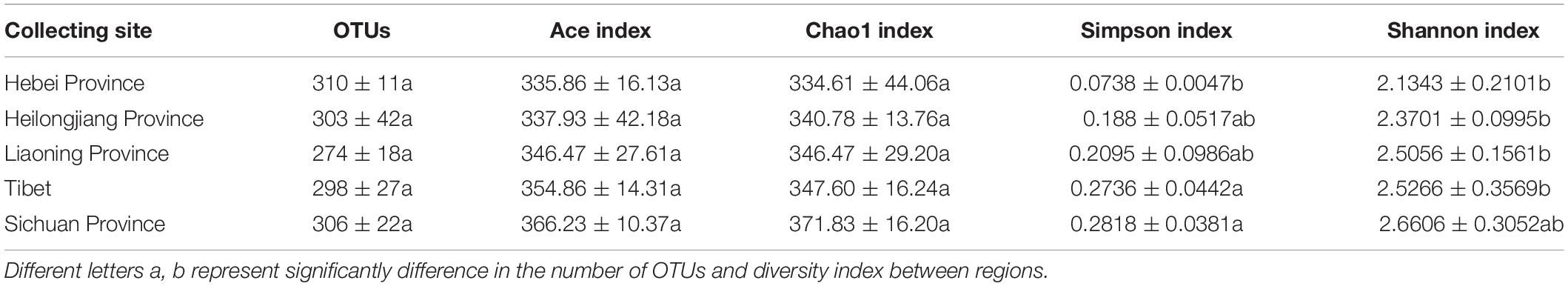
Table 1. Richness estimates and diversity indices of the bacterial communities in fruiting bodies of T. indicum produced from different regions of China.
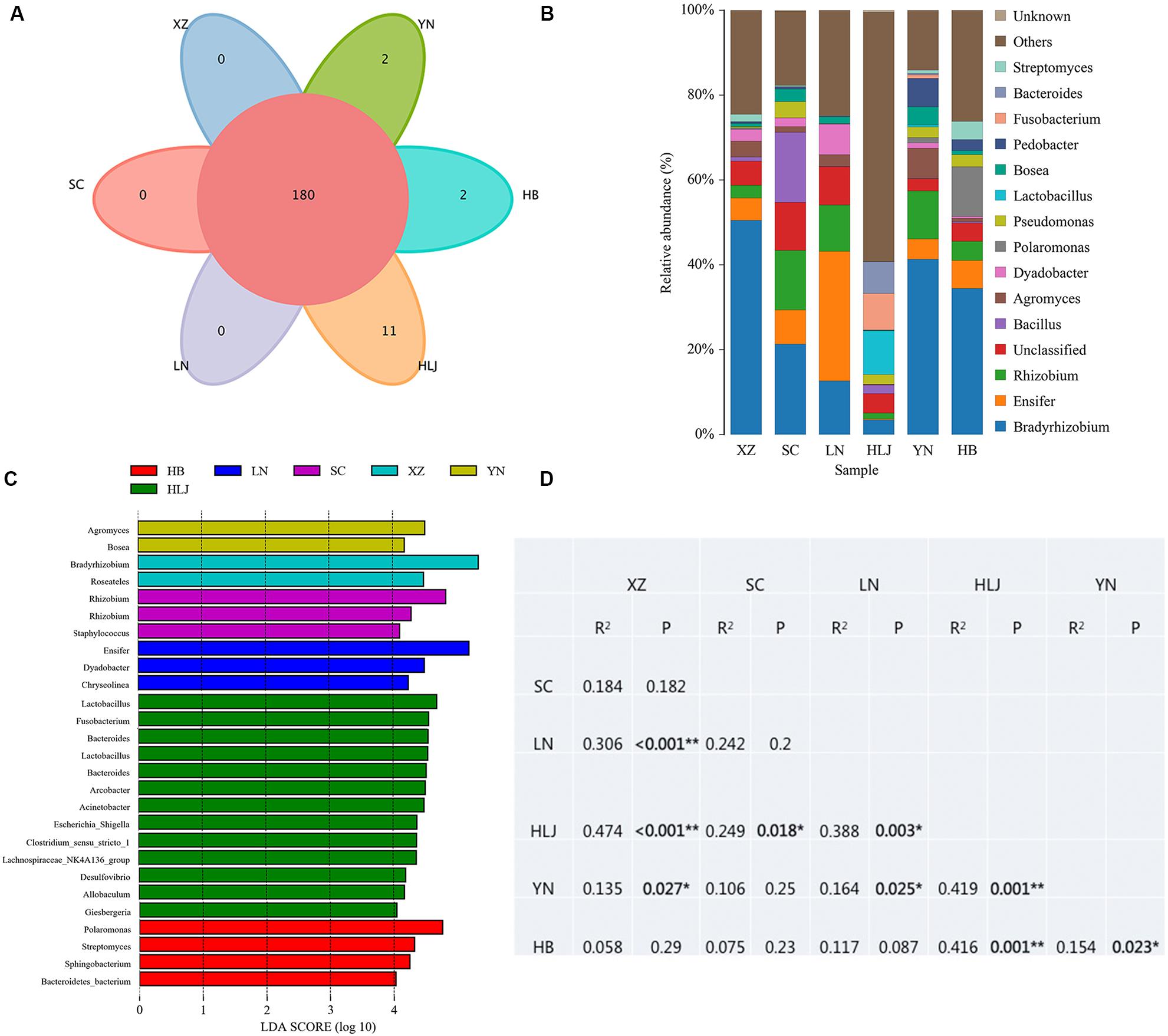
Figure 3. Effect of geographical location on bacterial composition of fruiting bodies of T. indicum. (A) Venn diagram showed the shared bacteria community across geographic locations at the genus level; (B) Bacteria relative abundance at the genus level across geographic regions; (C) Differentially abundant genus in fruiting bodies of T. indicum depending on geographical regions, as inferred by the LEfSe algorithm. The threshold for the logarithmic discriminant analysis (LDA) score was 4, with a P value of <0.05. (D) PERMANOVA analysis shows that the bacterial communities are significantly different in the fruiting bodies of T. indicum from different regions of China. HB, Hebei Province; LN, Liaoning Province; SC, Sichuan Province; XZ, Tibet; YN, Yunnan Province.
Impact of Geographical Location on Bacteria Composition of T. indicum Fruiting Bodies
The result of OTUs taxonomy at the phylum level showed that the relative abundance of Proteobacteria was the highest (36.90–80.79%) in all samples of T. indicum ascocarps from different geographic regions, followed by Firmicutes (0.04–37.20%), Bacteroidetes (4.44–14.85%), and Actinobacteria (1.82–10.36%). However, the richness of dominant bacteria was different in T. indicum ascocarps across different regions. For example, the relative abundance of Fusobacteria (8.64%) was significantly higher and Proteobacteria (36.90%) and Actinobacteria (1.82%) were lower in samples from Heilongjiang Province compared to those in other regions. Firmicutes was more abundant in samples from Heilongjiang (37.20%) and Sichuan (19.30%) than in other regions, and the difference was significant (P < 0.05).
The analysis of OTUs taxonomy at the genus level found that Bradyrhizobium (2.66–47.66%) was the most abundant genus in the bacterial communities of T. indicum fruiting bodies from different regions, followed by Ensifer (0.13–30.03%), Rhizobium (1.08–13.89%), Bacillus (1.21–16.87%), and Agromyces (0.83–7.05%) (Figure 3B). The 10 most observed genera in the fruiting bodies of T. indicum from 6 different regions demonstrated that Bradyrhizobium, Lactobacillus, Ensifer, Rhizobium, Agromyces, Pseudomonas, Streptomyces, Pedobacter, Acinetobacter, Bosea, and Pedobacter are significantly enriched in T. indicum fruiting bodies from different geographic regions (Table 2). LEfSe showed significant differences in bacterial groups between regions (Figure 3C). Agromyces and Bosea were more abundant in Yunnan samples, Bradyrhizobium and Bosea were more prevalent in Tibet samples, Rhizobium and Staphylococcus were more enriched in Sichuan samples, Ensifer, Dyadobacter, and Chryseolinea were more abundant in Liaoning samples, Fusobacterium, Lactobacillus, Bacteroides and Acinetobacter were more enriched in Heilongjiang samples, and Polaromonas, Streptomyces and Sphingobacterium were more prevalent in Hebei samples. Moreover, the effects of geographic location on the bacterial composition of T. indicum ascocarps were analyzed using permutational multivariate analysis of variance (PERMANOVA), and the result reflected that the bacterial communities of Tuber fruiting bodies were significantly different across geographical locations (P = 0.005) (Figure 3D).

Table 2. List of the ten most observed bacterial genera associated with T. indicum fruiting bodies from different regions.
Similar to the previous studies, we also confirmed that Bradyrhizobium spp. were dominant bacteria in the FBs of T. indicum regardless of geographic origin. To compare the phylogenetic relationship between the Bradyrhizobium 16S rDNA sequences generated in our study and those published by other authors (Benucci and Bonito, 2016), which were found in the Tuber ascomata on several different continents, we conducted a phylogenetic analysis, and the result suggested that the Bradyrhizobium 16S rDNA sequences detected in our study (OTU1) were 100% identical to those previously published (KU193064 OTU8), implying that the single Bradyrhizobium species is a specific microbe marker in true truffle microbiomes and their potential for tight symbiotic interactions (Figure 4).
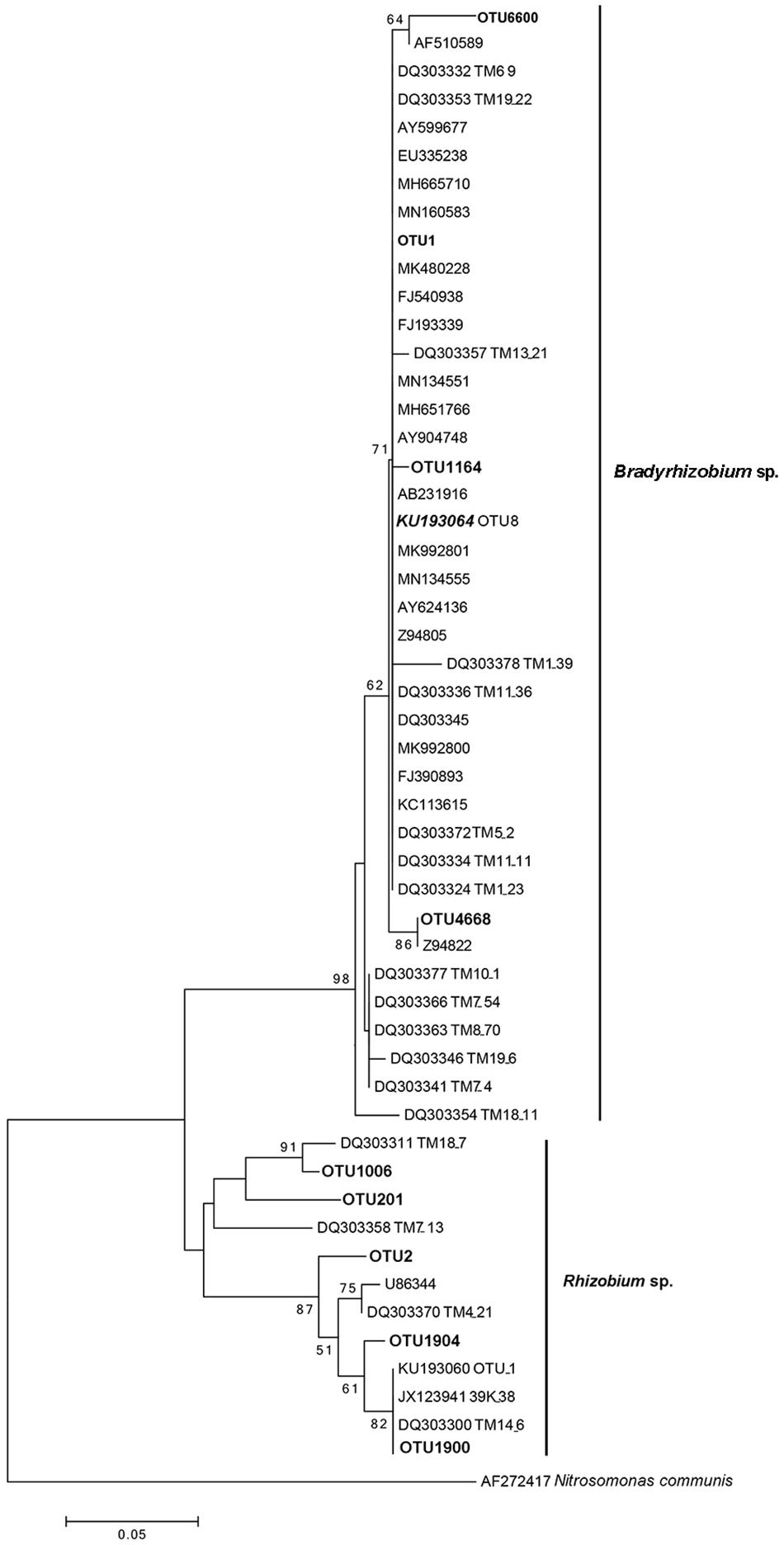
Figure 4. Neighbor-joining phylogenetic tree for 16S rDNA V3 + V4 region sequences of Bradyrhizobium spp. and Rhizobium spp. from T. indicum fruiting bodies. The number at each branch node are the bootstrap numbers from 1000 replicates. OTU1 (dominant taxa), OTU1164, OTU4668 and OTU6600 in bold, belonging to Bradyrhizobium and OTU2, OTU201, OTU1006, OTU 1900, and OTU 1904 belonging to Rhizobium in this study. KU193064 (OTU8) was regarded as marker Bradyrhizobium and specific present in Tuber species (Benucci and Bonito, 2016) and sequences of the accession number marked “DQ” were from T. magnatum (Barbieri et al., 2007).
Soil properties of the five sites from the four provinces were measured including organic matter (OM), total nitrogen (N), assimilable phosphorus (P), exchangeable potassium (K), calcium (Ca), magnesium (Mg), pH and soil granularity. The results showed that there were significant differences in the contents of Ca and Mg in the Heilongjiang samples but were not significant differences in the contents of K and pH between sites (Table 3). The highest contents of total nitrogen (5.53 ± 0.81), organic matter (113.52 ± 16.44) and effective phosphorus (31.9 ± 10.90) were observed in the Heilongjiang region. The content of changeable magnesium in Hebei (1.71 ± 0.10) was significantly lower than that in other sites (2.37–3.14). Redundancy analysis (RDA) of the correlation between microbial composition and soil property showed that Bradyrhizobium, Rhizobium, Ensifer, and Agromyces had a positive correlation with soil granularity (0.25 mm ≤ x < 2.00 mm and 0.002 mm ≤ x < 0.02 mm), Lactobacillus and Bacteroides had a positive correlation with the contents of N, Ca, K, Mg, and P and Acinetobacter and Pseudomonas displayed a positive correlation with the pH value (Supplementary Figure S2). In summary, bacterial compositions of T. indicum fruiting bodies were different from different geographical locations, and the soil parameters are possibly important contributors to the differences in the microbiome.
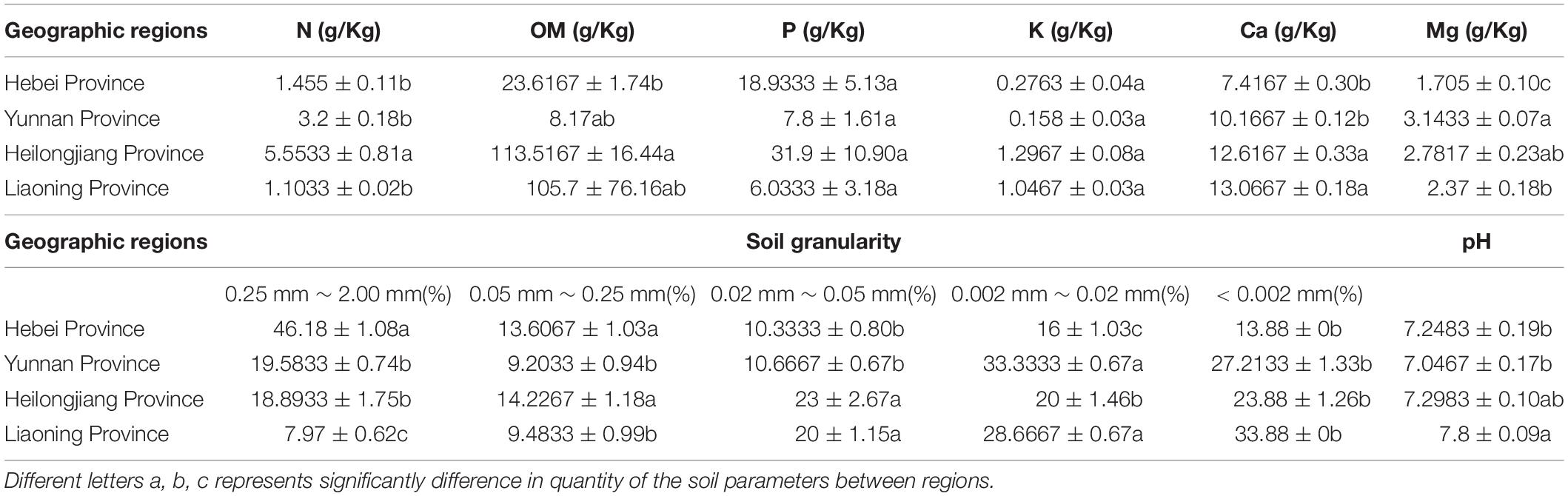
Table 3. Differences in soil characteristics of T. indicum among geographic sites after one-way ANOVA analysis.
Difference in the Bacterial Community Among Ascocarps, Ectomycorrhizae and Soil of T. indicum
In addition to exploring the bacterial composition of fruiting bodies of T. indicum from different geographical regions, we also determined the difference in bacterial composition colonizing the fruiting body (FB), ectomycorrhizae (ECM) and ectomycorrhizosphere soil of T. indicum on samples collected in September 2018 at the same time, from the same sites and from the same host tree (Castanea mollissima). Based on 16S rDNA V3 + V4 region sequencing, we sequenced 24 samples comprising 6 soils, 9 ectomycorrhizaes (more than 300 root tips) and 9 FBs samples, generating 2419871 high-quality reads. After filtering, we obtained 64693 clean tags and assigned 1503 OTUs, and the ectomycorrhizosphere soil samples contained more OTUs than the ECM and FB of samples T. indicum. The highest Shannon index value was observed in ectomycorrhizosphere soil (6.08 ± 0.03), followed by ectomycorrhizae (2.85 ± 0.19) and ascocarps (2.55 ± 0.16), indicating that the bacterial diversity was significantly different among the soil, fruiting bodies and ectomycorrhizae of T. indicum. All of the OTUs were distributed across 21 phyla, 218 families and 384 genera. A lot of 338 of 384 genera were shared between different compartments, and no specific genus was detected in the ECM. The numbers of specific genera in the FBs and soils were 7 and 11, respectively (Figure 5A).
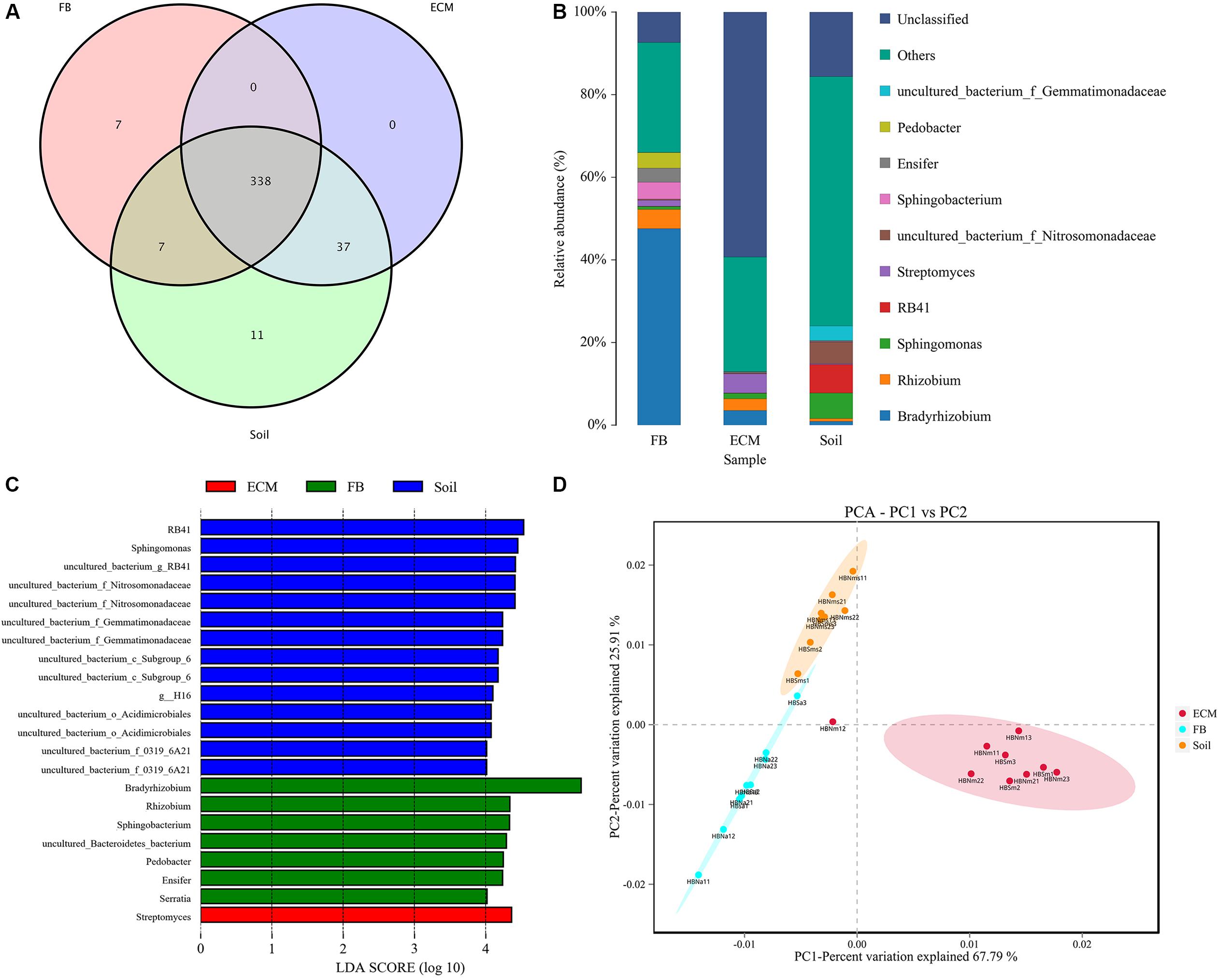
Figure 5. Effect of different compartments (Soil, ECM, and FB) on bacterial composition of T. indicum. (A) Venn diagram showed the shared bacterial community across three compartments at the genus level; (B) Bacterial relative abundance at the genus level across three compartments; (C) Differentially abundant genus in different compartments of T. indicum inferred by the LEfSe algorithm. The threshold for the logarithmic discriminant analysis (LDA) score was 4, with a P value of <0.05. (D) PCA analysis shows relationships in bacterial composition among samples belonging to different compartments and three compartments display significant different. ECM, Ectomycorrhiza; FB, Fruiting bodies.
The OTUs taxonomy results of three compartments of T. indicum at the phylum level showed that T. indicum FBs harbored a core bacterial community that was mainly composed of Proteobacteria (76.95%) and Bacteroidetes (12.90%); the ECM of T. indicum was dominated by Proteobacteria (30.17%) and Actinobacteria (12.25%), while soil samples contained mainly Proteobacteria (44.26%) and Acidobacteria (26.04%). At the genus level, all OTUs from three compartments were assigned to 384 genera. Bradyrhizobium (46.22%) and Rhizobium (5.29%) were significantly enriched in the FBs, Streptomyces (4.65%), Bradyrhizobium (3.60%) and Sphingomonas (1.35%) were prevalent in the ECM, while RB41 (7.97%), Sphingomonas (6.26%) and uncultured bacteria were dominated in the soil (Figure 5B). The result of LEfSe analysis showed significant differences in bacterial groups among the three compartments (Figure 5C). RB41, Sphingomonas and members of Nitrosomonadaceae and Gemmatimonadaceae were more enriched in the soil samples, Streptomyces were more abundant in the ECM samples, and Bradyrhizobium, Rhizobium, Pedobacter, Ensifer, and Serratia were more prevalent in the FB samples. PCA among three main compartments (FB, ECM, and soil) also revealed that the bacterial communities in soil were separated from the other two compartments according to the first axis, which explained 67.79% of the variability, and from those of the ectomycorrhzae according to the second axis, which explained 25.91% of the variability (Figure 5D).
The Cultural Bacterial Communities
A total of 60 bacterial colonies were isolated from ethanol-sterilized fruiting bodies of T. indicum by the tissue culture method. The 60 colonies were gathered from 26 different strains according to morphological and molecular characteristics. The 26 strains were divided into 4 phyla and 24 genera based on 16S rDNA sequencing (Supplementary Figure S3). Among them, the largest phylum was Proteobacteria (43 strains, 71.67%), including Pseudomonas (9 strains), Alcaligenes (5 strains), Ensifer (4 strains), Inquilinus (3 strains), Mesorhizobium (3 strains), Roseomonas (3 strains), Variovorax (3 strains), Rhizobium (2 strains), Acinetobacter (2 strains), Enterobacter sp. (2 strains), Stenotrophomonas (2 strains) and five other genera with only one strain. Moreover, the members of Actinobacteria were also dominant in the cultural bacteria from T. indicum fruiting bodies (21.67%), including Microbacterium (5 strains), Arthrobacter (5 strains) and three other genera. A total of three strains of Elizabethkingia (1 strain) and Sphingobacterium (2 strains) belonging to Bacteroidetes consisted of 5%, and Bacillus (1 strain) and Paenibacillus (1 strain) belonging to Firmicutes consisted of 3.33% (Table 4). All of these bacteria members were also found dominant in the fruiting bodies of T. indicum using high-throughput sequencing, confirming their presence in fruiting bodies. All bacterial strains were deposited in the Mycological lab of the Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences.
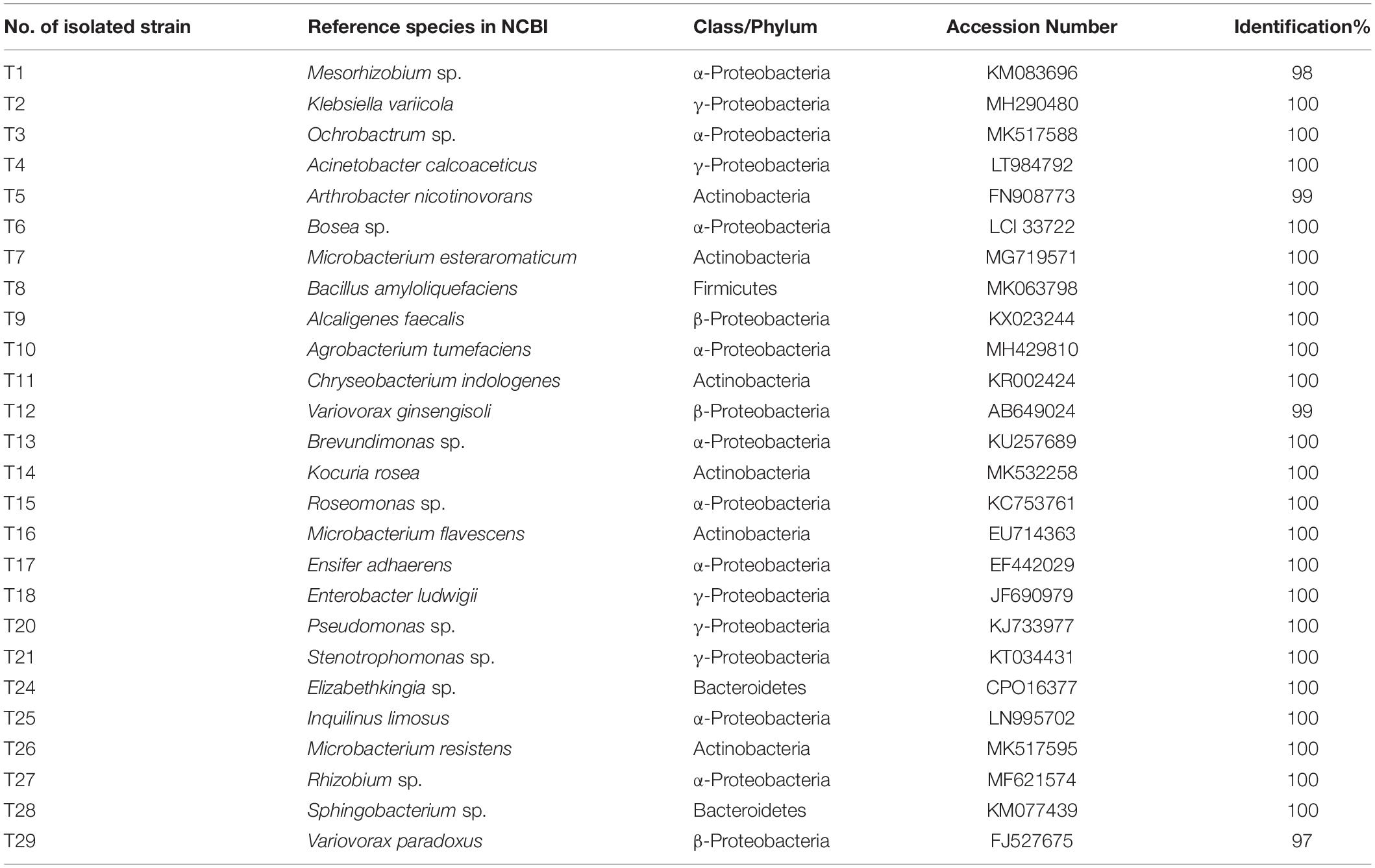
Table 4. Identification information about 26 strains cultural bacteria from fruiting bodies of T. indicum.
Nitrogen-Fixing Activity and Phosphorus-Solubilizing Traits
Out of the 26 tested strains, 13 strains (50%) were found to grow in Burk’s N-free medium including T1-T6, T8-T9, T15, T17-T18, T20, and T27, which belong to Proteobacteria and Firmicutes, respectively (Supplementary Figure S4). The nitrogen fixation activity of one strain (T20) was verified by enzymatic ARA test (Supplementary Figure S5). Eight strains harbored the nifH gene, which was determined by PCR amplification using specific primers, including strains T1, T2, T4, T8, T13, T18, T20, and T21 (Supplementary Figure S6). Only one strain, T4 (Acinetobacter calcoaceticus) belonging to γ-Proteobacteria showed the highest phosphate solubilization activity. Strain T4 exhibited two different activities: phosphate solubilization and nitrogen fixation bioactivity.
Discussion
Unlike other edible mushroom, truffles are produced and developed underground during the entire lifecycle. In recent years, soil microbes associated with mycorrhizae and fruiting bodies of several commercial species of Tuber have been investigated based on culture-dependent and high-throughput sequencing, including T. borchii, T. melanosporum, T. magnatum, T. aestivum, and T. brumale (Gazzanelli et al., 1999; Barbieri et al., 2005; Buzzini et al., 2005; Antony-Babu et al., 2014; Deveau et al., 2016). These results indicated that a considerable number of bacteria colonize truffle fruiting bodies, while Bradyrhizobium, Variovorax, Pedobacter, and Sphingobacterium are usually enriched in fruiting bodies of T. melanosporum (Antony-Babu et al., 2014). Similar to T. melanosporum, T. indicum was enrichment in a core set of bacterial taxa despite of the significant difference in composition of the six different geographical regions investigated. Bradyrhizobium, Ensifer, Rhizobium, Bacillus, Agromyces, Dyadobacter, and Polaromonas were dominant in T. indicum fruiting bodies. The enriched core bacteria in T. indicum fruiting bodies overlap with the reported core set for T. melanosporum (Antony-Babu et al., 2014), T. aestivum (Gryndler et al., 2013), T. magnatum (Barbieri et al., 2007), T. panzhihuaensis (Wan and Liu, 2014), T. lyonii and T. oregonense (Benucci et al., 2016), which placed in different clades in the phylogenetic tree, raising the possibility that a number of bacterial genera (e.g., Bradyrhizobium, Ensifer, Sphingobacterium, and Rhizobium) have had a long association with truffle, at least since the divergence of truffle from other fungal taxa in approximately 271 million years ago, implying some core bacterial communities most likely co-evolution with truffle (Jeandroz et al., 2008). Moreover, Pseudomonas has been reported to be enriched in white truffle T. borchii and T. magnatum (ascocarp surface white and no conspicuous warts) with high frequency (Bedini et al., 1999; Barbieri et al., 2007), but in black truffle, T. melanosporum is rarely observed (Mello et al., 2013; Antony-Babu et al., 2014). Similarly, a very low proportion of Pseudomonas was detected in T. indicum in our study with a culture-independent method.
Interestingly, the most representative Bradyrhizobium sp. (very closely related to B. elkanii) (OTU1) in T. indicum FBs in our study are identical to those described in other Tuber species (KU193064 OTU8) by Benucci and Bonito (2016), which were found to be dominant specifically in the genus Tuber irrespective of geographic origin but not in other truffle genera. Our results verified that Bradyrhizobium (OTU1) is consistently present in all T. indicum FBs collected from various locations although the proportion is different in each tested samples. Moreover, Bradyrhizobium and Rhizobium are important root-associated microbes known for their nitrogen-fixing capability (Hara et al., 2019). In this study, we detected that the T. indicum FBs have nitrogen-fixing activities and suspected that the activity is probably related to the presence of Bradyrhizobium spp. as previously reported in T. magnatum (Barbieri et al., 2010). Thus, the potential effects of nitrogen-fixing bacteria on the development of truffle in cultivation should be considered.
Deng et al. (2018) surveyed the bacterial community of T. indicum fruiting bodies from Yunnan and Sichuan Province using 454 pyrophosphate sequencing and found 220 OTUs belonging to 7 phyla and 43 genera. In this study, we explored more abundant bacterial taxa (20 phyla and 307 genera) in the fruiting bodies of T. indicum samples collected from different geographical regions, including samples collected from Yunnan and Sichuan Provinces. In addition to, Flavobacterium, Agromyces, Microbacterium, Ensifer, Sphingopyxis, and Variovorax are dominant in T. indicum fruiting bodies according to previous reports. In the present study, Bradyrhizobium (2.66–47.66%), Rhizobium (1.08–13.89%), and Bacillus (1.21–16.87%) etc., are also dominant in T. indicum fruiting bodies. Our survey substantially increases the number of bacterial taxa enriched in T. indicum fruiting bodies and emphasizes the important effect of the extensive sampling on bacterial diversity, as mentioned by Splivallo et al. (2019). Furthermore, PERMANOVA analysis also supported the difference in bacterial abundance in different samples from various geographic locations. The correlation analysis of RDA between soil propriety and microbial composition indicated that the enrichment of the bacterial community in the truffle was affected by soil physical and chemical characters, which probably contribute to the geographical difference of the microbes in the T. indicum fruiting bodies.
The microbial community are also different between different compartments of T. indicum, such as soil, ectomycorrhizae and fruiting bodies. Bradyrhizobium appeared to be more prevalent in the FBs (47.6%) than in the ECM (3.53%) or the soil (less than 1%). This result is consistent with previous result in other truffle species such as T. melanosporum (Antony-Babu et al., 2014) and T. aestivum (Gryndler et al., 2013). Similar to T. melanosporum, Rhizobium, and Streptomyces are dominant in the microbial composition of T. indicum ECM. A possible explanation is that FBs of truffle are site for accumulation of carbohydrate, protein and lipid, while ECM is a place where small molecules substances transfer and exchange; and the different metabolites and functions present in the ECM and FB possibly resulted in the difference in the microbial communities in the two parts. Overall, different compartments of T. indicum are enriched with different bacteria, indicating that T. indicum has selectivity for bacterial colonization during their development.
Plant growth-promoting rhizobacteria (PGPR) are acknowledged as new and promising biotechnology tools in agriculture field for enhancing nursery plant production, the growth of mycorrhizal plants, and sporocarp yield in recent years (Bhattacharyya and Jha, 2012; Navarroródenas et al., 2016). Their effect on the truffle mycorrhizal synthesis and mycelium growth were also reported (Dominguez et al., 2012; Gryndler et al., 2013; Navarroródenas et al., 2016). In our study, we isolated a total of 60 bacterial colonies from fruiting bodies of T. indicum and represented 26 different strains based on morphological traits and molecular sequences. Molecular identification using the 16S rDNA sequence divided the strains into 4 phyla and 24 genera, of which the largest were Pseudomonas of the Proteobacteria phylum (15% of the isolated strains), although a low abundance was detected in T. indicum FBs by 16S rDNA high-throughput assay. None of the members of Bradyrhizobium sp., the prevalent bacteria in the FBs of T. indicum using high-throughput sequencing, were isolated and cultured in artificial medium, confirming the complementary advantage of methodologies for deciphering the bacterial communities of truffle.
The bioactivity analysis displayed that 13 strains of 26 different taxa can grow in Burk’s N-free medium, implying the potential ability of nitrogen fixation, including Mesorhizobium sp., Klebsiella variicola, Acinetobacter calcoaceticus, Arthrobacter nicotinovorans, Roseomonas sp., Ensifer adhaerens, Pseudomonas sp., and Bacillus amyloliquefaciens etc., Pseudomonas and Bacillus have been the most reported as PGPR bacteria (Islam et al., 2014; Navarroródenas et al., 2016). Only one strain, Acinetobacter calcoaceticus, displayed the ability to solubilize phosphate and fix nitrogen in this study. These bacteria may be potential candidates for the formation and development of ectomycorrhizae and fruiting bodies of truffle, and the exact ecological function of these bacteria will be further validated in the next step by inoculating experiments on truffle mycorrhizal seedlings.
In conclusion, our results provide a comprehensive view of bacteria communities of T. indicum FBs from different geographic areas. We confirmed the dominance of Bradyrhizobium sp. in FBs of T. indicum and verified its close phylogenetic relationship with those in other Tuber species, regardless the geological location even across the continent. The core bacteria most likely coevolved with truffle and play an important role in the nutrition requirement and adaptation to the environment during truffle development. The diversity of bacterial communities is similar in FBs of T. indicum but relative abundance and richness are different from different geographical origins. The bacterial composition colonized in mycorrhizosphere soil, ectomycorrhizae, and fruiting bodies of T. indicum are significantly different. Partial representative bacterial strains in fruiting bodies of T. indicum, which were detected using a culture-independent method, were also isolated and cultured, confirming their occurrence during the development of fruiting bodies of T. indicum to some extent. Some cultural bacteria strains exhibited the potential abilities of nitrogen fixation and inorganic phosphorus solubilization, suspecting that these bacteria play important function in truffle seedling growth, fruiting body development and even truffle evolution process. Overall, this study provides important data for understanding the interaction between bacterial communities and fruiting bodies of T. indicum and provides useful microbe resource for T. indicum cultivation in future.
Data Availability Statement
The datasets generated for this study can be found in the NCBI database with GenBank accession numbers MK346212–MK346219.
Author Contributions
JC and S-XG designed the experiments. J-ML and Y-JT performed the experiments. PQ, P-GL, Y-MX, and YL collected the samples. JC and J-ML analyzed the data, and contributed to the text writing.
Funding
This study was supported by the National Natural Science Foundation of China (81573527 and 31500016), National Key R&D Programmes of China (2017YFC1701900), and National Basic Research Program of China (2014CB138304).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We are grateful to Prof. Francis Martin and Prof. Fu-Cheng Lin for critical discussion and suggestions. We thank Dr. Gregory M. Bonito for providing the microbial sequencing of Bradyrhizobium sp.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fmicb.2019.02515/full#supplementary-material
References
Amicucci, A., Barbieri, E., Sparvoli, V., Gioacchini, A. M., Calcabrini, C., Palma, F., et al. (2018). Microbial and pigment profile of the reddish patch occurring within Tuber magnatum ascomata. Fungal Biol. 122, 1134–1141. doi: 10.1016/j.funbio.2018.07.007
Anderson, M. J. (2010). A new method for non-parametric multivariate analysis of variance. Aust. Ecol. 26, 32–46. doi: 10.1111/j.1442-9993.2001.tb00081.x
Antony-Babu, S., Deveau, A., Van Nostrand, J. D., Zhou, J., Le Tacon, F., Robin, C., et al. (2014). Black truffle-associated bacterial communities during the development and maturation of Tuber melanosporum ascocarps and putative functional roles. Environ. Microbiol. 16, 2831–2847. doi: 10.1111/1462-2920.12294
Antony-Babu, S., Murat, C., Deveau, A., Le Tacon, F., Frey-Klett, P., and Uroz, S. (2013). An improved method compatible with metagenomic analyses to extract genomic DNA from soils in Tuber melanosporum orchards. J. Appl. Microbiol. 115, 163–170. doi: 10.1111/jam.12205
Balows, A. (1975). Bergey’s manual of determinative bacteriology. Eighth Edition. Am. J. Public Health 65:315. doi: 10.2105/AJPH.65.3.315-a
Barbieri, E., Bertini, L., Rossi, I., Ceccaroli, P., Saltarelli, R., Guidi, C., et al. (2005). New evidence for bacterial diversity in the ascoma of the ectomycorrhizal fungus Tuber borchii Vittad. FEMS Microbiol. Lett. 247, 23–35. doi: 10.1016/j.femsle.2005.04.027
Barbieri, E., Ceccaroli, P., Saltarelli, R., Guidi, C., Potenza, L., Basaglia, M., et al. (2010). New evidence for nitrogen fixation within the Italian white truffle Tuber magnatum. Fungal Biol. 114, 936–942. doi: 10.1016/j.funbio.2010.09.001
Barbieri, E., Guidi, C., Bertaux, J., Frey-Klett, P., and Garbaye, J. (2007). Occurrence and diversity of bacterial communities in Tuber magnatum during truffle maturation. Environ. Microbiol. 9, 2234–2246. doi: 10.1111/j.1462-2920.2007.01338.x
Bedini, S., Bagnoli, G., Sbrana, C., Leporini, C., Tola, E., Dunne, C., et al. (1999). Pseudomonads isolated from within fruit bodies of Tuber borchii are capable of producing biological control or phytostimulatory compounds in pure culture. Symbiosis 26, 223–236. doi: 10.1016/S0014-5793(00)02103-7
Belfiori, B., Riccioni, C., Tempesta, S., Pasqualetti, M., Paolocci, F., and Rubini, A. (2012). Comparison of ectomycorrhizal communities in natural and cultivated Tuber melanosporum truffle grounds. FEMS Microbiol. Ecol. 81, 547–561. doi: 10.1111/j.1574-6941.2012.01379.x
Benucci, G. M. N., Bonito, G., Falini, L. B., Bencivenga, M., and Donnini, D. (2012). “Truffles, timber, food, and fuel: sustainable approaches for multi-cropping truffles and economically important plants,” in Edible Ectomycorrhizal Mushrooms. Soil Biology, Vol. 34, eds A. Zambonelli, and G. Bonito (Heidelber: Springer), 265–280. doi: 10.1007/978-3-642-33823-6_15
Benucci, G. M. N., and Bonito, G. M. (2016). The truffle microbiome: species and geography effects on bacteria associated with fruiting bodies of hypogeous pezizales. Microb. Ecol. 72, 4–8. doi: 10.1007/s00248-016-0755-3
Benucci, G. M. N., Lefevre, C., and Bonito, G. (2016). Characterizing root-associated fungal communities soils of Douglas-fir (Pseudotsuga menziesii) stands that naturally produce Oregon white truffles (Tuber oregonense and Tuber gibbosum). Mycorrhiza 26, 367–376. doi: 10.1007/s00572-015-0677-9
Bhattacharyya, P. N., and Jha, D. K. (2012). Plant growth-promoting rhizobacteria (PGPR): emergence in agriculture. World J. Microbiol. Biotechnol. 28, 1327–1350. doi: 10.1007/s11274-011-0979-9
Bonet, J. A., Fischer, C. R., and Colinas, C. (2006). Cultivation of black truffle to promote reforestation and land-use stability. Agron. Sustain. Dev. 26, 69–76. doi: 10.1051/agro:2005059
Bonito, G., Smith, M. E., Nowak, M., Healy, R. A., Guevara, G., Cázares, E., et al. (2013). Historical biogeography and diversification of truffles in the Tuberaceae and their newly identified southern hemisphere sister lineage. PLoS One 8:e52765. doi: 10.1371/journal.pone.0052765
Bonito, G. M., and Smith, M. E. (2016). “General systematic position of the truffles: evolutionary theories,” in True truffle (Tuber spp.) in the World. Soil Biology, Vol. 47, eds A. Zambonelli, M. Iotti, and C. Murat (Cham: Springer).
Buzzini, P., Gasparetti, C., Turchetti, B., Cramarossa, M. R., Vaughan-Martini, A., Martini, A., et al. (2005). Production of volatile organic compounds (VOCs) by yeasts isolated from the ascocarps of black (Tuber melanosporum Vitt.) and white (Tuber magnatum Pico) truffles. Arch. Microbiol. 184, 187–193. doi: 10.1007/s00203-005-0043-y
Chao, A., Chazdon, R. L., and Colwell, R. K. (2005). A new statistical approach for assessing similarity of species composition with incidence and abundance data. Ecol. Lett. 8, 148–159. doi: 10.1111/j.1461-0248.2004.00707.x
Chen, J., Guo, S. X., and Liu, P. G. (2011). Species recognition and cryptic species in the Tuber indicum complex. PLoS One 6:e14625. doi: 10.1371/journal.pone.0014625
Das, S., and De Kumar, T. (2018). Microbial assay of N2 fixation rate, a simple alternate for acetylene reduction assay. MethodsX 5, 909–914. doi: 10.1016/j.mex.2017.11.010
Dastager, S. G., Deepa, C. K., and Pandey, A. (2011). Plant growth promoting potential of Pontibacter niistensis in cowpea (Vigna unguiculata (L.) Walp.). Appl. Soil Ecol. 49, 250–255. doi: 10.1016/j.apsoil.2011.04.016
Deng, X., Liu, J., Yan, X., and Liu, P. (2018). Community composition of bacteria associated with ascocarps of Tuber indicum using traditional culture method and Roche 454 high-throughput sequencing. Biodivers. Sci. 26, 1318–1324. doi: 10.17520/biods.2018184
Deveau, A., Antony-Babu, S., Le Tacon, F., Robin, C., Frey-Klett, P., and Uroz, S. (2016). Temporal changes of bacterial communities in the Tuber melanosporum ectomycorrhizosphere during ascocarp development. Mycorrhiza 26, 389–399. doi: 10.1007/s00572-015-0679-7
Dominguez, J. A., Martin, A., Anriquez, A., and Albanesi, A. (2012). The combined effects of Pseudomonas fluorescens and Tuber melanosporum on the quality of Pinus halepensis seedlings. Mycorrhiza 22, 429–436. doi: 10.1007/s00572-011-0420-0
Edgar, R. C. (2010). Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461. doi: 10.1093/bioinformatics/btq461
Feng, B., Zhao, Q., Xu, J., Qin, J., and Yang, Z. L. (2016). Drainage isolation and climate change-driven population expansion shape the genetic structures of Tuber indicum complex in the Hengduan Mountains region. Sci. Rep. 6:21811. doi: 10.1038/srep21811
Gazzanelli, G., Malatesta, M., Pianetti, A., Baffone, W., Stocchi, V., and Citterio, B. (1999). Bacteria associated to fruit bodies of the ectomycorrhizal fungus Tuber borchii Vittad. Symbiosis 26, 211–222.
Geng, L. Y., Wang, X. H., Yu, F. Q., Deng, X. J., Tian, X. F., Shi, X. F., et al. (2009). Mycorrhizal synthesis of Tuber indicum with two indigenous hosts, Castanea mollissima and Pinus armandii. Mycorrhiza 19, 461–467. doi: 10.1007/s00572-009-0247-240
Gryndler, M., Soukupová, L., Hršelová, H., Gryndlerová, H., Borovic̆ka, J., Streiblová, E., et al. (2013). A quest for indigenous truffle helper prokaryotes. Environ. Microbiol. Rep. 5, 346–352. doi: 10.1111/1758-2229.12014
Hara, S., Morikawa, T., Wasai, S., Kasahara, Y., Koshiba, T., Yamazaki, K., et al. (2019). Identification of nitrogen-fixing bradyrhizobium associated with roots of field-grown sorghum by metagenome and proteome analyses. Front. Microbiol. 10:407. doi: 10.3389/fmicb.2019.00407
Hughes, J. B., Hellmann, J. J., Ricketts, T. H., and Bohannan, B. J. (2001). Counting the uncountable: statistical approaches to estimating microbial diversity. Appl. Environ. Microbiol. 67, 4399–4406. doi: 10.1128/aem.67.10.4399-4406.2001
Islam, F., Yasmeen, T., Ali, Q., Ali, S., Arif, M. S., Hussain, S., et al. (2014). Influence of Pseudomonas aeruginosa as PGPR on oxidative stress tolerance in wheat under Zn stress. Ecotoxicol. Environ. Saf. 104, 285–293. doi: 10.1016/j.ecoenv.2014.03.008
Jeandroz, S., Murat, C., Wang, Y., Bonfante, P., and Tacon, F. L. (2008). Molecular phylogeny and historical biogeography of the genus Tuber, the ‘true truffles’. J. Biogeography 35, 815–829. doi: 10.1111/j.1365-2699.2007.01851.x
Kinoshita, A., Obase, K., and Yamanaka, T. (2018). Ectomycorrhizae formed by three Japanese truffle species (Tuber japonicum, T. longispinosum, and T. himalayense) on indigenous oak and pine species. Mycorrhiza 28, 679–690. doi: 10.1007/s00572-018-0860-x
Kõljalg, U., Nilsson, R. H., Abarenkov, K., Tedersoo, L., Taylor, A. F., Bahram, M., et al. (2013). Towards a unified paradigm for sequence-based identification of fungi. Mol. Ecol. 22, 5271–5277. doi: 10.1111/mec.12481
Leonardi, M., Iotti, M., Oddis, M., Lalli, G., Pacioni, G., Leonardi, P., et al. (2013). Assessment of ectomycorrhizal fungal communities in the natural habitats of Tuber magnatum (Ascomycota, Pezizales). Mycorrhiza 23, 349–358. doi: 10.1007/s00572-012-0474-7
Li, Q., Yan, L., Ye, L., Zhou, J., Zhang, B., Peng, W., et al. (2018). Chinese Black Truffle (Tuber indicum) alters the ectomycorrhizosphere and endoectomycosphere microbiome and metabolic profiles of the host tree Quercus aliena. Front. Microbiol. 9:2202. doi: 10.3389/fmicb.2018.02202
Linde, C. C., and Selmes, H. (2012). Genetic diversity and mating type distribution of Tuber melanosporum and their significance to truffle cultivation in artificially planted truffieres in Australia. Appl. Environ. Microbiol. 78, 6534–6539. doi: 10.1128/AEM.01558-12
Liu, B., and Tao, K. (1996). New species and new records of hypogeous fungi from China VI. J. Shanxi Univ. 19, 184–186.
Loaces, I., Ferrando, L., and Scavino, A. F. (2011). Dynamics, diversity and function of endophytic siderophore-producing bacteria in rice. Microb. Ecol. 61, 606–618. doi: 10.1007/s00248-010-9780-9
Mcmurdie, P. J., and Susan, H. (2013). phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8:e61217. doi: 10.1371/journal.pone.0061217
Mello, A., Ding, G. C., Piceno, Y. M., Napoli, C., Tom, L. M., Desantis, T. Z., et al. (2013). Truffle brûlés have an impact on the diversity of soil bacterial communities. PLoS One 8:e61945. doi: 10.1371/journal.pone.0061945
Mori, H., Maruyama, F., Kato, H., Toyoda, A., Dozono, A., and Ohtsubo, Y. (2014). Design and experimental application of a novel non-degenerate universal primer set that amplifies prokaryotic 16S rRNA genes with a low possibility to amplify eukaryotic rRNA genes. DNA Res. 21, 217–227. doi: 10.1093/dnares/dst052
Murat, C. (2015). Forty years of inoculating seedlings with truffle fungi: past and future perspectives. Mycorrhiza 25, 77–81. doi: 10.1007/s00572-014-0593-4
Navarroródenas, A., Berná, L. M., Lozanocarrillo, C., Andrino, A., and Morte, A. (2016). Beneficial native bacteria improve survival and mycorrhization of desert truffle mycorrhizal plants in nursery conditions. Mycorrhiza 26, 1–11. doi: 10.1007/s00572-016-0711-6
Oksanen, J., Blanchet, F. G., Friendly, F., Kindt, R., Legendre, P., McGlinn, D., et al. (2019). Vegan: Community Ecology Package. R Package Version 2.3-6. Available at: http://CRAN.R-project.org/package=vegan
Park, M., Kim, C., Yang, J., Lee, H., Shin, W., Kim, S., et al. (2005). Isolation and characterization of diazotrophic growth promoting bacteria from rhizosphere of agricultural crops of Korea. Microbiol. Res. 160, 127–133. doi: 10.1016/j.micres.2004.10.003
Pegler, D. N., Spooner, B. M., and Yong, T. W. K. (1993). British Truffles: A Revision of British Hypogeous Fungi. Kew: Royal Botanic Gardens.
Poly, F., Monrozier, L. J., and Bally, R. (2001). Improvement in the RFLP procedure for studying the diversity of nifH genes in communities of nitrogen fixers in soil. Res. Microbiol. 152, 95–103. doi: 10.1016/S0923-2508(00)01172-4
Qiao, P., Tian, W., Liu, P., Yu, F., Chen, J., Deng, X., et al. (2018). Phylogeography and population genetic analyses reveal the speciation of the Tuber indicum complex. Fungal Genet. Biol. 113, 14–23. doi: 10.1016/j.fgb.2018.02.001
Ramachandran, K., Srinivasan, V., Hamza, S., and Anandaraj, M. (2007). “Phosphate solubilizing bacteria isolated from the rhizosphere soil and its growth promotion on black pepper (Piper nigrum L.) cuttings,” in First International Meeting on Microbial Phosphate Solubilization, eds E. Velázquez, and C. Rodríguez-Barrueco (Dordrecht: Springer), doi: 10.1007/978-1-4020-5765-6_51
Rodrigues, V. D., Torres, T. T., and Ottoboni, L. M. M. (2014). Bacterial diversity assessment in soil of an active Brazilian copper mine using high-throughput sequencing of 16S rDNA amplicons. Antonie Van Leeuwenhoek 106, 879–890. doi: 10.1007/s10482-014-0257-6
Roux, C. L., Tournier, E., Lies, A., Sanguin, H., Chevalier, G., Duponnois, R., et al. (2016). Bacteria of the genus Rhodopseudomonas (Bradyrhizobiaceae): obligate symbionts in mycelial cultures of the black truffles Tuber melanosporum and Tuber brumale. Springerplus 5:1085. doi: 10.1186/s40064-016-2756-6
Sbrana, C., Agnolucci, M., Bedini, S., Lepera, A., Toffanin, A., Giovannetti, M., et al. (2002). Diversity of culturable bacterial populations associated to Tuber borchii ectomycorrhizas and their activity on T. borchii mycelial growth. FEMS Microbiol. Lett. 211, 195–201. doi: 10.1111/j.1574-6968.2002.tb11224.x
Schmieder, R., and Edwards, R. (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. doi: 10.1093/bioinformatics/btr026
Selosse, M. A., Schneider-Maunoury, L., Taschen, E., Rousset, F., and Richard, F. (2017). Black truffle, a hermaphrodite with forced unisexual behaviour. Trends Microbiol. 25, 784–787. doi: 10.1016/j.tim.2017.05.010
Splivallo, R., Deveau, A., Valdez, N., Kirchhoff, N., Frey-Klett, P., and Karlovsky, P. (2015). Bacteria associated with truffle-fruiting bodies contribute to truffle aroma. Environ. Microbiol. 17, 2647–2660. doi: 10.1111/1462-2920.12521
Splivallo, R., Ottonello, S., Mello, A., and Karlovsky, P. (2011). Truffle volatiles: from chemical ecology to aroma biosynthesis. New Phytol. 189, 688–699. doi: 10.2307/40983900
Splivallo, R., Vahdatzadeh, M., Maciá-Vicente, J. G., Molinier, V., Peter, M., Egli, S., et al. (2019). Orchard conditions and fruiting body characteristics drive the microbiome of the black truffle Tuber aestivum. Front. Microbiol. 10:1437. doi: 10.3389/fmicb.2019.01437
Tanja, M., and Salzberg, S. L. (2011). FLASH: fast length adjustment of short reads to improve genome assemblies. Bioinformatics 27, 2957–2963. doi: 10.1093/bioinformatics/btr507
Torregiani, E., Lorier, S., Sagratini, G., Maggi, F., Vittori, S., and Caprioli, G. (2017). Comparative analysis of the volatile profile of 20 commercial samples of truffles, truffle sauces, and truffle-flavored oils by using HS-SPME-GC-MS. Food Anal. Methods 10, 1857–1869. doi: 10.1007/s12161-016-0749-2
Vahdatzadeh, M., Deveau, A., and Splivallo, R. (2015). The role of the microbiome of truffles in aroma formation: a meta-analysis approach. Appl. Environ. Microbiol. 81, 6946–6952. doi: 10.1128/AEM.01098-15
Vahdatzadeh, M., Deveau, A., and Splivallo, R. (2019). Are bacteria responsible for aroma deterioration upon storage of the black truffle Tuber aestivum: a microbiome and volatilome study. Food Microbiol. 84:103251. doi: 10.1016/j.fm.2019.103251
Wan, S. P., and Liu, P. G. (2014). Diversity of culturable bacteria associated with ascocarps of a Chinese white truffle, Tuber panzhihuanense (Ascomycota). Plant Divers. Resour. 36, 29–36. doi: 10.7677/ynzwyj20141301
Wang, Q., Garrity, G. M., Tiedje, J. M., and Cole, J. R. (2007). Naive bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. doi: 10.1128/aem.00062-67
Wang, S., and Marcone, M. F. (2011). The biochemistry and biological properties of the world’s most expensive underground edible mushroom: truffles. Food Res. Int. 44, 2567–2581. doi: 10.1016/j.foodres.2011.06.008
Wang, Y., Sheng, H. F., He, Y., Wu, J. Y., Jiang, Y. X., Tam, N. F., et al. (2012). Comparison of the levels of bacterial diversity in freshwater, intertidal wetland, and marine sediments by using millions of illumina tags. Appl. Environ. Microbiol. 78, 8264–8271. doi: 10.1128/aem.01821-12
Wang, Y., Zhu, M. T., Da, C. Z., Murat, C., Jeandroz, S., and Tacon, F. L. (2006). Phylogenetic and populational study of the Tuber indicum complex. Mycol. Res. 110, 1034–1045. doi: 10.1016/j.mycres.2006.06.013
Wilson, P. W., and Knight, S. G. (1974). Experiments in bacterial physiology. Biochem. Educ. 2, 29–30. doi: 10.1016/0307-4412(74)90009-0
Wych, R. D., and Rains, D. W. (1978). Simultaneous measurements of nitrogen fixation estimated by the acetylene-ethylene assay and nitrate absorption by soybeans. Plant Physiol. 62, 442–448.
Ye, L., Li, Q., Fu, Y., Sourzat, P., Tan, H., Zou, J., et al. (2018). Host species effects on bacterial communities associated with the fruiting bodies of Tuber species from the Sichuan Province in Southwest China. Mycol. Prog. 17, 833–840. doi: 10.1007/s11557-018-1397-2
Zeppa, S., Guidi, C., Zambonelli, A., Potenza, L., Vallorani, L., and Pierleoni, R. (2002). Identification of putative genes involved in the development of Tuber borchii fruit body by mRNA differential display in agarose gel. Curr. Genet. 42, 161–168. doi: 10.1007/s00294-002-0343-6
Keywords: Chinese black truffle, bacterial composition, geographical region, high-throughput, tissue culture, nitrogen fixation activity
Citation: Chen J, Li J-M, Tang Y-J, Xing Y-M, Qiao P, Li Y, Liu P-G and Guo S-X (2019) Chinese Black Truffle-Associated Bacterial Communities of Tuber indicum From Different Geographical Regions With Nitrogen Fixing Bioactivity. Front. Microbiol. 10:2515. doi: 10.3389/fmicb.2019.02515
Received: 06 June 2019; Accepted: 18 October 2019;
Published: 05 November 2019.
Edited by:
Fu-Cheng Lin, Zhejiang University, ChinaReviewed by:
Alessandra Zambonelli, University of Bologna, ItalyAntonella Amicucci, University of Urbino Carlo Bo, Italy
Copyright © 2019 Chen, Li, Tang, Xing, Qiao, Li, Liu and Guo. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Juan Chen, a2liY2hlbmp1YW5AMTI2LmNvbQ==; Shun-Xing Guo, c3hndW8xOTg2QDE2My5jb20=